

Abstract
Glucagon-like peptide-1 (GLP-1) is a natural agonist for GLP-1R, a G protein-coupled receptor (GPCR) on the surface of pancreatic β cells. GLP-1R agoinsts are attractive for treatment of type 2 diabetes, but GLP-1 itself is rapidly degraded by peptidases in vivo. We describe a design strategy for retaining GLP-1-like activity while engendering prolonged activity in vivo, based on strategic replacement of native α residues with conformationally constrained β-amino acid residues. This backbone-modification approach may be useful for developing stabilized analogues of other peptide hormones.
Many vital physiological processes are modulated by G protein-coupled receptors (GPCRs) in the B family;1 their natural agonists are long polypeptide hormones (≥27 residues). Extensive efforts to develop small-molecule agonists or antagonists of these GPCRs have yielded little success, but peptide agonists have proven to be effective as therapeutic agents.2 Rapid proteolytic degradation of the natural hormones and functional analogues, however, can limit their utility. Here we describe the application of an unconventional strategy, backbone modification, for the development of protease-resistant analogues of glucagon-like peptide-1 (GLP-1) that retain full agonist potency and confer prolonged glucose control in vivo.
The C-terminal segments of B-family GPCR agonist peptides are α-helical when bound to their receptors.3 Our interest in these agonists arose from the discovery that information-bearing α-helices could be functionally mimicked with oligomers containing a combination of α- and β-amino acid residues (“α/β-peptides”).4 This finding has emerged from a broad effort to develop biopolymer-mimetic “foldamers” that differ from proteins or nucleic acids at the backbone level.5 Properly designed α/β-peptides retain the recognition properties of a prototype α-helical α-peptide, but the unnatural backbone confers protection from proteolysis. This approach is complementary to side chain cross-linking strategies that many groups have used for conformational and metabolic stabilization of bioactive α-helices.6−14 Cross-links have been generated via lactam formation, e.g., between Lys and Glu side chains,6−9,14 or with hydrocarbon connectors generated via alkene metathesis.10−13 Derivatives of several B-family GPCR agonists containing lactams in the C-terminal regions have been reported,6,7,9 including examples based on GLP-1.14 However, the cross-links themselves can make unintended contacts with partner proteins,15 and the extent of proteolytic stabilization may be limited. These factors motivated our interest in a backbone-focused rather than side-chain-focused approach.
Only a few prior studies have explored α → β replacement in B-family GPCR agonists, with mixed results. Replacements at one to three positions in the central portion of a parathyroid hormone receptor-1 (PTHR1) agonist were largely deleterious.16 Subsequent efforts to replace the entire C-terminal segment of a GLP-1R agonist with a β-peptide segment led to a million-fold decline in potency.17 However, we have recently shown that as many as seven α → β replacements are tolerated in the C-terminal portion of PTH(1–34), which is the osteoporosis drug teriparatide, without loss of PTHR1 agonist potency.18 In this case, α residues were replaced with their β3 homologues.
The present studies began with GLP-1(7–37)-NH2, which displays full activity at GLP-1R19 and can be viewed as a hybrid of the two natural hormone forms, GLP-1(7–36)-NH2 and GLP-1(7–37).20 (By convention, the N-terminal residue of native GLP-1 is designated position 7.) GLP-1(7–37)-NH2 has been used for systematic evaluation of i,i+4 side chain lactam placement;14 lactams involving positions 18, 22, 26, and 30 retain high potency, which is consistent with structural evidence indicating that side chains at these positions do not make direct contact with the receptor.3b Since the αααβ backbone pattern supports α-helix mimicry,4b,4e we made α → β3 modifications at positions 26, 30, and 34 (1, Figure 1). This compound maintains the side chain sequence of the prototype α-peptide, but each β3 residue contains an additional backbone CH2 unit relative to the original α residue. α/β-Peptide 1 was compared to GLP-1(7–36)-NH2 for the ability to activate human GLP-1R using a previously described cell-based assay in which agonist-stimulated cAMP production is monitored (Table 1).14b,21 GLP-1 is very potent (EC50 = 1.6 ± 0.2 nM), but analogue 1 was much less active (EC50 > 100 nM; modest cAMP production at 100 nM). This observation contrasts sharply with our recent finding that analogues of PTH(1–34) containing multiple α → β3 replacements retain native-like potency.18
Figure 1.
