

Abstract
Phospholipase D is a ubiquitous class of enzymes that generates phosphatidic acid as an intracellular signaling species. The phospholipase D superfamily plays a central role in a variety of functions in prokaryotes, viruses, yeast, fungi, plants, and eukaryotic species. In mammalian cells, the pathways modulating catalytic activity involve a variety of cellular signaling components, including G protein–coupled receptors, receptor tyrosine kinases, polyphosphatidylinositol lipids, Ras/Rho/ADP-ribosylation factor GTPases, and conventional isoforms of protein kinase C, among others. Recent findings have shown that phosphatidic acid generated by phospholipase D plays roles in numerous essential cellular functions, such as vesicular trafficking, exocytosis, autophagy, regulation of cellular metabolism, and tumorigenesis. Many of these cellular events are modulated by the actions of phosphatidic acid, and identification of two targets (mammalian target of rapamycin and Akt kinase) has especially highlighted a role for phospholipase D in the regulation of cellular metabolism. Phospholipase D is a regulator of intercellular signaling and metabolic pathways, particularly in cells that are under stress conditions. This review provides a comprehensive overview of the regulation of phospholipase D activity and its modulation of cellular signaling pathways and functions.
I. Background
Phospholipase D (PLD) catalyzes the hydrolysis of phosphatidylcholine (PC) into phosphatidic acid (PtdOH) and choline. The enzyme can use other amine-containing glycerophospholipids as substrates as well. In addition to hydrolyzing phospholipids, such as PC, PLD enzymes catalyze a transphosphatidylation reaction in the presence of primary alcohols in which the phosphatidyl group from PC is transferred to the alcohol instead of water to generate a phosphatidyl alcohol at the expense of PtdOH (Yang et al., 1967). This has occasionally led to an inaccurate statement that alcohols, such as ethanol or butanol, are inhibitors of PLD. Primary alcohols compete with water as nucleophiles in the PLD catalytic reaction and the production of a phosphatidylalcohol is promoted at the expense of generating PtdOH. Some confusion has been spawned over this misconception that primary alcohols inhibit PLD, in part, due to the fact that primary alcohols have broad nonspecific effects on cells and phosphatidyl alcohols, in some cases, may mimic biologic effects of PtdOH. The classic technique for measuring PLD activity in vivo is to measure production of metabolically and isotopically stable phosphatidylalcohol (Brown et al., 2007). Almost 30 years after the initial description of PLD in plants, Saito and Kanfer (1973, 1975) provided the first evidence of PLD activity in a mammalian tissue by partially purifying a PLD enzyme from rat brain particulate fractions. PLD enzymes are now known to be ubiquitously expressed and PLD activity has been described in almost all organisms from viruses and prokaryotes up to fungi, plants, and higher eukaryotes, such as humans (a detailed review on the enzymology of PLD enzymes may be found in Selvy et al., 2011).
PLD was first cloned from castor beans (Wang et al., 1994), and the sequence information enabled other groups to clone PLD enzymes from many other organisms. Over 4000 sequences for PLDs from various organisms have been deposited into the National Center for Biotechnology Information GenBank (Selvy et al., 2011). Two PLD isoforms have been cloned in humans and are commonly referenced as PLD1 (Hammond et al., 1995) and PLD2 (Lopez et al., 1998). PLD1 and PLD2 orthologs have also been cloned from mice (Colley et al., 1997a,b) and rats (Kodaki and Yamashita, 1997; Park et al., 1997). Although many PLD enzymes, both prokaryotic and eukaryotic, were initially described based on their ability to hydrolyze PC, cloning and subsequent sequence analyses revealed the truly diverse nature of these enzymes. The overall sequence homology between plant, yeast, and mammalian enzymes is quite low, with only four small regions of sequence similarity termed conserved regions (CRs) CRI, CRII, CRIII, and CRIV (Morris et al., 1996). CRII and CRIV contain duplicate catalytic sequences, termed the HKD domain (Koonin, 1996), characterized by the sequence HxKx4Dx6G(G/S), where x denotes amino acids between the histidine, lysine, and aspartic acid residues. Based on sequence analyses of PLDs from various organisms, enzymes with the characteristic HKD catalytic domain are categorized as part of a PLD “superfamily” and include PLD enzymes from prokaryotes, fungi, plants, and mammals (Koonin, 1996; Ponting and Kerr, 1996). In addition, non-PLD enzymes with HKD domains, such as endonucleases (Pohlman et al., 1993), cardiolipin (CL) synthase (Ivanisevic et al., 1995), and phosphatidylserine (PS) synthase (DeChavigny et al., 1991), are included in the PLD superfamily. Not all HKD enzymes share all four conserved regions because PLD from Streptomyces contains only CRI, CRII, and CRIV, with CRI and CRIV being the most similar to eukaryotic PLD, and the CL synthase/PS synthase bacterial enzymes contain only regions CRI and CRIV (Morris et al., 1996). These HKD enzymes are all believed to share a similar reaction mechanism.
In addition to the PC-hydrolyzing PLDs, several other PLDs have been identified and cloned in humans. Glycosylphosphatidylinositol (GPI)-PLD, a non-HKD PLD, hydrolyzes GPIs to produce an inositol glycan and PtdOH and functions primarily to release GPI-anchored proteins from membranes (Schofield and Rademacher, 2000). N-Acyl phosphatidylethanolamine (PE)–PLD is another non-HKD PLD that hydrolyzes N-acyl PE to produce PtdOH and N-acylethanolamine, which is further metabolized into anandamide, a ligand for cannabinoid receptors (Okamoto et al., 2004). Autotaxin, or lysophospholipase D, is a non-HKD PLD that hydrolyzes lysophospholipids such as lysophosphatidylcholine to produce the potent mitogen, lysophosphatidic acid (LPA) (Houben and Moolenaar, 2011). Besides these non-HKD PLD enzymes, a mitochondrial PLD (mitoPLD or PLD6) was recently cloned and shown to encode one copy of the HKD catalytic sequence (Choi et al., 2006). MitoPLD is believed to regulate mitochondrial fusion by hydrolyzing mitochondrial CL instead of PC to produce PtdOH (Choi et al., 2006). Although these non-HKD/noncanonical PLDs mediate important biologic events, this review focuses on the mammalian PLD1 and PLD2 isoenzymes.
A. Structure and Mechanism
Human PLD1 has three known splice variants termed PLD1a, PLD1b (Hammond et al., 1995), and PLD1c (Steed et al., 1998). PLD1b is 38 amino acids shorter than PLD1a and appears to have similar regulatory and catalytic properties (Hammond et al., 1995). By contrast, PLD1c contains an early truncation mutation and has been theorized to function as an inhibitor of endogenous PLD activity (Steed et al., 1998). Likewise, splice variants for PLD2 have been reported, although little is known about their functions in vivo (Steed et al., 1998). Full-length PLD1 and PLD2 share approximately 50% sequence identity and have similar domain structures. As such, PLD1 and PLD2 exhibit similar substrate preferences, namely mono- and diunsaturated PC (Pettitt et al., 2001). At their amino termini, PLD1 and PLD2 contain tandem phox homology (PX) and pleckstrin homology (PH) domains (Steed et al., 1998; Sung et al., 1999b), which are known to mediate interactions with lipid membranes (Lemmon, 2008) and are believed to regulate PLD localization within the cell (Fig. 1). The PX/PH domains were defined by predicted secondary structures, because there is little primary sequence similarity to other known PX/PH proteins. PLD from lower eukaryotes, such as yeast (Rose et al., 1995) and plants (Qin and Wang, 2002), also encode PX and PH domains. These domains are not required for catalytic activity because PX/PH truncation mutants are catalytically active in vitro (Sung et al., 1999a). CRI–CRIV, including the two catalytic HKD domains, are C terminal to the PX/PH domains and vary in length among the different PLD isoforms. The PLD1 isoforms contain a unique “loop” region between CRII and CRIII of approximately 120 amino acids in PLD1a; this loop is 38 amino acids shorter in PLD1b (Hammond et al., 1995). The function of this loop region is unknown, but it is speculated to inhibit the enzyme in vivo since deletion of this region results in a more highly active protein in vitro (Sung et al., 1999b). The function of the carboxy terminus of the protein is also unknown, but any mutations to this end of the enzyme result in a catalytically inactive protein, suggesting that this region participates in formation or stabilization of the active site (Liu et al., 2001).
Fig. 1.

Schematic of human PLD isoforms and splice variants. Human PLDs encode amino-terminal PX and PH domains followed by two catalytic HKD domains, characteristic of the PLD superfamily. PLD1a and PLD1b vary by 38 amino acids in a “loop” region between the two HKD domains. The PLD1c splice variant contains an early truncation mutation resulting in an inactive protein. Numbers indicate amino acid positions.
Early studies using strategically radiolabeled substrates and intermediates suggested that PLD enzymes use a two-step “ping pong” reaction mechanism in which a covalent phosphoenzyme intermediate is created before release of the product (Stanacev and Stuhne-Sekalec, 1970; Jiang et al., 1984). This reaction mechanism was supported by structural studies using Yersinia pestis murine toxin (Rudolph et al., 1999) and Nuc endonuclease (Gottlin et al., 1998), both members of the HKD-containing PLD superfamily. In the first step, a histidine residue from one HKD domain serves as a nucleophile to attack the phosphate group of substrate PC to form a phosphatidyl histidine. The histidine from the second HKD domain donates protons to the choline-leaving group to assist in formation of the phosphatidyl histidine. The second histidine then extracts protons from water and the activated water molecule hydrolyzes the phosphatidyl histidine intermediate to release PtdOH (Rudolph et al., 1999). In the presence of a primary alcohol, transphosphatidylation is the favored reaction since many short-chain primary alcohols are more nucleophilic than water. Crystal structures of the HKD-containing PLD from Streptomyces and the HKD-containing Nuc endonuclease (Stuckey and Dixon, 1999) have been solved and support these previously established biochemical insights into the molecular mechanisms of PLD hydrolase activity. The HKD-containing PLD from Streptomyces crystallized with the two HKD domains situated adjacent to each other along an axis of symmetry. Although Nuc contains only one HKD domain, it crystallized as a homodimer with the two HKD domains similarly situated along an axis of symmetry with the HKD domains coming together to form the active site. The crystal structures thus supported the mechanism of catalytic histidine residues. The exact functions of the lysines and aspartic acids are unknown, but they are believed to stabilize the phosphatidyl histidine intermediate as was reported with similar enzymes such as nucleoside diphosphate kinase (Moréra et al., 1995). A recent computation model by DeYonker and Webster (2013) strongly supports an associative-type mechanism for phosphoryl transfers within the PLD superfamily.
B. Localization and Tissue Distribution
The PLD enzymes are ubiquitously expressed and are found in almost all mammalian tissues. PLD1 is highly enriched in the human heart, brain, pancreas, uterus, and intestine and PLD2 is highly enriched in the brain, placenta, lung, thymus, prostate, and uterine tissue (Lopez et al., 1998). Early studies into PLD subcellular localization were limited to measurements of PLD enzymatic activity after subcellular fractionation (Edwards and Murray, 1995). Molecular cloning allowed for characterization of PLD1 and PLD2 subcellular localization through the use of fluorescent fusion proteins and isoform-specific antibodies to track endogenous protein. Many studies examining PLD subcellular localization have overexpressed PLD1 or PLD2 and this has led to some controversy over localization of endogenous proteins. However, the work of multiple, independent investigators has produced a relatively consistent pattern of PLD1 and PLD2 localization.
Under resting conditions, PLD1 resides on perinuclear, intracellular membranes of secretory vesicles, lysosomes, endosomes, Golgi, and endoplasmic reticulum (Brown et al., 1998a; Freyberg et al., 2001; Du et al., 2003). On the other hand, PLD1 is basally localized to the plasma membrane in neuroendocrine cells, such as chromaffin (Vitale et al., 2001) and PC12 cells (Du et al., 2003), although PC12 cells express very low levels of endogenous PLD1 (Gibbs and Meier, 2000). IgE stimulation of RBL-2H3 mast cells (Brown et al., 1998a; Cohen and Brown, 2001), or phorbol esters, such as phorbol-12-myristate-13-acetate (PMA), stimulation of fibroblasts (Kim et al., 1999b) and COS-7 cells (Du et al., 2003), results in PLD1 relocalization to the plasma membrane suggesting that PLD1 activation might require relocalization after extracellular stimulation. As such, mutations that prevent post-translational palmitoylation and plasma membrane recruitment reduce phorbol ester–stimulated PLD1 activity. The varied and dynamic subcellular localization patterns of PLD1 correlate with diverse functional roles.
In contrast with PLD1, PLD2 primarily localizes to plasma membranes under basal conditions. Studies measuring overexpressed (Colley et al., 1997b; O'Luanaigh et al., 2002; Du et al., 2004) and endogenous PLD2 (Park et al., 2000; Sarri et al., 2003; Du et al., 2004) support a plasma membrane localization pattern. PLD2 is enriched in detergent-insoluble, lipid-raft fractions (Czarny et al., 1999; Zheng and Bollinger Bollag, 2003), which contain clusters of cell surface receptors and other signal transduction molecules (Simons and Toomre, 2000). Agonist stimulation typically results in PLD2-receptor colocalization both at the plasma membrane and in endocytic vesicles and PLD2 has been proposed to participate in receptor endocytosis and recycling (Du et al., 2004). In addition, stimulation of fibroblasts with serum (Colley et al., 1997b) or mast cells with antigen (O'Luanaigh et al., 2002) results in PLD2 relocalization to filopodia and membrane ruffles, respectively. Thus, the subcellular localization patterns of PLD2 suggest functional roles ranging from signal transduction to cytoskeletal reorganization.
II. Regulation of Phospholipase D Activity by Lipids
A. Phosphoinositides
The gold standard in vitro PLD activity assay measures the release of a tritiated choline headgroup from an isotopically labeled PC substrate, typically as part of a vesicle containing PC, PE, and phosphatidylinositol-4,5-bisphosphate (PIP2). The development of this assay required optimization of lipid vesicle substrate compositions and PIP2 was absolutely required to detect choline hydrolysis from PLD purified from HL-60 membranes (Brown et al., 1993). Recombinant expression of mammalian PLD1 (Hammond et al., 1995; Min et al., 1998) and PLD2 (Colley et al., 1997b; Kodaki and Yamashita, 1997; Lopez et al., 1998) revealed that both enzymes require PIP2 for their activity in vitro. Besides PIP2, other phosphatidylinositol species can stimulate PLD activity. For example, phosphatidylinositol 3,4,5-trisphosphate (PIP3) stimulates PLD1b activity, although to about half that of the PIP2 species (Hammond et al., 1997; Hodgkin et al., 2000). Similarly, PLD1b displays approximately 2-fold higher binding affinity for PIP2 than PIP3. Phosphatidylinositol phosphate (PIP) and PI(3,5)P2 are much less effective stimulators of PLD activity and this fact suggests that the position of the phosphates on the inositol ring is critical for maximal activity (Hodgkin et al., 2000). For reasons not apparent, rat PLD1 is stimulated equally by PI(4,5)P2 and PIP3 but not by PI(3,4)P2 (Min et al., 1998), suggesting that different species might display some selectivity for PIPn species in vivo.
PIP2 is believed to interact with two distinct sites on PLD (one in the PH domain and one between the catalytic domains) and each site is believed to differentially regulate enzyme activity. Multiple groups have shown that the PX and PH domains of PLD1 (Sung et al., 1999b; Henage et al., 2006) and PLD2 (Sung et al., 1999a) are not required for enzyme activity. Since PIP2 is required for robust PLD activity, investigators deduced that there must be another PIP2 binding site outside of the PH domain. Mutational analysis narrowed PIP2 binding to a polybasic region between CRII and CRIII and when arginines 554 and 558 of PLD2 (corresponding residues are R691 and R695 for human PLD1a) were mutated, both the in vitro catalytic activity and PIP2 binding were severely compromised (Sciorra et al., 1999). Plant PLDs also require PIP2 and a similar binding region was established for plant-PLD1β. Mutation of lysines K437 and K440, also between CRII and CRIII, ablated PLDβ activity in vitro (Zheng et al., 2002). PIP2 binding induces a conformational shift in the catalytic domain of plant PLDβ that helps recruit PC to the active site (Zheng et al., 2002), suggesting that PIP2 binding to the region between catalytic domains might influence catalytic activity by promoting PC substrate binding. Later studies demonstrated that in the absence of PIP2, PLD was unable to bind PC vesicles, further supporting the role of PIP2 in PC binding.
Although mutation of the PIP2 binding region between CRII and CRIII significantly decreases PLD catalytic activity, these mutations do not affect PLD localization as measured by immunofluorescence or subcellular fractionation (Sciorra et al., 1999). PH domains are known to bind phosphatidylinositols and regulate subcellular localization (Lemmon, 2008). As such, PLD-PH domain binding to PIP2 is a means for regulating subcellular localization. Aligning the PH domain from PLD2 with PH domains from other proteins revealed several conserved residues believed to mediate PIPn binding. When arginine 236 and tryptophan 237 of PLD2 (corresponding residues are R317 and W318 in PLD1a) were mutated, the resulting protein was catalytically inactive in vivo but displayed similar catalytic activity to wild-type protein when PLD2 was immunoprecipitated and assayed in vitro (Sciorra et al., 2002). PLD2 resides primarily in detergent-insoluble membrane fractions but mutation of R237 and W238 resulted in relocalization of PLD2 to detergent-soluble membrane fractions. Mutation of PLD2 also resulted in a relocalization from plasma membranes to intracellular localizations (Sciorra et al., 2002). Parallel studies with PLD1b also demonstrated mislocalization after deletion of the PH domain (Hodgkin et al., 2000). The role of PIP2 in vivo might be to recruit PLD to a specific membrane and to enhance catalysis by promoting substrate binding to the active site.
Several lines of evidence support a role for PIP2 in regulating PLD in vivo. The rate-limiting step of PIP2 production is the phosphorylation of PI(4)P by PI(4)P 5-kinase (PIP5K) (Ling et al., 1989). When PIP5K was overexpressed in cells to increase levels of PIP2, PLD activity also increased, suggesting that higher PIP2 levels increase PLD activity in intact cells (Divecha et al., 2000). Modulation of PIP2 levels in the opposite direction also influences PLD activity. The antibiotic neomycin has a high affinity for PIP2 and is used for cell signaling studies due to its ability to sequester PIP2. As expected, treatment of cells with neomycin decreases PLD activity in a manner that is reversed when PIP2 is replenished in the system (Liscovitch et al., 1994). Similarly, synaptojanin, an inositol polyphosphate 5-phosphatase capable of dephosphorylating PIP2 (McPherson et al., 1996), was purified as a cytosolic factor capable of inhibiting PLD activity in vitro (Chung et al., 1997). Although the original characterization of PLD inhibition by synaptojanin was performed in vitro, synaptojanin might function as a mechanism for terminating PLD signaling in vivo. Similarly, the actin and PIP2-binding protein gelsolin inhibits PLD in vitro by binding and sequestering PIP2 (Banno et al., 1999). Overexpression of gelsolin also inhibits PLD transphosphatidylation activity, further supporting a role for PIP2 regulating PLD activity in vivo (Banno et al., 1999).
These studies create a model in which phosphoinositides (PIs), specifically PIP2, regulate PLD by influencing catalysis via an interaction in the PLD catalytic domains and by influencing membrane localization via an interaction in the PH domain. Furthermore, PX domains are also well established PI binding domains (Song et al., 2001). The PLD-PX domain appears to bind PIP3 quite selectively over mono- and diphosphorylated PIs and mutation of PLD1 arginine 179 substantially reduces PIP3 binding (Stahelin et al., 2004). Several studies have shown a dependence on PIP3 generation for PLD activation after cell surface receptor stimulation. Phosphoinositide 3-kinases (PI3Ks) produce PIP3 by phosphorylating the D3 position of the inositol ring (Cantley, 2002) and both PLD and PI3K activities are frequently stimulated after receptor tyrosine kinase stimulation. PI3K inhibitors decrease PLD activation after insulin receptor stimulation (Standaert et al., 1996) and mutation of the PIP3 binding site on PLD (PLD1-R179) prevents PLD activation and membrane recruitment after platelet-derived growth factor (PDGF) stimulation (Lee et al., 2005). Therefore, membrane recruitment of PLD is precisely regulated by both PIP2 and PIP3 through interactions with the PH and PX domains, respectively.
B. Fatty Acids
Purification of various mammalian and plant PLD enzymes revealed activities that were stimulated differently by unsaturated fatty acids. Before cloning of the PLD enzymes, a PLD was purified from pig lung that was stimulated by unsaturated fatty acids, such as oleic (18:1), linoleic (18:2), and arachidonic (20:4) acids (Okamura and Yamashita, 1994). Several lines of evidence suggest that PLD2 is the isoform stimulated by unsaturated fatty acids. PLD activity is highly stimulated by oleate in Jurkat T cells but not in HL-60 cells (Kasai et al., 1998). mRNA analysis suggests that Jurkat T cells express only PLD2, whereas HL-60 cells express PLD1. Likewise, PLD2 is highly enriched in the lung (Lopez et al., 1998). Finally, oleate stimulates PLD activity in RBL-2H3 mast cells when PLD2, but not PLD1, is overexpressed (Sarri et al., 2003). The in vivo relevance of unsaturated fatty acid stimulation of PLD2 is not fully understood.
C. Lipid Modifications
Both PLD1 and PLD2 undergo lipid modification. Labeling cells with tritiated fatty acids and subsequent measurement of lipid incorporation onto PLD protein shows that PLD1 contains a covalent palmitoylation (Manifava et al., 1999). Later studies concluded that cysteines 240 and 241 are the amino acids responsible for attachment (Sugars et al., 1999). The exact function of these lipid modifications is not known, but suggests that proper subcellular localization, and not catalytic activity, requires these palmitoylation events. In COS-7 cells, PLD1 is normally localized to punctate intracellular membranes. However, when the palmitoylated cysteines were mutated to alanine, the levels of punctate intracellular PLD1 decreased with a concomitant increase in plasma membrane localized protein (Sugars et al., 1999). The mutant protein was less active in vivo but showed no differences in activity compared with wild-type protein in vitro, suggesting that the palmitoylation promotes accessibility of substrate lipids to PLD in the cell (Sugars et al., 1999). Epidermal growth factor (EGF) stimulates PLD activity in a variety of cell lines. The cysteine-to-alanine mutants are much less responsive to EGF stimulation than wild-type protein, suggesting that palmitoylation is required for cell surface receptor activation of PLD1 (Han et al., 2002b). PLD2 is also palmitoylated on C223 and C224 (Xie et al., 2002a). Similarly to PLD1, mutation of the cysteine residues decreases in vivo activity and also results in a smaller fraction of membrane-associated PLD2 (Xie et al., 2002a). Therefore, palmitoylation is one of the mechanisms by which PLD is properly localized under basal and stimulated conditions.
III. Phosphorylation
A. Serine and Threonine Phosphorylation
Many groups have detected phosphorylation of PLD1 and PLD2 at tyrosine, serine, and threonine residues, yet the functional significance of these events is not clear and most likely depends on the cell system and stimulus under investigation. For example, protein kinase C (PKC) stimulates PLD1 activity through a direct protein–protein interaction and not via phosphorylation in fibroblast membranes (Conricode et al., 1992) (Table 1). By contrast, PKCα stimulation of PLD in neutrophil membranes requires ATP and is inhibited by treatment with staurosporine, a nonselective protein kinase inhibitor (Lopez et al., 1995), suggesting that PKCα phosphorylates PLD directly or phosphorylates an intermediate protein that activates PLD. A proteomic analysis revealed that PMA-stimulated PKC phosphorylates S2, T147, and S561 on PLD1 (Kim et al., 1999a). When these residues were mutated, the authors measured a slight decrease in PMA-stimulated PLD activity in vivo but no changes in in vitro activity, suggesting that phosphorylation of these residues is not required for catalytic activity (Kim et al., 1999a). Although not required for activity, one study has suggested that phosphorylation of S2 is required for association with the actin cytoskeleton after cell surface receptor activation (Farquhar et al., 2007). Similar proteomic analyses for PLD2 revealed that PMA stimulation of COS-7 cells increased phosphorylation of S134, S146, S243, T72, T99, T100, and T252 and that S243 and T252 were the predominant sites of phosphorylation (Chen and Exton, 2005). Mutation of S243 and T252 completely inhibited binding of phosphoserine and phosphothreonine antibodies to PMA-stimulated PLD2, but did not inhibit PMA-stimulated PLD activity (Chen and Exton, 2005). PMA treatment of COS-7 cells results in rapid increases in both PLD1 (Hu and Exton, 2003) and PLD2 (Chen and Exton, 2004) activity. PLD1 (Hu and Exton, 2003) and PLD2 (Chen and Exton, 2004) phosphorylation increases only after much longer PMA exposure and this correlates to a decrease in PLD activity. The functional consequence of PKC phosphorylation of PLD1 and PLD2 is probably to downregulate PLD activity in these cells. This hypothesis is substantiated by observations that staurosporine treatment prolongs PMA-stimulated PLD activity (Hu and Exton, 2003) and PKCα/β inhibitors block PLD phosphorylation but not PMA-stimulated PLD activity (Chen and Exton, 2004).
TABLE 1.
Modulators of PLD activity
| Protein Name | Isoform | Mechanism | Interaction Site | References |
|---|---|---|---|---|
| Activators | ||||
| PKC α,β1,β2 | PLD1 | Protein–protein interaction | N-terminal (AAs 50–115), C-terminal (AAs 325–582) | Conricode et al. (1992, 1994), Ohguchi et al. (1996), Hammond et al. (1997), Lee et al. (1997b), Min et al. (1998), Park et al. (1998), Sung et al. (1999b), Zhang et al. (1999), Siddiqi et al. (2000) |
| Arf | PLD1 | Protein–protein interaction | Unknown | Brown et al. (1993), Cockcroft et al. (1994), Hammond et al. (1997), Lopez et al. (1998), Min et al. (1998) |
| RhoA family | PLD1 | Protein–protein interaction | C-terminal (K946A, V950A, R955A, K962A) | Olson et al. (1991), Bowman et al. (1993), Kwak et al. (1995), Malcolm et al. (1994), Brown et al. (1995), Siddiqi et al. (1995), Singer et al. (1996), Hammond et al. (1997), Sung et al. (1997), Yamazaki et al. (1999), Cai and Exton (2001) |
| PKN | PLD1 | Protein–protein interaction | AAs 228–598 | Oishi et al. (2001) |
| Rheb | PLD1 | Protein–protein interaction | Unknown | Sun et al. (2008) |
| Ras | PLD1 | Indirect protein–protein interaction | Unknown | Jiang et al. (1995a) |
| RaIA | PLD1 | Indirect protein–protein interaction | Unknown | Jiang et al. (1995b), Kim and Wong (1998) |
| Cofilin | PLD1 | Protein–protein interaction | Region between loop and CRIII | Han et al. (2007) |
| CtBP1/BARS | PLD1 | Protein–protein interaction | Unknown | Haga et al. (2009) |
| AMPK | PLD1 | Phosphorylation | S505 | Kim et al. (2010) |
| p90 RSK | PLD1 | Phosphorylation | T147 | Zeniou-Meyer et al. (2008) |
| Cdk5 | PLD2 | Phosphorylation | S134 | Lee et al. (2008) |
| Grb2 | PLD2 | Protein–protein interaction | Y169/Y179 | Di Fulvio et al. (2006) |
| PKCδ | PLD2 | Phosphorylation | S566 | Han et al. (2002a), Chae et al. (2010) |
| Inhibitors | ||||
| AP3 | PLD1 | Protein–protein interaction | Unknown | Lee et al. (1997a) |
| Aldolase | PLD2 | Protein–protein interaction | PH domain | Kim et al. (2002) |
| α-Actinin | PLD2 | Protein–protein interaction | Unknown | Park et al. (2000) |
| CRMP-2 | PLD2 | Protein–protein interaction | N terminus | Lee et al. (2002) |
| Munc-18-1 | PLD1/PLD2 | Protein–protein interaction | PX domains | Lee et al. (2004) |
| Amphiphysin I and II | PLD1/PLD2 | Protein–protein interaction | Unknown | Lee et al. (2000) |
| F-actin | PLD1/PLD2 | Protein–protein interaction | Region between CRIII and CRIV | Lee et al. (2001), Kusner et al. (2002) |
| PKCα | PLD1/PLD2 | Phosphorylation | PLD1 (S2, T147, S561), PLD2 (S134, S146, S243, T72, T99, T100, T252) | Kim et al. (1999a), Chen and Exton (2005) |
AA, amino acid; BARS, brefeldin A ADP-ribosylated substrate; RSK, ribosomal S6 kinase.
Other lines of evidence suggest that phosphorylation of PLD by PKC may be required for cell surface receptor stimulation of PLD. EGF stimulation of COS-7 cells results in a rapid increase in both PLD1 T147 phosphorylation and activity that can be ablated by expression of dominant-negative PKCα or by mutating the PKC phosphorylation sites of PLD1 (Han et al., 2002b). The discrepancies between the functional consequences of PMA and EGF-stimulated PLD phosphorylation might be explained by differences in PKC isoforms being activated under each condition. For example, PMA activates many PKC isoforms and EGF might only activate PKC isoforms that positively regulate PLD activity, such as PKCα. In support of this hypothesis, PKCδ is believed to negatively regulate PLD1 activity in COS-7 cells (Han et al., 2002b). By contrast, PKCδ is believed to mediate the PMA activation of PLD2 in PC12 cells (Han et al., 2002a) and integrin-stimulated PLD2 activation in COS-7 cells requires phosphorylation of S566 by PKCδ (Chae et al., 2010). Therefore, the functional role of PKC phosphorylation of PLD1 and PLD2 largely depends on the cell background, stimulus, and PKC isoform under investigation.
In addition to PKC, other serine/threonine kinases are known to regulate downstream PLD functions by phosphorylation-dependent mechanisms. For example, the p90 ribosomal S6 kinase phosphorylates PLD1 at T147 and this phosphorylation event is required for K+-stimulated PLD activity and exocytosis in PC12 neuroendocrine cells (Zeniou-Meyer et al., 2008). In addition, AMP-activated protein kinase (AMPK) phosphorylates PLD1 at S505 (Kim et al., 2010). Glucose withdrawal stimulates AMPK and PLD activity and the phosphorylation of S505 by AMPK is required for glucose-stimulated PLD activity in vivo. Mutation of S505 has no effects on basal PLD activity, suggesting that the phosphorylation is required for catalytic activity (Kim et al., 2010). Likewise, PLD2 regulates insulin secretion after EGF stimulation in rat insulinoma cells via a mechanism that requires cyclin-dependent kinase 5 (Cdk5) phosphorylation of PLD2 S134 (Lee et al., 2008). Mutation of S134 to alanine partially decreases EGF-stimulated PLD2 activity, but does not inhibit basal PLD2 activity (Lee et al., 2008). Casein kinase II is found in complex with both PLD1 (Ganley et al., 2001) and PLD2 (Ahn et al., 2006) and phosphorylates both isozymes. The exact function of the casein kinase II phosphorylation events is unknown, but PLD catalytic activity is unaffected. Together, these studies suggest that phosphorylation of PLD most likely functions to alter PLD subcellular localization and availability to substrate since basal activity in vivo and PLD activity in vitro are not sensitive to mutation of phosphorylation sites.
B. Tyrosine Phosphorylation
Tyrosine phosphorylation as a mechanism of PLD regulation was first proposed after the observation that vanadate, a tyrosine phosphatase inhibitor, increased PLD activity in HL-60 granulocytes (Bourgoin and Grinstein, 1992). In later studies, PLD was shown to be tyrosine phosphorylated after immunoprecipitation of PLD and immunoblotting with a phosphotyrosine antibody (Gomez-Cambronero, 1995). Stimulation of the G protein–coupled receptor (GPCR) for the chemotactic peptide fMLP in neutrophils increased PLD activity and tyrosine phosphorylation in a manner that was inhibited by general tyrosine kinase inhibitors, suggesting that tyrosine phosphorylation directly regulates PLD activity (Gomez-Cambronero, 1995). These studies, however, did not discriminate between direct activation of PLD by tyrosine phosphorylation and activation of PLD by upstream regulatory proteins. Later studies indicated that tyrosine phosphorylation of PLD2 influences catalytic activity to some extent since dephosphorylation of immunoprecipitated PLD2 with purified tyrosine phosphatases can decrease (Henkels et al., 2009) or increase (Horn et al., 2005) catalytic activity in vitro, suggesting that the two phosphatases used in these studies are dephosphorylating distinct tyrosine residues.
EGF stimulation results in tyrosine phosphorylation of PLD2 (Min et al., 2001a) at Y11 (Slaaby et al., 1998) and Y296 (Henkels et al., 2010), presumably through epidermal growth factor receptor (EGFR)–mediated phosphorylation of PLD2, although the possibility of an intermediate kinase was not completely eliminated. When Y11 was mutated to alanine, a 2-fold increase in EGF-stimulated PLD activity was observed, suggesting that phosphorylation of Y11 functions to downregulate the PLD2 response (Slaaby et al., 1998). Likewise, mutation of Y296 to phenylalanine also increased in vivo PLD2 activity (Henkels et al., 2010), suggesting that like Y11, Y296 also functions to downregulate PLD2 activity. Other tyrosine kinases, such as Janus kinase 3 (JAK3) and Src, have been reported to phosphorylate PLD2 at Y415 and Y511, respectively (Henkels et al., 2010). In this study, purified JAK3 or Src was incubated with cell lysate and PLD2 was immunoprecipitated to determine which tyrosine sites were phosphorylated. The authors do not address the possibility that JAK3 or Src might be activating an intermediate kinase that could phosphorylate PLD2 and so caution must be exercised when interpreting these results. Regardless, the findings suggest that phosphorylation of Y415 stimulates PLD activity, whereas phosphorylation of Y511 is inhibitory (Henkels et al., 2010). These studies suggest that the level of PLD2 activity between various cell lines might result from different ratios of tyrosine phosphorylation on both activating and inhibitory residues.
Besides modulating catalytic activity, tyrosine phosphorylation is known to regulate PLD2–protein interactions. PLD2 couples EGF stimulation to activation of the small G protein Ras by binding the adaptor protein Grb2 and activating son of sevenless (SOS), a guanine nucleotide exchange factor (GEF) for Ras. Grb2 interacts with PLD2 at Y169 and mutation of nearby tyrosine Y179 to phenylalanine increases the interaction of Grb2 with Y169 and enhances activation of Ras signaling pathways (Di Fulvio et al., 2006). The interaction of Grb2 with Y169 increases PLD2 catalytic activity and mutation of Y169 to phenylalanine results in a catalytically inactive protein (Di Fulvio et al., 2006). Furthermore, Akt was suggested to phosphorylate threonine 175 of PLD2 (Di Fulvio et al., 2008). A relatively nonspecific Akt-kinase substrate antibody was used to probe immunoprecipitated PLD2 instead of using purified, recombinant Akt kinase. Regardless, mutation of T175 to alanine inhibited the Y179F mutant from stimulating Ras, suggesting that T175 and Y179 function to fine-tune Ras signaling through PLD2 (Di Fulvio et al., 2008).
Because tyrosine kinases are frequently upregulated in cancer, discovering precise molecular targets of tyrosine kinases further enhances our understanding of cancer biology. A recent proteomic study examined changes in global phosphotyrosine residues in cells overexpressing the constitutively active and transforming the nucleophosmin-anaplastic lymphoma kinase. PLD1 and PLD2 had increased phosphorylation at Y711 and Y573, respectively, in cells overexpressing the nucleophosmin-anaplastic lymphoma kinase (Wu et al., 2010). Further studies will be required to determine the functional significance of these and other novel phosphorylation events on PLD.
IV. Protein Activators of Phospholipase D
A. Protein Kinase C
Phorbol esters such as PMA potently stimulate PLD activity in many cell lines and tissues (Exton, 1999). These compounds function as diacylglycerol (DAG) mimetics and potently stimulate the conventional (DAG and Ca2+ responsive; α, β1, β2, and γ) and novel (DAG responsive; δ, ε, η, and θ) PKC isoforms (Nishizuka, 1984), thus suggesting that PMA stimulates PLD through a PKC-dependent mechanism. In early studies linking PMA, PKC, and PLD, investigators separated fibroblast membranes (containing PLD) from cytosol (containing PKC), and showed that PMA did not stimulate membrane PLD activity unless purified PKC was included in the reaction (Conricode et al., 1992). In the same study, PKC was shown to stimulate PLD activity in a kinase-independent manner. In addition, PKC stimulation of PLD was potentiated by PMA, leading to the hypothesis that activated PKC stimulates PLD through a protein–protein interaction, independent of kinase activity. Multiple groups confirmed a direct interaction using purified proteins (Lee et al., 1997b) and by coimmunoprecipitation of PLD and PKC from cell lysates (Siddiqi et al., 2000). Interestingly, the interaction is enhanced after PMA stimulation, suggesting that activated PKC is a better stimulator of PLD than nonactivated PKC. The requirement for activated PKC suggests that cell surface receptors might stimulate PLD through a PKC-dependent mechanism and many reports have indeed demonstrated a requirement for PKC in PLD activation after receptor stimulation.
The conventional PKC isoforms (namely α, β1, and β2, but not PKCδ) stimulate PLD from fibroblast and HL-60 membranes (Conricode et al., 1994; Ohguchi et al., 1996) and purified PLD from heterologous expression systems (Min et al., 1998). Purified conventional PKC stimulates PLD1a and PLD1b indistinguishably in vitro (Hammond et al., 1997), whereas PLD2 is unresponsive to PKC stimulation (Colley et al., 1997b). The interaction between PKC and PLD1 is complex because several lines of evidence suggest that at least two sites on PLD1 mediate the interaction with PKC. Expression of a PLD1 deletion mutant lacking the first 325 amino acids results in a construct that is catalytically active but is unresponsive to PMA stimulation, which suggests that PKC stimulates PLD activity through an interaction with the N terminus (Sung et al., 1999b). However, the N-terminally truncated PLD1 still coimmunoprecipitates with PKC, suggesting another site of interaction somewhere in the C terminus (Sung et al., 1999b). The N-terminal binding site was narrowed to a region between amino acids 50 and 115 (Park et al., 1998; Zhang et al., 1999) and the C-terminal binding region on PLD1 was determined to be between amino acids 325 and 582 (Park et al., 1998).
The mechanism by which PKC stimulates PLD1 activity is also complex and requires interaction with both the N- and C-terminal interaction sites. A detailed kinetic analysis of the activation of PLD1 by PKC revealed that PKC is a mixed activator of PLD1. PKC increases substrate binding (decrease in Km value) and also increases catalytic activity (increased kcat value). An N-terminally truncated PLD1 lacking the first 311 amino acids was much less responsive to PKC stimulation than full-length PLD1 protein in vitro. The kinetic analysis suggested that PKC still increased kcat of the N-terminally truncated protein but had no effect on substrate binding (Henage et al., 2006). Thus, PKC modulates individual kinetic parameters through distinct interactions on PLD1. The unique kinetic properties of PKC explain how other activators of PLD, such as the small GTPase ADP-ribosylation factor (Arf), synergize with PKC to robustly stimulate PLD1 activity (Singer et al., 1996; Henage et al., 2006).
Although PMA and PKC stimulate PLD1 activity in vitro through a direct protein–protein interaction, the regulation of PLD by PKC in vivo is more complex. As discussed above, PKC phosphorylates both PLD1 and PLD2 and these phosphorylation events are generally believed to inhibit PLD activity and may function to terminate PLD signaling after cell surface receptor activation. In addition, PMA stimulation can also increase PLD2 activity in vivo (Colley et al., 1997b; Siddiqi et al., 2000; Han et al., 2002a), although the fold stimulation of PLD2 by PMA is much less than the fold stimulation of PLD1. Given the wide range of PKC effectors, PMA stimulation of PLD2 may be explained by other intermediate proteins instead of a direct protein–protein interaction. In certain cells, such as PC12 cells, PLD2 stimulation by PMA appears to require the activity of PKCδ because a dominant-negative PKCδ construct inhibits PMA-induced PLD2 activation (Han et al., 2002a). Since purified PKCδ did not stimulate PLD1 (Conricode et al., 1994), more evidence is needed to determine whether PKCδ stimulates PLD2 directly or through an intermediate protein. In addition to PKCδ, PKCε was also shown to regulate PLD2 activation after sphingosine 1-phosphate (S1P) stimulation in lung epithelial cells (Gorshkova et al., 2008). A direct protein–protein interaction was not measured and an intermediate protein is likely mediating the effects of PKCε on PLD2.
B. ADP-Ribosylation Factor GTPases
Arf proteins are small-molecular-mass GTPases (Mr = 21 kDa) first identified as the factors needed for ADP-ribosylation of the Gαs heterotrimeric G protein by cholera toxin (Kahn and Gilman, 1984). Arfs have since been implicated in a variety of cellular events but are predominantly involved in vesicle formation and trafficking along with the cytoskeletal and membrane rearrangements that accompany these events (Moss and Vaughan, 1998). Arf was originally identified as a cytosolic factor capable of stimulating PLD activity in HL-60 cell membranes. In those experiments, the addition of cytosol and a nonhydrolyzable GTP analog (GTPγS) to HL-60 cell membranes robustly stimulated PLD activity. The observation that GTPγS was required for this stimulatory activity strongly implicated a GTPase as the activating factor. The factor was purified, sequenced, and identified as Arf1/Arf3 (Brown et al., 1993; Cockcroft et al., 1994). There are six known Arf family members in humans (Arf1–Arf6) and are divided into three classes based on size, sequence, and gene structure: class I (Arf1– Arf3), class II (Arf4 to Arf5), and class III (Arf 6) (Moss and Vaughan, 1993). All six Arf proteins are capable of directly stimulating PLD activity, although with varying efficiencies (Massenburg et al., 1994). Arf proteins also stimulate PLD activity indirectly by stimulating PIP5K activity to increase local PIP2 levels (Honda et al., 1999). The Arf-stimulated increase in PIP2 and PtdOH levels through PIP5K and PLD, respectively, might function to terminate Arf signaling as the GTPase activating protein (GAP) activities of several Arf GAPs are synergistically stimulated by PIP2 and PtdOH.
