

Background: Thermal stress affects inheritance of prion amyloids in budding yeast.
Results: Heat shock-induced processing of actin assembly protein Lsb1 is involved in yeast prion inheritance.
Conclusion: Components of the actin cytoskeleton affect prion dynamics in response to heat shock.
Significance: Understanding the mechanisms of aggregate segregation in response to stress may present new opportunities for pharmacological interventions aimed at protein aggregation disorders.
Keywords: Actin, Endoplasmic Reticulum (ER), Prion, Proteasome, Ubiquitylation (Ubiquitination), Rsp5, Sup35, Heat Shock
Abstract
Yeast prions are self-propagating amyloid-like aggregates of Q/N-rich protein that confer heritable traits and provide a model of mammalian amyloidoses. [PSI+] is a prion isoform of the translation termination factor Sup35. Propagation of [PSI+] during cell division under normal conditions and during the recovery from damaging environmental stress depends on cellular chaperones and is influenced by ubiquitin proteolysis and the actin cytoskeleton. The paralogous yeast proteins Lsb1 and Lsb2 bind the actin assembly protein Las17 (a yeast homolog of human Wiskott-Aldrich syndrome protein) and participate in the endocytic pathway. Lsb2 was shown to modulate maintenance of [PSI+] during and after heat shock. Here, we demonstrate that Lsb1 also regulates maintenance of the Sup35 prion during and after heat shock. These data point to the involvement of Lsb proteins in the partitioning of protein aggregates in stressed cells. Lsb1 abundance and cycling between actin patches, endoplasmic reticulum, and cytosol is regulated by the Guided Entry of Tail-anchored proteins pathway and Rsp5-dependent ubiquitination. Heat shock-induced proteolytic processing of Lsb1 is crucial for prion maintenance during stress. Our findings identify Lsb1 as another component of a tightly regulated pathway controlling protein aggregation in changing environments.
Introduction
Environmental stresses such as heat shock (HS) lead to an increased misfolding of a wide range of proteins (1–4). Recent data suggest that the spread of large quantities of misfolded proteins throughout the cell could be toxic, whereas their assembly into the large aggregate deposits plays, to a certain extent, a protective role (5–8). Large aggregates of stress-damaged proteins accumulate asymmetrically in the older mother cell upon cell division and this serves as a mechanism of protecting “rejuvenated” daughter cells from toxic effects of protein aggregation in the budding yeast (9–11). This mechanism is clearly not restricted to organisms with the morphologically asymmetric cell division and is proposed to also play a role in differentiation between the “mortal” somatic and “immortal” stem cells (12). Decreased diffusion of larger aggregates through the bud neck, coupled with more efficient solubilization of aggregates in the bud, was suggested as an explanation for the asymmetry (13). However, other evidence indicates that at least some aggregates either are trapped in a scaffold of actin cables in the mother cell or are subject to active retrograde transport back to the mother from the growing bud, in a process requiring the polarisome and the actin cytoskeletal network (10, 14). Although the relative contributions of these alternative mechanisms are still being debated, it is clear that asymmetric accumulation of aggregates in the yeast mother cells depends on the chaperone Hsp104 and the actin cytoskeleton (9, 15, 16). Other data confirm that the actin cytoskeleton is crucial for mediating responses of a eukaryotic cell to environmental stresses (17). How the actin machinery selectively binds to the damaged protein aggregates is a yet unresolved question.
Self-perpetuating ordered β-rich protein aggregates (amyloids) are associated with a variety of human diseases, including such widespread disorders as Huntington, Alzheimer, and Parkinson diseases (18). Transmissible amyloids (prions) can be spread by extracellular infection in mammals (19, 20). In lower eukaryotes such as yeast, prions are inherited via the cytoplasm (19, 20). Yeast prions are Q/N-rich proteins broadly used as an experimental model for the aggregation-related disorders (19, 20). Recent data demonstrate that the yeast prion [PSI+], a self-perpetuating amyloid form of the translation termination (release) factor Sup35 (eRF3), exhibits asymmetric mother-specific distribution in the cell divisions after heat stress, leading to increased prion loss (21). Therefore, segregation of this yeast prion parallels the segregation of heat-damaged aggregated proteins. As the [PSI+] prion exhibits a detectable phenotype, this allows for the phenotypic monitoring of aggregate distribution and loss in cell divisions following stress treatment. Destabilization of some [PSI+] variants after disruption of the actin cytoskeleton by latrunculin A (22) reinforces the connection between [PSI+] homeostasis and the actin cytoskeleton.
