

Abstract
The occurrence and frequency of outcrossing in homothallic fungal species in nature is an unresolved question. Here we report detection of frequent outcrossing in the homothallic fungus Sclerotinia sclerotiorum. In using multilocus linkage disequilibrium (LD) to infer recombination among microsatellite alleles, high mutation rates confound the estimates of recombination. To distinguish high mutation rates from recombination to infer outcrossing, 8 population samples comprising 268 S. sclerotiorum isolates from widely distributed agricultural fields were genotyped for 12 microsatellite markers, resulting in multiple polymorphic markers on three chromosomes. Each isolate was homokaryotic for the 12 loci. Pairwise LD was estimated using three methods: Fisher's exact test, index of association (IA) and Hedrick's D′. For most of the populations, pairwise LD decayed with increasing physical distance between loci in two of the three chromosomes. Therefore, the observed recombination of alleles cannot be simply attributed to mutation alone. Different recombination rates in various DNA regions (recombination hot/cold spots) and different evolutionary histories of the populations could explain the observed differences in rates of LD decay among the chromosomes and among populations. The majority of the isolates exhibited mycelial incompatibility, minimizing the possibility of heterokaryon formation and mitotic recombination. Thus, the observed high intrachromosomal recombination is due to meiotic recombination, suggesting frequent outcrossing in these populations, supporting the view that homothallism favors universal compatibility of gametes instead of traditionally believed haploid selfing in S. sclerotiorum. Frequent outcrossing facilitates emergence and spread of new traits such as fungicide resistance, increasing difficulties in managing Sclerotinia diseases.
Introduction
Fungi, as in many other organisms, can reproduce asexually (hereafter clonally) and sexually. Clonal reproduction in fungi includes production of mitotic propagules such as conidia, vegetative structure like sclerotia and mycelial fragments. In sexual reproduction, fungi can be heterothallic or homothallic. In heterothallic species, syngamy occurs between haploid cells with different alleles at mating type loci. Such regulatory mechanisms prevent haploid selfing and promote outcrossing (Billiard et al., 2012). In homothallic species, on the other hand, syngamy can occur between genetically identical haploid cells (clones) resulting in same clone mating or haploid selfing (Billiard et al., 2012). It is often believed that homothalism means predominantly, if not exclusively, haploid selfing because of proximity of compatible gametes from the same haploid individual (Kohn, 1995). Haploid selfing is functionally equivalent to clonal reproduction and thus haploid selfing populations exhibit a population structure indistinguishable from those of clonal reproduction. In contrast, outcrossing populations have recombining population structure, and recombination is the hallmark of outcrossing sexual reproduction (Tibayrenc et al., 1991; Smith et al., 1993; Milgroom, 1996). Phylogenetic studies suggest that homothallism evolved from ancient heterothallic species (Billiard et al., 2012). An unsolved question is whether homothallism has evolved to promote haploid selfing or to ensure universal mating compatibility under outcrossing (Billiard et al., 2012). Answering this question requires an accurate estimate of outcrossing frequency in natural populations of homothallic species that is rarely investigated (López-Villavicencio et al., 2013). However, high mutation rates can yield patterns looking like footprint of recombination. Thus, it is also important to ascertain that the observed recombination is not due to mutation before making an inference about outcrossing.
In addition to the conceptual issue of homothallism, whether a population has clonal or recombining population structure is a fundamental question in fungal biology, and has important implications in medicine, plant pathology and evolutionary biology. Most of the medically important fungi are asexual, resulting in clonal population structures (Taylor et al., 1999b). However, genetic recombination (or outcrossing sexual reproduction) provides a selective advantage for adaptation to new environments, especially under stressful conditions (Goddard et al., 2005). Fungal recombination could be sexual or parasexual. Parasexual cycle is a nonsexual process, peculiar to fungi and single-celled organisms, of transferring genetic material without meiosis or the development of sexual structures. It requires formation of heterokaryons through protoplast fusion or mycelial anastomosis. Mycelial anastomosis is possible only between members of the same mycelial compatibility groups (MCGs). Parasexual recombination is relatively rare and limited in nature (Clutterbuck, 1996), and sexual recombination is considered to be the main source of genotypic variations. Recombination can be detected by studying the linkage disequilibrium (LD) that measures the nonrandom association of alleles among loci. LD has been frequently used to distinguish clonality from outcrossing in microbial population genetics studies (Tibayrenc et al., 1991; Milgroom, 1996).
The most common LD measures are D′ and r2 (Lewontin, 1964; Ardlie et al., 2002). Hedrick (1987) compared several coefficients of LD and found that D′ performs better over several other estimates primarily because D′ is independent of allele frequencies and with no negative values (Hedrick, 1987). Another measure of LD at a multilocus scale is the index of association (IA) that has been frequently applied to microbial populations (Smith et al., 1993). In the IA test, the observed variance of distances (estimated by the number of different loci) between all pairs of individuals is compared with the expected variance of a simulated data set with unlimited recombination. The null hypothesis is complete panmixia and it is tested with randomization of the data set.
Among various molecular marker systems available, microsatellite markers have been the choice of marker systems in many recent population genetics studies because of their ubiquitous existence and the compatibility with high-throughput analysis. IA tests have been used to infer recombination (Taylor et al., 1999a; Bihon et al., 2012; Klaassen et al., 2012) despite high mutation rates of microsatellite loci (Li et al., 2002; Lynch et al., 2008). However, if the marker system has a high mutation rate, it might confound the inferences of recombination. This has been precisely the situation we found in the Ascomycete Sclerotinia sclerotiorum.
S. sclerotiorum, a plant pathogen of more than 400 plant species, is homothallic (Bolton et al., 2006). Its genome contains both mating-type idiomorphs (Amselem et al., 2011) and it can reproduce both clonally and sexually. Lacking a conidial state in its life cycle, its clonal reproduction is through mycelial fragments and sclerotia, recalcitrant melanized aggregates of haploid mycelium (Kohn, 1979; Bolton et al., 2006). Under appropriate (prolonged cool and moist) conditions, single sclerotia can germinate meiotically (carpogenically) possibly through haploid selfing with a transient diploid phase, producing apothecia bearing ascospores that are released through air current (Kohn, 1979; Bolton et al., 2006). Because of homothallism, the airborne ascospores are thought to propagate clonal genotypes and are functionally equivalent to clonal reproduction (Anderson and Kohn, 1995; Kohn, 1995). In environments where the prolonged cool and moist conditions are absent, sclerotia can germinate mitotically (myceliogenically), generating new mycelium to initiate a new disease cycle (Bolton et al., 2006). Earlier population studies of S. sclerotiorum found association of independent traits like DNA fingerprints hybridized with a repetitive DNA sequence probe and mycelial compatibility (anastomosing) groups (MCGs), and widespread distribution of a few haplotypes over time and space, and concluded that S. sclerotiorum had a clonal population structure (Kohli et al., 1992, 1995; Anderson and Kohn, 1995; Hambleton et al., 2002) even when considerable recombinations of alleles were detected (Kohli and Kohn, 1998). It was believed that the clonal population structure resulted from combination of asexual reproduction of sclerotia and predominantly, if not exclusively, haploid selfing of sexual reproduction (Kohn, 1995). However, studies using independent microsatellite markers found high genotypic diversity, discordance between multilocus haplotypes and MCGs and random association of alleles, and inferred occurrence of recombination, implicitly outcrossing, in S. sclerotiorum populations in potato fields in Washington State, USA (Atallah et al., 2004), bean fields in Brazil (Gomes et al., 2011) and canola fields in Australia (Sexton et al., 2006), Turkey (Mert-Turk et al., 2007), Iran (Hemmati et al., 2009), China and United States (Attanayake et al., 2013). As parasexuality in S. sclerotiorum has been shown only once under laboratory conditions (Ford et al., 1995), outcrossing is supposed to play a key role in generating recombinant genotypes.
