

Abstract
The native bases of RNA and DNA are prominent examples of the narrow selection of organic molecules upon which life is based. How did nature “decide” upon these specific heterocycles? Evidence suggests that many types of heterocycles could have been present on the early Earth. It is therefore likely that the contemporary composition of nucleobases is a result of multiple selection pressures that operated during early chemical and biological evolution. The persistence of the fittest heterocycles in the prebiotic environment towards, for example, hydrolytic and photochemical assaults, may have given some nucleobases a selective advantage for incorporation into the first informational polymers. The prebiotic formation of polymeric nucleic acids employing the native bases remains, however, a challenging problem to reconcile. Hypotheses have proposed that the emerging RNA world may have included many types of nucleobases. This is supported by the extensive utilization of non-canonical nucleobases in extant RNA and the resemblance of many of the modified bases to heterocycles generated in simulated prebiotic chemistry experiments. Selection pressures in the RNA world could have therefore narrowed the composition of the nucleic acid bases. Two such selection pressures may have been related to genetic fidelity and duplex stability. Considering these possible selection criteria, the native bases along with other related heterocycles seem to exhibit a certain level of fitness. We end by discussing the strength of the N-glycosidic bond as a potential fitness parameter in the early DNA world, which may have played a part in the refinement of the alphabetic bases.
Keywords: DNA, molecular evolution, nucleobases, prebiotic chemistry, RNA world
1. Introduction
The Native Bases of the Genetic Alphabet
Just five nucleobases, also termed the genetic alphabet, are known to dominate the composition of deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) (Figure 1). They appear simple and robust, and their well-known canonical or Watson-Crick (WC) base-pairing properties (Figure 1) are so elegant that considering nature’s use of any other heterocycles seems almost unlikely. Yet, we submit here that the composition of the genetic alphabet may not have emerged in one pivotal epoch before or at the time of the origin of life. Instead, the alphabetic composition is the product of a continual process of refinement that evolved to its current state. We begin this journey by illustrating the widespread occurrence of modified bases in the biological world.[1]
Figure 1.
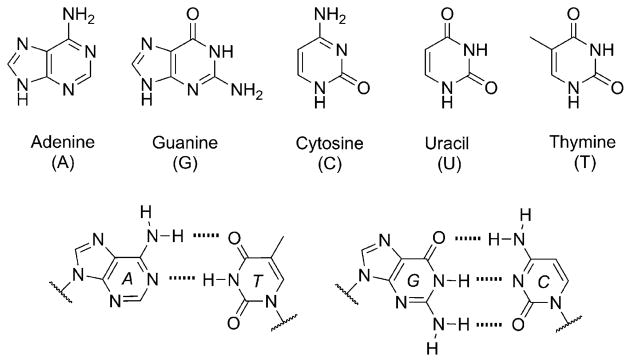
The native nucleobases of the genetic alphabet (top) and the Watson-Crick base-pairing relationship of the DNA nucleo-bases (bottom). In RNA the base uracil (U) assumes the place of thymine (T).
The Occurrence of Modified Bases
Nucleobase modification is a ubiquitous post-transcriptional activity found across all domains of life. These transformations are vital to cellular function since they modulate genetic expression, provide the means for editing and proper translation, and impact the folding and stability of nucleic acids, to name a few essential outcomes.[1] The diversity of modifications found in nucleic acids can range from extensive alterations, which almost disguise the base entirely (Figure 2A), to more modest transformations that closely resemble the native bases and retain their canonical base pairing (Figure 2B). When did the practice of base modification originate in biotic evolution? Were they always post-transcriptional modifications or do they have a more ancient origin?[2]
Figure 2.

A) Selected examples of extensive RNA nucleobase modification identified in biological nucleic acids. i) Adenine is most often alkylated at its N6 amino group, but here it has also been altered at the C2 position with an alkylthio group. ii) Guanine has been heavily reworked by the replacement of its heterocyclic N7 with a carbon, which is further substituted with an amidine group. iii) Cytosine has been tautomerized and aminated at the C1 position with a lysine residue. iv) Uracil substituted at the C5 position with a secondary aminoalkyl group and a thiocarbonyl at C2. B) Selected examples of conservative base modifications that are known to occur more often in RNA. The modified pyrimidines, with the exception of s2U, are also utilized in exotic DNA genomes. Frequently, the modifications include alkylation of the purine exocyclic amino groups (as for N2 of guanine and N6 of adenine) or modifications at C5 of the pyrimidines. Also shown is the deamination of adenine leading to hypoxanthine (called inosine in RNA), an important modification used as a guanine surrogate in edited RNA transcripts.
As shown in Figure 2B, it is often overlooked that thymine itself is a base modification of uracil.[3] Interestingly, of all the DNA nucleotides biosynthesized, thymidine is the only one that is made from another deoxynucleotide, where thymidine monophosphate (TMP) is obtained via the methylation of deoxyuridine monophosphate (dUMP).[3] The roundabout way in which cells generate TMP, coupled to the frequent misincorporation of UMP into DNA, has led to the idea that DNA was based, at one time, on A, G, C and U.[3,4] There is direct evidence for uracil-based DNA in extant biochemistry; it is one of the more common native base substitutions found in bacteriophage DNA.[5] More intriguing is that other nucleobases, which are nowadays considered altered bases, have also been found to completely replace some of the native bases in bacteriophage DNA. 5-Methylcytosine, 5-hydrox-ymethylcytosine, and 5-hydroxymethyluracil are the most prevalent examples.[6] The occurrence of these “modified” bases and others that can serve as viable genetic DNA surrogates, and the idea that thymine itself is a product of chemical modification that replaced uracil in early DNA evolution, has influenced how investigators think about the origin of the nucleobase composition in the extant alphabet.
A major contribution to our understanding of this evolutionary perception was made by Benner in the late 1980s and early 1990s. His laboratory demonstrated that RNA and DNA polymerases could selectively accept non-native bases and base pairs in the presence of the native ones, thus exhibiting faithful recognition and replication for all bases included.[7] This illustrated that contemporary polymerases could, in principle, utilize an expanded genetic alphabet. Additionally, it suggested that the native bases do not necessarily exhibit any unique genetic property over the non-native ones, outside of the fact that contemporary polymerases have evolved alongside the native bases, making the latter easier to incorporate. Further insight came from the identity of the artificial bases used, because they appeared to be just as elegant and simple in structure as the native ones. One pair in particular consisted of isomeric structures of guanine and cytosine (called isoG and isoC, respectively) that had previously been hypothesized by Rich in 1962, as plausible alphabetic components in early life given the prebiotic synthetic feasibility of these heterocyclic structures.[8] Many types of bases and base pairs have since been considered prebiotically viable, which has added further intrigue to questions surrounding the specific nature of the genetic alphabet (see below). A short and modest list of alternative bases and base pairs, relevant to this discussion of the genetic alphabet, is given in Figure 3. Many of the alternative nucleobases and pairs are drawn from their occurrence in contemporary biology or demonstrated utility in synthetic biology. Also are highlighted some of the most intriguing examples for alternative choices that may have been contenders during the course of chemical and early biological evolution of nucleic acids.[9] Note that we have not included newer synthetic base pairs that have recently been promoted for diverse molecular biological applications but are likely prebiotically irrelevant.[10]
Figure 3.

Modified or alternative bases that have been shown to be capable of genetic functions. The left column shows bases that are known to be reliable surrogates for adenine or thymine either in vivo or in vitro. The middle column contains guanine or cytosine surrogates. The right column represents base-pairing relationships that are not known to occur in nature. The base pairs colored gray in this figure were among those first demonstrated to be enzymatically incorporated into RNA and DNA by Benner.[7]
Why the Native Bases and Not the Others?