Prior to cloning PLD1 and PLD2, chromatographic separation of PLD activities from rat brain revealed two distinct PLD activities in which one activity was stimulated by Arf and the other by oleate, most likely PLD1 and PLD2, respectively (Massenburg et al., 1994). In vitro studies using purified proteins later showed that Arf1 and Arf3 are capable of stimulating PLD1a and PLD1b between 10- and 40-fold compared with unstimulated PLD1 (Hammond et al., 1997; Lopez et al., 1998; Min et al., 1998). PLD2 is either completely unresponsive (Colley et al., 1997b), modestly responsive (less than 2-fold), or responsive (Lopez et al., 1998; Sung et al., 1999a), suggesting that Arf1 and Arf3 stimulate PLD1 specifically. Arf4 and Arf6 have been implicated in regulating PLD2 activity in vivo (Caumont et al., 1998; Kim et al., 2003b). The sites of Arf interaction on PLD have not been determined conclusively, although the amino terminus of PLD1 is dispensable for Arf stimulation and kinetic analyses of an N-terminal deletion mutation revealed that Arf was a catalytic activator of PLD (Sung et al., 1999b; Henage et al., 2006). Interestingly, when the amino terminus of PLD2 is deleted, Arf stimulates PLD activity nearly as much as PLD1 (Sung et al., 1999a). This suggests that sequestration of the PLD2 N terminus, by lipid or protein binding or by post-translational modification, might free an Arf binding site that is otherwise sterically hindered on PLD2. As an example, the GM2 ganglioside activating protein binds PLD1 and PLD2 and significantly enhances the responsiveness to Arf (Nakamura et al., 1998; Sarkar et al., 2001). Likewise, proteins such as Arfaptin, which bind and sequester Arf (Tsai et al., 1998), inhibit PLD activity and emphasize the importance of Arf for regulating PLD activity.
Although the Arf interaction site has not been determined for PLD, several groups have screened Arf mutants that still bind GTP and downstream effectors but do not stimulate PLD activity. Deletion of the first 17 amino acids of Arf1 or mutation of asparagine 52 to arginine in the switch I region renders the protein unable to stimulate PLD (Liang et al., 1997; Jones et al., 1999). The analogous asparagine mutation in Arf6 (N48R) also inhibits PLD stimulation (Jovanovic et al., 2006) and these mutants have been used to discriminate PLD-dependent and -independent functions of Arf.
C. Rho GTPases
Rho family members were originally cloned from marine snails as homologs of Ras GTPases that shared approximately 35% sequence identity (Madaule and Axel, 1985). Over 20 Rho family members have been identified and the best-characterized family members include RhoA, Rac1, and Cdc42 (Heasman and Ridley, 2008). Rho proteins are frequently activated after receptor stimulation and control cytoskeletal dynamics such as formation of stress fibers, lamellipodia, and filopodia, along with controlling membrane ruffling and cell polarity. Rho proteins have also been shown to regulate transcription of certain genes and promote cell proliferation, which underscores the importance of Rho proteins in cancer (Ellenbroek and Collard, 2007). Like other GTPases, Rho proteins are regulated by GEFs and GAPs. However, Rho proteins are uniquely controlled by guanine nucleotide dissociation inhibitors (GDIs), which promote the inactive, GDP-bound state, and prevent Rho-membrane association by sequestering their lipid moieties (Ellenbroek and Collard, 2007).
For some time, it was known that the combination of cytosol and nonhydrolyzable GTP analogs such as GTPγS stimulated PLD activity from human neutrophil membranes, implicating the participation of small-molecular-mass GTPases (Olson et al., 1991). By including GEFs and GDIs specific for Rho proteins in the neutrophil membrane PLD assay, investigators measured stimulation and inhibition, respectively, of cytosol and GTPγS-stimulated PLD activity (Bowman et al., 1993; Kwak et al., 1995; Siddiqi et al., 1995). Although these experiments strongly suggested that the Rho family of GTPases mediated the cytosol-GTPγS stimulation of PLD activity in neutrophils, later studies with rat liver membranes confirmed the involvement of RhoA. Treatment with RhoGDI resulted in the loss of membrane-bound Rho and inhibition of the cytosol-GTPγS stimulation of rat liver PLD. Reconstitution with recombinant RhoA fully restored the PLD response after treatment with RhoGDI confirming that Rho proteins stimulate PLD activity (Malcolm et al., 1994). In addition, independent studies identified the stimulating factor as Rho due to sensitivity of PLD activity to the C3 toxin from Clostridium botulinum, which mediates the ADP-ribosylation of Rho proteins (Kuribara et al., 1995). In these studies, other Rho proteins, such as Rac1 and Cdc42, were partially able to rescue PLD activity after RhoA depletion or inactivation.
Later studies using purified PLD1a and PLD1b demonstrated a direct stimulation of PLD1a and PLD1b activity by RhoA and associated family members Rac1 and Cdc42 (Hammond et al., 1997; Min et al., 1998; Walker and Brown, 2002), where RhoA stimulates PLD activity more than other family members. By contrast, PLD2 is relatively unresponsive to Rho family members (Colley et al., 1997b; Lopez et al., 1998; Sung et al., 1999a). Mutational analysis has mapped the Rho interaction site on PLD1 to the C terminus (Sung et al., 1997; Yamazaki et al., 1999) and mutation of several nonconserved residues (K946A, V950A, R955A, and K962A) inhibits RhoA binding and stimulation of PLD1 activity, potentially explaining the specificity for PLD1 versus PLD2 (Cai and Exton, 2001). In addition, PKC, Arf, and RhoA are believed to bind distinct sites on PLD1 and modulate different kinetic parameters. Rho family members are thought to function primarily as binding activators and enhance substrate binding (Henage et al., 2006). As such, PKC, Arf, and Rho synergistically activate PLD1 in vitro (Ohguchi et al., 1996; Hammond et al., 1997) and most likely converge on PLD1 to precisely regulate activity in vivo. The mechanism by which Rho activates PLD in vivo is complicated by the observation that Rho also stimulates PIP5K activity to increase PIP2 levels (Chong et al., 1994). In fact, some studies suggest that the decrease in PLD activity after removal of Rho proteins might be due to a decrease in PIP2 because the addition of PIP2 is able to fully restore PLD activity in some circumstances (Schmidt et al., 1996). Thus, the role of Rho proteins in vivo might be to stimulate PIP2 synthesis via an interaction with PIP5K and then to increase PLD substrate binding and subsequent activity.
D. Ras and Ral GTPases
In addition to Arf and Rho, other small GTPases regulate PLD activity. Early investigations into the mechanisms of cellular transformation demonstrated that PLD activity was elevated in v-Ras and v-Src transformed fibroblasts compared with nontransformed cells (Song et al., 1991; Carnero et al., 1994). Maximal PLD activity in v-Src transformed fibroblasts required both cytosol and GTPγS and depletion of Ras from cytosol significantly decreased PLD activity, suggesting that Ras was required for the v-Src–induced increase in PLD activity (Jiang et al., 1995a). Activated Ras has many downstream effectors including the guanine nucleotide dissociation stimulator for RalA (Hofer et al., 1994; Spaargaren and Bischoff, 1994). As such, later studies demonstrated that RalA was required for the v-Src–induced increase in PLD activity since PLD formed a complex with RalA, but not Ras (Jiang et al., 1995b). The stimulatory properties of RalA are not direct, however, and experiments with purified proteins demonstrated that RalA does not directly stimulate PLD activity (Kim and Wong, 1998). However, RalA potentiates the stimulation of PLD1 by Arf proteins in vitro and Arf was found in a ternary complex with PLD1 and RalA when immunoprecipitated from cell lysates (Kim and Wong, 1998). Indeed, inhibition of Arf GEFs in v-Src–transformed fibroblasts reduces PLD activity and suggests that Arf is a key mediator of the v-Src/Ras/RalA/PLD transformation pathway.
E. Rheb GTPases
Regulation of the mammalian target of rapamycin (mTOR) pathways is complex and involves input signals from many sources. PLD and PtdOH positively regulate mTOR (Fang et al., 2001). Rheb, a small Ras-like GTPase, also positively regulates mTOR activity through a mechanism that was thought to involve direct binding of Rheb to the mTOR complex (Long et al., 2005). Rheb coimmunoprecipitates with PLD1 from cell lysate and stimulates PLD1 activity in vitro (Sun et al., 2008) and it was proposed that Rheb positively regulates mTOR by modulating PLD1 activity. These in vitro experiments used immunoprecipitated PLD1 from human cell lysate and bacterially expressed Rheb to demonstrate the regulation of PLD1. Subsequent studies from our laboratory with highly purified PLD1 from insect cells did not reproduce the findings of the original article suggesting that the regulation of PLD1 by Rheb likely involves an intermediate protein (data not shown). Rheb is negatively regulated by the GAP activity of the tuberous sclerosis 2 protein (TSC2), a component of the tuberous sclerosis complex (Manning and Cantley, 2003). Cells deficient in TSC2 show higher PLD and mTOR activity (Sun et al., 2008) consistent with the idea of Rheb positively regulating mTOR through PLD.
F. Non–G Protein/Protein Kinase C Regulators of Phospholipase D
Although PKC and small G proteins appear to be the major regulators of PLD1 activity, a small number of additional PLD-stimulating factors have been identified although their role in the regulation of PLD is far less studied.
1. Protein Kinase N.
The protein kinase N (PKN) family of kinases are structurally related to PKC, yet are calcium independent and not activated by phorbol esters (Morrice et al., 1994). These kinases are ubiquitously expressed (Kitagawa et al., 1995) and share many of the same intracellular locations as PLD1 (Kawamata et al., 1998). PKN is activated by GTP-bound RhoA and participates in cytoskeleton dynamics, cell migration, and tumor invasion (Watanabe et al., 1996). PKN directly stimulates PLD1 activity by binding to a region between amino acids 228 and 598 (Oishi et al., 2001). In addition, PtdOH, CL, and fatty acids are potent activators of PKN in vitro (Khan et al., 1994; Morrice et al., 1994). Thus, PLD may participate in a feed-forward activation scheme for PKN in which Rho stimulates both PKN and PLD and subsequent PtdOH production further upregulates PKN activity.
2. Cofilin.
Cofilin proteins sever filamentous actin to generate free actin, an important process for cytoskeleton rearrangement (DesMarais et al., 2005). The activity of cofilin is regulated through a phosphorylation-dependent mechanism in which phosphorylation by LIM kinases results in cofilin inactivation and a reduction in actin severing resulting in actin polymerization. Rho and Rac/Cdc42 activate LIM kinases through Rho kinase and p21-activated protein kinase (PAK), respectively (Sells and Chernoff, 1997; Edwards and Gill, 1999; Kaibuchi et al., 1999). Phosphorylated cofilin stimulates PLD1 activity by directly binding to a region between the loop and CRIII (Han et al., 2007). The activation of PLD1 by phosphocofilin underscores the importance of PLD1 in regulating cytoskeletal dynamics since Rho, Rac, and Cdc42 can directly bind and stimulate PLD1 and also indirectly activate PLD1 through a LIM kinase-cofilin pathway.
3. C-Terminal Binding Protein 1.
The C-terminal binding protein 1 (CtBP1) is a dual function protein that regulates gene transcription in the nucleus and also mediates membrane fission during intracellular trafficking events (Corda et al., 2006). CtBP1 specifically colocalizes with PLD1 after cell stimulation either with serum or EGF and purified CtBP1 was shown to stimulate PLD1 activity in an in vitro activity assay using purified proteins (Haga et al., 2009). Interestingly, CtBP1 stimulated PLD1 activity in an additive fashion with Arf and RhoA, suggesting that CtBP1 binds a distinct site on PLD1. Although further research is necessary, the membrane fission functions of CtBP1 might be mediated through PLD and PtdOH.
V. Phospholipase D Inhibitory Proteins
Most cells exhibit low basal PLD activity until stimulated by extracellular agonists or cellular stress (Zheng et al., 2006; Bruntz et al., 2014). In vivo, this stimulation most likely results from activation of stimulatory proteins and also inactivation of inhibitory proteins. As such, a number of PLD inhibitory factors have been purified over the years and a surprising number fall into the category of actin-binding proteins or proteins involved with vesicular trafficking.
A. Synaptic Vesicle Proteins
Munc-18-1 is a syntaxin binding protein enriched in neurons and it plays a critical role in synaptic vesicle exocytosis (Harrison et al., 1994; Wu et al., 1998). Munc-18-1 interacts directly with the PX domain of PLD1 and PLD2 and inhibits PLD activity in vitro. When overexpressed in cells, Munc-18-1 and PLD both colocalize and coimmunoprecipitate under basal conditions, but dissociate once stimulated with EGF when Munc-18-1 relocalizes from plasma membranes to the cytosol and no longer inhibits PLD activity (Lee et al., 2004). Munc-18-1 is thus one of the few examples of signaling pathway components that activates PLD via repression of inhibition. In addition to Munc-18-1, clathrin assembly protein 3 (AP3) is another example of a trafficking protein known to regulate PLD activity. AP3 binds to clathrin triskelia and promotes assembly of clathrin-coated vesicles, which are dynamic organelles that participate in intracellular membrane trafficking (Ahle and Ungewickell, 1986; Prasad and Lippoldt, 1988) including recycling of synaptic vesicles. Like Munc-18-1, AP3 is highly enriched in neuronal tissue (Ahle and Ungewickell, 1990; Keen, 1990) and was purified as a factor from rat brain cytosol capable of inhibiting purified PLD1 activity in vitro (Lee et al., 1997a). Even though AP3 is known to bind PIP2 (Norris et al., 1995; Ye and Lafer, 1995), AP3 appears to bind and inhibit PLD1 in a PIP2-independent manner, suggesting a direct inhibition of PLD1 activity (Lee et al., 1997a). Like AP3, amphiphysins are nerve terminal proteins that participate in clathrin-mediated synaptic vesicle endocytosis. Amphiphysins are believed to function in concert with dynamin to promote vesicle budding (Wigge and McMahon, 1998). As with AP3, amphiphysin I and II were purified from rat brain cytosol as factors capable of inhibiting purified PLD1 and PLD2 activity in vitro and inhibit PMA-stimulated PLD activity when overexpressed in cells. Although the exact physiologic relevance of PLD inhibition by these synaptic vesicle proteins is unknown, they most likely function to terminate PtdOH generation during the early stages of vesicle formation.
B. Miscellaneous Phospholipase D Inhibitors
In addition to the vesicular and actin-associated PLD inhibitors, several proteins have been identified as PLD inhibitors that do not fit into broad categories. Aldolase is a glycolytic enzyme that mediates the reversible cleavage of fructose-1,6-bisphosphate into dihydroxyacetone phosphate and glyceraldehyde-3-phosphate. Aldolase was identified as a cytosolic factor that directly inhibits PLD2. Mutational analysis narrowed the site of interaction to the PH domain of PLD2 (Kim et al., 2002). The physiologic function of the aldolase–PLD2 interaction is unknown, but this interaction might contribute to the role of PLD in regulating cellular bioenergetics. Another protein known to inhibit PLD is the Gβγ subunit from heterotrimeric G proteins. GPCR stimulation results in dissociation of Gβγ from Gα and Gβγ regulates downstream effector molecules containing PH domains such as G protein–coupled receptor kinases (GRKs) (Carman et al., 2000) and phospholipase C (PLC)-β (Wang et al., 2000). Recombinant Gβγ inhibits PLD1 or PLD2 activity in vitro, presumably through an interaction with the PH domain since an N-terminal PLD truncation mutant was resistant to Gβγ inhibition (Preininger et al., 2006). Thus, Gβγ inhibition of PLD serves as one of the ways PLD signaling is terminated after GPCR stimulation.
Finally, two neuronal proteins have been identified as PLD2 inhibitors. The first protein, α-synuclein, is a small, highly conserved protein of unknown function that is highly enriched in the brain presynaptic terminals. α-Synuclein is a major component of Lewy bodies and Lewy neuritis, which are neuropathological hallmarks of familial and sporadic Parkinson disease (Spillantini et al., 1997). α-Synuclein was purified as a factor capable of inhibiting purified PLD2 activity in vitro (Jenco et al., 1998). This observation was later hypothesized as a mechanism by which neurons are protected from the toxicity associated with elevated PLD activity (Gorbatyuk et al., 2010). In these studies, PLD2 was purified by an immunoaffinity technique and no gels of purity are shown. A subsequent study with highly purified PLD2 was unable to replicate the original findings (Rappley et al., 2009), suggesting the presence of an intermediate protein in the original article. Regardless of the interaction between α-synuclein and PLD2, no associations have been made in vivo to support a role for PLD2 in the pathophysiology of α-synuclein (Ahn et al., 2002). Therefore, the relevance and nature of this interaction is still a matter for investigation. The second neuronal protein capable of directly inhibiting PLD2 is collapsin response mediator protein-2 (CRMP-2). CRMP-2 is a critical component of neuron outgrowth during development and is believed to participate in early stages of Alzheimer’s disease (Hensley et al., 2011). Like α-synuclein, CRMP-2 was purified as a cytosolic brain factor capable of inhibiting purified PLD2 in vitro and is believed to directly inhibit activity by binding to the N terminus (Lee et al., 2002). Future studies will no doubt shed further light on the role of PLD2-related pathologies such as Alzheimer’s disease.
VI. Cell Surface Receptor Regulation of Phospholipase D
A. Receptor Tyrosine Kinases
Ligand binding to cell surface receptors is known to stimulate PLD activity in many cell lines and tissues. EGF stimulates PLD activity (Fisher et al., 1991) and this pathway has been used as a model system for characterizing the intermediate signaling events between receptor tyrosine kinases (RTKs) and PLD over the past 25 years. EGF binding to the EGFR results in receptor dimerization and autophosphorylation of several tyrosine residues within the cytoplasmic tails via an intrinsic kinase activity (Schlessinger and Ullrich, 1992). These phosphorylated tyrosine residues create high affinity binding sites for proteins containing Src homology 2 (SH2) domains, such as PLC-γ1, Grb2, the p85 subunit of PI3K, Src, Src homology region 2 domain-containing phosphatase (SHP)-2, and Bruton’s tyrosine kinase, which mediate a variety of signaling events including those that regulate DNA synthesis, cell division and proliferation, cytoskeletal rearrangements, and a variety of other biologic functions (Yaffe, 2002). As such, several mechanisms of PLD activation by EGF have been described.
Early studies of PC hydrolysis or phosphatidyl alcohol production after EGF stimulation in intact cells did not discriminate between PLD1 and PLD2 activation, although later studies have shown that both isoforms couple to the EGFR (Slaaby et al., 1998). The most-studied EGFR-PLD signaling pathway begins with activation of PLC-γ1 by binding to the activated EGFR via its SH2 domain (Anderson et al., 1990; Margolis et al., 1990). Activation of PLC-γ1 results in the rapid hydrolysis of PIP2 into inositol triphosphate (IP3) and DAG. IP3 increases intracellular calcium levels, which together with DAG activates PKC (Rana and Hokin, 1990). As discussed, PKC is a well established activator of PLD and multiple studies have demonstrated an EGFR/PLC-γ1/PKC axis for PLD activation. By using small-molecule PKC inhibitors or by downregulating PKC levels with prolonged phorbol ester treatment, multiple groups have shown decreased EGF-stimulated PLD activation after inactivation of PKC in various cell types (Yeo and Exton, 1995; Voss et al., 1999). Later studies using mouse embryonic fibroblasts demonstrated compromised EGF stimulation of PLD activity in PLC-γ1 null cells versus wild-type cells, supporting the requirement of PLC-γ1 and PKC for EGF stimulation of PLD activity (Hess et al., 1998). Although PKC is believed to primarily activate the PLD1 isoform (Colley et al., 1997b; Sung et al., 1999b), the PX domain of PLD2 interacts with the Src homology 3 (SH3) domain of PLC-γ1 after EGF stimulation and is believed to increase PLC-γ1–mediated hydrolysis of PIP2 (Jang et al., 2003). When the PLD2–PLC-γ1 interaction is disrupted by mutating specific proline residues, EGF no longer stimulates PLD2 activity, indicating a complex mode of PLD2 regulation by PLC-γ1 (Jang et al., 2003).
Although EGFR activation stimulates PLD activity through a PLC-γ1–PKC axis in many cell types, EGFR activation does not always stimulate PIP2 hydrolysis and PKC activation in other cell lines (Cook and Wakelam, 1992; Hess et al., 1997). Alternative EGFR-PLD coupling mechanisms have been described and usually involve activation of small GTPases. Ras participates in a complex regulatory mechanism with RTK-stimulated PLD. The first step of the canonical Ras activation sequence involves recruitment and activation of SOS, which stimulates GDP-GTP exchange on Ras and allows Ras to activate downstream effectors (Schlessinger, 2000). GEFs for the small GTPase RalA are stimulated by activated Ras (Matsubara et al., 1999) and RalA has been implicated in the activation of PLD. RalA interacts with PLD1 (Luo et al., 1997) and is believed to enhance PLD catalytic activity in the presence of other activators, such as Arf GTPase (Kim and Wong, 1998). As such, PLD stimulation by the EGFR requires Ras-activated RalA in multiple cell lines (Voss et al., 1999; Lu et al., 2000), although the intermediate signaling proteins between RalA and PLD are not fully characterized. In addition to RalA, Ras activates the p110 subunit of PI3K by binding to a Ras binding domain (Rodriguez-Viciana et al., 1996). The product of PI3K, PIP3, can recruit and activate PLD1 after RTK stimulation via the PLD-PX domain (Standaert et al., 1996; Lee et al., 2005), and may offer an alternative mechanism by which Ras regulates PLD activity.
By contrast, PLD and PtdOH influence EGF-stimulated Ras activation by regulating membrane recruitment and activation of SOS. The adaptor molecule Grb2 contains two SH3 domains and one SH2 domain. Receptor stimulation recruits Grb2 via an interaction with the SH2 domain. Once receptor-bound, Grb2 binds SOS via one of the SH3 domains and this has been a well established mechanism for SOS recruitment (Schlessinger, 2000). PLD2 binds the other SH3 domain of Grb2 and this interaction was necessary for EGF stimulation of PLD2 activity (Di Fulvio et al., 2006). Interestingly, PLD-generated PtdOH also directly binds and recruits SOS to membranes after EGFR activation (Zhao et al., 2007). Thus, a potential feed-forward mechanism emerges in which PLD activates Ras, which leads to activation of downstream effectors, such as RalA, further stimulating PLD activity. The regulation of Ras signaling by PLD has important clinical implications and is further discussed in the section below on mechanisms of cancer involving PLD. A schematic illustrating the signaling pathways leading to PLD activation as well as the highly integrated involvement of PLD in well characterized cancer pathways is shown in Fig. 2.
Fig. 2.
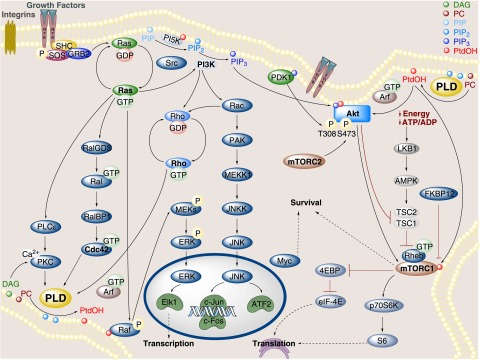
Cellular signaling modulating PLD activity from receptor tyrosine kinases and pathways downstream of PLD involved in oncogenic transformation.
In addition to Ras, Arf proteins also participate in PLD1 and PLD2 activation after RTK stimulation. RTK activation of PI3K recruits certain GEFs for Arf proteins (Venkateswarlu et al., 1998) and results in the formation of an active ArfGEF-RTK complex (Li et al., 2003). Studies using Brefeldin A, an inhibitor of certain ArfGEFs (Peyroche et al., 1999), demonstrated that the ArfGEF “ARF nucleotide-binding site opener” and Arf1 are required for PLD1 activation after stimulation of the insulin receptor in rat fibroblasts (Shome et al., 1997; Li et al., 2003). The requirement of Arf in insulin signaling appears to be cell line dependent because PLD activation by insulin does not require Arf in Chinese hamster ovary T cells (Emoto et al., 2000). In addition, Arf4 was identified as an EGFR binding partner in a yeast two-hybrid screen that interacts with EGFR upon agonist stimulation (Kim et al., 2003b). Coexpression of Arf4 with PLD2 but not PLD1 resulted in a substantial increase in EGF-stimulated PLD2 activity compared with the expression of PLD2 alone (Kim et al., 2003b). Although further studies are required to delineate the role of Arf proteins in RTK-PLD activation, current research suggests that Arf proteins may play nonredundant roles in regulating PLD1 versus PLD2.
The Rho family of small GTPases (Rho/Rac/Cdc42) also regulates RTK stimulation of PLD activity in some cell systems. Like Ras and Arf, Rho GTPases are usually activated by upstream GEFs through a variety of mechanisms (Buchsbaum, 2007). In fibroblasts, Rac1 and RhoA were required for PLD activation by PDGF and EGF, respectively (Hess et al., 1997). However, RTK stimulation of PLD in vascular smooth muscle cells requires Arf proteins, but not Rho proteins (Shome et al., 2000). Rho proteins are particularly important for regulating cytoskeleton rearrangements needed for processes, such as stress fiber and lamellipodia formation in motile cells (Nobes and Hall, 1995). The role of RTK signaling to PLD through Rho family proteins may thus be dependent on the motile nature of the cell.
Recent studies have elucidated new regulators of RTK-PLD signaling in addition to the “classic” PLD inhibitors previously discussed. Cdk5 is a multifunctional serine/threonine kinase that mediates a variety of signaling events including exocytosis of insulin in pancreatic β cells (Lilja et al., 2004). Upon stimulation with EGF in rat insulinoma cells, Cdk5 phosphorylates PLD2 at serine 134 and this phosphorylation event was required for EGF-stimulated PLD activity and subsequent release of insulin from these cells (Lee et al., 2008). Future studies should determine whether the phosphorylation of PLD2 by Cdk5 is required for RTK-mediated function in other cell types. The multitude of pathways in which RTKs stimulate PLD activity underscores the importance of PLD and PtdOH in mediating the biologic effects of RTK ligands.
B. G Protein–Coupled Receptors
GPCRs represent another large class of cell surface receptors that frequently lead to PLD activation. Under inactive conditions, GPCRs bind different classes of heterotrimeric G proteins composed of an α subunit and dimer composed of β and γ subunits. Ligand binding at the extracellular face of the GPCR leads to a conformational change in the receptor that promotes GDP to GTP exchange on the α subunit and GTP-bound Gα then dissociates from βγ (Oldham and Hamm, 2008). Each class of activated Gα subunit is associated with a distinct cellular signaling pathway. The Gαs G proteins activate adenylyl cyclase (AC) to increase intracellular levels of the second messenger cAMP, which activates protein kinase A (PKA). The Gαi/o G proteins are associated with a decrease in AC activity; however, the liberated Gβγ dimer has AC-independent roles such as recruiting and activating proteins with PH domains (Touhara et al., 1994). Gαq/11 G proteins stimulate PLC activity, which subsequently leads to activation of PKC and downstream effectors (McCudden et al., 2005). Finally, Gα12/13 proteins are associated with activation of Rho proteins and effectors by activating Rho GEFs (Kozasa et al., 1998). The list of GPCR ligands that activates PLD is quite extensive and GPCRs of all classes have been reported to activate PLD (Exton, 1999). Therefore, the known mechanisms of PLD activation by each class of Gα G protein are discussed in the following section.
Once activated, Gαq binds and recruits PLC-β to membranes where it hydrolyzes PIP2 to generate IP3 and DAG, leading to activation of PKC (Rhee, 2001). Several lines of evidence suggest that PKC mediates the activation of PLD by Gαq. Cells that express constitutively active Gαq have elevated PLD activity (Plonk et al., 1998). Coexpression of a PKC-resistant PLD mutant significantly reduces the fold activation of PLD activity due to constitutively active Gαq, suggesting that PKC mediates Gαq activation of PLD (Xie et al., 2002b). Likewise, stimulation of a Gαq-coupled receptor failed to stimulate another PKC-resistant PLD mutant. This mutant was responsive to non-Gαq GPCR stimuli, confirming proper protein folding and underscoring the importance of PKC in mediating Gαq-PLD signaling (Zhang et al., 1999). Small-molecule PKC inhibitors block PLD activation by a number of GPCR agonists including bradykinin (Pyne and Pyne, 1995), thrombin (Vasta et al., 1998), and S1P (Meacci et al., 1999) and overexpression of PKC-β is known to potentiate the PLD response to endothelin-1 stimulation in Rat6 fibroblasts (Pai et al., 1991). Similarly, downregulation of PKC with prolonged phorbol ester treatment inhibits bradykinin stimulation of PLD in SF3271 fibroblasts (Clark and Murray, 1995). Data from our laboratory suggest that the combination knockdown of PKCα and PKCβ significantly decreases M1 muscarinic receptor stimulation of PLD in HEK293 cells (data not shown), confirming the importance of PKC in mediating Gαq signaling to PLD. However, many Gαq-coupled GPCRs couple to additional G proteins, including members of the Gα12/13 family.
Gα12/13 G proteins have long been known to stimulate cytoskeletal rearrangements through activation of the Rho family of proteins and some GPCRs that predominantly couple to Gαq also couple to Gα12/13. Expression of a dominant-negative RhoA construct or treatment with C3 exoenzyme inhibits M3 muscarinic receptor (M3R) stimulation of PLD, suggesting that RhoA participates in M3R activation of PLD (Schmidt et al., 1996; Mitchell et al., 1998). Likewise, expression of a RhoA-resistant PLD mutant is less responsive to M3R stimulation (Du et al., 2000). C3 exoenzyme treatment inhibits the PLD response to other GPCR agonists such as bradykinin (Meacci et al., 2003) and S1P (Meacci et al., 2001). Expression of constitutively active Gα13, like Gαq, stimulates PLD activity. This active Gα13 mutant stimulates PKC-resistant PLD constructs, suggesting that Gα13 stimulates PLD independently of PKC. C3 exoenzyme treatment inhibits Gα13 activation of PLD (Plonk et al., 1998; Xie et al., 2002a), suggesting that RhoA mediates Gα13 stimulation of PLD. In addition to a direct protein–protein interaction between RhoA and PLD, several other activation mechanisms have been proposed. RhoA is known to stimulate synthesis of PIP2 (Chong et al., 1994) and exogenous addition of PIP2 after RhoA inactivation is known to restore PLD activity (Schmidt et al., 1996). Besides direct stimulation of PLD activity and increasing PIP2 levels, a recent study suggested that LIM1 kinase, a RhoA effector, stimulates PLD activity after M3R stimulation. As discussed previously, LIM1 kinase phosphorylates cofilin, and phosphocofilin stimulates PLD activity downstream of M3 activation (Han et al., 2007). Phosphocofilin depletion had no effect on PMA-stimulated PLD activity, suggesting a RhoA-dependent mechanism. These effects may also be a consequence of upstream actin remodeling. On the other hand, several studies suggest that RhoA and PKC synergize to stimulate PLD downstream of GPCRs. For example, the C3 exoenzyme reduces PLD activity after S1P stimulation in C212 myoblasts, but PLD activity is further reduced after treatment with PKC inhibitors (Meacci et al., 2001). In addition, overexpression of RhoA partially rescues M3R-mediated activation of PKC-resistant PLD and together these results suggest that both RhoA and PKC can mediate signals from the same GPCR to PLD (Zhang et al., 1999).
The signaling pathways from Gαs-coupled receptors to PLD remain somewhat elusive. Stimulation of Gαs-coupled GPCRs or treatment of cells with AC-activating compounds, such as forskolin and dibutyryl-cAMP, results in PLD activation (Ginsberg et al., 1997). Under these conditions, treatment of cells with PKA inhibitors reduces PLD activity, suggesting that PKA somehow mediates PLD activation (Yoon et al., 2005). In addition, transfection of a dominant-negative Src and dominant-negative Ras also decreases PLD activation downstream of AC, suggesting that Src and Ras participate in Gαs coupling to PLD (Yoon et al., 2005). Exchange protein directly activated by cAMP 1 (Epac1) is a cAMP-activated GEF for small G proteins, such as Rap1. Recent studies have shown that depletion of Epac1 inhibits Gαs signaling to PLD. Epac1 also promotes GDP to GTP exchange on R-Ras and this AC/Epac1/R-Ras pathway has been implicated in PLD activation (López De Jesús et al., 2006)
Several GPCRs activate PLD in a pertussis toxin–sensitive manner, suggesting that Gαi/o proteins also stimulate PLD activity. For example, stimulation with fMLP in neutrophils or S1P in A549 lung adenocarcinoma cells and in human airway epithelial cells results in pertussis toxin–sensitive stimulation of PLD (Fensome et al., 1998; Ghelli et al., 2002; Meacci et al., 2003). In addition, known Gαi/o receptors, such as M2 and M4 muscarinic receptors, stimulate PLD activity in HEK293 cells (Sandmann et al., 1991). The pathways by which Gαi/o stimulate PLD are relatively uncharacterized, but are thought to require the activity of Arf and Rho (Fensome et al., 1998) and participation by tyrosine kinases, such as Src (Ghelli et al., 2002). Likewise, Arf activity is required for stimulation of PLD activity by pertussis toxin–insensitive GPCRs (Rümenapp et al., 1995; Mitchell et al., 1998, 2003). Liberated Gβγ subunits are known to activate certain PI3Ks and increase local concentrations of PIP3 after receptor activation (Stephens et al., 1997). Thus, one potential mechanism for Arf activation appears to be recruitment of PI3K-dependent ArfGEFs downstream of Gβγ (Touhara et al., 1994). Whether other mechanisms besides Gβγ stimulation of GEFs mediate Gαi/o stimulation of PLD should be the subject of future investigations.
The PtdOH derived from GPCR-stimulated PLD may function to terminate GPCR signaling by regulating the activity of proteins required for GPCR inactivation. GRKs phosphorylate GPCRs and create a high-affinity binding site for arrestin proteins, which uncouple GPCRs from G proteins and promote receptor internalization and desensitization (Moore et al., 2007). PtdOH, along with PS, CL, and PI, are stimulators of GRK2/3 activity in vitro as determined by measuring phosphorylation of GPCRs in micelles containing phospholipids and purified GPCRs incubated with purified GRK. Kinase activity toward muscarinic (DebBurman et al., 1995) and adrenergic (Onorato et al., 1995) receptors was much higher in the presence of PtdOH, suggesting that PLD may promote GPCR internalization and desensitization through GRKs.
Termination of AC signaling requires the activity of cyclic nucleotide phosphodiesterases (PDEs) that hydrolyze 3′,5′-cyclic nucleotides to a nucleoside 5′-monophosphate. Several families of cAMP-specific PDEs have been identified that are responsible for terminating signaling events mediated by this important second messenger (Conti and Beavo, 2007). As such, multiple variants of the cAMP-specific PDE4 family are regulated by phosphatidic acid. Screening recombinant variants of PDE4 enzymes for PtdOH sensitivity revealed that PDE4A5, PDE4D3, and PDE4EB1 were all stimulated by PtdOH in an in vitro enzyme assay (Némoz et al., 1997). PDE4D3 was shown to bind PtdOH directly and the binding site was mapped to a region enriched in basic amino acids from 31 to 59 (Grange et al., 2000). Another PDE4 family member, PDE4A1, is not activated in vitro by PtdOH (Némoz et al., 1997) but requires an interaction with PtdOH at its amino terminus for proper membrane localization (Baillie et al., 2002). Thus, by activating PDEs, PtdOH may function to terminate Gαs signaling by promoting metabolism of the cAMP second messenger.
VII. Functions of Phospholipase D Contributing to Cancer
Over the last half century, PLD enzymes have been implicated in a variety of pathologies ranging from neurodegenerative diseases (Lindsley and Brown, 2012) to blood disorders (Elvers et al., 2010). A recent report identified a risk variant in the PLD3 gene for late-onset Alzheimer’s disease (Cruchaga et al., 2014), although the virtual absence of biochemical characterization of this gene product limits what can be summarized with regard to underlying mechanisms. The best-characterized, disease-relevant roles for PLD relate to the requirement of PLD activity for oncogenesis and cancer progression. Early indications of the importance of PLD and PtdOH in cancer came from observations that cells transformed by viral oncogenes, such as v-Src (Song et al., 1991), v-Fps (Jiang et al., 1994) v-Ras (Carnero et al., 1994; Jiang et al., 1995a), and v-Raf (Frankel et al., 1999), all showed elevated PLD activity relative to nontransformed control cells. Furthermore, PLD mRNA and protein analyses from tumors of breast (Uchida et al., 1997; Noh et al., 2000), renal (Zhao et al., 2000), colorectal (Yoshida et al., 1998), gastric (Uchida et al., 1999), thyroid (Kim et al., 2008), and brain (Park et al., 2009) origin show elevated PLD1 and/or PLD2 expression relative to normal surrounding tissue. As such, overexpression of PLD isoforms is associated with enhanced tumorigenesis in cultured cells as measured by increased anchorage-independent growth (Min et al., 2001b; Ahn et al., 2003), the most important measure of tumorigenicity (Shin et al., 1975). Recent genomic analyses of human cancers have revealed several unique PLD mutations in breast (Wood et al., 2007), stomach (Zang et al., 2012), and brain (Molenaar et al., 2012; Pugh et al., 2012) cancers, although most of the reported mutations remain to be functionally characterized. Due to the common upregulation of PLD expression and activity along with the growing list of PLD mutations, researchers have long assumed that PLD and PtdOH confer growth advantages to transformed cells.
Definitive characterization of the molecular mechanisms behind these growth advantages has been difficult due to the lack of specific, small-molecule inhibitors. The three most common techniques for studying PLD function have been the use of primary alcohols to produce phosphatidyl alcohols at the expense of PtdOH via the PLD transphosphatidylation reaction (Yang et al., 1967), RNA interference (RNAi) to silence the expression of PLD mRNA, and expression of catalytically inactive PLD mutant proteins that function in a dominant-negative manner. Each technique has its own set of flaws including nonspecific enzyme inhibition, incomplete knockdown of target proteins, and retention of lipase-independent activities. The recent development of isoform-selective PLD inhibitors (Scott et al., 2009; Lavieri et al., 2010) and PLD knockout (KO) mice (Elvers et al., 2010; Oliveira et al., 2010) are now being integrated and incorporated into the arsenal of tools to study PLD function, and these will certainly assist in defining the exact roles of PLD in cancer.
The path to malignancy requires several unique steps. Hanahan and Weinberg (2011) proposed eight “hallmarks” of cancer that comprise eight biologic capabilities acquired during the development of human tumors. These include sustaining proliferative signaling, evading growth suppression, activating invasion and metastasis, enabling replicative immortality, inducing angiogenesis, resisting cell death, avoiding immune destruction, and deregulating cellular energetics. Interestingly, PLD has been implicated in almost every hallmark of cancer and the following discussion will highlight these various cancer-related PLD functions.
A. Sustaining Proliferative Signaling
In the absence of growth factors, cells enter a state of quiescence and no longer proliferate. This state of cellular quiescence is usually reached after terminal differentiation and is critical for maintaining proper tissue function and structure. Cancer cells acquire mutations that allow abnormal proliferation in the absence of normal growth signals. Mitogenic signals, or signals that allow passage through cell cycle checkpoints, are usually mediated by cell surface receptors, such as EGFR, and their downstream effectors, such as the mitogen-activated protein kinase (MAPK) pathways and PI3K/Akt/mTOR pathways. As such, mutated or overexpressed growth factors such as the PDGF receptor and the EGFR are commonly observed in malignancies such as glioblastoma and breast cancer, respectively (Slamon et al., 1987; Shih and Holland, 2006). Cancer cells also upregulate and secrete growth factors to stimulate growth in an autocrine fashion (Sporn and Roberts, 1985). As discussed, PLD is frequently activated as a consequence of receptor stimulation and participates in the pathways of cellular proliferation. Early evidence that PLD might directly regulate cell proliferation came from observations that exogenously added PtdOH or highly active bacterial PLD increased levels of c-Protein and c-Myc transcription factors, and stimulated thymidine uptake, a marker of DNA synthesis and cell proliferation, when added exogenously to cultured cells (Moolenaar et al., 1986). Since these early studies, PLD has been shown to participate in several cell proliferation pathways (see Fig. 2).
B. Mitogen-Activated Protein Kinase
One of the most commonly deregulated proliferative pathways in human cancers is the MAPK pathway. Growth factors, hormones, and chemokines activate the MAPK pathway by signaling through their cognate receptors and initiating a series of signaling events that leads to activation of multiple protein kinases (Shaw and Cantley, 2006). Under the canonical activation sequence, receptor activation recruits exchange factors for Ras to the membrane, and stimulates GDP to GTP exchange to activate Ras. Raf kinases subsequently bind activated Ras at the membrane and phosphorylate an intermediate kinase, generally termed mitogen-activated protein kinase kinase (MEK). Activated MEK phosphorylates and activates a terminal MAPK termed extracellular signal–regulated kinase (ERK)1/2 (De Luca et al., 2012). Activated ERK1/2 upregulates expression of many genes important for cell cycle progression, metabolism, and proliferation by activating a plethora of transcription factors, such as nuclear factor of activated T cells, ETS domain–containing protein Elk-1, myocyte enhancer factor-2, c-Protein, c-Myc, and signal transducer and activator of transcription 3 (STAT3) (Roux and Blenis, 2004). ERK1/2 can regulate protein translation by activating p90 ribosomal S6 kinase, MAPK-activated protein kinases, and MAPK-interacting kinases, which are known to directly regulate the activity of ribosomal translation machinery (Chen et al., 2001). In addition, ERK1/2 increases expression of antiapoptotic genes and Ras can directly inhibit the apoptotic machinery at the mitochondrial membrane, independently of ERK (Alejandro and Johnson, 2008). PLD and PtdOH have been intricately linked to multiple steps within the MAPK pathway and overexpression of PLD has been linked to upregulated ERK activity as measured by increased gene transcription downstream of ERK-activated transcription factors such as STAT3 (Choi and Han, 2012).
