

Abstract
The lungs can undergo irreversible damage from chronic alcohol consumption. Herein, we developed an animal model predisposed for edematous lung injury following chronic ingestion of alcohol to better understand the etiology of alcohol-related disorders. Using animal modeling, alongside high-throughput proteomic and microarray assays, we identified changes in lung protein and transcript in mice and rats, respectively, following chronic alcohol ingestion or a caloric control diet. Liquid chromatography-mass spectrometry identified several mitochondrial-related proteins in which the expression was upregulated following long-term alcohol ingestion in mice. Consistent with these observations, rat gene chip microarray analysis of alveolar cells obtained from animals maintained on a Lieber-DeCarli liquid alcohol diet confirmed significant changes in mitochondrial-related transcripts in the alcohol lung. Transmission electron microscopy revealed significant changes in the mitochondrial architecture in alcohol mice, particularly following lipopolysaccharide exposure. Chronic alcohol ingestion was also shown to worsen mitochondrial respiration, mitochondrial membrane polarization, and NAD+-to-NADH ratios in alveolar type 2 cells. In summary, our studies show causal connection between chronic alcohol ingestion and mitochondrial dysfunction, albeit the specific role of each of the mitochondrial-related proteins and transcripts identified in our study requires additional study.
Keywords: mitochondrial stress, chronic alcohol ingestion, ENaC, gene chip array, mass spectrometry
chronic ethanol ingestion changes the molecular landscape of the lung (17). Serious changes in the lung proteome of alcoholics are often masked, given that the effects of chronic alcohol ingestion remain benign until secondary injury occurs (40–42). Despite recent significant advances made in understanding the etiology of alcohol-related disorders (7, 14, 17, 33), the molecular mechanisms that predispose alcoholics to acute respiratory distress syndrome are complex and remain unclear. For example, it has only been recently appreciated that intracellular zinc levels in alveolar macrophages are significantly attenuated in chronic alcoholics, despite normal serum zinc bioavailability (39). Alcohol-induced oxidative stress has also been shown to play a pivotal role in acute lung injury (17, 33). In particular, subcellular ethanol-induced oxidation is reflected in organelles where there is a +60-mV increase in oxidation of the GSH/GSSG redox potential in the mitochondria of alveolar type 2 cells (33). Significant changes in the alcohol lung's ability to maintain alveolar fluid homeostasis have also been reported. Acute exposure to chronic alcohol ingestion significantly alters the gating properties of apically located epithelial sodium channels (ENaC) (18) and basolaterally expressed Na-K-ATPase transporters (14). Taken together, these studies highlight the fact that molecular changes associated with chronic alcohol ingestion are complex and can target many cell types, organelles, and specific membrane proteins. As such, high-throughput assays are necessary to provide novel insight into the pathogenesis and adaptations to chronic alcohol ingestion.
Herein, we utilized liquid chromatography-mass spectrometry and Affymetrix gene chip assays to assess significant change in alveolar cell protein and gene expression following chronic alcohol ingestion in both mice and rats. In line with our previous studies (7, 33), our present findings indicate that a large number of the differentially expressed proteins in the alcohol lung are related to mitochondrial dysfunction.
METHODS
Reagents.
Ethanol was obtained from AAPER Alcohol and Chemical (Shelbyville, KY). Buffer and solutions were prepared in high-quality deionized, distilled water (≥18 MΩ).
Animals.
The use of animals was approved by the Institutional Care and Use of Animals Committee at Emory University School of Medicine.
Ethanol feeding.
A chronic alcohol model was studied in young adult female C57Bl/6 mice purchased from Jackson Laboratory (Bar Harbor, ME). To start, 6-wk-old mice were fed 5% wt/vol ethanol for the first week, with subsequent 5% incremental increases each week until animals reached 20% wt/vol ethanol. Animals remained on a 20% wt/vol ethanol diet for an additional 6 wk to model chronic alcohol consumption. An age-matched, control group of animals was fed an isocaloric maltodextrin diet. Male Sprague-Dawley rats were maintained on the Lieber-DeCarli liquid ethanol diet as described and reviewed in Ref. 17. Pair-fed control rats were maintained on an isocaloric mixture of liquid diet without ethanol. All animals were given standard chow ad libitum; dietary intake was comparable in both groups. With these general approaches, the blood alcohol levels of animals following chronic ethanol consumption has been reported to be 0.08% (38, 54).
Isolation of AT2.
Primary rat and mouse alveolar type 2 cells (AT2) cells were isolated as previously described (10) by an immunoabsorption approach. Briefly, excised lungs were lavaged with warm Elastase then minced and shaken in warm FBS and DNAse. The cell suspension was filtered sequentially through 100-μm and 40-μm filters into a 50-ml conical tube. The cells were centrifuged and resuspended in selection media. They were then plated on rat or mouse IgG plates, as appropriate, and incubated for 1 h in 5% CO2 at 37°C. After incubation period, the nonadherent cells were removed, centrifuged, and plated in supplemented DMEM/F12 growth media in a 5% CO2 incubator maintained at 37°C.
LPS inoculation.
Mice chronically ingesting alcohol were inoculated with 35 μl of lipopolysaccharide (LPS; 1 mg/ml) or vehicle (35 μl) as shown in Ref. 25. The instillate vehicle for all experiments consisted of 140 mM NaCl, 5 mM KCl, 1 mM CaCl2, and 10 mM HEPES with pH = 7.4 (saline). All animals were allowed to recover from this procedure overnight.
Lung fluid clearance assays.
Following overnight inoculation of LPS or vehicle, mice were tracheally instilled with 5 μl/g body wt of saline (defined above) to model alveolar flooding in control- and ethanol-fed animals following a “second hit.” Briefly, animals were placed in an in vivo animal imaging system (Bruker BioSpin, Billerica, MA) with continuous anesthesia (1.5 l/min isoflurane) and supplemental oxygen (100%). Mice were X-ray imaged at 5-min intervals up to 240 min with an acquisition period of 120 s. X-ray settings were as described in Ref. 22. In this way, we are able to measure the rate of alveolar fluid clearance in freely breathing, anesthetized mice. X-ray intensities of flooded areas of the lung were averaged over 30-min intervals, and data fitted to the following model: F(t) = K(1 − e−kat), where F(t) represents the amount of surface fluid in the lung at time t, K is the steady-state or peak amount of lung fluid, and ka is the rate of fluid absorption, the rates of secretion can be determined by dividing the peak fluid volume by the rate of absorption (K/ka) (50). Parameter estimation from curve fitting and statistical evaluations were done with SigmaPlot (San Rafael, CA).
2D gel electrophoresis.