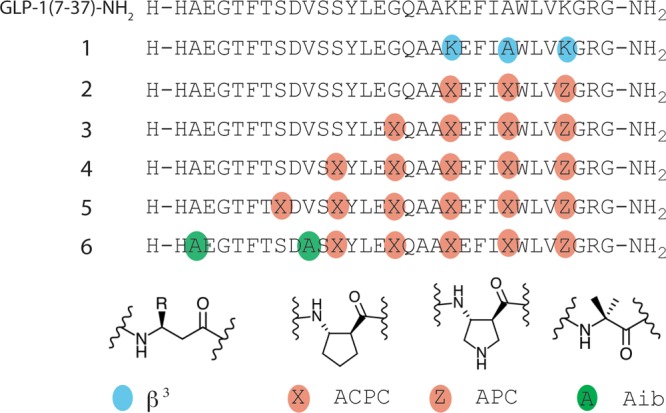
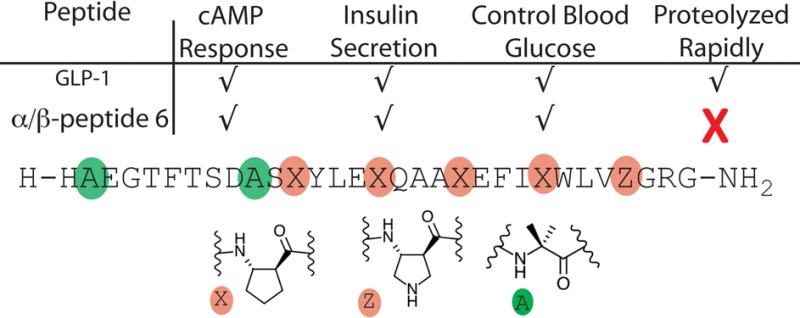
GLP-1(7–37)-NH2 and α/β-peptide analogues. Each molecule has a free N-terminus and a primary amide at the C-terminus. The single-letter code identifies proteinogenic α residues. Colored circles indicate positions of other types of residues, including the nonproteinogenic α residue Aib (green), β3 homologues of proteingenic α residues (blue), and ring-constrained β residues (tan; X = ACPC, Z = APC).
Table 1. GLP-1R Activation, Monitored by cAMP Production for GLP-1(7-36)-NH2 and Analogues.
| EC50 (nM) | max response (% GLP-1 max) | |
|---|---|---|
| GLP-1 | 1.6 ± 0.2 | 100 |
| 1 | >100 | – |
| 2 | 0.7 ± 0.1 | 100 |
| 3 | 2.0 ± 0.1 | 85 |
| 4 | 4.0 ± 0.5 | 103 |
| 5 | >100 | – |
| 6 | 3.2 ± 0.3 | 88 |
When α → β3 modifications within an α-helix-forming sequence weaken binding to a target protein, affinity can sometimes be restored via use of ring-constrained β residues such as ACPC or APC (Figure 1, X and Z).4c This trend is attributed to the extra degree of torsional flexibility within a β3 residue relative to the homologous α residue, which presumably increases the entropic cost of helix formation.22 We prepared α/β-peptide 2, an analogue of 1 containing three cyclic β residues, in an effort to improve agonist activity. Lys34 of GLP-1 was replaced with APC in 2 to maintain the cationic charge, but Ala30 and Lys26 were replaced with uncharged ACPC. Since Lys26 can be used to generate potent lactam derivatives of GLP-1,14a we concluded that cationic charge was not necessary at this position. α/β-Peptide 2 proved to be a full agonist of GLP-1R with native-like potency.
We extended the αααβ pattern toward the N-terminus via progressive introduction of ACPC residues in place of Gly22, Ser18, and Ser14. The α/β-peptides containing four or five α → β modifications (3 and 4) were full agonists of GLP-1R with activities comparable to that of GLP-1. In contrast, introduction of a sixth α → β replacement drastically reduced the activity (α/β-peptide 5). Ser14 → Ala modification is reported not to affect GLP-1 potency,23 so loss of the native side chain at this position, in 5, does not explain this α/β-peptide’s lack of agonist activity. Because the receptor-bound conformation of the N-terminal segment of GLP-1 is unknown,3b the origin of this dramatic impact of α → ACPC replacement is difficult to explain.