Perturbations of the actin cytoskeleton are linked to physiological and environmental signals by actin nucleation-promoting factors (17). The human Wiskott-Aldrich syndrome protein and its yeast homolog Las17 are the crucial components (23). The activity of Las17 is regulated by multiple interacting partners that bind to its proline-rich regions via SH34 domains and form dynamic protein complexes at actin patches, major sites of endocytic vesicle assembly. Regulation of these complexes by altered protein-protein interaction and protein turnover involves ubiquitination by the HECT-type ubiquitin ligase Rsp5, and/or binding to ubiquitin (Ub) (24). Lsb2, a yeast Las17-binding protein with a Ub-binding domain modulates prion dynamic, and both, Lsb1 and paralogous Lsb2 protein, have been identified as negative regulators of Las17-induced actin polymerization (25).
In this work, we use the yeast prion [PSI+] as a model system for investigation of the role of actin assembly proteins in aggregation-induced damage. We have previously demonstrated that the Las17-binding protein Lsb2 promotes the initial nucleation of the [PSI+] prion aggregates and aids in [PSI+] maintenance after heat shock (26). Lsb1 is highly homologous to Lsb2, however, it does not promote [PSI+] nucleation. Here, we show that, similar to the deletion of LSB2, the deletion of LSB1 increases destabilization of [PSI+] by short-term heat shock, whereas a combination of both deletions has a cumulative effect. Moreover, the proteolytic processing of Lsb1 at the C-proximal region is triggered by heat shock and is required for its role in prion maintenance. Cycling of Lsb1 between actin patches, cytosol, and the nuclear-ER rim is controlled by Rsp5-dependent ubiquitination, proteolytic processing, and the Guided Entry of Tail-anchored proteins (GET) pathway. Our data show that both Lsb1 and Lsb2 are involved in the cellular machinery of aggregate segregation in the stressed cells.
EXPERIMENTAL PROCEDURES
Strains and Plasmids
Saccharomyces cerevisiae strains used in this study are listed in the Table 1. Disruptions, tagging, and mutations of chromosomal genes were generated by PCR-mediated gene replacement (27) and delitto perfetto, in vivo site-directed mutagenesis (28) and verified by sequencing.
TABLE 1.
Yeast strains
| Strain | Genotype | Source |
|---|---|---|
| GT81-1C | MATa ade1-14 (UGA), his3-Δ200, leu2-3,112 lys2-801, trp1-289 ura3-52 [PSI+ PIN+] | 62 |
| GT982 | get2Δ::HISMX6 derivative of GT81-C | 31 |
| GT1929-1C | kanMX6::PGAL-GFP-SBH1 in GT81-C | 31 |
| MHY501 | MATα his3- Δ200 leu2-3,112 lys2-801, trp1-1 ura3-52 | 63 |
| MHY3646 | doa3-1 derivative of MHY501 | 63 |
| WTY623 | lsb2Δ::kanMX6 disruption of MHY501 | 26 |
| WTY659 | lsb1Δ::HIS3MX6 disruption of MHY501 | This study |
| WTY660 | lsb2Δ::kanMX6, lsb1Δ::HIS3MX6 in MHY501 | This study |
| pJ69-4A | PGAL-ADE2 [psi−] | 37 |
| FW1808 | MATα, rsp5-1, his4-912δR5, lys2-128δ, ura3-52 | 42 |
| FY56 | MATα, his4-912δR5, lys2-128δ, ura3-52 | 42 |
| OT55 | MATa, ade1-14 (UGA), his3-Δ200 leu2-3, 112 ura3-52, trp1-289 [PSI+] [PIN+] | 64 |
| WTY682 | lsb2Δ::kanMX6 disruption of OT55 | 26 |
| WTY730 | lsb1Δ::HIS3MX6 disruption of OT55 | This study |
| WTY731 | lsb1Δ::HIS3MX6 disruption of WTY682 | This study |
| WTY637 | lsb1Δ::KlURA3, hyg, GAL1-I-SceI disruption of WTY 682 | This study |
| WTY643 | lsb1 Y182A, Y183A replacement of WTY 637 | This study |
| WTY649 | lsb1 K79R replacement of WTY 637 | This study |
| WGC4a | MATa leu2-3, 112 ura3, his3-11, 15CanS, GAL2 | 44 |
| YUS5 | pup1-T30A, pre3-T20A mutant of WGC4a | 44 |