The base pairs shown in Figure 3 appear to be just as plausible as the native WC pairs. But as organic chemists know all too well, what appears feasible on paper does not necessarily translate into practice. Even minute structural changes can have substantial consequences, impacting the intermolecular, intramolecular and macromolecular “chemical physiology” of nucleic acids.[11] However, since modified bases are utilized in nature, often to a high degree[12] and in some cases completely replacing a native letter,[5] it does suggest that not all bases or base pairs would be precluded from an alphabetic role.
Numerous and insightful investigations have been pursued to identify plausible selection pressures and evolutionary processes that might have operated.[9,13] We discuss below contributions that have enhanced our appreciation for the “fitness” of the native bases in comparison to those that are largely highlighted in Figure 3. The selection pressures we will consider are segmented into periods or events (Figure 4) that are currently viewed as essential to the chemical and biological evolution of the genetic alphabet. Nevertheless, these pressures are not necessarily stationed in one hypothetical period.
Figure 4.

Periods of possible selection pressures. Evolutionary arrow from the formation of the Earth ~4.5 billion years ago and prebiotic chemistry to LUCA-based life and extant biology today, marked by major “events” that could have been crucial to hosting multiple selection pressures that shaped the composition of the genetic alphabet (LUCA = Last Universal Common Ancestor).
2. Prebiotic Chemistry and Environmental Selection Pressures
Results from simulated prebiotic chemistry experiments conducted over the past fifty years[14] and the ongoing analysis of meteorites[15] provide evidence that not only the native bases were likely present on the early Earth, but so were many others (Figure 5). Furthermore, a diverse population does not have to originate from separate formation scenarios. Purines and pyrimidines are known to undergo chemical reactions of prebiotic relevance that can enrich their population diversity. Hydrolytic deamination is probably one of the most ubiquitous reaction pathways that augments the population. As shown in Figure 6, the entire genetic alphabet and close relatives can be generated from just three simple “molecular ancestors”.[16] Oxidation, via Fenton chemistry (Fe2+ and H2O2),[17] at the C8 or C2 position of the purines and C5 position of pyrimidines is another way that modified bases can result. Outside of deamination, electrophilic aromatic substitution at the pyrimidine C5 position is probably the second most important way that modified pyrimidines could have populated the landscape.[18] This pathway has also been hypothesized to facilitate the methylation of uracil in a prebiotic reaction.[19] Many more prebiotic scenarios for the generation of modified bases, either from the native bases or independently, have been explored but are not shown here.[20]
Figure 5.

Numerous heterocycles have been identified in prebiotic chemistry simulation experiments and in meteorites. Shown here are some of the purine and pyrimidine nucleobases relevant to our discussion. The structures shown in black have been identified in both meteorites and observed in prebiotic chemistry experiments. The structures in red have only been observed in simulated prebiotic chemistry experiments. The structure in green has been recently identified in meteorites.
Figure 6.

Reaction genealogy of purines and pyrimidines starting from three simple amino-substituted heterocycles. Shown are 2,6-diaminopurine (Dap), adenine (A), and 2,4-diaminopyrimidine (Dpy), all considered to be prebiotically plausible heterocycles, which can in principle give rise to many different types of nucleobases, including the native alphabet and its surrogates. The reactions shown here are all well known and occur in contemporary biochemistry.
Clearly, the considerations applicable to potential prebiotic synthetic pathways do not appear to provide much insight into the selection of the bases of interest. We have not taken into account, however, stability, which is considered by many to be the more important factor regarding prebiotic abundance. In the context of prebiotic chemistry, the persistence of a nucleobase in a particular hypothetical environment reflects a balance between synthetic production (or deposition), side reactions (productive or deterrent), and degradation (Figure 7). Comparing the stability of the native bases to others when confronted with reactions that might challenge their persistence has thus been a useful way of understanding their relative fitness. There is a limited record of what the early Earth was like,[21] and so it is often difficult to identify the most important environmental conditions that could have generated a selection pressure (temperature and temperature cycling, pH, etc.). However, it does seem that at some point hydrolysis and UV irradiation, two potent environmental factors, would have likely challenged the persistence of nucleobases on the early Earth. We discuss both and assess their significance.
Figure 7.
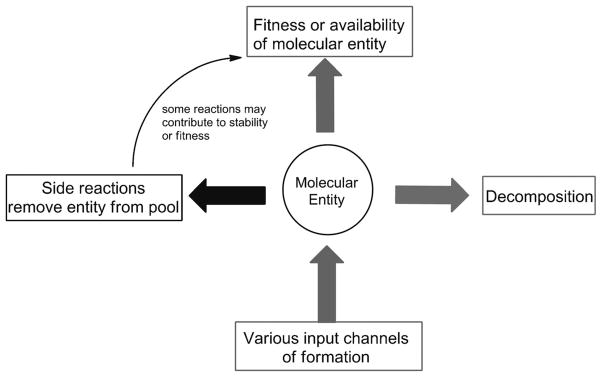
The prebiotic abundance of a nucleobase (or any molecular entity) can be viewed as the net result of synthetic (input channels) and degradation (decomposition or side reactions) pathways. While decomposition removes the nucleobase from the pool, some side reactions (such as alkylation of amino groups) could actually enhance the stability of a particular base in a prebiotic environment.
Hydrolytic Degradation of the Bases
The amino groups of the nucleobases in the native alphabet (Figure 1) and the ones shown in Figure 3 are absolutely essential to maintaining the fidelity of genetic information. Spontaneous deamination reactions, replacing an NH2 group, a H donating moiety, with a carbonyl, a H-bond acceptor, are thus highly deleterious and can lead to genetic mutations.[22] It would seem reasonable to hypothesize that the bases used by nature would have been selected to exhibit some of the highest stabilities against these spontaneous deamination reactions in comparison to alternative nucleobases. In a prebiotic scenario though, it could also be possible that the native bases exhibited the most robust heterocyclic stability in an aqueous environment against the deamination and ring degradation. Greater persistence in this environment would have given the native bases an advantage over others, possibility facilitating their selective incorporation into the first primitive genetic polymers. While much work has been done on the stability of nucleobases under a variety of conditions,[20a,23] Stanley Miller’s comparative studies of the deamination and ring-opening reaction rates are the most relevant to this discussion, as they provide clues to the relative stability of the native heterocycles (see Table 1 for a descending list of half-life values).[20a,24]
Table 1.
Half-life values of hydrolytic deamination and ring degradation/opening at pH 7 and 100°.[a]
| Deamination | Half-life value | Ring degradation or opening | Half-life value |
|---|---|---|---|
| 2,6-diaminopurine | 2.1 yr | Thymine | 56 yr |
| Adenine | 1 yr | Uracil | 12 yr |
| Guanine | 0.8 yr | Xanthine | 146 days |
| 2,4-Diaminopyrimidine | 42 days | Hypoxanthine | 12 days |
| N4-Methylcytosine | 38 days | 5,6-Dihydrouracil | 9.1 hours |
| Isocytosine | 21 days | ||
| Isoguanine | 20 days | ||
| Cytosine | 19 days | ||
| 5-Hydroxymethylcytosine | 13 days | ||
| 2-Thiocytosine | 11 days | ||
| 5-Methylcytosine | 9 days |
With regards to hydrolytic deamination reactions, it is clear that the native purines (A and G) are among the most stable. However, the longer half-life value reported for deamination of 2,6-diaminopurine shows that the native purines are not the most stable under these reaction conditions. In ring-opening or general degradation pathways, it is apparent that thymine and uracil are significantly more stable than any of the deaminated purines or 5,6-dihydrouracil. The true quandary has always been cytosine, as it is the one native base that is highly susceptible to deamination. Comparing its half-life value to those of other pyrimidines (Table 1) suggests that there are not any decent alternatives with dramatically enhanced stability outside of the modestly stabilizing N4-methylation.