We previously observed random association of alleles between pairs of loci (Attanayake et al., 2012). Close examination found that microsatellite markers located on the same supercontig (physically linked loci) were in linkage equilibrium, suggesting random association of alleles among physically linked loci. Such random association could be either because of crossover between two loci during outcrossing or because of high mutation rates at these loci or both. Ascertainment of the cause has important implications because it may either reinforce or invalidate the previous inference of outcrossing in S. sclerotiorum. This is very important as many recent studies were conducted using microsatellite marker loci that are known to have high mutation rates (Li et al., 2002; Lynch et al., 2008). Ideally, crossover is best studied in controlled populations with two parents (biallelic systems), and is commonly practiced in quantitative trait locus mapping and linkage mapping in plant and animal breeding experiments, but little has been done in fungi (Christians et al., 2011). Because of the homothallic nature, techniques for controlled crosses of S. sclerotiorum are not available. Therefore, we chose an approach based on LD and physical distances among markers to test whether crossover events take place in fungal populations, choosing S. sclerotiorum as a model in which outcrossing is cryptic and is only inferred.
The frequency of crossover events is used to estimate recombinational distances as a measure of genetic distances between loci. In a given DNA region, physical distance is proportional to the amount of recombination (via crossover) and inversely proportionate to the LD. In other words, LD would decay with increasing physical distance between markers. Therefore, decay of LD with physical distance is a direct result of meiotic recombination via crossover and is highly unlikely because of mutations alone. Because S. sclerotiorum is haploid and homokaryotic (identical nuclei in multinucleate cells of mycelium; natural heterokaryons have not been found and rigorously proven), the source of variation must have come from outcrossing followed by crossover, and mutations. Sexual reproduction (meiosis) itself may introduce mutations (Ni et al., 2013) but such mutations should be independent of physical distance between loci.
We wanted to assess that homologous recombination events take place in S. sclerotiorum populations and can be detected in some regions of the genome. The first objective of this study was to develop additional microsatellite markers in such a way that at least three markers were on the same chromosome. The second objective was to test pairwise LD between microsatellite marker loci on each chromosome and detect the relationship of LD with the physical distances among markers. The third objective was to see whether the recombination rates differed among different populations.
Materials and methods
Isolate collection and MCGs
A total of 238 isolates of S. sclerotiorum were collected from the United States from the commercial fields of canola, gourd, lentil, pea and potato. Another 30 isolates were collected from a commercial field of canola in Anhui Province, China (Table 1 and Figure 1). Sclerotia were collected from infected plants that were at a minimum of 6 feet (∼2 m) apart to avoid collecting clones originating from the same infections. Isolates were obtained from the sclerotia as previously described (Attanayake et al., 2013) and kept at 4 °C for short-term storage. All the isolates were tested for MCGs by pairing isolates in all possible combinations among isolates from a field (Kohn et al., 1990). Each pair was tested at least twice. MCG richness in each field was estimated by G/N where G is the number of MCGs and N is the total number of isolates.
Table 1. Host, year collected, location and number of Sclerotinia sclerotiorum isolates and the number of MCGs included in this study.
| Sample number | Host/population abbreviation | Year collected | Location | Number of isolates | Number of MCGsa | MCG richness (G/N)b | Number of haplotypec | Haplotype richness (G/N) |
|---|---|---|---|---|---|---|---|---|
| 1 | Pea | 2007 | Walla Walla, WA | 34 | 22 | 0.64 | 32 | 0.94 |
| 2 | Gourd | 2007 | Prosser, WA | 41 | 19 | 0.46 | 28 | 0.68 |
| 3 | Potato (D) | 2009 | Hermiston, OR | 32 | 16 | 0.5 | 23 | 0.72 |
| 4 | Potato (E) | 2009 | Hermiston, OR | 32 | 19 | 0.6 | 29 | 0.91 |
| 5 | Lentil | 2003 | Colton, WA | 40 | 18 | 0.45 | 28 | 0.70 |
| 6 | Lentil | 2005 | Spangle, WA | 33 | 17 | 0.57 | 26 | 0.87 |
| 7 | Canola (USA) | 2009 | Fargo, ND | 29 | 19 | 0.65 | 24 | 0.83 |
| 8 | Canola (China) | 2010 | Anhui province, China | 30 | 21 | 0.7 | 29 | 0.97 |
Abbreviations: G, number of MCGs; MCG, mycelial compatibility group; N, total number of isolates.
MCGs in each population.
Ratio of the number of MCGs over population size.
Unique combination of microsatellite alleles of all loci.
Figure 1.

(a) Locations where population samples (triangles labeled 1 to 8) were taken from. Numbers correspond to sample numbers in Table 1. (b) Relative locations on supercontigs (corresponding chromosomes in parenthesis) of microsatellite loci tested in this study. Locus with an asterisk (*) is polymorphic and used final LD analyses and locus without an asterisk was either monomorphic or hypervariable (9D) and was not suitable for population analyses.
Development of new microsatellite loci located on the same chromosomes of the S. sclerotiorum genome
The genome sequences of S. sclerotiorum (http://www.broadinstitute.org/annotation/genome/sclerotinia_sclerotiorum/MultiHome.html) were used to develop 12 microsatellite markers with the aid of the software WebSat (Martins et al., 2009). These new microsatellite markers together with seven existing microsatellite markers developed by Sirjusingh and Kohn (2001) allowed us to use multiple loci on each of four supercontigs (each supercontig on a separate chromosome) for further analysis. Physical location of each marker on a supercontig is determined based on the genome data base. Newly designed primers were tested for PCR amplification and screened for polymorphisms using at least 70 isolates from two fields of S. sclerotiorum. PCR amplification conditions were as previously described (Attanayake et al., 2012). Nucleotide sequences of amplified alleles were determined from both strands using an ABI PRISM 377 automatic sequencer (Applied Biosystems, Foster City, CA, USA) at the Sequencing Core Facility at Washington State University to confirm the presence of tandem repeats.
Screening the 19 microsatellite loci identified 12 loci that were polymorphic on the subset of the isolates. These 12 polymorphic loci were used to genotype all of the 268 isolates using an ABI3730xl DNA Analyzer (Applied Biosystems) at the USDA-ARS Western Regional Small Grain Genotyping Laboratory (Pullman, WA, USA) as described in Attanayake et al. (2012). Briefly, PCR reactions were performed with one of the four fluorophores (Vic, Pet, Ned and 6-Fam) and multiplexed. For fragment analysis, GeneMarker software (SoftGenetics, State College, PA, USA) was used to determine fragment sizes. Each isolate was genotyped at least twice for each locus.