GLP-1 is rapidly degraded in vivo.24 Proteolysis is mediated by at least two enzymes, dipeptidyl peptidase-4 (DPP-4),24a which specifically cleaves after Ala8, and neprilysin (NEP 24.11), which cleaves after Asp15, Ser18, Tyr19, Glu27, Phe28, and Trp31.24b Based on previous observations,4 we anticipated that the β residues in 4 would suppress neprilysin action at all sites except perhaps the one closest to the N-terminus (Asp15-Val16). Replacement of Ala8 of GLP-1with Aib suppresses DPP-4 activity without affecting GLP-1R agonist activity,25 and we predicted that replacement of Val16 with Aib would exert a comparable suppression of cleavage at the adjacent neprilysin site without affecting potency at GLP-1R (Supplementary Figure 18).
α/β-Peptide 6, which contains the five α → cyclic β backbone modifications of 4 along with Aib modifications at positions 8 and 16, proved to be a full agonist of GLP-1R, with native-like potency. In addition, this α/β-peptide analogue of GLP-1(7–37)-NH2 is highly resistant to in vitro degradation by the enzymes that cleave GLP-1 itself (Supplementary Figures 13, 14, and 16). No cleavage of 6 by DPP-4 could be detected over 7 days under conditions that result in a 13.5 min half-life of GLP-1(7–37)-NH2, as expected.25 More significantly, 6 displayed a half-life of 83 h in the presence of neprilysin under conditions14b that lead to a 20 min half-life of GLP-1.
A critical physiological role of GLP-1 is to augment glucose-stimulated insulin secretion (GSIS) from pancreatic islet β cells. We compared α/β-peptide 6 and GLP-1(7–36)-NH2 for the ability to promote GSIS from freshly isolated mouse islets (Figure 2). Administration of both compounds resulted in a dose-dependent increase in GSIS, yielding ∼2-fold greater insulin secretion at 90 nM agonist (p < 0.03). It is noteworthy that the dose-dependence observed for the α/β-peptide matched that of GLP-1, which is consistent with the similarity in EC50 values for activation of GLP-1R reported in Table 1. Residual islet insulin content values were not significantly different between islets treated with GLP-1 and those treated with 6 (Supplementary Figure 19), which indicates that the effect of the α/β-peptide is not simply due to GLP-1 release via damaging of β cell membranes.
Figure 2.

α/β-Peptide 6 and GLP-1 are equally effective insulin segretagogues. Insulin secretion was monitored from primary mouse islets in response to low glucose alone (3 mM, ∇) or high glucose (HG, 16 mM) in the absence (▼) or presence of varying concentrations of the α/β-peptide 6 (○) or GLP-1(7–36)-NH2 (●). Measurements show the mean of five independent experiments (±SEM). Secretion is plotted as the percentage of total insulin content per islet. *, P < 0.05 for α/β-peptide 6 or GLP-1(7–36)-NH2 vs HG alone.
We evaluated α/β-peptide 6in vivo via glucose tolerance tests (GTT) (Figure 3). In addition to the α/β-peptide and GLP-1(7–37)-NH2, these studies included exendin-4 (39 residues). All three compounds were tested for the ability to normalize circulating glucose levels. For mice injected with vehicle rather than peptide (negative control), the subsequent intraperitoneal glucose challenge caused a rapid rise in blood glucose concentration that peaked at 30 min (Figure 3A). Mice injected with GLP-1, exendin-4, or α/β-peptide 6 at 1 mg/kg showed a dramatic suppression in the rise of blood glucose concentration relative to vehicle-treated mice during the GTT; the three compounds were equally effective at this dose. Dose–response behavior was observed for 6, with glucose control maintained at 0.1 mg/kg, but not at 0.01 mg/kg (Figure 3B).
Figure 3.