In spite of its hydrolytic instability,[23c] the production of cytosine might have resulted from formation pathways that balanced out the decomposition one (Figure 7). Degradation of other heterocycles could serve as plausible enrichment pathways. The deamination of 2,4-diaminopyrimidine (Dpy, Figure 6 and Table 1), which could lead to a slower continuous release of cytosine (and isocytosine) into the prebiotic environment, is one such example. This is supported by an earlier study, where Miller reported that the degradation rate of 2,4-diaminopyrimidine under these conditions is largely attributed to the C4 deamination reaction that generates cytosine.[20a]
Photochemical Considerations
The action of UV irradiation on prebiotic mixtures has been historically considered a driver of prebiotic chemistry, but also as a selection pressure for the most photostable molecules.[14c,25] The nucleobases are strong ultraviolet-absorbing chromophores with a combined absorbance range of 230–280 nm, and a molar absorptivity range in water at λmax of ~ 8,000–15,000 M−1cm−1.[26] As such, the native bases are not inert to photochemical damage. Indeed, one of the most recognized and significant photo-chemical reactions that can occur is the photodimerization of uracil or thymine residues in RNA and DNA.[27] Yet, as free nucleobases, they exhibit outstanding photo-stability in comparison to other heterocycles.[26]
The last 15 years have seen detailed investigations of the photophysical properties of the native bases.[26] It has been established that, upon radiative excitation (via a π-π* transition), the native nucleobases exhibit most effective non-radiative decay pathways back to the ground state, resulting in exceptionally short excited-state lifetimes.[26] It is this attribute that explains their extremely low fluorescence and photochemical quantum yields.[26] The ultrafast excited-state decay rates shared by the native bases have prompted investigators to hypothesize that photostability was a viable selection pressure in the prebiotic environment, enriching the native bases, the clear winners.[28] Assessing the plausibility of this selection pressure has only recently become possible given the published investigations on excited-state lifetimes of alternative and modified nucleobases. Table 2 lists these contributions. Most were conducted in aqueous solution, although a few references to gas-phase experiments are included. The lifetime values of τ=0.1–2.8 ps reported for the native bases under neutral pH are indeed extremely short for simple organic chromophores, but it is apparent that they are not unique among such heterocycles. Xanthine derivatives also appear to display very short lifetimes, but it is hypoxanthine that has recently been shown to exhibit the fastest excited-state decay rates.[29]
Table 2.
Comparison of excited-state lifetime values of native and alternative nucleobases in aqueous solution.[a]
| Nucleobase | Excited-state lifetimes (τ, ps) | Reference |
|---|---|---|
| Uncharged bases (neutral pH 6.8–7.3) | ||
| Hypoxathine | 0.13 ± 0.3 | [29] |
| Adenine | 0.18 ± 0.3 | [34] |
| Guanine(as Guanosine) | 0.16[b] | [35] |
| Xanthine | 0.28–0.50[c] | [29] |
| Cytosine | 1.0 ± 0.2, 2.9, 12 | [36] |
| Uracil | 1.9 ± 0.1, 24 ± 0.2 | [36b] |
| Thymine | 2.8 ± 0.1, 30 ± 13 | [36b] |
| 5-Methylcytosine | 7.2 ± 0.4 | [36a] |
| N4-Acetylcytosine | 280 ± 30 | [36a] |
| 2,4-Diaminopyrimidine | 10–1000[a] (gas) | [37] |
| 5-Hydroxyuracil | 1800[a] | [38] |
| 2,6-Diaminopurine | 6300 ± 400[a] (gas) | [37] |
| 2-Aminopurine | 11800 | [39] |
| Cation charged bases (pH 0–2) via protonatation | ||
| Guanine | 191 ± 4[b] | [40] |
| Hypoxanthine | <0.2 | [35] |
| 5-Methylcytosine | 2.57 ± 0.22 | [36a] |
| Anion charged bases (pH 13) | ||
| Hypoxanthine (pH 10) | 19 | [35] |
| Cytosine | 13.3 ± 0.4 | [36a] |
| 5-Methylcytosine | 250 ± 30 | [36a] |
Gas-phase experimental studies.
Due to solubility problems with guanine, the community has relied on the value obtained from deoxyguanosine and guanosine to model/approximate nucleobase values.
Expected value range based on the derivatives used in the study.
More intriguing is how relatively simple structural changes create drastic differences in the excited-state properties of the heterocycles. 2-Aminopurine (2AP), a highly fluorescent adenine isomer (ϕ=0.68) with an excited-state lifetime of τ=11.8 ns (compared to 0.18 ps for adenine), displays one of the most dramatic differences ever reported.[30] 2,6-Diaminopurine, another adenine analog, is also known for its enhanced fluorescence properties (ϕ~0.01) compared to adenosine.[30b] It was recently reported to also display a relatively long excited-state lifetime (τ=6.3 ns) in gas-phase experiments. Much smaller, yet meaningful, differences are the photophysical changes observed upon methylation at the C5 position or modification to amino substituents in the native pyrimidines. Whereas uracil and thymine are close in their excited-state lifetime values (τ=1.9 and 2.8 ps for U and T, respectively), the analogous cytosine and 5-methylcytosine (5mC) display significantly larger differences (τ=1.0 and 7.2 ps, respectively).
The neutral native bases exhibit fast decay times, but a change in pH can substantially impact these values as a result of protonation or deprotonation events. Such alterations are especially germane to a prebiotic environment and selection pressures, because early oceans may have displayed significant fluctuations in their pH values.[31] Guanine (in this case as guanosine) under acidic conditions (pH 1.5) has a dramatic lifetime change from τ=0.16 to 191 ps upon protonation. Interestingly, the alternative base hypoxanthine was reported to not exhibit any significant lifetime alterations, and 5mC actually exhibited a decrease from τ=7.2 to 2.57 ps. Alkaline pH can also have an effect, with major differences for cytosine (from τ=1.0 to 13.3 ps) but a rather dramatic disparity for 5 mC (from τ=7.2 to 250 ps).
Many of the non-native bases listed in Table 2 with longer lifetime values are found in RNA as base modifications (e.g., 5 mC, N4-acetylcytosine, Hyp). 2,4-Diamino-pyrimidine is the core heterocycle formed in lysidine (Figure 2A). The occurrence of these “photophysically unfit” bases in nature may seem to contradict the selection pressure proposed here, as photostability may have been an advantage in the prebiotic environment for free nucleo-bases. However, the inclusion of alternative bases with heightened photochemical activity may actually have offered an advantage for the first genetic polymers, or more importantly, the first biotic systems. Recent work by Burrows and coworkers has contributed a fresh angle to the importance of these photophysically interactive bases in the context of early RNA genomes.[32] 8-Oxoguanosine (8oxoG, Figure 6), a base widely known as a oxidatively damaged lesion in DNA, has been shown to serve as a photo-induced repair catalyst, capable of reversing the photodimerization products of thymine and uracil residues within oligomeric RNA and DNA.[33] This activity has been attributed to the enhanced photophysical and redox ability of 8oxoG compared to G.[32] The excited-state lifetime of 8oxoG has not yet been reported, but that of 5-hydroxyuracil is known (τ=1.8 ns). This is also a base proposed to exhibit similar repair properties given its redox activity.[32] Conversely, Burrows’ attempts to employ xanthine as a photorepair nucleobase were unsuccessful despite its lower redox potential compared to G. These observations are consistent with the ultrashort excited-state lifetime data obtained for xanthine derivatives (Table 2), supporting their inability to display favorable photorepair activity.