Population structure and isolate grouping
For LD estimations, initially isolates from each field were considered as one population sample. Previous studies showed that isolates collected from the same field at different times were not differentiated (Sexton et al., 2006; Attanayake, 2012), but isolates collected from different fields showed various levels of genetic differentiation (Attanayake, 2012). The canola–China and canola–USA population samples were genotypically and phenotypically distinct (Attanayake et al., 2013). To remove any potential effects of clones on LD decay analysis, isolates of identical haplotypes (presumably clones) were removed so that each haplotype was represented by one isolate (clone correction) before LD analyses. In addition, the effect of any hidden genetic structure on LD analysis was examined. Genetic structure of the isolates was assessed using a posterior probability of Bayesian clustering approach implemented in the software STRUCTURE ver. 2.2 (Pitchard et al., 2000). The clustering method assigns isolates into K genetic clusters, each characterized by allele frequency data at each locus. Each individual isolate is assigned to one of the K clusters without consideration of its geographic origin. Although not all the markers satisfy the assumption of STRUCTURE because of close linkage of some markers, the loci are on three separate chromosomes and some linked loci are still in linkage equilibrium. A population admixture model was used and each simulation consisted of 100 000 Markov Chain Monte Carlo iterations with a burn-in period of 50 000 iterations. Five independent runs of 1 to 10 clusters (K=1–10) were performed to estimate the most probable number (K) of genetically homogenous clusters, as determined by the largest estimate of the posterior probability of the data for a given K, using ‘Ln P(D)' of the STRUCTURE output (Evanno et al., 2005). Pairwise LD analyses were then carried out within clusters defined by the STRUCTURE analyses. Isolates that did not belong to a genetic cluster with >50% probability were not included in the analyses.
Detection of LD between loci
LD between loci on each supercontig was calculated using three methods. First, the P-values of Fisher's exact test were used as proxy for LD. The null hypothesis is independent association of alleles or linkage equilibrium. Statistical significance of the LD was tested with Markov Chain algorithm with 1000 iterations as implemented in GenePop (Raymond and Rousset, 1995) for all possible pairs of loci on each supercontig. It is assumed that the smaller the P-value, the greater the evidence for rejecting the null hypothesis, and thus the stronger the linkage.
The second LD measure was Hedrick's D′ (Hedrick, 1987) that is based on Lewontin's D (Lewontin, 1964). Hedrick's D′ is a normalized multiallelic LD coefficient with properties of independency of allele frequencies and lack of negative values (Hedrick, 1987). Contingency tables for genotypes of each pair of loci on each supercontig were generated using GenePop and Hedrick's D′ for the total disequilibrium was estimated as follows
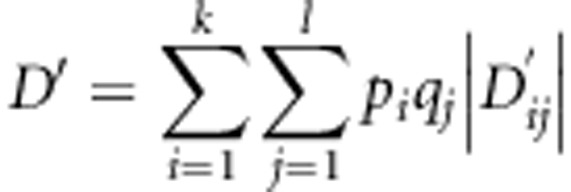 |
where, is Lewontin's D (Hedrick, 1987). An Excel spreadsheet with built-in functions for the calculation of Hedrick's D′ was developed for this purpose (Supplementary Spreadsheet 1).
The third LD measure was the pairwise LD, the multilocus IA test implemented in Multilocus (Agapow and Burt, 2001). Multilocus estimates IA, a traditional measure of multilocus LD (Brown et al., 1980), using the variances of distances generated by the number of different alleles among all pairs of isolates compared with a variance of distances of a hypothetical population with random association of the alleles. Hypothesis testing was done with 1000 randomizations of the data set.
To test the relationship and the reliability of the three LD measures, Pearson's correlation analyses among Hedrick's D′ (hereafter referred as D′), IA and P-values from Fisher's exact test were performed.
Results
MCG richness
Within each population sample, many isolates were mycelially incompatible. Minimum number of MCGs was 16 in the potato D population sample, whereas the highest number of MCGs was 22 in the pea population sample (Table 1). MCG richness, measured by the number of MCGs per isolate in each population (G/N), ranged from 0.45 to 0.7 (Table 1). A caution should be taken in directly comparing the number of MCGs among the population samples because of the unequal sample sizes.
Polymorphism of newly developed microsatellite markers
All the isolates showed single alleles for every microsatellite locus. Of the 19 microsatellite markers, 12 were found to be polymorphic in the initial screening and were selected for further analyses (5 of them were developed in this study and 7 were from Sirjusingh and Kohn, 2001). These 12 loci were located on four supercontigs each on a separate chromosome (four on supercontig 9, three each on supercontigs 3 and 19 and two on supercontig 15; Figure 1 and Table 2). Number of haplotypes (unique combinations of alleles among all loci) and haplotypes richness of the population samples are shown in Table 1. The sequences of these five new microsatellite markers were deposited in GenBank (GenBank accession numbers JX181754, JX181755, JX181756, JX181757 and JX181758). Except locus SC9-2, the number of alleles in each locus ranged from four to seven. Sequencing different alleles of each locus confirmed that the differences were because of the number of repeat motifs. However, locus SC9-2 had an unusually large (up to 24) number of alleles, and sequencing the alleles from eight isolates found that the different allele sizes were because of sequence variations within and outside the microsatellite repeats. Consequently, locus SC9-2 was excluded from further analysis, rendering three effective loci on supercontig 9. For supercontig 15, eight newly designed primers were tested and only one (SC15-9) was polymorphic. Primer sequences, nucleotide locations in supercontigs, microsatellite repeat motifs and annealing temperatures for the markers used in this study are shown in Table 2.
Table 2. Primer sequences, chromosomal locations, microsatellite repeat motifs, annealing temperatures and number of alleles for the markers used in this study.