Ras was one of the earliest identified oncogenes, first discovered as a viral protein capable of inducing sarcoma in rats (Malumbres and Barbacid, 2003). Around 30% of human tumors contain mutations in Ras genes that result in constitutive Ras activation (Dunn et al., 2005). A growing body of literature suggests that PLD and PtdOH directly regulate Ras activation through several mechanisms. A well established Ras activation pathway involves recruitment of the adaptor protein Grb2 to phosphorylated tyrosine residues on RTKs via its SH2 domain (Lowenstein et al., 1992). Grb2 contains two SH3 domains (Matuoka et al., 1992) that bind proteins with proline-rich motifs, such as the Ras GEF SOS (Simon et al., 1991; Chardin et al., 1993). Ras family members undergo farnesylation or geranylgeranylation that results in constitutive membrane association. Thus, formation of the RTK/Grb2/SOS complex allows GDP to GTP exchange and activation of membrane-associated Ras. In addition to RTKs, Grb2 can bind PLD2 via the Grb2 SH2 domain at PLD2-Y169 and Y179 (Di Fulvio et al., 2006). The PLD2-Grb2 complex recruits SOS and stimulates activation of Ras and MAPK pathways (Di Fulvio et al., 2006, 2008) in a manner that does not require phospholipase activity. Furthermore, the PX domain of PLD2 has been shown to act as a GEF for Ras directly (Henkels et al., 2013b), providing an alternative route for lipase-independent Ras activation. On the other hand, PtdOH directly regulates Ras activation by serving as a recruitment signal for the SOS exchange factor (Zhao et al., 2007). SOS contains a PH domain that binds PtdOH, and residues H475 and R479 mediate this interaction. When PtdOH binding residues are mutated, SOS is no longer recruited to the plasma membrane after EGFR stimulation. PLD2 was found to colocalize with SOS and genetic silencing of PLD2 prevented the EGF-stimulated recruitment of SOS to the membrane and subsequent activation of Ras (Zhao et al., 2007). In addition, PtdOH can inhibit the activity of neurofibromin 1 (NF1), a Ras GAP, in vitro. NF1 was identified as one the earliest tumor suppressor genes (Cichowski and Jacks, 2001) and although the in vivo relevance of PtdOH inhibition of NF1 remains to be established, upregulated PLD activity could potentially activate Ras by inhibiting NF1. The diverse mechanisms by which PLD can regulate Ras activity certainly underscore the importance of PLD and PtdOH for Ras activation.
Interestingly, Ras activates PLD via a mechanism involving RalA, Arf, and the guanine nucleotide dissociation stimulator for RalA. The importance of PLD and PtdOH in Ras-mediated tumorigenesis was demonstrated in an elegant study in which the introduction of dominant-negative PLD into rat fibroblasts blocked the transforming ability of constitutively active Ras. When the Ras-transformed fibroblasts were injected into immunocompromised mice, fibroblasts coexpressing dominant-negative PLD failed to form tumors. However, when PtdOH was delivered to the mice via an osmotic pump, Ras-transformed fibroblasts expressing dominant-negative PLD formed tumors similarly to Ras-transformed fibroblasts that did not express dominant-negative PLD (Buchanan et al., 2005). Other studies have also demonstrated the importance of PLD as an oncogenic Ras signal transduction molecule. One of the negative regulators of ERK is a small, 15-kDa protein known as protein enriched in astrocytes (PEA15). PEA15 was originally identified as a substrate for PKC in astrocytes (Araujo et al., 1993), and was later identified in a yeast two-hybrid screen as a binding partner of PLD1 (Zhang et al., 2000). PEA15 positively regulates PLD activity, potentially by acting as a chaperone protein to increase protein stability and decrease degradation (Zhao et al., 2000). PEA15 also directly inhibits ERK by sequestration in the cytoplasm and blocking ERK nuclear import (Formstecher et al., 2001; Pastorino et al., 2010). When PEA15 is phosphorylated at S116 by Akt kinase (Trencia et al., 2003), a downstream effector of Ras and PLD, ERK is no longer sequestered by PEA15 and can activate downstream targets. PEA15 overexpressed in epithelial cells that express constitutively active Ras is hyperphosphorylated at S116 and the overexpression potentiates Ras-mediated transformation. Interestingly, disruption of the PEA15/PLD1 complex or PLD1 enzyme inhibition with butanol or small-molecule PLD inhibitors blocks the PEA15 potentiation of Ras-mediated tumorigenesis, suggesting that the PEA15 enhancement of PLD activity in this system drives ERK activation and anchorage-independent growth downstream of Ras (Sulzmaier et al., 2012). These studies strongly suggest that PLD and PtdOH transduce the signals required for Ras-induced oncogenesis, potentially through downstream activation of Raf and the MAPK pathway.
The three members of the Raf family (A-Raf, B-Raf and Raf-1) all activate the MAPK pathway to varying degrees, with B-Raf and Raf-1 being the most potent stimulators of MEK (McCubrey et al., 2007). Raf mutations have been reported in many cancers, including colorectal, ovarian, and especially melanoma, with around 60% of melanomas containing activating Raf mutations (Pollock and Meltzer, 2002). As such, inhibitors of mutant Raf, such as vemurafenib, are highly efficacious in treating malignant melanoma (Bollag et al., 2012). The activation of Raf kinases is complex and generally requires membrane recruitment and subsequent phosphorylation at a number of activating residues (Yan et al., 1998). Raf contains a Ras binding domain that allows requisite membrane recruitment through interaction with activated Ras. Raf-1 can also translocate to membranes by directly interacting with PtdOH (Ghosh et al., 1996). Through the use of butanol and catalytically inactive PLD mutants, investigators determined that PLD-derived PtdOH bound and recruited the Raf-1 kinase to facilitate ERK activation, but the interaction did not influence Raf-1 activity directly (Rizzo et al., 1999). The PtdOH binding domain was narrowed to a region of 35 amino acids and mutation of specific arginine residues abolished PtdOH binding to Raf-1. All three Raf family members contain this PtdOH binding domain (Ghosh et al., 1996, 2003). Although the canonical Raf activation sequence involves membrane recruitment by Ras, other studies have suggested that PtdOH mediates the translocation event and Ras somehow mediates subsequent Ras activation. Evidence for this claim comes from observations that mutation of the PtdOH binding site of Raf-1 completely prevents membrane binding, whereas expression of a dominant-negative Ras construct inhibits Raf-1 activation but not translocation (Rizzo et al., 2000). The validation that PtdOH mediates Raf-1 translocation and Ras mediates Raf-1 activation is supported by later studies showing that Raf-1 binding to liposomes containing PtdOH is not further enhanced by Ras (Hekman et al., 2002). However, Ras binding to the same liposomes was significantly enhanced in the presence of Raf-1, suggesting that Ras binds lipid-bound Raf-1 (Hekman et al., 2002). Therefore, PtdOH appears to directly recruit Raf-1 to membranes and possibly facilitate activation by Ras.
In addition to directly regulating Raf translocation to the membrane, PLD may indirectly affect ERK activation by regulating receptor endocytosis. Like RTKs, agonist binding to many GPCRs leads to a rapid increase in ERK activation and many of the same components that mediate RTK activation of ERK also mediate GPCR activation of ERK. However, inhibitors of endocytosis are known to also inhibit GPCR-stimulated ERK activation (Luttrell et al., 1997; Daaka et al., 1998). Arrestins are proteins that mediate GPCR internalization and serve as scaffolds to nucleate components of the MAPK pathways (Shenoy et al., 2006). PLD activity has been shown to regulate receptor endocytosis for a variety of cell surface receptors, including RTKs such as EGFR (Shen et al., 2001) as well as GPCRs, such as the μ-opioid receptor (Koch et al., 2003) and angiotensin II receptor (Du et al., 2004). The cone-shaped PtdOH has been suggested to participate in membrane fission events, such as endocytosis (Barr and Shorter, 2000). However, a recent study suggested that PLD regulates receptor endocytosis independently of PtdOH. The small GTPase dynamin is critical participant in endocytosis and functions by circling the neck of an invaginated membrane and pinching off the vesicle by constricting the neck in a manner that requires GTP hydrolysis (Ferguson and De Camilli, 2012). PLD1 and PLD2 stimulate GTPase activity of dynamin through a PtdOH-independent GAP activity in the PLD-PX domain. By using EGFR as an example, mutation of the catalytic GAP residue in the PLD-PX domain or overexpression of the PX domain fragment decreased and increased EGFR endocytosis, respectively (Lee et al., 2006). Therefore, in addition to PtdOH regulation of Ras and Raf activation, PLD may regulate MAPK signaling through GEF and GAP activities for Ras and dynamin, respectively.
C. Mammalian Target of Rapamycin
The mechanistic, or mammalian, target of rapamycin (mTOR) is a large serine/threonine protein kinase that serves as a sensor of cellular homeostasis and responds to amino acids, stress, oxygen, energy levels, and growth factors and in turn regulates cell growth, proliferation, survival, and metabolism by phosphorylating a plethora of downstream targets (Laplante and Sabatini, 2012). Canonical activation of mTOR involves PI3K, which stimulates Akt through a PIP3-dependent mechanism. Akt then activates mTOR by phosphorylating and inhibiting TSC2, an upstream inhibitor of mTOR (Manning and Cantley, 2003; Shaw and Cantley, 2006). In addition to growth factor receptor mutations (Libermann et al., 1985), PI3K activity is upregulated in many cancers through inactivation of phosphatase and tensin homolog, the lipid phosphatase that hydrolyzes PIP3 (Haas-Kogan et al., 1998), and also through activating mutations in the PI3K catalytic domains (Engelman et al., 2006). Thus, cancers that have elevated PI3K activity frequently have elevated mTOR activity.
Growth factor activation of mTOR results in protein translation through several known substrates including p70 S6 kinase 1 (p70S6K1) and eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1). mTOR phosphorylates p70S6K1, which in turn phosphorylates and activates the S6 ribosomal protein (Chung et al., 1992). 4E-BP1 interacts with the eukaryotic translation initiation factor 4E and inhibits translation until mTOR phosphorylation relieves the repressive function of 4E-BP1 on protein translation (Richter and Sonenberg, 2005). The bacterial macrolide rapamycin binds FK506 binding protein 12 (FKBP12) and the rapamycin-FKBP12 complex binds and inhibits mTOR (Koltin et al., 1991; Sabatini et al., 1994) at the FKBP12-rapamycin binding (FRB) domain (Choi et al., 1996). PtdOH was shown to bind R2109 in the FRB domain and an NMR structure of FRB-bound PtdOH suggested that binding of PtdOH to mTOR competes with rapamycin binding (Fang et al., 2001; Veverka et al., 2008). Some have suggested that PtdOH stabilizes mTOR complexes and that high levels of PtdOH may outcompete rapamycin and explain why some systems resist rapamycin induced cell death (Chen et al., 2003; Toschi et al., 2009). Consistent with the importance of PtdOH in mTOR activation, the three PtdOH-generating enzymes PLD (Fang et al., 2001), DAG kinase (Ávila-Flores et al., 2005), and lysophosphatidic acid acyl transferase (Tang et al., 2006) have all been shown to regulate mTOR activity. Although the literature is somewhat contradictory, both PLD1 (Fang et al., 2003; Sun et al., 2008) and PLD2 (Ha et al., 2006) have been reported as regulators of mTOR. However, small-molecule PLD inhibitors do not inhibit mTOR activity in all cells (Bruntz et al., 2014), which calls into question some of the previous studies that relied on butanol as a PLD “inhibitor.”
Although PtdOH can regulate p70S6K1 activity through mTOR, recent studies demonstrated that PtdOH also directly binds and activates p70S6K1 independently of mTOR, as measured by recombinant p70S6K1 binding to PtdOH in enzyme-linked immunosorbent assays and lipid-sedimentation–based assays (Lehman et al., 2007). To demonstrate direct activation of p70S6K1, the authors treated cells with rapamycin to inhibit mTOR and then with PtdOH before immunoprecipitation of p70S6K1 and measurement of activity. Although they measured increased p70S6K1 activity after PtdOH treatment, the authors did not address the potential displacement of rapamycin from mTOR by PtdOH, thus calling into question a direct stimulation by PtdOH. Further studies must address whether the binding of PtdOH to p70S6K1 has physiologic relevance.
D. Evading Growth Suppression
Most cells enter a state of cellular quiescence upon terminal differentiation or contact with other cells within a tissue or culture. The signals that prevent cell division and replication are diverse and complex, but are largely mediated by soluble factors and cell surface proteins that activate inhibitors of cell cycle progression through restriction points into S (DNA synthesis) or M (mitosis) phases (Fagotto and Gumbiner, 1996). As discussed in the previous section, mitogenic signals, such as those from growth factor receptors, evoke passage through cell cycle checkpoints to replicate DNA, undergo mitosis, and ultimately divide. To establish a transformed primary cell line in culture, at least two cooperating oncogenes or tumor suppressors must be mutated. These include, for example, mutations allowing constitutive mitogenic signaling, mutations that allow cells to overcome restrictions on cell cycle checkpoints, or mutations that allow cells to overcome programmed cell death (Land et al., 1983). As such, excessive proliferative signaling alone often leads to cell cycle arrest as a normal cellular response to preventing neoplastic growth. Overexpression of Raf (Woods et al., 1997) and oncogenic Ras (Serrano et al., 1997) proteins in nonimmortalized, primary cells leads to cell cycle arrest. Likewise, oncogenic Ras, which does not normally transform primary cells, transforms cells with the introduction of a protein that sequesters and inactivates cell cycle checkpoint proteins ( Serrano et al., 1997; Hahn et al., 1999). Although PLD overexpression alone does not appear to be transforming, PLD overexpression cooperates with overexpressed proto-oncogenes, such as c-Src or EGFR, in rat fibroblasts (Joseph et al., 2001), suggesting that PLD might also function at the level of cell cycle progression.
The transcription factor p53 is one of the most important tumor suppressors involved in cell cycle control and over 50% of human tumors contain inactivating mutations in the p53 gene (Hollstein et al., 1994). As cells pass through the cell cycle, DNA integrity is constantly monitored. Cellular stressors such as DNA damage can activate a number of proteins including ataxia telangiectasia mutated, a well known activator of p53 (Xu and Baltimore, 1996). Once activated, p53 either initiates a series of events triggering a cell cycle arrest or events triggering programmed cell death. By enforcing a G1 checkpoint, p53 prevents entry into the S phase, allowing time for DNA repair to proceed. If DNA damage is sufficient, p53 can initiate programmed cell death to prevent replication and passage of damaged genes (Evan and Littlewood, 1998). p53 enforces a cell cycle arrest by upregulating a number of genes that prevent cell cycle progression. Although there are many, the p21 cyclin-dependent kinase inhibitor (p21Cip1) is a well characterized p53 target (el-Deiry et al., 1993). p21Cip1 binds to cyclin-dependent kinases and blocks their activity to enforce a G1 cell cycle arrest. p53 can initiate apoptosis by upregulating proapoptotic genes such as Bax (Miyashita and Reed, 1995) and death receptors (Owen-Schaub et al., 1995), and p53 can stimulate apoptosis independently of its transcriptional activity by directly activating the mitochondrial apoptotic machinery (Chipuk et al., 2004). Therefore, inactivation or suppression of p53 is often a key step in neoplastic growth and PLD has been linked to the p53 pathway in several studies.
In normal proliferating cells, DNA-damaging agents cause apoptosis through a mechanism that involves increased expression of p53. In rat fibroblasts and MDA-MB-231 breast cancer cells, overexpression of PLD1 results in decreased p53 levels and decreased apoptosis after treatment with DNA-damaging agents, suggesting that PLD activity promotes p53 degradation (Hui et al., 2004, 2006). p53 levels are usually low as the protein is continuously degraded via a mechanism that involves ubiquitination and proteasomal degradation. The E3 ubiquitin-protein ligase mouse double minute 2 homolog functions as a negative regulator of p53 by targeting the protein for proteasomal degradation (Moll and Petrenko, 2003). Overexpression of PLD correlates with increased levels of mouse double minute 2 homolog in rat (Hui et al., 2004) and human (Hui et al., 2006) cells, potentially explaining the mechanism by which PLD decreases p53 levels. Furthermore, the possibility that PLD activity promotes p53 degradation was later supported in etoposide-treated HCT116 colorectal cancer cells when small interfering RNA (siRNA) knockdown of PLD1 or treatment of cells with exogenous PtdOH increased and decreased p53 levels, respectively (Jang et al., 2008b). In addition, overexpression of PLD1 and PLD2 inhibits etoposide-induced increase in p53 expression (Kwun et al., 2003; Hui et al., 2004). Similar results were observed in fibroblasts with high-intensity Raf signaling, normally prone to cell cycle arrest (Woods et al., 1997). Overexpression of either PLD1 or PLD2 in these cells led to a dramatic decrease in p21Cip1 (Joseph et al., 2002). In one study, PLD2 controlled p21Cip1 expression independently of p53, raising the possibility that the PLD1 may regulate p21Cip1 levels through p53, whereas PLD2 may regulate p21Cip1 levels through other transcription factors such as Sp1 (Kwun et al., 2003). The molecular mechanism by which PLD regulates p53 and p21Cip1 levels largely remains to be determined. However, MAPK pathways are known to regulate p53 (Wu, 2004) and inhibitors of ERK partially negate the effects of PLD overexpression on p53 degradation (Hui et al., 2004), potentially implicating a PLD-MAPK pathway.
The retinoblastoma protein (pRB) is another frequently mutated tumor suppressor protein that functions as a cell cycle restriction point guardian to allow passage from the G1 gap phase into the S phase. Accordingly, loss of pRB allows unrestricted passage from the G1 phase to the S phase. pRB normally binds E2F transcription factors that, when liberated from pRB, upregulate genes necessary for G1 to S phase transition (Dyson, 1998). The regulation of pRB is complex, but multiple phosphorylation events determine whether pRB restricts S phase entry. Before cells reach the G1 restriction point, pRB is hypophosphorylated and levels of phosphorylated pRB increase during the last few hours of the G1 phase. Hyperphosphorylated pRB then allows passage through the G1 checkpoint (Weinberg, 1995) and hyperphosphorylated pRB is associated with cell cycle progression. Cyclins of the D class regulate pRB activity (Ewen et al., 1993; Kato et al., 1993) by activating cyclin-dependent kinases, which phosphorylate and activate pRB (Tamrakar et al., 2000). Although no studies to date have directly implicated PLD in pRB activation, several putative links exist. First, when PLD1 or PLD2 is overexpressed in mouse fibroblasts, cyclin D3 levels increase and a greater population of cells enter the S phase relative to cells that do not overexpress PLD (Min, 2001). In addition, p21Cip1 is a potent inhibitor of cyclin-dependent kinases that phosphorylate pRB (Harper et al., 1993), offering another mechanism by which PLD may regulate pRB activation since PLD activity is negatively correlated with p21Cip1 expression. Finally, protein phosphatase 1 (PP1), a serine/threonine phosphatase, dephosphorylates pRB and can inhibit the G1 to S phase transition (Rubin et al., 1998). PP1 is a ubiquitously expressed phosphatase in all eukaryotic cells with a broad spectrum of functions including cell division, apoptosis, protein synthesis, metabolism, cytoskeletal reorganization, and regulation of membrane receptors (Shi, 2009). PP1 is a holoenzyme consisting of catalytic and regulatory subunits that target phosphatase specificity. PP1 activity is regulated by inhibitory proteins in vivo (Cohen, 2002) and PtdOH was shown to inhibit the catalytic subunit of PP1γ with an IC50 value of 15 nM (Jones and Hannun, 2002). The PtdOH binding site was mapped to a small stretch of amino acids between 274 and 299 and mutation of serine 292 reduced PtdOH binding by about 50% (Jones et al., 2013). Theoretically, PLD activity could inhibit PP1 leading to hyperphosphorylated pRB and G1 cell cycle arrest since PP1-induced hypophosphorylation of pRB is associated with a G1 arrest (Kwon et al., 1997) and conditions that favor pRB phosphorylation favor cell proliferation (Cobrinik et al., 1992).
E. Resisting Cell Death
1. Apoptosis.
Along the journey to malignancy, cancer cells must overcome intrinsic programmed cell death mechanisms that exist to destroy genetically unstable or cells undergoing deregulated, autonomous proliferation. DNA-damaging agents, death receptor stimulation, inhibition of oncogenic kinases, and nutrient deprivation are established stimuli of apoptotic cell death (Kelly and Strasser, 2011). As cells within a solid tumor proliferate, the nutrient supply temporarily decreases until cells can stimulate angiogenesis and restore blood flow via neovascularization. In cell culture, viability is normally compromised after serum or nutrient withdrawal. PLD activity is frequently upregulated in cultured cells after serum withdrawal (Zheng et al., 2006) and many cancer types require PLD and its product, PtdOH, for sustained survival under stress conditions (Foster and Xu, 2003). Several groups have thus proposed the idea that PLD and PtdOH provide some sort of survival signal to prevent programmed cell death. For example, when serum is withdrawn in rat fibroblasts overexpressing the proto-oncogene c-Src, cells undergo apoptosis unless PLD1 or PLD2 is co-overexpressed (Zhong et al., 2003). Interestingly, the same rat fibroblast line transformed with oncogenic v-Src has elevated basal PLD activity and does not undergo apoptosis after serum withdrawal, illustrating a correlation with PLD activity and cell survival. Established cancer cell lines also require PLD activity for survival in the serum-withdrawal paradigm. For example, when PLD activity is inhibited in T24 bladder, Calu-1 lung, 786-O renal, HCT116 colorectal, and MDA-MB-231 breast cancer cells after serum withdrawal, the established cancer lines undergo apoptosis (Zhong et al., 2003; Shi et al., 2007; Kang et al., 2008; Toschi et al., 2008). These studies used butanol, dominant-negative PLD, and PLD siRNA to inhibit PLD activity and expression. The importance of PLD in the serum-withdrawal survival pathway has since been corroborated using small-molecule PLD inhibitors in MDA-MA-231 cells (Lavieri et al., 2010).
Although the molecular mechanisms by which PLD protects against apoptotic cell death are not fully understood, several mechanisms have been proposed. The first mechanism involves upregulation of antiapoptotic proteins. B-cell lymphoma 2 (Bcl-2) is an antiapoptotic protein that prevents apoptosis by binding proapoptotic proteins, such as Bax and Bak (Tsujimoto et al., 1984; Cheng et al., 2001). Not surprisingly, overexpression of Bcl-2 is associated with apoptosis resistance in many cancers (Adams and Cory, 2007). Several cell types increase Bcl-2 expression as a function of PLD activity. PLD overexpression and exogenous PtdOH treatment in HeLa cells increases Bcl-2 transcription (Choi and Han, 2012). H19-7 rat embryonic hippocampal cells increase Bcl-2 expression when stimulated by fibroblast growth factor (FGF). Dominant-negative PLD and PLD siRNA inhibit the FGF-induced increase in Bcl-2, suggesting that PLD activity is required for Bcl-2 expression (Yoon et al., 2012). Both studies demonstrate that Bcl-2 transcription increases through the STAT3 transcription factor in a mechanism that requires MAPK activity. Therefore, Bcl-2 expression by PLD may be a byproduct of PLD-induced MAPK activation.
Another mechanism by which PLD may protect against apoptosis involves the regulation of mTOR. Several cell types undergo apoptosis when treated with the mTOR inhibitor rapamycin (Woltman et al., 2001). As mentioned previously, PtdOH is believed to bind and stimulate mTOR directly in a manner that competes with rapamycin binding (Chen et al., 2003). Along these lines, the prosurvival transcription factor c-Myc (Dang, 2012) is translated downstream of mTOR (Gera et al., 2004). Inhibition of PLD using butanol or dominant-negative PLD is associated with a decrease in c-Myc protein in MCF7 cells and it has been suggested that PLD promotes c-Myc expression through mTOR activation (Rodrik et al., 2005). In some circumstances, c-Myc overexpression can force the cell to initiate apoptosis in a manner that requires the early growth response protein 1 (Egr1) transcription factor (Sherr, 2001; Boone et al., 2011). Egr1 is a tumor suppressor involved in differentiation, proliferation, and apoptosis (Yu et al., 2007; Boyle et al., 2009) and enhances expression of the phosphatase and tensin homolog (Virolle et al., 2001). Apoptotic stimuli increase Egr1 expression in NIH3T3 and C6 glioma cells. However, PLD overexpression or treatment with exogenous PtdOH prevents Egr1 induction and subsequent apoptosis (Kim et al., 2006). These results suggest that PLD may regulate apoptosis by regulating expression of proapoptotic and antiapoptotic proteins, potentially through MAPK and mTOR pathways, although determination of the exact pathways should be the subject of future investigation.
Apoptosis initiation is a stepwise process involving mitochondrial permeabilization, cytochrome c release, and activation of several caspase proteases (Parrish et al., 2013). As such, treatment of cells with apoptotic stimuli, such as small-molecule PLD inhibitors is associated with an increase in caspase activity (Lavieri et al., 2010). PLD1 and PLD2 are targets for activated caspases and are cleaved at several sites. PLD1 is cleaved at D545 and PLD2 is cleaved at D13, D16, and D28 (Jang et al., 2008a; Riebeling et al., 2008). Although a functional significance of PLD2 cleavage is unknown, cleavage of PLD1 generates a protein fragment that inhibits endogenous PLD activity and renders cells more susceptible to apoptosis-inducing agents (Jang et al., 2008b). Likewise, expression of a caspase-resistant mutant, D545A, renders cells resistant to apoptotic stimuli (Jang et al., 2008b). These studies suggest that caspase cleavage of PLD1 leads to generation of protein fragments that enable apoptosis by inhibiting PLD activity. These studies link the intrinsic apoptotic machinery to PLD activity and further underscore the importance of PLD as a survival factor.
2. Autophagy.
Eukaryotic cells encounter a variety of environmental stressors ranging from nutrient and growth factor deprivation to chemical stressors and infectious agents. Macroautophagy (hereafter referred to as autophagy) is an evolutionarily conserved process whereby cytoplasmic constituents are enveloped in double-membrane vesicles called autophagosomes and delivered to lysosomes for degradation and nutrient recycling in times of stress (Yang and Klionsky, 2010). Autophagy is the only mechanism to degrade large bulky structures, such as organelles, and serves a housekeeping function, under nonstress conditions, to clear the cell of damaged and potentially toxic components (Rabinowitz and White, 2010). In addition to providing recycled nutrients, such as amino acids and nucleic acids, autophagy is critical for immune function (Puleston and Simon, 2014) and preventing neurodegenerative diseases such as Huntington and Parkinson diseases by clearing toxic protein aggregates (Sarkar et al., 2007; Wang and Mandelkow, 2012). The relationship between autophagy and cancer is complex and autophagy has been shown to serve tumor-suppressing and tumor-promoting roles. Mice with genetic deletions for essential autophagy genes have increased incidences of cancer, most likely due to increased genotoxic stress after buildup of damaged organelles (Edinger and Thompson, 2003). On the other hand, autophagy is required for the metabolic shift toward aerobic glycolysis in Ras-transformed fibroblasts (Kim et al., 2011b) and autophagy inhibitors decrease Ras-induced transformation (Kim et al., 2011c). Autophagy also supports tumor cell growth by clearing damaged organelles and toxic metabolites after chemotherapy treatment; thus, pharmacological modulation of autophagy is clinically important (Rubinsztein et al., 2012).
Autophagy is a multistage process coordinated by distinct protein complexes. In yeast, approximately 30 autophagy-related genes control the various autophagic stages, which include initiation, elongation, and maturation. Approximately one-half of these genes are conserved in mammals (Xie and Klionsky, 2007). Autophagosome initiation begins with the budding of a preautophagosomal structure (PAS) from a membrane within the cell. Although there is debate as to the membrane of origin, autophagosomes may contain membrane components from the endoplasmic reticulum (Axe et al., 2008), mitochondria (Hailey et al., 2010), and plasma membrane (Ravikumar et al., 2010). PAS formation requires recruitment of several proteins including Vps34, Vps15, Atg14L, and beclin1 (Itakura et al., 2008). Atg14L targets the complex to PAS structures (Matsunaga et al., 2010) where beclin1 can stimulate the activity of the class III phosphatidylinositol 3-kinase Vps34 to increase local concentrations of PI(3)P (Liang et al., 1999; Kihara et al., 2001). Increased concentrations of PI(3)P contribute to the negative curvature of the PAS isolation membrane and recruit proteins containing PI(3)P-binding domains, such as PX domains (Knævelsrud and Simonsen, 2012). Vps34 activity results in the formation of another complex containing Atg5, Atg12, and Atg16 to promote the elongation phase. Atg5 and Atg12 are conjugated to each other through the actions of Atg7 and Atg10, which function similarly to E1 and E2 ubiquitin-activating and ubiquitin-carrier proteins, respectively (Mizushima et al., 1998; Shintani et al., 1999). As the nascent autophagosome elongates, Atg7, Atg3, and Atg5–Atg12 function as E1, E2, and E3 ubiquitin-like conjugation proteins to covalently attach PE to microtubule-associated protein/light chain 3 (LC3) to facilitate LC3 attachment to autophagosomal membranes (Hanada et al., 2007; Noda et al., 2011). Once LC3 attaches to the autophagosome, the Atg5/Atg12/Atg16 complex dissociates and LC3 assists in the final fusion of the PAS membranes into an autophagosome containing the engulfed cytoplasmic constituents (Nakatogawa et al., 2007). As autophagosomes mature, they fuse with endosomes to create amphisomes before ultimately fusing with lysosomes. The maturation process is less understood, but is partially mediated by the formation of beclin1 complexes distinct from those formed during autophagosome initiation (Matsunaga et al., 2009; Zhong et al., 2009) and by components of the endosomal and lysosomal membranes such as Rab7 and lysosome-associated membrane glycoprotein 2 (Eskelinen, 2005).
mTOR is a classic negative regulator of autophagy and inhibits autophagosome formation by directly inhibiting components of the autophagosome initiation complex (Chang et al., 2009; Jung et al., 2010). When nutrients are widely available, mTOR inhibits autophagosome initiation by directly regulating components of the autophagy initiation complex. The formation of the Vsp34/Beclin1 complex and the subsequent autophagosome initiation is subject to regulation by proteins such as Atg1/Unc-51–like kinase (ULK), another key component of the autophagosome initiation complex (Mizushima, 2010). When nutrients are abundant, mTOR phosphorylates and binds the ULK complex, preventing autophagosome initiation (Ganley et al., 2009; Hosokawa et al., 2009). When nutrients become limiting or cells are treated with rapamycin, mTOR activity decreases and dissociates from the ULK complex, enabling autophagosome formation. ULK is also regulated by the AMPK when AMP levels increase due to metabolic demand (Hardie, 2007; Kim et al., 2011a). In addition to mTOR and AMPK, PLD is emerging as an important, multifaceted regulator of autophagy. Vps34 directly regulates PLD1 localization and activity in response to amino acid availability or deprivation (Xu et al., 2011; Yoon et al., 2011). PLD binds to PI(3)P-containing membranes via the PX domain where it colocalizes with mTOR and promotes mTOR activity in the presence of amino acids. Amino acid deprivation decreases PLD activity (Yoon et al., 2011) and could trigger an increase in autophagosome biogenesis through decreasing mTOR activity. As such, PLD KO mice produce fewer autophagosomes in liver slices and in embryonic fibroblasts after autophagic stimuli compared with wild-type littermates (Dall'Armi et al., 2010). Furthermore, Arf6, which is known to localize to the plasma and endosomal membranes (Donaldson, 2003; D'Souza-Schorey and Chavrier, 2006), stimulates autophagosome biogenesis from plasma membranes in a manner that requires PLD activity (Moreau et al., 2012). These observations were not directly correlated with mTOR and follow-up studies suggest that PLD may independently regulate autophagy. In cells derived from polycystic kidney disease, which show upregulated mTOR activity, PLD inhibitor treatment results in a large increase in autophagosome numbers. However, rapamycin treatment of the same cells does not increase autophagosome numbers and suggests that PLD may be uncoupled from mTOR in these cells (Liu et al., 2013). Increased autophagosome numbers can result from increased biogenesis or decreased degradation, and the investigators of this study did not perform the experiments to make the distinction.
Agents that inhibit lysosomal acidification prevent autophagosome fusion and ultimate degradation. By pretreating cells with lysosomal proton pump inhibitors, such as bafilomycin A1, investigators can clamp autophagosome degradation and determine whether a compound increases autophagosome biogenesis or inhibits autophagic flux (Yamamoto et al., 1998b). PLD inhibitor treatments induce a robust increase in autophagosome numbers in glioblastoma cells, but do not increase autophagosome numbers more than bafilomycin treatment alone (Bruntz et al., 2014). These results suggest that PLD promotes autophagosome flux and degradation. Similarly, when HeLa cells are cultured in balanced saline solutions lacking amino acids, PLD1 localizes with LC3-containing, late endosomal/lysosomal structures, but not PAS membranes, suggesting that PLD promotes the later stages of autophagy as opposed to the initiation stages (Dall'Armi et al., 2010). Furthermore, PLD inhibitors increase levels of autophagy substrates in both nutrient-deprived Chinese hamster ovary cells (Dall'Armi et al., 2010) and serum-deprived glioblastoma cells (Bruntz et al., 2014), consistent with a blockade in autophagosome degradation. Beclin1 promotes autophagosome maturation and degradation by interacting with a variety of partners throughout the process. Rubicon is a Beclin1-interacting protein that inhibits autophagosome maturation by inhibiting Vps34 lipid kinase activity (Matsunaga et al., 2009; Zhong et al., 2009; Sun et al., 2011). Akt phosphorylates Beclin1 and promotes autophagic flux by preventing the interaction with Rubicon (Bruntz et al., 2014). PLD promotes Akt activity in glioblastoma cells and promotes autophagic flux by preventing the binding of Rubicon to Beclin1 (Bruntz et al., 2014). This PLD2-Akt–mediated access to autophagy via modulation of the Beclin1-Rubicon complex may be essential to cell survival in human glioblastomas. This signaling axis appears to become a pathway of central importance under conditions of metabolic stress.
Although most of the current data are consistent with PLD promoting autophagic flux, PLD may function to promote autophagosome biogenesis in some systems. For example, the decrease in autophagosome numbers in PLD1 KO mice is consistent with a role of PLD in biogenesis and differs from the effects seen with PLD inhibitors (Dall'Armi et al., 2010; Liu et al., 2013; Bruntz et al., 2014). These differences may be explained by the various treatment paradigms themselves or by differences in the cell lines or tissues under investigation. Another likely possibility is that the PLD protein provides a nonlipase function that is required for autophagosome biogenesis. In addition, data obtained from KO animals may expose the nonlipase functions that small-molecule inhibitors may obscure. PLD may also promote autophagy through production of other lipids, such as DAG. Autophagy is upregulated when pathogens such as Salmonella typhimurium infect cells. PLD-derived DAG was recently shown to be required for autophagy after infection (Shahnazari et al., 2010). Since PLD was only recently discovered to modulate autophagy (Liu et al., 2009; Dall’Armi et al., 2010; Moreau et al., 2012; Bae et al., 2014; Bruntz et al., 2014), future studies are needed clarify the detailed mechanism of autophagy regulation by PLD. It will also be important to ascertain whether other PtdOH-generating pathways (see Fig. 6) are able to compensate for blocks of PLD activity by supplying alternate sources of PtdOH to critical signaling nodes such as Akt or mTORC1. It will also be important to assess whether such ability to compensate differs between more acute treatments, such as small-molecule inhibitors and primary alcohol-mediated transphosphatidylation, or sustained knockdowns of gene products using RNAi.
Fig. 6.
PtdOH biosynthesis and metabolism. PtdOH is an intermediate in de novo triacylglycerol (TAG) biosynthetic pathway (i.e., the Kennedy pathway). It starts with glycerol-3-phosphate produced in the cell either via glycerol phosphorylation by glycerol kinase (GK) or from glucose via dihydroxyacetone phosphate and glycerol-3-phosphate dehydrogenase (G3P dehydrogenase). Glycerol-3-phosphate is sequentially acylated first at the sn-1 position by the enzyme glycerol-3-phosphate acyltransferase (GPAT; there are four known isoforms) using Acyl-CoA as a fatty acid donor to form LPA. This reaction is reversible and is catalyzed by PLA. LPA can be dephosphorylated to monoacylglycerol via lipid phosphate phosphatase (LPP), or as part of the Kennedy pathway, is further acylated at the sn-2 position again using Acyl-CoA as the fatty acid donor and carried out by 1-acylglycerol-3-phosphate acyltransferase, also known as lysophosphatidic acid acyltransferase (LPAAT) to generate PtdOH. This reaction is also reversible and catalyzed by LPA. PtdOH and DAG are “partners” in a synthetic route involving LPP also known as lipin or phosphatidic acid phosphatase, to generate DAG, which could be converted into PtdOH by diacylglycerol kinase (DGK; 10 known isoforms). Alternatively, DAG can be synthesized via acylation of monoacylglycerol by monoacylglycerol acyltransferase (MGAT) (also a reversible reaction). The final step is DAG esterification by diacylglycerol acyltransferase (DGAT; two known isoforms) to triacylglycerol. The reverse reaction is carried out by adipose triacylglycerol lipase (ATGL). PtdOH is also a product of PC hydrolysis catalyzed by PLD (two known isoforms). The remodeling pathway (Lands’ cycle) depends on the coordinated actions of PLA2 and lysophospholipid acyltransferases (LPLATs). Thus, PC can be hydrolyzed by PLA2 to form LPC, which can feed back into LPA via autotaxin (ATX)–carried out hydrolysis. Conversely, LPC can be reacylated back into PC with different fatty acyl substituents, supplied by Acyl-CoA and the action of lysophosphatidylcholine acyltransferase (LPCAT).
F. Activating Invasion and Metastasis
Once a tumor reaches a critical size, nutrient availability and space become limiting factors for continued tumor growth. The solution usually involves invasion of surrounding tissues and metastasis to distant sites where nutrients and space are not initially limiting (Hanahan and Weinberg, 2000). Invasion is a multistep process involving dissolution of the extracellular matrix (ECM) and surrounding tissue by protease secretion, detachment from neighboring cells and ECM, and finally cellular migration into the surrounding tissue. Many studies have implicated PLD in the promotion of cancer cell invasion. In early studies investigating invasive properties of human small-cell lung cancer cells, a proinvasion role for PLD was suggested after observations that exogenously added bacterial PLD or PtdOH dramatically stimulated lung cancer cell invasion in vitro (Imamura et al., 1993). Later studies revealed a positive correlation between PLD activity and invasive potential. Overexpression of PLD in breast, glioblastoma, or lymphoma cells stimulates invasion (Zheng et al., 2006; Knoepp et al., 2008; Park et al., 2009), whereas expression of dominant-negative PLD prevents invasion (Zheng et al., 2006). Similarly, small-molecule PLD inhibitors and PLD siRNA decrease breast cancer cell invasion and further implicate the importance of PLD in invasive processes (Scott et al., 2009).
Prior to leaving their tissues of origin, cancer cells must degrade ECM components and surrounding tissues to provide a path for migration and intravasation. This process is usually mediated by production and secretion of collagenases/gelatinases termed matrix metalloproteinases (MMPs) (Rao, 2003). MMP activity is the best predictor of invasiveness in some cancers, such as glioblastoma (Wild-Bode et al., 2001), and PLD activity is highly correlated with increased MMP activity in many cancer types. Simulation of HT1080 fibrosarcoma cells with activators of PLD, such as laminin (Reich et al., 1995) or PMA (Williger et al., 1999) resulted in increased MMP2 and MMP9 secretion. By using primary alcohols and exogenously added PtdOH to decrease and increase MMP secretion, respectively, the investigators established a role for PLD in MMP secretion (Reich et al., 1995; Williger et al., 1999). Since these original studies, the requirement of PLD in MMP secretion has been established for multiple cell types including melanoma (Kato et al., 2005), colorectal (Kang et al., 2008), glioblastoma (Park et al., 2009), and breast (Kang et al., 2011) cancer cells. These studies used a combination of primary alcohol, dominant-negative PLDs, PLD siRNA, and small-molecule PLD inhibitors to block MMP secretion from cancer cells, firmly supporting the requirement for PLD activity. The most widely accepted mechanism for PLD regulation of MMP secretion is through a transcription-dependent mechanism. A Ras-MAPK–dependent pathway likely mediates MMP transcription because dominant-negative constructs or inhibitors of multiple proteins within the MAPK pathway block MMP secretion in response to stimuli such as PDGF, EGF, and PMA. Nuclear factor κ-light chain enhancer of activated B cells is a transcription factor downstream of ERK (Kurland et al., 2003) and has been named as the ERK-dependent transcription factor responsible for inducing MMP2 and MMP9 transcription (Kato et al., 2005; Kang et al., 2008, 2011; Park et al., 2009).