3. Selection during the Formation of the First Informational Polymers
One of the more enigmatic and difficult problems confronting the prebiotic chemistry community is identifying how the monomers of RNA, or pre-RNA, or even non-related polymeric components selectively formed and self-assembled out of the presumed random prebiotic mixtures.[41] It is in this assembly into informational polymers (Figure 4) where significant selection processes must have occurred not only for the base composition but also for the other components of nucleic acids (or nucleic acid alternatives and precursors).[31,42] Focusing on just a narrow view of RNA precursors, the linking of a nucleo-base to a ribose sugar is one such pressure. There are multiple ways in which a nucleobase can be attached to ribose via an N-glycosidic bond, but only one is found in contemporary nucleic acids (via the N9 of purines and N1 of pyrimidines).
Achieving regio- and stereochemical selectivity of glycosylation reactions under simulated prebiotic conditions has plagued the community ever since Orgel and others began working on this problem (Figure 8A).[14b,c,41a] The challenge with prebiotic glycosylation has consequently led Sutherland and coworkers to circumvent this reaction by attempting to build the nucleobase heterocycles and sugars together,[43] intriguingly demonstrating preferential formation of the native ribonucleotides.[44] These routes have been criticized for their “external” or directed nature of synthetic utility,[45] in addition to abandoning the presumed abundance of nucleobases that populated the early Earth environment. Additionally, the selective formation of native ribonucleotides does not preclude the base itself from further modification. Nevertheless, Sutherland’s contributions are highly creative and do employ simple prebiotic precursors.
Figure 8.

Generation of nucleosides under prebiotic conditions using the nucleobases and D-ribose. A) Orgel and coworkers demonstrated that of all the native bases, only adenine formed the relevant glycosidic bond to produce adenosine, but still with only low yields. B) Using a concentrated solution of magnesium sulfate and magnesium chloride (mimicking seawater) and an excess of ribose, hypoxanthine has been observed to regio- and stereoselectively produce the corresponding nucleoside. C) Under similar conditions, the Hud laboratory was the first to report a successful prebiotic glycosylation reaction using an alternative pyrimidine heterocycle.
While the contemporary native bases themselves are less prone to forming the “correct” glycosidic bonds, success came from the use of modified or alternative purines and pyrimidines. Hypoxanthine was one of the first non-native bases used in the 1970s by Orgel (Figure 8B), to exhibit some of the highest yields of ribonucleoside formation with the “correct” anomeric stereochemistry.[14b] Previous work from the Miller laboratory successfully illustrated the use of non-pyrimidine or non-purine base heterocycles as pre-RNA world surrogates.[20c] More recently, Hud and coworkers demonstrated that 2-pyrimidinone, a non-native pyrimidine, could undergo effective glycosylation reactions with D-ribose (Figure 8C) to give the zebularine ribonucleoside in high yield.[46] The higher regio- and stereoselective formation of glycosidic bonds between non-native bases and ribose might have played a role in the assembly of the first informational polymers.[41a] In such a scenario, eventual chemical modifications to generate the native bases during the course of evolution could have then given rise to the extant alphabet.
Related to the prebiotic formation of ribonucleosides is the topic of hydrolytic stability of the N-glycosyl bonds. The N-glycosyl stabilities of the native ribonucleosides have been well studied but little has been done, outside of the recent work from the Hud laboratory, to specifically compare how prebiotically relevant nucleosides measure up to the native ones.[46,47] Investigations in this area could provide much insight into identifying other selection pressures in this particular period of chemical evolution. We will return to the role of hydrolytic stability of N-glycosyl bonds in Section 5 and consider what is known about relevant nucleosides in that context.
4. Base Selection in an Early RNA World?
The RNA world is an assumed period near or at the origin of life where RNA was the genomic and catalytic center of early cells.[48] The native nucleobases, while exceptionally good in their genetic capabilities, do not appear to be catalytically useful in comparison to the functionality-rich side chains of amino acids. It is, however, the presence of base modifications in extant RNA that has encouraged many to ponder that the RNA world may have utilized an expanded set of heterocycles.[49] Many of the modifications found in extant RNA do have striking similarities to the functional groups of the amino acids (see Figure 2A and 2B).[18,50] While the utility of modified bases in the RNA world may seem to compound the problem of narrowing the alphabetic composition, there are still plausible pressures that could have occurred in this epoch. This is especially true as RNA began to partition its role more towards the genomic rather than the catalytic, or relinquish its catalytic function via the recruitment of primitive proteins. One such pressure is based on the reliability of genetic information storage and transfer, and another might have been the contributions to the stability of duplex polymers. Both are inherently related to individual bases.
Genetic Fidelity Considerations
Genetic fidelity is likely to be among the most important functions of a nucleobase when part of a living (however primitive) system. Spontaneous hydrolytic deamination reactions that challenge the integrity of base-pairing faces, as previously discussed, can certainly be considered a viable pressure in the RNA world. However, even with the integrity of the heterocycles intact, not all bases are equal in maintaining faithful coding properties.
A well-known example comes from evaluating isoguanine (isoG). Isoguanine, when incorporated into nucleic acids, can base pair with its natural complement isoC (Figure 9A), but also with uracil (Figure 9B). This is due to the propensity of isoG to tautomerize to its enol form, which enables the alternative pairing.[51] Furthermore, it was observed that in the presence of polymerases isoG can also direct the incorporation of U and vice versa.[52] This problem, along with their deamination susceptibilities (Table 1), has been previously hypothesized as one reason for the unfitness of the isoG :isoC pair to permanently become a part of a long-lived genetic alphabet.[53]
Figure 9.

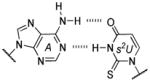
Genetic fidelity pressures. A) A WC-like isoG : isoC base pair that utilizes the dominant tautomer. B) A minor tautomeric form of isoG paired with U. C) Guanine can also base pair with uracil, generating a wobble pair. D) The 2-thiouracil base is highly specific for adenine since it cannot wobble pair with guanine.
The extant native alphabet is, however, not entirely free of promiscuous pairing. Guanine also recognizes uracil through the generation of a wobble base pair (Figure 9C). The G :U wobble pair is quite ubiquitous in extant biology and is considered to be one of the most important non-canonical pairing interactions used by RNA for its structural and functional diversity.[54] Because of the inherent stability and pervasive nature of this interaction, it might be pondered how life in the RNA world maintained genetic fidelity without the assistance of evolved polymerases to ensure proper replication. One possibility comes from clues in extant biochemistry, where life ingeniously found ways around this “little problem” by utilizing modified bases such as 2-thiouracil (s2U) and its derivatives. It has been reported that these thiolated modifications occur most often in the wobble position of tRNA, where proper pairing of A :U is needed for translation.[1,55] The substitution of oxygen for sulfur in the C2 position of uracil creates a highly specific base pair with adenine because it destabilizes the putative wobble G :s2U pair (Figure 9D).[56] The occurrence of 2-thiouracil and derivatives in extant biology suggests that alternative bases could have been advantageous for replication fidelity in early RNAs.