| ACY1568 | MATα MLP1-RFP-kanMX6 leu2, his3, ura3,lys2 | 47 |
The LSB1 coding sequence was cloned under copper inducible promoter PCUP1 either in pRS316 or pRS315, pGAD, and pGBDU. Mutations and fusions of LSB1 with GFP, mCherry, FLAG, HA, or yeast two-hybrid DNA-binding/activating domains were achieved via the overlapping PCR cloning approach. The following plasmids were also used in this study: pGBDU and pACTII with Sup35N fragment, which includes the first 113 codons of SUP35 (6), pGAD-Lsb1 and pGBDU-Lsb2 wild type and mutant (26) for yeast two-hybrid analysis; pYep105-Myc-Ub for analysis of Lsb1 ubiquitination (29); pYes-HA-Rsp5ΔC for analysis of Lsb1 stability (30); pRS315-Sup35-dsRED (31) and pRS316-Lsb1-GFP (26) to study protein co-localization.
Growth Conditions
Standard yeast media, cultivation conditions, procedures for transformation, phenotype scoring, and yeast two-hybrid analysis were used (32).
[PSI+] Detection
The presence of [PSI+] was monitored by its ability to suppress the reporter ade1-14 (containing the premature stop codon, UGA), resulting in a light-pink color on complete YPD medium, as opposed to the deep red color of a [psi−] strain growth on media lacking adenine (−Ade) (33).
Heat Shock Experiment
Various exponential yeast cultures grown at 25 °C were shifted to 39 °C. Aliquots were collected at the specified time points. For protein analysis cells were lysed by boiling and analyzed by Western blotting. For [PSI+] curing experiments, aliquots were plated onto YPD medium, and incubated at 25 °C (21). [PSI+] (pink), [psi−] (red), and mosaic [PSI+]/[psi−] colonies were detected by visual inspection.
Analysis of [PSI+] Segregation by Micromanipulation
Individual cells from heat-shocked culture were placed onto YPD, spaced apart by using the Singer micromanipulator MSM system Series 300, and allowed to form buds. The first bud (“daughter”) was separated from the original cell (“mother”) by micromanipulation. After each cell formed a colony, the presence of [PSI+] was monitored by color (21). The probability of random deviation of the observed results from those expected in case of equal segregation of [PSI+] between the mother and daughter cells has been calculated by using the χ-square approach and table (34).
Protein Analysis
Cells were lysed by boiling in SDS loading buffer or by vortexing with glass beads (for immunoprecipitation (IP) and cellular fractionation) (26). The IP experiments were performed using FLAG M2 affinity gel (Sigma) or anti-HA-agarose (Thermo Scientific). Cell extracts were fractionated by centrifugation at 25,000 × g. The sucrose step gradient was performed as outlined (35). The cycloheximide chase experiment was performed as described (26). To examine the effect of proteasome inhibitors on processing of Lsb1, MG132 was added to the cells growing in a synthetic medium supplemented with 0.1% proline and 0.003% SDS to increase an uptake of MG132 (Sigma) as described previously (36). An equal amount of cells was collected at the specific time point and lysed by boiling to isolate protein. Protein extracts and IPs were examined by Western analysis with specific antibodies. We used the following specific antibodies: anti-HA HA.11 (Covance, Inc., Emerville, California), anti-Myc 9B11 (Cell Signaling Technology), anti-Pgk (Molecular Probes, Inc., Eugene, OR), anti-Rpt5 (Enzo Life Science (Plymouth Meeting, PA), anti-Lsb1/2 generated by ProSci, Inc., (Poway, Ca), anti-Pma1 (Abcam), and anti-Dpm1 (Molecular Probes). In all experiments, we used appropriate secondary antibodies from GE Healthcare Ltd.