α/β-Peptide 6 demonstrates long-lasting improvement of in vivo glucose tolerance. (A) Plasma glucose values during a glucose tolerance test (GTT) for mice treated with GLP-1(7–37)-NH2 (1 mg/kg), exendin-4 (Ex-4, 1 mg/kg), varying doses (0.01 to 1 mg/kg) of α/β-peptide 6 or vehicle. Upward arrow indicates timing of the peptide treatments delivered via IP injection. Results show mean (±SEM) of 4 separate mice per condition. (B) Average area under the curve (AUC) values for the GTT data shown in part A. (C) Plasma glucose values at 30 min following a second GTT that was conducted 5 h following that shown in A. *, P < 0.05 vs vehicle.
The GTT was repeated 5 h after agonist administration (Figure 3C). Mice treated with GLP-1(7–37)-NH2 showed no significant difference from those treated with vehicle 30 min after the second glucose challenge; this result is expected based on the rapid enzymatic inactivation of GLP-1 in vivo. In contrast, the glucose-lowering effect of exendin-4 was maintained at 5 h, presumably because exendin-4 is not cleaved by DPP-4 and is only very slowly degraded by neprilysin or other peptidases.26 α/β-Peptide 6 exerted a glucose-lowering effect at 5 h comparable to that of exendin-4 (each at 1 mg/kg). The ability of 6 to control blood glucose levels at 5 h was manifested even at 0.1 mg/kg. These results presumably reflect the resistance of the α/β-peptide to proteolysis.
Our results show that a GLP-1-derived oligomer containing multiple replacements of α-amino acid residues with β residues can maintain native-like agonist activity and mimic hormone function in vivo. Replacement of native α residues with flexible β3-homologues proved to be highly deleterious to agonist activity (α/β-peptide 1), perhaps because of β3 residue flexibility; however, use of β residues preorganized to support an α-helix-like conformation provided potent GLP-1R agonists. These findings reveal a striking contrast between two B-family GPCRs, because we previously found that potent PTHR1 agonist activity could be maintained after multiple α → β3 replacements,18 while the present study shows that GLP-1R agonist potency requires constrained β residues.
Despite the variations in preferred β residue type between GLP-1R and PTHR1, our successful implementations of the α → β replacement strategy with both receptors suggest that this approach may offer a general route to highly potent B-family GPCR agonists that display favorable pharmacokinetic profiles in vivo. The backbone-modification strategy can be readily implemented because the necessary α/β-peptides are accessible via conventional solid-phase synthesis, and many protected β-amino acids are commercially available.
Acknowledgments
This work was supported by the U.S. National Institutes of Health (GM056414 (S.H.G.), DK58037 (A.D.A.), and DK56593 (A.D.A.)) and the American Diabetes Association (ADA #7-12-IN-38 (A.S.)). L.M.J. and M.V.H. were supported in part by a Chemistry-Biology Interface Training Grant from NIGMS (T32 GM008505).
Supporting Information Available
Experimental procedures, peptide synthesis and purification, proteolysis assays, additional receptor activation assays, and insulin content measurements. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interests: L.M.J., M.P.K., A.D.D., A.S., and S.H.G. are co-inventors on a patent application covering the GLP-1 analogues reported here. S.H.G. is a co-founder of Longevity Biotech, Inc., which is pursuing biomedical applications of α/β-peptides.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Fredriksson R.; Lagerstrom M. C.; Lundin L. G.; Schioth H. B. Mol. Pharmacol. 2003, 63, 1256–72. [DOI] [PubMed] [Google Scholar]; b Lagerstrom M. C.; Schioth H. B. Nat. Rev. Drug Discovery 2008, 7, 339–57. [DOI] [PubMed] [Google Scholar]
