

Background: SMC proteins organize chromosome architecture, whereas microtubules form the structural framework for chromosome movement in mitosis.
Results: SMC proteins bind to and stabilize microtubules to promote proper mitotic spindle formation.
Conclusion: SMC proteins can provide a functional link between microtubules and DNA to ensure faithful chromosome segregation.
Significance: SMC-microtubule interactions are essential to establish a robust system for maintaining genome integrity.
Keywords: Cell Division, Chromosomes, Microtubule, Mitosis, Mitotic Spindle, SMC Proteins
Abstract
Structural maintenance of chromosome (SMC) proteins are key organizers of chromosome architecture and are essential for genome integrity. They act by binding to chromatin and connecting distinct parts of chromosomes together. Interestingly, their potential role in providing connections between chromatin and the mitotic spindle has not been explored. Here, we show that yeast SMC proteins bind directly to microtubules and can provide a functional link between microtubules and DNA. We mapped the microtubule-binding region of Smc5 and generated a mutant with impaired microtubule binding activity. This mutant is viable in yeast but exhibited a cold-specific conditional lethality associated with mitotic arrest, aberrant spindle structures, and chromosome segregation defects. In an in vitro reconstitution assay, this Smc5 mutant also showed a compromised ability to protect microtubules from cold-induced depolymerization. Collectively, these findings demonstrate that SMC proteins can bind to and stabilize microtubules and that SMC-microtubule interactions are essential to establish a robust system to maintain genome integrity.
Introduction
During cell division, genomic DNA must be partitioned to daughter cells in a highly regulated fashion to ensure the maintenance of genome integrity from each generation to the next. High fidelity segregation of eukaryotic genomes requires precise coordination between the multiple components of the cellular segregation machinery. For instance, replicated sister chromatids must be connected to each other and reconfigured into highly condensed chromosomes to allow effective segregation in anaphase (1, 2). This reorganization of chromatin depends in large part on a family of proteins known as the structural maintenance of chromosome (SMC)6 proteins (2–4). SMC proteins assemble into large multisubunit complexes (cohesin, condensin, and the Smc5-Smc6 complex) that bind to chromatin in mitosis, where they promote the assembly of functional chromosomes. Once mitotic chromosomes are assembled, they become competent for attachment to a microtubule-based structure, the mitotic spindle, and for subsequent segregation in anaphase.
The microtubule-based cytoskeleton also undergoes dramatic changes and intricate regulation during the cell cycle. Microtubules switch from providing a structural role for the maintenance of cell shape and for intracellular transport in interphase to building the underlying framework for chromosome segregation during mitosis. Mitotic chromosome formation and spindle assembly are often considered to be two precisely coordinated but separate events because proteins that are involved in chromatin organization and remodeling are often distinct from those that regulate cytoskeletal structures. Prime examples include histones, which are not linked to any polymer structures other than chromatin. Similarly, many known microtubule-associated proteins, including the XMAP215 family of TOG domain-containing proteins and the EB family of proteins, are not known to interact with chromatin (5, 6), and yet, during cell division, the mitotic spindle microtubules need to make contact with the chromosomes in order to move and segregate them to opposite poles of the cell. Therefore, proteins linking chromosomes to the spindle apparatus are of great interest because they can provide fundamental insights into the mechanisms responsible for high fidelity genome segregation in eukaryotes. Microtubule-chromosome connections are thought to be mediated in part through the kinetochore (7–9), a large multi-protein complex assembled on the centromeric region of chromosomes. Microtubules can also make direct contact with chromosome arms via proteins endowed with dual affinity for chromatin and microtubules (e.g. motor proteins of the chromokinesin family (10–12)). It is currently unclear how many different protein families can mediate direct interaction between chromatin and microtubules and what their specific contributions are to high fidelity chromosome segregation.
There have been a number of proteomic studies on microtubule-interacting proteins reported in the literature (13–16). Interestingly, some chromatin-binding proteins, most notably SMC family members, were consistently identified as microtubule-interacting proteins in such studies (13, 15). The fact that these microtubule interactomes are devoid of other abundant chromatin-associated proteins, such as histones, suggests that the SMC family members identified in these studies are not simply contaminants or artifacts. This raises the possibility that SMC proteins directly bind to microtubules, an unexpected property for this family of chromatin-binding proteins. However, formal proof of this possibility is still lacking. Given the biological significance of proteins linking microtubules and chromatin, we set out to determine whether SMC proteins interact directly with microtubules and to understand the potential roles and implications of these interactions in vivo.
EXPERIMENTAL PROCEDURES
Protein Purification
Full-length Smc5-STH was expressed and purified in yeast, as described previously (17). Smc5 fragments were expressed in bacteria from pET-series vectors (Novagen) and purified by nickel chelate and/or Strep-Tactin chromatography (Qiagen), according to the manufacturers' suggested conditions. The condensin complex containing Brn1, Smc2, Smc4, Ycg1, and Ycs4 subunits was purified from budding yeast by sequential nickel chelate, Strep-Tactin, and gel exclusion chromatography, as described previously (18). All proteins were purified to ≥95% purity, as judged by Coomassie Blue staining. Purification of bovine tubulin and the labeling of tubulin with X-Rhodamine dye were performed as described (19).
Microtubule Binding Experiments
Co-sedimentation Assay
The co-sedimentation assay was performed as described previously with some modifications (20). Briefly, microtubules were prepared by polymerizing 25–40 μm tubulin in 1× BRB80 (80 mm PIPES, 1 mm EGTA, 1 mm MgCl2), 1 mm GTP, 1 mm DTT in the presence of 10% DMSO at 37 °C for 30 min and then diluted to 10 or 20 μm working stocks in 1× BRB80 with 20 μm paclitaxel. Binding reactions were done by mixing SMC proteins with microtubules in 1× BRB80 supplemented with the indicated salt concentrations (75–125 mm NaCl), 1 mm DTT, 0.01% Tween 20, and 20 μm paclitaxel. After a 20-min incubation, mixtures were spun at 45,000–60,000 rpm in a Sorvall S120AT3 rotor at 25 °C for 5 min. Samples from supernatant and pellet fractions were resolved by SDS-PAGE. Gels were stained by Coomassie Blue and scanned with a digital scanner. Band intensities were quantified using ImageJ (NIH). Kd measurements were done as described previously (20).
Microscopy-based Microtubule Binding Assays
Microscopy-based microtubule binding assays were performed as described previously (20). For detection of the microtubule-Smc5-DNA ternary complex, samples of Smc5-bound X-Rhodamine-labeled microtubules were first generated by mixing of the two components at a molar ratio of ∼1:4 (Smc5/tubulin dimer) and then attached them onto a tubulin antibody-coated (rat α-tubulin antibody from Serotec, at 10 ng/ml) coverslip in a flow chamber. Fluorescently labeled DNA (6-carboxyfluorescein (FAM)-labeled 60-bp single-stranded DNA, Integrated DNA Technologies) was then introduced into the chamber. After a 5-min incubation, unbound DNA was flushed out, and images of the bound DNA and microtubules were visualized and recorded on a Zeiss Axio-Imager Z1 microscope equipped with a 1.4 numerical aperture Plan-APOCHROMAT 63× objective using the Zeiss AxioVision release 4.8 software. The TIFF files of the acquired images were then processed by MetaMorph (Molecular Devices, LLC).
Image Analysis for Microtubule-Smc5-DNA Interactions
The quantitative analysis of the microtubules and the associated FAM-labeled DNA was done using MetaMorph® (Molecular Devices). To identify microtubules, the images of the X-Rhodamine-labeled microtubules for the no Smc5 control were first thresholded to a signal intensity that would allow the detection of individual microtubules in the control image while minimizing background noise. The objects were identified using the “integrated morphometry analysis” functionality with exclusion of objects that touched the image edge and those with an area smaller than 50 square pixels (non-microtubule signals or X-Rhodamine aggregates). The same thresholding and exclusion filtering were applied to the acquired images with Smc5-WT and Smc5-3KE. The average area and average signal intensity of all of the objects were obtained automatically using the integrated morphometry analysis. A conservative cut-off of area occupied by microtubules was set to distinguish single microtubules from bundles. Object identification was verified by visual inspection. To analyze the extent of the FAM-oligonucleotide binding to microtubules, regions were created around the identified objects in the microtubule images and were transferred onto the corresponding images containing the FAM signal. This allows the determination of the average signal intensity within the microtubule delineated areas. For all calculations, the average background signal intensities (from unoccupied areas) were subtracted from the average signal intensity of microtubules and FAM, respectively. Using this analytical method, the average area occupied by single microtubules in the control, the average number of microtubules within bundles and the FAM intensity per microtubule were calculated.