Proteins harvested from lungs of ethanol-fed and control-fed mice were analyzed for differential protein expression as previously described (3), but with the following modifications. The ReadyPrep 2-D cleanup kit (Bio-Rad) was used to remove contaminants that would impede isoelectric focusing (IEF). Two-hundred and fifty micrograms of total protein, measured by the BCA protein assay (Thermo Scientific Pierce), was prepared in rehydration/sample (5 M urea, 2 M thiourea, 2% CHAPS, 2% SB 3–10, 40 mM Tris, 0.2% Bio-Lyte 3/10 ampholyte) and then applied to immobilized pH gradient (IPG) strips (pH 4–7 ReadyStrip, 7 cm, Bio-Rad). The IPG strips were subject to rehydration for 16 h at room temperature before being transferred to an IEF focusing tray. Electrode wicks were applied to the IPG strips to act as receptacles for nonamphoteric constituents. The IPG strips were covered with mineral oil and then subjected to IEF in the first dimension under nonreducing conditions by use of a Protean IEF cell (Bio-Rad) while maintaining the following parameters: 250 V, linear ramp, 20 min; 4,000 V, linear ramp, 2 h; 4,000 V, rapid ramp, 10,000 V-h; 20°C, 50 μA/strip. After being transferred to an equilibration tray the focused IPG strips were equilibrated in 2.5 ml of equilibration buffer 1 [6 M urea, 2% SDS, 0.375 M Tris·HCl (pH 8.8), 20% glycerol, and 2% (wt/vol) DTT] for 10 min on an orbital shaker at room temperature. The IPG strips were then equilibrated in 2.5 ml of equilibration buffer 2 [6 M urea, 2% SDS, 0.375 M Tris·HCl (pH 8.8), 20% glycerol, and with 2.5% (wt/vol) iodoacetamide] for 10 min on an orbital shaker at room temperature. The focused (murine) proteins were subject to SDS-PAGE in the second dimension. Densitometric analyses of 2D blots were done with PDQuest software (Bio-Rad).
Colloidal Coomassie staining and mass spectrometry.
After SDS-PAGE gels were fixed in colloidal Coomassie fix solution (45% methanol, 1% acetic acid) for 1 h with gentle agitation. The gels were then stained in colloidal Coomassie stain solution (170 g/l ammonium sulfate, 1 g/l Coomassie G250, 0.5% acetic acid, 34% methanol) for 8 h with gentle agitation. The background of the gels was removed by three consecutive washes with destaining solution (5% methanol) each for 1 h with gentle agitation. A small region of the Coomassie-stained gel that contained a majority of proteins that appeared to be either upregulated or downregulated was divided into four quadrants and the protein spots from each quadrant were pooled together. The spots were subject to in-gel tryptic digestion for identification of peptide surrogates as previously described (2, 3).
Genome chip analysis.
RNA purified from rat AT2 cells (n = 4 caloric control fed; 3 ethanol fed) were hybridized to Rat Genome U34 Arrays (Affymetrix) for characterization of gene expression. Hybridization mixtures were prepared, revealed, and washed according to the Affymetrix protocol. GeneChips were scanned by using the Affymetrix 3000 scanner and images were converted to digital data files. Data were analyzed by GeneSpring GX (Agilent) compatible with Affymetrix chips. The Emory Integrated Genomics Core submitted all relevant data to the NCBI Gene Expression Omnibus database [accession number GSE55243] in compliance with MIAME standards for microarray data.
Transmission electron microscopy.
Mouse lungs from ethanol-fed and maltodextrin-fed animals were perfused with 2.5% glutaraldehyde (in PBS) fixation solution and then minced into 1- to 2-mm2 tissue blocks and incubated in fixation solution for 2 h at 4°C. Then, tissue blocks were washed in 0.2 M cacodylate buffer prepared in PBS prior to subsequent fixation with 1% osmium fixative for 1 h at room temperature. Lung tissue blocks were then dehydrated through as series of ethanol washes, embedded in epoxy resin, and polymerized overnight at 60–70°C. All transmission electron microscopy (TEM) studies were performed at the Robert P. Apkarian Integrated Electron Microscopy Core at Emory University, Atlanta, GA.
Mitochondrial respiration.
Oxygen consumption was measured in a closed, stirred Perspex chamber fitted with a Clark oxygen electrode. Isolated mitochondria (0.5 mg protein/ml) were incubated in a respiratory reaction medium consisting of 120 mM KCl, 5 mM KH2PO4, 1 mM EGTA, 2 mM MgCl2, 5 μM rotenone, 80 ng/ml nigericin, and 3 mM HEPES (pH 7.4, 30°C) supplemented with 0.3% (wt/vol) BSA and saturated with room air. State 3 respiration was measured with 0.15 mM ADP. State 4 respiration was measured with 5 mM succinate. Data represent means ± SE for n = 6 rats/group.
Mitochondrial membrane potential.
The mitochondrial membrane potential (ΔΨm) was analyzed by use of JC-1 (Invitrogen). AT2 cells from control or ethanol-fed rats were seeded onto 12-well plates in triplicate overnight. Cells were then stained with JC-1 for 30 min. This cell-permeable, positively charged, red-orange fluorescent dye is readily sequestered by active mitochondria owing to the relative negative charge of the fluorophore. However, there is no accumulation of JC-1 when the mitochondria become depolarized or inactive. RFUs per cell were determined by computer analysis of confocal microscopic images and the fluorescence of the ethanol group (n = 6) compared with the control group (n = 6).
Mitochondrial ratio of NAD+ to NADH.
EnzyChrom NAD+/NADH assay kit (Bioassay Systems, Hayward, CA) was used to determine the mitochondrial ratio of nicotinamide adenine dinucleotide (NAD+) to NADH. In brief, mitochondria were isolated from AT2 cells derived from control or ethanol-fed rats by using a Mitochondria Isolation Kit (Thermo Fisher Scientific, Rockford, IL). NAD+ and NADH were then extracted with the extraction buffer provided in the assay kit, mixed with assay buffer, and absorbance was read at 565 nm.
RESULTS
Effect of LPS on alveolar fluid clearance in the chronic alcohol lung.
It is clear that chronic ethanol ingestion predisposes the human lung to acute respiratory distress syndrome (40–42). To model secondary injury in mice chronically ingesting alcohol or isocaloric maltodextrin, 35 μg LPS was inoculated via tracheal instillation (to serve as the second hit and thereby model lung injury). The following day, both groups of animals were tracheally instilled with 5 μl/g body wt saline to assess the effect of LPS on alveolar fluid clearance in the alcohol vs. control lungs. Figure 1 shows that chronic (>18 h of) LPS exposure significantly decreases the rate of alveolar fluid clearance in animals that had been maintained on a chronic alcohol diet compared with age-matched isocaloric-fed control animals. This study indicates that, after overnight exposure of chronic ethanol animals to LPS, a proinjury environment was created, hindering clearance and resulting in alveolar flooding. Thus alcohol-fed animals are indeed more susceptible to edematous lung injury compared with control animals.
Fig. 1.
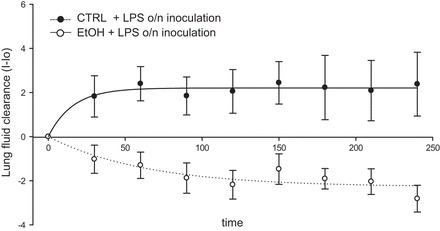
Alcohol and LPS-induced lung injury model. Radiographic determination of the rate of lung fluid clearance in mice chronically ingesting alcohol [ethanol (EtOH); n = 11] vs. age-matched caloric control animals (CTRL; n = 9) following overnight (o/n) exposure to LPS. The lungs of alcohol-fed animals +LPS stayed flooded, whereas control animals + LPS maintained balance between secretion and absorption after prolonged LPS exposure. *P < 0.01. I-Io, lung fluid clearance measured as X-ray intensity − intensity at time 0.
High-throughput analysis of changes in protein and transcript following chronic ingestion of alcohol.