Once the surrounding tissue environment has been appropriately remodeled to permit tumor cell escape, the cells must detach from their surrounding cells and ECM before beginning the process of migration. In epithelial cells, this process is usually termed the epithelial–mesenchymal transition (EMT) in which cells lose polarity and cell–cell adhesion and transition into migratory mesenchymal-like cells (Hanahan and Weinberg, 2000). During EMT, cells reorganize adhesion proteins that promote interaction with the ECM. E-cadherin is a cell surface protein that couples to the actin cytoskeleton, regulates adhesion-dependent signaling, and interacts with other cells to transmit antigrowth signals (De Craene and Berx, 2013). Downregulation of E-cadherin is one the earliest steps in EMT, usually achieved through transcriptional regulation by a variety of pathways. Growth factors, such as EGF, FGF, and PDGF, stimulate EMT through a Ras-MAPK pathway leading to activation of transcription factors, such as SLUG, which represses E-cadherin and promotes expression of EMT genes (Savagner et al., 1997; Bolós et al., 2003; Yang and Weinberg, 2008). Because PLD is intimately involved in RTK/Ras/MAPK signaling, it stands to reason that PLD may also regulate EMT and loss of cell adhesion. Indeed, a positive correlation between PLD activity and loss of E-cadherin exists in certain cells. Wounded corneal epithelial cells undergo an EMT as cells migrate at the leading edges of the wound. Treatment with exogenous PtdOH or overexpression of PLD2 caused a dramatic reduction of cell surface E-cadherin in these cells (Mazie et al., 2006). However, butanol did not decrease EGF stimulation of ERK in this system, suggesting that PLD may regulate E-cadherin through different means. In addition to the Ras-ERK pathway, the Wnt/β-catenin pathway also promotes EMT (Yang and Weinberg, 2008; Sánchez-Tilló et al., 2011). The Wnt pathway is critical in tissue development and is frequently mutated in many cancers (Kikuchi, 2003). Recent studies have shown that PLD1 is a transcriptional product of Wnt activation and participates in a feed-forward mechanism in which PLD activity is required for transcription of a number of Wnt-responsive genes (Kang et al., 2010). Although EMT gene expression was not measured in the study, PLD may potentially regulate EMT and loss of adhesion through Wnt. Small-molecule PLD inhibitors block Wnt-mediated transformation of NIH3T3 cells and underscore the importance of PLD in Wnt-signal transduction (Kang et al., 2010). Whether PLD directly participates in EMT and loss of cell–cell and ECM adhesion remains a point of conjecture.
After ECM remodeling and loss of adhesion, invading cells must migrate into surrounding tissues. Migration events occur under normal conditions such as the chemotaxis of cells toward chemical stimuli during infection and development. The process of migration requires significant cytoskeletal rearrangements and PLD is an important regulator of these events. PLD activity is required for neutrophil migration toward chemotactic peptides and cytokines, such as fMLP and interleukin-8 (Lehman et al., 2006; Carrigan et al., 2007), and for macrophage migration toward colony stimulating factor 1 (Knapek et al., 2010). Cancer cells can secrete chemoattractants to recruit immune cells into tumors where they produce MMP and other factors that promote invasion (Condeelis and Pollard, 2006). In addition, PLD activity is required for fibroblast migration toward LPA (Pilquil et al., 2006) and endothelial cell migration toward S1P (Gorshkova et al., 2008). Like nontransformed cells, cancer cells, such as MDA-MB-231, also require PLD activity for migration (Zheng et al., 2006; Scott et al., 2009). Cellular migration is a multistep process consisting of cycles of cells protrusion, attachment to the ECM, and retraction. The first step of the cycle requires polarization of the cell into leading and retracting edges with the development of protrusions, such as lamellipodia or filopodia, at the leading edges. These protrusions must then be stabilized by formation of focal adhesions to the surrounding matrix, and finally, the trailing edge of the cell must detach and contract to move the cell forward (Etienne-Manneville, 2004; Ananthakrishnan and Ehrlicher, 2007). PLD and PtdOH have been linked to each step of the process.
PLD enzymes, especially PLD2, are frequently localized to the leading edge of motile cells in membrane ruffles and lamellipodia, depending on the cell type (Colley et al., 1997b; O'Luanaigh et al., 2002; Nagasaki et al., 2008). PLD activity is required for leading edge formation (Santy and Casanova, 2001) and overexpression of PLD promotes leading edge characteristics (Shen et al., 2002). Actin-rich leading edge formation requires a series of cytoskeletal rearrangements coordinated by members of the Rho family such as RhoA, Cdc42, and Rac (Ridley et al., 2003) along with other proteins, such as nonreceptor tyrosine kinases (NRTKs) of the Src family (Kanda et al., 2007). Rac proteins appear to control lamellipodia formation, whereas Cdc42 controls filopodia formation (Etienne-Manneville, 2004). As discussed previously, the RhoA family members stimulate PLD1 activity by a direct protein–protein interaction. Several studies have proposed mechanisms in which Arf6 and Rac1 converge on PLD1 to promote PtdOH production and membrane ruffling in epithelial cells and mast cells (Santy and Casanova, 2001; Powner et al., 2002). However, the PLD2 isoform is perhaps more important for cell migration and an intriguing mechanism of Rac regulation by PLD2 is emerging.
In endothelial cells, Rac activation appears to require PLD2 activity because RNAi silencing of PLD2 decreases Rac activation and cell migration in response to S1P (Gorshkova et al., 2008). The PH domain of PLD2 encodes a putative Cdc42/Rac interactive binding domain that directly interacts with Rac proteins, located between amino acids 255 and 269 (Mahankali et al., 2011; Peng et al., 2011). In vitro, purified PLD2 potently stimulates GDP-GTP exchange on Rac2 and silencing of PLD2 leads to decreased Rac2 activation and chemotaxis in neutrophils (Mahankali et al., 2011). PLD2 also stimulates GEF activity on Rac1, although the rate of PLD2-catalyzed GDP-GTP exchange for Rac1 is less than for Rac2 (Henkels et al., 2013b). Although these studies describe a lipase-independent mechanism of Rac activation by PLD2, other studies suggest that PLD catalytic activity is required for Rac activation (Gorshkova et al., 2008) and several lipase-dependent mechanisms have been described. In one mechanism, PtdOH directly binds the C-terminal polybasic motif of Rac1 and promotes membrane translocation (Chae et al., 2008). In the description of the PLD2-GEF activity, inclusion of PtdOH in the reaction mixture increases GEF activity (Mahankali et al., 2011), suggesting a putative mechanism in which PtdOH recruits Rac1 binding to the membrane and the combination of PtdOH and PLD2-GEF activity stimulates Rac activation. Another mechanism for PtdOH-mediated Rac activation involves direct modulation of the Rac GEF, dedicator of cytokinesis 2 (DOCK2). DOCK2 contains a DOCK homology region that binds PIP3 and controls membrane localization. Full recruitment of DOCK2 to leading edges of migrating cells was shown to require PtdOH in addition to PIP3 and the PtdOH binding site was narrowed to a polybasic amino acid stretch in the C terminus (Nishikimi et al., 2009). DOCK2 translocation required PtdOH generated by PLD because the expression of catalytically inactive PLD, treatment with butanol, and treatment with small-molecule PLD inhibitors all reduced DOCK2 accumulation at the leading edges of neutrophils undergoing chemotaxis (Nishikimi et al., 2009). In nonactivated neutrophils, Rac is sequestered by GDIs. Upon activation, Rac dissociates and allows GEFs to stimulate GDP-GTP exchange leading to membrane ruffling. In vitro, PtdOH and other lipids such as arachidonic acid and PIPns stimulate dissociation of Rac from the Rac-GDI (Chuang et al., 1993). The number of mechanisms by which PLD and PtdOH stimulate Rac activity certainly highlights Rac as an important mediator of PLD-mediated leading edge formation.
Just beneath the plasma membrane lies a region rich in cortical actin known as the cortex. During leading edge formation, the cortical cytoskeleton undergoes a series of events that requires de novo synthesis of F-actin filaments, which provide structure and force to the protruding end of the cell (Small et al., 1999). A complex network of actin-binding proteins severs, caps, and nucleates actin monomers to promote formation of F-actin filaments. Cytoskeletal proteins, such as actin (Lee et al., 2001; Kusner et al., 2002), and actin-binding proteins, such as fodrin (Lukowski et al., 1996), spectrin (Lukowski et al., 1998), α-actinin (Park et al., 2000), and gelsolin (Banno et al., 1999), directly regulate PLD activity. Cortactin is an actin-binding protein that promotes nucleation and branching of actin filaments (Ammer and Weed, 2008). Cortactin is subject to regulation by NRTKs and phosphorylated cortactin promotes polymerization of actin filaments (Wu et al., 1991; Kim and Wong, 1998). In pulmonary epithelial cells, Src phosphorylation of cortactin requires PLD activity because PLD siRNA prevented Src and cortactin redistribution to the cell periphery (Usatyuk et al., 2009). In addition to Src, PLD has been shown to regulate the activity of Fer. The NRTK Fer is a cytosolic protein implicated in cell adhesion and cell migration (Greer, 2002). Cortactin is the best-characterized substrate of Fer and this phosphorylation is critical for fibroblast migration (Sangrar et al., 2007). PtdOH was recently shown to bind and activate Fer by binding to R417 (Itoh et al., 2009). Activation of Fer was inhibited by butanol but not DGK inhibitors, suggesting that PLD-produced PtdOH was critical for Fer activation. Migration of rat kidney epithelial cells was inhibited by butanol or by genetic knockdown of PLD1/PLD2. Knockdown of Fer in addition to PLD1/PLD2 did not result in further reduction of migration, suggesting that PLD is a critical upstream regulator of Fer and cortactin phosphorylation (Itoh et al., 2009). Fes is an NRTK that shares structural similarity to Fer and is also implicated in cell migration (Kanda et al., 2007). PLD2 interacts with Fes and overexpression of PLD2 increases Fes activity (Di Fulvio et al., 2012). Treatment of cells with exogenous PtdOH stimulates Fes activity, possibly through a direct interaction, based on sequence homology to Fer kinase (Ye et al., 2013). In addition to NRTKs, other upstream kinases, such as PAKs and MAPKs, can activate cortactin (Campbell et al., 1999; Webb et al., 2006).
The PAKs are serine/threonine protein kinases that are activated by the small G proteins Cdc42 and Rac. PAKs mediate many intracellular functions including cytoskeleton rearrangement (Sells and Chernoff, 1997), stress signaling through the p38/c-Jun N-terminal kinase pathways (Zhang et al., 1995), and regulation of NADPH oxidase activity by direct phosphorylation of p47phox (Knaus et al., 1995). Although the PAKs were originally identified based on the ability of Cdc42/Rac to stimulate autophosphorylation and kinase activity, sphingosine and PtdOH also potently stimulate PAK1 in vitro (Bokoch et al., 1998). The activation of PAK1 by Cdc42/Rac and sphingosine or PtdOH is not additive or synergistic, suggesting a common binding site on PAK1. In vivo, PAK1 activity is inhibited following butanol treatment, which suggests that PLD-derived PtdOH regulates PAK1 activity (Chae et al., 2008). Along with Src, Fer, and MAPK, PAK1 provides another potential downstream effector of PLD capable of activating cortactin.
One the ways in which cortactin promotes actin nucleation is to bind and activate the actin-related protein complex 2/3, which binds to a preexisting actin filament and nucleates branching of a new filament (Weed et al., 2000; Ammer and Weed, 2008). The Wiskott–Aldrich syndrome protein (WASp) family of proteins cooperates with cortactin to activate Arf2/3 (Mizutani et al., 2002; Martinez-Quiles et al., 2004). As mentioned previously, PLD2 forms a complex with the Grb2 adaptor protein (Di Fulvio et al., 2006). WASp forms a complex with Grb2 and PLD2 and this protein complex promotes the formation of phagocytic cups in macrophages, a process requiring similar actin rearrangements to lamellipodia formation (Kantonen et al., 2011). When the Grb2 site on PLD2 is mutated, the complex fails to form properly and phagocytosis is decreased. Although a direct relationship between the PLD2/Grb2/WASp complex and cancer cell migration remains to be established, PLD is poised as a central regulator of several proteins that cooperate to promote cortical actin polymerization.
As cell protrusions extend forward, focal adhesions are formed that serve as mechanical anchors to the ECM and provide contractile forces that propel the cell forward. These adhesions are mediated in large part by the integrin family of proteins that bind ECM components and relay information about the cellular environment to intracellular components (Hood and Cheresh, 2002). Integrins attach to the cytoskeleton through a complex set of adaptor proteins including vinculin (Geiger, 1979) and talin (Burridge and Connell, 1983) and induce F-actin polymerization. Integrins are known to stimulate PLD and the integrin-induced increase in F-actin requires PLD activity. As such, butanol treatment or knockdown of PLD is associated with defects in the ability to form adhesions (Aguirre Ghiso et al., 1997; Iyer et al., 2006). The speed of migration is largely controlled by the speed at which cells form and detach from these adhesions (Lauffenburger and Horwitz, 1996) and PLD regulates migration speed in wound-healing assays (Mazie et al., 2006). PLD activity is associated with an increase in vinculin levels (Mazie et al., 2006) as well as activation of focal adhesion kinase, a key regulator of focal adhesion turnover (Llić et al., 1995; Knoepp et al., 2008). Mechanistically, PLD can regulate adhesion dynamics by regulating the activity of integrin adaptor proteins. PIP2 binding to the adaptor protein talin enables bindings to the integrin β subunit and changes integrin conformation to a high-affinity state for ECM components (Calderwood et al., 2002; García-Alvarez et al., 2003; Tadokoro et al., 2003). PLD can enhance binding of talin to integrins through regulation of PIP2 levels by activating PIP5K. Early investigations revealed that PtdOH stimulated PIP5K activity from bovine brain membranes (Moritz et al., 1992) and later investigations revealed that only the class I PIP5Ks are stimulated by PtdOH (Jenkins et al., 1994). In vivo regulation of PIP5K by PtdOH has been documented as a significant decrease in PIP2 levels in lysosomal membranes following butanol treatment (Arneson et al., 1999) and a decrease in PIP5K-dependent actin stress fiber formation after PLD inhibition in murine fibroblasts (Jarquin-Pardo et al., 2007). Recent studies demonstrated reduced PIP5K activity after treatment with small-molecule PLD inhibitors (Roach et al., 2012), further underscoring the importance of PtdOH in PIP5K regulation. In mast cells, PLD inhibition was associated with decreased PIP2 levels and reduced binding of talin to integrins (Powner et al., 2005). Thus, by regulating PIP5K and PIP2 levels, PLD is directly able to control cell adhesion.
After protruding forward and forming focal adhesions, the retracting edge of a migrating cell must detach from the ECM and contract. This process is assisted by the formation of actin stress fibers in some systems (Kovac et al., 2013), which connect to focal adhesions (Geiger et al., 2009; Parsons et al., 2010). Stress fibers are bundles of actin filaments that provide contractile force for actively moving cells (Pellegrin and Mellor, 2007). Agents that elevate PLD activity frequently result in stress fiber formation. These include thrombin stimulation of fibroblasts (Ha and Exton, 1993), LPA stimulation of porcine aortic endothelial cells and rat fibroblasts (Cross et al., 1996; Kam and Exton, 2001), and S1P stimulation of human airway epithelial cells (Porcelli et al., 2002). In these cases, inhibition of PLD activity with butanol or dominant-negative PLD expression prevented the agonist-mediated stress fiber formation and exogenous PtdOH-stimulated stress fiber formation in the absence of agonist. Overexpression of PLD is able to induce stress fiber formation in L6 myoblasts (Komati et al., 2005) and expression of dominant-negative PLD prevents LPA-stimulated stress fiber formation in fibroblasts (Kam and Exton, 2001). RhoA is a master regulator of stress fiber formation in many cells (Mackay and Hall, 1998; Kaibuchi et al., 1999) and RhoA-stimulated PLD activity has been implicated in stress fiber formation. The observations that butanol and dominant-negative PLD inhibit stress fiber formation and that exogenous PtdOH stimulates stress fiber formation strongly implicate a PtdOH-dependent mechanism for stress fiber formation. However, the PX domain of PLD2 was recently shown to stimulate GDP-GTP exchange for RhoA directly, offering a lipase-independent mechanism for stress fiber formation (Jeon et al., 2011). In this study, overexpression of the isolated PX domain was sufficient to trigger stress fiber formation. Therefore, PLD appears to regulate stress fiber formation through lipase-dependent and -independent mechanisms.
G. Inducing Angiogenesis
Angiogenesis is the process of sprouting new blood vessels from existing vessels and is required to provide nutrients, remove metabolic waste, and promote metastasis. In this process, vascular endothelial cells are either recruited into the tumor mass or undifferentiated tumor stem cells already present undergo differentiation into epithelial-like cells to contribute to the formation of new blood vessels (Hanahan and Folkman, 1996; Wang et al., 2010b). Initiation of angiogenesis usually begins with secretion of pro-angiogenic factors that interact with cell surface receptors on endothelial cells and stimulate migration into the tumor (Colville-Nash and Willoughby, 1997; Ferrara and Alitalo, 1999). Many of these factors, such as vascular endothelial growth factor (VEGF) (Seymour et al., 1996), EGF, FGF, hepatocyte growth factor (Adachi et al., 1996), and interleukin-8 (Sozzani et al., 1994), stimulate PLD activity, and PLD activity is required for VEGF-induced angiogenesis (Zhang et al., 2011). In zebrafish, PLD activity is required for angiogenesis during embryonic development of vasculature (Zeng et al., 2009), further emphasizing the importance of PLD in the blood vessel development.
PLD may contribute to angiogenesis in several ways. As a downstream effector of angiogenic growth factors, PLD and PtdOH may mediate growth factor signaling. Cells with elevated Raf signals demonstrate elevated VEGF expression (Akula et al., 2005). Similarly, oncogenic Ras can upregulate VEGF expression (Rak et al., 1995) and PLD may contribute to the Ras-MAPK signaling cascades in these cells. Latent forms of VEGF can be sequestered in the ECM and become bioavailable after proteolytic processing by proteases, such as MMP9 (Bergers et al., 2000; Kessenbrock et al., 2010). As such, PLD regulation of MMP9 activity may directly contribute to VEGF secretion.
Angiogenesis requires migration of endothelial cells, and PLD likely regulates the migratory processes required during angiogenesis. When mouse melanoma or lung cancer cells were implanted into wild-type or PLD1 KO mice, tumors in PLD1 KO mice showed a much lower density of microvascular cells (Chen et al., 2012). When VEGF-coated matrigel plugs were inserted into the same mice, endothelial cells failed to migrate to the plugs in the PLD1 KO mice, suggesting inherent defects in the migration of PLD1 KO-derived endothelial cells. Consistent with this observation, PLD1 KO mice showed impaired integrin signaling as manifested by a failure to properly adhere to ECM integrin ligands, such as fibronectin, vitronectin, and collagen (Elvers et al., 2010; Chen et al., 2012). Therefore, the role of PLD in angiogenesis is most likely to mediate endothelial cell migration.
H. Deregulating Cellular Energetics
In the 1920s, the German physiologist Otto Warburg pioneered the study of cancer metabolism by characterizing fundamental differences in glucose utilization between tumors and normal tissue. These differences included avid glucose consumption and lactate production by tumor cells resulting in increased ATP production via the less efficient glycolysis pathways compared with the highly efficient mitochondrial oxidative phosphorylation pathways, even in the presence of ample oxygen (Warburg, 1956). This surprising discovery, termed the “Warburg effect,” violated the established ideas of the Pasteur effect in which O2 was known to suppress glucose consumption and glycolysis (Krebs, 1972). Subsequent studies have confirmed and broadened Warburg’s initial discoveries and most cancer cells today have altered metabolic profiles consistent with the Warburg effect (Hanahan and Weinberg, 2011). When tumors outgrow the diffusion limits of nutrients and oxygen from the blood supply, a metabolic shift occurs in which glycolytic gene expression increases along with increases in cell surface expression of glucose transporters. Thus, oxygen dependence is reduced compared with the more vascularized surrounding tissue. This glycolytic shift is advantageous for the cancer cell because it allows for growth and survival in the tumor microenvironment (Hsu and Sabatini, 2008). Proliferating cells must accumulate biomass such as lipids and ribosomes, and the high rate of glucose and glutamine (DeBerardinis et al., 2007) uptake provides anabolic carbons for pathways such as the pentose phosphate pathway and the Krebs cycle (Vander Heiden et al., 2009). Therefore, deregulated cellular energetics is emerging as a fundamental process in tumorigenesis.
The molecular mechanisms behind the Warburg effect are not entirely characterized, but several key components have been identified including established oncogenes. For example, expression of oncogenic Ras or Myc results in an upregulation of glucose transporter activity and glycolytic gene expression (Dang and Semenza, 1999; Osthus et al., 2000; Ramanathan et al., 2005; Ahuja et al., 2010). Activation of the PI3K/Akt pathway stimulates glucose uptake and a shift toward aerobic glycolysis by regulating the activities of multiple glycolytic enzymes (Deprez et al., 1997; Elstrom et al., 2004; Majewski et al., 2004). Because PLD is a signaling component in these pathways, the aerobic glycolysis induced by Ras, Myc, or PI3K may require PLD activity. As such, PLD can participate in the development of the Warburg phenotype through several different mechanisms. PLD1 is phosphorylated at S505 and activated by AMPK, a protein responsible for sensing and responding to changes in cellular energy status (Hardie, 2007; Kim et al., 2010). Glucose deprivation stimulates PLD1 activity via AMPK activation (Kim et al., 2010), suggesting that PLD may be one of the early responders to energetic perturbations as seen in a growing tumor. Glycolytic enzymes such as aldolase and glyceraldehyde-3-phosphate dehydrogenase also directly regulate PLD activity, supporting the idea that PLD may serve as sensor of cellular energy homeostasis (Kim et al., 2002, 2003a). The downstream effect of AMPK-mediated PLD activation is an increase in glucose transporters at the plasma membrane (Kim et al., 2010) to facilitate glucose uptake. Likewise, several studies have shown a requirement for PLD activity for glucose transporter translocation to the plasma membrane after insulin stimulation (Bandyopadhyay et al., 2001; Sajan et al., 2002; Huang et al., 2005). PtdOH is a key component of the glucose transporter vesicular membrane and the authors demonstrated that PtdOH contributes to the late-stage fusion events. The role of PLD in exocytic events is well established in neuroendocrine cells (Vitale et al., 2001; Zeniou-Meyer et al., 2008) and similar mechanisms may be at play for glucose transporter vesicle fusion. Besides directly contributing to the biophysics of glucose transporter translocation, PLD also controls expression of glycolytic genes.
The hypoxia-inducible factors (HIFs) are transcription factors that control expression of many glycolytic enzymes (Kaelin and Ratcliffe, 2008). HIF regulation is complex and involves several upstream components. Prolyl hydroxylases sense O2 and hydroxylate residues on the α subunit of HIF in the presence of O2 (Dann and Bruick, 2005). Ubiquitin is then coupled to these modified residues by the Von Hippel–Lindau (VHL) E3 ubiquitin ligase and HIF is targeted for proteasomal destruction (Schofield and Ratcliffe, 2005). Thus, loss of the VHL tumor suppressor leads to constitutively high HIF expression resulting in upregulation of glycolytic gene transcription seen in many cancers (Zhong et al., 1999). For example, 786-O renal carcinoma cells are VHL null and have constitutively high HIF levels. Butanol, PLD siRNA, and small-molecule inhibitors suppress HIF expression in these cells (Garcia et al., 2008; Toschi et al., 2008). After restoring VHL function in these cells, HIF expression decreases and can be induced again by treatment with hypoxia mimetic compounds. PLD inhibition blocks the hypoxia mimetic-induced HIF expression and the authors attribute these results to PLD regulating HIF expression through mTOR (Toschi et al., 2008). However, PLD may serve an mTOR-independent role as a general sensor of cellular bioenergetics.
VIII. Phospholipase D Small-Molecule Inhibitors
For many years, the investigation of PLD-mediated functions relied on biochemical and genetic approaches such as overexpression of catalytically active or inactive forms of either PLD1 or PLD2 in vivo, or employed RNAi for the individual isoforms in an effort to discern discrete roles for PLD1 and PLD2. Undoubtedly the use of primary alcohols has led to some dubious findings regarding PLD functionality. To assess the therapeutic potential of the inhibition of PLD1, PLD2, and/or both isoforms, the implications from biochemical and genetic studies must be verified with small-molecule probes, suitable for further development into therapeutics. Until 2007, small molecules employed to study PLD function (Fig. 3) were either indirect inhibitors, such as resveratrol and honokiol (compounds 1 and 2, respectively); direct inhibitors, such as tungstate (compound 3); lipid mimetics such as (Z)-PSDP, the (Z)-isomer of presqualene diphosphate (compound 4); selective estrogen receptor modulators, such as raloxifene (compound 5); or alternative substrates to inhibit the ability of PLD to make the lipid product PtdOH, such as n-butanol (compound 6), which leads to the formation of phosphatidylbutanol [reviewed in Selvy et al. (2011) and Scott et al. (2014)]. Indeed, the lack of small-molecule ligands to use as tools to probe both the cellular and the in vivo roles of each PLD isoform has arguably hindered the target validation for PLD because these ligands do not discriminate between the PLD isoforms. Moreover, the vast majority of studies have employed n-butanol, which is incorrectly referred to as a PLD inhibitor.
Fig. 3.

Ligands reported to modulate PLD function either indirectly (e.g., 1 and 2), directly (e.g., 3–5) or alternative substrates to compete in a transphosphatidylation reaction with water (e.g., 6).
In 2007, a brief report from Novartis (Monovich et al., 2007) reported that the Janssen atypical antipsychotic agent halopemide (compound 7) was an inhibitor of PLD (Fig. 4), with an IC50 of 1.5 μM against PLD2, and 13 additional analogs were reported, including what has later been coined FIPI [N-(2-(4-(2,3-dihydro-2-oxo-1H-benzimidazol-1-yl)-1-piperidinyl)ethyl)-5-fluoro-1H-indole-2-carboxamide] (compound 8; PLD2 IC50 = 200 nM). However, there was no mention of PLD1 inhibition by the Novartis group, but it was subsequently found that halopemide (compound 7) potently inhibits both PLD1 (cellular IC50 = 21 nM, biochemical IC50 = 220 nM) and PLD2 (cellular IC50 = 300 nM, biochemical IC50 = 310 nM) as does FIPI 7 (PLD1 cellular IC50 = 1 nM, biochemical IC50 = 9.5 nM; PLD2 cellular IC50 = 44 nM, biochemical IC50 = 17 nM). Thus, compound 7 and all of the halopemide analogs described were not PLD2-selective inhibitors, but rather proved to be dual PLD1/PLD2 inhibitors or modestly preferring PLD1 inhibitors. Not only was halopemide an attractive lead molecule, but compound 7 had also been evaluated by Janssen in five separate, positive clinical trials with over 100 schizophrenic, oligophrenic, and autistic patients receiving the drug. No adverse side effects or toxicities were noted, despite achieving plasma exposures above the IC50s for PLD inhibition, suggesting inhibition of PLD by this chemotype is safe in humans and a therapeutically viable mechanism. Although this finding was exciting, it was far from a panacea for the PLD field. Like most atypical antipsychotic agents, halopemide (7) (as well as FIPI 8) possessed a promiscuous ancillary pharmacology profile (significant activity at > 30 biogenic amine targets); thus, to enable the PLD field, isoform-selective PLD inhibitors would need to be developed with improved ancillary pharmacology.
Fig. 4.
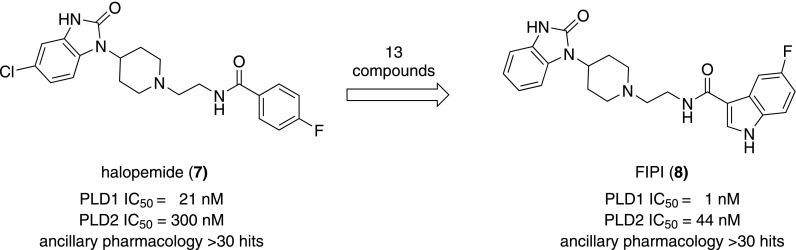
Structures and cellular PLD activity of halopemide (7) and the related analog FIPI (8), dual PLD1/PLD2 inhibitors with classic atypical antipsychotic promiscuous ancillary pharmacology.
To rapidly explore the PLD structure–activity relationship for halopemide (compound 7), our laboratories employed both a diversity-oriented synthesis approach and an iterative parallel synthesis strategy to rapidly synthesize and screen over 900 analogs of compound 7 (Fig. 4). This effort identified key structural elements that increased PLD1 selectivity, such as the (S)-chiral methyl group, that led to the discovery of the 1700-fold PLD1 selective inhibitor VU359595 [(1R,2R)-N-((S)-1-(4-(5-bromo-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-1-yl)piperidin-1-yl)propan-2-yl)-2-phenylcyclopropanecarboxamide] (compound 9), as well as other members within this functionalized benzimidazolone series that displayed 200- to 900-fold selectivity for inhibition of PLD1 (Fig. 5) (Lewis et al., 2009). Moreover, these benzimidazolone-based PLD1-selective inhibitors possessed favorable drug metabolism and pharmacokinetic (DMPK) properties [fu 2–6%, low to moderate Cl in rats, >20 μM versus cytochrome P450 (P450) enzymes] and subtle changes provided both peripherally restricted and central nervous system (CNS) penetrant inhibitors. Importantly, ancillary pharmacology was significantly improved over compound 7. Also from the diversity-oriented synthesis effort was the identification of an N-aryl triazaspirone moiety that effectively replaced the benzimidazolone, but engendered, for the first time, preference for inhibition of PLD2. Optimization through a matrix library resulted in the important 75-fold selective PLD2 inhibitor VU0364739 [N-(2-(1-(3-fluorophenyl)-4-oxo-1,3,8-triazaspiro[4.5]decan-8-yl)ethyl)-2-naphthamide] (compound 10), which was CNS penetrant (B:P of 0.73), displayed a good DMPK profile (fu 2%, low to moderate Cl in rats, >20 μM versus P450s) and improved ancillary pharmacology (Lavieri et al., 2010). Further optimization of compound 10 through an iterative parallel synthesis approach provided the Molecular Libraries Probe Production Centers Network probe ML298 [3,4-difluoro-N-(2-(1-(3-fluorophenyl)-4-oxo-1,3,8-triazaspiro[4.5]decan-8-yl)ethyl)benzamide] (compound 11), a >50-fold PLD2-selective inhibitor (O'Reilly et al., 2013). ML298 was an important tool compound, because it afforded no inhibition of PLD1 (PLD1 IC50 >20 μM), yet further improved ancillary pharmacology (only three hits >50% at 10 μM in a radioligand binding panel). Introduction of the (S)-methyl group, that enhanced PLD1 inhibition on the benzimidazolone series 9 similarly enhanced PLD1 inhibitory activity within the N-aryl triazaspirone series, as well as PLD2 activity. This minor structural modification led to the discovery of the potent, dual PLD1/2 inhibitor ML299 [(S)-4-bromo-N-(1-(1-(3-fluorophenyl)-4-oxo-1,3,8-triazaspiro[4.5]decan-8-yl)propan-2-yl)benzamide] (compound 12), an inhibitor with good CNS exposure (B:P of 0.48), a good DMPK profile (fu 3%, low to moderate Cl in rats, >20 μM versus P450s), and improved ancillary pharmacology (only three hits) (O'Reilly et al., 2013). Interestingly, incorporation of the (R)-methyl group led to a diminution in potency at both PLD1 and PLD2. Thus, a tool kit of PLD1-selective, PLD2-selective, and dual PLD1/2 inhibitors with good DMPK profiles that display either peripheral restriction or CNS exposure are now available to dissect the therapeutic potential and physiologic roles of direct and selective inhibition of the PLD isoforms both in vitro and in vivo (Lavieri et al., 2009, 2010; Lewis et al., 2009; Scott et al., 2009).
Fig. 5.
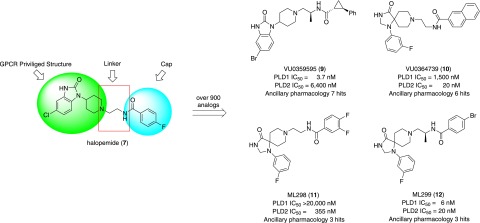
Chemical optimization strategy for halopemide (7) employing a combination of diversity-oriented synthesis, and more focused iterative parallel synthesis. This effort led to the development of the 1700-fold PLD1-preferring inhibitor VU0359595 (9), the 75-fold PLD2-preferring VU0364739 (10), the >50-fold PLD2-selective MLPCN probe ML298 (11), and the potent dual PLD1/PLD2 inhibitor MLPCN probe ML299 (12). Importantly, ancillary pharmacology was improved significantly over compound 7 as well as PLD isoform selectivity.
IX. Functions of Phosphatidic Acid
Phosphatidic acid is a multifunctional lipid that is generated by two other pathways besides PC hydrolysis by PLD (Fig. 6). In the first pathway, PtdOH is synthesized de novo by a two-step mechanism involving the transfer of two fatty acyl chains to the sn-1 and sn-2 positions of glycerol-3-phosphate, situating PtdOH as the precursor to all other glycerophospholipids (Kennedy, 1987). In addition to de novo synthesis and PC hydrolysis by PLD, diacylglycerol kinase can transfer a phosphate group to DAG to generate PtdOH, although these PtdOH species are believed to be distinct in acyl composition compared with PLD-derived PtdOH (reviewed in Shulga et al., 2011). Once generated, PtdOH can mediate signaling events by indirect mechanisms through metabolism into other bioactive lipids such as LPA by phospholipase A (Aoki, 2004) and DAG by lipid phosphate phosphatase (Brindley, 2004). PtdOH constitutes between 2 and 6% in the cellular membranes and that accounts for about 10% of the PC content (typically 30–50% of the total glycerophospholipids, depending on the biologic source) of the membranes (Andreyev et al., 2010; Shulga et al., 2013). By comparison, LPA is typically found to be less than 1% (nanomolar quantities) (Sugiura et al., 1999; Jesionowska et al., 2014), whereas total PIPn represents less than 1% of the membrane phospholipids (Czech, 2000). The amount of PtdOH in basal conditions depends on the cell, tissue, or organ where it is measured, and it may double under certain types of stimulation. For example, in astrocytoma cells after UDP stimulation, we measured PtdOH production increases of 70% over unstimulated levels (Scott et al., 2013).
Many signaling cascades begin with the generation of bioactive lipids such as PtdOH or phosphorylated PIs that recruit or modulate enzymatic activities of downstream effectors. These transient protein–lipid interactions are mediated by families of lipid-binding domains that share some degree of sequence or structural homology (Lemmon, 2008). The C1 and C2 domains (CR1 and CR2) were first identified in PKC and are responsible for binding DAG and Ca2+/PS, respectively (Cho and Stahelin, 2006; Colón-González and Kazanietz, 2006). PH domains were identified as PIPn binding domains from pleckstrin, the major substrate for PKC in platelets (Lemmon, 2008). Proteins with PH domains show some degree of specificity for PIPn species. For example, the PH domain of Akt kinase has a high affinity for PIs phosphorylated at the D3 and D4 positions, such as PI(3,4)P2 and PI(3,4,5)P3, but has much weaker affinity for PI(4,5)P2 (Thomas et al., 2002). The PH domain of PLCδ1, on the other hand, has a high affinity for PI(4,5,)P2 and this interaction is required for its catalytic activity (Ferguson et al., 1995). Monophosphorylated PI(3)P is recognized by several binding motifs, including FYVE and PX domains, named after the phagocyte NADPH oxidase (Kanai et al., 2001; Kutateladze, 2006). Although most FYVE and PX domains bind PI(3)P, some have affinity for PIP2 and PIP3 (Song et al., 2001). Annexin domains are calcium-dependent phospholipid-binding domains that are thought to predominantly bind PS, although binding to other phospholipids has been reported (Gerke et al., 2005). Besides interacting with specific lipids, other domains are known to detect or promote membrane curvature. Domains such as F-BAR, BAR, epsin N-terminal homology, and AP180 N-terminal homology participate in functions that require membrane deformation such as endocytosis and cytokinesis (Itoh and De Camilli, 2006). The specificity and conservation of these lipid binding domains has helped to identify proteins involved with lipid-mediated signal transduction. However, no specific domain has been identified for PtdOH binding; thus, the identification of proteins modulated by PLD-generated PtdOH has been especially challenging (Stace and Ktistakis, 2006). Many of the known PtdOH binding proteins were described in relevant sections of this review. Thus, a survey of other known PtdOH binding proteins along with the functional significance of the interactions is presented in the following section and a summary of all PtdOH binding proteins described in this introduction is included in Table 2.
TABLE 2.
Phosphatidic acid binding proteins
| Protein Name | PtdOH Function | Binding Site | References |
|---|---|---|---|
| Kinases | |||
| Raf1 | Membrane recruitment | AAs 339–423 | Ghosh et al. (1996, 2003), Rizzo et al. (1999, 2000) |
| PKCε | Membrane recruitment | C2 domain | Corbalan-Garcia et al. (2003), Jose Lopez-Andreo et al. (2003) |
| PKCζ | Activation | Unknown | Nakanishi and Exton (1992), Limatola et al. (1994) |
| PKCα | Activation | Unknown | Yokozeki et al. (1998), Nakashima (2002) |
| PKCδ | Activation | Unknown | Aris et al. (1993) |
| mTOR | Activation | FRB domain - R2109 | Fang et al. (2001), Veverka et al. (2008) |
| p70S6K1 | Activation | Unknown | Lehman et al. (2007) |
| PIP5K | Activation | Unknown | Jenkins et al. (1994), Arneson et al. (1999), Roach et al. (2012) |
| Fer | Activation | R417, R425, and H426 | Itoh et al. (2009) |
| GRK | Activation | Unknown | DebBurman et al. (1995) |
| Akt | Membrane recruitment | PH domain | Mahajan et al. (2010), Bruntz et al. (2014) |
| PAK1 | Activation | Unknown | Bokoch et al. (1998) |
| PKN | Activation | Unknown | Khan et al. (1994), Morrice et al. (1994) |
| SPHK1 | Membrane recruitment | C terminus | Jose Lopez-Andreo et al. (2003) |
| Phosphatases | |||
| SHP1 | Activation | C terminus - last 41 AAs | Tomic et al. (1995), Frank et al. (1999) |
| PP1cγ | Inhibition | AAs 274–299 | Jones and Hannun (2002), Jones et al. (2013) |
| Lipin1β | Membrane localization | Polybasic region around AA 153 | Corbalan-Garcia et al. (2003), Jose Lopez-Andreo et al. (2003) |
| G protein regulators | |||
| Rac-GDI | Inhibition | Unknown | Chuang et al. (1993) |
| n-Chimaerin | Activation | Unknown | Ahmed et al. (1993) |
| NF1 | Inhibition | Unknown | Bollag and McCormick (1991) |
| ASAP1 | Activation | Unknown | Randazzo (1997), Brown et al. (1998b) |
| AGAP1 | Activation | Unknown | Nie et al. (2002) |
| ACAP1/2 | Activation | Unknown | Jackson et al. (2000) |
| ArfGAP1/2 | Activation | Unknown | Randazzo (1997) |
| RA-RhoGAP | Activation | R399 | Kurooka et al. (2011) |
| RSS4 | Inhibition | First 57 AAs | Ouyang et al. (2003) |
| SOS | Membrane recruitment | R475 and R479 | Zhao et al. (2007) |
| DOCK2 | Membrane recruitment | C terminus | Nishikimi et al. (2009) |
| Phosphodiesterases | |||
| PDE4D3 | Activation | AAs 31–59 | Némoz et al. (1997), Grange et al. (2000) |
| PDE4B1 | Activation | Unknown | Némoz et al. (1997) |
| PDE4A5 | Activation | Unknown | Bawab et al. (1997), Némoz et al. (1997) |
| PDE4A1 | Membrane recruitment | W19 and W20 | Baillie et al. (2002) |
| Phospholipase C | |||
| PLCβ1 | Activation | Y952, 1955 | Litosch (2003) |
| PLCγ1 | Activation | Unknown | Jones and Carpenter (1993) |
| PLCε | Activation | Unknown | Murthy et al. (2013) |
| PLCδ3 | Activation | Unknown | Pawelczyk and Matecki (1999) |
| Miscellaneous | |||
| NADPH oxidase | Activation | R70, K55 in p47phox | McPhail et al. (1995, 1999), Palicz et al. (2001), Karathanassis et al. (2002) |
| Ng | Membrane recruitment | Unknown | Baudier et al. (1991) |
| β-Cotomer | Unknown | Unknown | Manifava et al. (2001) |
| Af1/6 | Unknown | Unknown | Manifava et al. (2001) |
| NSF | Unknown | Unknown | Manifava et al. (2001) |
| Kinesin | Unknown | Unknown | Manifava et al. (2001) |
| Sin1 | Membrane localization | PH domain | Schroder et al. (2007) |
| ANT | Unknown | Unknown | Epand et al. (2009) |
| Rac1 | Membrane recruitment | C-terminal polybasic motif | Chae et al. (2008) |
AA, amino acid; ACAP, ArfGAP with coiled-coil, ankyrin repeat, and PH domains; ANT, adenine nucleotide translocase.
A. Kinases
1. Sphingosine Kinase 1.
Sphingosine kinase (SPHK) phosphorylates sphingosine to produce the bioactive S1P. A family of GPCRs, known as Edg receptors, mediates a variety of functions including cell growth, cytoskeleton rearrangement, and vascular maturation (Spiegel and Milstien, 2002). Membrane recruitment of SPHK1 to perinuclear regions requires PtdOH (Delon et al., 2004). The translocation of SPHK1 is enhanced by overexpression of PLD or stimulation with phorbol esters and is inhibited by treatment with butanol, thus suggesting that PtdOH derived from PLD regulates SPHK1 membrane localization (Delon et al., 2004).