Determination of Candidate Microtubule Binding Sites
To prepare biotin-labeled Smc5 protein that has impaired microtubule binding ability, purified Smc5 proteins were chemically modified in the presence of 0.1–0.5 mm NHS-LC-biotin (Pierce/Thermo Fisher). Biotinylation reactions were performed at room temperature for 5 min and quenched by the addition of potassium glutamate. Modification was verified by streptavidin-HRP Western blot (streptavidin-HRP from Pierce/Thermo Fisher, 1:2000 dilution). Biotinylated Smc5 proteins were allowed to bind to microtubules and subjected to a sedimentation assay. This allows the separation of the fraction that associated with microtubules (pellet) and the fraction that remains in the supernatant (enriched for protein with biotinylated sites that interfere with binding). Optimal biotinylation and microtubule binding conditions were determined experimentally. Smc5 proteins in the supernatant fraction and the pellet fraction were analyzed by mass spectrometry. Intensities of the trypsin-digested peptides were normalized and compared between the two fractions. Biased enrichment of any biotinylated peptides in the supernatant fraction indicated lysine sites that were potentially essential for microtubule binding.
Cold-induced Microtubule Depolymerization Assay
The assay was performed as previously described (21). Briefly, stock microtubules were diluted 10-fold in 1× BRB80 with 2 μm paclitaxel and 75 mm NaCl either in the absence or presence of the Smc5 proteins. The mixtures were then chilled to 2 °C in a PCR machine and incubated for 30 min to induce microtubule depolymerization while the control sample was placed at room temperature. The samples were then subjected to ultracentrifugation to separate the depolymerized tubulin dimers from microtubules and processed as described above in the co-sedimentation assay.
Yeast Genetics and Growth Conditions
Yeast culture and genetic modifications were performed according to St-Pierre et al. (18). The genotypes of yeast strains used in this study are described in Table 1. smc5 mutants defective in microtubule-binding residues were created by site-directed mutagenesis (QuikChange, Stratagene) and integrated into the yeast genome, as described previously (17). The presence of the appropriate mutations in the yeast genome was confirmed by sequencing the SMC5 locus. Cell proliferation at various temperatures and/or in the presence of DNA-damaging agents was determined by spotting 5-fold dilution series of yeast cultures on solid medium containing the appropriate supplements and/or drugs. Cells were grown until individual colonies became visible on the surface of the solid media (2–3 days between 23 and 37 °C; 4–5 days at 18 °C). A previously characterized temperature-sensitive mutant in the SMC5 gene, smc5-6, was included in some experiments as a control for the effect associated with losing Smc5-Smc6 complex function (22).
TABLE 1.
Yeast strains used in this study
| Name | Relevant genotype |
|---|---|
| D1 | MATa SMC5 |
| D3 | MATa/MATα SMC5/SMC5 SMC6/SMC6 |
| D224 | MATa smc5-6 |
| D253 | MATa tub2-401 |
| D3465 | MATa smc5-950-STH::HIS3MX6 |
| D4052 | MATa SMC5-STH::HIS3MX6 |
| D4119 | MATa smc5-950-STH::HIS3MX6 tub2-401 |
| D4121 | MATa SMC5-STH::HIS3MX6 tub2-401 |
| D4134 | MATa SMC5-STH::HIS3MX6 mad1::URA3 |
| D4136 | MATa smc5-950::HIS3MX6 mad1::URA3 |
| D4240 | MATa/MATα SMC5-STH::HIS3MX6/SMC5 |
| D4241 | MATa/MATα smc5-950-STH::HIS3MX6/SMC5 |
| D4258 | MATa/MATα SMC5-STH::HIS3MX6/SMC5 SMC6-13MYC::HIS3MX6/SMC6 |
| D4259 | MATa/MATα smc5-950-STH::HIS3MX6/SMC5 SMC6-13MYC::HIS3MX6/SMC6 |
Yeast Cell Cycle Experiments
Cell cultures were arrested in the G1 phase of the cell cycle using α-factor (50 ng/ml for 180 min) and synchronously released from this arrest at 18 °C. Samples were taken at intervals and processed for microscopic characterization of yeast bud, spindle, and nuclear morphology, as described previously (23). Yeast images were acquired on a Leica DM5500B microscope equipped with an EM-CCD camera (Hamamatsu). Volocity software (PerkinElmer Life Sciences) was used to acquire images. Brightness and contrast adjustments were applied to primary micrographs using Photoshop software (Adobe). During time course experiments, yeast culture samples were also processed for determination of DNA content by flow cytometry using published procedures (23).
Immunoprecipitation and Western Blotting
Cell lysates were prepared in lysis buffer (50 mm Tris-HCl, pH 7.5, 100 mm KCl, 100 mm NaF, 10% glycerol, 0.1% Tween 20, 1 mm DTT, 10 μm 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride, 10 μm pepstatin A, 10 μm E-64), as described previously (18). During the immunoprecipitation process, 5 μg of anti-Myc 9E-10 (GenTex) or anti-Strep-tagII (Qiagen) were allowed to bind to GammaBind Plus Sepharose beads (GE Healthcare). Protein samples were resolved by electrophoresis on 8% polyacrylamide gels for 16 h and then transferred onto polyvinylidene difluoride (PVDF) membranes. For Western blotting, antibodies were used at the following concentrations: mouse monoclonal anti-Strep-tagII (Qiagen, 1:1000), mouse monoclonal anti-PGK-22C5 (Abcam, 1:1000), mouse monoclonal anti-Myc 9E-10 (GenTex, 1:1000).
Detection of Smc5-Microtubule Interactions in Yeast
Proximity ligation assay (PLA) experiments were performed as described previously (20), with minor modifications. Briefly, images were acquired on a DeltaVision microscope (Olympus), controlled by softWoRx software (Applied Precision). Antibodies were used at the following concentrations: anti-α-tubulin (DM1α, Sigma) at 1:1000; anti-Streptag (GenScript) at 1:400; Alexa-Fluor 488-conjugated secondary antibody (Molecular Probes) at 1:500.
RESULTS
Smc5 Binds Directly to Microtubules in Vitro
To test the possibility that SMC proteins bind to microtubules directly, we first tested the purified condensin complex in a microtubule co-sedimentation assay and found it to be associated with the polymers. Indeed, Fig. 1A shows that the five subunits of the yeast condensin complex (i.e. Brn1, Smc2, Smc4, Ycg1, and Ycs4) (18) are enriched in the pellet fraction containing microtubules after sedimentation. We next sought to determine whether this microtubule-binding behavior is also typical of other SMC complexes. Specifically, we focused on Smc5 because this protein can be purified in recombinant form (17). Smc5 is a member of the Smc5-Smc6 complex related structurally to the condensin complex but completely distinct in subunit composition. The fact that it can be purified in monomeric form enabled us to determine whether SMC subunits are directly responsible for the interaction with microtubules within their own complexes. We found that Smc5 also associated with microtubules in the co-sedimentation assay, indicating a direct interaction between these proteins (Fig. 1B). Judging by the observation that more than half of the SMC proteins associated with microtubules (at 0.5 μm tubulin dimer concentration) in these experiments, we concluded that SMC-microtubule interaction was of high affinity, with a dissociation constant (kd) of about or below 0.5 μm. It should be noted that the ionic strength of the buffer condition used in these experiments (80 mm PIPES with ∼125 mm NaCl) was much higher than those used traditionally (ranging typically from 12 to 80 mm PIPES or 20 mm sodium phosphate with 0–50 mm KCl or NaCl) (24–26). Although the condensin complex is an ATPase (2), its association with microtubules did not appear to be affected by the presence of different nucleotides (ADP, ATP, or the non-hydrolyzable ATP analog AMP-PNP). The condensin complex did, however, appear to bind more to microtubules in the absence of nucleotide (Fig. 1C). This is distinct from the nucleotide dependence of many prototypical kinesin motor proteins, where ATP hydrolysis or ADP releases motors from microtubules (24, 27). Interestingly, although Smc5 is capable of hydrolyzing ATP as a monomer (17), possible differences in the levels of ATPase activity present in the purified Smc5 and condensin fractions may explain their differential sensitivity to nucleotides in microtubule-binding assays (Fig. 1C).
FIGURE 1.