- a Davidson M. B.; Bate G.; Kirkpatrick P. Nat. Rev. Drug Discovery 2005, 4, 713–4. [DOI] [PubMed] [Google Scholar]; b Drucker D. J.; Dritselis A.; Kirkpatrick P. Nat. Rev. Drug Discovery 2010, 9, 267–8. [DOI] [PubMed] [Google Scholar]
- a Hollenstein K.; de Graaf C.; Bortolato A.; Wang M.-W.; Marshall F. H.; Stevens R. C. Trends Pharmacol. Sci. 2014, 35, 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Underwood C. R.; Garibay P.; Knudsen L. B.; Hastrup S.; Peters G. H.; Rudolph R.; Reedtz-Runge S. J. Biol. Chem. 2010, 285, 723–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Horne W. S.; Boersma M. D.; Windsor M. A.; Gellman S. H. Angew. Chem., Int. Ed. 2008, 47, 2853–6. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Horne W. S.; Price J. L.; Gellman S. H. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 9151–6. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Horne W. S.; Johnson L. M.; Ketas T. J.; Klasse P. J.; Lu M.; Moore J. P.; Gellman S. H. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 14751–6. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Johnson L. M.; Mortenson D. E.; Yun H. G.; Horne W. S.; Ketas T. J.; Lu M.; Moore J. P.; Gellman S. H. J. Am. Chem. Soc. 2012, 134, 7317–20. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Boersma M. D.; Haase H. S.; Peterson-Kaufman K. J.; Lee E. F.; Clarke O. B.; Colman P. M.; Smith B. J.; Horne W. S.; Fairlie W. D.; Gellman S. H. J. Am. Chem. Soc. 2012, 134, 315–23. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Johnson L. M.; Gellman S. H. Meth. Enzymol. 2013, 523, 407–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Azzarito V.; Long K.; Murphy N. S.; Wilson A. J. Nat. Chem. 2013, 5, 161–73. [DOI] [PubMed] [Google Scholar]; b Guichard H.; Huc I. Chem. Commun. 2011, 47, 5933–41. [DOI] [PubMed] [Google Scholar]; c Horne W. S.; Gellman S. H. Acc. Chem. Res. 2008, 41, 1399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Goodman C. M.; Choi S.; Shandler S.; DeGrado W. F. Nat. Chem. Biol. 2007, 3, 252–62. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Yin H.; Hamilton A. D. Angew. Chem., Int. Ed. 2005, 44, 4130–63. [DOI] [PubMed] [Google Scholar]
- Felix A. M.; Heimer E. P.; Wang G. T.; Lambros T. J.; Fournier A. J.; Mowles T. F.; Maines S.; Campbell R. M.; Wegrzynski B. B.; Toomer V.; Fry D.; Madison V. S. Int. J. Peptide Protein Res. 1988, 21, 441–54. [DOI] [PubMed] [Google Scholar]
- Chorev M.; Roubini E.; McKee R. L.; Gibbons S. W.; Goldman M. E.; Caufield M. P.; Rosenblatt M. Biochemistry 1991, 30, 5968–74. [DOI] [PubMed] [Google Scholar]
- Judice J. K.; Tom J. Y. K.; Huang W.; Wrin T.; Vennari J.; Petropoulos C. J.; McDowell R. S. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 13426–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi D.; Lin Y.; Ahn J. M.; Siegel M.; Mollova N. N.; Schram K. H.; Hruby V. J. J. Med. Chem. 2000, 43, 1714–22. [DOI] [PubMed] [Google Scholar]
- Blackwell H. E.; Grubbs R. H. Angew. Chem., Int. Ed. 1998, 37, 3281–4. [DOI] [PubMed] [Google Scholar]