Liquid Chromatography Coupled to Tandem Mass Spectrometry (LC-MS/MS)
To identify the processing site of Lsb1, full-length and processed Lsb1 (tagged with HA) were immunoprecipitated using anti-HA-agarose, separated by SDS-PAGE, and analyzed by LC-MS/MS after in-gel tryptic digestion. For this purpose, protein samples were run on a 10% SDS-PAGE gel and stained with Coomassie Blue. Slices containing the respective bands were cut off the gel, distained, and subjected to in-gel digestion (12.5 ng/μl of chymotrypsin). Extracted peptides were loaded onto a C18 column (100-μm inner diameter, 15 cm long, ∼300 nl/min flow rate, 3 μm resin from Michrom Bioresources, Auburn, CA) and eluted during a 10–30% gradient (Buffer A: 0.4% acetic acid, 0.005% heptafluorobutyric acid, and 5% AcN; Buffer B: 0.4% acetic acid, 0.005% heptafluorobutyric acid, and 95% AcN). The eluted peptides were detected by Orbitrap (300–1600 m/z, 1,000,000 automatic gating control target, 1,000-ms maximum ion time, resolution 30,000). MS/MS scans in an LTQ linear-ion trap mass spectrometer (2 m/z isolation width, 35% collision energy, 5,000 automatic gating control target, 150 ms maximum ion time) (Thermo Finnigan, San Jose, CA) were acquired by data-dependent acquisition. All data were converted from raw files to the .dta format using ExtractMS version 2.0 (Thermo Electron) and searched against Yeast Reference Database downloaded from the National Center for Biotechnology Information using the SEQUEST Sorcerer algorithm (version 3.11, SAGE-N). Searching parameters included mass tolerance of precursor ions (±50 ppm) and product ion (±0.5 m/z), semichymotryptic restriction, with a dynamic mass shift for oxidized Met (+15.9949). Only b and y ions were considered during the database match. To evaluate the false discovery rate, all original protein sequences were reversed to generate a decoy database that was concatenated to the original database. The false discovery rate was estimated by the number of decoy matches (nd), and total number of assigned matches (nt). False discovery rate = 2 × nd/nt, assuming that mismatches in the original database were the same as in the decoy database. All MS/MS spectra for proteins identified by a single peptide were manually inspected.
Fluorescence Microscopy
Proteins with the indicated fluorescent tag were imaged in living cells with a ×100 oil immersion objective on the Olympus IX81 microscope (Olympus America, Inc., Melville, NY), equipped with a Hamamatsu digital camera (Hamamatsu photomics, Japan). Time lapse experiments (supplemental Movies S1 and S2) were carried out on Applied Precision Delta Vision Core system (Applied Precision, Issaquah, WA) with Olympus ×100 1.35 NA oil immersion objective (Olympus America, Inc. Melville, NY) and deconvolved with SoftWoRx software. Colocalization of Lsb1-GFP with Sup35-NM-dsRED were analyzed on the Zeiss LSM510 confocal microscope (Carl Zeiss, Inc., Thornwood, NY) with a ×100 1.3 NA Zeiss oil immersion objective. GFP was excited with a 488-nm laser line attenuated to 1.25% of laser intensity (Argon 40 milliwatt), Ds Red was excited with 543 nm attenuated to 34% of laser intensity (1 milliwatt HeNe).