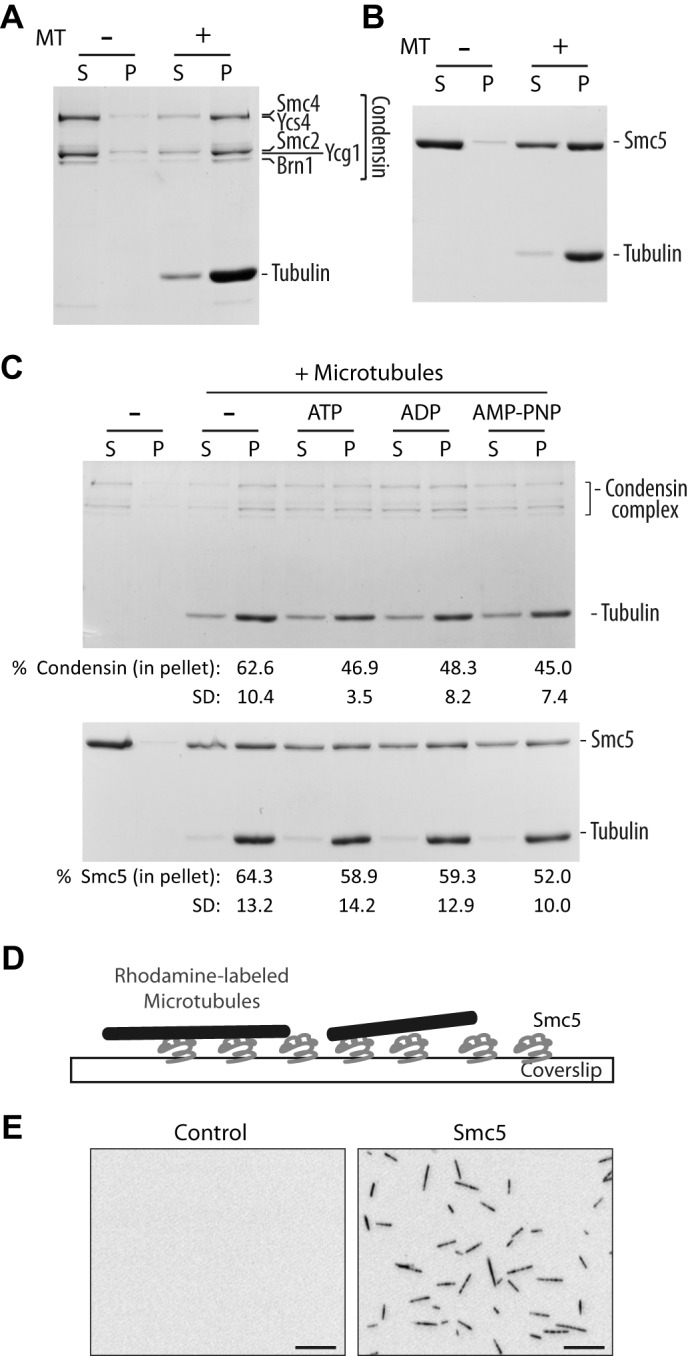
SMC proteins bind directly to microtubules in vitro. A and B, microtubule co-sedimentation assay with condensin complex (A) and monomeric Smc5 (B). Coomassie Blue-stained gels of supernatant (S) and pellet (P) fractions are shown. Microtubules were used at 0.5 μm (calculated for tubulin dimer concentration). Note that due to the large differences in the molecular mass of tubulin and condensin subunits, it is difficult to resolve all individual components of this complex in a single gel while simultaneously visualizing tubulin subunits (hence the appearance of only three major bands at the position of the condensin complex in the gel (18)). The weak band above the major Ycg1-Smc2 band corresponds to phosphorylated Ycg1, as reported previously (18). C, microtubule co-sedimentation assay with condensin complex (top) and Smc5 (bottom) in the presence of buffer alone (−), ATP, ADP, and a non-hydrolyzable analog, AMP-PNP. Microtubules were used at 0.25 and 0.5 μm for condensin complex and Smc5, respectively. The percentage of SMC proteins in the pellet under each condition is shown below the corresponding lane below the Coomassie Blue-stained gel. Averages of four independent experiments are shown, with S.D. D, an illustration depicting the microscopy-based microtubule binding assay. E, representative images of X-Rhodamine-labeled microtubules captured in control (uncoated) or Smc5-coated coverslip. Scale bars, 10 μm.
To simplify our investigation, we decided to follow the SMC component of the Smc5-Smc6 complex and chose purified Smc5 for subsequent experiments (17). We reconfirmed the Smc5-microtubule interaction using a microscopy-based flow chamber assay (Fig. 1D). In this assay, Smc5 protein was nonspecifically absorbed onto a glass coverslide in a flow chamber. Fluorescently labeled microtubules were then infused into the chamber to allow binding to the surface-immobilized Smc5. As observed in Fig. 1E, microtubules were efficiently captured only in the chamber that had been coated with Smc5 protein, again indicating a direct interaction.
Mapping and Assessment of the Smc5-Microtubule Interaction
Having demonstrated the ability of Smc5 to bind directly to microtubules, we next asked what the functional significance of this interaction would be. One way to address this question is to identify and characterize a mutant with impaired microtubule binding ability. However, without a crystal structure or a recognizable microtubule-binding motif in Smc5, locating the interaction interface in a ∼125-kDa protein is not a trivial task. To narrow our search, we decided to map the minimal binding domain using truncated forms of Smc5 containing only the ATPase heads, or the hinge domain with medium (HM) or long (HL) coiled-coil extensions (Fig. 2A). By a co-sedimentation assay, we found that the hinge and the coiled-coil domains, but not the ATPase domain, contain most if not all of the microtubule-binding sites (Fig. 2, B and C). This finding is consistent with our finding that Smc5-microtubule interaction is not sensitive to the nucleotide-binding states of Smc5 (Fig. 1C). Next we determined whether Smc5, like many microtubule-associated proteins, interacts with microtubules via the C-terminal negatively charged tails of tubulins known as the E-hooks (28–31). To achieve this, we used the protease subtilisin to cleave off the E-hooks from microtubules and found that this treatment did indeed drastically reduce Smc5 binding (Fig. 2D). This shows that Smc5 interacts with microtubules largely by electrostatic interactions via the C-terminal tails of tubulins.
FIGURE 2.

Smc5 binds to microtubules via electrostatic interaction with its hinge domain and adjacent coiled-coil arm. A, domain structure of Smc5 constructs. FL, full length; HM, Hinge domain with medium coiled-coil extensions; HL, Hinge domain with long coiled-coil extensions. B, microtubule co-sedimentation with truncation constructs of Smc5. A representative Coomassie Blue-stained gel of supernatant (S) and pellet (P) fractions is shown (n = 3). C, quantification of the percentage of Smc5 constructs associated with microtubules. Averages of three independent experiments are shown. Error bars, S.D. D, co-sedimentation assay with full-length Smc5 using untreated paclitaxel-stabilized microtubules (MT) and subtilisin-treated microtubules (sMT).
Identification and Characterization of Microtubule-binding Mutants of Smc5
Although we had narrowed down the domains that contain the bulk of the microtubule binding activity of Smc5, we still needed to pinpoint the key regions or amino acid residues that mediate the interaction. To achieve this, we took advantage of the observation from the subtilisin experiment that negative charges played a role in the microtubule-Smc5 interaction to develop a strategy to obtain a microtubule-binding mutant (Fig. 3A). We hypothesized that positively charged residues on Smc5 are important to interact with the E-hooks of tubulins, so we chemically modified lysine residues in Smc5 with biotin under non-saturating conditions. Our rationale was that the labeling of lysine residues essential for microtubule interaction would impair binding to the polymers, whereas that of unrelated lysine residues would not. By combining this approach with the microtubule co-sedimentation assay, it is possible to obtain a biased distribution of differentially labeled Smc5 protein molecules. Smc5 with those lysine residues “essential” for microtubule-binding biotinylated will be enriched in the supernatant fraction (i.e. non-microtubule binding), whereas those with lysines not relevant to microtubule-binding biotinylated will remain associated with the microtubule pellet, behaving similarly to the non-modified protein (Fig. 3B). Subsequent comparative mass spectrometry analysis of the supernatant and the pellet fractions will then determine which biotinylated lysines are specifically enriched in the individual fractions.
FIGURE 3.

Generation of a microtubule-binding mutant of SMC5. A, schematic depicting the method used to identify surface-exposed lysine residues on Smc5 that are essential (red +) or non-essential (blue +) for microtubule binding. Upon chemical labeling by biotin, modified lysines essential for binding will be enriched in the supernatant fraction. Relative abundances of tryptic peptides containing the modified lysines in the supernatant and pellet fractions were quantified by mass spectrometric analysis. B, Coomassie Blue-stained gel and streptavidin-HRP blot of the supernatant and pellet fractions of mock-labeled or biotin-labeled Smc5-HM proteins from the microtubule co-sedimentation assay. The enrichment of labeled Smc5-HM in the supernatant indicates the perturbation of microtubule binding upon lysine modifications. C, three lysine residues in the hinge region of Smc5 identified by the method illustrated above in A and B were mutated into glutamic acid to generate the microtubule-binding mutant called Smc5-3KE. Coomassie Blue-stained microtubule titration gels of full-length Smc5-WT (wild-type) or Smc5-3KE from the co-sedimentation assay are shown. D, fractions of microtubule-bound Smc5-WT (red) and Smc5-3KE from four independent microtubule co-sedimentation experiments were plotted against microtubule concentrations and fitted to a hyperbola to determine their dissociation constants (Kd).