Duplex Stability Considerations
From observations of DNA damage in extant biology, it would seem that a strong selection pressure for cells to maintain double-stranded nucleic acids would have occurred early in biotic evolution. More than providing two copies of genetic information, duplex systems are highly advantageous in protecting genetic material from chemical or physical assaults. Just about every type of damage that can occur to DNA components (deamination, deglycosylation, oxidation, alkylation, and even some photo-chemical reactions) is known to be diminished when double stranded.[22,57] Many factors contribute to duplex stability, such as an anionic sugar-phosphate backbone, hydration, and metal ions,[13,58] but two important contributions directly related to nucleobases are their stacking propensity and the base-pairing strength.[11] Of these two contributions, the base-pairing strength seems to be inherently related to the specific nature of the heterocycle.
Assuming the same sugar-phosphate backbone, do the native bases contribute to the highest stabilities of RNA and DNA duplexes? Building upon his work on non-natural bases, Benner undertook a systematic study of measuring how alternative bases and their pairing relationships affected the thermal denaturation of short DNA duplexes.[59] The differences in melting temperatures (Tm), directly correlated to the relative stability of the base pairs, are summarized in Table 3 for two orientations.[59,60] In this short list of closely related nucleobases and base pairs, the native bases do frequently form some of the strongest base pairs, albeit not by much. This observation has guided Benner to the conclusion that the bases of nucleic acids are quite interchangeable and can be manipulated to a high degree.[58]
Table 3.
Base pair stability ranking from relative thermal denaturation at pH 7.9.[a]
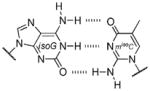
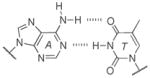
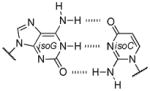
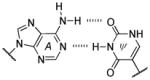
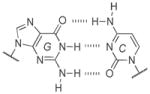
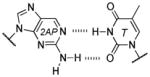
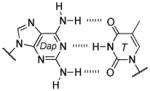
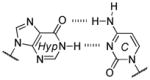
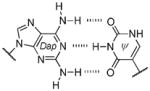
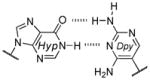
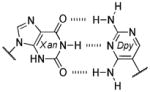
Among the interesting examples are the bases or base pairs that are more stable than the native ones. Most striking is the A :s2U interaction, one that occurs in nature, because even with just two hydrogen bonds it approaches the strength of a three H-bonded G :C pair. The reason for the enhanced stabilization has been studied and identified to originate from the better base-pair stacking of the s2U with the adjacent base-pair groups compared to U.[61] The isoG :5-methylisoC pair is also intriguing since it is probably one of the strongest reported for bases that appear to be prebiotically plausible. Other studies measuring the original isoG :isoC pair also find that it is either at or above the base-pair strength of the G :C interaction.[51,62] The data reveal considerably lower Tm values for the Hyp :Dpy and Xan :Dpy base pairs and thus suggest the presence of destabilizing effects, which may indicate that not all of the bases could have been viable contenders.
Some of the observations discussed above are supportive of a recent hypothesis presented by Krishnamurthy, where the pKa values of the bases are correlated with base-pairing strength and overall duplex stability.[63] Optimum base-pair strength is observed when the difference between the pKa values of the hydrogen-bonding faces, ΔpKa, is ≥ 5 units, suggesting highly polarized intermolecular interactions. Duplex stability is greater when the pKa values of individual bases are >2 units away from the pH of the aqueous medium, denoted as the pKa–pH correlation. This aligns with the assumption that bases with pKa values closer to the pH of the aqueous medium (<2 units) are likely to be ionized to a great extent, thus disrupting stacking within the duplex. Table 4 lists the values using this empirical correlation along with some of the experimental data from Table 3. While not completely congruent, a general trend may be found.[63] In particular, the Xan :Dpy and Hyp :Dpy base pairs in Table 3 displayed the lowest stability, in agreement with the calculated ΔpKa and pKa–pH values. Was base-pairing strength then a viable selection pressure? Base pairs such as the Xan :Dpy example may have posed a liability to duplex stability,[63] but in comparison to some of the other base pairs listed in Table 3, the native bases do not appear to exhibit any clear advantage.
Table 4.
| Base pair | Melting temp. (°C) | ΔpKa | pKa 3pH for each base pH was conducted at 7.9 |
|---|---|---|---|
| 3 H-bonds: | |||
| isoG :isoC | 61.5, 63.3 | ~4.8 | 1 (isoG), 3.7 (isoC) |
| G :C | 58.5, 59.5 | ~5.1 | 1.8 (G), 3.5 (C) |
| Dap : T | 56.9, 58.4 | ~4.6 | 2.4 (Dap), 1.9 (T) |
| Dap : Psi | 56.7 | ~4.5 | 2.4 (Dap), 1.8 (Psi) |
| Xan :Dpy[b] | 47.5 | ~2 | ~1 (Xan), ~1.6 (Dpy) |
| 2 H-bonds: | |||
| A :T | 52.9, 55.9 | ~6.1 | 4.2 (A), 1.8 (T) |
| A :Psi | 54.4 | ~6 | 4.2 (A), 1.8 (Psi) |
| Hyp : C | 52.8, 54.6 | ~4.5 | ~1 (Hyp), 3.5 (C) |
| Hyp : Dpy | 47.5 | ~1.8 | 1 (Hyp), 1.6 (Dpy) |
Calculations were made using model presented in reference [63].
The pH in this experiment was measured at 5.4.
5. Base Refinement in an Early DNA World?
The transition from RNA to DNA as the genetic repository in early cells and its potential mechanisms have been of interest.[64] Surprisingly, however, little attention has been given to the possibility of nucleobase refinement during or after this transition. As previously discussed regarding uracil-based DNA, the possibility exists that life in the early DNA world may still have been “settling” on the specific composition of the genetic alphabet.[4] Could there have been other selection pressures during this period? A recent hypothesis has detailed the possibility of one such selection pressure that implicates the role of N-glycosyl bonds in the early DNA world.[65] While the polymeric stability of DNA is far superior to that of RNA,[66] it is well established that DNA suffers from weaker N-glycosyl bonds.[22,67] The hydrolysis of these linkages (i.e., depurination/depyrimidination) leaves an abasic site in the sugar-phosphate backbone of DNA that, unless repaired, is well known to not only be mutagenic, but also to lead to the cleavage of genetic material (Figure 10).[68] It was proposed that, given the vital role N-glycosyl bonds have on the overall stability of the genetic material, a strong selection pressure might have played out in the early DNA world that facilitated the selection of bases exhibiting the most hydrolytically resistant glycosidic bonds.[65]
Figure 10.
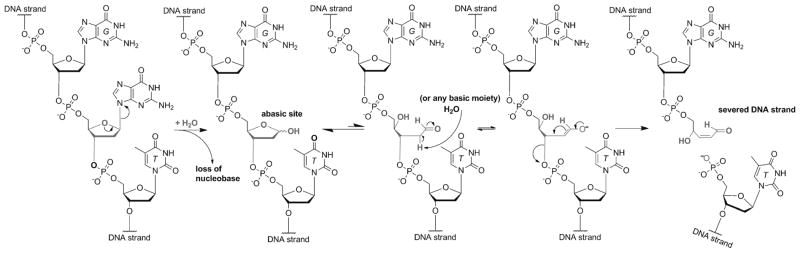
A general mechanism for DNA strand cleavage that originates from a spontaneous deglycosylation reaction.