RESULTS
Roles of Lsb1 and Lsb2 in Viability and [PSI+] Maintenance during and after Stress
There are many different variants of the [PSI+] prion found in yeast (19, 20). In this work, we employ a weak [PSI+] variant, which is more sensitive to stress. In the yeast strain OT55 (21), bearing a weak [PSI+] variant, the prion is destabilized if cell divisions are resumed after short term, mild HS (30–60 min, 39 °C). However, if cells are incubated at higher temperature for a longer time (several hours) and then resume cell divisions, prion destabilization is not observed, i.e. it is not lost after longer incubation at high temperature (21). As Sup35 is a release factor, its inactive prion form exhibits a translation termination defect, leading to the readthrough of a stop codon. In the strains bearing an ade1–14 (UGA) reporter allele, this defect is phenotypically detectable by growth on −Ade medium and a light-pink color on complete YPD medium, as opposed to the deep red color of a [psi−] strain (33).
A [PSI+] culture treated by short-term HS produces a few red [psi−] colonies and a larger fraction of mosaic [PSI+]/[psi−] (pink/red) colonies (Fig. 1A), indicative of a prion loss occurring preferentially upon cell division during recovery from HS. However, such a destabilization of the [PSI+] prion is not detected if cells are incubated at 39 °C for longer times (up to 4 h) before cell division is allowed to resume (21). Under normal growth conditions, strains with deletions of LSB1 and/or LSB2 propagated the weak [PSI+] identically to the wild-type strain. However, deletion of LSB2 significantly increased the [PSI+]-destabilizing effect of HS (26). We have now demonstrated that lsb1Δ also increases heat shock-induced [PSI+] destabilization, although less dramatically than lsb2Δ (Fig. 1B). Notably, the double lsb1Δ lsb2Δ strain showed the same magnitude of [PSI+] destabilization as the single lsb2Δ strain after short exposures to HS, but exhibited increased prion loss after longer exposure to HS, in comparison to both wild type and single deletion strains. Each deletion (and most dramatically the double lsb1Δ lsb2Δ) significantly increased the proportion of red versus mosaic colonies, indicating that lack of Lsb protein(s) impairs the ability of the [PSI+] prion to survive HS treatment more than it affects segregation in cell divisions following HS (data not shown). Although loss of viability in the wild type, lsb1Δ and/or lsb2Δ exponential cultures at 39 °C is very modest (Fig. 1C), it can be detected by micromanipulation analysis of heat-shocked cells; moreover, HS-induced cell death is increased in the lsb1Δ lsb2Δ strain compared with the wild type strain (Table 2). In addition, the heat-shocked lsb1Δ lsb2Δ culture contained more cells producing only one viable cell in the first cell division after HS. Morphological asymmetry of the yeast cell division allowed us to determine that the daughter cell (originated from the bud) was more frequently viable in these cases. This cannot completely explain an increase in the proportion of red (completely “cured”) colonies, as the proportion of cell divisions where both cells were viable but have lost a prion was also increased in the lsb1Δ lsb2Δ strain. In cases when both products of the first post-HS division were viable and only one of them had lost a prion, heat-shocked lsb1Δ lsb2Δ cells showed a bias toward prion retention in the mother cell as observed previously (21) and now confirmed for the wild type strain (Fig. 1D). The lsb1Δ lsb2Δ cells retained this bias after 4-h exposure to heat stress, whereas [PSI+] prion in the wild type strain was almost never lost. These data confirm a tendency to segregate the prion into the mother cell in the cell divisions following HS reported previously (21), and show that Lsb depletion enhances this asymmetry.
FIGURE 1.