The representative 2D gels of proteins derived from maltodextrin- and ethanol-fed mice are shown in Fig. 2, A and B, respectively. A centrally located rectangular area is enlarged to show protein migration at higher resolution. There were 20 unique protein spots in the maltodextrin (control) group of mice, 44 unique spots in the ethanol-fed group of mice, and 73 protein spots that were expressed in both groups (Fig. 2C).
Fig. 2.
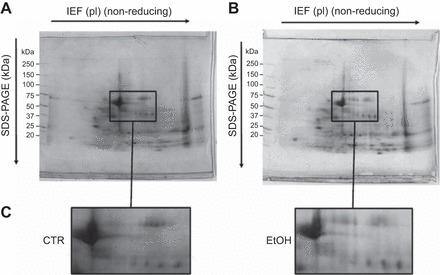
2D proteome profiling of alcohol lung. Two-dimensional gel electrophoresis analysis of lung tissue homogenate from mice chronically ingesting maltodextrin, serving as a caloric control-fed group (CTR; A), or ethanol (EtOH; B). Protein samples (2 mg) were separated by isoelectric focusing (IEF) under nonreducing conditions by using 17-cm, pH 4–7 immobilized pH gradient (IPG) strips, followed by SDS-PAGE on 12% gels, and stained with Coomassie brilliant blue under reducing conditions. The approximate molecular weights are shown on the left of the gel, and the enlarged rectangular areas (C) show gel regions in which spots corresponding to differentially expressed proteins were identified. pI, isoelectric points.
The molecular identity of the protein spots positively identified is reported in Table 1. These proteins were classified into several categories based on their functional significance, including but not limited to enzymatic, kinase, peptidase activity, ATP synthesis, posttranslational modification, transcriptional regulation, transport, intracellular signaling, and myelination. As Table 1 shows, 6 of the 43 positively identified proteins are related to mitochondrial function (boldfaced proteins are related to mitochondrial function). Further PDQuest software analysis of the densimetric spots revealed that the boldfaced proteins in Table 1 (ALDH_MOUSE; ATPA_MOUSE; IVD_MOUSE; THIM_MOUSE; SCOT1_MOUSE; EFTU_MOUSE) differed significantly (by at least 2-fold) compared with control-fed mice.
Table 1.
Proteins identified by LC-MS/MS
| SwissProt | Accession Number | Subcellular Location | Function | pI | Unique Peptides | Protein Description |
|---|---|---|---|---|---|---|
| ALDH2_MOUSE | P47738 | MT | enzyme | 7.53 | 10 | Aldehyde dehydrogenase, mitochondrial |
| MMSA_MOUSE | Q9EQ20 | MT | enzyme | 5.45 | 14 | Methylmalonate-semialdehyde dehydrogenase [acylating], mitochondrial |
| FUMH_MOUSE | P97807 | MT | enzyme | 9.12 | 11 | Fumerate hydratase |
| ATPA_MOUSE | Q03265 | MT | ATP synthesis, transport | 7.3 | 17 | ATP synthase subunit alpha, mitochondrial |
| IVD_MOUSE | Q9JHI5 | MT | enzyme | 6.43 | 8 | Isovaleryl-CoA dehydrogenase, mitochondrial |
| THIM_MOUSE | Q8BWT1 | MT | enzyme | 7.32 | 13 | 3-ketoacyl-CoA thiolase, mitochondrial |
| SCOT1_MOUSE | Q9DOK2 | MT | enzyme | 8.78 | 14 | Succinyl-CoA:3-ketoacid-coenzyme A transferase 1, mitochondrial |
| DHE3_MOUSE | P26443 | MT | enzyme | 8 | 21 | Glutamate dehydrogenase 1, mitochondrial |
| EFTU_MOUSE | Q8BFR5 | MT | translation regulator | 7.23 | 9 | Elongation factor Tu, mitochondrial |
| ACADM_MOUSE | P45952 | MT | enzyme | 8.6 | 9 | Medium-chain specific acyl-CoA dehydrogenase, mitochondrial |
| THIL_MOUSE | Q8QZT1 | MT | enzyme | 8.7 | 9 | Acetyl-CoA acetyltransferase, mitochondrial |
| NONO_MOUSE | Q99K48 | N | other | 9.01 | 8 | Non-POU domain-containing octamer-binding protein |
| KCRM_MOUSE | P07310 | CT | enzyme | 6.56 | 9 | Creatine kinase M-type |
| SFPQ_MOUSE | Q8VIJ6 | N | other | 9.45 | 9 | Splicing factor, proline- and glutamine-rich |
| CATA_MOUSE | P24270 | CT | enzyme | 6.5 | 16 | Catalase OS |
| SDPR_MOUSE | Q63918 | PM | other | 5.2 | 10 | Serum deprivation-response protein |
| AMPL_MOUSE | Q9CPY7 | CT | peptidase | 7 | 7 | Cytosol aminopeptidase |
| PCBP1_MOUSE | P60335 | N | translation regulator | 7.1 | 8 | Poly(rC)-binding protein 1 |
| PGK1_MOUSE | P09411 | CT | kinase | 7.6 | 17 | Phosphoglycerate kinase 1 |
| KPYM_MOUSE | P52480 | CT | kinase | 7.5 | 11 | Pyruvate kinase PKM |
| NUCL_MOUSE | P09405 | N | other | 4.8 | 13 | Nucleolin |
| ALDOA_MOUSE | P05064 | CT | enzyme | 8.3 | 9 | fructose-bisphosphate aldolase |
| RAGE_MOUSE | Q62151 | CT | regulatory | 5.78 | 9 | Advanced glycosylation end product-specific receptor |
| PTRF_MOUSE | O54724 | N | transcription regulator | 5.5 | 8 | Polymerase I and transcript release factor |
| NHRF2_MOUSE | Q9JHL1 | PM | transporter | 7.6 | 9 | Na+/H+ exchange regulatory cofactor NHE-RF2 |
| ENOA_MOUSE | P17182 | CT | transcription regulator | 6.8 | 19 | Alpha-enolase |
| SEP11_MOUSE | Q8C1B7 | N | other | 6.7 | 12 | Septin-11 |
| IDHC_MOUSE | O88844 | CT | enzyme | 6.9 | 18 | Isocitrate dehydrogenase [NADP] cytoplasmic |
| ZO1_MOUSE | P39447 | PM | other | 6.7 | 9 | Tight junction protein ZO-1 |
| SYWC_MOUSE | P32921 | CT | regulatory | 5.83 | 8 | Tryptophan–tRNA ligase, cytoplasmic |
| AL1A1_MOUSE | P24549 | CT | enzyme | 7.8 | 23 | Retinal dehydrogenase 1 |
| AMRP_MOUSE | P55302 | PM | transmembrane receptor | 7.9 | 9 | Alpha-2-macroglobulin receptor-associated protein |
| MOES_MOUSE | P26041 | PM | other | 6.1 | 17 | Moesin |
| HEMO_MOUSE | Q91X72 | ES | transporter | 7.8 | 9 | Hemopexin |
| ALBU_MOUSE | P07724 | ES | transporter | 6.1 | 27 | Serum albumin |
| TRFE_MOUSE | Q921I1 | ES | other | 7.2 | 40 | Serotransferrin |
| FLNA_MOUSE | Q8BTM8 | CT | other | 6 | 20 | Filamin-A |
| BCAM_MOUSE | Q9R069 | PM | intracellular signaling | 9 | Basal cell adhesion molecule | |
| STIP1_MOUSE | Q60864 | CT | other | 6.8 | 12 | Stress-induced-phosphoprotein 1 |
| PRAX_MOUSE | O55103 | CT | myelination | 8.14 | 14 | Periaxin |
| DPYL2_MOUSE | O08553 | CT | enzyme | 6.4 | 9 | Dihydropyrimidinase-related protein 2 |
| EZRI_MOUSE | P26040 | PM | other | 6.1 | 14 | Ezrin |
Identification of the proteins from LC-MS/MS data was achieved using the MASCOT search engine. The number of unique peptides, isoelectric point (pI), molecular weight (MW), and accession number are given for each identified (mouse) protein. The boldface mitochondrial proteins were significantly upregulated in alcohol lung vs. caloric control-fed animals. Subcellular location:
MT, mitochondria; CT, cytoplasm; PM, plasma membrane; ES, extracellular space; N, nucleus.