| Locusa | Code | Primer sequences (5′ to 3′) | Start position | End position | Repeat motifb | Ta °C | Allele fragment size (bp)b | No. of alleles |
|---|---|---|---|---|---|---|---|---|
| Supercontig 3 (chromosome 4) | ||||||||
| 17-3 | 3A | F: TCATAGTGAGTGCATGATGCC | NAc | NAc | (TTA) | 55 | 371 | 9 |
| R: CAGGGATGACTTTGGAATGG | 92 | 111 | ||||||
| 114-4 | 3B | F: GCTCCTGTATACCATGTCTTG | 55 958 | 55 978 | (TAGA) | 55 | 388 | 11 |
| R: GGACTTTCGGACATGATGAT | 55 610 | 55 629 | ||||||
| 7-2 | 3C | F: TTTGCGTATTATGGTGGGC | 1 612 091 | 1 612 109 | (GA) | 55 | 191 | 4 |
| R: ATGGCGCAACTCTCAATAGG | 1 612 243 | 1 612 262 | ||||||
| Supercontig 19 (chromosome 5) | ||||||||
| SC19-1 | 19A | F: AAGAGAAGGGATGTGACTACG | 515 485 | 515 505 | (GATTT)(G | 57 | 397 | 9 |
| R: CGTTCGTTACTCAGGAGTCAGT | 515 841 | 515 862 | ATT) | |||||
| SC19-4 | 19B | F: CAAAATATCATCATCCCATCCC | 763 733 | 763 754 | (TCTCA) | 57 | 316 | 4 |
| R: ATCAAGCAAGCGCCACTAAT | 764 010 | 764 029 | ||||||
| 12-2 | 19C | F: CGATAATTTCCCCTCACTTGC | 777 981 | 778 001 | (CA) | 55 | 236 | 5 |
| R: GGAAGTCCTGATATCGTTGAGG | 778 176 | 778 197 | ||||||
| Supercontig 9 (chromosome 6) | ||||||||
| 110-4 | 9A | F: ATCCCTAACATCCCTAACGC | 1 083 935 | 1 083 954 | (TATG) | 55 | 395 | 6 |
| R: GGAGAATTGAAGAATTGAATGC | 1 084 289 | 1 084 310 | ||||||
| 13-2 | 9B | F: TCTACCCAAGCTTCAGTATTCC | 1 492 929 | 1 492 944 | (GTGGT) | 55 | 317 | 8 |
| R: GAACTGGTTAATTGTCTCGG | 1 492 647 | 1 492 666 | ||||||
| SC9-1 | 9C | F: CGGTCTTTTACTCTCCCATCTG | 1 503 300 | 1 503 321 | (TGGA) | 55 | 361 | 5 |
| R: TCACCATTCCTCTTTTACTCCC | 1 503 620 | 1 503 641 | ||||||
| SC9-2 | 9D | F: TCGCAATGTAGTAAACCGACAC | 1 590 634 | 1 590 655 | (TAGA) | 55 | 388 | 24 |
| R: ATTTGGGTCGTTGAGATGAGAT | 1 590 981 | 1 591 002 | ||||||
| Supercontig 15 (chromosome 15) | ||||||||
| SC15-1 | 15A | F: GCACAAATTCAAGCTCAAACTG | 73 594 | 73 615 | (GTAT) | 55 | 327 | 1d |
| R: TGGGAAGAGTGTTATGCAGAGA | 73 880 | 73 901 | ||||||
| SC15-2 | 15B | F: ATGAAGATATGCAAACGCACAC | 114 553 | 114 574 | (GAG) | 55 | 361 | 1d |
| R: AACCTATCGACCAGCAACAAAC | 114 857 | 114 878 | ||||||
| SC15-3 | 15C | F: CATTGCTGGAGTATGTAGCGAA | 115 460 | 115 481 | (TCA) | 55 | 261 | 1d |
| R: TCAACTCAACTTATCCCAACCC | 115 680 | 115 701 | ||||||
| SC15-4 | 15D | F: AGTCCCAAGTCCCAAGTCCAG | 184 464 | 184 484 | (AAT) | 55 | 257 | 2d |
| R: AGCGAATGGAATGACAGG | 18 4684 | 184 701 | ||||||
| SC15-5 | 15E | F: CTCGTCTCGTGCCTAGAATTG | 187 084 | 187 104 | (TAA) | 55 | 223 | 1d |
| R:GCTTTACAGGCTTGGGTTTGTA | 187 266 | 187 287 | ||||||
| SC15-9 | 15F | F: TCTCCTGTGTGTATGCTTTGCT | 188 832 | 188 853 | (CCTA)A | 55 | 240 | 5 |
| R:TGTCTGGTCTGAGGGTTATTCA | 189 031 | 189 052 | (TAA) | |||||
| 55-4 | 15G | F: GTTTTCGGTTGTGTGCTGG | 1 056 466 | 1 056 485 | (TACA) | 55 | 191 | 9 |
| R: GCTCGTTCAAGCTCAGCAAG | 1 056 617 | 1 056 637 | ||||||
| SC15–6 | 15H | F: TCGGTTTCTTCTCTGGATTGAT | 1 116 513 | 1 116 534 | (TGTA) | 55 | 211 | 2d |
| R: ATAGGTGAACCACACAGCTTGG | 1 116 683 | 1 116 704 | ||||||
| SC15–7 | 15I | F: TTAGTTTACCAACACCAAAGCC | 1 185 114 | 1 185 135 | (TAA) | 55 | 375 | 2d |
| R: TATATTAGGGCGCAAACGG | 1 185 451 | 1 185 469 | ||||||
Loci were either defined by Sirjusingh and Kohn (2001) or developed in this study. Polymorphic loci that were developed in this study are in bold.
Repeat motif and fragment size are based on the genome sequence of isolate 1980, for locus 17-3 based on Sirjusingh and Kohn (2001), and the fragment size includes a 19-bp M13 tail sequence.
NA, genomic designation of the primer is not available in the genome sequence database.
One allele accounted for >98% of the frequency and the locus was considered monomorphic.
Relationship of the three LD measures
Pairwise LD estimates were calculated in three ways: (1) all the isolates in the population samples based on geographic locations, (2) clone correction of the isolate in the geographic population samples and (3) genetic clusters defined by STRUCTURE analysis. In Bayesian clustering using STRUCTURE with K=1 to 10, the log likelihood values ‘Ln P(D)' increased when K values increased from 1 to 7 and plateaued at K=7. The log likelihood values then declined and its variance increased considerably when K>7 (Supplementary Figure S1), indicating there is strong likelihood of 7 genetic clusters among the 268 isolates. Pairwise LD analyses were then carried out within these seven clusters after clone correction. Two of the seven clusters corresponded with the canola–USA and canola–China population samples. The isolates in the other six population samples (1 to 6 in Table 1 and Figure 1) from the western United States were assigned into five genetic clusters showing no clear relationship with the fields where the samples were taken (Supplementary Figure S1). Because the effect of population structure on multiallelic LD analysis is not known, we calculated pairwise LDs based on the genetic clusters defined by STRUCTURE (clone corrected). The results obtained using the three methods were very similar and a decay of LD with increasing distance between loci was observed in each of the isolate-grouping methods.
Significant correlations among the three LD measures were found for the markers on supercontigs 3 and 9 (chromosomes 4 and 6; Figure 2). D′ and IA were significantly correlated (r=0.44, P=0.02 and r=0.84, P<0.001 for supercontigs 3 and 9, respectively). A negative correlation was found between D′ and P-values of Fisher's exact test (r=−0.75, P<0.001 and r=−0.72, P<0.001 for supercontigs 3 and 9, respectively) as well as IA and P-values of Fisher's exact test (r=−0.52, P=0.005 and r=−0.58, P=0.002 for supercontigs 3 and 9, respectively). However, for supercontig 19 (chromosome 5), no significant correlations were detected except the negative correlation coefficient between IA and P-values of Fisher's exact test (r=−0.55, P=0.004). Significant correlations among the three LD measures were also found when data from the three supercontigs were pooled together (Figure 2).
Figure 2.

Scatter plots and regressions showing the relationships of the three LD estimates for three supercontigs. (a) IA vs P-values of Fisher's exact test; (b) Hedrick's D′ vs IA; and (c) Hedrick's D′ vs P-values of Fisher's exact test.