Using the strategy described above, we identified three residues (Lys-624, Lys-631, and Lys-667) in the microtubule-binding region of Smc5 (i.e. Smc5-HM) that were consistently enriched in the supernatant fraction in the microtubule co-sedimentation assay (n > 3). Using site-directed mutagenesis, we generated a lysine to glutamic acid mutant (called Smc5-3KE) to determine the effect of these mutations on microtubule binding. We purified Smc5-3KE protein from yeast and tested the mutant in a microtubule co-sedimentation assay. We found that, in comparison with its wild-type counterpart, Smc5-3KE had an ∼2-fold decrease in binding affinity for microtubules (Kd Smc5-3KE = 1.19 μm versus Kd Smc5-WT = 0.59 μm; Fig. 3, C and D).
DNA Binding Activity of Microtubule-binding Mutants of Smc5
Because Smc5 is a known DNA-binding protein (17), we wished to determine whether the 3KE mutations affect the DNA-binding affinity of this protein. The ability of purified wild-type and 3KE mutant forms of Smc5 to bind single-stranded DNA (ssDNA) was determined using an electrophoretic mobility shift assay (EMSA). As shown previously (17), we find that wild-type Smc5 is capable of fully binding/shifting ssDNA in the well of the gel at a protein/DNA ratio of 25-fold molar excess (Fig. 4A). The Smc5-3KE mutant could also fully shift the ssDNA to the well of the gel although at a slightly higher protein/DNA molar ratio (Fig. 4B). We thus concluded from these experiments that the Smc5-3KE mutant largely retains its DNA binding activity, although the mutations may have a slight effect on this activity. It should be noted that the DNA binding activity of SMC complexes is contributed by multiple independent subunits of these complexes (32–34) and that a modest loss in the activity of a single component is likely to be compensated by the DNA binding activity of other subunits.
FIGURE 4.

Smc5 protein can bind to DNA and microtubules simultaneously. A, DNA binding assay with Smc5-WT and Smc5-3KE. B, quantification of the DNA binding assay. Error bars, S.D. C, schematic depicting the microscopy-based assay to visualize the formation of the microtubule-Smc5-DNA ternary complex. D, representative images of the surface-anchored X-Rhodamine-labeled microtubules (red) that were untreated (microtubules alone) or preincubated with either Smc5-WT or Smc5-3KE. Right panels, corresponding images of the associated FAM-labeled single-stranded DNA oligonucleotides (ssDNA). Scale bars, 10 μm. E and F, quantification of the data set shown in D. E, counts of single microtubules (gray bars) and bundled objects (black bars) for each condition are shown. Dashed unfilled bars representing estimated numbers of averaged microtubules (MTs) in bundled objects, as described under “Experimental Procedures,” are overlaid onto the corresponding black bars to provide a measure on the sizes of the bundles. Note that there are more microtubules in the Smc5-WT bundles than in Smc5-3KE or control. F, to quantify the associated FAM-labeled DNA on microtubules, average FAM signal intensity in arbitrary fluorescence units (AFU) under each experimental condition was determined and shown in the histogram. Microscopy-based experiments shown in C and D were repeated more than three times in similar settings. A representative set is quantified and shown here in E and F.
Smc5 Can Bind to DNA and Microtubules Simultaneously
To determine whether Smc5 can bind to both DNA and microtubules simultaneously, we developed a microscopy-based assay to visualize ternary complex formation. In this assay, surface-anchored X-Rhodamine-labeled microtubules precoated with Smc5 were allowed to interact with fluorescently labeled (FAM-labeled) DNA in solution (Fig. 4C). Using this method, we observed the capturing of fluorescently labeled DNA by Smc5-coated but not naked microtubules, indicating the formation of microtubule-Smc5-DNA ternary complex (Fig. 4D). It is worth noting that this capturing of FAM-labeled DNA was much reduced on Smc5-3KE-coated microtubules, as evidenced by the weak FAM signal that was only barely above that of the control condition. In addition, we found that microtubules were more efficiently bundled by Smc5 than by Smc5-3KE. Based on our quantification (Fig. 4E), Smc5-induced bundles represented ∼90% of the input microtubules (versus 72% with Smc5-3KE), and each bundle consisted of ∼17 microtubules on average (versus ∼12 microtubules with Smc5-3KE). In addition, the average FAM intensity on Smc5-induced microtubules is substantially higher than that on Smc5-3KE bundles or control (Fig. 4F). Together, our results show that Smc5 can interact with both microtubules and DNA simultaneously, and 3KE mutations compromise the formation of the ternary complex.
Smc5 Interacts with Microtubules in Dividing Yeast Cells
Having demonstrated the interaction between Smc5 and microtubules in vitro, we next sought to determine whether this interaction occurs in vivo. To test this notion, we performed a proximity ligation assay (PLA), an established method to detect protein-protein interactions based on their proximity within 40 nm in distance (20, 35, 36). For this, we used antibodies against tubulin and the StreptagII-fused Smc5. Their close proximity was detected and amplified by the secondary antibody-conjugated fluorescently labeled Duolink® probes (Fig. 5A). Specifically, we observed a significant increase in the number of cells showing a PLA-positive spot (indicating an in vivo Smc5-microtubule interaction) in the SMC5-STH yeast strains, as compared with that in the no-tag control (SMC5; Fig. 5, B and C). In addition, there was a specific enrichment of PLA signals in the nucleus and on microtubules in the StreptagII-fused Smc5 strain (Fig. 5D). Together, these results suggest a direct Smc5-microtubule interaction in dividing yeast cells.
FIGURE 5.
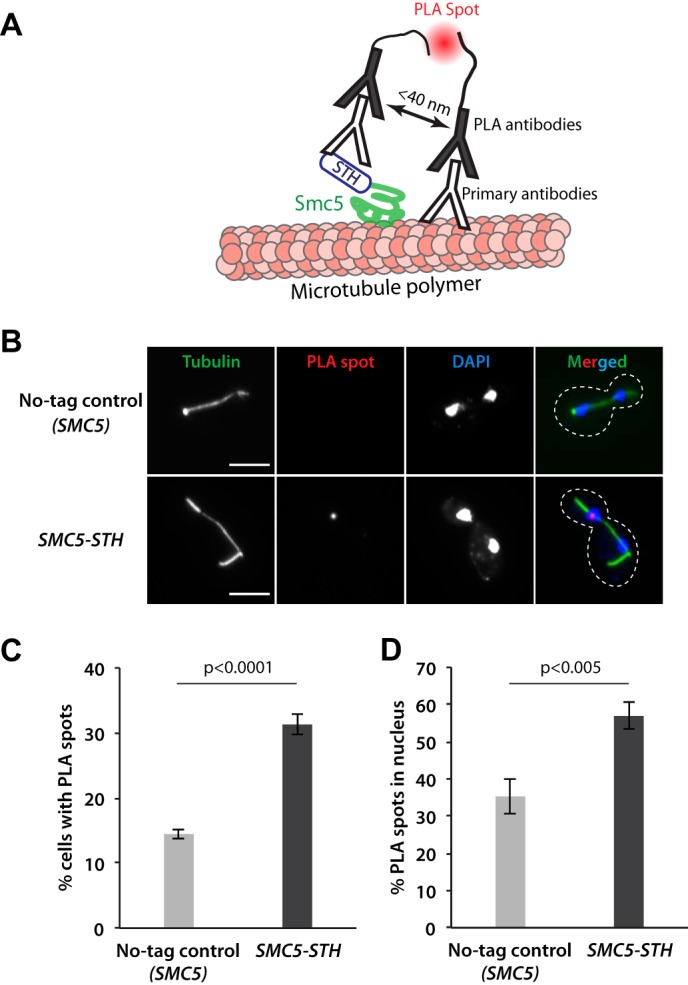
Smc5 protein interacts directly with microtubules in dividing yeast cells. A, the proximal ligation assay (PLA) used to detect Smc5-microtubule interactions. B, representative fluorescence microscopy images of cells in yeast strains expressing the untagged SMC5 (no-tag control) and the SMC5-STH in the PLA experiment. The peripheries of yeast cells shown in the merged panels are outlined by dashed lines. Scale bars, 5 μm. C, quantification of the percentage of cells with PLA spots for the SMC5 (no-tag control) and the SMC5-STH yeast strains. D, quantification of the percentage of PLA spots appearing in the nucleus for the SMC5 (no-tag control) and the SMC5-STH yeast strains. Data in C and D were obtained from three independent experiments with at least 179 yeast cells counted per condition. Error bars, S.D. p values were calculated by unpaired two-tailed t tests.