On the basis of the outcome of proteomic analysis of the lungs of mice chronically ingesting alcohol, we restricted our analysis of Affymetrix rat gene chip data to mitochondrial-related genes (Fig. 3); however, all gene chip data have been deposited into the Gene Expression Omnibus database online (accession number GSE55243).
Fig. 3.
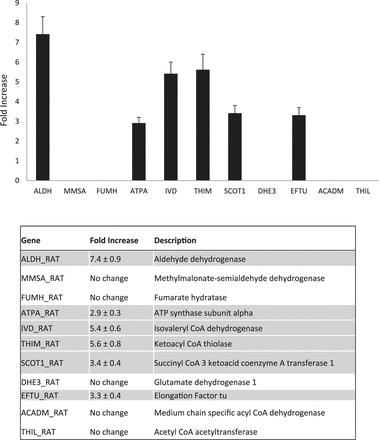
Genome Affymetrix chip analysis. RNA purified from primary isolated rat alveolar type 2 cells was hybridized to Rat Genome Affymetrix chips. Differences in the expression of mitochondrial gene were assessed. The data represent significant fold increase of genes presented (EtOH-fed, n = 3 vs. caloric control-fed, n = 4). P < 0.05 by unpaired t-test.
In agreement with our alcohol-fed mouse data obtained via mass spectrometry, gene chip data confirm a significant increase in the following mitochondrial related transcripts (highlighted) in alcohol-fed rats: aldehyde dehydrogenase; ATP synthase subunit alpha, isovaleryl CoA dehydrogenase, ketoacyl CoA thiolase; succinyl CoA 3 ketoacid coenzyme A transferase 1; and elongation factor Tu. Together, the proteomic and gene chip data, obtained from mouse and rat, respectively, support the hypothesis that mitochondrial dysfunction contributes to the pathogenesis of alcohol-induced lung injury.
Mitochondrial damage following chronic alcohol ingestion.
In Fig. 4, TEM images of distal lung sections obtained from animals chronically ingesting alcohol showed enlarged mitochondria compared with control animals (top). Severe damage to the mitochondria, with cristae degradation, could be seen following overnight inoculation with 35 μg LPS (delivered tracheally in a 35-μl PBS solution 24 h prior to TEM analysis) at bottom. Fragmented cristae or completely missing cristae that leave an electron-lucent matrix, as seen in Fig. 4, following LPS inoculation, are hallmarks of mitochondrial dysfunction (21). TEM imaging also shows that dual ethanol and LPS exposure caused lysosomal degradation within autophagic vacuoles in the mouse lung (bottom right). The significant changes in mitochondrial morphology (measured length-to-width ratio) induced by ethanol and/or LPS are quantified in Fig. 4B. Significant change in the length-to-width ratio of mitochondrial structure has been reported to be indicative of serious mitochondrial swelling and ultrastructure damage (36). These observations are indeed in line with the clinical finding that alcoholics have seemingly normal lung function and that a secondary infection (modeled by overnight LPS exposure in our studies) leads to deleterious complications in the lungs (40). The structural integrity of mitochondria following chronic alcohol ingestion (alone) remains intact. However, chronic ethanol and LPS treatment lead to significantly increased levels of cristae fragmentation and lysosomal degradation.
Fig. 4.
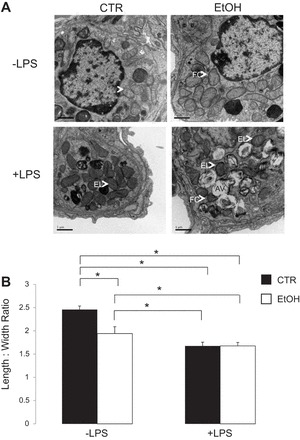
Transmission electron microscopy (TEM) imaging of mitochondria (A). Top: representative TEM images of CTR vs. EtOH mouse lung. Bottom: representative CTR and EtOH lung following 35 μg LPS (24 h) exposure. Arrows indicate mitochondria. AV, autophagic vacuoles; FC, fragmented cristae; EL, electron lucent (missing cristae) (36). Original magnification ×20,000. Quantification of change in mitochondria morphology (B) shows that chronic ethanol ingestion significantly lowers the length-to-width ratio of mitochondria. For each treatment, a minimum of 20 observations were analyzed from 3 independently treated mice (n = 60). *P < 0.05.
On the basis of the significant change in protein and transcript expression profiles, and significant decline in mitochondria structure following chronic alcohol ingestion observed, we assessed mitochondrial function in alveolar type 2 cells to support the hypothesis that mitochondrial dysfunction contributes to the pathogenesis of alcohol-induced lung injury.
Mitochondrial dysfunction in alcohol lung.
Mitochondria generate ATP by utilizing the proton electrochemical gradient potential, or electrochemical proton motive force (Δp). This is generated by a series of reduction of electrons through the respiratory electron transport chain. Reduction of electrons occurs in the inner mitochondrial membrane, which provides the energy needed to drive protons against a concentration gradient out of the mitochondrial cytoplasm. As a result, there is net accumulation of H+ outside the membrane, which then flows back into the mitochondria in an ATP-dependent manner, thus completing the electron transport chain. The total force driving H+ into the mitochondria is a combination of both the mitochondrial membrane potential (ΔΨm, a charge or electrical gradient) and a H+ gradient (ΔpH). Both the mitochondrial rate of respiration and membrane potential can be measured to assess the decline of mitochondrial function in the alcohol lung. Oxygen consumption (during state 3 and 4 respiration) was measured in a closed oxygen chamber and the respiratory control ratio (RCR) was determined by using 0.15 mM ADP and 5 mM succinate, respectively. Figure 5 shows that chronic ethanol ingestion significantly decreased state 3 respiration (0.58 ± 0.07 nmol O2·mg protein−1·min−1) and RCR (4.21 ± 0.48) compared with the control (0.95 ± 0.07 nmol O2·mg protein−1·min−1 and 5.87 ± 0.52) rates, respectively. In the absence of ADP (state 4 respiration) very little oxygen is consumed. However, chronic ethanol ingestion continued to significantly attenuate oxygen consumption (0.11 ± 0.01 nmol O2·mg protein−1·min−1) and RCR (5.09 ± 0.36) compared with mitochondria obtained from control-fed animals (0.18 ± 0.01 nmol O2·mg protein−1·min−1 and 7.74 ± 0.67), respectively.