Relationship between the P-values of Fisher's exact test of pairwise LD and physical distance
Pairwise LD as measured by Fisher's exact tests for each pairs of loci on supercontigs 3, 9 and 19 (chromosomes 4, 5 and 6) were generated. Significance of the exact test and physical distances between markers are shown in Table 3. Overall, more population samples showed significant LD (low P-values) between pairs of loci close to each other than between pairs of loci further apart on the same supercontig, particularly for supercontigs 3 and 9 (Table 3). On supercontig 3, the closest pair of loci (56 kb between loci 3A and 3B) showed significant LD in six of the eight population samples (P<0.001), whereas for the loci that were further apart (212 kb between loci 3A and 3C) only two of the eight population samples had significant LD. This relationship showed that P-values (recombination rate) increased as the distance between loci increased (Table 3).
Table 3. Physical distance and significance of Fisher's exact test (pairwise linkage disequilibrium test) between alleles on three supercontigs of Sclerotinia sclerotiorum in eight population samples.
| Locus pair | Distance (kb) | Pea | Gourds | Potato (E) | Potato (D) | Lentil 2003 | Lentil 2005 | Canola (USA) | Canola (China) |
|---|---|---|---|---|---|---|---|---|---|
| Supercontig 3 (chromosome 4) | |||||||||
| 3A vs 3B | 56 | 0.002 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.016 |
| 3B vs 3C | 156 | 0.027 | 0.0 | 0.2 | 0.11 | 0.03 | 0.03 | 0.009 | 1.00 |
| 3A vs 3C | 212 | 0.30 | 0.0 | 0.6 | 0.16 | 0.02 | 0.0 | 0.77 | 1.00 |
| Supercontig 9 (chromosome 6) | |||||||||
| 9B vs 9C | 1000 | 0.0 | 0.0 | 0.005 | 0.005 | 0.0 | 0.0 | 0.002 | 0.12 |
| 9A vs 9B | 4000 | 0.3 | 0.003 | 0.28 | 0.2 | 0.05 | 0.09 | 0.16 | 0.19 |
| 9A vs 9C | 5000 | 0.77 | 0.04 | 0.28 | 0.08 | 0.007 | 0.77 | 0.72 | 0.86 |
| Supercontig 19 (chromosome 5) | |||||||||
| 19B vs 19C | 14.2 | 0.3 | 0.15 | 0.07 | 0.0 | 0.0 | 0.0 | 0.0 | NAa |
| 19A vs 19B | 247 | 0.6 | 0.01 | 0.08 | 0.03 | 0.0 | 0.004 | 0.7 | 1.0 |
| 19A vs 19C | 261.2 | 0.03 | 0.06 | 0.79 | 0.28 | 0.0 | 0.0 | 0.1 | NAa |
NA, not applicable, not tested because locus 19C was monomorphic in this population.
Similarly, on supercontig 9, the closest pair of loci (1000 kb between loci 9B and 9C) showed significant (P<0.001) LD in four of the eight population samples (Table 3), and pairwise comparisons of loci that were further apart (4000 kb between loci 9A and 9B, and 5000 kb between markers 9A and 9C) showed no significant LD (large P-values) in any of the eight population samples, failing to reject the null hypothesis of random association (Table 3). Again, a trend of increasing P-values of Fisher's exact test with increasing distance between loci on supercontig 9 was observed in six of the eight population samples (Table 3).
However, the three loci on supercontig 19 behaved differently. One locus was monomorphic in one population sample (canola–China sample). When a locus has allele frequency=0 or 1, no disequilibrium can be estimated. The two closest loci (14.2 kb between loci 19B and 19C) showed significant LD in four of the remaining seven populations, whereas the two loci that were furthest apart (261.2 kb between loci 19A and 19C) showed significant LD in only two of the seven populations. A trend of increasing P-values with increasing distance between loci was detected only in two of the seven populations (Table 3).
Similar trend in increasing P-values of Fisher's exact test with increasing distance between loci was also observed after clone correction (Supplementary Table S1). Actually, the trend was enhanced (became more obvious) after clone correction, for example, loci on supercontig 19 in the potato E population sample (Supplementary Table S1). Similarly, the same trend was also observed in genetic clusters inferred by STRCUTURE (Supplementary Table S2). The Bayesian posterior probability clustering made some loci monomorphic within some clusters and rendered LD calculations impossible, for example, loci on supercontig 9 in the red and blue genetic clusters (Supplementary Table S2).
Relationship between Hedrick's D′ and physical distance
In general, LD measured by Hedrick's D′ decayed with increasing distances on two of the three supercontigs (Figure 3).
Figure 3.

Relationship between pairwise LD and the physical distance between markers on three supercontigs. Trend lines are shown only for r2>0.5. (a) Supercontig 3 (chromosome 4); (b) supercontig 9 (chromosome 6); and (c) supercontig 19 (chromosome 5).
For supercontig 3, four (canola–China, canola–USA, pea and potato E) of the eight population samples showed a clear relationship of reduced D′ values with increasing physical distances. The remaining four populations showed no apparent strong relationship between D′ values and physical distance. Similarly, for supercontig 9, six (canola–USA, gourds, lentil 2003, lentil 2005, pea and potato E) of the eight population samples displayed a decay of LD (decreasing D′ values) with increasing physical distance. The remaining two (canola–China and potato D) population samples showed no apparent relationship between D′ value and physical distance. However, for supercontig 19, only two (gourds and potato E) out of eight population samples showed such relationship of LD decay with increasing physical distance (Figure 3).
Similar trend of decreasing D′ value with increasing distance between loci was also observed on two of the three supercontigs when the isolates were grouped based on Bayesian posterior probability clustering (Supplementary Figure S2). On supercontig 3, four of the seven clusters showed decreasing D′ value with increasing distance between loci (Supplementary Figure S2). On supercontig 9, four loci became monomorphic in two of the seven genetic clusters that rendered LD test inapplicable. Three of the remaining five genetic clusters showed the trend of LD decay (Supplementary Figure S2). However, only one of the seven genetic clusters showed such a trend in supercontig 19.
Discussion
Whether homothallism has evolved to promote haploid selfing or to favor universal compatibility of gametes is an unresolved question (Billiard et al., 2012). Answering this question not only allows proper understanding of the evolutionary cause underlying the evolution of homothallism, but also affects choice of approaches to investigate homothallic fungi. Lack of a sound understanding of homothallism has hampered investigations of the important plant pathogen S. sclerotiorum. A view of homothallism promoting haploid selfing has undoubtedly played a role in earlier investigations that concluded that S. sclerotiorum had a clonal genetic structure and was functionally clonal (Kohli et al., 1992, 1995; Anderson and Kohn, 1995; Kohn 1995; Kohli and Kohn, 1998; Hambleton et al., 2002). However, more recent studies using microsatellite markers in several countries (Atallah et al., 2004; Sexton et al., 2006; Mert-Turk et al., 2007; Hemmati et al., 2009; Gomes et al., 2011; Attanayake et al., 2013) have detected random association, and inferred outcrossing, in S. sclerotiorum. Microsatellite markers are known to have high mutation rates (Li et al., 2002; Lynch et al., 2008) and subject to PCR artifacts, and mutations and PCR artifacts contribute to the observed polymorphisms and may increase estimation bias toward recombination because common statistic analyses of recombination cannot differentiate mutation from recombination.