Functional Significance of Smc5-Microtubule Interaction
To assess the physiological consequence of impaired microtubule binding on Smc5 functions, we integrated the smc5-3KE mutant at the endogenous SMC5 locus in yeast. We found that the smc5-3KE mutant could substitute endogenous Smc5 function for viability at 30 and 37 °C. However, the mutant strain exhibited compromised growth kinetics at lower temperatures (Fig. 6A), consistent with a conditional cold-sensitive (cs) phenotype. The absence of detectable phenotype in diploid cells expressing smc5-3KE and wild-type SMC5 indicates that the mutant allele is recessive for cs behavior (Fig. 6A, bottom panels). smc5-3KE is the first identified mutant allele in any members of the SMC family that shows a conditional cs phenotype in budding yeast. Cold-sensitive phenotypes are relatively rare in essential proteins in eukaryotes and typically associate with defects in structural components of the cell (37–39). Consistent with this, tubulin mutants represent one of the largest classes of essential proteins for which cs mutations have been isolated (e.g. see the tub2-401 allele in Fig. 6A (38)). Microtubule mutants are known to be sensitive to lower temperatures at least in part because this condition can lead to microtubule depolymerization (40–42). This, together with the shared cs phenotypes of tubulin and smc5-3KE mutants, led us to wonder whether Smc5 acts by stabilizing microtubule polymers. To test this idea, we developed an in vitro assay to reconstitute cold-induced depolymerization. In this assay, paclitaxel-stabilized microtubules were diluted to low concentration and subjected to low temperature (i.e. 2 °C) to induce depolymerization. Compared with the identical sample at room temperature, the cold-treated sample had substantially more depolymerization, evident by the presence of more tubulins in the supernatant fraction in the sedimentation assay. Remarkably, substoichiometric ratio of Smc5 effectively stabilized microtubules at 2 °C (Fig. 6, B and C). In contrast, Smc5-3KE had a significantly reduced ability to protect microtubules from cold-induced depolymerization. This result led us to hypothesize that the cs phenotype and growth arrest we observed in smc5-3KE mutant yeast were due to microtubule instability and activation of the spindle assembly checkpoint. To test this in vivo, we investigated whether double mutants defective in smc5-3KE and microtubule components (38) or spindle assembly checkpoint activity (43, 44) show synthetic defects relative to single mutants. Remarkably, combining tub2-401 or mad1Δ with the smc5-3KE allele in yeast resulted in a significant decrease in the proliferative properties of the double mutant, relative to the single mutants, at lower temperatures (Fig. 6, D and E). Importantly, the microtubule-related phenotype of the smc5 mutant was not the consequence of mutation-induced disassembly of the Smc5-Smc6 complex or abnormal abundance of the Smc5-3KE protein in cells (Fig. 7). Together, these data suggest that Smc5 plays a direct role in stabilizing microtubules in yeast.
FIGURE 6.

Smc5 stabilizes microtubules in vivo and in vitro. A, the smc5-3KE mutant exhibits a recessive cold-sensitive growth defect in yeast. 5-Fold dilution series of wild-type haploid cells, smc5-6 mutant (22), tubulin mutant tub2-401 (38), wild-type SMC5, or smc5-3KE, were spotted on solid medium to evaluate growth at the indicated temperatures of 18, 23, 30, and 37 °C. Bottom panels, proliferation of diploid yeast heterozygous for SMC5-STH or smc5-3KE-STH on solid medium at various temperatures. The absence of cold-sensitive growth defect in the diploids indicates that the smc5-3KE allele is recessive. B, Smc5 protects microtubules from cold-induced depolymerization. After incubation at the indicated temperature (2 °C or room temperature (RT)), reaction mixtures containing buffer control (−), Smc5-WT, or Smc5-3KE were subjected to ultracentrifugation to separate the dissociated tubulin dimers and the remaining microtubule polymers into supernatant (S) and pellet (P) fractions, respectively. Coomassie Blue-stained gels of the samples are shown. The relative quantity of the Smc5 proteins in arbitrary units (a.u.) in each sample is shown below the corresponding lanes on the gel. C, quantification of the percentage of protection provided by Smc5 proteins (average of three independent experiments; error bars, S.D.). Percentage protection is defined as percentage of excess microtubules retained in the pellet relative to the control at 2 °C. D and E, genetic interactions between smc5-3KE and the tubulin-defective mutant tub2-401 (D) or the spindle assembly checkpoint mutant mad1Δ (E). Growth phenotypes of the indicated yeast strains were evaluated as described in A. F, proliferation of smc5 mutants and wild-type yeast strains on solid medium containing methyl methanesulfonate (MMS) at 0.005%, hydroxyurea (HU) at 100 mm, and 4-nitroquinoline 1-oxide (4NQO) at 0.2 μm.
FIGURE 7.
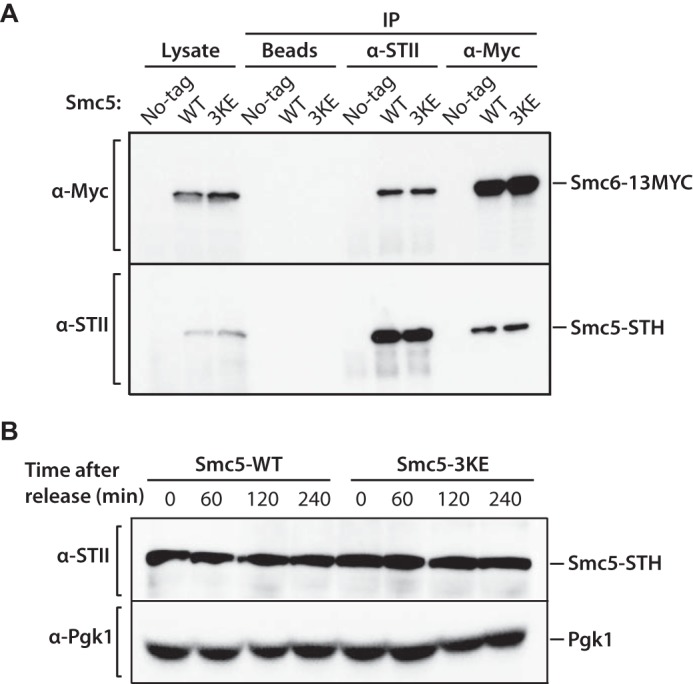
Mutations in Smc5-3KE do not affect Smc5-Smc6 complex formation and protein abundance. A, Smc5 (or Smc5-3KE) and Smc6 proteins were immunoprecipitated (IP) from whole cell extract using anti-StreptagII (STII) and anti-Myc antibodies to evaluate the association between Smc5-STH (or Smc5-3KE) and Smc6-13xMYC by Western blotting with the reciprocal antibodies, as indicated. Cell lysate (Lysate) and beads alone (Beads) were used as controls. The strains used in this experiment are as follows. No-tag, SMC5/SMC5; SMC6/SMC6; WT, SMC5-STH/SMC5; SMC6-13MYC/SMC6; 3KE, smc5-3KE-STH/SMC5; SMC6-13MYC/SMC6. B, Western blotting analysis of Smc5-3KE protein abundance in yeast upon α-factor arrest and release into a synchronous cell cycle at 18 °C, using a StreptagII antibody. Pgk1 protein was used as a loading control.
We next wondered whether loss of microtubule binding affected the known functions of the Smc5-Smc6 complex in the DNA damage response (3). To test this notion, we monitored the cellular proliferation of smc5-3KE mutants in the presence of various DNA-damaging agents, including methyl methanesulfonate (MMS), 4-nitroquinoline 1-oxide (4NQO), and hydroxyurea (HU). Fig. 6F shows that loss of microtubule binding ability significantly reduced the capacity of the smc5-3KE mutant to grow in the presence of all three DNA-damaging agents tested. As observed previously for the cs phenotype, the DNA damage sensitivity of the smc5-3KE mutant was complemented by a single wild-type copy of SMC5, indicating that the mutant allele is recessive for its sensitivity to DNA-damaging agents (data not shown). Together, these observations indicate that Smc5 depends at least in part on its interaction with microtubules and/or on a normal microtubule network (see below) to maintain genome integrity in cells exposed to DNA damage.