Effects of Lsb proteins on [PSI+]. A, [PSI+] destabilization by HS. B, lsb1Δ affects [PSI+] destabilization during HS. In panels A and B, yeast cultures were grown to early exponential stage at 25 °C, shifted to 39 °C for the specified periods of time, plated on YPD and incubated at 25 °C. [PSI+] (pink), [psi−] (red), and mosaic [PSI+]/[psi−] colonies were detected by visual inspection. The percentages of colonies in which prion was destabilized by heat shock (red [psi−] and [PSI+]/[psi−] mosaic) are indicated. Averages of six experiments are shown with error bars correspond to standard deviations. C, the lsb1Δ and/or lsb2Δ do not affect viability of the exponential [PSI+] culture at 39 °C. The control strain LSB1, LSB2 (OT55), and isogenic lsb1Δ and/or lsb2Δ strains were pre-grown at 25 °C, and aliquots were placed at 25 and 39 °C. Serial decimal dilutions were prepared after the indicated periods of time, and 2.5 μl of each dilution were spotted onto YPD medium. Plates were photographed after 6 days at 30 °C. D, analysis of the first cell divisions after HS at 39 °C. After resumption of cell division, mother and daughter cells were separated by micromanipulation. Only pairs where both mother and daughter were viable are presented (for viability data, see Table 2). For each type of mother/daughter [PSI+] distribution, numbers are shown. The probability of random deviation of the observed results from those expected in case of equal segregation of [PSI+] between the mother and daughter cells, calculated by using the χ-square approach, is indicated.
TABLE 2.
Viability of dissected mother and daughter cells in yeast cultures treated with mild heat shock
Exponential yeast cultures were incubated at 39 °C for the specified periods of time. Mother and daughter cells were separated from each other by micromanipulation. Numbers are shown for each type of mother/daughter.
| Time at 39 °C |
|||
|---|---|---|---|
| 30 min | 60 min | 240 min | |
| WT | |||
| Viable mother and daughter | 265 (64%) | 427 (68%) | 75 (69%) |
| Viable mother | 5 (1%) | 9 (1%) | 0 (0%) |
| Viable daughter | 17 (4%) | 12 (2%) | 3 (3%) |
| Delay in budding | 21 | 26 | 3 |
| Petitte | 1 | 5 | 4 |
| Total viable | 309 (75%) | 479 (77%) | 85 (79%) |
| Total cells analyzed | 412 | 624 | 108 |
| lsb1Δ lsb2Δ | |||
| Viable mother and daughter | 318 (52%) | 371 (49%) | 207 (41%) |
| Viable mother | 18 (3%) | 24 (3%) | 13 (3%) |
| Viable daughter | 23 (4%) | 26 (3%) | 36 (7%) |
| Delay in budding | 32 | 36 | 29 |
| Petitte | 15 | 14 | 6 |
| Total viable | 406 (66%) | 471 (62%) | 296 (59%) |
| Total cells analyzed | 612 | 757 | 499 |
Lsb1 and Lsb2 Bind to Each Other via N-terminal Domains, Which Are Also Required for Their Interactions with Sup35
Previously we have shown that Lsb1 and Lsb2 interact with the Sup35 prion domain (26). To identify domains of Lsb proteins involved in interaction with Sup35, we performed yeast two-hybrid analysis (37) of various truncated constructs of Lsb1 and Lsb2 (Fig. 2, A and B). Our data show that in each Lsb2 and Lsb1, the C-terminal 32 (Lsb2) or 59 (Lsb1) amino acids are dispensable, whereas the N-terminal 53 amino acids are essential (although not sufficient) for interactions with the Sup35 prion domain. We have also found that Lsb1 and Lsb2 interact with each other in yeast two-hybrid assays, and that their conserved N-terminal domains are both essential and sufficient for Lsb1/Lsb2 association (Fig. 2C). In contrast, mutations at conserved Trp-90 (Lsb1) or Trp-91 (Lsb2) position, essential for most other SH3-mediated protein interactions (38), did not abolish interaction of Lsb proteins with each other (Fig. 2C). We have confirmed the Lsb1-Lsb2 interaction in cell extracts by pulldown experiments (Fig. 2D).
FIGURE 2.