Mutation is inevitable and we need an approach to measure mutation rates relative to recombination. In this study, we used physically linked microsatellite markers to estimate chromosomal scale LD and tested whether LD decays with increasing physical distance between markers. This allows us to detect mutation rates relative to recombination due to crossover at homologous chromosomes. If the observed random association of alleles between linked loci is largely due to random mutation, the measured LD should be independent of physical distance between loci. The concept of LD decay with the physically distant markers (increasing the likelihood of crossover events) is widely applied in linkage mapping in controlled breeding experiments and in association genetics. To our knowledge, this is the first time that the reverse concept of linkage mapping is used in fungal population genetics to infer outcrossing. LD decay with increasing distances between markers detected in this study proves that the observed recombination is due to outcrossing and not mutation. The results provide another line of evidence that S. sclerotiorum, in addition to haploid selfing and clonal sclerotial production, undergoes frequent outcrossing in nature, supporting the view that homothallism has evolved in S. sclerotiorum to favor universal compatibility of gametes (Billiard et al., 2012).
Syngamy in S. sclerotiorum occurs in or on sclerotia that is haploid and presumably homokaryotic. It is unclear when outcrossing occurs, but it is likely through fertilization with microconidia that have no other known functions (Kohn, 1979), similar to that found in its sister, heterothallic species S. trifoliorum (Uhm and Fujii, 1983). Sexton et al. (2006) suggested the possibility of formation of heterokaryotic sclerotia when the disease pressure is high and simultaneous coinfections might occur, producing sclerotia with multiple genotypes facilitating outcrossing. Indeed, outcrossing was observed in forced heterokaryonic sclerotia of S. sclerotiorum (Ford et al., 1995). Ekins et al. (2006) observed ascospore dimorphism in S. sclerotiorum, reminiscent to the morphological feature found in the heterothallic species S. trifoliorum.
Estimating LD for biallelic systems in populations from controlled crosses has been extensively investigated (Lewontin, 1964; Hedrick, 1987; Ardlie et al., 2002). However, for multiallelic systems such as microsatellite markers, estimating LD is more complicated and different coefficients have been proposed for measuring the extent of overall disequilibrium among all possible pairs of alleles (Hedrick, 1987; Zhao et al., 2005). Population structure is thought to affect LD estimate, but the precise effect is not certain (Ardlie et al., 2002; Zhao et al., 2005). More simulation and empirical studies are needed to assess the effect of population structure on LD estimates. In this study we used two approaches to population grouping for LD estimates: a population grouping based on geography where isolates collected from a field were grouped as a population sample, and a Bayesian posterior probability clustering where isolates were grouped based on allele frequencies regardless of their geographic origin. A clear relationship of LD decay with increasing physical distance between markers was observed when either approach was used (Table 3 and Figure 3; Supplementary Table S1 and Supplementary Figure S2), ruling out the possibility that the detected LD decay was because of artificial isolate grouping.
We detected pairwise LD decay with increasing physical distance between loci in several populations on two of the three chromosomes. Potato E population had LD decay for all three chromosomes indicating high recombination rates. Similarly, Conway et al. (1999) observed significant (measured with Fisher's exact test) decline in LD (Lewontin's D) with the increasing distance between nucleotide pairs of the merozoite surface protein 1 antigen (msp1) gene and concluded that meiotic recombination is frequent in certain populations of Plasmodium falciparum. Fungal recombination can be meiotic (sexual), mitotic or parasexual. However, the majority of the isolates were mycelially incompatible, likely minimizing the possibility of heterokaryon formation and mitotic recombination in nature that is rare (Clutterbuck, 1996). Heterokaryon formation has been reported only in laboratory conditions for S. sclerotiorum (Ford et al., 1995). All isolates in this study showed single alleles for each and every locus, suggesting the isolates are homokaryotic at least for the loci concerned. Thus, the recombination of alleles of loci on the same chromosomes observed in this study cannot be attributed to mitotic recombination or to mutation alone. Therefore, we conclude that the LD decay is because of outcrossing in this homothallic fungus.
A clear relationship between LD decay and physical distance was detected on chromosomes 4 and 6 for most of the populations, but not on chromosome 5 in which only two out of eight population samples had such a relationship. The DNA region on chromosome 5 covered by the three microsatellite markers could be either a recombination cold or hot spot, or the three microsatellite markers have high mutation rates relative to crossover events. It appears that this DNA region of the chromosome 5 had some level of recombination or high mutation rate, as seven of the eight populations showed linkage equilibrium between markers 19A and 19B (Table 3). Recombination hot spots have been detected in the human genome (Chakravarti et al., 1984; Ardlie et al., 2002; Arnheim et al., 2003; Jeffreys et al., 2004), and also found in many other genomes including Cryptococcus spp. (Hsueh et al., 2006), P. falciparum (Mu et al., 2005) and Schizosaccharomyces pombe (Steiner and Smith, 2005). In DNA regions of recombination hot spots, crossover occurs frequently and even closely linked loci do not show LD. In addition, the frequent crossover events in the recombination hot spots may interfere with each other, known as interference. Thus, the relationship between LD decay and physical distance cannot be observed in DNA regions of recombination hot spots or in mutation hot spots. In such instances, markers in close proximity may not be in LD (Jeffreys et al., 2004).
LD decay with increasing marker distance was not observed in every population sample. This could be because of different histories and environmental conditions of the populations (Arnheim et al., 2003). It is obvious that the crossover rates are influenced by many other factors. The eight agricultural fields have different histories, and have faced different environmental conditions. As an example, pea and lentil fields were nonirrigated in Washington state, whereas potato and gourd fields were frequently irrigated. Canola fields in North Dakota state have severe winters compared with the Columbia Basin in Washington State. Fungicide application is a common practice in canola and potato production systems, whereas fungicides are usually not applied in pea and lentil cropping systems. Thus, a combination of ecological, molecular and evolutionary forces such as mutation events, natural selection, genetic drift, population bottleneck and migration in different populations could explain the lack of linear relationship between LD and the distance on a chromosome in all the populations studied (Jeffreys et al., 2004; Hsueh et al., 2006). For example, low levels of polymorphisms at certain loci (because of population bottleneck) could reduce the chance of detecting crossover. There are reported recombination activators in other fungal genomes (Hsueh et al., 2006) and similar recombination activators may be in different on and off states in various natural populations of S. sclerotiorum, presenting another possible explanation of the observed differences among the populations.
We also detected different mutation rates in S. sclerotiorum in this study. Locus SC9-2 located on supercontig 9 had unusually high numbers of alleles, indicating a mutation hot spot. Many factors could affect mutation rates of microsatellite loci including sequence composition, repeat unit size, repeat unit purity and copy number. Occurrence of mutation hot spots is a norm for many organisms and has been detected in the human genome (Itsara et al., 2009). On the other hand, only two polymorphic loci were found among nine different microsatellite markers tested on supercontig 15 (Table 2), indicating that this DNA region is resistant to mutation and could be a mutation cold spot within the S. sclerotiorum genome. Lack of mutations indicate strong selection pressure, suggesting highly conserved and likely critical functions of this DNA region for S. sclerotiorum.