If the binding of Smc5 to microtubule affects the stability and/or functionality of the microtubule cytoskeleton in vivo, one would expect that the smc5-3KE mutant should have mitotic phenotypes resembling those of tub2 mutants. To test this hypothesis, we performed a cell cycle experiment in which smc5-3KE mutants and wild-type yeasts where synchronized in G1 at 30 °C using α-factor and then released from this arrest into a synchronous cell cycle at non-permissive temperature for the smc5-3KE mutant (i.e. 18 °C). Samples were taken at 30-min intervals, and cell cycle progression was monitored using flow cytometry and bud morphology. Yeast carrying the tub2-401 allele or an epitope-tagged version of wild-type SMC5 (i.e. the STH epitope that is also present in the smc5-3KE allele (17)) were included as controls. Wild-type cells and SMC5-STH controls progressed normally in the cell cycle, accumulating small buds concomitantly with the initiation of DNA synthesis (at 90 min after release; Fig. 8A) and entering M phase 30–60 min thereafter (as evidenced by the increased population of large budded cells in the culture; Fig. 8A). The subsequent appearance of unbudded cells and return to a 1n genomic DNA content marked an exit from mitosis and return to G1 210 min after the release (Fig. 8, A and B). In parallel experiments, the tub2-401 and smc5-3KE mutant populations showed similar kinetics of DNA replication and mitotic entry compared with wild-type cells (Fig. 8B). However, at later time points, the two mutant populations failed to exit mitosis and return to G1 with normal kinetics. Indeed, tub2-401 and smc5-3KE cells accumulated with a 2n/large budded phenotype and remained in this state until the end of the experiment (Fig. 8, A and B), suggesting a persistent mitotic arrest in the absence of the TUB2 and SMC5 functions. A small fraction of the smc5-3KE mutant population rebudded toward the end of the experiment (i.e. at 240 min), whereas tub2-401 mutant cells remained mostly arrested with no bud under identical conditions (Fig. 8A). The population of rebudded smc5-3KE mutants may represent cells that have adapted to the spindle checkpoint arrest, as observed previously (45).
FIGURE 8.

Reducing Smc5 binding to microtubules in vivo induces a mitotic arrest. A, cell cycle progression of yeast carrying SMC5, SMC5-STH, tub2-401, and smc5-3KE after a G1 arrest using α-factor. After release at 18 °C, samples of the cell cultures were taken every 30 min to evaluate the budding index and DNA content. Small budded cells were scored when the bud was smaller than one-third of the mother cell, whereas big budded cells were scored when the bud was bigger than one-third of the mother cell. B, flow cytometry analysis showing the DNA content of the yeast strains used in the time course experiment.
The mitotic arrest phenotype of smc5-3KE cells is similar to that of tubulin mutants (46), raising the possibility that Smc5 might affect microtubule functions in mitosis. To test this possibility, we monitored microtubule morphology by immunofluorescence in SMC5 and smc5-3KE yeast cultures progressing synchronously in mitosis (Fig. 9A). Although smc5-3KE cells were clearly capable of building a microtubule cytoskeleton, our analysis revealed a significant proportion of mitotic cells with aberrant or defective microtubule morphology at 18 °C (Fig. 9B). Moreover, there was a noticeable fraction of smc5-3KE mutants carrying anaphase spindles with unsegregated nuclear masses, indicating a defect in microtubule-driven separation of chromosomes (Fig. 9, B and C). Furthermore, relative to wild-type controls, a significant fraction of smc5-3KE cells contained nuclear microtubules that differed from their normal morphology, with spindles containing more than one continuous nuclear structure (referred to as “branched”; Fig. 9B) by immunofluorescence. Likewise, another fraction of smc5-3KE cells contained spindles that were either mispositioned or misoriented, being entirely localized in the daughter cells or having initiated anaphase in the mother cell instead of at the bud neck (Fig. 9, B and C). Collectively, these results indicate that inactivation of the microtubule binding activity of Smc5 gives rise to defects in mitotic spindle morphology and positioning in vivo.
FIGURE 9.
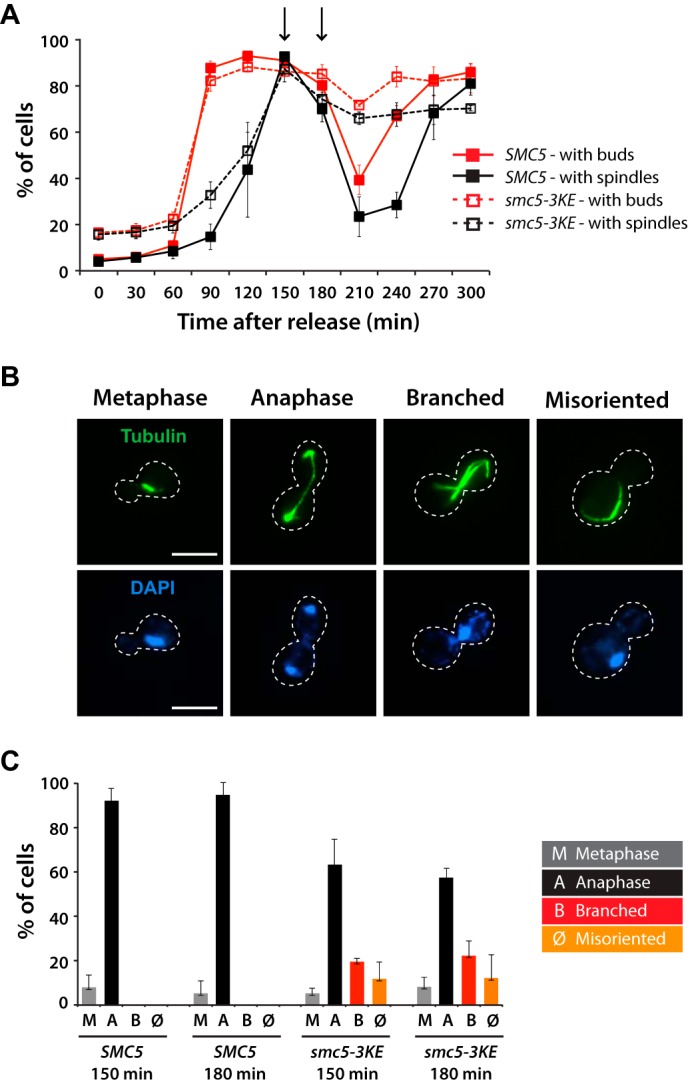
The microtubule-binding mutant of Smc5 exhibits temperature-dependent spindle defects in yeast. A, quantification of cells with buds (red lines) and spindles (black lines) in SMC5 (solid lines) and smc5-3KE (dashed lines) yeast strains after release from G1 arrest at 18 °C. Arrows, time points that were analyzed by immunofluorescence. B, representative mitotic phenotypes observed for smc5-3KE are shown, in comparison with the wild-type morphology (metaphase and anaphase). The peripheries of the yeast cells shown in the pictures are outlined by dashed lines. Scale bars, 5 μm. C, quantification of the different spindle structures observed in mitosis. Data in A and C are shown as averages of three independent experiments; error bars, S.D.
DISCUSSION
The ability of cells to maintain genomic integrity from generation to generation depends on faithful segregation of chromosomes. This is achieved, in large part, by ensuring that chromosomes are properly connected to the spindle microtubules during mitosis and meiosis. Proteins linking chromatin to microtubules play a crucial role in this respect (9, 47). Here, we uncover the existence of an unexpected new activity in a known regulator of chromosome stability, the ability to bind both microtubules and DNA. This, together with previous mass spectrometry characterization of the microtubule interactome (13, 15), suggests that Smc5 and other SMC family members represent a novel class of evolutionarily conserved proteins that can directly connect spindle microtubules to chromosomes during cell division. Furthermore, we provide evidence that this novel activity can contribute to the functional stability of microtubules and help sustain robust cell cycle progression in vivo.
The unique cold-sensitive phenotype of the smc5-3KE mutant relative to other alleles affecting Smc5-Smc6 complex components raises an interesting question: why a few point mutations in a single SMC protein have such a profound effect on microtubule stability? This question is particularly relevant in light of the fact that proteomic studies have shown that there are hundreds of proteins in the microtubule-associated proteome (13–16). One explanation for this is that not all proteins in this group are likely to be direct regulators of microtubule dynamics and/or stability. In particular, we think that the phenotype of smc5-3KE mutant reflects the fact that budding yeast is a highly sensitized system to observe microtubule-related defects. Indeed, this organism has an unusually low concentration of tubulin compared with other eukaryotic species. Although tubulin concentration in other systems, such as human cultured cell or Xenopus oocyte, is about 20 μm (48–51), yeast may have less than 2 μm tubulin.7 Because low tubulin concentration will favor microtubule depolymerization rather than its polymerization, yeast may be particularly reliant on microtubule stabilizers to build a functional spindle structure. Separate from a role in stabilization, SMC proteins, such as those of the Smc5-Smc6 or cohesin complexes, could provide anchorage points on the chromosomes for spindle microtubules to attach during mitosis. Given the high affinity of SMC proteins for microtubules, we postulate that SMC proteins are key stabilizing factors for microtubule polymers. Therefore, impairment of one major component in this system could have a noticeable consequence on mitotic spindle integrity, especially when cells are exposed to microtubule stressors, such as low temperature (41, 42).