A central observation to this proposal lies with the known variability of N-glycosyl stabilities that exists among the DNA nucleosides and related heterocycles. In the native set, the purines are more susceptible to deglycosylation reactions than the pyrimidines. Modifications or damage made to the DNA bases, such as alkylation, deamination, and oxidation, are known to destabiliize the glycosidic bonds even further.[22,69] With respect to prebiotically significant bases, it is known that the glycosidic bonds in d-isoG and d-isoC are significantly weaker than the native counterparts.[70] The pyrimidine nucleosides zebularine and deoxyzebularine have also been reported to exhibit extremely labile glycosidic bonds.[46,47,71] While available literature data appears to support the notion that native DNA nucleosides exhibit some of the strongest N-glycosyl bonds, further investigations with bases of prebiotic interest compared to the native ones are needed. It was also mentioned that while DNA might be vulnerable to alterations made to the native bases, cells intentionally utilize some of the very same heterocycles as post-transcriptional modifications in RNA (Figure 11). Congruent to the premise that the RNA world contained an expanded alphabet, the higher N-glycosyl stability associated with RNA may have been an advantageous feature in the RNA world that utilized functionalized nucleobases. This scenario might also offer a reason why the selection pressure did not surface until the invention of DNA.
Figure 11.

Damaged DNA bases often contain weaker glycosidic bonds (shown in red) in comparison to the native bases. Interestingly, many of these bases arise in cells as post-transcriptional RNA modifications. The greater N-glycosyl stability asssociated with ribonucleosides may have endowed the RNA world with the flexibility to utilize a wider variety of heterocycles.
6. Summary and Outlook
A Continuous Process of Refinement
From the topics discussed in this less-than-exhaustive review, it should be apparent that diverse selection pressures could have shaped the composition of the nucleic acid bases. However, it is also likely that no single pressure (at least from the topics introduced) could have designated the native bases as clear winners. We share the idea that many bases became part of the first genetic polymers, and contributed to the viability of the RNA world.[42b] As early life evolved, so did the nature of the bases as new environmental and functional pressures emerged.[65] Alternative viewpoints have been put forward suggesting that selection of the native bases coincided with the emergence of RNA from the prebiotic environment.[10c,44] This scenario appears to offer a simpler explanation for the origin of the nucleic acid bases. However, in addition to the challenges mentioned in this review, the direct selection of the native bases from a prebiotic environment seems a little too elegant for evolutionary processes that were likely much more messy, complex and prolonged.[72]
Acknowledgments
We are grateful to the NIH, via grant number GM 069773, for support.
Biographies
 Andro Rios received his B. S. in chemistry from California State University, Sacramento, in 2006 and recently his Ph.D. from the University of California, San Diego in 2012. As a doctoral student in the laboratory of Professor Yitzhak Tor, Andro became interested in using the tools of physical organic chemistry for application to questions associated with chemical evolution and astrobiology. He is currently pursuing a postdoctoral appointment using a similar combination of research tools and themes.
Andro Rios received his B. S. in chemistry from California State University, Sacramento, in 2006 and recently his Ph.D. from the University of California, San Diego in 2012. As a doctoral student in the laboratory of Professor Yitzhak Tor, Andro became interested in using the tools of physical organic chemistry for application to questions associated with chemical evolution and astrobiology. He is currently pursuing a postdoctoral appointment using a similar combination of research tools and themes.
 Yitzhak Tor carried out his doctorate work at the Weizmann Institute of Science, earning his Ph.D. in 1990. After a postdoctoral stay at the California Institute of Technology (1990–1993), he took his first faculty position at the University of Chicago. In 1994, he moved to the University of California, San Diego, where he is currently a Professor of Chemistry and Biochemistry. He was the Traylor Scholar in Organic Chemistry between 2006 and 2011. His research interests are diverse and include the chemistry and biology of nucleosides, nucleotides and nucleic acids, and the discovery of novel antiviral and antibacterial agents, as well as the development of cellular delivery agents and biomolecular fluorescent probes. He is currently the Editor in Chief of Perspectives in Medicinal Chemistry and Organic Chemistry Insights.
Yitzhak Tor carried out his doctorate work at the Weizmann Institute of Science, earning his Ph.D. in 1990. After a postdoctoral stay at the California Institute of Technology (1990–1993), he took his first faculty position at the University of Chicago. In 1994, he moved to the University of California, San Diego, where he is currently a Professor of Chemistry and Biochemistry. He was the Traylor Scholar in Organic Chemistry between 2006 and 2011. His research interests are diverse and include the chemistry and biology of nucleosides, nucleotides and nucleic acids, and the discovery of novel antiviral and antibacterial agents, as well as the development of cellular delivery agents and biomolecular fluorescent probes. He is currently the Editor in Chief of Perspectives in Medicinal Chemistry and Organic Chemistry Insights.
References
- 1.Carell T, Brandmayr C, Hienzsch A, Müller M, Pearson D, Reiter V, Thoma I, Thumbs P, Wagner M. Angew Chem, Int Ed. 2012;51:7110. doi: 10.1002/anie.201201193. [DOI] [PubMed] [Google Scholar]
- 2.a) Martínez Giménez JA, Sáez GT, Seisdedos RT. J Theor Biol. 1998;194:485. doi: 10.1006/jtbi.1998.0770. [DOI] [PubMed] [Google Scholar]; b) Forterre P, Grosjean H. In: DNA and RNA Modification Enzymes: Structure, Mechanism, Function and Evolution. Grosjean H, editor. Landes Bioscience; Austin, Texas: 2009. p. 259. [Google Scholar]; c) Cermakian N, Cedergren R. In: Modification and Editing of RNA. Grosjean H, Benne R, editors. ASM Press; Washington, D. C: 1998. p. 535. [Google Scholar]