Fig. 5.
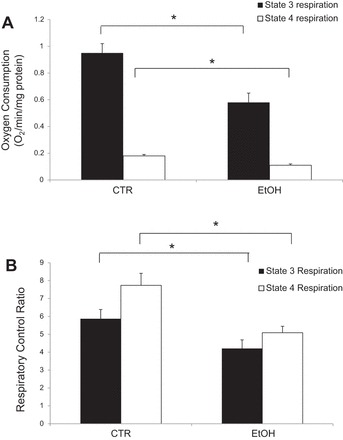
Mitochondrial respiration. Chronic alcohol ingestion significantly decreased respiration rates in isolated rat mitochondria. A: state 3 and state 4 oxygen consumption. B: state 3 and state 4 respiratory control ratio. *P < 0.05; n = 6 independent observations.
Next, we used a carbocyanine dye, JC-1, as a reliable determinant of ΔΨm (46). JC-1 is a reliable measure of ΔΨm because of its dual emission properties. At low concentrations, monomeric JC-1 emits a green fluorescence (535 nm; λ). High concentrations of JC-1 dye uptake will aggregate, which emits orange-red fluorescence (610 λ). Since the amount of JC-1 dye uptake depends on its transmembrane potential, at low ΔΨm a primarily green emission will be detected, whereas at high ΔΨm increases in orange-red relative light units can be quantified. Representative confocal analysis of alveolar type 2 cells freshly isolated from alcohol- or control-fed animals labeled with JC-1 are shown in Fig. 6A. Computer analysis of confocal images (Fig. 6B) indicates that chronic ethanol ingestion significantly decreases the mitochondrial membrane potential from 1.0 ± 0.08 (610 λ RLUs measured from alveolar type 2 cells isolated from control-fed mice) to 0.32 ± 0.05 RLUs in alcohol-fed animals. Data were obtained from six independent animals, with isolated cells seeded onto 12-well plates for triplicate JC-1 labeling.
Fig. 6.
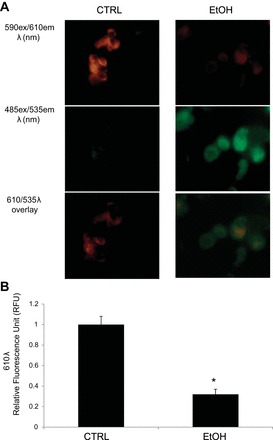
Mitochondrial membrane potential. Chronic alcohol ingestion significantly decreased membrane polarization in mitochondria derived from rat alveolar type 2 cells as measured from JC-1 labeling. A: chronic alcohol consumption results in predominantly green JC-1 fluorescence (due to low mitochondrial membrane potential; right), whereas the dye aggregates and fluoresces red under control conditions (due to high mitochondrial membrane potential; left). Bottom: overlay of red and green emission profiles of JC-1 labeled cells. B: average 610 λ excitation values of JC-1 labeled cells performed in triplicates from 6 animals (*P < 0.05) indicating that chronic ethanol consumption significantly decreases mitochondrial membrane potential.
NAD+ plays key roles in nearly all aspects of cellular metabolism, and several studies indicate that the NAD+-to-NADH ratio is a useful indicator of cellular redox state and cell health (reviewed in Ref. 55). In Fig. 7, we measured the NAD+-to-NADH ratio to assess the redox state of mitochondria. The bioassay utilized relies on NADH reduction of formazan (MTT) reagent. The intensity of the reduced MTT compound, measured at 565 nm, is proportional to the NAD+-to-NADH ratio (37, 56). Consistent with our other findings, chronic ethanol consumption significantly decreased the mitochondrial NAD+-to-NADH ratio (Control: 1.0 ± 0.11, Ethanol: 0.39 ± 0.06; P < 0.05 from 6 independent animals studied).
Fig. 7.
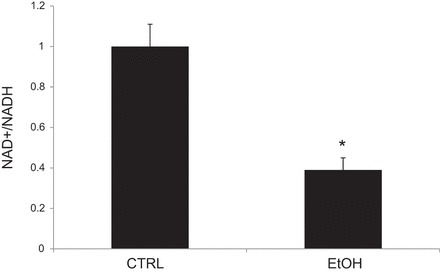
Chronic ethanol ingestion decreases mitochondrial NAD+-to-NADH ratio. *P < 0.05; n = 6.
DISCUSSION
Proteomic analysis of the lung following chronic ethanol exposure.
One aim of our study was to investigate the differential expression of the entire lung proteome by using 2D gel electrophoresis and mass spectrometry with animal models of chronic ethanol ingestion predisposed for edematous lung injury. There are some inherent limitations related to taking this proteomics approach, particularly related to protein solubility, that warrant further discussion. Since there are limitations to protein solubility, we focused our effort on examining differentially expressed proteins with isoelectric points (pI) ranging from 3–10 and with molecular masses between 15 and 250 kDa. Additionally, it is difficult to detect proteins that are not abundantly expressed. As such, proteins that are insoluble, have extreme pI, or are expressed in low quantity may be beyond the detection limit of the methodologies described herein. Using 2D analysis, however, we were able to unambiguously characterize the differential expression of mitochondrial and other proteins that are differentially regulated by ethanol. Coomassie staining of protein was utilized in our studies (as opposed to alternative methods such as silver staining) to facilitate and ensure abundant protein amounts for subsequent mass spectrometry analysis. The proteomics data obtained in this way are in agreement with the observed effects of alcohol on transcript levels of mitochondrial protein assayed by use of gene chip technology. These are important findings, given that the underlying causes of alcohol-induced tissue damage and various diseases, such as acute lung injury and acute pulmonary distress syndrome, remain unclear. Conducting our studies in both mouse and rat models of chronic alcohol ingestion indicates that similar changes in protein and transcript expression can be appreciated regardless of the species investigated.
Chronic ethanol ingestion and mitochondrial dysfunction.
When studying alcohol abuse in animal models of chronic alcohol ingestion, it is important to normalize for the excess calorie intake from consuming alcohol. To ensure that baseline metabolic demands are consistent between groups, we and others (14, 19, 20) administer an isocaloric diet to an age-matched control group. High-caloric diets have indeed been shown to affect mitochondrial function (34). By controlling for possible effects of a high-calorie diet, we ensure that the excess reactive oxygen species (ROS) production and observed mitochondrial damage can be attributed to ethanol alone.
Table 1 provides a list of differentially expressed proteins identified by LC MS/MS. We focused our attention on proteins (and genes in Fig. 3) related to mitochondria, in part, because chronic alcohol ingestion has been implicated in several metabolic disorders (4, 20, 23) and oxidative stress (1, 9, 19, 33). Alcohol metabolism results in the generation of acetaldehyde, a highly reactive and toxic by-product that has deleterious effects. As such, it was not surprising that mitochondrial aldehyde dehydrogenase (ALDH2) protein and transcript increased in the alcohol lung. Alongside alcohol dehydrogenase, mitochondrial ALDH2 is also responsible for metabolizing and eliminating the majority of ethanol consumed (12). Interestingly, Xu et al. (53) found that activation of the ERK/MAPK and PI3K-Akt signaling pathways after mitochondrial ALDH overexpression protected lung epithelial cells from hyperoxia-induced cell death. Although the beneficial role of ALDH2 overexpression in the alcohol lung is apparent, the molecular mechanisms that predispose the alcohol lung for acute respiratory disorder remain unclear. Additional studies are needed to characterize the role of ATP synthase, isovaleryl CoA dehydrogenase, ketoacyl CoA thiolase, succinyl CoA 3 ketoacid coenzyme A transferase, and elongation factor Tu in lung injury, since these proteins were also upregulated in a mouse model of chronic alcohol ingestion.