- Walensky L. D.; Kung A. L.; Escher I.; Malia T. J.; Barbuto S.; Wright R. D.; Wagner G.; Verdine G. L.; Korsmeyer S. J. Science 2004, 305, 1466–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman R. N.; Dimartino G.; Arora P. S. J. Am. Chem. Soc. 2004, 126, 12252–3. [DOI] [PubMed] [Google Scholar]
- Okamoto T.; Zobel K.; Fedorova A.; Quan C.; Yang H.; Fairbrother W. J.; Huang D. C. S.; Smith B. J.; Keshayes K.; Czabotar P. E. ACS Chem. Biol. 2013, 8, 297–302. [DOI] [PubMed] [Google Scholar]
- a Miranda L. P.; Winters K. A.; Gegg C. V.; Patel A.; Aral J.; Long J.; Zhang J.; Diamond S.; Guido M.; Stanislaus S.; Ma M.; Li H.; Rose M. J.; Poppe L.; Veniant M. M. J. Med. Chem. 2008, 51, 2758–65. [DOI] [PubMed] [Google Scholar]; b Murage E. N.; Gao G. Z.; Bisello A.; Ahn J. M. J. Med. Chem. 2010, 53, 6412–20. [DOI] [PubMed] [Google Scholar]
- a Stewart M. L.; Fire E.; Keating A. E.; Walensky L. D. Nat. Chem. Biol. 2010, 6, 595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Phillips C.; Roberts L. R.; Schade M.; Bazin R.; Bent A.; Davies N. L.; Moore R.; Pannifer A. D.; Pickford A. R.; Prior S. H.; Read C. M.; Scott A.; Brown D. G.; Xu B.; Irving S. L. J. Am. Chem. Soc. 2011, 133, 9696–9. [DOI] [PubMed] [Google Scholar]; c Baek S.; Kutchukian P. S.; Verdine G. L.; Huber R.; Holak T. A.; Lee K. W.; Popowicz G. M. J. Am. Chem. Soc. 2012, 134, 103–6. [DOI] [PubMed] [Google Scholar]
- a Peggion E.; Mammi S.; Schievano E.; Silvestri L.; Schiebler L.; Bisello A.; Rosenblatt M.; Chorev M. Biochemistry 2002, 41, 8162–8175. [DOI] [PubMed] [Google Scholar]; b Schievano E.; Mammi S.; Carretta E.; Fiori N.; Corich N.; Bisello A.; Rosenblatt M.; Chorev M.; Peggion E. Biopolymers 2003, 70, 534–547. [DOI] [PubMed] [Google Scholar]
- Denton E. V.; Craig C. J.; Pongratz R. L.; Appelbaum J. S.; Doerner A. E.; Narayanan A.; Shulman G. I.; Gline G. W.; Schepartz A. Org. Lett. 2013, 15, 5318–5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheloha R. W.; Maeda A.; Dean T.; Gardella T. J.; Gellman S. H. Nat. Biotechnol. 2014, 32, 653–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Menthiere C. S.; Chavanieu A.; Grassy G.; Dalle S.; Salazar G.; Kervran A.; Pfieiffer B.; Renard P.; Delagrange P.; Manechez D.; Bakes D.; Ktorza A.; Calas B. Eur. J. Med. Chem. 2004, 39, 473–80. [DOI] [PubMed] [Google Scholar]
- Hansen L.; Deacon C. F.; Orskov C.; Holst J. J. Endocrinology 1999, 140, 5356–63. [DOI] [PubMed] [Google Scholar]
- Salomon Y.; Londos C.; Rodbell M. Anal. Biochem. 1974, 58, 541–8. [DOI] [PubMed] [Google Scholar]
- Price J. L.; Hadley E. B.; Steinkruger J. D.; Gellman S. H. Angew. Chem., Int. Ed. 2010, 49, 368–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adelhorst K.; Hedegaard B. B.; Knudsen L. B.; Kirk O. J. Biol. Chem. 1994, 269, 6275–8. [PubMed] [Google Scholar]
- a Jessen L.; Aulinger B. A.; Hassel J. L.; Roy K. J.; Smith E. P.; Greer T. M.; Woods S. C.; Seeley R. J.; D’Alessio D. A. Endocrinology 2012, 153, 5735–45. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hupe-Sodmann K.; McGregor G. P.; Bridenbaugh R.; Goke R.; Goke B.; Thole H.; Zimmermann B.; Voigt K. Regul. Pept. 1995, 58, 149–56. [DOI] [PubMed] [Google Scholar]
- Deacon C. F.; Knudsen L. B.; Madsen K.; Wiberg F. C.; Jacobsen O.; Holst J. J. Diabetologia 1998, 41, 271–8. [DOI] [PubMed] [Google Scholar]
- a Gao W.; Jusko W. J. Drug Metab. Dispos. 2012, 40, 990–7. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Parkes D.; Jodka C.; Smith P.; Nayak S.; Rinehart L.; Gingerich R.; Chen K.; Young A. Drug Dev. Res. 2001, 53, 260–7. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.