Mechanistically, how might Smc5 contribute to microtubule stability? We think this could be achieved simply through high affinity association with microtubules. The buffer condition that we used for measuring kd has much higher ionic strength than what was traditionally used for a microtubule-binding experiment, suggesting a very strong interaction between SMC proteins and microtubules. In fact, under this condition, most kinesins would barely interact with microtubules (29, 52, 53). Additionally, Smc5 might have multiple microtubule-binding sites and thus allow bundling of adjacent microtubules, as we have observed in our microscopy experiments (Fig. 4D). However, it remains unclear how much this would contribute to spindle integrity in yeast and whether it could partly account for the occurrence of the “branched” phenotypes observed in cells expressing smc5-3KE (Fig. 9, B and C). This is further complicated by the presence of other microtubule-regulating factors in vivo. Given the small size of the yeast spindle, we could not resolve microtubule bundles (or the lack of them) by fluorescence microscopy. Separate from this bundling activity, high affinity binding by Smc5 could physically hinder tubulin disassembly at microtubule ends, which would effectively slow down catastrophe and promote rescue.
Functionally, besides preserving spindle integrity, we have considered other impacts of the newly described SMC-microtubule interaction. It is conceivable that microtubule binding could affect other Smc5 functions. For instance, this binding event could act to allosterically regulate the affinity of Smc5 with other binding partners, such as chromatin or other members of the Smc5-Smc6 complex (3, 54). Alternatively, microtubule binding could sequester the pool of free Smc5 or Smc5-Smc6 complexes in cells and in such a way modulate the dynamics and availability of Smc5 for specific functions in cells, such as DNA repair (55). This is consistent with our observation that smc5-3KE is sensitive to DNA-damaging agents, although the precise regulatory mechanism in this process has yet to be determined.
Our work has uncovered a novel and unexpected function of SMC proteins in the regulation of microtubule stability. This discovery raises many interesting questions. For example, are other chromatin binding proteins also contributing to this function? If so, do these proteins collectively contribute to the formation and maintenance of microtubule-based structures, such as the mitotic spindle? In this regard, it is interesting to note that SMC proteins, including the Smc5-Smc6 complex, have been shown to be enriched at centromeres in yeast, suggesting a possible link between this complex and the regulation of kinetochore function (55–59). It is tempting to speculate that SMC proteins can take part in promoting kinetochore-microtubule interactions by capturing and stabilizing microtubules in close proximity of the centromeres. Regardless, our data suggest that SMC proteins could provide a linkage between chromatin and microtubules, and this could in turn directly contribute to faithful segregation of chromosomes during mitosis. In light of our finding, it will also be interesting to determine in the future whether microtubules can have a direct effect on the known functions of the SMC complexes, including chromosome cohesion, condensation, and DNA repair.
This work was supported by Canadian Institutes of Health Research (CIHR) Grants MOP-82912 (to D. D.) and MOP-97928 (to B. H. K.), Canadian Cancer Society Research Institute Grant 020304 (to D. D.), Fonds de Recherche du Québec en Santé Grant 22131 (to B. H. K.), and the Natural Sciences and Engineering Research Council (to P. T.). The Institute for Research in Immunology and Cancer (IRIC) is supported in part by the Canadian Center of Excellence in Commercialization and Research (CECR), the Canada Foundation for Innovation, and Fonds de Recherche Santé (FRQS).
Based on the number of Tub1 molecules per yeast cell, 5590, reported by Ghaemmaghami et al. (60) and a haploid cell volume of 42 fl (61), the calculated concentration of tubulin in a yeast cell is ∼0.22 μm. However, we think this probably represents an underestimation of the actual tubulin concentration in cells based on our estimated minimal tubulin subunits needed to assemble the yeast mitotic spindle (∼39,000 tubulin dimers needed to assemble 16 × 1.5-μm-long microtubules, which makes the tubulin concentration close to 1.5 μm).
- SMC
- structural maintenance of chromosome
- FAM
- 6-carboxyfluorescein
- PLA
- proximity ligation assay
- STH
- 3xStreptagII-9xHIS fusion tag
- AMP-PNP
- 5′-adenylyl-β,γ-imidodiphosphate
- cs
- cold-sensitive.
REFERENCES
- 1. Bazile F., St-Pierre J., D'Amours D. (2010) Three-step model for condensin activation during mitotic chromosome condensation. Cell Cycle 9, 3243–3255 [DOI] [PubMed] [Google Scholar]
- 2. Hirano T. (2012) Condensins: universal organizers of chromosomes with diverse functions. Genes Dev. 26, 1659–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. De Piccoli G., Torres-Rosell J., Aragón L. (2009) The unnamed complex: what do we know about Smc5-Smc6? Chromosome Res. 17, 251–263 [DOI] [PubMed] [Google Scholar]
- 4. Hirano T. (2006) At the heart of the chromosome: SMC proteins in action. Nat. Rev. Mol. Cell Biol. 7, 311–322 [DOI] [PubMed] [Google Scholar]
- 5. Kinoshita K., Habermann B., Hyman A. A. (2002) XMAP215: a key component of the dynamic microtubule cytoskeleton. Trends Cell Biol. 12, 267–273 [DOI] [PubMed] [Google Scholar]
- 6. Tirnauer J. S., Bierer B. E. (2000) EB1 proteins regulate microtubule dynamics, cell polarity, and chromosome stability. J. Cell Biol. 149, 761–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Akiyoshi B., Nelson C. R., Ranish J. A., Biggins S. (2009) Quantitative proteomic analysis of purified yeast kinetochores identifies a PP1 regulatory subunit. Genes Dev. 23, 2887–2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheeseman I. M., Chappie J. S., Wilson-Kubalek E. M., Desai A. (2006) The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell 127, 983–997 [DOI] [PubMed] [Google Scholar]
- 9. Cheeseman I. M., Desai A. (2008) Molecular architecture of the kinetochore-microtubule interface. Nat. Rev. Mol. Cell Biol. 9, 33–46 [DOI] [PubMed] [Google Scholar]
- 10. Antonio C., Ferby I., Wilhelm H., Jones M., Karsenti E., Nebreda A. R., Vernos I. (2000) Xkid, a chromokinesin required for chromosome alignment on the metaphase plate. Cell 102, 425–435 [DOI] [PubMed] [Google Scholar]
- 11. Funabiki H., Murray A. W. (2000) The Xenopus chromokinesin Xkid is essential for metaphase chromosome alignment and must be degraded to allow anaphase chromosome movement. Cell 102, 411–424 [DOI] [PubMed] [Google Scholar]
- 12. Lee Y. M., Lee S., Lee E., Shin H., Hahn H., Choi W., Kim W. (2001) Human kinesin superfamily member 4 is dominantly localized in the nuclear matrix and is associated with chromosomes during mitosis. Biochem. J. 360, 549–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gache V., Waridel P., Winter C., Juhem A., Schroeder M., Shevchenko A., Popov A. V. (2010) Xenopus meiotic microtubule-associated interactome. PLoS One 5, e9248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mack G. J., Compton D. A. (2001) Analysis of mitotic microtubule-associated proteins using mass spectrometry identifies astrin, a spindle-associated protein. Proc. Natl. Acad. Sci. U.S.A. 98, 14434–14439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sauer G., Körner R., Hanisch A., Ries A., Nigg E. A., Silljé H. H. (2005) Proteome analysis of the human mitotic spindle. Mol. Cell Proteomics 4, 35–43 [DOI] [PubMed] [Google Scholar]
- 16. Skop A. R., Liu H., Yates J., 3rd, Meyer B. J., Heald R. (2004) Dissection of the mammalian midbody proteome reveals conserved cytokinesis mechanisms. Science 305, 61–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roy M. A., Siddiqui N., D'Amours D. (2011) Dynamic and selective DNA-binding activity of Smc5, a core component of the Smc5-Smc6 complex. Cell Cycle 10, 690–700 [DOI] [PubMed] [Google Scholar]
- 18. St-Pierre J., Douziech M., Bazile F., Pascariu M., Bonneil E., Sauvé V., Ratsima H., D'Amours D. (2009) Polo kinase regulates mitotic chromosome condensation by hyperactivation of condensin DNA supercoiling activity. Mol. Cell 34, 416–426 [DOI] [PubMed] [Google Scholar]
- 19. Desai A., Verma S., Mitchison T. J., Walczak C. E. (1999) Kin I kinesins are microtubule-destabilizing enzymes. Cell 96, 69–78 [DOI] [PubMed] [Google Scholar]