- 3.Berg JM, Tymoczko JL, Stryer L. Biochemistry. 6. W. H. Freeman; New York: 2007. [Google Scholar]
- 4.Poole A, Penny D, Sjöberg BM. Nat Rev Mol Cell Biol. 2001;2:147. doi: 10.1038/35052091. [DOI] [PubMed] [Google Scholar]
- 5.Warren RAJ. Annu Rev Microbiol. 1980;34:137. doi: 10.1146/annurev.mi.34.100180.001033. [DOI] [PubMed] [Google Scholar]
- 6.a) Rae PMM, Steele RE. Biosystems. 1978;10:37. [Google Scholar]; b) Gommers-Ampt J, Borst P. FASEB J. 1995;9:1034. doi: 10.1096/fasebj.9.11.7649402. [DOI] [PubMed] [Google Scholar]
- 7.a) Piccirilli JA, Benner SA, Krauch T, Moroney SE. Nature. 1990;343:33. doi: 10.1038/343033a0. [DOI] [PubMed] [Google Scholar]; b) Switzer C, Moroney SE, Benner SA. J Am Chem Soc. 1989;111:8322. [Google Scholar]
- 8.Rich A. In: Horizons in Biochemistry. Kasha M, Pullman B, editors. Academic Press; New York: 1962. p. 103. [Google Scholar]
- 9.Eschenmoser A. Science. 1999;284:2118. doi: 10.1126/science.284.5423.2118. [DOI] [PubMed] [Google Scholar]
- 10.a) Khakshoor O, Kool ET. Chem Commun. 2011;47:7018. doi: 10.1039/c1cc11021g. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chiba J, Inouye M. Chem Biodiversity. 2010;7:259. doi: 10.1002/cbdv.200900282. [DOI] [PubMed] [Google Scholar]; c) Hirao I, Kimoto M, Yamashige R. Acc Chem Res. 2012;45:2055. doi: 10.1021/ar200257x. [DOI] [PubMed] [Google Scholar]
- 11.Saenger W. Principles of Nucleic Acid Structure. Springer-Verlag; New York: 1984. [Google Scholar]
- 12.Nabel CS, Manning SA, Kohli RM. ACS Chem Biol. 2011;7:20. doi: 10.1021/cb2002895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benner SA, Sismour AM. Nat Rev Genet. 2005;6:533. doi: 10.1038/nrg1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Borquez E, Cleaves HJ, Lazcano A, Miller SL. Origins Life Evol Biospheres. 2005;35:79. doi: 10.1007/s11084-005-5945-9. [DOI] [PubMed] [Google Scholar]; b) Orgel LE. Crit Rev Biochem Mol Biol. 2004;39:99. doi: 10.1080/10409230490460765. [DOI] [PubMed] [Google Scholar]; c) Miller SL, Orgel LE. The Origins of Life on the Earth. Prentice-Hall; Englewood Cliffs, N. J: 1974. [Google Scholar]; d) Cleaves HJ, II, Nelson KE, Miller SL. Naturwissenschaften. 2006;93:228. doi: 10.1007/s00114-005-0073-y. [DOI] [PubMed] [Google Scholar]; e) Barks HL, Buckley R, Grieves GA, Di Mauro E, Hud NV, Orlando TM. ChemBioChem. 2010;11:1240. doi: 10.1002/cbic.201000074. [DOI] [PubMed] [Google Scholar]; f) Saladino R, Botta G, Pino S, Costanzo G, Di Mauro E. Chem Soc Rev. 2012;41:5526. doi: 10.1039/c2cs35066a. [DOI] [PubMed] [Google Scholar]
- 15.a) Botta O, Bada JL. Surv Geophys. 2002;23:411. [Google Scholar]; b) Callahan MP, Smith KE, Cleaves HJ, Ruzicka J, Stern JC, Glavin DP, House CH, Dworkin JP. Proc Natl Acad Sci USA. 2011;108:13995. doi: 10.1073/pnas.1106493108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siegel JS, Tor Y. Org Biomol Chem. 2005;3:1591. doi: 10.1039/b500921a. [DOI] [PubMed] [Google Scholar]
- 17.Walling C. Acc Chem Res. 1975;8:125. [Google Scholar]
- 18.Robertson M, Miller S. Science. 1995;268:702. doi: 10.1126/science.7732378. [DOI] [PubMed] [Google Scholar]
- 19.a) Choughuley ASU, Subbaraman AS, Kazi ZA, Chadha MS. Biosystems. 1977;9:73. doi: 10.1016/0303-2647(77)90014-4. [DOI] [PubMed] [Google Scholar]; b) Saladino R, Ciambecchini U, Crestini C, Costanzo G, Negri R, Di Mauro E. ChemBioChem. 2003;4:514. doi: 10.1002/cbic.200300567. [DOI] [PubMed] [Google Scholar]
- 20.a) Robertson MP, Levy M, Miller SL. J Mol Evol. 1996;43:543. doi: 10.1007/BF02202102. [DOI] [PubMed] [Google Scholar]; b) Levy M, Miller SL. J Mol Evol. 1999;48:631. doi: 10.1007/pl00006506. [DOI] [PubMed] [Google Scholar]; c) Kolb VM, Dworkin JP, Miller SL. J Mol Evol. 1994;38:549. doi: 10.1007/BF00175873. [DOI] [PubMed] [Google Scholar]; d) Menor-Salvan C, Marin-Yaseli MR. Chem Soc Rev. 2012;41:5404. doi: 10.1039/c2cs35060b. [DOI] [PubMed] [Google Scholar]
- 21.a) Sleep NH. Cold Spring Harbor Perspect Biol. 2010;2:a002527. doi: 10.1101/cshperspect.a002527. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zahnle K, Schaefer L, Fegley B. Cold Spring Harbor Perspect Biol. 2010;2:a004895. doi: 10.1101/cshperspect.a004895. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Nisbet EG, Sleep NH. Nature. 2001;409:1083. doi: 10.1038/35059210. [DOI] [PubMed] [Google Scholar]
- 22.Gates KS. Chem Res Toxicol. 2009;22:1747. doi: 10.1021/tx900242k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a) Garrett ER, Tsau J. J Pharm Sci. 1972;61:1052. doi: 10.1002/jps.2600610703. [DOI] [PubMed] [Google Scholar]; b) Shapiro R. Origins Life Evol Biospheres. 1995;25:83. doi: 10.1007/BF01581575. [DOI] [PubMed] [Google Scholar]; c) Shapiro R. Proc Natl Acad Sci USA. 1999;96:4396. doi: 10.1073/pnas.96.8.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.a) House CH, Miller SL. Biochemistry. 1996;35:315. doi: 10.1021/bi951577+. [DOI] [PubMed] [Google Scholar]; b) Levy M, Miller SL. Proc Natl Acad Sci USA. 1998;95:7933. doi: 10.1073/pnas.95.14.7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sagan C. J Theor Biol. 1973;39:195. doi: 10.1016/0022-5193(73)90216-6. [DOI] [PubMed] [Google Scholar]
- 26.Middleton CT, de La Harpe K, Su C, Law YK, Crespo-Hernández CE, Kohler B. Annu Rev Phys Chem. 2009;60:217. doi: 10.1146/annurev.physchem.59.032607.093719. [DOI] [PubMed] [Google Scholar]
- 27.Friedberg EC. Nature. 2003;421:436. doi: 10.1038/nature01408. [DOI] [PubMed] [Google Scholar]
- 28.Gustavsson T, Improta R, Markovitsi D. J Phys Chem Lett. 2010;1:2025. [Google Scholar]
- 29.Chen JQ, Kohler B. Phys Chem Chem Phys. 2012;14:10677. doi: 10.1039/c2cp41296a. [DOI] [PubMed] [Google Scholar]
- 30.a) Ward DC, Reich E, Stryer L. J Biol Chem. 1969;244:1228. [PubMed] [Google Scholar]; b) Sinkeldam RW, Greco NJ, Tor Y. Chem Rev. 2010;110:2579. doi: 10.1021/cr900301e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kua J, Bada J. Origins Life Evol Biospheres. 2011;41:553. doi: 10.1007/s11084-011-9250-5. [DOI] [PubMed] [Google Scholar]
- 32.Nguyen KV, Burrows CJ. Acc Chem Res. 2012;45:2151. doi: 10.1021/ar300222j. [DOI] [PubMed] [Google Scholar]
- 33.Nguyen KV, Burrows CJ. J Am Chem Soc. 2011;133:14586. doi: 10.1021/ja2072252. [DOI] [PubMed] [Google Scholar]
- 34.Cohen B, Hare PM, Kohler B. J Am Chem Soc. 2003;125:13594. doi: 10.1021/ja035628z. [DOI] [PubMed] [Google Scholar]
- 35.Villabona-Monsalve JP, Noria R, Matsika S, Peon J. J Am Chem Soc. 2012;134:7820. doi: 10.1021/ja300546x. [DOI] [PubMed] [Google Scholar]