Mitochondrial swelling and a second hit.
Both control and ethanol fed animals were inoculated with LPS overnight to evaluate mitochondrial damage using a “two-hit” model of alcohol induced lung injury. Figure 1 shows that LPS exposure significantly attenuates the alcohol lungs' ability to clear fluid, resulting in pulmonary edema. This finding shows that the animal studies conducted are indeed relevant to the clinical observation that patients with positive history of alcohol abuse have increased risk for lung injury following a secondary infection. Moreover, Fig. 4A indicates that lysosomal degradation within autophagic vacuoles was observed only in the alcohol- and LPS-treated lungs. This observation is indeed in line with the second-hit hypothesis, where ethanol exposure alongside a secondary assault has severe deleterious effects on the lung.
In addition to the significant morphological changes, chronic alcohol ingestion also induces mitochondrial oxidative stress and even compromises the mitochondria's ability to defend against oxidative stress by decreasing GSH (28, 33). This increase in ROS also leads to a buildup of damaged mitochondrial DNA and, thus, compromised ATP synthesis. With a decreased ability to make ATP, the cell may still be able to function normally owing to residual levels of ATP, but when confronted with a second hit the cell is not equipped to recover (48). Depleted ATP stores have been shown to lead to necrosis and gross tissue damage (28). Many individuals smoke cigarettes while consuming alcohol (chronically). Together, cigarette smoke and alcohol are two extremely perilous stressors. Cigarette smoke extract has recently been shown to significantly increase ROS production and cause mitochondrial fragmentation and subsequent cellular senescence (24). Whether protecting the mitochondrial integrity can improve health outcomes for chronic alcoholics who develop acute respiratory disorder is an intriguing possibility that has not been completely studied.
Mitochondrial dysfunction and altered cell function.
Since chronic ethanol ingestion predisposes the lung to edematous injury, it is important to consider putative effects of mitochondrial damage on the alveolar cells ability to transport salt and water across the epithelium. In alveolar type 2 cells, ENaCs play an important role in transporting Na+ across the epithelium by creating the osmotic gradient for water reabsorption. Generally speaking, prooxidants serve as important signaling molecules that regulate lung ENaC activity (11, 15, 16, 22, 27, 47, 49). More specifically, we have recently studied the effect of chronic ethanol ingestion on ENaC activity in AT2 cells and found that acute ethanol induces an increase in ROS production, which significantly increases the open probability of ENaC prior to a second hit (18). The changes in cell signaling and regulation of ENaC activity following a secondary insult ultimately leads to alveolar flooding, albeit the precise mechanisms remain unclear.
Results from our present study, alongside our previous reports, suggest that mitochondrial damage could play a major role in altering apical channel activity. Rotenone is a high-affinity inhibitor of complex I of the mitochondrial electron transport chain (6, 26). Inhibition of this complex could lead to an increase in ENaC activity by several possible mechanisms. First, complex I is a major physiologically and pathologically important site for ROS production, and inhibition by rotenone has been shown to increase ROS production (30, 34). Increases in ROS, such as H2O2, have been shown to increase ENaC activity, possibly through altered NOX signaling (18, 22, 49). Second, inhibition of complex I results in a disruption of the electron transport chain and a decrease in ATP production, resulting in depolarization of the mitochondrial membrane potential. The presence of ATP inhibits ENaC activity via a purinergic signaling cascade (13, 31, 35, 51). ATP binds to P2 receptors and activates PLC, decreasing the amount of membrane-bound PI(4,5)P2. This is done by either activating PKC or by direct hydrolysis of PI(4,5)P2. The open probability of ENaC is increased in the presence of PI(4,5)P2 (45); therefore, in the absence of ATP, PI(4,5)P2 levels would be preserved and ENaC activity would be increased. A third method may involve a suppression of hypoxic signaling. ENaC activity is normally decreased under hypoxic conditions, possibly owing to a decrease in surface expression of the channel (8, 29, 44, 52). However, complex I inhibition with rotenone has been shown to induce rapid hyperphosphorylation and inhibition of translation initiation factor eIF2α and elongation factor eEF2 (32). These proteins play a regulatory role in the hypoxia-induced reduction of ENaC activity; therefore, their inhibition by rotenone may ultimately prevent the hypoxia-induced suppression of ENaC activity.
Our research group has also recently examined the role of ethanol's metabolic product, acetaldehyde, in regulation of ion channel activity. In A6 distal nephron cells, acetaldehyde significantly increased ENaC activity (5). Moreover, it has recently been established that serum- and glucocorticoid- induced protein kinase-1 (Sgk-1), a kinase that phosphorylates Neural precursor cell-expressed developmentally downregulated (Nedd) 4-2 (preventing Nedd4-2 from transferring its ubiquitin proteins to ENaC, thus marking ENaC to be destroyed) localizes to the mitochondrial membrane (43). However, mitochondria under chronic ethanol conditions show significant swelling, especially when confronted with a secondary insult (Fig. 4), and a decreased polarization of the mitochondrial membrane (Fig. 6), indicating a compromised membrane. If the mitochondrial membrane is not functioning properly, it is possible that Sgk is not able to phosphorylate NEDD 4-2, leading to the ubiquitination and destruction of ENaC. This could possibly be an explanation for the decreased ENaC activity that our group observed under chronic ethanol and LPS conditions, when mitochondrial swelling is also present and is consistent with the second-hit hypothesis. Together, these observations provide novel correlation between mitochondrial function and the sodium transport properties of the lung. The exact signaling mechanisms that ensue following secondary injury and mitochondrial dysfunction, that ultimately lead to a significant change in ENaC activity, remain unknown and are a topic for future investigation.
GRANTS
Support for this research was provided by grants from Children's Healthcare of Atlanta (to M. N. Helms and L. A. Brown); NIAAA P50 AA 135757 pilot award to M. N. Helms, F32 KD093255-01 to A. A. Alli; and R37 DK037963 to D. C. Eaton.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
A.A.A., L.A.S.B., and M.N.H. conception and design of research; A.A.A., E.M.B., D.S.M., and M.S.G. performed experiments; A.A.A., L.A.S.B., and M.N.H. analyzed data; A.A.A., D.C.E., L.A.S.B., and M.N.H. interpreted results of experiments; A.A.A. and E.M.B. prepared figures; A.A.A., E.M.B., and M.N.H. drafted manuscript; A.A.A., E.M.B., D.C.E., L.A.S.B., and M.N.H. edited and revised manuscript; A.A.A., E.M.B., D.S.M., M.S.G., D.C.E., L.A.S.B., and M.N.H. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Preston Goodson and Nicholle M. Johnson for excellent technical assistance. We thank the Proteomics Core Facility at the Moffitt Cancer Center, Tampa, FL, for mass spectrometry services.