2. Protein Kinase Cε.
The ε isoform of PKC is a member of the calcium-independent, DAG-dependent PKC family. PKCε has been implicated physiologic functions such as neurite outgrowth, inflammatory or immune responses, tumorigenesis, myocardial development, and protection against ischemic damage (Akita, 2002). Membrane recruitment of PKCε requires DAG and PLD-generated PtdOH, as determined by the use of butanol (Jose Lopez-Andreo et al., 2003). To address the possibility that PtdOH was metabolized to DAG and did not directly influence membrane recruitment of PKCε, the authors demonstrated synergistic PKCε activation by costimulation with DAG and PtdOH (Jose Lopez-Andreo et al., 2003).
3. Protein Kinase Cδ.
Like PKCε, the δ isoform belongs to the novel PKC family of calcium-independent, DAG-dependent PKCs. The PKCδ isoform shares partial functional redundancy to other PKC isoforms, but PKCδ mediates specific roles in apoptosis, cell cycle progression, and transcriptional regulation (Steinberg, 2004). PtdOH can stimulate PKCδ in vitro although the fold stimulation is much less than PS, suggesting a general requirement for anionic phospholipids (Aris et al., 1993). In vivo relevance of PtdOH regulation of PKCδ remains to be determined.
4. Protein Kinase Cζ.
The ζ isoform of PKC is a member of the atypical family of PKCs that do not respond to calcium or DAG and appear to regulated by PIP3 and by protein–protein interactions (Ways et al., 1992; Hirai and Chida, 2003). PKCζ is believed to participate in the MAPK cascade, nuclear factor κB transcription, the p70S6-kinase cascade, and development of cell motility (Hirai and Chida, 2003). In vitro, PKCζ is activated by PtdOH (Nakanishi and Exton, 1992; Limatola et al., 1994). However, mono- and polyunsaturated fatty acids, PS, and CL also stimulate PKCζ in vitro (Nakanishi and Exton, 1992) and an in vivo regulation of PKCζ by PLD-derived PtdOH remains to be established.
5. Protein Kinase Cα.
The α isoform of PKC belongs to the conventional family of PKCs and requires both calcium and DAG for full activation. PKCα is activated by many extracellular stimuli and plays important roles in proliferation, apoptosis, differentiation, motility, and inflammation (Nakashima, 2002). PtdOH has been shown to regulate PKCα in multiple studies. PtdOH added to PKCα-transfected COS-7 cells results in a significant upregulation of PKCα autophosphorylation (Limatola et al., 1994). In addition, PtdOH stimulates PKCα kinase activity when added to bovine brain cytosol (Yokozeki et al., 1998). Neither study ruled out the possibility of an intermediate PtdOH-stimulated protein or metabolism of PtdOH to DAG as a potential explanation for the increase in PKCα activation following PtdOH treatment. Therefore, the exact role of PtdOH remains unclear.
6. Akt.
A recent study demonstrated that Akt could be phosphorylated at tyrosine 176, distinct from the canonical activating phosphorylation sites of threonine 308 and serine 473. It was speculated that Akt1 itself may be the upstream kinase. When phosphorylated at tyrosine 176, the lipid-binding profile of Akt changed such that the kinase preferred to bind PtdOH, yet no functional consequence of Akt-PtdOH binding was determined (Mahajan et al., 2010). Binding of PtdOH directly to Akt has recently been described (Bruntz et al., 2014)
B. Phosphatases
1. Src Homology Region 2 Domain-Containing Phosphatase-1.
SHP1 is a nonreceptor tyrosine phosphatase, mainly expressed in hematopoietic cells, with a wide spectrum of substrates (Soulsby and Bennett, 2009). SHP1, but not SHP2, was activated by PtdOH in vitro (Tomic et al., 1995). In addition, PtdOH enhanced the association between SHP1 and EGFR and increased EGFR dephosphorylation. The high-affinity PtdOH binding site on SHP1 was later mapped to the C-terminal 41 amino acids (Frank et al., 1999). Although PLD was not directly shown to participate in the modulation of SHP1, PLD-generated PtdOH could act as part of a negative feedback loop involving SHP1 to terminate receptor tyrosine kinase signaling.
2. Lipin1β.
Lipin1β is a type 1 (magnesium-dependent and N-ethylmaleimide–sensitive) PtdOH phosphatase that plays important roles in adipocyte differentiation by regulating triacylglycerol production and storage. Mutations in Lipin1β lead to lipodystrophy in mice (Péterfy et al., 2001). In addition to its phosphatase activity, Lipid1β translocates to the nucleus to activate a subset of peroxisome proliferator-activated receptor γ coactivator 1a target pathways including fatty acid oxidation and mitochondrial oxidative phosphorylation (Finck et al., 2006). A PtdOH binding site was recently mapped to a polybasic motif of Lipin1β, independent of the catalytic site. Interestingly, when these residues were mutated or when cells were treated with small-molecule PLD inhibitors, Lipin1β localized to the nucleus, suggesting that PLD and PtdOH may serve to modulate specific gene transcription (Ren et al., 2010).
C. Phospholipase C
1. Phospholipase Cβ1.
PI-specific PLC enzymes hydrolyze PIP2 to generate IP3 and DAG, which increase intracellular calcium stores and activate PKC. PtdOH directly binds purified PLCβ1 and stimulates its activity in vitro. The activation of PLCβ1 is unique to PtdOH because other phospholipids were unable to stimulate PIP2 hydrolysis (Litosch, 2000). In addition, primary butanol significantly rightward shifts muscarinic receptor–stimulated PIP2 hydrolysis, implicating a role for PLD regulating in PLC in vivo (Litosch et al., 2009). Tyrosine 952 and isoleucine 955 mediate PtdOH binding, suggesting distinct modes of PLC regulation by PtdOH and Gαa (Litosch et al., 2009). PLCβ stimulation by PtdOH is unique to PLCβ1 because other β isoforms were unresponsive to PtdOH stimulation (Litosch, 2003).
2. Phospholipase Cγ1.
Receptor tyrosine kinase stimulation increases PIP2 hydrolysis through the activation of PLCγ (Meisenhelder et al., 1989) and PtdOH was shown to stimulate the activity of PLCγ1 in vitro (Jones and Carpenter, 1993). The hypothesized mechanism for PtdOH regulation of PLCγ1 is through allosterically increasing enzyme activity rather than increasing the equilibrium dissociation constant, Ks (Jones and Carpenter, 1993).
3. Phospholipase Cε.
The epsilon isoform of PLC is activated through the actions of small G proteins, such as Ras, and through heterotrimeric G proteins, such as Gα12 and Gα13 (Song et al., 2001). When purified to homogeneity, PLCε had very low basal activity that was stimulated by arachidonic acid, PtdOH, and to a lesser extent by PS and LPA (Murthy et al., 2013). Interestingly, PtdOH was a more potent stimulator of PLCε hydrolysis of PI than PIP2 whereas arachidonic acid stimulated PLCε activity to wards both substrates equally. These results suggest that lipids may regulate substrate specificity of PLCε.
4. Phospholipase Cδ3.
The delta isoforms of PLC are activated by increased Ca2+, which promotes association with PIP2 (Rhee, 2001). Purified PLCδ3 was shown to bind PtdOH vesicles through its PH domain (Pawelczyk and Matecki, 1999) and the interaction with PtdOH increased PLCδ3 activity in vitro.
D. G Protein Regulatory Proteins
1. RA-Rho GTPase Activating Protein.
As key regulators of cytoskeleton dynamics, Rho GTPases are known to control neurite development and decreased Rho activity is associated with enhanced neurite sprouting, extension, and branching (Luo, 2000). RA-RhoGAP is a Rho GAP that binds PtdOH via its PH domain (Kurooka et al., 2011). As with several other PtdOH–protein interactions, mutation of a basic residue, arginine 399, abolished PtdOH binding. The interaction of RA-RhoGAP with PtdOH-stimulated GAP activity in vitro (Kurooka et al., 2011). Overexpression of DAG kinase increased neurite length in a manner that depended on PtdOH binding to RA-RhoGAP. Contributions of PLD-derived PtdOH were not measured in this study.
2. ADP-Ribosylation Factor GTPase Activating Proteins.
Several Arf GAPs have been identified that are modulated by PtdOH in vitro. Two of the earliest ArfGAPs discovered, ArfGAP1 and ArfGAP2, are both stimulated by phospholipids. ArfGAP1 activity is stimulated about 7-fold by PIP2, PtdOH, and PS. ArfGAP2 is stimulated 2-fold by PIP2, but not by PtdOH or PS. However, PIP2 and PtdOH synergize to stimulate ArfGAP2 activity about 20-fold (Randazzo, 1997). Arf-GAP with SH3 domain, ANK repeat and PH domain–containing protein 1 (ASAP1), like ArfGAP2, is weakly stimulated by PtdOH alone, but synergistic activity is observed in the presence of PtdOH and PIP2. Like ArfGAP2, PtdOH and PIP2 synergize to activate another ArfGAP, ASAP1. ASAP1 contains a PH domain that most likely mediates the phospholipid interactions (Brown et al., 1998b). AGAP1 is another PH domain-containing ArfGAP that is synergistically stimulated by PIP2 and PtdOH. Although the synergy was most robust with PtdOH, other anionic lipids such as PS and PI also synergized with PIP2 (Nie et al., 2002). Arf6-specific GAPs, ACAP1 and ACAP2, display similar synergy with PIP2 and PtdOH (Jackson et al., 2000). The requirement of PIP2 for PLD and ArfGAP activities along with the ability of Arf to stimulate PLD presents a potential negative feedback loop whereby PLD-generated PtdOH may serve to terminate Arf signaling.
3. n-Chimaerin.
n-Chimaerin was identified as a phorbol ester binding GAP for the small G-protein Rac. n-Chimaerin is stimulated by PtdOH and, to lesser extent, PS. Interestingly, arachidonic acid inhibits n-chimaerin GAP activity (Ahmed et al., 1993).
4. Regulator of G Protein Signaling 4.
The Gα subunits are regulated by a special class of GAPs known as regulators of G protein signaling (RGSs) (Ross and Wilkie, 2000). RGS proteins function to attenuate GPCR signaling by stimulating Gα GTP hydrolysis. When M1 muscarinic acetylcholine receptors were reconstituted in lipid vesicles and incubated with RGS4, vesicles containing PtdOH inhibited RGS4 GAP activity after M1-agonist stimulation. PtdOH was then determined to directly bind the first 57 amino acids of RGS4 (Ouyang et al., 2003). Because muscarinic receptors are well established activators of PLD (Sandmann et al., 1991), the inhibition of RGS4 activity by PLD-generated PtdOH might potentiate muscarinic receptor signaling.
E. Miscellaneous Proteins
1. NADPH Oxidase.
Host defense systems against invading pathogens involve phagocytosis and subsequent destruction by the generation of superoxide anions, hydrogen peroxide, and hypochlorous acid via the NADPH oxidase complex in phagocytes. This complex contains multiple membrane (p22Phox and gp91Phox) and cytosolic subunits (p47Phox, p40Phox, p67Phox, and Rac1/2) that are subject to regulation by PLD and PtdOH (DeLeo and Quinn, 1996). Upon engagement of cell surface receptors by pathogens, cytokines, or chemoattractants, PLD activity and PtdOH production increase (McPhail et al., 1999). PtdOH stimulates an unknown kinase that phosphorylates the p47Phox and p22Phox subunits (McPhail et al., 1995, 1999). Later studies demonstrated that PtdOH was able to stimulate NADPH oxidase activity by directly interacting with different subunits of the complex (Palicz et al., 2001). Crystal structures of the p47Phox subunit revealed a high-affinity binding site for PIP2 and separate binding site for anionic phospholipids, such as PtdOH. Binding of p47Phox to PIP2 vesicles was synergistically increased when PtdOH was also as a vesicle component, suggesting that PtdOH increases p47Phox association to PIP2 (Karathanassis et al., 2002). Studies with small-molecule PLD inhibitors recently confirmed the involvement of PLD in regulating NADPH oxidase (Chang et al., 2011).
2. Neurogranin.
Neurogranin (Ng) is a protein originally identified as a PKC substrate in the brain and is involved in synaptic plasticity (Baudier et al., 1991). Ng specifically binds PtdOH in vitro in a site that overlaps with its calmodulin-binding motif. Overexpression of PLD increased localization of Ng at dendritic spines, suggesting that PLD-generated PtdOH regulates Ng localization.
3. Sin1.
mTOR exists in two distinct complexes (mTORC1 and mTORC2) distinguished by different binding partners and unique substrate specificities. Sin1 is an essential component of the mTORC2 complex and is required for complex stability and kinase activity toward downstream effectors (Yang et al., 2006). Sin1 contains a PH domain that binds PIPns and PtdOH in vitro (Schroder et al., 2007). The relevance of Sin1-PtdOH binding is unknown, but PLD may regulate mTORC2 activity by activating or localizing Sin1.
4. ADP/ATP Translocase.
The ADP/ATP translocase is an inner mitochondrial membrane protein that transports newly synthesized ATP from the mitochondria to the cytosol (Klingenberg, 2008). When purified from bovine heart and analyzed for bound lipids, PtdOH was tightly bound to the ADP/ATP translocase protein (Epand et al., 2009). The physiologic relevance of this interaction is unknown.
5. β-Coatomer, ADP-Ribosylation Factor, N-Ethylmaleimide–Sensitive Factor, and Kinesin.
In an attempt to isolate novel PtdOH binding proteins, investigators coupled PtdOH or PIP2 to affinity beads to isolate proteins specifically bound to PtdOH from brain cytosol. β-Coatomer, Arf, N-ethylmaleimide–sensitive factor, and kinesin, all traffic-related proteins, were identified as specifically binding PtdOH beads. Recombinant Arf and N-ethylmaleimide–sensitive factor were shown to directly bind PtdOH, but the physiologic relevance remains unknown (Manifava et al., 2001).
X. Closing Points
Over the last 2 decades, there has been a proliferation in our understanding of the regulation of cellular PLD activity and the roles these enzymes play in a variety of physiologic functions. The discovery that the gene products of both PLD1 and PLD2 can be ablated in mice has led to a growing appreciation that this signaling enzyme provides an attractive therapeutic target for a variety of pathophysiological conditions in humans. Development of potent and isoenzyme-selective, small-molecule inhibitors of PLD is contributing to a renaissance in our understanding of how PLD activity can be modulated to provide novel therapeutic agents to some of our most challenging diseases. The finding that some inhibitors modulate a PLD-Akt signal axis opens an interesting new pharmacological target in the treatment of certain cancers that have previously been recalcitrant to chemotherapeutics. Many important discoveries are yet to be made in fully understanding the role of PLD in cellular signaling and tumorigenesis. Among these is a more comprehensive understanding of the interactions among different phosphatidic acid–generating pathways and how these pathways compensate and respond to perturbations.
Acknowledgments
The authors thank Pavlina Ivanova, Michelle Armstrong, and Sarah Scott for assistance with figures, editing, and insightful conversations during the writing of this review.
Abbreviations
- 4E-BP1
4E binding protein 1
- AC
adenylyl cyclase
- AMPK
AMP-activated protein kinase
- AP3
assembly protein 3
- Arf
ADP-ribosylation factor
- ASAP1
Arf-GAP with SH3 domain, ANK repeat and PH domain–containing protein 1
- Bcl-2
B-cell lymphoma 2
- Cdk5
cyclin-dependent kinase 5
- CL
cardiolipin
- CNS
central nervous system
- CR
conserved region
- CRMP-2
collapsin response mediator protein-2
- CtBP1
C-terminal binding protein 1
- DAG
diacylglycerol
- DMPK
drug metabolism and pharmacokinetics
- DOCK2
dedicator of cytokinesis 2
- ECM
extracellular matrix
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- Egr1
early growth response protein 1
- EMT
epithelial–mesenchymal transition
- Epac1
exchange protein directly activated by cAMP 1
- ERK
extracellular signal-regulated kinase
- FIPI
N-(2-(4-(2,3-dihydro-2-oxo-1H-benzimidazol-1-yl)-1-piperidinyl)ethyl)-5-fluoro-1H-indole-2-carboxamide
- FGF
fibroblast growth factor
- FKBP12
FK506 binding protein 12
- FRB
FKBP12-rapamycin binding
- GAP
GTPase activating protein
- GDI
guanine nucleotide dissociation inhibitor
- GEF
guanine nucleotide exchange factor
- GPCR
G protein–coupled receptor
- GPI
glycosylphosphatidylinositol
- GRK
G protein–coupled receptor kinase
- GTPγS
nonhydrolyzable GTP analog
- HIF
hypoxia-inducible factor
- IP3
inositol triphosphate
- JAK3
Janus kinase 3
- KO
knockout
- LC3
light chain 3
- LPA
lysophosphatidic acid
- M3R
M3 muscarinic receptor
- MAPK
mitogen-activated protein kinase
- MEK
mitogen-activated protein kinase kinase
- ML298
3,4-difluoro-N-(2-(1-(3-fluorophenyl)-4-oxo-1,3,8-triazaspiro[4.5]decan-8-yl)ethyl)benzamide
- ML299
(S)-4-bromo-N-(1-(1-(3-fluorophenyl)-4-oxo-1,3,8-triazaspiro[4.5]decan-8-yl)propan-2-yl)benzamide
- MMP
matrix metalloproteinase
- mTOR
mammalian target of rapamycin
- NF1
neurofibromin 1
- Ng
neurogranin
- NRTK
nonreceptor tyrosine kinase
- p21Cip1
p21 cyclin-dependent kinase inhibitor
- P450
cytochrome P450
- p70S6K1
p70 S6 kinase 1
- PAK
p21-activated protein kinase
- PAS
preautophagosomal structure
- PC
phosphatidylcholine
- PDE
phosphodiesterase
- PDGF
platelet-derived growth factor
- PE
phosphatidylethanolamine
- PEA15
protein enriched in astrocytes
- PH
pleckstrin homology
- PI
phosphoinositide
- PI3K
phosphoinositide 3-kinase
- PIP
phosphatidylinositol phosphate
- PIP2
phosphatidylinositol bisphosphate
- PIP3
phosphatidylinositol trisphosphate
- PIP5K
PI(4)P 5-kinase
- PKA
protein kinase A
- PKC
protein kinase C
- PKN
protein kinase N
- PLC
phospholipase C
- PLD
phospholipase D
- PMA
phorbol-12-myristate-13-acetate
- PP1
protein phosphatase 1
- pRB
retinoblastoma protein
- PS
phosphatidylserine
- PtdOH
phosphatidic acid
- PX
phox homology
- RGS
regulator of G protein signaling
- RNAi
RNA interference
- RTK
receptor tyrosine kinase
- S1P
sphingosine 1-phosphate
- SH2
Src homology 2
- SH3
Src homology 3
- SHP
Src homology region 2 domain-containing phosphatase
- siRNA
small interfering RNA
- SOS
son of sevenless
- SPHK
sphingosine kinase
- STAT3
signal transducer and activator of transcription 3
- TSC2
tuberous sclerosis 2 protein
- ULK
UNC-52–like kinase
- VEGF
vascular endothelial growth factor
- VU0364739
N-(2-(1-(3-fluorophenyl)-4-oxo-1,3,8-triazaspiro[4.5]decan-8-yl)ethyl)-2-naphthamide
- VU359595
(1R,2R)-N-((S)-1-(4-(5-bromo-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-1-yl)piperidin-1-yl)propan-2-yl)-2-phenylcyclopropanecarboxamide
- VHL
Von Hippel–Lindau
- WASp
Wiskott–Aldrich syndrome protein
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Bruntz, Lindsley, Brown.
Footnotes
This research was supported in part by the National Institutes of Health National Institute of Environmental Health Sciences [Grant P01-ES013125]; the National Institutes of Health National Institute of Mental Health [Grant U54-MH084659]; the McDonnell Foundation for Brain Cancer Research; Voices Against Brain Cancer; and the Vanderbilt Institute of Chemical Biology [Lai Sulin scholarship (to R.C.B.), Bixler-Johnson-Mayes Endowed Chair (to H.A.B.), and the William K. Warren Jr. Chair in Medicine (to C.W.L.)]. Vanderbilt University is a member of the National Institutes of Health Molecular Libraries Probe Production Centers Network and houses the Vanderbilt Specialized Chemistry Center for Accelerated Probe Development.
Portions of this review appeared in the following: Bruntz RC (2013) Insights into the Molecular Mechanisms of Phospholipase D–Mediated Cancer Cell Survival. Doctoral dissertation, Vanderbilt University, Nashville, TN.
References
- Adachi T, Nakashima S, Saji S, Nakamura T, Nozawa Y. (1996) Phospholipase D activation in hepatocyte growth factor-stimulated rat hepatocytes mediates the expressions of c-jun and c-protein: involvement of protein tyrosine kinase, protein kinase C, and Ca2+. Hepatology 24:1274–1281 [DOI] [PubMed] [Google Scholar]
- Adams JM, Cory S. (2007) The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 26:1324–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre Ghiso JA, Farías EF, Alonso DF, Arregui C, Bal de Kier Joffé E. (1997) A phospholipase D and protein kinase C inhibitor blocks the spreading of murine mammary adenocarcinoma cells altering f-actin and beta1-integrin point contact distribution. Int J Cancer 71:881–890 [DOI] [PubMed] [Google Scholar]
- Ahle S, Ungewickell E. (1986) Purification and properties of a new clathrin assembly protein. EMBO J 5:3143–3149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahle S, Ungewickell E. (1990) Auxilin, a newly identified clathrin-associated protein in coated vesicles from bovine brain. J Cell Biol 111:19–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed S, Lee J, Kozma R, Best A, Monfries C, Lim L. (1993) A novel functional target for tumor-promoting phorbol esters and lysophosphatidic acid. The p21rac-GTPase activating protein n-chimaerin. J Biol Chem 268:10709–10712 [PubMed] [Google Scholar]
- Ahn BH, Kim SY, Kim EH, Choi KS, Kwon TK, Lee YH, Chang JS, Kim MS, Jo YH, Min DS. (2003) Transmodulation between phospholipase D and c-Src enhances cell proliferation. Mol Cell Biol 23:3103–3115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn BH, Min G, Bae YS, Bae YS, Min DS. (2006) Phospholipase D is activated and phosphorylated by casein kinase-II in human U87 astroglioma cells. Exp Mol Med 38:55–62 [DOI] [PubMed] [Google Scholar]
- Ahn BH, Rhim H, Kim SY, Sung YM, Lee MY, Choi JY, Wolozin B, Chang JS, Lee YH, Kwon TK, et al. (2002) α-Synuclein interacts with phospholipase D isozymes and inhibits pervanadate-induced phospholipase D activation in human embryonic kidney-293 cells. J Biol Chem 277:12334–12342 [DOI] [PubMed] [Google Scholar]
- Ahuja P, Zhao P, Angelis E, Ruan H, Korge P, Olson A, Wang Y, Jin ES, Jeffrey FM, Portman M, et al. (2010) Myc controls transcriptional regulation of cardiac metabolism and mitochondrial biogenesis in response to pathological stress in mice. J Clin Invest 120:1494–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akita Y. (2002) Protein kinase C-ε (PKC-ε): its unique structure and function. J Biochem 132:847–852 [DOI] [PubMed] [Google Scholar]
- Akula SM, Ford PW, Whitman AG, Hamden KE, Bryan BA, Cook PP, McCubrey JA. (2005) B-Raf-dependent expression of vascular endothelial growth factor-A in Kaposi sarcoma-associated herpesvirus-infected human B cells. Blood 105:4516–4522 [DOI] [PubMed] [Google Scholar]
- Alejandro EU, Johnson JD. (2008) Inhibition of Raf-1 alters multiple downstream pathways to induce pancreatic β-cell apoptosis. J Biol Chem 283:2407–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammer AG, Weed SA. (2008) Cortactin branches out: roles in regulating protrusive actin dynamics. Cell Motil Cytoskeleton 65:687–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananthakrishnan R, Ehrlicher A. (2007) The forces behind cell movement. Int J Biol Sci 3:303–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson D, Koch CA, Grey L, Ellis C, Moran MF, Pawson T. (1990) Binding of SH2 domains of phospholipase C γ 1, GAP, and Src to activated growth factor receptors. Science 250:979–982 [DOI] [PubMed] [Google Scholar]
- Andreyev AY, Fahy E, Guan Z, Kelly S, Li X, McDonald JG, Milne S, Myers D, Park H, Ryan A, et al. (2010) Subcellular organelle lipidomics in TLR-4-activated macrophages. J Lipid Res 51:2785–2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki J. (2004) Mechanisms of lysophosphatidic acid production. Semin Cell Dev Biol 15:477–489 [DOI] [PubMed] [Google Scholar]
- Araujo H, Danziger N, Cordier J, Glowinski J, Chneiweiss H. (1993) Characterization of PEA-15, a major substrate for protein kinase C in astrocytes. J Biol Chem 268:5911–5920 [PubMed] [Google Scholar]
- Aris JP, Basta PV, Holmes WD, Ballas LM, Moomaw C, Rankl NB, Blobel G, Loomis CR, Burns DJ. (1993) Molecular and biochemical characterization of a recombinant human PKC-δ family member. Biochim Biophys Acta 1174:171–181 [DOI] [PubMed] [Google Scholar]
- Arneson LS, Kunz J, Anderson RA, Traub LM. (1999) Coupled inositide phosphorylation and phospholipase D activation initiates clathrin-coat assembly on lysosomes. J Biol Chem 274:17794–17805 [DOI] [PubMed] [Google Scholar]
- Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. (2008) Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 182:685–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ávila-Flores A, Santos T, Rincón E, Mérida I. (2005) Modulation of the mammalian target of rapamycin pathway by diacylglycerol kinase-produced phosphatidic acid. J Biol Chem 280:10091–10099 [DOI] [PubMed] [Google Scholar]
- Bae EJ, Lee HJ, Jang YH, Michael S, Masliah E, Min DS, Lee SJ. (2014) Phospholipase D1 regulates autophagic flux and clearance of α-synuclein aggregates. Cell Death Differ 21:1132–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillie GS, Huston E, Scotland G, Hodgkin M, Gall I, Peden AH, MacKenzie C, Houslay ES, Currie R, Pettitt TR, et al. (2002) TAPAS-1, a novel microdomain within the unique N-terminal region of the PDE4A1 cAMP-specific phosphodiesterase that allows rapid, Ca2+-triggered membrane association with selectivity for interaction with phosphatidic acid. J Biol Chem 277:28298–28309 [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay G, Sajan MP, Kanoh Y, Standaert ML, Quon MJ, Reed BC, Dikic I, Farese RV. (2001) Glucose activates protein kinase C-zeta /lambda through proline-rich tyrosine kinase-2, extracellular signal-regulated kinase, and phospholipase D: a novel mechanism for activating glucose transporter translocation. J Biol Chem 276:35537–35545 [DOI] [PubMed] [Google Scholar]
- Banno Y, Fujita H, Ono Y, Nakashima S, Ito Y, Kuzumaki N, Nozawa Y. (1999) Differential phospholipase D activation by bradykinin and sphingosine 1-phosphate in NIH 3T3 fibroblasts overexpressing gelsolin. J Biol Chem 274:27385–27391 [DOI] [PubMed] [Google Scholar]
- Barr FA, Shorter J. (2000) Membrane traffic: do cones mark sites of fission? Curr Biol 10:R141–R144 [DOI] [PubMed] [Google Scholar]
- Baudier J, Deloulme JC, Van Dorsselaer A, Black D, Matthes HW. (1991) Purification and characterization of a brain-specific protein kinase C substrate, neurogranin (p17). Identification of a consensus amino acid sequence between neurogranin and neuromodulin (GAP43) that corresponds to the protein kinase C phosphorylation site and the calmodulin-binding domain. J Biol Chem 266:229–237 [PubMed] [Google Scholar]
- Bawab El S, Macovschi O, Sette C, Conti M, Lagarde M, Nemoz G, Prigent AF. (1997) Selective stimulation of a cAMP-specific phosphodiesterase (PDE4A5) isoform by phosphatidic acid molecular species endogenously formed in rat thymocytes. European Journal of Biochemistry 247:1151–1157 [DOI] [PubMed] [Google Scholar]
- Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, et al. (2000) Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol 2:737–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokoch GM, Reilly AM, Daniels RH, King CC, Olivera A, Spiegel S, Knaus UG. (1998) A GTPase-independent mechanism of p21-activated kinase activation. Regulation by sphingosine and other biologically active lipids. J Biol Chem 273:8137–8144 [DOI] [PubMed] [Google Scholar]
- Bollag G, McCormick F. (1991) Differential regulation of rasGAP and neurofibromatosis gene product activities. Nature 351:576–579 [DOI] [PubMed] [Google Scholar]
- Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K, Hirth P. (2012) Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat Rev Drug Discov 11:873–886 [DOI] [PubMed] [Google Scholar]
- Bolós V, Peinado H, Pérez-Moreno MA, Fraga MF, Esteller M, Cano A. (2003) The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci 116:499–511 [DOI] [PubMed] [Google Scholar]
- Boone DN, Qi Y, Li Z, Hann SR. (2011) Egr1 mediates p53-independent c-Myc-induced apoptosis via a noncanonical ARF-dependent transcriptional mechanism. Proc Natl Acad Sci USA 108:632–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgoin S, Grinstein S. (1992) Peroxides of vanadate induce activation of phospholipase D in HL-60 cells. Role of tyrosine phosphorylation. J Biol Chem 267:11908–11916 [PubMed] [Google Scholar]
- Bowman EP, Uhlinger DJ, Lambeth JD. (1993) Neutrophil phospholipase D is activated by a membrane-associated Rho family small molecular weight GTP-binding protein. J Biol Chem 268:21509–21512 [PubMed] [Google Scholar]
- Boyle KB, Hadaschik D, Virtue S, Cawthorn WP, Ridley SH, O’Rahilly S, Siddle K. (2009) The transcription factors Egr1 and Egr2 have opposing influences on adipocyte differentiation. Cell Death Differ 16:782–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brindley DN. (2004) Lipid phosphate phosphatases and related proteins: signaling functions in development, cell division, and cancer. J Cell Biochem 92:900–912 [DOI] [PubMed] [Google Scholar]
- Brown FD, Thompson N, Saqib KM, Clark JM, Powner D, Thompson NT, Solari R, Wakelam MJO. (1998a) Phospholipase D1 localises to secretory granules and lysosomes and is plasma-membrane translocated on cellular stimulation. Curr Biol 8:835–838 [DOI] [PubMed] [Google Scholar]
- Brown HA, Gutowski S, Kahn RA, Sternweis PC. (1995). Partial purification and characterization of Arf-sensitive phospholipase D from porcine brain. J Biol Chem 270:14935–14943 [DOI] [PubMed] [Google Scholar]
- Brown HA, Gutowski S, Moomaw CR, Slaughter C, Sternweis PC. (1993) ADP-ribosylation factor, a small GTP-dependent regulatory protein, stimulates phospholipase D activity. Cell 75:1137–1144 [DOI] [PubMed] [Google Scholar]
- Brown HA, Henage LG, Preininger AM, Xiang Y, Exton JH. (2007) Biochemical analysis of phospholipase D. Methods Enzymol 434:49–87 [DOI] [PubMed] [Google Scholar]
- Brown MT, Andrade J, Radhakrishna H, Donaldson JG, Cooper JA, Randazzo PA. (1998b) ASAP1, a phospholipid-dependent arf GTPase-activating protein that associates with and is phosphorylated by Src. Mol Cell Biol 18:7038–7051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruntz RC, Taylor HE, Lindsley CW, Brown HA. (2014) Phospholipase D2 mediates survival signaling through direct regulation of Akt in glioblastoma cells. J Biol Chem 289:600–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan FG, McReynolds M, Couvillon A, Kam Y, Holla VR, Dubois RN, Exton JH. (2005) Requirement of phospholipase D1 activity in H-RasV12-induced transformation. Proc Natl Acad Sci USA 102:1638–1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchsbaum RJ. (2007) Rho activation at a glance. J Cell Sci 120:1149–1152 [DOI] [PubMed] [Google Scholar]
- Burridge K, Connell L. (1983) Talin: a cytoskeletal component concentrated in adhesion plaques and other sites of actin-membrane interaction. Cell Motil 3:405–417 [DOI] [PubMed] [Google Scholar]
- Cai S, Exton JH. (2001) Determination of interaction sites of phospholipase D1 for RhoA. Biochem J 355:779–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood DA, Yan B, de Pereda JM, Alvarez BG, Fujioka Y, Liddington RC, Ginsberg MH. (2002) The phosphotyrosine binding-like domain of talin activates integrins. J Biol Chem 277:21749–21758 [DOI] [PubMed] [Google Scholar]
- Campbell DH, Sutherland RL, Daly RJ. (1999) Signaling pathways and structural domains required for phosphorylation of EMS1/cortactin. Cancer Res 59:5376–5385 [PubMed] [Google Scholar]
- Cantley LC. (2002) The phosphoinositide 3-kinase pathway. Science 296:1655–1657 [DOI] [PubMed] [Google Scholar]
- Carman CV, Barak LS, Chen C, Liu-Chen LY, Onorato JJ, Kennedy SP, Caron MG, Benovic JL. (2000) Mutational analysis of Gbetagamma and phospholipid interaction with G protein-coupled receptor kinase 2. J Biol Chem 275:10443–10452 [DOI] [PubMed] [Google Scholar]
- Carnero A, Cuadrado A, del Peso L, Lacal JC. (1994) Activation of type D phospholipase by serum stimulation and ras-induced transformation in NIH3T3 cells. Oncogene 9:1387–1395 [PubMed] [Google Scholar]
- Carrigan SO, Pink DBS, Stadnyk AW. (2007) Neutrophil transepithelial migration in response to the chemoattractant fMLP but not C5a is phospholipase D-dependent and related to the use of CD11b/CD18. J Leukoc Biol 82:1575–1584 [DOI] [PubMed] [Google Scholar]
- Caumont AS, Galas MC, Vitale N, Aunis D, Bader MF. (1998) Regulated exocytosis in chromaffin cells. Translocation of ARF6 stimulates a plasma membrane-associated phospholipase D. J Biol Chem 273:1373–1379 [DOI] [PubMed] [Google Scholar]
- Chae YC, Kim JH, Kim KL, Kim HW, Lee HY, Heo WD, Meyer T, Suh PG, Ryu SH. (2008) Phospholipase D activity regulates integrin-mediated cell spreading and migration by inducing GTP-Rac translocation to the plasma membrane. Mol Biol Cell 19:3111–3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae YC, Kim KL, Ha SH, Kim J, Suh PG, Ryu SH. (2010) Protein kinase Cdelta-mediated phosphorylation of phospholipase D controls integrin-mediated cell spreading. Mol Cell Biol 30:5086–5098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang LC, Huang TH, Chang CS, Tsai YR, Lin RH, Lee PW, Hsu MF, Huang LJ, Wang JP. (2011) Signaling mechanisms of inhibition of phospholipase D activation by CHS-111 in formyl peptide-stimulated neutrophils. Biochem Pharmacol 81:269–278 [DOI] [PubMed] [Google Scholar]
- Chang YY, Juhász G, Goraksha-Hicks P, Arsham AM, Mallin DR, Muller LK, Neufeld TP. (2009) Nutrient-dependent regulation of autophagy through the target of rapamycin pathway. Biochem Soc Trans 37:232–236 [DOI] [PubMed] [Google Scholar]
- Chardin P, Camonis JH, Gale NW, van Aelst L, Schlessinger J, Wigler MH, Bar-Sagi D. (1993) Human Sos1: a guanine nucleotide exchange factor for Ras that binds to GRB2. Science 260:1338–1343 [DOI] [PubMed] [Google Scholar]
- Chen JS, Exton JH. (2004) Regulation of phospholipase D2 activity by protein kinase C alpha. J Biol Chem 279:22076–22083 [DOI] [PubMed] [Google Scholar]
- Chen JS, Exton JH. (2005) Sites on phospholipase D2 phosphorylated by PKCalpha. Biochem Biophys Res Commun 333:1322–1326 [DOI] [PubMed] [Google Scholar]
- Chen Q, Hongu T, Sato T, Zhang Y, Ali W, Cavallo JA, van der Velden A, Tian H, Di Paolo G, Nieswandt B, et al. (2012) Key roles for the lipid signaling enzyme phospholipase d1 in the tumor microenvironment during tumor angiogenesis and metastasis. Sci Signal 5:ra79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zheng Y, Foster DA. (2003) Phospholipase D confers rapamycin resistance in human breast cancer cells. Oncogene 22:3937–3942 [DOI] [PubMed] [Google Scholar]
- Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Xu B, Wright A, Vanderbilt C, Cobb MH. (2001) MAP kinases. Chem Rev 101:2449–2476 [DOI] [PubMed] [Google Scholar]
- Cheng EHYA, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ. (2001) BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell 8:705–711 [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR. (2004) Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303:1010–1014 [DOI] [PubMed] [Google Scholar]
- Cho W, Stahelin RV. (2006) Membrane binding and subcellular targeting of C2 domains. Biochim Biophys Acta 1761:838–849 [DOI] [PubMed] [Google Scholar]
- Choi HJ, Han JS. (2012) Overexpression of phospholipase D enhances Bcl-2 expression by activating STAT3 through independent activation of ERK and p38MAPK in HeLa cells. Biochim Biophys Acta 1823:1082–1091 [DOI] [PubMed] [Google Scholar]
- Choi J, Chen J, Schreiber SL, Clardy J. (1996) Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science 273:239–242 [DOI] [PubMed] [Google Scholar]
- Choi SY, Huang P, Jenkins GM, Chan DC, Schiller J, Frohman MA. (2006) A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat Cell Biol 8:1255–1262 [DOI] [PubMed] [Google Scholar]
- Chong LD, Traynor-Kaplan A, Bokoch GM, Schwartz MA. (1994) The small GTP-binding protein Rho regulates a phosphatidylinositol 4-phosphate 5-kinase in mammalian cells. Cell 79:507–513 [DOI] [PubMed] [Google Scholar]
- Chuang TH, Bohl BP, Bokoch GM. (1993) Biologically active lipids are regulators of Rac.GDI complexation. J Biol Chem 268:26206–26211 [PubMed] [Google Scholar]
- Chung J, Kuo CJ, Crabtree GR, Blenis J. (1992) Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell 69:1227–1236 [DOI] [PubMed] [Google Scholar]
- Chung JK, Sekiya F, Kang HS, Lee C, Han JS, Kim SR, Bae YS, Morris AJ, Rhee SG. (1997) Synaptojanin inhibition of phospholipase D activity by hydrolysis of phosphatidylinositol 4,5-bisphosphate. J Biol Chem 272:15980–15985 [DOI] [PubMed] [Google Scholar]
- Cichowski K, Jacks T. (2001) NF1 tumor suppressor gene function: narrowing the GAP. Cell 104:593–604 [DOI] [PubMed] [Google Scholar]
- Clark KJ, Murray AW. (1995) Evidence that the bradykinin-induced activation of phospholipase D and of the mitogen-activated protein kinase cascade involve different protein kinase C isoforms. J Biol Chem 270:7097–7103 [DOI] [PubMed] [Google Scholar]
- Cobrinik D, Dowdy SF, Hinds PW, Mittnacht S, Weinberg RA. (1992) The retinoblastoma protein and the regulation of cell cycling. Trends Biochem Sci 17:312–315 [DOI] [PubMed] [Google Scholar]
- Cockcroft S, Thomas GM, Fensome A, Geny B, Cunningham E, Gout I, Hiles I, Totty NF, Truong O, Hsuan JJ. (1994) Phospholipase D: a downstream effector of ARF in granulocytes. Science 263:523–526 [DOI] [PubMed] [Google Scholar]
- Cohen JS, Brown HA. (2001) Phospholipases stimulate secretion in RBL mast cells. Biochemistry 40:6589–6597 [DOI] [PubMed] [Google Scholar]
- Cohen PTW. (2002) Protein phosphatase 1—targeted in many directions. J Cell Sci 115:241–256 [DOI] [PubMed] [Google Scholar]
- Colley WC, Altshuller YM, Sue-Ling CK, Copeland NG, Gilbert DJ, Jenkins NA, Branch KD, Tsirka SE, Bollag RJ, Bollag WB, et al. (1997a) Cloning and expression analysis of murine phospholipase D1. Biochem J 326:745–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colley WC, Sung TC, Roll R, Jenco J, Hammond SM, Altshuller Y, Bar-Sagi D, Morris AJ, Frohman MA. (1997b) Phospholipase D2, a distinct phospholipase D isoform with novel regulatory properties that provokes cytoskeletal reorganization. Curr Biol 7:191–201 [DOI] [PubMed] [Google Scholar]
- Colón-González F, Kazanietz MG. (2006) C1 domains exposed: from diacylglycerol binding to protein-protein interactions. Biochim Biophys Acta 1761:827–837 [DOI] [PubMed] [Google Scholar]
- Colville-Nash PR, Willoughby DA. (1997) Growth factors in angiogenesis: current interest and therapeutic potential. Mol Med Today 3:14–23 [DOI] [PubMed] [Google Scholar]
- Condeelis J, Pollard JW. (2006) Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 124:263–266 [DOI] [PubMed] [Google Scholar]