- 36.a) Malone RJ, Miller AM, Kohler B. Photochem Photobiol. 2003;77:158. doi: 10.1562/0031-8655(2003)077<0158:sesloc>2.0.co;2. [DOI] [PubMed] [Google Scholar]; b) Hare PM, Crespo-Hernández CE, Kohler B. Proc Natl Acad Sci USA. 2007;104:435. doi: 10.1073/pnas.0608055104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gengeliczki Z, Callahan MP, Svadlenak N, Pongor CI, Sztaray B, Meerts L, Nachtigallova D, Hobza P, Barbatti M, Lischka H, de Vries MS. Phys Chem Chem Phys. 2010;12:5375. doi: 10.1039/b917852j. [DOI] [PubMed] [Google Scholar]
- 38.Nachtigallova D, Lischka H, Szymczak JJ, Barbatti M, Hobza P, Gengeliczki Z, Pino G, Callahan MP, de Vries MS. Phys Chem Chem Phys. 2010;12:4924. doi: 10.1039/b925803p. [DOI] [PubMed] [Google Scholar]
- 39.Neely RK, Magennis SW, Dryden DTF, Jones AC. J Phys Chem B. 2004;108:17606. [Google Scholar]
- 40.Pecourt JML, Peon J, Kohler B. J Am Chem Soc. 2001;123:10370. doi: 10.1021/ja0161453. [DOI] [PubMed] [Google Scholar]
- 41.a) Bean HD, Lynn DG, Hud NV. In: Chemical Evolution II: From the Origins of Life to Modern Society. Zaikowski L, Friedrich JM, Seidel SR, editors. American Chemical Society; Washington, D. C: 2009. p. 109. [Google Scholar]; b) Benner SA, Kim HJ, Yang Z. Cold Spring Harbor Perspect Biol. 2012;4:a003541. doi: 10.1101/cshperspect.a003541. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Robertson MP, Joyce GF. Cold Spring Harbor Perspect Biol. 2012;4:a003608. doi: 10.1101/cshperspect.a003608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.a) Cleaves HJ, II, Bada J. In: Genesis – In The Beginning. Seckbach J, editor. Vol. 22. Springer; Netherlands: 2012. p. 3. [Google Scholar]; b) Engelhart AE, Hud NV. Cold Spring Harbor Perspect Biol. 2010;2:a002196. doi: 10.1101/cshperspect.a002196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.a) Powner MW, Gerland B, Sutherland JD. Nature. 2009;459:239. doi: 10.1038/nature08013. [DOI] [PubMed] [Google Scholar]; b) Powner MW, Sutherland JD, Szostak JW. J Am Chem Soc. 2010;132:16677. doi: 10.1021/ja108197s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Powner MW, Sutherland JD, Szostak JW. Synlett. 2011:1956. [Google Scholar]
- 45.a) Benner SA, Kim HJ, Carrigan MA. Acc Chem Res. 2012;45:2025. doi: 10.1021/ar200332w. [DOI] [PubMed] [Google Scholar]; b) Eschenmoser A. Angew Chem, Int Ed. 2011;50:12412. doi: 10.1002/anie.201103672. [DOI] [PubMed] [Google Scholar]
- 46.Bean HD, Sheng Y, Collins JP, Anet FAL, Leszczynski J, Hud NV. J Am Chem Soc. 2007;129:9556. doi: 10.1021/ja072781a. [DOI] [PubMed] [Google Scholar]
- 47.Sheng Y, Bean HD, Mamajanov I, Hud NV, Leszczynski J. J Am Chem Soc. 2009;131:16088. doi: 10.1021/ja900807m. [DOI] [PubMed] [Google Scholar]
- 48.Joyce GF. Nature. 1989;338:217. doi: 10.1038/338217a0. [DOI] [PubMed] [Google Scholar]
- 49.Benner SA, Burgstaller P, Battersby TR, Jurczyk S. In: The RNA World. Gesteland RF, Cech TR, Atkins JF, editors. Vol. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1999. p. 163. [Google Scholar]
- 50.Rozenski J, Crain PF, McCloskey JA. Nucleic Acids Res. 1999;27:196. doi: 10.1093/nar/27.1.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roberts C, Bandaru R, Switzer C. J Am Chem Soc. 1997;119:4640. [Google Scholar]
- 52.Tor Y, Dervan PB. J Am Chem Soc. 1993;115:4461. [Google Scholar]
- 53.Switzer CY, Moroney SE, Benner SA. Biochemistry. 1993;32:10489. doi: 10.1021/bi00090a027. [DOI] [PubMed] [Google Scholar]
- 54.Varani G, McClain WH. EMBO Rep. 2000;1:18. doi: 10.1093/embo-reports/kvd001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.a) Yarian C, Townsend H, Czestkowski W, Sochacka E, Malkiewicz AJ, Guenther R, Miskiewicz A, Agris PF. J Biol Chem. 2002;277:16391. doi: 10.1074/jbc.M200253200. [DOI] [PubMed] [Google Scholar]; b) Ajitkumar P, Cherayil JD. Microbiol Rev. 1988;52:103. doi: 10.1128/mr.52.1.103-113.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.a) Donohue J. J Mol Biol. 1969;45:231. doi: 10.1016/0022-2836(69)90102-8. [DOI] [PubMed] [Google Scholar]; b) Siegfried NA, Kierzek R, Bevilacqua PC. J Am Chem Soc. 2010;132:5342. doi: 10.1021/ja9107726. [DOI] [PubMed] [Google Scholar]; c) Sismour AM, Benner SA. Nucleic Acids Res. 2005;33:5640. doi: 10.1093/nar/gki873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. 2. ASM Press; Washington, D. C: 2006. [Google Scholar]
- 58.Benner SA. Acc Chem Res. 2004;37:784. doi: 10.1021/ar040004z. [DOI] [PubMed] [Google Scholar]
- 59.Geyer CR, Battersby TR, Benner SA. Structure. 2003;11:1485. doi: 10.1016/j.str.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 60.Testa SM, Disney MD, Turner DH, Kierzek R. Biochemistry. 1999;38:16655. doi: 10.1021/bi991187d. [DOI] [PubMed] [Google Scholar]
- 61.a) Diop-Frimpong B, Prakash TP, Rajeev KG, Manoharan M, Egli M. Nucleic Acids Res. 2005;33:5297. doi: 10.1093/nar/gki823. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Vendeix FAP, Munoz AM, Agris PF. RNA. 2009;15:2278. doi: 10.1261/rna.1734309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen X, Kierzek R, Turner DH. J Am Chem Soc. 2001;123:1267. doi: 10.1021/ja002623i. [DOI] [PubMed] [Google Scholar]
- 63.Krishnamurthy R. Acc Chem Res. 2012;45:2035. doi: 10.1021/ar200262x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.a) Forterre P. Biochimie. 2005;87:793. doi: 10.1016/j.biochi.2005.03.015. [DOI] [PubMed] [Google Scholar]; b) Burton AS, Lehman N. Astrobiology. 2009;9:125. doi: 10.1089/ast.2008.0240. [DOI] [PubMed] [Google Scholar]
- 65.Rios AC, Tor Y. Astrobiology. 2012;12:884. doi: 10.1089/ast.2011.0789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schroeder GK, Lad C, Wyman P, Williams NH, Wolfenden R. Proc Natl Acad Sci USA. 2006;103:4052. doi: 10.1073/pnas.0510879103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lindahl T. Nature. 1993;362:709. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 68.Lhomme J, Constant JF, Demeunynck M. Biopolymers. 1999;52:65. doi: 10.1002/1097-0282(1999)52:2<65::AID-BIP1>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 69.Schroeder GK, Wolfenden R. Biochemistry. 2007;46:13638. doi: 10.1021/bi701480f. [DOI] [PubMed] [Google Scholar]
- 70.a) Kazimierczuk Z, Mertens R, Kawczynski W, Seela F. Helv Chim Acta. 1991;74:1742. [Google Scholar]; b) Seela F, He Y. Helv Chim Acta. 2000;83:2527. [Google Scholar]
- 71.Iocono JA, Gildea B, McLaughlin LW. Tetrahedron Lett. 1990;31:175. [Google Scholar]
- 72.Benner SA. Curr Opin Chem Biol. 2012;16:581. doi: 10.1016/j.cbpa.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]