In this study we applied LD decay, a method widely used in association genetics and linkage mapping in controlled breeding experiments, to detect cryptic sexual outcrossing in a homothallic fungal species. LD decay analysis is robust in allowing detection of mutation rate relative to crossover under outcrossing that cannot be accomplished in IA analyses. In addition, we found that the three measures of multiallelic pairwise LD (Hedrick's D′, IA and Fisher's exact test) were correlated. Measuring pairwise LD in multiallelic situations has been problematic and a subject of several studies (see Zhao et al., 2005). This study also provides baseline information about recombination cold spots and hot spots in S. sclerotiorum genome. Mutation cold spots could suggest critical functions of the DNA region. Finally, we provide strong evidence of frequent outcrossing in the homothallic species S. sclerotiorum, supporting the view that homothallism has evolved to ensure universal mating-type compatibility under outcrossing (Billiard et al., 2012).
Data archiving
The sequence sets containing all the microsatellites have been deposited at Genbank (accession numbers: JX181754–JX181758). Data for genotypes of the microsatellite loci used in analysis of all the isolates are available from the Dryad Digital Repository: 10.5061/dryad.cd48f.
Acknowledgments
We thank the editor and two anonymous referees for their insightful and constructive comments that improved an earlier version of this manuscript. We thank Drs Austin Burt (Imperial Collage, London), Michael G Milgroom (Cornell University, USA) and Tobin Peever (Washington State University, USA) for suggestions and frequent communications during the study. This work was supported in part by the USDA National Sclerotinia Initiative.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on Heredity website (http://www.nature.com/hdy)
Supplementary Material
References
- Agapow P-M, Burt A. Indices of multilocus linkage disequilibrium. Mol Ecol Notes. 2001;1:101–102. [Google Scholar]
- Amselem J, Cuomo CA, van Kan JAL, Viaud M, Benito EP, Couloux A, et al. Genetic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet. 2011;7:e1002230. doi: 10.1371/journal.pgen.1002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JB, Kohn LM. Clonality in soilborne, plant-pathogenic fungi. Annu Rev Phytopathol. 1995;33:369–391. doi: 10.1146/annurev.py.33.090195.002101. [DOI] [PubMed] [Google Scholar]
- Ardlie KG, Kruglyak L, Seielstad M. Patterns of linkage disequilibrium in the human genome. Nat Rev Genet. 2002;3:299–309. doi: 10.1038/nrg777. [DOI] [PubMed] [Google Scholar]
- Arnheim N, Calabrese P, Nordborg M. Hot and cold spots of recombination in the human genome: the reason we should find them and how this can be achieved. Am J Hum Genet. 2003;73:5–16. doi: 10.1086/376419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atallah ZK, Larget B, Chen X, Johnson DA. High genetic diversity, phenotypic uniformity, and evidence of outcrossing in Sclerotinia sclerotiorum in the Columbia Basin of Washington State. Phytopathology. 2004;94:737–742. doi: 10.1094/PHYTO.2004.94.7.737. [DOI] [PubMed] [Google Scholar]
- Attanayake RN.2012. Genetic diversity, population differentiation and interachromosomal recombination inferring outcrossing in natural populations of Sclerotinia sclerotiorum. PhD dissertation, Washington State University.
- Attanayake RN, Carter PA, Jiang D, del Río-Mendoza L, Chen W. Sclerotinia sclerotiorum populations infecting canola from China and the United States are genetically and phenotypically distinct. Phytopathology. 2013;103:750–761. doi: 10.1094/PHYTO-07-12-0159-R. [DOI] [PubMed] [Google Scholar]
- Attanayake RN, Porter L, Johnson DA, Chen W. Genetic and phenotypic diversity and random association of DNA markers of isolates of the fungal plant pathogen Sclerotinia sclerotiorum from soil on a fine geographic scale. Soil Biol Biochem. 2012;55:28–36. [Google Scholar]
- Bihon W, Burgess T, Slippers B, Wingfield M, Wingfield B. High levels of genetic diversity and cryptic recombination is widespread in introduced Diplodia pinea populations. Australasian Plant Pathol. 2012;41:41–46. [Google Scholar]
- Billiard S, Lopez-Villavicencio M, Hood ME, Giraud T. Sex, outcrossing and mating types: unsolved questions in fungi and beyond. J Evol Biol. 2012;25:1020–1038. doi: 10.1111/j.1420-9101.2012.02495.x. [DOI] [PubMed] [Google Scholar]
- Bolton MD, Thomma BPHJ, Nelson BD. Sclerotinia sclerotiorum (Lib.) de Bary: biology and molecular traits of a cosmopolitan pathogen. Mol Plant Pathol. 2006;7:1–16. doi: 10.1111/j.1364-3703.2005.00316.x. [DOI] [PubMed] [Google Scholar]
- Brown AHD, Feldman MW, Nevo E. Multilocus structure of natural populations of Hordeum spontaneum. Genetics. 1980;96:523–536. doi: 10.1093/genetics/96.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarti A, Buetow KH, Antonarakis SE, Waber PG, Boehm CD, Kazazian HH. Nonuniform recombination within the human beta-globin gene cluster. Am J Hum Genet. 1984;36:1239–1258. [PMC free article] [PubMed] [Google Scholar]
- Christians JK, Cheema MS, Vergara IA, Watt CA, Pinto LJ, Chen N, et al. Quantitative trait locus (QTL) mapping reveals a role for unstudied genes in Aspergillus virulence. PLoS ONE. 2011;6:e19325. doi: 10.1371/journal.pone.0019325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clutterbuck A. Parasexual recombination in fungi. J Genet. 1996;75:281–286. [Google Scholar]
- Conway DJ, Roper C, Oduola AMJ, Arnot DE, Kremsner PG, Grobusch MP, et al. High recombination rate in natural populations of Plasmodium falciparum. Proc Natl Acad Sci USA. 1999;96:4506–4511. doi: 10.1073/pnas.96.8.4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins M, Aitken EA, Coulter KC. Homothallism in Sclerotinia minor. Mycol Res. 2006;110:1193–1199. doi: 10.1016/j.mycres.2006.07.014. [DOI] [PubMed] [Google Scholar]
- Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of indivuduals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- Ford EJ, Miller RV, Gray H, Sherwood JE. Heterokaryon formation and vegetative compatibility in Sclerotinia sclerotiorum. Mycol Res. 1995;99:241–247. [Google Scholar]
- Goddard MR, Godfray HCJ, Burt A. Sex increases the efficacy of natural selection in experimental yeast populations. Nature. 2005;434:636–640. doi: 10.1038/nature03405. [DOI] [PubMed] [Google Scholar]
- Gomes EV, do Nascimento LB, de Freitas MA, Nasser LCB, Petrofeza S. Microsatellite markers reveal genetic variation within Sclerotinia sclerotiorum populations in irrigated dry bean crops in Brazil. J Phytopathol. 2011;159:94–99. [Google Scholar]
- Hambleton S, Walker C, Kohn LM. Clonal lineages of Sclerotinia sclerotiorum previously known from other crops predominate in 1999-2000 samples from Ontario and Quebec soybean. Can J Plant Pathol. 2002;24:309–315. [Google Scholar]