REFERENCES
- 1.Albano E. Alcohol, oxidative stress and free radical damage. Proc Nutr Soc 65: 278–290, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Alli AA, Gower WR., Jr. Molecular approaches to examine the phosphorylation state of the C type natriuretic peptide receptor. J Cell Biochem 110: 985–994, 2010 [DOI] [PubMed] [Google Scholar]
- 3.Alli AA, Song JZ, Al-Khalili O, Bao HF, Ma HP, Alli AA, Eaton DC. Cathepsin B is secreted apically from Xenopus 2F3 cells and cleaves the epithelial sodium channel (ENaC) to increase its activity. J Biol Chem 287: 30073–30083, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baik I, Shin C. Prospective study of alcohol consumption and metabolic syndrome. Am J Clin Nutr 87: 1455–1463, 2008 [DOI] [PubMed] [Google Scholar]
- 5.Bao HF, Song JZ, Duke BJ, Ma HP, Denson DD, Eaton DC. Ethanol stimulates epithelial sodium channels by elevating reactive oxygen species. Am J Physiol Cell Physiol 303: C1129–C1138, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barrientos A, Moraes CT. Titrating the effects of mitochondrial complex I impairment in the cell physiology. J Biol Chem 274: 16188–16197, 1999 [DOI] [PubMed] [Google Scholar]
- 7.Brown LA, Harris FL, Bechara R, Guidot DM. Effect of chronic ethanol ingestion on alveolar type II cell: glutathione and inflammatory mediator-induced apoptosis. Alcohol Clin Exp Res 25: 1078–1085, 2001 [PubMed] [Google Scholar]
- 8.Carpenter TC, Schomberg S, Nichols C, Stenmark KR, Weil JV. Hypoxia reversibly inhibits epithelial sodium transport but does not inhibit lung ENaC or Na-K-ATPase expression. Am J Physiol Lung Cell Mol Physiol 284: L77–L83, 2003 [DOI] [PubMed] [Google Scholar]
- 9.Cederbaum AI, Lu Y, Wu D. Role of oxidative stress in alcohol-induced liver injury. Arch Toxicol 83: 519–548, 2009 [DOI] [PubMed] [Google Scholar]
- 10.Chen J, Chen Z, Narasaraju T, Jin N, Liu L. Isolation of highly pure alveolar epithelial type I and type II cells from rat lungs. Lab Invest 84: 727–735, 2004 [DOI] [PubMed] [Google Scholar]
- 11.Chen L, Fuller CM, Kleyman TR, Matalon S. Mutations in the extracellular loop of α-rENaC alter sensitivity to amiloride and reactive species. Am J Physiol Renal Physiol 286: F1202–F1208, 2004 [DOI] [PubMed] [Google Scholar]
- 12.Crabb DW, Matsumoto M, Chang D, You M. Overview of the role of alcohol dehydrogenase and aldehyde dehydrogenase and their variants in the genesis of alcohol-related pathology. Proc Nutr Soc 63: 49–63, 2004 [DOI] [PubMed] [Google Scholar]
- 13.Cuffe JE, Bielfeld-Ackermann A, Thomas J, Leipziger J, Korbmacher C. ATP stimulates Cl− secretion and reduces amiloride-sensitive Na+ absorption in M-1 mouse cortical collecting duct cells. J Physiol 524: 77–90, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dada L, Gonzalez AR, Urich D, Soberanes S, Manghi TS, Chiarella SE, Chandel NS, Budinger GR, Mutlu GM. Alcohol worsens acute lung injury by inhibiting alveolar sodium transport through the adenosine A1 receptor. PLoS One 7: e30448, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Downs CA, Helms MN. Regulation of ion transport by oxidants. Am J Physiol Lung Cell Mol Physiol 305: (9) L595–L603, 2013 [DOI] [PubMed] [Google Scholar]
- 16.Downs CA, Kumar A, Kreiner LH, Johnson NM, Helms MN. H2O2 regulates lung epithelial sodium channel (ENaC) via ubiquitin-like protein Nedd8. J Biol Chem 288: 8136–8145, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Downs CA, Trac D, Brewer EM, Brown LA, Helms MN. Chronic alcohol ingestion changes the landscape of the alveolar epithelium. Biomed Res Int 2013: 470217, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Downs CA, Trac DQ, Kreiner LH, Eaton AF, Johnson NM, Brown LA, Helms MN. Ethanol alters alveolar fluid balance via Nadph oxidase (NOX) signaling to epithelial sodium channels (ENaC) in the lung. PLoS One 8: e54750, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El-Assal O, Hong F, Kim WH, Radaeva S, Gao B. IL-6-deficient mice are susceptible to ethanol-induced hepatic steatosis: IL-6 protects against ethanol-induced oxidative stress and mitochondrial permeability transition in the liver. Cell Mol Immunol 1: 205–211, 2004 [PubMed] [Google Scholar]
- 20.Fan AZ, Russell M, Dorn J, Freudenheim JL, Nochajski T, Hovey K, Trevisan M. Lifetime alcohol drinking pattern is related to the prevalence of metabolic syndrome. The Western New York Health Study (WNYHS). Eur J Epidemiol 21: 129–138, 2006 [DOI] [PubMed] [Google Scholar]
- 21.Golomb E, Matza D, Cummings CA, Schwalb H, Kodavanti UP, Schneider A, Houminer E, Korach A, Nyska A, Shapira OM. Myocardial mitochondrial injury induced by pulmonary exposure to particulate matter in rats. Toxicol Pathol 40: 779–788, 2012 [DOI] [PubMed] [Google Scholar]
- 22.Goodson P, Kumar A, Jain L, Kundu K, Murthy N, Koval M, Helms MN. Nadph oxidase regulates alveolar epithelial sodium channel activity and lung fluid balance in vivo via O2− signaling. Am J Physiol Lung Cell Mol Physiol 302: L410–L419, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goude D, Fagerberg B, Hulthe J. Alcohol consumption, the metabolic syndrome and insulin resistance in 58-year-old clinically healthy men (AIR study). Clin Sci (Lond) 102: 345–352, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Hara H, Araya J, Ito S, Kobayashi K, Takasaka N, Yoshii Y, Wakui H, Kojima J, Shimizu K, Numata T, Kawaishi M, Kamiya N, Odaka M, Morikawa T, Kaneko Y, Nakayama K, Kuwano K. Mitochondrial fragmentation in cigarette smoke induced-bronchial epithelial cell senescence. Am J Physiol Lung Cell Mol Physiol 305: (10) L737–L746, 2013 [DOI] [PubMed] [Google Scholar]
- 25.Helms MN, Torres-Gonzalez E, Goodson P, Rojas M. Direct tracheal instillation of solutes into mouse lung. J Vis Exp 42: 1941, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hensley K, Pye QN, Maidt ML, Stewart CA, Robinson KA, Jaffrey F, Floyd RA. Interaction of alpha-phenyl-N-tert-butyl nitrone and alternative electron acceptors with complex I indicates a substrate reduction site upstream from the rotenone binding site. J Neurochem 71: 2549–2557, 1998 [DOI] [PubMed] [Google Scholar]