- 20. Solinet S., Mahmud K., Stewman S. F., Ben El Kadhi K., Decelle B., Talje L., Ma A., Kwok B. H., Carreno S. (2013) The actin-binding ERM protein Moesin binds to and stabilizes microtubules at the cell cortex. J. Cell Biol. 202, 251–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Arora K., Talje L., Asenjo A. B., Andersen P., Atchia K., Joshi M., Sosa H., Allingham J. S., Kwok B. H. (2014) KIF14 binds tightly to microtubules and adopts a rigor-like conformation. J. Mol. Biol. 426, 2997–3015 [DOI] [PubMed] [Google Scholar]
- 22. Torres-Rosell J., Machín F., Farmer S., Jarmuz A., Eydmann T., Dalgaard J. Z., Aragón L. (2005) SMC5 and SMC6 genes are required for the segregation of repetitive chromosome regions. Nat. Cell Biol. 7, 412–419 [DOI] [PubMed] [Google Scholar]
- 23. Ratsima H., Ladouceur A. M., Pascariu M., Sauvé V., Salloum Z., Maddox P. S., D'Amours D. (2011) Independent modulation of the kinase and polo-box activities of Cdc5 protein unravels unique roles in the maintenance of genome stability. Proc. Natl. Acad. Sci. U.S.A. 108, E914–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Crevel I. M., Lockhart A., Cross R. A. (1996) Weak and strong states of kinesin and ncd. J. Mol. Biol. 257, 66–76 [DOI] [PubMed] [Google Scholar]
- 25. Kwok B. H., Yang J. G., Kapoor T. M. (2004) The rate of bipolar spindle assembly depends on the microtubule-gliding velocity of the mitotic kinesin Eg5. Curr. Biol. 14, 1783–1788 [DOI] [PubMed] [Google Scholar]
- 26. Woehlke G., Ruby A. K., Hart C. L., Ly B., Hom-Booher N., Vale R. D. (1997) Microtubule interaction site of the kinesin motor. Cell 90, 207–216 [DOI] [PubMed] [Google Scholar]
- 27. Vale R. D., Reese T. S., Sheetz M. P. (1985) Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell 42, 39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hayashi I., Ikura M. (2003) Crystal structure of the amino-terminal microtubule-binding domain of end-binding protein 1 (EB1). J. Biol. Chem. 278, 36430–36434 [DOI] [PubMed] [Google Scholar]
- 29. Helenius J., Brouhard G., Kalaidzidis Y., Diez S., Howard J. (2006) The depolymerizing kinesin MCAK uses lattice diffusion to rapidly target microtubule ends. Nature 441, 115–119 [DOI] [PubMed] [Google Scholar]
- 30. Okada Y., Hirokawa N. (2000) Mechanism of the single-headed processivity: diffusional anchoring between the K-loop of kinesin and the C terminus of tubulin. Proc. Natl. Acad. Sci. U.S.A. 97, 640–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ramey V. H., Wang H. W., Nakajima Y., Wong A., Liu J., Drubin D., Barnes G., Nogales E. (2011) The Dam1 ring binds to the E-hook of tubulin and diffuses along the microtubule. Mol. Biol. Cell 22, 457–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Akhmedov A. T., Frei C., Tsai-Pflugfelder M., Kemper B., Gasser S. M., Jessberger R. (1998) Structural maintenance of chromosomes protein C-terminal domains bind preferentially to DNA with secondary structure. J. Biol. Chem. 273, 24088–24094 [DOI] [PubMed] [Google Scholar]
- 33. Chiu A., Revenkova E., Jessberger R. (2004) DNA interaction and dimerization of eukaryotic SMC hinge domains. J. Biol. Chem. 279, 26233–26242 [DOI] [PubMed] [Google Scholar]
- 34. Yoshimura S. H., Hizume K., Murakami A., Sutani T., Takeyasu K., Yanagida M. (2002) Condensin architecture and interaction with DNA: regulatory non-SMC subunits bind to the head of SMC heterodimer. Curr. Biol. 12, 508–513 [DOI] [PubMed] [Google Scholar]
- 35. Fredriksson S., Gullberg M., Jarvius J., Olsson C., Pietras K., Gústafsdóttir S. M., Ostman A., Landegren U. (2002) Protein detection using proximity-dependent DNA ligation assays. Nat. Biotechnol. 20, 473–477 [DOI] [PubMed] [Google Scholar]
- 36. Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K. J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L. G., Landegren U. (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 [DOI] [PubMed] [Google Scholar]
- 37. Broach J. R. (1986) New approaches to a genetic analysis of mitosis. Cell 44, 3–4 [DOI] [PubMed] [Google Scholar]
- 38. Huffaker T. C., Thomas J. H., Botstein D. (1988) Diverse effects of β-tubulin mutations on microtubule formation and function. J. Cell Biol. 106, 1997–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jarvik J., Botstein D. (1975) Conditional-lethal mutations that suppress genetic defects in morphogenesis by altering structural proteins. Proc. Natl. Acad. Sci. U.S.A. 72, 2738–2742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Johnson K. A., Borisy G. G. (1977) Kinetic analysis of microtubule self-assembly in vitro. J. Mol. Biol. 117, 1–31 [DOI] [PubMed] [Google Scholar]
- 41. Rieder C. L. (1981) The structure of the cold-stable kinetochore fiber in metaphase PtK1 cells. Chromosoma 84, 145–158 [DOI] [PubMed] [Google Scholar]
- 42. Salmon E. D., Begg D. A. (1980) Functional implications of cold-stable microtubules in kinetochore fibers of insect spermatocytes during anaphase. J. Cell Biol. 85, 853–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hoyt M. A., Totis L., Roberts B. T. (1991) S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell 66, 507–517 [DOI] [PubMed] [Google Scholar]
- 44. Li R., Murray A. W. (1991) Feedback control of mitosis in budding yeast. Cell 66, 519–531 [DOI] [PubMed] [Google Scholar]
- 45. Rossio V., Galati E., Ferrari M., Pellicioli A., Sutani T., Shirahige K., Lucchini G., Piatti S. (2010) The RSC chromatin-remodeling complex influences mitotic exit and adaptation to the spindle assembly checkpoint by controlling the Cdc14 phosphatase. J. Cell Biol. 191, 981–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Reijo R. A., Cooper E. M., Beagle G. J., Huffaker T. C. (1994) Systematic mutational analysis of the yeast β-tubulin gene. Mol. Biol. Cell 5, 29–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cleveland D. W., Mao Y., Sullivan K. F. (2003) Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell 112, 407–421 [DOI] [PubMed] [Google Scholar]
- 48. Hiller G., Weber K. (1978) Radioimmunoassay for tubulin: a quantitative comparison of the tubulin content of different established tissue culture cells and tissues. Cell 14, 795–804 [DOI] [PubMed] [Google Scholar]
- 49. Gard D. L., Kirschner M. W. (1987) Microtubule assembly in cytoplasmic extracts of Xenopus oocytes and eggs. J. Cell Biol. 105, 2191–2201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Heidemann S. R., Kirschner M. W. (1975) Aster formation in eggs of Xenopus laevis. Induction by isolated basal bodies. J. Cell Biol. 67, 105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Elinson R. P. (1985) Changes in levels of polymeric tubulin associated with activation and dorsoventral polarization of the frog egg. Dev. Biol. 109, 224–233 [DOI] [PubMed] [Google Scholar]
- 52. Kapitein L. C., Kwok B. H., Weinger J. S., Schmidt C. F., Kapoor T. M., Peterman E. J. (2008) Microtubule cross-linking triggers the directional motility of kinesin-5. J. Cell Biol. 182, 421–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Weinger J. S., Qiu M., Yang G., Kapoor T. M. (2011) A nonmotor microtubule binding site in kinesin-5 is required for filament crosslinking and sliding. Curr. Biol. 21, 154–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kegel A., Sjögren C. (2010) The Smc5/6 complex: more than repair? Cold Spring Harb. Symp. Quant. Biol. 75, 179–187 [DOI] [PubMed] [Google Scholar]
- 55. Lindroos H. B., Ström L., Itoh T., Katou Y., Shirahige K., Sjögren C. (2006) Chromosomal association of the Smc5/6 complex reveals that it functions in differently regulated pathways. Mol. Cell 22, 755–767 [DOI] [PubMed] [Google Scholar]
- 56. Nakazawa N., Nakamura T., Kokubu A., Ebe M., Nagao K., Yanagida M. (2008) Dissection of the essential steps for condensin accumulation at kinetochores and rDNAs during fission yeast mitosis. J. Cell Biol. 180, 1115–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ono T., Fang Y., Spector D. L., Hirano T. (2004) Spatial and temporal regulation of Condensins I and II in mitotic chromosome assembly in human cells. Mol. Biol. Cell 15, 3296–3308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pebernard S., Schaffer L., Campbell D., Head S. R., Boddy M. N. (2008) Localization of Smc5/6 to centromeres and telomeres requires heterochromatin and SUMO, respectively. EMBO J. 27, 3011–3023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stephens A. D., Haase J., Vicci L., Taylor R. M., 2nd, Bloom K. (2011) Cohesin, condensin, and the intramolecular centromere loop together generate the mitotic chromatin spring. J. Cell Biol. 193, 1167–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ghaemmaghami S., Huh W. K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., Weissman J. S. (2003) Global analysis of protein expression in yeast. Nature 425, 737–741 [DOI] [PubMed] [Google Scholar]
- 61. Jorgensen P., Nishikawa J. L., Breitkreutz B. J., Tyers M. (2002) Systematic identification of pathways that couple cell growth and division in yeast. Science 297, 395–400 [DOI] [PubMed] [Google Scholar]