- Conricode KM, Brewer KA, Exton JH. (1992) Activation of phospholipase D by protein kinase C. Evidence for a phosphorylation-independent mechanism. J Biol Chem 267:7199–7202 [PubMed] [Google Scholar]
- Conricode KM, Smith JL, Burns DJ, Exton JH. (1994) Phospholipase D activation in fibroblast membranes by the α and β isoforms of protein kinase C. FEBS Lett 342:149–153 [DOI] [PubMed] [Google Scholar]
- Conti M, Beavo J. (2007) Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem 76:481–511 [DOI] [PubMed] [Google Scholar]
- Cook SJ, Wakelam MJ. (1992) Epidermal growth factor increases sn-1,2-diacylglycerol levels and activates phospholipase D-catalysed phosphatidylcholine breakdown in Swiss 3T3 cells in the absence of inositol-lipid hydrolysis. Biochem J 285:247–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbalán-Garcia S, Sánchez-Carrillo S, García-García J, Gómez-Fernández JC. (2003) Characterization of the membrane binding mode of the C2 domain of PKCε. Biochemistry 42:11661–11668 [DOI] [PubMed] [Google Scholar]
- Corda D, Colanzi A, Luini A. (2006) The multiple activities of CtBP/BARS proteins: the Golgi view. Trends Cell Biol 16:167–173 [DOI] [PubMed] [Google Scholar]
- Cross MJ, Roberts S, Ridley AJ, Hodgkin MN, Stewart A, Claesson-Welsh L, Wakelam MJ. (1996) Stimulation of actin stress fibre formation mediated by activation of phospholipase D. Curr Biol 6:588–597 [DOI] [PubMed] [Google Scholar]
- Cruchaga C, Karch CM, Jin SC, Benitez BA, Cai Y, Guerreiro R, Harari O, Norton J, Budde J, Bertelsen S, et al. UK Brain Expression Consortium. Alzheimer’s Research UK Consortium (2014) Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature 505:550–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czarny M, Lavie Y, Fiucci G, Liscovitch M. (1999) Localization of phospholipase D in detergent-insoluble, caveolin-rich membrane domains. Modulation by caveolin-1 expression and caveolin-182-101. J Biol Chem 274:2717–2724 [DOI] [PubMed] [Google Scholar]
- Czech MP. (2000) PIP2 and PIP3: complex roles at the cell surface. Cell 100:603–606 [DOI] [PubMed] [Google Scholar]
- D’Souza-Schorey C, Chavrier P. (2006) ARF proteins: roles in membrane traffic and beyond. Nat Rev Mol Cell Biol 7:347–358 [DOI] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM, Ahn S, Della Rocca GJ, Ferguson SS, Caron MG, Lefkowitz RJ. (1998) Essential role for G protein-coupled receptor endocytosis in the activation of mitogen-activated protein kinase. J Biol Chem 273:685–688 [DOI] [PubMed] [Google Scholar]
- Dall’Armi C, Hurtado-Lorenzo A, Tian H, Morel E, Nezu A, Chan RB, Yu WH, Robinson KS, Yeku O, Small SA, et al. (2010) The phospholipase D1 pathway modulates macroautophagy. Nat Commun 1:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV. (2012) MYC on the path to cancer. Cell 149:22–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV, Semenza GL. (1999) Oncogenic alterations of metabolism. Trends Biochem Sci 24:68–72 [DOI] [PubMed] [Google Scholar]
- Dann CE, 3rd, Bruick RK. (2005) Dioxygenases as O2-dependent regulators of the hypoxic response pathway. Biochem Biophys Res Commun 338:639–647 [DOI] [PubMed] [Google Scholar]
- De Craene B, Berx G. (2013) Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer 13:97–110 [DOI] [PubMed] [Google Scholar]
- De Luca A, Maiello MR, D’Alessio A, Pergameno M, Normanno N. (2012) The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets 16 (Suppl 2):S17–S27 [DOI] [PubMed] [Google Scholar]
- DebBurman SK, Ptasienski J, Boetticher E, Lomasney JW, Benovic JL, Hosey MM. (1995) Lipid-mediated regulation of G protein-coupled receptor kinases 2 and 3. J Biol Chem 270:5742–5747 [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. (2007) Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA 104:19345–19350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeChavigny A, Heacock PN, Dowhan W. (1991) Sequence and inactivation of the pss gene of Escherichia coli. Phosphatidylethanolamine may not be essential for cell viability. J Biol Chem 266:5323–5332 [PubMed] [Google Scholar]
- DeLeo FR, Quinn MT. (1996) Assembly of the phagocyte NADPH oxidase: molecular interaction of oxidase proteins. J Leukoc Biol 60:677–691 [DOI] [PubMed] [Google Scholar]
- Delon C, Manifava M, Wood E, Thompson D, Krugmann S, Pyne S, Ktistakis NT. (2004) Sphingosine kinase 1 is an intracellular effector of phosphatidic acid. J Biol Chem 279:44763–44774 [DOI] [PubMed] [Google Scholar]
- Deprez J, Vertommen D, Alessi DR, Hue L, Rider MH. (1997) Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the insulin signaling cascades. J Biol Chem 272:17269–17275 [DOI] [PubMed] [Google Scholar]
- DesMarais V, Ghosh M, Eddy R, Condeelis J. (2005) Cofilin takes the lead. J Cell Sci 118:19–26 [DOI] [PubMed] [Google Scholar]
- DeYonker NJ, Webster CE. (2013) Phosphoryl transfers of the phospholipase D superfamily: a quantum mechanical theoretical study. J Am Chem Soc 135:13764–13774 [DOI] [PubMed] [Google Scholar]
- Di Fulvio M, Frondorf K, Gomez-Cambronero J. (2008) Mutation of Y179 on phospholipase D2 (PLD2) upregulates DNA synthesis in a PI3K-and Akt-dependent manner. Cell Signal 20:176–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fulvio M, Frondorf K, Henkels KM, Grunwald WC, Jr, Cool D, Gomez-Cambronero J. (2012) Phospholipase D2 (PLD2) shortens the time required for myeloid leukemic cell differentiation: mechanism of action. J Biol Chem 287:393–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fulvio M, Lehman N, Lin X, Lopez I, Gomez-Cambronero J. (2006) The elucidation of novel SH2 binding sites on PLD2. Oncogene 25:3032–3040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divecha N, Roefs M, Halstead JR, D’Andrea S, Fernandez-Borga M, Oomen L, Saqib KM, Wakelam MJ, D’Santos C. (2000) Interaction of the type Ialpha PIPkinase with phospholipase D: a role for the local generation of phosphatidylinositol 4, 5-bisphosphate in the regulation of PLD2 activity. EMBO J 19:5440–5449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson JG. (2003) Multiple roles for Arf6: sorting, structuring, and signaling at the plasma membrane. J Biol Chem 278:41573–41576 [DOI] [PubMed] [Google Scholar]
- Du G, Altshuller YM, Kim Y, Han JM, Ryu SH, Morris AJ, Frohman MA. (2000) Dual requirement for rho and protein kinase C in direct activation of phospholipase D1 through G protein-coupled receptor signaling. Mol Biol Cell 11:4359–4368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du G, Altshuller YM, Vitale N, Huang P, Chasserot-Golaz S, Morris AJ, Bader MF, Frohman MA. (2003) Regulation of phospholipase D1 subcellular cycling through coordination of multiple membrane association motifs. J Cell Biol 162:305–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du G, Huang P, Liang BT, Frohman MA. (2004) Phospholipase D2 localizes to the plasma membrane and regulates angiotensin II receptor endocytosis. Mol Biol Cell 15:1024–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn KL, Espino PS, Drobic B, He S, Davie JR. (2005) The Ras-MAPK signal transduction pathway, cancer and chromatin remodeling. Biochem Cell Biol 83:1–14 [DOI] [PubMed] [Google Scholar]
- Dyson N. (1998) The regulation of E2F by pRB-family proteins. Genes Dev 12:2245–2262 [DOI] [PubMed] [Google Scholar]
- Edinger AL, Thompson CB. (2003) Defective autophagy leads to cancer. Cancer Cell 4:422–424 [DOI] [PubMed] [Google Scholar]
- Edwards DC, Gill GN. (1999) Structural features of LIM kinase that control effects on the actin cytoskeleton. J Biol Chem 274:11352–11361 [DOI] [PubMed] [Google Scholar]
- Edwards YS, Murray AW. (1995) Accumulation of phosphatidylalcohol in cultured cells: use of subcellular fractionation to investigate phospholipase D activity during signal transduction. Biochem J 308:473–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75:817–825 [DOI] [PubMed] [Google Scholar]
- Ellenbroek SI, Collard JG. (2007) Rho GTPases: functions and association with cancer. Clin Exp Metastasis 24:657–672 [DOI] [PubMed] [Google Scholar]
- Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, et al. (2004) Akt stimulates aerobic glycolysis in cancer cells. Cancer Res 64:3892–3899 [DOI] [PubMed] [Google Scholar]
- Elvers M, Stegner D, Hagedorn I, Kleinschnitz C, Braun A, Kuijpers MEJ, Boesl M, Chen Q, Heemskerk JWM, Stoll G, et al. (2010) Impaired alpha(IIb)beta(3) integrin activation and shear-dependent thrombus formation in mice lacking phospholipase D1. Sci Signal 3:ra1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emoto M, Klarlund JK, Waters SB, Hu V, Buxton JM, Chawla A, Czech MP. (2000) A role for phospholipase D in GLUT4 glucose transporter translocation. J Biol Chem 275:7144–7151 [DOI] [PubMed] [Google Scholar]
- Engelman JA, Luo J, Cantley LC. (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet 7:606–619 [DOI] [PubMed] [Google Scholar]
- Epand RM, Epand RF, Berno B, Pelosi L, Brandolin G. (2009) Association of phosphatidic acid with the bovine mitochondrial ADP/ATP carrier. Biochemistry 48:12358–12364 [DOI] [PubMed] [Google Scholar]
- Eskelinen EL. (2005) Maturation of autophagic vacuoles in Mammalian cells. Autophagy 1:1–10 [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S. (2004) Actin and microtubules in cell motility: which one is in control? Traffic 5:470–477 [DOI] [PubMed] [Google Scholar]
- Evan G, Littlewood T. (1998) A matter of life and cell death. Science 281:1317–1322 [DOI] [PubMed] [Google Scholar]
- Ewen ME, Sluss HK, Sherr CJ, Matsushime H, Kato J, Livingston DM. (1993) Functional interactions of the retinoblastoma protein with mammalian D-type cyclins. Cell 73:487–497 [DOI] [PubMed] [Google Scholar]
- Exton JH. (1999) Regulation of phospholipase D. Biochim Biophys Acta 1439:121–133 [DOI] [PubMed] [Google Scholar]
- Fagotto F, Gumbiner BM. (1996) Cell contact-dependent signaling. Dev Biol 180:445–454 [DOI] [PubMed] [Google Scholar]
- Fang Y, Park IH, Wu AL, Du G, Huang P, Frohman MA, Walker SJ, Brown HA, Chen J. (2003) PLD1 regulates mTOR signaling and mediates Cdc42 activation of S6K1. Curr Biol 13:2037–2044 [DOI] [PubMed] [Google Scholar]
- Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. (2001) Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 294:1942–1945 [DOI] [PubMed] [Google Scholar]
- Farquhar MJ, Powner DJ, Levine BA, Wright MH, Ladds G, Hodgkin MN. (2007) Interaction of PLD1b with actin in antigen-stimulated mast cells. Cell Signal 19:349–358 [DOI] [PubMed] [Google Scholar]
- Fensome A, Whatmore J, Morgan C, Jones D, Cockcroft S. (1998) ADP-ribosylation factor and Rho proteins mediate fMLP-dependent activation of phospholipase D in human neutrophils. J Biol Chem 273:13157–13164 [DOI] [PubMed] [Google Scholar]
- Ferguson KM, Lemmon MA, Schlessinger J, Sigler PB. (1995) Structure of the high affinity complex of inositol trisphosphate with a phospholipase C pleckstrin homology domain. Cell 83:1037–1046 [DOI] [PubMed] [Google Scholar]
- Ferguson SM, De Camilli P. (2012) Dynamin, a membrane-remodelling GTPase. Nat Rev Mol Cell Biol 13:75–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N, Alitalo K. (1999) Clinical applications of angiogenic growth factors and their inhibitors. Nat Med 5:1359–1364 [DOI] [PubMed] [Google Scholar]
- Finck BN, Gropler MC, Chen Z, Leone TC, Croce MA, Harris TE, Lawrence JC, Jr, Kelly DP. (2006) Lipin 1 is an inducible amplifier of the hepatic PGC-1α/PPARalpha regulatory pathway. Cell Metab 4:199–210 [DOI] [PubMed] [Google Scholar]
- Fisher GJ, Henderson PA, Voorhees JJ, Baldassare JJ. (1991) Epidermal growth factor-induced hydrolysis of phosphatidylcholine by phospholipase D and phospholipase C in human dermal fibroblasts. J Cell Physiol 146:309–317 [DOI] [PubMed] [Google Scholar]
- Formstecher E, Ramos JW, Fauquet M, Calderwood DA, Hsieh JC, Canton B, Nguyen XT, Barnier JV, Camonis J, Ginsberg MH, et al. (2001) PEA-15 mediates cytoplasmic sequestration of ERK MAP kinase. Dev Cell 1:239–250 [DOI] [PubMed] [Google Scholar]
- Foster DA, Xu L. (2003) Phospholipase D in cell proliferation and cancer. Mol Cancer Res 1:789–800 [PubMed] [Google Scholar]
- Frank C, Keilhack H, Opitz F, Zschörnig O, Böhmer FD. (1999) Binding of phosphatidic acid to the protein-tyrosine phosphatase SHP-1 as a basis for activity modulation. Biochemistry 38:11993–12002 [DOI] [PubMed] [Google Scholar]
- Frankel P, Ramos M, Flom J, Bychenok S, Joseph T, Kerkhoff E, Rapp UR, Feig LA, Foster DA. (1999) Ral and Rho-dependent activation of phospholipase D in v-Raf-transformed cells. Biochem Biophys Res Commun 255:502–507 [DOI] [PubMed] [Google Scholar]
- Freyberg Z, Sweeney D, Siddhanta A, Bourgoin S, Frohman M, Shields D. (2001) Intracellular localization of phospholipase D1 in mammalian cells. Mol Biol Cell 12:943–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley IG, Lam H, Wang J, Ding X, Chen S, Jiang X. (2009) ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 284:12297–12305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley IG, Walker SJ, Manifava M, Li D, Brown HA, Ktistakis NT. (2001) Interaction of phospholipase D1 with a casein-kinase-2-like serine kinase. Biochem J 354:369–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia A, Zheng Y, Zhao C, Toschi A, Fan J, Shraibman N, Brown HA, Bar-Sagi D, Foster DA, Arbiser JL. (2008) Honokiol suppresses survival signals mediated by Ras-dependent phospholipase D activity in human cancer cells. Clin Cancer Res 14:4267–4274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Alvarez B, de Pereda JM, Calderwood DA, Ulmer TS, Critchley D, Campbell ID, Ginsberg MH, Liddington RC. (2003) Structural determinants of integrin recognition by talin. Mol Cell 11:49–58 [DOI] [PubMed] [Google Scholar]
- Geiger B. (1979) A 130K protein from chicken gizzard: its localization at the termini of microfilament bundles in cultured chicken cells. Cell 18:193–205 [DOI] [PubMed] [Google Scholar]
- Geiger B, Spatz JP, Bershadsky AD. (2009) Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol 10:21–33 [DOI] [PubMed] [Google Scholar]
- Gera JF, Mellinghoff IK, Shi Y, Rettig MB, Tran C, Hsu JH, Sawyers CL, Lichtenstein AK. (2004) AKT activity determines sensitivity to mammalian target of rapamycin (mTOR) inhibitors by regulating cyclin D1 and c-myc expression. J Biol Chem 279:2737–2746 [DOI] [PubMed] [Google Scholar]
- Gerke V, Creutz CE, Moss SE. (2005) Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol 6:449–461 [DOI] [PubMed] [Google Scholar]
- Ghelli A, Porcelli AM, Facchini A, Hrelia S, Flamigni F, Rugolo M. (2002) Phospholipase D1 is threonine-phosphorylated in human-airway epithelial cells stimulated by sphingosine-1-phosphate by a mechanism involving Src tyrosine kinase and protein kinase Cdelta. Biochem J 366:187–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Moore S, Bell RM, Dush M. (2003) Functional analysis of a phosphatidic acid binding domain in human Raf-1 kinase: mutations in the phosphatidate binding domain lead to tail and trunk abnormalities in developing zebrafish embryos. J Biol Chem 278:45690–45696 [DOI] [PubMed] [Google Scholar]
- Ghosh S, Strum JC, Sciorra VA, Daniel L, Bell RM. (1996) Raf-1 kinase possesses distinct binding domains for phosphatidylserine and phosphatidic acid. Phosphatidic acid regulates the translocation of Raf-1 in 12-O-tetradecanoylphorbol-13-acetate-stimulated Madin-Darby canine kidney cells. J Biol Chem 271:8472–8480 [DOI] [PubMed] [Google Scholar]
- Gibbs TC, Meier KE. (2000) Expression and regulation of phospholipase D isoforms in mammalian cell lines. J Cell Physiol 182:77–87 [DOI] [PubMed] [Google Scholar]
- Ginsberg J, Gupta S, Matowe WC, Kline L, Brindley DN. (1997) Activation of phospholipase D in FRTL-5 thyroid cells by forskolin and dibutyryl-cyclic adenosine monophosphate. Endocrinology 138:3645–3651 [DOI] [PubMed] [Google Scholar]
- Gomez-Cambronero J. (1995) Immunoprecipitation of a phospholipase D activity with antiphosphotyrosine antibodies. J Interferon Cytokine Res 15:877–885 [DOI] [PubMed] [Google Scholar]
- Gorbatyuk OS, Li S, Nguyen FN, Manfredsson FP, Kondrikova G, Sullivan LF, Meyers C, Chen W, Mandel RJ, Muzyczka N. (2010) α-Synuclein expression in rat substantia nigra suppresses phospholipase D2 toxicity and nigral neurodegeneration. Mol Ther 18:1758–1768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorshkova I, He D, Berdyshev E, Usatuyk P, Burns M, Kalari S, Zhao Y, Pendyala S, Garcia JGN, Pyne NJ, et al. (2008) Protein kinase C-ϵ regulates sphingosine 1-phosphate-mediated migration of human lung endothelial cells through activation of phospholipase D2, protein kinase C-ζ, and Rac1. J Biol Chem 283:11794–11806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlin EB, Rudolph AE, Zhao Y, Matthews HR, Dixon JE. (1998) Catalytic mechanism of the phospholipase D superfamily proceeds via a covalent phosphohistidine intermediate. Proc Natl Acad Sci USA 95:9202–9207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grange M, Sette C, Cuomo M, Conti M, Lagarde M, Prigent AF, Némoz G. (2000) The cAMP-specific phosphodiesterase PDE4D3 is regulated by phosphatidic acid binding. Consequences for cAMP signaling pathway and characterization of a phosphatidic acid binding site. J Biol Chem 275:33379–33387 [DOI] [PubMed] [Google Scholar]
- Greer P. (2002) Closing in on the biological functions of Fps/Fes and Fer. Nat Rev Mol Cell Biol 3:278–289 [DOI] [PubMed] [Google Scholar]
- Ha KS, Exton JH. (1993) Activation of actin polymerization by phosphatidic acid derived from phosphatidylcholine in IIC9 fibroblasts. J Cell Biol 123:1789–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha SH, Kim DH, Kim IS, Kim JH, Lee MN, Lee HJ, Kim JH, Jang SK, Suh PG, Ryu SH. (2006) PLD2 forms a functional complex with mTOR/raptor to transduce mitogenic signals. Cell Signal 18:2283–2291 [DOI] [PubMed] [Google Scholar]
- Haas-Kogan D, Shalev N, Wong M, Mills G, Yount G, Stokoe D. (1998) Protein kinase B (PKB/Akt) activity is elevated in glioblastoma cells due to mutation of the tumor suppressor PTEN/MMAC. Curr Biol 8:1195–1198 [DOI] [PubMed] [Google Scholar]
- Haga Y, Miwa N, Jahangeer S, Okada T, Nakamura S. (2009) CtBP1/BARS is an activator of phospholipase D1 necessary for agonist-induced macropinocytosis. EMBO J 28:1197–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. (1999) Creation of human tumour cells with defined genetic elements. Nature 400:464–468 [DOI] [PubMed] [Google Scholar]
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. (2010) Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141:656–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond SM, Altshuller YM, Sung TC, Rudge SA, Rose K, Engebrecht J, Morris AJ, Frohman MA. (1995) Human ADP-ribosylation factor-activated phosphatidylcholine-specific phospholipase D defines a new and highly conserved gene family. J Biol Chem 270:29640–29643 [DOI] [PubMed] [Google Scholar]
- Hammond SM, Jenco JM, Nakashima S, Cadwallader K, Gu Q, Cook S, Nozawa Y, Prestwich GD, Frohman MA, Morris AJ. (1997) Characterization of two alternately spliced forms of phospholipase D1. Activation of the purified enzymes by phosphatidylinositol 4,5-bisphosphate, ADP-ribosylation factor, and Rho family monomeric GTP-binding proteins and protein kinase C-alpha. J Biol Chem 272:3860–3868 [DOI] [PubMed] [Google Scholar]
- Han JM, Kim JH, Lee BD, Lee SD, Kim Y, Jung YW, Lee S, Cho W, Ohba M, Kuroki T, et al. (2002a) Phosphorylation-dependent regulation of phospholipase D2 by protein kinase C delta in rat Pheochromocytoma PC12 cells. J Biol Chem 277:8290–8297 [DOI] [PubMed] [Google Scholar]
- Han JM, Kim Y, Lee JS, Lee CS, Lee BD, Ohba M, Kuroki T, Suh PG, Ryu SH. (2002b) Localization of phospholipase D1 to caveolin-enriched membrane via palmitoylation: implications for epidermal growth factor signaling. Mol Biol Cell 13:3976–3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Stope MB, de Jesús ML, Oude Weernink PA, Urban M, Wieland T, Rosskopf D, Mizuno K, Jakobs KH, Schmidt M. (2007) Direct stimulation of receptor-controlled phospholipase D1 by phospho-cofilin. EMBO J 26:4189–4202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. (2007) The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem 282:37298–37302 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Folkman J. (1996) Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86:353–364 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. (2000) The hallmarks of cancer. Cell 100:57–70 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. (2011) Hallmarks of cancer: the next generation. Cell 144:646–674 [DOI] [PubMed] [Google Scholar]
- Hardie DG. (2007) AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol 8:774–785 [DOI] [PubMed] [Google Scholar]
- Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. (1993) The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75:805–816 [DOI] [PubMed] [Google Scholar]
- Harrison SD, Broadie K, van de Goor J, Rubin GM. (1994) Mutations in the Drosophila Rop gene suggest a function in general secretion and synaptic transmission. Neuron 13:555–566 [DOI] [PubMed] [Google Scholar]
- Heasman SJ, Ridley AJ. (2008) Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9:690–701 [DOI] [PubMed] [Google Scholar]
- Hekman M, Hamm H, Villar AV, Bader B, Kuhlmann J, Nickel J, Rapp UR. (2002) Associations of B- and C-Raf with cholesterol, phosphatidylserine, and lipid second messengers: preferential binding of Raf to artificial lipid rafts. J Biol Chem 277:24090–24102 [DOI] [PubMed] [Google Scholar]
- Henage LG, Exton JH, Brown HA. (2006) Kinetic analysis of a mammalian phospholipase D: allosteric modulation by monomeric GTPases, protein kinase C, and polyphosphoinositides. J Biol Chem 281:3408–3417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkels KM, Mahankali M, Gomez-Cambronero J. (2013b) Increased cell growth due to a new lipase-GEF (Phospholipase D2) fastly acting on Ras. Cell Signal 25:198–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkels KM, Peng HJ, Frondorf K, Gomez-Cambronero J. (2010) A comprehensive model that explains the regulation of phospholipase D2 activity by phosphorylation-dephosphorylation. Mol Cell Biol 30:2251–2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkels KM, Short S, Peng HJ, Di Fulvio M, Gomez-Cambronero J. (2009) PLD2 has both enzymatic and cell proliferation-inducing capabilities, that are differentially regulated by phosphorylation and dephosphorylation. Biochem Biophys Res Commun 389:224–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley K, Venkova K, Christov A, Gunning W, Park J. (2011) Collapsin response mediator protein-2: an emerging pathologic feature and therapeutic target for neurodisease indications. Mol Neurobiol 43:180–191 [DOI] [PubMed] [Google Scholar]
- Hess JA, Ji QS, Carpenter G, Exton JH. (1998) Analysis of platelet-derived growth factor-induced phospholipase D activation in mouse embryo fibroblasts lacking phospholipase C-gamma1. J Biol Chem 273:20517–20524 [DOI] [PubMed] [Google Scholar]
- Hess JA, Ross AH, Qiu RG, Symons M, Exton JH. (1997) Role of Rho family proteins in phospholipase D activation by growth factors. J Biol Chem 272:1615–1620 [DOI] [PubMed] [Google Scholar]
- Hirai T, Chida K. (2003) Protein kinase Czeta (PKCzeta): activation mechanisms and cellular functions. J Biochem 133:1–7 [DOI] [PubMed] [Google Scholar]
- Hodgkin MN, Masson MR, Powner D, Saqib KM, Ponting CP, Wakelam MJ. (2000) Phospholipase D regulation and localisation is dependent upon a phosphatidylinositol 4,5-biphosphate-specific PH domain. Curr Biol 10:43–46 [DOI] [PubMed] [Google Scholar]
- Hofer F, Fields S, Schneider C, Martin GS. (1994) Activated Ras interacts with the Ral guanine nucleotide dissociation stimulator. Proc Natl Acad Sci USA 91:11089–11093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollstein M, Rice K, Greenblatt MS, Soussi T, Fuchs R, Sørlie T, Hovig E, Smith-Sørensen B, Montesano R, Harris CC. (1994) Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res 22:3551–3555 [PMC free article] [PubMed] [Google Scholar]
- Honda A, Nogami M, Yokozeki T, Yamazaki M, Nakamura H, Watanabe H, Kawamoto K, Nakayama K, Morris AJ, Frohman MA, et al. (1999) Phosphatidylinositol 4-phosphate 5-kinase α is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell 99:521–532 [DOI] [PubMed] [Google Scholar]
- Hood JD, Cheresh DA. (2002) Role of integrins in cell invasion and migration. Nat Rev Cancer 2:91–100 [DOI] [PubMed] [Google Scholar]
- Horn J, Lopez I, Miller MW, Gomez-Cambronero J. (2005) The uncovering of a novel regulatory mechanism for PLD2: formation of a ternary complex with protein tyrosine phosphatase PTP1B and growth factor receptor-bound protein GRB2. Biochem Biophys Res Commun 332:58–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al. (2009) Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 20:1981–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houben AJS, Moolenaar WH. (2011) Autotaxin and LPA receptor signaling in cancer. Cancer Metastasis Rev 30:557–565 [DOI] [PubMed] [Google Scholar]
- Hsu PP, Sabatini DM. (2008) Cancer cell metabolism: Warburg and beyond. Cell 134:703–707 [DOI] [PubMed] [Google Scholar]
- Hu T, Exton JH. (2003) Mechanisms of regulation of phospholipase D1 by protein kinase Calpha. J Biol Chem 278:2348–2355 [DOI] [PubMed] [Google Scholar]
- Huang P, Altshuller YM, Hou JC, Pessin JE, Frohman MA. (2005) Insulin-stimulated plasma membrane fusion of Glut4 glucose transporter-containing vesicles is regulated by phospholipase D1. Mol Biol Cell 16:2614–2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui L, Abbas T, Pielak RM, Joseph T, Bargonetti J, Foster DA. (2004) Phospholipase D elevates the level of MDM2 and suppresses DNA damage-induced increases in p53. Mol Cell Biol 24:5677–5686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui L, Zheng Y, Yan Y, Bargonetti J, Foster DA. (2006) Mutant p53 in MDA-MB-231 breast cancer cells is stabilized by elevated phospholipase D activity and contributes to survival signals generated by phospholipase D. Oncogene 25:7305–7310 [DOI] [PubMed] [Google Scholar]
- Imamura F, Horai T, Mukai M, Shinkai K, Sawada M, Akedo H. (1993) Induction of in vitro tumor cell invasion of cellular monolayers by lysophosphatidic acid or phospholipase D. Biochem Biophys Res Commun 193:497–503 [DOI] [PubMed] [Google Scholar]
- Itakura E, Kishi C, Inoue K, Mizushima N. (2008) Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 19:5360–5372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, De Camilli P. (2006) BAR, F-BAR (EFC) and ENTH/ANTH domains in the regulation of membrane-cytosol interfaces and membrane curvature. Biochim Biophys Acta 1761:897–912 [DOI] [PubMed] [Google Scholar]
- Itoh T, Hasegawa J, Tsujita K, Kanaho Y, Takenawa T. (2009) The tyrosine kinase Fer is a downstream target of the PLD-PA pathway that regulates cell migration. Sci Signal 2:ra52. [DOI] [PubMed] [Google Scholar]
- Ivanisevic R, Milić M, Ajdić D, Rakonjac J, Savić DJ. (1995) Nucleotide sequence, mutational analysis, transcriptional start site, and product analysis of nov, the gene which affects Escherichia coli K-12 resistance to the gyrase inhibitor novobiocin. J Bacteriol 177:1766–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer SS, Agrawal RS, Thompson CR, Thompson S, Barton JA, Kusner DJ. (2006) Phospholipase D1 regulates phagocyte adhesion. J Immunol 176:3686–3696 [DOI] [PubMed] [Google Scholar]
- Jackson TR, Brown FD, Nie Z, Miura K, Foroni L, Sun J, Hsu VW, Donaldson JG, Randazzo PA. (2000) ACAPs are arf6 GTPase-activating proteins that function in the cell periphery. J Cell Biol 151:627–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang IH, Lee S, Park JB, Kim JH, Lee CS, Hur EM, Kim IS, Kim KT, Yagisawa H, Suh PG, et al. (2003) The direct interaction of phospholipase C-γ 1 with phospholipase D2 is important for epidermal growth factor signaling. J Biol Chem 278:18184–18190 [DOI] [PubMed] [Google Scholar]
- Jang YH, Ahn BH, Namkoong S, Kim YM, Jin JK, Kim YS, Min S. (2008a) Differential regulation of apoptosis by caspase-mediated cleavage of phospholipase D isozymes. Cell Signal 20:2198–2207 [DOI] [PubMed] [Google Scholar]
- Jang YH, Namkoong S, Kim YM, Lee SJ, Park BJ, Min DS. (2008b) Cleavage of phospholipase D1 by caspase promotes apoptosis via modulation of the p53-dependent cell death pathway. Cell Death Differ 15:1782–1793 [DOI] [PubMed] [Google Scholar]
- Jarquin-Pardo M, Fitzpatrick A, Galiano FJ, First EA, Davis JN. (2007) Phosphatidic acid regulates the affinity of the murine phosphatidylinositol 4-phosphate 5-kinase-Ibeta for phosphatidylinositol-4-phosphate. J Cell Biochem 100:112–128 [DOI] [PubMed] [Google Scholar]
- Jenco JM, Rawlingson A, Daniels B, Morris AJ. (1998) Regulation of phospholipase D2: selective inhibition of mammalian phospholipase D isoenzymes by α- and β-synucleins. Biochemistry 37:4901–4909 [DOI] [PubMed] [Google Scholar]
- Jenkins GH, Fisette PL, Anderson RA. (1994) Type I phosphatidylinositol 4-phosphate 5-kinase isoforms are specifically stimulated by phosphatidic acid. J Biol Chem 269:11547–11554 [PubMed] [Google Scholar]
- Jeon H, Kwak D, Noh J, Lee MN, Lee CS, Suh PG, Ryu SH. (2011) Phospholipase D2 induces stress fiber formation through mediating nucleotide exchange for RhoA. Cell Signal 23:1320–1326 [DOI] [PubMed] [Google Scholar]
- Jesionowska A, Cecerska E, Dolegowska B. (2014) Methods for quantifying lysophosphatidic acid in body fluids: a review. Anal Biochem 453:38–43 [DOI] [PubMed] [Google Scholar]
- Jiang H, Lu Z, Luo JQ, Wolfman A, Foster DA. (1995a) Ras mediates the activation of phospholipase D by v-Src. J Biol Chem 270:6006–6009 [DOI] [PubMed] [Google Scholar]
- Jiang H, Luo JQ, Urano T, Frankel P, Lu Z, Foster DA, Feig LA. (1995b) Involvement of Ral GTPase in v-Src-induced phospholipase D activation. Nature 378:409–412 [DOI] [PubMed] [Google Scholar]
- Jiang RT, Shyy YJ, Tsai MD. (1984) Phospholipids chiral at phosphorus. Absolute configuration of chiral thiophospholipids and stereospecificity of phospholipase D. Biochemistry 23:1661–1667 [DOI] [PubMed] [Google Scholar]
- Jiang YW, Song J, Zang Q, Foster DA. (1994) Phosphatidylcholine-specific phospholipase D activity is elevated in v-Fps-transformed cells. Biochem Biophys Res Commun 203:1195–2003 [DOI] [PubMed] [Google Scholar]
- Jones DH, Bax B, Fensome A, Cockcroft S. (1999) ADP ribosylation factor 1 mutants identify a phospholipase D effector region and reveal that phospholipase D participates in lysosomal secretion but is not sufficient for recruitment of coatomer I. Biochem J 341:185–192 [PMC free article] [PubMed] [Google Scholar]
- Jones GA, Carpenter G. (1993) The regulation of phospholipase C-gamma 1 by phosphatidic acid. Assessment of kinetic parameters. J Biol Chem 268:20845–20850 [PubMed] [Google Scholar]
- Jones JA, Hannun YA. (2002) Tight binding inhibition of protein phosphatase-1 by phosphatidic acid. Specificity of inhibition by the phospholipid. J Biol Chem 277:15530–15538 [DOI] [PubMed] [Google Scholar]
- Jones JA, Rawles R, Hannun YA. (2013) Identification of a novel phosphatidic acid binding domain in protein phosphatase-1. Biochemistry 44:13235–13245 [DOI] [PubMed] [Google Scholar]
- Jose Lopez-Andreo M, Gomez-Fernandez JC, Corbalan-Garcia S. (2003) The simultaneous production of phosphatidic acid and diacylglycerol is essential for the translocation of protein kinase Cepsilon to the plasma membrane in RBL-2H3 cells. Mol Biol Cell 14:4885–4895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph T, Bryant A, Frankel P, Wooden R, Kerkhoff E, Rapp UR, Foster DA. (2002) Phospholipase D overcomes cell cycle arrest induced by high-intensity Raf signaling. Oncogene 21:3651–3658 [DOI] [PubMed] [Google Scholar]
- Joseph T, Wooden R, Bryant A, Zhong M, Lu Z, Foster DA. (2001) Transformation of cells overexpressing a tyrosine kinase by phospholipase D1 and D2. Biochem Biophys Res Commun 289:1019–1024 [DOI] [PubMed] [Google Scholar]
- Jovanovic OA, Brown FD, Donaldson JG. (2006) An effector domain mutant of Arf6 implicates phospholipase D in endosomal membrane recycling. Mol Biol Cell 17:327–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CH, Ro SH, Cao J, Otto NM, Kim DH. (2010) mTOR regulation of autophagy. FEBS Lett 584:1287–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG, Jr, Ratcliffe PJ. (2008) Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 30:393–402 [DOI] [PubMed] [Google Scholar]
- Kahn RA, Gilman AG. (1984) Purification of a protein cofactor required for ADP-ribosylation of the stimulatory regulatory component of adenylate cyclase by cholera toxin. J Biol Chem 259:6228–6234 [PubMed] [Google Scholar]
- Kaibuchi K, Kuroda S, Amano M. (1999) Regulation of the cytoskeleton and cell adhesion by the Rho family GTPases in mammalian cells. Annu Rev Biochem 68:459–486 [DOI] [PubMed] [Google Scholar]
- Kam Y, Exton JH. (2001) Phospholipase D activity is required for actin stress fiber formation in fibroblasts. Mol Cell Biol 21:4055–4066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai F, Liu H, Field SJ, Akbary H, Matsuo T, Brown GE, Cantley LC, Yaffe MB. (2001) The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat Cell Biol 3:675–678 [DOI] [PubMed] [Google Scholar]
- Kanda S, Miyata Y, Kanetake H, Smithgall TE. (2007) Non-receptor protein-tyrosine kinases as molecular targets for antiangiogenic therapy (Review). Int J Mol Med 20:113–121 [PubMed] [Google Scholar]
- Kang DW, Lee SH, Yoon JW, Park WS, Choi KY, Min S. (2010) Phospholipase D1 drives a positive feedback loop to reinforce the Wnt/beta-catenin/TCF signaling axis. Cancer Res 70:4233–4242 [DOI] [PubMed] [Google Scholar]
- Kang DW, Park MH, Lee YJ, Kim HS, Kwon TK, Park WS, Min S. (2008) Phorbol ester up-regulates phospholipase D1 but not phospholipase D2 expression through a PKC/Ras/ERK/NFkappaB-dependent pathway and enhances matrix metalloproteinase-9 secretion in colon cancer cells. J Biol Chem 283:4094–4104 [DOI] [PubMed] [Google Scholar]
- Kang DW, Park MH, Lee YJ, Kim HS, Lindsley CW, Alex Brown H, Min S. (2011) Autoregulation of phospholipase D activity is coupled to selective induction of phospholipase D1 expression to promote invasion of breast cancer cells. Int J Cancer 128:805–816 [DOI] [PubMed] [Google Scholar]
- Kantonen S, Hatton N, Mahankali M, Henkels KM, Park H, Cox D, Gomez-Cambronero J. (2011) A novel phospholipase D2-Grb2-WASp heterotrimer regulates leukocyte phagocytosis in a two-step mechanism. Mol Cell Biol 31:4524–4537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karathanassis D, Stahelin RV, Bravo J, Perisic O, Pacold CM, Cho W, Williams RL. (2002) Binding of the PX domain of p47(phox) to phosphatidylinositol 3,4-bisphosphate and phosphatidic acid is masked by an intramolecular interaction. EMBO J 21:5057–5068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai T, Ohguchi K, Nakashima S, Ito Y, Naganawa T, Kondo N, Nozawa Y. (1998) Increased activity of oleate-dependent type phospholipase D during actinomycin D-induced apoptosis in Jurkat T cells. J Immunol 161:6469–6474 [PubMed] [Google Scholar]
- Kato J, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ. (1993) Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev 7:331–342 [DOI] [PubMed] [Google Scholar]
- Kato Y, Lambert CA, Colige AC, Mineur P, Noël A, Frankenne F, Foidart JM, Baba M, Hata R, Miyazaki K, et al. (2005) Acidic extracellular pH induces matrix metalloproteinase-9 expression in mouse metastatic melanoma cells through the phospholipase D-mitogen-activated protein kinase signaling. J Biol Chem 280:10938–10944 [DOI] [PubMed] [Google Scholar]
- Kawamata T, Taniguchi T, Mukai H, Kitagawa M, Hashimoto T, Maeda K, Ono Y, Tanaka C. (1998) A protein kinase, PKN, accumulates in Alzheimer neurofibrillary tangles and associated endoplasmic reticulum-derived vesicles and phosphorylates tau protein. J Neurosci 18:7402–7410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen JH. (1990) Clathrin and associated assembly and disassembly proteins. Annu Rev Biochem 59:415–438 [DOI] [PubMed] [Google Scholar]
- Kelly PN, Strasser A. (2011) The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ 18:1414–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy EP. (1987) Metabolism and function of membrane lipids. Klin Wochenschr 65:205–212 [DOI] [PubMed] [Google Scholar]
- Kessenbrock K, Plaks V, Werb Z. (2010) Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141:52–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan WA, Blobe GC, Richards AL, Hannun YA. (1994) Identification, partial purification, and characterization of a novel phospholipid-dependent and fatty acid-activated protein kinase from human platelets. J Biol Chem 269:9729–9735 [PubMed] [Google Scholar]
- Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. (2001) Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep 2:330–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi A. (2003) Tumor formation by genetic mutations in the components of the Wnt signaling pathway. Cancer Sci 94:225–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. (2011a) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13:132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lee YH, Kwon TK, Chang JS, Chung KC, Min DS. (2006) Phospholipase D prevents etoposide-induced apoptosis by inhibiting the expression of early growth response-1 and phosphatase and tensin homologue deleted on chromosome 10. Cancer Res 66:784–793 [DOI] [PubMed] [Google Scholar]
- Kim JH, Kim HY, Lee YK, Yoon YS, Xu WG, Yoon JK, Choi SE, Ko YG, Kim MJ, Lee SJ, et al. (2011b) Involvement of mitophagy in oncogenic K-Ras-induced transformation: overcoming a cellular energy deficit from glucose deficiency. Autophagy 7:1187–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Lee S, Kim JH, Lee TG, Hirata M, Suh PG, Ryu SH. (2002) Phospholipase D2 directly interacts with aldolase via Its PH domain. Biochemistry 41:3414–3421 [DOI] [PubMed] [Google Scholar]
- Kim JH, Lee S, Park JB, Lee SD, Kim JH, Ha SH, Hasumi K, Endo A, Suh PG, Ryu SH. (2003a) Hydrogen peroxide induces association between glyceraldehyde 3-phosphate dehydrogenase and phospholipase D2 to facilitate phospholipase D2 activation in PC12 cells. J Neurochem 85:1228–1236 [DOI] [PubMed] [Google Scholar]