- Hedrick PW. Gametic disequilibrium measures: proceed with caution. Genetics. 1987;117:331–341. doi: 10.1093/genetics/117.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmati R, Javan-Nikkhah M, Linde CC. Population genetic structure of Sclerotinia sclerotiorum on canola in Iran. Eur J Plant Pathol. 2009;125:617–628. [Google Scholar]
- Hsueh Y-P, Idnurm A, Heitman J. Recombination hotspots flank the Cryptococcus mating-type locus: implications for the evolution of a fungal sex chromosome. PLoS Genet. 2006;2:e184. doi: 10.1371/journal.pgen.0020184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itsara A, Cooper GM, Baker C, Girirajan S, Li J, Absher D, et al. Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet. 2009;84:148–161. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys AJ, Holloway JK, Kauppi L, May CA, Neumann R, Slingsby MT, et al. Meiotic recombination hot spots and human DNA diversity. Phil Trans R Soc Lond B. 2004;359:141–152. doi: 10.1098/rstb.2003.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassen CHW, Gibbons JG, Fedorova ND, Meis JF, Rokas A. Evidence for genetic differentiation and variable recombination rates among Dutch populations of the opportunistic human pathogen Aspergillus fumigatus. Mol Ecol. 2012;21:57–70. doi: 10.1111/j.1365-294X.2011.05364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli Y, Brunner LJ, Yoell H, Milgroom MG, Anderson JB, Morrall RAA, et al. Clonal dispersal and spatial mixing in populations of the plant pathogenic fungus Sclerotinia sclerotiorum. Mol Ecol. 1995;4:69–77. [Google Scholar]
- Kohli Y, Kohn LM. Random association among alleles in clonal populations of Sclerotinia sclerotiorum. Fungal Genet Biol. 1998;23:139–149. doi: 10.1006/fgbi.1997.1026. [DOI] [PubMed] [Google Scholar]
- Kohli Y, Morrall R, Anderson JB, Kohn LM. Local and trans canadian clonal distribution of Sclerotinia sclerotiorum on canola. Phytopathology. 1992;82:875–880. [Google Scholar]
- Kohn LM. A monographic revision of the genus Sclerotinia. Mycotaxon. 1979;9:365–444. [Google Scholar]
- Kohn LM. The clonal dynamics in wild and agricultural plant-pathogen populations. Can J Bot. 1995;73:S1231–S1240. [Google Scholar]
- Kohn LM, Carbone I, Anderson JB. Mycelial interactions in Sclerotinia sclerotiorum. Exp Mycol. 1990;14:255–267. [Google Scholar]
- Lewontin RC. The interaction of selection and linkage. I. General considerations; heterotic models. Genetics. 1964;49:49–67. doi: 10.1093/genetics/49.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YC, Korol AB, Fahima T, Beiles A, Nevo E. Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review. Mol Ecol. 2002;11:2453–2465. doi: 10.1046/j.1365-294x.2002.01643.x. [DOI] [PubMed] [Google Scholar]
- López-Villavicencio ML, Debets AJM, Slakhorst M, Giraud T, Schoustra SE. Deleterious effects of recombination and possible nonrecopmbinatorial advantages of sex in a fungal model. J Evol Biol. 2013;26:1968–1978. doi: 10.1111/jeb.12196. [DOI] [PubMed] [Google Scholar]
- Lynch M, Sung W, Morris K, Coffey N, Landry CR, Dopman EB, et al. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci USA. 2008;105:9272–9277. doi: 10.1073/pnas.0803466105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins WS, Lucas DC, Neves KF, Bertioli DJ. WebSat - a web software for microsatellite marker development. Bioinformation. 2009;3:282–283. doi: 10.6026/97320630003282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mert-Turk F, Ipek M, Mermer D, Nicholson P. Microsatellite and morphological markers reveal genetic variation within a population of Sclerotinia sclerotiorum from oilseed rape in the Canakkale Province of Turkey. J Phytopathol. 2007;155:182–187. [Google Scholar]
- Milgroom MG. Recombination and the multilocus structure of fungal populations. Ann Rev Phytopathol. 1996;34:457–477. doi: 10.1146/annurev.phyto.34.1.457. [DOI] [PubMed] [Google Scholar]
- Mu J, Awadalla P, Duan J, McGee KM, Joy DA, McVean GAT, et al. Recombination hotspots and population structure in Plasmodium falciparum. PLoS Biol. 2005;3:e335. doi: 10.1371/journal.pbio.0030335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni M, Feretzaki M, Li W, Floyd-Averette A, Mieczkowski P, Dietrich FS, et al. Unisexual and heterosexual meiotic reproduction generate aneuploidy and phenotypic diversity de novo in the yeast Cryptococcus neoformans. PLoS Biol. 2013;11:e1001653. doi: 10.1371/journal.pbio.1001653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitchard JK, Stephens M, Donnelly P. Inference for population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond M, Rousset F. GENEPOP (Version 1.2): Population genetics software for exact tests and ecumenicism. J Hered. 1995;86:248–249. [Google Scholar]
- Sexton AC, Whitten AR, Howlett BJ. Population structure of Sclerotinia sclerotiorum in an Australian canola field at flowering and stem-infection stages of the disease cycle. Genome. 2006;49:1408–1415. doi: 10.1139/g06-101. [DOI] [PubMed] [Google Scholar]
- Sirjusingh C, Kohn LM. Characterization of microsatellites in the fungal plant pathogen, Sclerotinia sclerotiorum. Mol Ecol Notes. 2001;1:267–269. [Google Scholar]
- Smith JM, Smith NH, O'Rourke M, Spratt BG. How clonal are bacteria. Proc Natl Acad Sci USA. 1993;90:4384. doi: 10.1073/pnas.90.10.4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner WW, Smith GR. Natural meiotic recombination hot spots in the Schizosaccharomyces pombe genome successfully predicted from the simple sequence motif M26. Mol Cell Biol. 2005;25:9054–9062. doi: 10.1128/MCB.25.20.9054-9062.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JW, Jacobson DJ, Fisher MC. The evolution of asexual fungi: reproduction, speciation and classification. Annu Rev Phytopathol. 1999;37:197–246. doi: 10.1146/annurev.phyto.37.1.197. [DOI] [PubMed] [Google Scholar]
- Taylor JW, Geiser DM, Burt A, Koufopanou V. The evolutionary biology and population genetics underlying fungal strain typing. Clin Microbiol Rev. 1999;12:126–146. doi: 10.1128/cmr.12.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibayrenc M, Kjellberg F, Arnaud J, Oury B, Brenière SF, Dardé ML, et al. Are eukaryotic microorganisms clonal or sexual? A population genetics vantage. Proc Natl Acad Sci USA. 1991;88:5129–5133. doi: 10.1073/pnas.88.12.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhm JY, Fujii J. Heterothallism and mating type mutation in Sclerotinia trifoliorum. Phytopathology. 1983;73:569–572. [Google Scholar]
- Zhao H, Nettleton D, Soller M, Dekkers JCM. Evaluation of linkage disequilibrium measures between multi-allelic markers as predictors of linkage disequilibrium between markers and QTL. Genet Res. 2005;86:77–87. doi: 10.1017/S001667230500769X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.