- 27.Hickman-Davis JM, McNicholas-Bevensee C, Davis IC, Ma HP, Davis GC, Bosworth CA, Matalon S. Reactive species mediate inhibition of alveolar type II sodium transport during mycoplasma infection. Am J Respir Crit Care Med 173: 334–344, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoek JB, Cahill A, Pastorino JG. Alcohol and mitochondria: a dysfunctional relationship. Gastroenterology 122: 2049–2063, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Husted RF, Lu H, Sigmund RD, Stokes JB. Oxygen regulation of the epithelial Na channel in the collecting duct. Am J Physiol Renal Physiol 300: F412–F424, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kushnareva Y, Murphy AN, Andreyev A. Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem J 368: 545–553, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leipziger J. Control of epithelial transport via luminal P2 receptors. Am J Physiol Renal Physiol 284: F419–F432, 2003 [DOI] [PubMed] [Google Scholar]
- 32.Li J, Mahdi F, Du L, Datta S, Nagle DG, Zhou YD. Mitochondrial respiration inhibitors suppress protein translation and hypoxic signaling via the hyperphosphorylation and inactivation of translation initiation factor eIF2alpha and elongation factor eEF2. J Nat Prod 74: 1894–1901, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang Y, Yeligar SM, Brown LA. Chronic-alcohol-abuse-induced oxidative stress in the development of acute respiratory distress syndrome. ScientificWorldJournal 2012: 740308, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem 80: 780–787, 2002 [DOI] [PubMed] [Google Scholar]
- 35.Ma HP, Li L, Zhou ZH, Eaton DC, Warnock DG. ATP masks stretch activation of epithelial sodium channels in A6 distal nephron cells. Am J Physiol Renal Physiol 282: F501–F505, 2002 [DOI] [PubMed] [Google Scholar]
- 36.Manygoats KR, Yazzie M, Stearns DM. Ultrastructural damage in chromium picolinate-treated cells: a TEM study. Transmission electron microscopy. J Biol Inorg Chem 7: 791–798, 2002 [DOI] [PubMed] [Google Scholar]
- 37.Matsumura H, Miyachi S. Cycling assay for nicotinamide adenine dinucleotides. Methods Enzymol 69: 465–470, 1980 [Google Scholar]
- 38.McCaskill ML, Romberger DJ, DeVasure J, Boten J, Sisson JH, Bailey KL, Poole JA, Wyatt TA. Alcohol exposure alters mouse lung inflammation in response to inhaled dust. Nutrients 4: 695–710, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mehta AJ, Yeligar SM, Elon L, Brown LA, Guidot DM. Alcoholism causes alveolar macrophage zinc deficiency and immune dysfunction. Am J Respir Crit Care Med 188: 716–723, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moss M, Bucher B, Moore FA, Moore EE, Parsons PE. The role of chronic alcohol abuse in the development of acute respiratory distress syndrome in adults. JAMA 275: 50–54, 1996 [PubMed] [Google Scholar]
- 41.Moss M, Guidot DM, Wong-Lambertina M, Ten HT, Perez RL, Brown LA. The effects of chronic alcohol abuse on pulmonary glutathione homeostasis. Am J Respir Crit Care Med 161: 414–419, 2000 [DOI] [PubMed] [Google Scholar]
- 42.Moss M, Steinberg KP, Guidot DM, Duhon GF, Treece P, Wolken R, Hudson LD, Parsons PE. The effect of chronic alcohol abuse on the incidence of ARDS and the severity of the multiple organ dysfunction syndrome in adults with septic shock: an interim and multivariate analysis. Chest 116: 97S–98S, 1999 [PubMed] [Google Scholar]
- 43.O'Keeffe BA, Cilia S, Maiyar AC, Vaysberg M, Firestone GL. The serum- and glucocorticoid-induced protein kinase-1 (Sgk-1) mitochondria connection: identification of the IF-1 inhibitor of the F1F0-ATPase as a mitochondria-specific binding target and the stress-induced mitochondrial localization of endogenous Sgk-1. Biochimie 95: 1258–1265, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Planes C, Blot-Chabaud M, Matthay MA, Couette S, Uchida T, Clerici C. Hypoxia and beta 2-agonists regulate cell surface expression of the epithelial sodium channel in native alveolar epithelial cells. J Biol Chem 277: 47318–47324, 2002 [DOI] [PubMed] [Google Scholar]
- 45.Pochynyuk O, Bugaj V, Vandewalle A, Stockand JD. Purinergic control of apical plasma membrane PI(4,5)P2 levels sets ENaC activity in principal cells. Am J Physiol Renal Physiol 294: F38–F46, 2008 [DOI] [PubMed] [Google Scholar]
- 46.Salviolia S, Ardizzoni A, Franceschi C, Cossarizza A. JC-1, but not DiOC6(3) or rhodamine 123, is a reliable fluorescent probe to assess DY changes in intact cells: implications for studies on mitochondrial functionality during apoptosis. FEBS Lett 411: 77–82, 1997 [DOI] [PubMed] [Google Scholar]
- 47.Song W, Liu G, Bosworth CA, Walker JR, Megaw GA, Lazrak A, Abraham E, Sullender WM, Matalon S. Respiratory syncytial virus inhibits lung epithelial Na+ channels by up-regulating inducible nitric-oxide synthase. J Biol Chem 284: 7294–7306, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spach PI, Herbert JS, Cunningham CC. The interaction between chronic ethanol consumption and oxygen tension in influencing the energy state of rat liver. Biochim Biophys Acta 1056: 40–46, 1991 [DOI] [PubMed] [Google Scholar]
- 49.Takemura Y, Goodson P, Bao HF, Jain L, Helms MN. Rac1-mediated NADPH oxidase release of O− regulates epithelial sodium channel (ENaC) activity in the alveolar epithelium. Am J Physiol Lung Cell Mol Physiol 298: L509–L520, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takemura Y, Helms MN, Eaton AF, Self J, Ramosevac S, Jain L, Bao HF, Eaton DC. Cholinergic regulation of epithelial sodium channels in rat alveolar type 2 epithelial cells. Am J Physiol Lung Cell Mol Physiol 304: L428–L437, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thomas J, Deetjen P, Ko WH, Jacobi C, Leipziger J. P2Y(2) receptor-mediated inhibition of amiloride-sensitive short circuit current in M-1 mouse cortical collecting duct cells. J Membr Biol 183: 115–124, 2001 [DOI] [PubMed] [Google Scholar]
- 52.Wodopia R, Ko HS, Billian J, Wiesner R, Bartsch P, Mairbaurl H. Hypoxia decreases proteins involved in epithelial electrolyte transport in A549 cells and rat lung. Am J Physiol Lung Cell Mol Physiol 279: L1110–L1119, 2000 [DOI] [PubMed] [Google Scholar]
- 53.Xu D, Guthrie JR, Mabry S, Sack TM, Truog WE. Mitochondrial aldehyde dehydrogenase attenuates hyperoxia-induced cell death through activation of ERK/MAPK and PI3K-Akt pathways in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol 291: L966–L975, 2006 [DOI] [PubMed] [Google Scholar]
- 54.Yeligar SM, Harris FL, Hart CM, Brown LA. Ethanol induces oxidative stress in alveolar macrophages via upregulation of NADPH oxidases. J Immunol 188: 3648–3657, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ying W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal 10: 179–206, 2008 [DOI] [PubMed] [Google Scholar]
- 56.Zhao Z, Hu X, Ross CW. Comparison of tissue preparation methods for assay of nicotinamide coenzymes. Plant Physiol 84: 987–988, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]