- Kim JH, Park JM, Yea K, Kim HW, Suh PG, Ryu SH. (2010) Phospholipase D1 mediates AMP-activated protein kinase signaling for glucose uptake. PLoS ONE 5:e9600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim L, Wong TW. (1998) Growth factor-dependent phosphorylation of the actin-binding protein cortactin is mediated by the cytoplasmic tyrosine kinase FER. J Biol Chem 273:23542–23548 [DOI] [PubMed] [Google Scholar]
- Kim MJ, Woo SJ, Yoon CH, Lee JS, An S, Choi YH, Hwang SG, Yoon G, Lee SJ. (2011c) Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J Biol Chem 286:12924–12932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SW, Hayashi M, Lo JF, Yang Y, Yoo JS, Lee JD. (2003b) ADP-ribosylation factor 4 small GTPase mediates epidermal growth factor receptor-dependent phospholipase D2 activation. J Biol Chem 278:2661–2668 [DOI] [PubMed] [Google Scholar]
- Kim Y, Han JM, Park JB, Lee SD, Oh YS, Chung C, Lee TG, Kim JH, Park SK, Yoo JS, et al. (1999a) Phosphorylation and activation of phospholipase D1 by protein kinase C in vivo: determination of multiple phosphorylation sites. Biochemistry 38:10344–10351 [DOI] [PubMed] [Google Scholar]
- Kim Y, Kim JE, Lee SD, Lee TG, Kim JH, Park JB, Han JM, Jang SK, Suh PG, Ryu SH. (1999b) Phospholipase D1 is located and activated by protein kinase C alpha in the plasma membrane in 3Y1 fibroblast cell. Biochim Biophys Acta 1436:319–330 [DOI] [PubMed] [Google Scholar]
- Kim YR, Byun HS, Won M, Park KA, Kim JM, Choi BL, Lee H, Hong JH, Park J, Seok JH, et al. (2008) Modulatory role of phospholipase D in the activation of signal transducer and activator of transcription (STAT)-3 by thyroid oncogenic kinase RET/PTC. BMC Cancer 8:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa M, Mukai H, Shibata H, Ono Y. (1995) Purification and characterization of a fatty acid-activated protein kinase (PKN) from rat testis. Biochem J 310:657–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg M. (2008) The ADP and ATP transport in mitochondria and its carrier. Biochim Biophys Acta 1778:1978–2021 [DOI] [PubMed] [Google Scholar]
- Knævelsrud H, Simonsen A. (2012) Lipids in autophagy: constituents, signaling molecules and cargo with relevance to disease. Biochim Biophys Acta 1821:1133–1145 [DOI] [PubMed] [Google Scholar]
- Knapek K, Frondorf K, Post J, Short S, Cox D, Gomez-Cambronero J. (2010) The molecular basis of phospholipase D2-induced chemotaxis: elucidation of differential pathways in macrophages and fibroblasts. Mol Cell Biol 30:4492–4506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus UG, Morris S, Dong HJ, Chernoff J, Bokoch GM. (1995) Regulation of human leukocyte p21-activated kinases through G protein—coupled receptors. Science 269:221–223 [DOI] [PubMed] [Google Scholar]
- Knoepp SM, Chahal MS, Xie Y, Zhang Z, Brauner DJ, Hallman MA, Robinson SA, Han S, Imai M, Tomlinson S, et al. (2008) Effects of active and inactive phospholipase D2 on signal transduction, adhesion, migration, invasion, and metastasis in EL4 lymphoma cells. Mol Pharmacol 74:574–584 [DOI] [PubMed] [Google Scholar]
- Koch T, Brandenburg LO, Schulz S, Liang Y, Klein J, Höllt V. (2003) ADP-ribosylation factor-dependent phospholipase D2 activation is required for agonist-induced μ-opioid receptor endocytosis. J Biol Chem 278:9979–9985 [DOI] [PubMed] [Google Scholar]
- Kodaki T, Yamashita S. (1997) Cloning, expression, and characterization of a novel phospholipase D complementary DNA from rat brain. J Biol Chem 272:11408–11413 [DOI] [PubMed] [Google Scholar]
- Koltin Y, Faucette L, Bergsma DJ, Levy MA, Cafferkey R, Koser PL, Johnson RK, Livi GP. (1991) Rapamycin sensitivity in Saccharomyces cerevisiae is mediated by a peptidyl-prolyl cis-trans isomerase related to human FK506-binding protein. Mol Cell Biol 11:1718–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komati H, Naro F, Mebarek S, De Arcangelis V, Adamo S, Lagarde M, Prigent AF, Némoz G. (2005) Phospholipase D is involved in myogenic differentiation through remodeling of actin cytoskeleton. Mol Biol Cell 16:1232–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV. (1996) A duplicated catalytic motif in a new superfamily of phosphohydrolases and phospholipid synthases that includes poxvirus envelope proteins. Trends Biochem Sci 21:242–243 [PubMed] [Google Scholar]
- Kovac B, Teo JL, Mäkelä TP, Vallenius T. (2013) Assembly of non-contractile dorsal stress fibers requires α-actinin-1 and Rac1 in migrating and spreading cells. J Cell Sci 126:263–273 [DOI] [PubMed] [Google Scholar]
- Kozasa T, Jiang X, Hart MJ, Sternweis PM, Singer WD, Gilman AG, Bollag G, Sternweis PC. (1998) p115 RhoGEF, a GTPase activating protein for Galpha12 and Galpha13. Science 280:2109–2111 [DOI] [PubMed] [Google Scholar]
- Krebs HA. (1972) The Pasteur effect and the relations between respiration and fermentation. Essays Biochem 8:1–34 [PubMed] [Google Scholar]
- Kuribara H, Tago K, Yokozeki T, Sasaki T, Takai Y, Morii N, Narumiya S, Katada T, Kanaho Y. (1995) Synergistic activation of rat brain phospholipase D by ADP-ribosylation factor and rhoA p21, and its inhibition by Clostridium botulinum C3 exoenzyme. J Biol Chem 270:25667–25671 [DOI] [PubMed] [Google Scholar]
- Kurland JF, Voehringer DW, Meyn RE. (2003) The MEK/ERK pathway acts upstream of NF kappa B1 (p50) homodimer activity and Bcl-2 expression in a murine B-cell lymphoma cell line. MEK inhibition restores radiation-induced apoptosis. J Biol Chem 278:32465–32470 [DOI] [PubMed] [Google Scholar]
- Kurooka T, Yamamoto Y, Takai Y, Sakisaka T. (2011) Dual regulation of RA-RhoGAP activity by phosphatidic acid and Rap1 during neurite outgrowth. J Biol Chem 286:6832–6843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusner DJ, Barton JA, Wen KK, Wang X, Rubenstein PA, Iyer SS. (2002) Regulation of phospholipase D activity by actin. Actin exerts bidirectional modulation of Mammalian phospholipase D activity in a polymerization-dependent, isoform-specific manner. J Biol Chem 277:50683–50692 [DOI] [PubMed] [Google Scholar]
- Kutateladze TG. (2006) Phosphatidylinositol 3-phosphate recognition and membrane docking by the FYVE domain. Biochim Biophys Acta 1761:868–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak JY, Lopez I, Uhlinger DJ, Ryu SH, Lambeth JD. (1995) RhoA and a cytosolic 50-kDa factor reconstitute GTP gamma S-dependent phospholipase D activity in human neutrophil subcellular fractions. J Biol Chem 270:27093–27098 [DOI] [PubMed] [Google Scholar]
- Kwon YG, Lee SY, Choi Y, Greengard P, Nairn AC. (1997) Cell cycle-dependent phosphorylation of mammalian protein phosphatase 1 by cdc2 kinase. Proc Natl Acad Sci USA 94:2168–2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwun HJ, Lee JH, Min DS, Jang KL. (2003) Transcriptional repression of cyclin-dependent kinase inhibitor p21 gene by phospholipase D1 and D2. FEBS Lett 544:38–44 [DOI] [PubMed] [Google Scholar]
- Land H, Parada LF, Weinberg RA. (1983) Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 304:596–602 [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. (2012) mTOR signaling in growth control and disease. Cell 149:274–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffenburger DA, Horwitz AF. (1996) Cell migration: a physically integrated molecular process. Cell 84:359–369 [DOI] [PubMed] [Google Scholar]
- Lavieri R, Scott SA, Lewis JA, Selvy PE, Armstrong MD, Alex Brown H, Lindsley CW. (2009) Design and synthesis of isoform-selective phospholipase D (PLD) inhibitors. Part II. Identification of the 1,3,8-triazaspiro[4,5]decan-4-one privileged structure that engenders PLD2 selectivity. Bioorg Med Chem Lett 19:2240–2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavieri RR, Scott SA, Selvy PE, Kim K, Jadhav S, Morrison RD, Daniels JS, Brown HA, Lindsley CW. (2010) Design, synthesis, and biological evaluation of halogenated N-(2-(4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)ethyl)benzamides: discovery of an isoform-selective small molecule phospholipase D2 inhibitor. J Med Chem 53:6706–6719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Kang HS, Chung JK, Sekiya F, Kim JR, Han JS, Kim SR, Bae YS, Morris AJ, Rhee SG. (1997a) Inhibition of phospholipase D by clathrin assembly protein 3 (AP3). J Biol Chem 272:15986–15992 [DOI] [PubMed] [Google Scholar]
- Lee C, Kim SR, Chung JK, Frohman MA, Kilimann MW, Rhee SG. (2000) Inhibition of phospholipase D by amphiphysins. J Biol Chem 275:18751–18758 [DOI] [PubMed] [Google Scholar]
- Lee CS, Kim IS, Park JB, Lee MN, Lee HY, Suh PG, Ryu SH. (2006) The phox homology domain of phospholipase D activates dynamin GTPase activity and accelerates EGFR endocytosis. Nat Cell Biol 8:477–484 [DOI] [PubMed] [Google Scholar]
- Lee HY, Jung H, Jang IH, Suh PG, Ryu SH. (2008) Cdk5 phosphorylates PLD2 to mediate EGF-dependent insulin secretion. Cell Signal 20:1787–1794 [DOI] [PubMed] [Google Scholar]
- Lee HY, Park JB, Jang IH, Chae YC, Kim JH, Kim IS, Suh PG, Ryu SH. (2004) Munc-18-1 inhibits phospholipase D activity by direct interaction in an epidermal growth factor-reversible manner. J Biol Chem 279:16339–16348 [DOI] [PubMed] [Google Scholar]
- Lee JS, Kim JH, Jang IH, Kim HS, Han JM, Kazlauskas A, Yagisawa H, Suh PG, Ryu SH. (2005) Phosphatidylinositol (3,4,5)-trisphosphate specifically interacts with the phox homology domain of phospholipase D1 and stimulates its activity. J Cell Sci 118:4405–4413 [DOI] [PubMed] [Google Scholar]
- Lee S, Kim JH, Lee CS, Kim JH, Kim Y, Heo K, Ihara Y, Goshima Y, Suh PG, Ryu SH. (2002) Collapsin response mediator protein-2 inhibits neuronal phospholipase D(2) activity by direct interaction. J Biol Chem 277:6542–6549 [DOI] [PubMed] [Google Scholar]
- Lee S, Park JB, Kim JH, Kim Y, Kim JH, Shin KJ, Lee JS, Ha SH, Suh PG, Ryu SH. (2001) Actin directly interacts with phospholipase D, inhibiting its activity. J Biol Chem 276:28252–28260 [DOI] [PubMed] [Google Scholar]
- Lee TG, Park JB, Lee SD, Hong S, Kim JH, Kim Y, Yi KS, Bae S, Hannun YA, Obeid LM, et al. (1997b) Phorbol myristate acetate-dependent association of protein kinase C alpha with phospholipase D1 in intact cells. Biochim Biophys Acta 1347:199–204 [DOI] [PubMed] [Google Scholar]
- Lehman N, Di Fulvio M, McCray N, Campos I, Tabatabaian F, Gomez-Cambronero J. (2006) Phagocyte cell migration is mediated by phospholipases PLD1 and PLD2. Blood 108:3564–3572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman N, Ledford B, Di Fulvio M, Frondorf K, McPhail LC, Gomez-Cambronero J. (2007) Phospholipase D2-derived phosphatidic acid binds to and activates ribosomal p70 S6 kinase independently of mTOR. FASEB J 21:1075–1087 [DOI] [PubMed] [Google Scholar]
- Lemmon MA. (2008) Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol 9:99–111 [DOI] [PubMed] [Google Scholar]
- Lewis JA, Scott SA, Lavieri R, Buck JR, Selvy PE, Stoops SL, Armstrong MD, Brown HA, Lindsley CW. (2009) Design and synthesis of isoform-selective phospholipase D (PLD) inhibitors. Part I: Impact of alternative halogenated privileged structures for PLD1 specificity. Bioorg Med Chem Lett 19:1916–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HS, Shome K, Rojas R, Rizzo MA, Vasudevan C, Fluharty E, Santy LC, Casanova JE, Romero G. (2003) The guanine nucleotide exchange factor ARNO mediates the activation of ARF and phospholipase D by insulin. BMC Cell Biol 4:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang JO, Sung TC, Morris AJ, Frohman MA, Kornfeld S. (1997) Different domains of mammalian ADP-ribosylation factor 1 mediate interaction with selected target proteins. J Biol Chem 272:33001–33008 [DOI] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402:672–676 [DOI] [PubMed] [Google Scholar]
- Libermann TA, Nusbaum HR, Razon N, Kris R, Lax I, Soreq H, Whittle N, Waterfield MD, Ullrich A, Schlessinger J. (1985) Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature 313:144–147 [DOI] [PubMed] [Google Scholar]
- Lilja L, Johansson JU, Gromada J, Mandic SA, Fried G, Berggren PO, Bark C. (2004) Cyclin-dependent kinase 5 associated with p39 promotes Munc18-1 phosphorylation and Ca(2+)-dependent exocytosis. J Biol Chem 279:29534–29541 [DOI] [PubMed] [Google Scholar]
- Limatola C, Schaap D, Moolenaar WH, van Blitterswijk WJ. (1994) Phosphatidic acid activation of protein kinase C-zeta overexpressed in COS cells: comparison with other protein kinase C isotypes and other acidic lipids. Biochem J 304:1001–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley CW, Brown HA. (2012) Phospholipase D as a therapeutic target in brain disorders. Neuropsychopharmacology 37:301–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling LE, Schulz JT, Cantley LC. (1989) Characterization and purification of membrane-associated phosphatidylinositol-4-phosphate kinase from human red blood cells. J Biol Chem 264:5080–5088 [PubMed] [Google Scholar]
- Liscovitch M, Chalifa V, Pertile P, Chen CS, Cantley LC. (1994) Novel function of phosphatidylinositol 4,5-bisphosphate as a cofactor for brain membrane phospholipase D. J Biol Chem 269:21403–21406 [PubMed] [Google Scholar]
- Litosch I. (2000) Regulation of phospholipase C-β(1) activity by phosphatidic acid. Biochemistry 39:7736–7743 [DOI] [PubMed] [Google Scholar]
- Litosch I. (2003) Regulation of phospholipase C-β activity by phosphatidic acid: isoform dependence, role of protein kinase C, and G protein subunits. Biochemistry 42:1618–1623 [DOI] [PubMed] [Google Scholar]
- Litosch I, Pujari R, Lee SJ. (2009) Phosphatidic acid regulates signal output by G protein coupled receptors through direct interaction with phospholipase C-β(1). Cell Signal 21:1379–1384 [DOI] [PubMed] [Google Scholar]
- Liu MY, Gutowski S, Sternweis PC. (2001) The C terminus of mammalian phospholipase D is required for catalytic activity. J Biol Chem 276:5556–5562 [DOI] [PubMed] [Google Scholar]
- Liu L, Xie R, Nguyen S, Ye M, McKeehan WL. (2009) Robust autophagy/mitophagy persists during mitosis. Cell Cycle 8:1616–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Käch A, Ziegler U, Ong ACM, Wallace DP, Arcaro A, Serra AL. (2013) The role of phospholipase D in modulating the MTOR signaling pathway in polycystic kidney disease. PLoS ONE 8:e73173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llić DK, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T, et al. (1995) Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature 377:539–544 [DOI] [PubMed] [Google Scholar]
- Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. (2005) Rheb binds and regulates the mTOR kinase. Curr Biol 15:702–713 [DOI] [PubMed] [Google Scholar]
- Lopez I, Arnold RS, Lambeth JD. (1998) Cloning and initial characterization of a human phospholipase D2 (hPLD2). ADP-ribosylation factor regulates hPLD2. J Biol Chem 273:12846–12852 [DOI] [PubMed] [Google Scholar]
- Lopez I, Burns DJ, Lambeth JD. (1995) Regulation of phospholipase D by protein kinase C in human neutrophils. Conventional isoforms of protein kinase C phosphorylate a phospholipase D-related component in the plasma membrane. J Biol Chem 270:19465–19472 [DOI] [PubMed] [Google Scholar]
- López De Jesús M, Stope MB, Oude Weernink PA, Mahlke Y, Börgermann C, Ananaba VN, Rimmbach C, Rosskopf D, Michel MC, Jakobs KH, et al. (2006) Cyclic AMP-dependent and Epac-mediated activation of R-Ras by G protein-coupled receptors leads to phospholipase D stimulation. J Biol Chem 281:21837–21847 [DOI] [PubMed] [Google Scholar]
- Lowenstein EJ, Daly RJ, Batzer AG, Li W, Margolis B, Lammers R, Ullrich A, Skolnik EY, Bar-Sagi D, Schlessinger J. (1992) The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 70:431–442 [DOI] [PubMed] [Google Scholar]
- Lu Z, Hornia A, Joseph T, Sukezane T, Frankel P, Zhong M, Bychenok S, Xu L, Feig LA, Foster DA. (2000) Phospholipase D and RalA cooperate with the epidermal growth factor receptor to transform 3Y1 rat fibroblasts. Mol Cell Biol 20:462–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukowski S, Lecomte MC, Mira JP, Marin P, Gautero H, Russo-Marie F, Geny B. (1996) Inhibition of phospholipase D activity by fodrin. An active role for the cytoskeleton. J Biol Chem 271:24164–24171 [DOI] [PubMed] [Google Scholar]
- Lukowski S, Mira JP, Zachowski A, Geny B. (1998) Fodrin inhibits phospholipases A2, C, and D by decreasing polyphosphoinositide cell content. Biochem Biophys Res Commun 248:278–284 [DOI] [PubMed] [Google Scholar]
- Luo JQ, Liu X, Hammond SM, Colley WC, Feig LA, Frohman MA, Morris AJ, Foster DA. (1997) RalA interacts directly with the Arf-responsive, PIP2-dependent phospholipase D1. Biochem Biophys Res Commun 235:854–859 [DOI] [PubMed] [Google Scholar]
- Luo L. (2000) Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci 1:173–180 [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Daaka Y, Della Rocca GJ, Lefkowitz RJ. (1997) G protein-coupled receptors mediate two functionally distinct pathways of tyrosine phosphorylation in rat 1a fibroblasts. Shc phosphorylation and receptor endocytosis correlate with activation of Erk kinases. J Biol Chem 272:31648–31656 [DOI] [PubMed] [Google Scholar]
- Mackay DJ, Hall A. (1998) Rho GTPases. J Biol Chem 273:20685–20688 [DOI] [PubMed] [Google Scholar]
- Madaule P, Axel R. (1985) A novel ras-related gene family. Cell 41:31–40 [DOI] [PubMed] [Google Scholar]
- Mahajan K, Coppola D, Challa S, Fang B, Chen YA, Zhu W, Lopez AS, Koomen J, Engelman RW, Rivera C, et al. (2010) Ack1 mediated AKT/PKB tyrosine 176 phosphorylation regulates its activation. PLoS ONE 5:e9646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahankali M, Peng HJ, Henkels KM, Dinauer MC, Gomez-Cambronero J. (2011) Phospholipase D2 (PLD2) is a guanine nucleotide exchange factor (GEF) for the GTPase Rac2. Proc Natl Acad Sci USA 108:19617–19622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, Chandel NS, Thompson CB, Robey RB, Hay N. (2004) Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell 16:819–830 [DOI] [PubMed] [Google Scholar]
- Malcolm KC, Ross AH, Qiu RG, Symons M, Exton JH. (1994) Activation of rat liver phospholipase D by the small GTP-binding protein RhoA. J Biol Chem 269:25951–25954 [PubMed] [Google Scholar]
- Malumbres M, Barbacid M. (2003) RAS oncogenes: the first 30 years. Nat Rev Cancer 3:459–465 [DOI] [PubMed] [Google Scholar]
- Manifava M, Sugars J, Ktistakis NT. (1999) Modification of catalytically active phospholipase D1 with fatty acid in vivo. J Biol Chem 274:1072–1077 [DOI] [PubMed] [Google Scholar]
- Manifava M, Thuring JWJF, Lim ZY, Packman L, Holmes AB, Ktistakis NT. (2001) Differential binding of traffic-related proteins to phosphatidic acid- or phosphatidylinositol (4,5)- bisphosphate-coupled affinity reagents. J Biol Chem 276:8987–8994 [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. (2003) Rheb fills a GAP between TSC and TOR. Trends Biochem Sci 28:573–576 [DOI] [PubMed] [Google Scholar]
- Margolis B, Li N, Koch A, Mohammadi M, Hurwitz DR, Zilberstein A, Ullrich A, Pawson T, Schlessinger J. (1990) The tyrosine phosphorylated carboxyterminus of the EGF receptor is a binding site for GAP and PLC-gamma. EMBO J 9:4375–4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Quiles N, Ho HYH, Kirschner MW, Ramesh N, Geha RS. (2004) Erk/Src phosphorylation of cortactin acts as a switch on-switch off mechanism that controls its ability to activate N-WASP. Mol Cell Biol 24:5269–5280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massenburg D, Han JS, Liyanage M, Patton WA, Rhee SG, Moss J, Vaughan M. (1994) Activation of rat brain phospholipase D by ADP-ribosylation factors 1,5, and 6: separation of ADP-ribosylation factor-dependent and oleate-dependent enzymes. Proc Natl Acad Sci USA 91:11718–11722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara K, Kishida S, Matsuura Y, Kitayama H, Noda M, Kikuchi A. (1999) Plasma membrane recruitment of RalGDS is critical for Ras-dependent Ral activation. Oncogene 18:1303–1312 [DOI] [PubMed] [Google Scholar]
- Matsunaga K, Morita E, Saitoh T, Akira S, Ktistakis NT, Izumi T, Noda T, Yoshimori T. (2010) Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol 190:511–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, et al. (2009) Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 11:385–396 [DOI] [PubMed] [Google Scholar]
- Matuoka K, Shibata M, Yamakawa A, Takenawa T. (1992) Cloning of ASH, a ubiquitous protein composed of one Src homology region (SH) 2 and two SH3 domains, from human and rat cDNA libraries. Proc Natl Acad Sci USA 89:9015–9019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazie AR, Spix JK, Block ER, Achebe HB, Klarlund JK. (2006) Epithelial cell motility is triggered by activation of the EGF receptor through phosphatidic acid signaling. J Cell Sci 119:1645–1654 [DOI] [PubMed] [Google Scholar]
- McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, et al. (2007) Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta 1773:1263–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCudden CR, Hains MD, Kimple RJ, Siderovski DP, Willard FS. (2005) G-protein signaling: back to the future. Cell Mol Life Sci 62:551–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhail LC, Qualliotine-Mann D, Waite KA. (1995) Cell-free activation of neutrophil NADPH oxidase by a phosphatidic acid-regulated protein kinase. Proc Natl Acad Sci USA 92:7931–7935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhail LC, Waite KA, Regier DS, Nixon JB, Qualliotine-Mann D, Zhang WX, Wallin R, Sergeant S. (1999) A novel protein kinase target for the lipid second messenger phosphatidic acid. Biochim Biophys Acta 1439:277–290 [DOI] [PubMed] [Google Scholar]
- McPherson PS, Garcia EP, Slepnev VI, David C, Zhang X, Grabs D, Sossin WS, Bauerfeind R, Nemoto Y, De Camilli P. (1996) A presynaptic inositol-5-phosphatase. Nature 379:353–357 [DOI] [PubMed] [Google Scholar]
- Meacci E, Donati C, Cencetti F, Oka T, Komuro I, Farnararo M, Bruni P. (2001) Dual regulation of sphingosine 1-phosphate-induced phospholipase D activity through RhoA and protein kinase C-alpha in C2C12 myoblasts. Cell Signal 13:593–598 [DOI] [PubMed] [Google Scholar]
- Meacci E, Nuti F, Catarzi S, Vasta V, Donati C, Bourgoin S, Bruni P, Moss J, Vaughan M. (2003) Activation of phospholipase D by bradykinin and sphingosine 1-phosphate in A549 human lung adenocarcinoma cells via different GTP-binding proteins and protein kinase C delta signaling pathways. Biochemistry 42:284–292 [DOI] [PubMed] [Google Scholar]
- Meacci E, Vasta V, Donati C, Farnararo M, Bruni P. (1999) Receptor-mediated activation of phospholipase D by sphingosine 1-phosphate in skeletal muscle C2C12 cells. A role for protein kinase C. FEBS Lett 457:184–188 [DOI] [PubMed] [Google Scholar]
- Meisenhelder J, Suh PG, Rhee SG, Hunter T. (1989) Phospholipase C-gamma is a substrate for the PDGF and EGF receptor protein-tyrosine kinases in vivo and in vitro. Cell 57:1109–1122 [DOI] [PubMed] [Google Scholar]
- Min DS, Ahn BH, Jo YH. (2001a) Differential tyrosine phosphorylation of phospholipase D isozymes by hydrogen peroxide and the epidermal growth factor in A431 epidermoid carcinoma cells. Mol Cells 11:369–378 [PubMed] [Google Scholar]
- Min DS, Kwon TK, Park WS, Chang JS, Park SK, Ahn BH, Ryoo ZY, Lee YH, Lee YS, Rhie DJ, et al. (2001b) Neoplastic transformation and tumorigenesis associated with overexpression of phospholipase D isozymes in cultured murine fibroblasts. Carcinogenesis 22:1641–1647 [DOI] [PubMed] [Google Scholar]
- Min DS, Park SK, Exton JH. (1998) Characterization of a rat brain phospholipase D isozyme. J Biol Chem 273:7044–7051 [DOI] [PubMed] [Google Scholar]
- Mitchell R, McCulloch D, Lutz E, Johnson M, MacKenzie C, Fennell M, Fink G, Zhou W, Sealfon SC. (1998) Rhodopsin-family receptors associate with small G proteins to activate phospholipase D. Nature 392:411–414 [DOI] [PubMed] [Google Scholar]
- Mitchell R, Robertson DN, Holland PJ, Collins D, Lutz EM, Johnson MS. (2003) ADP-ribosylation factor-dependent phospholipase D activation by the M3 muscarinic receptor. J Biol Chem 278:33818–33830 [DOI] [PubMed] [Google Scholar]
- Miyashita T, Reed JC. (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80:293–299 [DOI] [PubMed] [Google Scholar]
- Mizushima N. (2010) The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 22:132–139 [DOI] [PubMed] [Google Scholar]
- Mizushima N, Sugita H, Yoshimori T, Ohsumi Y. (1998) A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J Biol Chem 273:33889–33892 [DOI] [PubMed] [Google Scholar]
- Mizutani K, Miki H, He H, Maruta H, Takenawa T. (2002) Essential role of neural Wiskott-Aldrich syndrome protein in podosome formation and degradation of extracellular matrix in src-transformed fibroblasts. Cancer Res 62:669–674 [PubMed] [Google Scholar]
- Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, Hamdi M, van Nes J, Westerman BA, van Arkel J, et al. (2012) Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483:589–593 [DOI] [PubMed] [Google Scholar]
- Moll UM, Petrenko O. (2003) The MDM2-p53 interaction. Mol Cancer Res 1:1001–1008 [PubMed] [Google Scholar]
- Monovich L, Mugrage B, Quadros E, Toscano K, Tommasi R, LaVoie S, Liu E, Du Z, LaSala D, Boyar W, et al. (2007) Optimization of halopemide for phospholipase D2 inhibition. Bioorg Med Chem Lett 17:2310–2311 [DOI] [PubMed] [Google Scholar]
- Moolenaar WH, Kruijer W, Tilly BC, Verlaan I, Bierman AJ, de Laat SW. (1986) Growth factor-like action of phosphatidic acid. Nature 323:171–173 [DOI] [PubMed] [Google Scholar]
- Moore CAC, Milano SK, Benovic JL. (2007) Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol 69:451–482 [DOI] [PubMed] [Google Scholar]
- Moreau K, Ravikumar B, Puri C, Rubinsztein DC. (2012) Arf6 promotes autophagosome formation via effects on phosphatidylinositol 4,5-bisphosphate and phospholipase D. J Cell Biol 196:483–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moréra S, Chiadmi M, LeBras G, Lascu I, Janin J. (1995) Mechanism of phosphate transfer by nucleoside diphosphate kinase: X-ray structures of the phosphohistidine intermediate of the enzymes from Drosophila and Dictyostelium. Biochemistry 34:11062–11070 [PubMed] [Google Scholar]
- Moritz A, De Graan PN, Gispen WH, Wirtz KW. (1992) Phosphatidic acid is a specific activator of phosphatidylinositol-4-phosphate kinase. J Biol Chem 267:7207–7210 [PubMed] [Google Scholar]
- Morrice NA, Gabrielli B, Kemp BE, Wettenhall RE. (1994) A cardiolipin-activated protein kinase from rat liver structurally distinct from the protein kinases C. J Biol Chem 269:20040–20046 [PubMed] [Google Scholar]
- Morris AJ, Engebrecht J, Frohman MA. (1996) Structure and regulation of phospholipase D. Trends Pharmacol Sci 17:182–185 [DOI] [PubMed] [Google Scholar]
- Moss J, Vaughan M. (1993) ADP-ribosylation factors, 20,000 M(r) guanine nucleotide-binding protein activators of cholera toxin and components of intracellular vesicular transport systems. Cell Signal 5:367–379 [DOI] [PubMed] [Google Scholar]
- Moss J, Vaughan M. (1998) Molecules in the ARF orbit. J Biol Chem 273:21431–21434 [DOI] [PubMed] [Google Scholar]
- Murthy SNP, Chung PH, Lin L, Lomasney JW. (2013) Activation of phospholipase Cepsilon by free fatty acids and cross talk with phospholipase D and phospholipase A2. Biochemistry 45:10987–10997 [DOI] [PubMed] [Google Scholar]
- Nagasaki A, Inotsume K, Kanada M, Uyeda TQ. (2008) Phospholipase D is essential for keratocyte-like migration of NBT-II cells. Cell Struct Funct 33:27–33 [DOI] [PubMed] [Google Scholar]
- Nakamura S, Akisue T, Jinnai H, Hitomi T, Sarkar S, Miwa N, Okada T, Yoshida K, Kuroda S, Kikkawa U, et al. (1998) Requirement of GM2 ganglioside activator for phospholipase D activation. Proc Natl Acad Sci USA 95:12249–12253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi H, Exton JH. (1992) Purification and characterization of the zeta isoform of protein kinase C from bovine kidney. J Biol Chem 267:16347–16354 [PubMed] [Google Scholar]
- Nakashima S. (2002) Protein kinase C α (PKC α): regulation and biological function. J Biochem 132:669–675 [DOI] [PubMed] [Google Scholar]
- Nakatogawa H, Ichimura Y, Ohsumi Y. (2007) Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 130:165–178 [DOI] [PubMed] [Google Scholar]
- Némoz G, Sette C, Conti M. (1997) Selective activation of rolipram-sensitive, cAMP-specific phosphodiesterase isoforms by phosphatidic acid. Mol Pharmacol 51:242–249 [DOI] [PubMed] [Google Scholar]
- Nie Z, Stanley KT, Stauffer S, Jacques KM, Hirsch DS, Takei J, Randazzo PA. (2002) AGAP1, an endosome-associated, phosphoinositide-dependent ADP-ribosylation factor GTPase-activating protein that affects actin cytoskeleton. J Biol Chem 277:48965–48975 [DOI] [PubMed] [Google Scholar]
- Nishikimi A, Fukuhara H, Su W, Hongu T, Takasuga S, Mihara H, Cao Q, Sanematsu F, Kanai M, Hasegawa H, et al. (2009) Sequential regulation of DOCK2 dynamics by two phospholipids during neutrophil chemotaxis. Science 324:384–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizuka Y. (1984) The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature 308:693–698 [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A. (1995) Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81:53–62 [DOI] [PubMed] [Google Scholar]
- Noda NN, Satoo K, Fujioka Y, Kumeta H, Ogura K, Nakatogawa H, Ohsumi Y, Inagaki F. (2011) Structural basis of Atg8 activation by a homodimeric E1, Atg7. Mol Cell 44:462–475 [DOI] [PubMed] [Google Scholar]
- Noh DY, Ahn SJ, Lee RA, Park IA, Kim JH, Suh PG, Ryu SH, Lee KH, Han JS. (2000) Overexpression of phospholipase D1 in human breast cancer tissues. Cancer Lett 161:207–214 [DOI] [PubMed] [Google Scholar]
- Norris FA, Ungewickell E, Majerus PW. (1995) Inositol hexakisphosphate binds to clathrin assembly protein 3 (AP-3/AP180) and inhibits clathrin cage assembly in vitro. J Biol Chem 270:214–217 [DOI] [PubMed] [Google Scholar]
- O’Luanaigh N, Pardo R, Fensome A, Allen-Baume V, Jones D, Holt MR, Cockcroft S. (2002) Continual production of phosphatidic acid by phospholipase D is essential for antigen-stimulated membrane ruffling in cultured mast cells. Mol Biol Cell 13:3730–3746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohguchi K, Banno Y, Nakashima S, Nozawa Y. (1996) Regulation of membrane-bound phospholipase D by protein kinase C in HL60 cells. Synergistic action of small GTP-binding protein RhoA. J Biol Chem 271:4366–4372 [DOI] [PubMed] [Google Scholar]
- Oishi K, Takahashi M, Mukai H, Banno Y, Nakashima S, Kanaho Y, Nozawa Y, Ono Y. (2001) PKN regulates phospholipase D1 through direct interaction. J Biol Chem 276:18096–18101 [DOI] [PubMed] [Google Scholar]
- Okamoto Y, Morishita J, Tsuboi K, Tonai T, Ueda N. (2004) Molecular characterization of a phospholipase D generating anandamide and its congeners. J Biol Chem 279:5298–5305 [DOI] [PubMed] [Google Scholar]
- Okamura S, Yamashita S. (1994) Purification and characterization of phosphatidylcholine phospholipase D from pig lung. J Biol Chem 269:31207–31213 [PubMed] [Google Scholar]
- Oldham WM, Hamm HE. (2008) Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol 9:60–71 [DOI] [PubMed] [Google Scholar]
- Oliveira TG, Chan RB, Tian H, Laredo M, Shui G, Staniszewski A, Zhang H, Wang L, Kim TW, Duff KE, et al. (2010) Phospholipase d2 ablation ameliorates Alzheimer’s disease-linked synaptic dysfunction and cognitive deficits. J Neurosci 30:16419–16428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson SC, Bowman EP, Lambeth JD. (1991) Phospholipase D activation in a cell-free system from human neutrophils by phorbol 12-myristate 13-acetate and guanosine 5′-O-(3-thiotriphosphate). Activation is calcium dependent and requires protein factors in both the plasma membrane and cytosol. J Biol Chem 266:17236–17242 [PubMed] [Google Scholar]
- Onorato JJ, Gillis ME, Liu Y, Benovic JL, Ruoho AE. (1995) The beta-adrenergic receptor kinase (GRK2) is regulated by phospholipids. J Biol Chem 270:21346–21353 [DOI] [PubMed] [Google Scholar]
- O'Reilly MC, Scott SA, Brown KA, Oguin T, Thomas PG, Daniels JS, Morrison R, Brown HA, Lindsley CW. (2013) Development of dual PLD1/2 and PLD2 selective inhibitors from a common 1,3,8-triazaspiro[4.5]decane core: discovery of ML298 and ML299 that decrease invasive migration in U87-MG glioblastoma cells. J Med Chem 56:2695–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, Xu Y, Wonsey D, Lee LA, Dang CV. (2000) Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem 275:21797–21800 [DOI] [PubMed] [Google Scholar]
- Ouyang YS, Tu Y, Barker SA, Yang F. (2003) Regulators of G-protein signaling (RGS) 4, insertion into model membranes and inhibition of activity by phosphatidic acid. J Biol Chem 278:11115–11122 [DOI] [PubMed] [Google Scholar]
- Owen-Schaub LB, Zhang W, Cusack JC, Angelo LS, Santee SM, Fujiwara T, Roth JA, Deisseroth AB, Zhang WW, Kruzel E, et al. (1995) Wild-type human p53 and a temperature-sensitive mutant induce Fas/APO-1 expression. Mol Cell Biol 15:3032–3040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai JK, Dobek EA, Bishop WR. (1991) Endothelin-1 activates phospholipase D and thymidine incorporation in fibroblasts overexpressing protein kinase C beta 1. Cell Regul 2:897–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palicz A, Foubert TR, Jesaitis AJ, Marodi L, McPhail LC. (2001) Phosphatidic acid and diacylglycerol directly activate NADPH oxidase by interacting with enzyme components. J Biol Chem 276:3090–3097 [DOI] [PubMed] [Google Scholar]
- Park JB, Kim JH, Kim Y, Ha SH, Yoo JS, Du G, Frohman MA, Suh PG, Ryu SH. (2000) Cardiac phospholipase D2 localizes to sarcolemmal membranes and is inhibited by alpha-actinin in an ADP-ribosylation factor-reversible manner. J Biol Chem 275:21295–21301 [DOI] [PubMed] [Google Scholar]
- Park MH, Ahn BH, Hong YK, Min S. (2009) Overexpression of phospholipase D enhances matrix metalloproteinase-2 expression and glioma cell invasion via protein kinase C and protein kinase A/NF-kappaB/Sp1-mediated signaling pathways. Carcinogenesis 30:356–365 [DOI] [PubMed] [Google Scholar]
- Park SK, Min DS, Exton JH. (1998) Definition of the protein kinase C interaction site of phospholipase D. Biochem Biophys Res Commun 244:364–367 [DOI] [PubMed] [Google Scholar]
- Park SK, Provost JJ, Bae CD, Ho WT, Exton JH. (1997) Cloning and characterization of phospholipase D from rat brain. J Biol Chem 272:29263–29271 [DOI] [PubMed] [Google Scholar]
- Parrish AB, Freel CD, Kornbluth S. (2013) Cellular mechanisms controlling caspase activation and function. Cold Spring Harb Perspect Biol 5:a008672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons JT, Horwitz AR, Schwartz MA. (2010) Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol 11:633–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorino S, Renganathan H, Caliva MJ, Filbert EL, Opoku-Ansah J, Sulzmaier FJ, Gawecka JE, Werlen G, Shaw AS, Ramos JW. (2010) The death effector domain protein PEA-15 negatively regulates T-cell receptor signaling. FASEB J 24:2818–2828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawelczyk T, Matecki A. (1999) Phospholipase C-delta3 binds with high specificity to phosphatidylinositol 4,5-bisphosphate and phosphatidic acid in bilayer membranes. Eur J Biochem 262:291–298 [DOI] [PubMed] [Google Scholar]
- Pellegrin S, Mellor H. (2007) Actin stress fibres. J Cell Sci 120:3491–3499 [DOI] [PubMed] [Google Scholar]
- Peng HJ, Henkels KM, Mahankali M, Dinauer MC, Gomez-Cambronero J. (2011) Evidence for two CRIB domains in phospholipase D2 (PLD2) that the enzyme uses to specifically bind to the small GTPase Rac2. J Biol Chem 286:16308–16320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettitt TR, McDermott M, Saqib KM, Shimwell N, Wakelam MJ. (2001) Phospholipase D1b and D2a generate structurally identical phosphatidic acid species in mammalian cells. Biochem J 360:707–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyroche A, Antonny B, Robineau S, Acker J, Cherfils J, Jackson CL. (1999) Brefeldin A acts to stabilize an abortive ARF-GDP-Sec7 domain protein complex: involvement of specific residues of the Sec7 domain. Mol Cell 3:275–285 [DOI] [PubMed] [Google Scholar]
- Péterfy M, Phan J, Xu P, Reue K. (2001) Lipodystrophy in the fld mouse results from mutation of a new gene encoding a nuclear protein, lipin. Nat Genet 27:121–124 [DOI] [PubMed] [Google Scholar]
- Pilquil C, Dewald J, Cherney A, Gorshkova I, Tigyi G, English D, Natarajan V, Brindley DN. (2006) Lipid phosphate phosphatase-1 regulates lysophosphatidate-induced fibroblast migration by controlling phospholipase D2-dependent phosphatidate generation. J Biol Chem 281:38418–38429 [DOI] [PubMed] [Google Scholar]
- Plonk SG, Park SK, Exton JH. (1998) The alpha-subunit of the heterotrimeric G protein G13 activates a phospholipase D isozyme by a pathway requiring Rho family GTPases. J Biol Chem 273:4823–4826 [DOI] [PubMed] [Google Scholar]
- Pohlman RF, Liu F, Wang L, Moré MI, Winans SC. (1993) Genetic and biochemical analysis of an endonuclease encoded by the IncN plasmid pKM101. Nucleic Acids Res 21:4867–4872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock PM, Meltzer PS. (2002) A genome-based strategy uncovers frequent BRAF mutations in melanoma. Cancer Cell 2:5–7 [DOI] [PubMed] [Google Scholar]
- Ponting CP, Kerr ID. (1996) A novel family of phospholipase D homologues that includes phospholipid synthases and putative endonucleases: identification of duplicated repeats and potential active site residues. Protein Sci 5:914–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcelli AM, Ghelli A, Hrelia S, Rugolo M. (2002) Phospholipase D stimulation is required for sphingosine-1-phosphate activation of actin stress fibre assembly in human airway epithelial cells. Cell Signal 14:75–81 [DOI] [PubMed] [Google Scholar]
- Powner DJ, Hodgkin MN, Wakelam MJO. (2002) Antigen-stimulated activation of phospholipase D1b by Rac1, ARF6, and PKCalpha in RBL-2H3 cells. Mol Biol Cell 13:1252–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powner DJ, Payne RM, Pettitt TR, Giudici ML, Irvine RF, Wakelam MJO. (2005) Phospholipase D2 stimulates integrin-mediated adhesion via phosphatidylinositol 4-phosphate 5-kinase Igamma b. J Cell Sci 118:2975–2986 [DOI] [PubMed] [Google Scholar]
- Prasad K, Lippoldt RE. (1988) Molecular characterization of the AP180 coated vesicle assembly protein. Biochemistry 27:6098–6104 [DOI] [PubMed] [Google Scholar]
- Preininger AM, Henage LG, Oldham WM, Yoon EJ, Hamm HE, Brown HA. (2006) Direct modulation of phospholipase D activity by Gbetagamma. Mol Pharmacol 70:311–318 [DOI] [PubMed] [Google Scholar]
- Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, Carneiro MO, Carter SL, Cibulskis K, Erlich RL, et al. (2012) Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 488:106–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puleston DJ, Simon AK. (2014) Autophagy in the immune system. Immunology 141:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyne S, Pyne NJ. (1995) Bradykinin-stimulated phosphatidylcholine hydrolysis in airway smooth muscle: the role of Ca2+ and protein kinase C. Biochem J 311:637–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin C, Wang X. (2002) The Arabidopsis phospholipase D family. Characterization of a calcium-independent and phosphatidylcholine-selective PLD zeta 1 with distinct regulatory domains. Plant Physiol 128:1057–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz JD, White E. (2010) Autophagy and metabolism. Science 330:1344–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rak J, Mitsuhashi Y, Bayko L, Filmus J, Shirasawa S, Sasazuki T, Kerbel RS. (1995) Mutant ras oncogenes upregulate VEGF/VPF expression: implications for induction and inhibition of tumor angiogenesis. Cancer Res 55:4575–4580 [PubMed] [Google Scholar]
- Ramanathan A, Wang C, Schreiber SL. (2005) Perturbational profiling of a cell-line model of tumorigenesis by using metabolic measurements. Proc Natl Acad Sci USA 102:5992–5997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana RS, Hokin LE. (1990) Role of phosphoinositides in transmembrane signaling. Physiol Rev 70:115–164 [DOI] [PubMed] [Google Scholar]
- Randazzo PA. (1997) Resolution of two ADP-ribosylation factor 1 GTPase-activating proteins from rat liver. Biochem J 324:413–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao JS. (2003) Molecular mechanisms of glioma invasiveness: the role of proteases. Nat Rev Cancer 3:489–501 [DOI] [PubMed] [Google Scholar]
- Rappley I, Gitler AD, Selvy PE, LaVoie MJ, Levy BD, Brown HA, Lindquist S, Selkoe DJ. (2009) Evidence that α-synuclein does not inhibit phospholipase D. Biochemistry 48:1077–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. (2010) Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 12:747–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich R, Blumenthal M, Liscovitch M. (1995) Role of phospholipase D in laminin-induced production of gelatinase A (MMP-2) in metastatic cells. Clin Exp Metastasis 13:134–140 [DOI] [PubMed] [Google Scholar]
- Ren H, Federico L, Huang H, Sunkara M, Drennan T, Frohman MA, Smyth SS, Morris AJ. (2010) A phosphatidic acid binding/nuclear localization motif determines lipin1 function in lipid metabolism and adipogenesis. Mol Biol Cell 21:3171–3181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SG. (2001) Regulation of phosphoinositide-specific phospholipase C. Annu Rev Biochem 70:281–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD, Sonenberg N. (2005) Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 433:477–480 [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. (2003) Cell migration: integrating signals from front to back. Science 302:1704–1709 [DOI] [PubMed] [Google Scholar]
- Riebeling C, Bourgoin S, Shields D. (2008) Caspase cleavage of phospholipase D1 in vitro alters its regulation and reveals a novel property of the “loop” region. Biochim Biophys Acta 1781:376–382 [DOI] [PubMed] [Google Scholar]
- Rizzo MA, Shome K, Vasudevan C, Stolz DB, Sung TC, Frohman MA, Watkins SC, Romero G. (1999) Phospholipase D and its product, phosphatidic acid, mediate agonist-dependent raf-1 translocation to the plasma membrane and the activation of the mitogen-activated protein kinase pathway. J Biol Chem 274:1131–1139 [DOI] [PubMed] [Google Scholar]
- Rizzo MA, Shome K, Watkins SC, Romero G. (2000) The recruitment of Raf-1 to membranes is mediated by direct interaction with phosphatidic acid and is independent of association with Ras. J Biol Chem 275:23911–23918 [DOI] [PubMed] [Google Scholar]
- Roach AN, Wang Z, Wu P, Zhang F, Chan RB, Yonekubo Y, Di Paolo G, Gorfe AA, Du G. (2012) Phosphatidic acid regulation of PIPKI is critical for actin cytoskeletal reorganization. J Lipid Res 53:2598–2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Vanhaesebroeck B, Waterfield MD, Downward J. (1996) Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. EMBO J 15:2442–2451 [PMC free article] [PubMed] [Google Scholar]
- Rodrik V, Zheng Y, Harrow F, Chen Y, Foster DA. (2005) Survival signals generated by estrogen and phospholipase D in MCF-7 breast cancer cells are dependent on Myc. Mol Cell Biol 25:7917–7925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose K, Rudge SA, Frohman MA, Morris AJ, Engebrecht J. (1995) Phospholipase D signaling is essential for meiosis. Proc Natl Acad Sci USA 92:12151–12155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross EM, Wilkie TM. (2000) GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu Rev Biochem 69:795–827 [DOI] [PubMed] [Google Scholar]
- Roux PP, Blenis J. (2004) ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev 68:320–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin E, Tamrakar S, Ludlow JW. (1998) Protein phosphatase type 1, the product of the retinoblastoma susceptibility gene, and cell cycle control. Front Biosci 3:D1209–D1219 [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Codogno P, Levine B. (2012) Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 11:709–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph AE, Stuckey JA, Zhao Y, Matthews HR, Patton WA, Moss J, Dixon JE. (1999) Expression, characterization, and mutagenesis of the Yersinia pestis murine toxin, a phospholipase D superfamily member. J Biol Chem 274:11824–11831 [DOI] [PubMed] [Google Scholar]
- Rümenapp U, Geiszt M, Wahn F, Schmidt M, Jakobs KH. (1995) Evidence for ADP-ribosylation-factor-mediated activation of phospholipase D by m3 muscarinic acetylcholine receptor. Eur J Biochem 234:240–244 [DOI] [PubMed] [Google Scholar]
- Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. (1994) RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 78:35–43 [DOI] [PubMed] [Google Scholar]
- Saito M, Kanfer J. (1973) Solubilization and properties of a membrane-bound enzyme from rat brain catalyzing a base-exchange reaction. Biochem Biophys Res Commun 53:391–398 [DOI] [PubMed] [Google Scholar]
- Saito M, Kanfer J. (1975) Phosphatidohydrolase activity in a solubilized preparation from rat brain particulate fraction. Arch Biochem Biophys 169:318–323 [DOI] [PubMed] [Google Scholar]
- Sajan MP, Bandyopadhyay G, Kanoh Y, Standaert ML, Quon MJ, Reed BC, Dikic I, Farese RV. (2002) Sorbitol activates atypical protein kinase C and GLUT4 glucose transporter translocation/glucose transport through proline-rich tyrosine kinase-2, the extracellular signal-regulated kinase pathway and phospholipase D. Biochem J 362:665–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Tilló E, de Barrios O, Siles L, Cuatrecasas M, Castells A, Postigo A. (2011) β-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc Natl Acad Sci USA 108:19204–19209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandmann J, Peralta EG, Wurtman RJ. (1991) Coupling of transfected muscarinic acetylcholine receptor subtypes to phospholipase D. J Biol Chem 266:6031–6034 [PubMed] [Google Scholar]
- Sangrar W, Gao Y, Scott M, Truesdell P, Greer PA. (2007) Fer-mediated cortactin phosphorylation is associated with efficient fibroblast migration and is dependent on reactive oxygen species generation during integrin-mediated cell adhesion. Mol Cell Biol 27:6140–6152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santy LC, Casanova JE. (2001) Activation of ARF6 by ARNO stimulates epithelial cell migration through downstream activation of both Rac1 and phospholipase D. J Cell Biol 154:599–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Miwa N, Kominami H, Igarashi N, Hayashi S, Okada T, Jahangeer S, Nakamura S. (2001) Regulation of mammalian phospholipase D2: interaction with and stimulation by G(M2) activator. Biochem J 359:599–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Perlstein EO, Imarisio S, Pineau S, Cordenier A, Maglathlin RL, Webster JA, Lewis TA, O’Kane CJ, Schreiber SL, et al. (2007) Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat Chem Biol 3:331–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarri E, Pardo R, Fensome-Green A, Cockcroft S. (2003) Endogenous phospholipase D2 localizes to the plasma membrane of RBL-2H3 mast cells and can be distinguished from ADP ribosylation factor-stimulated phospholipase D1 activity by its specific sensitivity to oleic acid. Biochem J 369:319–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savagner P, Yamada KM, Thiery JP. (1997) The zinc-finger protein slug causes desmosome dissociation, an initial and necessary step for growth factor-induced epithelial-mesenchymal transition. J Cell Biol 137:1403–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger J. (2000) Cell signaling by receptor tyrosine kinases. Cell 103:211–225 [DOI] [PubMed] [Google Scholar]
- Schlessinger J, Ullrich A. (1992) Growth factor signaling by receptor tyrosine kinases. Neuron 9:383–391 [DOI] [PubMed] [Google Scholar]
- Schmidt M, Rümenapp U, Nehls C, Ott S, Keller J, Von Eichel-Streiber C, Jakobs KH. (1996) Restoration of Clostridium difficile toxin-B-inhibited phospholipase D by phosphatidylinositol 4,5-bisphosphate. Eur J Biochem 240:707–712 [DOI] [PubMed] [Google Scholar]
- Schofield CJ, Ratcliffe PJ. (2005) Signalling hypoxia by HIF hydroxylases. Biochem Biophys Res Commun 338:617–626 [DOI] [PubMed] [Google Scholar]
- Schofield JN, Rademacher TW. (2000) Structure and expression of the human glycosylphosphatidylinositol phospholipase D1 (GPLD1) gene. Biochim Biophys Acta 1494:189–194 [DOI] [PubMed] [Google Scholar]
- Schroder WA, Buck M, Cloonan N, Hancock JF, Suhrbier A, Sculley T, Bushell G. (2007) Human Sin1 contains Ras-binding and pleckstrin homology domains and suppresses Ras signalling. Cell Signal 19:1279–1289 [DOI] [PubMed] [Google Scholar]
- Sciorra VA, Rudge SA, Prestwich GD, Frohman MA, Engebrecht J, Morris AJ. (1999) Identification of a phosphoinositide binding motif that mediates activation of mammalian and yeast phospholipase D isoenzymes. EMBO J 18:5911–5921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciorra VA, Rudge SA, Wang J, McLaughlin S, Engebrecht J, Morris AJ. (2002) Dual role for phosphoinositides in regulation of yeast and mammalian phospholipase D enzymes. J Cell Biol 159:1039–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott SA, Mathews TP, Ivanova PT, Lindsley CW, Brown HA. (2014) Chemical modulation of glycerolipid signaling and metabolic pathways. Biochim Biophys Acta 1841:1060–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott SA, Selvy PE, Buck JR, Cho HP, Criswell TL, Thomas AL, Armstrong MD, Arteaga CL, Lindsley CW, Brown HA. (2009) Design of isoform-selective phospholipase D inhibitors that modulate cancer cell invasiveness. Nat Chem Biol 5:108–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott SA, Xiang Y, Mathews TP, Cho HP, Myers DS, Armstrong MD, Tallman KA, O’Reilly MC, Lindsley CW, Brown HA. (2013) Regulation of phospholipase D activity and phosphatidic acid production after purinergic (P2Y6) receptor stimulation. J Biol Chem 288:20477–20487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sells MA, Chernoff J. (1997) Emerging from the Pak: the p21-activated protein kinase family. Trends Cell Biol 7:162–167 [DOI] [PubMed] [Google Scholar]
- Selvy PE, Lavieri RR, Lindsley CW, Brown HA. (2011) Phospholipase D: enzymology, functionality, and chemical modulation. Chem Rev 111:6064–6119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88:593–602 [DOI] [PubMed] [Google Scholar]
- Seymour LW, Shoaibi MA, Martin A, Ahmed A, Elvin P, Kerr DJ, Wakelam MJ. (1996) Vascular endothelial growth factor stimulates protein kinase C-dependent phospholipase D activity in endothelial cells. Lab Invest 75:427–437 [PubMed] [Google Scholar]
- Shahnazari S, Yen WL, Birmingham CL, Shiu J, Namolovan A, Zheng YT, Nakayama K, Klionsky DJ, Brumell JH. (2010) A diacylglycerol-dependent signaling pathway contributes to regulation of antibacterial autophagy. Cell Host Microbe 8:137–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Cantley LC. (2006) Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 441:424–430 [DOI] [PubMed] [Google Scholar]
- Shen Y, Xu L, Foster DA. (2001) Role for phospholipase D in receptor-mediated endocytosis. Mol Cell Biol 21:595–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Zheng Y, Foster DA. (2002) Phospholipase D2 stimulates cell protrusion in v-Src-transformed cells. Biochem Biophys Res Commun 293:201–206 [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, Lefkowitz RJ. (2006) beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem 281:1261–1273 [DOI] [PubMed] [Google Scholar]
- Sherr CJ. (2001) The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol 2:731–737 [DOI] [PubMed] [Google Scholar]
- Shi M, Zheng Y, Garcia A, Xu L, Foster DA. (2007) Phospholipase D provides a survival signal in human cancer cells with activated H-Ras or K-Ras. Cancer Lett 258:268–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. (2009) Serine/threonine phosphatases: mechanism through structure. Cell 139:468–484 [DOI] [PubMed] [Google Scholar]
- Shih AH, Holland EC. (2006) Platelet-derived growth factor (PDGF) and glial tumorigenesis. Cancer Lett 232:139–147 [DOI] [PubMed] [Google Scholar]
- Shin SI, Freedman VH, Risser R, Pollack R. (1975) Tumorigenicity of virus-transformed cells in nude mice is correlated specifically with anchorage independent growth in vitro. Proc Natl Acad Sci USA 72:4435–4439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani T, Mizushima N, Ogawa Y, Matsuura A, Noda T, Ohsumi Y. (1999) Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. EMBO J 18:5234–5241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shome K, Vasudevan C, Romero G. (1997) ARF proteins mediate insulin-dependent activation of phospholipase D. Curr Biol 7:387–396 [DOI] [PubMed] [Google Scholar]
- Shome K, Rizzo MA, Vasudevan C, Andresen B, Romero G. (2000) The activation of phospholipase D by endothelin-1, angiotensin II, and platelet-derived growth factor in vascular smooth muscle A10 cells is mediated by small G proteins of the ADP-ribosylation factor family. Endocrinology 141:2200–2208 [DOI] [PubMed] [Google Scholar]
- Shulga YV, Loukov D, Ivanova PT, Milne SB, Myers DS, Hatch GM, Umeh G, Jalan D, Fullerton MD, Steinberg GR, et al. (2013) Diacylglycerol kinase delta promotes lipogenesis. Biochemistry 52:7766–7776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulga YV, Topham MK, Epand RM. (2011) Regulation and functions of diacylglycerol kinases. Chem Rev 111:6186–6208 [DOI] [PubMed] [Google Scholar]
- Siddiqi AR, Smith JL, Ross AH, Qiu RG, Symons M, Exton JH. (1995) Regulation of phospholipase D in HL60 cells. Evidence for a cytosolic phospholipase D. J Biol Chem 270:8466–8473 [DOI] [PubMed] [Google Scholar]
- Siddiqi AR, Srajer GE, Leslie CC. (2000) Regulation of human PLD1 and PLD2 by calcium and protein kinase C. Biochim Biophys Acta 1497:103–114 [DOI] [PubMed] [Google Scholar]
- Simon MA, Bowtell DDL, Dodson GS, Laverty TR, Rubin GM. (1991) Ras1 and a putative guanine nucleotide exchange factor perform crucial steps in signaling by the sevenless protein tyrosine kinase. Cell 67:701–716 [DOI] [PubMed] [Google Scholar]
- Simons K, Toomre D. (2000) Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 1:31–39 [DOI] [PubMed] [Google Scholar]
- Singer WD, Brown HA, Jiang X, Sternweis PC. (1996) Regulation of phospholipase D by protein kinase C is synergistic with ADP-ribosylation factor and independent of protein kinase activity. J Biol Chem 271:4504–4510 [DOI] [PubMed] [Google Scholar]
- Slaaby R, Jensen T, Hansen HS, Frohman MA, Seedorf K. (1998) PLD2 complexes with the EGF receptor and undergoes tyrosine phosphorylation at a single site upon agonist stimulation. J Biol Chem 273:33722–33727 [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. (1987) Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235:177–182 [DOI] [PubMed] [Google Scholar]
- Small JV, Rottner K, Kaverina I. (1999) Functional design in the actin cytoskeleton. Curr Opin Cell Biol 11:54–60 [DOI] [PubMed] [Google Scholar]
- Song C, Hu CD, Masago M, Kariyai K, Yamawaki-Kataoka Y, Shibatohge M, Wu D, Satoh T, Kataoka T. (2001) Regulation of a novel human phospholipase C, PLCepsilon, through membrane targeting by Ras. J Biol Chem 276:2752–2757 [DOI] [PubMed] [Google Scholar]
- Song JG, Pfeffer LM, Foster DA. (1991) v-Src increases diacylglycerol levels via a type D phospholipase-mediated hydrolysis of phosphatidylcholine. Mol Cell Biol 11:4903–4908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulsby M, Bennett AM. (2009) Physiological signaling specificity by protein tyrosine phosphatases. Physiology (Bethesda) 24:281–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sozzani S, Agwu DE, Ellenburg MD, Locati M, Rieppi M, Rojas A, Mantovani A, McPhail LC. (1994) Activation of phospholipase D by interleukin-8 in human neutrophils. Blood 84:3895–3901 [PubMed] [Google Scholar]
- Spaargaren M, Bischoff JR. (1994) Identification of the guanine nucleotide dissociation stimulator for Ral as a putative effector molecule of R-ras, H-ras, K-ras, and Rap. Proc Natl Acad Sci USA 91:12609–12613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel S, Milstien S. (2002) Sphingosine 1-phosphate, a key cell signaling molecule. J Biol Chem 277:25851–25854 [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. (1997) Alpha-synuclein in Lewy bodies. Nature 388:839–840 [DOI] [PubMed] [Google Scholar]
- Sporn MB, Roberts AB. (1985) Autocrine growth factors and cancer. Nature 313:745–747 [DOI] [PubMed] [Google Scholar]
- Stace CL, Ktistakis NT. (2006) Phosphatidic acid- and phosphatidylserine-binding proteins. Biochim Biophys Acta 1761:913–926 [DOI] [PubMed] [Google Scholar]
- Stahelin RV, Ananthanarayanan B, Blatner NR, Singh S, Bruzik KS, Murray D, Cho W. (2004) Mechanism of membrane binding of the phospholipase D1 PX domain. J Biol Chem 279:54918–54926 [DOI] [PubMed] [Google Scholar]
- Stanacev NZ, Stuhne-Sekalec L. (1970) On the mechanism of enzymatic phosphatidylation. Biosynthesis of cardiolipin catalyzed by phospholipase D. Biochim Biophys Acta 210:350–352 [DOI] [PubMed] [Google Scholar]
- Standaert ML, Avignon A, Yamada K, Bandyopadhyay G, Farese RV. (1996) The phosphatidylinositol 3-kinase inhibitor, wortmannin, inhibits insulin-induced activation of phosphatidylcholine hydrolysis and associated protein kinase C translocation in rat adipocytes. Biochem J 313:1039–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steed PM, Clark KL, Boyar WC, Lasala DJ. (1998) Characterization of human PLD2 and the analysis of PLD isoform splice variants. FASEB J 12:1309–1317 [DOI] [PubMed] [Google Scholar]
- Steinberg SF. (2004) Distinctive activation mechanisms and functions for protein kinase Cdelta. Biochem J 384:449–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens LR, Eguinoa A, Erdjument-Bromage H, Lui M, Cooke F, Coadwell J, Smrcka AS, Thelen M, Cadwallader K, Tempst P, et al. (1997) The G beta gamma sensitivity of a PI3K is dependent upon a tightly associated adaptor, p101. Cell 89:105–114 [DOI] [PubMed] [Google Scholar]
- Stuckey JA, Dixon JE. (1999) Crystal structure of a phospholipase D family member. Nat Struct Biol 6:278–284 [DOI] [PubMed] [Google Scholar]
- Sugars JM, Cellek S, Manifava M, Coadwell J, Ktistakis NT. (1999) Fatty acylation of phospholipase D1 on cysteine residues 240 and 241 determines localization on intracellular membranes. J Biol Chem 274:30023–30027 [DOI] [PubMed] [Google Scholar]
- Sugiura T, Nakane S, Kishimoto S, Waku K, Yoshioka Y, Tokumura A, Hanahan DJ. (1999) Occurrence of lysophosphatidic acid and its alkyl ether-linked analog in rat brain and comparison of their biological activities toward cultured neural cells. Biochim Biophys Acta 1440:194–204 [DOI] [PubMed] [Google Scholar]
- Sulzmaier FJ, Valmiki MKG, Nelson DA, Caliva MJ, Geerts D, Matter ML, White EP, Ramos JW. (2012) PEA-15 potentiates H-Ras-mediated epithelial cell transformation through phospholipase D. Oncogene 31:3547–3560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Zhang J, Fan W, Wong KN, Ding X, Chen S, Zhong Q. (2011) The RUN domain of rubicon is important for hVps34 binding, lipid kinase inhibition, and autophagy suppression. J Biol Chem 286:185–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Fang Y, Yoon MS, Zhang C, Roccio M, Zwartkruis FJ, Armstrong M, Brown HA, Chen J. (2008) Phospholipase D1 is an effector of Rheb in the mTOR pathway. Proc Natl Acad Sci USA 105:8286–8291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung TC, Altshuller YM, Morris AJ, Frohman MA. (1999a) Molecular analysis of mammalian phospholipase D2. J Biol Chem 274:494–502 [DOI] [PubMed] [Google Scholar]
- Sung TC, Roper RL, Zhang Y, Rudge SA, Temel R, Hammond SM, Morris AJ, Moss B, Engebrecht J, Frohman MA. (1997) Mutagenesis of phospholipase D defines a superfamily including a trans-Golgi viral protein required for poxvirus pathogenicity. EMBO J 16:4519–4530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung TC, Zhang Y, Morris AJ, Frohman MA. (1999b) Structural analysis of human phospholipase D1. J Biol Chem 274:3659–3666 [DOI] [PubMed] [Google Scholar]
- Tadokoro S, Shattil SJ, Eto K, Tai V, Liddington RC, de Pereda JM, Ginsberg MH, Calderwood DA. (2003) Talin binding to integrin beta tails: a final common step in integrin activation. Science 302:103–106 [DOI] [PubMed] [Google Scholar]
- Tamrakar S, Rubin E, Ludlow JW. (2000) Role of pRB dephosphorylation in cell cycle regulation. Front Biosci 5:D121–D137 [DOI] [PubMed] [Google Scholar]
- Tang W, Yuan J, Chen X, Gu X, Luo K, Li J. (2006) Identification of a novel human lysophosphatidic acid acyltransferase, LPAAT-theta, which activates mTOR pathway. J Biochem Mol Biol 39:626–635 [DOI] [PubMed] [Google Scholar]
- Thomas CC, Deak M, Alessi DR, van Aalten DM. (2002) High-resolution structure of the pleckstrin homology domain of protein kinase b/akt bound to phosphatidylinositol (3,4,5)-trisphosphate. Curr Biol 12:1256–1262 [DOI] [PubMed] [Google Scholar]
- Tomic S, Greiser U, Lammers R, Kharitonenkov A, Imyanitov E, Ullrich A, Böhmer FD. (1995) Association of SH2 domain protein tyrosine phosphatases with the epidermal growth factor receptor in human tumor cells. Phosphatidic acid activates receptor dephosphorylation by PTP1C. J Biol Chem 270:21277–21284 [DOI] [PubMed] [Google Scholar]
- Toschi A, Edelstein J, Rockwell P, Ohh M, Foster DA. (2008) HIF α expression in VHL-deficient renal cancer cells is dependent on phospholipase D. Oncogene 27:2746–2753 [DOI] [PubMed] [Google Scholar]
- Toschi A, Lee E, Xu L, Garcia A, Gadir N, Foster DA. (2009) Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: competition with rapamycin. Mol Cell Biol 29:1411–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touhara K, Inglese J, Pitcher JA, Shaw G, Lefkowitz RJ. (1994) Binding of G protein beta gamma-subunits to pleckstrin homology domains. J Biol Chem 269:10217–10220 [PubMed] [Google Scholar]
- Trencia A, Perfetti A, Cassese A, Vigliotta G, Miele C, Oriente F, Santopietro S, Giacco F, Condorelli G, Formisano P, et al. (2003) Protein kinase B/Akt binds and phosphorylates PED/PEA-15, stabilizing its antiapoptotic action. Mol Cell Biol 23:4511–4521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SC, Adamik R, Hong JX, Moss J, Vaughan M, Kanoh H, Exton JH. (1998) Effects of arfaptin 1 on guanine nucleotide-dependent activation of phospholipase D and cholera toxin by ADP-ribosylation factor. J Biol Chem 273:20697–20701 [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM. (1984) Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 226:1097–1099 [DOI] [PubMed] [Google Scholar]
- Uchida N, Okamura S, Kuwano H. (1999) Phospholipase D activity in human gastric carcinoma. Anticancer Res 19 (1B):671–675 [PubMed] [Google Scholar]
- Uchida N, Okamura S, Nagamachi Y, Yamashita S. (1997) Increased phospholipase D activity in human breast cancer. J Cancer Res Clin Oncol 123:280–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usatyuk PV, Gorshkova IA, He D, Zhao Y, Kalari SK, Garcia JGN, Natarajan V. (2009) Phospholipase D-mediated activation of IQGAP1 through Rac1 regulates hyperoxia-induced p47phox translocation and reactive oxygen species generation in lung endothelial cells. J Biol Chem 284:15339–15352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasta V, Meacci E, Romiti E, Farnararo M, Bruni P. (1998) A role for phospholipase D activation in the lipid signalling cascade generated by bradykinin and thrombin in C2C12 myoblasts. Biochim Biophys Acta 1391:280–286 [DOI] [PubMed] [Google Scholar]
- Venkateswarlu K, Gunn-Moore F, Oatey PB, Tavaré JM, Cullen PJ. (1998) Nerve growth factor- and epidermal growth factor-stimulated translocation of the ADP-ribosylation factor-exchange factor GRP1 to the plasma membrane of PC12 cells requires activation of phosphatidylinositol 3-kinase and the GRP1 pleckstrin homology domain. Biochem J 335:139–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veverka V, Crabbe T, Bird I, Lennie G, Muskett FW, Taylor RJ, Carr MD. (2008) Structural characterization of the interaction of mTOR with phosphatidic acid and a novel class of inhibitor: compelling evidence for a central role of the FRB domain in small molecule-mediated regulation of mTOR. Oncogene 27:585–595 [DOI] [PubMed] [Google Scholar]
- Virolle T, Adamson ED, Baron V, Birle D, Mercola D, Mustelin T, de Belle I. (2001) The Egr-1 transcription factor directly activates PTEN during irradiation-induced signalling. Nat Cell Biol 3:1124–1128 [DOI] [PubMed] [Google Scholar]
- Vitale N, Caumont AS, Chasserot-Golaz S, Du G, Wu S, Sciorra VA, Morris AJ, Frohman MA, Bader MF. (2001) Phospholipase D1: a key factor for the exocytotic machinery in neuroendocrine cells. EMBO J 20:2424–2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss M, Weernink PA, Haupenthal S, Möller U, Cool RH, Bauer B, Camonis JH, Jakobs KH, Schmidt M. (1999) Phospholipase D stimulation by receptor tyrosine kinases mediated by protein kinase C and a Ras/Ral signaling cascade. J Biol Chem 274:34691–34698 [DOI] [PubMed] [Google Scholar]
- Walker SJ, Brown HA. (2002) Specificity of Rho insert-mediated activation of phospholipase D1. J Biol Chem 277:26260–26267 [DOI] [PubMed] [Google Scholar]
- Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, Fligelman B, Leversha M, Brennan C, Tabar V. (2010b) Glioblastoma stem-like cells give rise to tumour endothelium. Nature 468:829–833 [DOI] [PubMed] [Google Scholar]
- Wang T, Dowal L, El-Maghrabi MR, Rebecchi M, Scarlata S. (2000) The pleckstrin homology domain of phospholipase C-beta(2) links the binding of gbetagamma to activation of the catalytic core. J Biol Chem 275:7466–7469 [DOI] [PubMed] [Google Scholar]
- Wang X, Xu L, Zheng L. (1994) Cloning and expression of phosphatidylcholine-hydrolyzing phospholipase D from Ricinus communis L. J Biol Chem 269:20312–20317 [PubMed] [Google Scholar]
- Wang Y, Mandelkow E. (2012) Degradation of tau protein by autophagy and proteasomal pathways. Biochem Soc Trans 40:644–652 [DOI] [PubMed] [Google Scholar]
- Warburg O. (1956) On the origin of cancer cells. Science 123:309–314 [DOI] [PubMed] [Google Scholar]
- Watanabe G, Saito Y, Madaule P, Ishizaki T, Fujisawa K, Morii N, Mukai H, Ono Y, Kakizuka A, Narumiya S. (1996) Protein kinase N (PKN) and PKN-related protein rhophilin as targets of small GTPase Rho. Science 271:645–648 [DOI] [PubMed] [Google Scholar]
- Ways DK, Cook PP, Webster C, Parker PJ. (1992) Effect of phorbol esters on protein kinase C-zeta. J Biol Chem 267:4799–4805 [PubMed] [Google Scholar]
- Webb BA, Zhou S, Eves R, Shen L, Jia L, Mak AS. (2006) Phosphorylation of cortactin by p21-activated kinase. Arch Biochem Biophys 456:183–193 [DOI] [PubMed] [Google Scholar]
- Weed SA, Karginov AV, Schafer DA, Weaver AM, Kinley AW, Cooper JA, Parsons JT. (2000) Cortactin localization to sites of actin assembly in lamellipodia requires interactions with F-actin and the Arp2/3 complex. J Cell Biol 151:29–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA. (1995) The retinoblastoma protein and cell cycle control. Cell 81:323–330 [DOI] [PubMed] [Google Scholar]
- Wigge P, McMahon HT. (1998) The amphiphysin family of proteins and their role in endocytosis at the synapse. Trends Neurosci 21:339–344 [DOI] [PubMed] [Google Scholar]
- Wild-Bode C, Weller M, Wick W. (2001) Molecular determinants of glioma cell migration and invasion. J Neurosurg 94:978–984 [DOI] [PubMed] [Google Scholar]
- Williger BT, Ho WT, Exton JH. (1999) Phospholipase D mediates matrix metalloproteinase-9 secretion in phorbol ester-stimulated human fibrosarcoma cells. J Biol Chem 274:735–738 [DOI] [PubMed] [Google Scholar]
- Woltman AM, de Fijter JW, Kamerling SWA, van Der Kooij SW, Paul LC, Daha MR, van Kooten C. (2001) Rapamycin induces apoptosis in monocyte- and CD34-derived dendritic cells but not in monocytes and macrophages. Blood 98:174–180 [DOI] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. (2007) The genomic landscapes of human breast and colorectal cancers. Science 318:1108–1113 [DOI] [PubMed] [Google Scholar]
- Woods D, Parry D, Cherwinski H, Bosch E, Lees E, McMahon M. (1997) Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol Cell Biol 17:5598–5611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Wang P, Zhang J, Young LC, Lai R, Li L. (2010) Studies of phosphoproteomic changes induced by nucleophosmin-anaplastic lymphoma kinase (ALK) highlight deregulation of tumor necrosis factor (TNF)/Fas/TNF-related apoptosis-induced ligand signaling pathway in ALK-positive anaplastic large cell lymphoma. Mol Cell Proteomics 9:1616–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GS. (2004) The functional interactions between the p53 and MAPK signaling pathways. Cancer Biol Ther 3:156–161 [DOI] [PubMed] [Google Scholar]
- Wu H, Reynolds AB, Kanner SB, Vines RR, Parsons JT. (1991) Identification and characterization of a novel cytoskeleton-associated pp60src substrate. Mol Cell Biol 11:5113–5124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MN, Littleton JT, Bhat MA, Prokop A, Bellen HJ. (1998) ROP, the Drosophila Sec1 homolog, interacts with syntaxin and regulates neurotransmitter release in a dosage-dependent manner. EMBO J 17:127–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Ho WT, Exton JH. (2002a) Functional implications of post-translational modifications of phospholipases D1 and D2. Biochim Biophys Acta 1580:9–21 [DOI] [PubMed] [Google Scholar]
- Xie Z, Ho WT, Spellman R, Cai S, Exton JH. (2002b) Mechanisms of regulation of phospholipase D1 and D2 by the heterotrimeric G proteins G13 and Gq. J Biol Chem 277:11979–11986 [DOI] [PubMed] [Google Scholar]
- Xie Z, Klionsky DJ. (2007) Autophagosome formation: core machinery and adaptations. Nat Cell Biol 9:1102–1109 [DOI] [PubMed] [Google Scholar]
- Xu L, Salloum D, Medlin PS, Saqcena M, Yellen P, Perrella B, Foster DA. (2011) Phospholipase D mediates nutrient input to mammalian target of rapamycin complex 1 (mTORC1). J Biol Chem 286:25477–25486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Baltimore D. (1996) Dual roles of ATM in the cellular response to radiation and in cell growth control. Genes Dev 10:2401–2410 [DOI] [PubMed] [Google Scholar]
- Yaffe MB. (2002) Phosphotyrosine-binding domains in signal transduction. Nat Rev Mol Cell Biol 3:177–186 [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. (1998b) Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct 23:33–42 [DOI] [PubMed] [Google Scholar]
- Yamazaki M, Zhang Y, Watanabe H, Yokozeki T, Ohno S, Kaibuchi K, Shibata H, Mukai H, Ono Y, Frohman MA, et al. (1999) Interaction of the small G protein RhoA with the C terminus of human phospholipase D1. J Biol Chem 274:6035–6038 [DOI] [PubMed] [Google Scholar]
- Yan J, Roy S, Apolloni A, Lane A, Hancock JF. (1998) Ras isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. J Biol Chem 273:24052–24056 [DOI] [PubMed] [Google Scholar]
- Yang J, Weinberg RA. (2008) Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 14:818–829 [DOI] [PubMed] [Google Scholar]
- Yang Q, Inoki K, Ikenoue T, Guan KL. (2006) Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev 20:2820–2832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SF, Freer S, Benson AA. (1967) Transphosphatidylation by phospholipase D. J Biol Chem 242:477–484 [PubMed] [Google Scholar]
- Yang Z, Klionsky DJ. (2010) Eaten alive: a history of macroautophagy. Nat Cell Biol 12:814–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Kantonen S, Henkels KM, Gomez-Cambronero J. (2013) A new signaling pathway (JAK-Fes-phospholipase D) that is enhanced in highly proliferative breast cancer cells. J Biol Chem 288:9881–9891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye W, Lafer EM. (1995) Clathrin binding and assembly activities of expressed domains of the synapse-specific clathrin assembly protein AP-3. J Biol Chem 270:10933–10939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo EJ, Exton JH. (1995) Stimulation of phospholipase D by epidermal growth factor requires protein kinase C activation in Swiss 3T3 cells. J Biol Chem 270:3980–3988 [DOI] [PubMed] [Google Scholar]
- Yokozeki T, Homma K, Kuroda S, Kikkawa U, Ohno S, Takahashi M, Imahori K, Kanaho Y. (1998) Phosphatidic acid-dependent phosphorylation of a 29-kDa protein by protein kinase Calpha in bovine brain cytosol. J Neurochem 71:410–417 [DOI] [PubMed] [Google Scholar]
- Yoon MS, Du G, Backer JM, Frohman MA, Chen J. (2011) Class III PI-3-kinase activates phospholipase D in an amino acid-sensing mTORC1 pathway. J Cell Biol 195:435–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon MS, Koo JB, Hwang JH, Lee KS, Han JS. (2005) Activation of phospholipase D by 8-Br-cAMP occurs through novel pathway involving Src, Ras, and ERK in human endometrial stromal cells. FEBS Lett 579:5635–5642 [DOI] [PubMed] [Google Scholar]
- Yoon SN, Kim KS, Cho JH, Ma W, Choi HJ, Kwon SJ, Han JS. (2012) Phospholipase D1 mediates bFGF-induced Bcl-2 expression leading to neurite outgrowth in H19-7 cells. Biochem J 441:407–416 [DOI] [PubMed] [Google Scholar]
- Yoshida M, Okamura S, Kodaki T, Mori M, Yamashita S. (1998) Enhanced levels of oleate-dependent and Arf-dependent phospholipase D isoforms in experimental colon cancer. Oncol Res 10:399–406 [PubMed] [Google Scholar]
- Yu J, Baron V, Mercola D, Mustelin T, Adamson ED. (2007) A network of p73, p53 and Egr1 is required for efficient apoptosis in tumor cells. Cell Death Differ 14:436–446 [DOI] [PubMed] [Google Scholar]
- Zang ZJ, Cutcutache I, Poon SL, Zhang SL, McPherson JR, Tao J, Rajasegaran V, Heng HL, Deng N, Gan A, et al. (2012) Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet 44:570–574 [DOI] [PubMed] [Google Scholar]
- Zeng XXI, Zheng X, Xiang Y, Cho HP, Jessen JR, Zhong TP, Solnica-Krezel L, Brown HA. (2009) Phospholipase D1 is required for angiogenesis of intersegmental blood vessels in zebrafish. Dev Biol 328:363–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeniou-Meyer M, Liu Y, Béglé A, Olanich ME, Hanauer A, Becherer U, Rettig J, Bader MF, Vitale N. (2008) The Coffin-Lowry syndrome-associated protein RSK2 is implicated in calcium-regulated exocytosis through the regulation of PLD1. Proc Natl Acad Sci USA 105:8434–8439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Wang D, Singh NK, Kundumani-Sridharan V, Gadiparthi L, Rao ChM, Rao GN. (2011) Activation of cytosolic phospholipase A2 downstream of the Src-phospholipase D1 (PLD1)-protein kinase C γ (PKCγ) signaling axis is required for hypoxia-induced pathological retinal angiogenesis. J Biol Chem 286:22489–22498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Han J, Sells MA, Chernoff J, Knaus UG, Ulevitch RJ, Bokoch GM. (1995) Rho family GTPases regulate p38 mitogen-activated protein kinase through the downstream mediator Pak1. J Biol Chem 270:23934–23936 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Altshuller YM, Hammond SM, Hayes F, Morris AJ, Frohman MA. (1999) Loss of receptor regulation by a phospholipase D1 mutant unresponsive to protein kinase C. EMBO J 18:6339–6348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Redina O, Altshuller YM, Yamazaki M, Ramos J, Chneiweiss H, Kanaho Y, Frohman MA. (2000) Regulation of expression of phospholipase D1 and D2 by PEA-15, a novel protein that interacts with them. J Biol Chem 275:35224–35232 [DOI] [PubMed] [Google Scholar]
- Zhao C, Du G, Skowronek K, Frohman MA, Bar-Sagi D. (2007) Phospholipase D2-generated phosphatidic acid couples EGFR stimulation to Ras activation by Sos. Nat Cell Biol 9:706–712 [DOI] [PubMed] [Google Scholar]
- Zhao Y, Ehara H, Akao Y, Shamoto M, Nakagawa Y, Banno Y, Deguchi T, Ohishi N, Yagi K, Nozawa Y. (2000) Increased activity and intranuclear expression of phospholipase D2 in human renal cancer. Biochem Biophys Res Commun 278:140–143 [DOI] [PubMed] [Google Scholar]
- Zheng L, Shan J, Krishnamoorthi R, Wang X. (2002) Activation of plant phospholipase Dbeta by phosphatidylinositol 4,5-bisphosphate: characterization of binding site and mode of action. Biochemistry 41:4546–4553 [DOI] [PubMed] [Google Scholar]
- Zheng X, Bollinger Bollag W. (2003) Aquaporin 3 colocates with phospholipase d2 in caveolin-rich membrane microdomains and is downregulated upon keratinocyte differentiation. J Invest Dermatol 121:1487–1495 [DOI] [PubMed] [Google Scholar]
- Zheng Y, Rodrik V, Toschi A, Shi M, Hui L, Shen Y, Foster DA. (2006) Phospholipase D couples survival and migration signals in stress response of human cancer cells. J Biol Chem 281:15862–15868 [DOI] [PubMed] [Google Scholar]
- Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, Buechler P, Isaacs WB, Semenza GL, Simons JW. (1999) Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res 59:5830–5835 [PubMed] [Google Scholar]
- Zhong M, Shen Y, Zheng Y, Joseph T, Jackson D, Foster DA. (2003) Phospholipase D prevents apoptosis in v-Src-transformed rat fibroblasts and MDA-MB-231 breast cancer cells. Biochem Biophys Res Commun 302:615–619 [DOI] [PubMed] [Google Scholar]
- Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z. (2009) Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol 11:468–476 [DOI] [PMC free article] [PubMed] [Google Scholar]