Lsb proteins interact with each other and with the prion domain of Sup35. A, structural organization of the Lsb1 and Lsb2 proteins. Conserved lysine (K), tryptophan (W), glutamine (Q), and tyrosine (Y) as well as putative Rsp5 binding sites PPSY (Lsb1) and PPQY (Lsb2) are shown. Superscript and subscript numbers correspond to amino acid positions and the number of repeated residues in a stretch, respectively. Black bars with numbers indicate the numbers of amino acids used to make truncations. B and C, yeast two-hybrid assay. Lsb proteins (full size or truncated versions) and the Sup35 prion domain consisting of the first 113 codons of SUP35 fused to activation (AD) and DNA binding (BD) domains of Gal4 were used. Two-hybrid interaction is detected by activation of the GAL-ADE2 reporter construct, resulting in growth on −Ade. B, the C-terminal region of Lsb1/Lsb2 is not required for interaction with Sup35. C, the N-terminal region of Lsb1/Lsb2 is required and sufficient for their interactions with each other. Mutation of the conservative tryptophan residue does not affect this interaction. D, biochemical detection of Lsb1/Lsb2s interaction. Yeast lysates prepared from cells expressing HA-Lsb1 and FLAG-Lsb2-HA from plasmid PCUP1 promoter were incubated with anti-FLAG-agarose. Bound proteins were analyzed by SDS-PAGE and anti-HA immunoblotting. Lysate of cells expressing HA-Lsb1 and Myc-Lsb2-HA proteins was used as a control. 10% of total protein used for immunoprecipitation was loaded as the “lysate.” The processed form of Lsb1 (see Fig. 3) is not distinguishable because of the overlap with more abundant Lsb2.
Lsb1 Protein Undergoes Stress-inducible Processing at the C-terminal
Lsb1 protein, expressed from the endogenous chromosomal promoter, exists as two bands reacting with Lsb1/2 antibody on Western blots (Fig. 3A), suggesting that it undergoes proteolytic processing. This result was reproduced in two different genotypic and prion backgrounds, OT55 ([PSI+]) (see Fig. 3A) and MHY501 ([psi−]) (data not shown). To determine which portion of the protein is lost on processing, we have constructed Lsb1 derivatives tagged with either HA or GFP at either the N or C terminus, and used tag-specific antibodies for protein detection. For N-tagged Lsb1, two bands were detected that correspond to the full size and processed protein (Fig. 3B). In the case of C terminally tagged Lsb1, tag-specific antibody detected only a full-length band. This indicated that the lower molecular weight (Mr) Lsb1′ band lacks the C-terminal region. Processing of Lsb1 did not depend upon the presence of Lsb2 protein, as it occurred efficiently in the lsb2Δ strains (Fig. 3A).
FIGURE 3.

Processing of Lsb1. A, Lsb1, but not Lsb2, is processed. Lsb1 and Lsb2 expressed from endogenous promoter at the chromosomal foci were identified in cell lysates by anti-Lsb Ab. B, Lsb1 undergoes processing at the C terminus as detected by HA-tag/anti-HA Ab (left) and GFP-tag/anti-GFP Ab (right). N terminally or C terminally tagged proteins were expressed from plasmid PCUP1 promoter in cells growing at 30 °C. C, HS induces processing of Lsb1 expressed from chromosomal endogenous promoter and detected by anti-Lsb Ab. Pgk1 protein detected by anti-Pgk1 Ab was used as a loading control (A–C). D, densitometry was used to determine the relative levels of total, processed, and unprocessed form of Lsb1 protein after 60 min at 39 °C. Average measurements of levels of Lsb1 forms at 0 min at 39 °C for each blot were set to a value 1.0 and compared with measurement of the same form of protein after 60 min at 39 °C. The error bar represents mean ± S.D. for 6 independent experiments in each case.
We have shown previously that levels of Lsb2 protein, but not full-length Lsb1 protein, are dramatically increased during HS (26). However, we have now found that processed Lsb1 protein (Lsb1′) is increased in abundance (up to more than 5-fold) after 45–60 min at 39 °C, despite only a slight increase in abundance of full-length Lsb1 (Fig. 3, C and D). Therefore Lsb1 is actually induced by HS, however, Lsb1 processing is also induced, so that the level of the full size protein is not increased dramatically.
To determine the precise site of processing, full-length and processed HA-Lsb1 protein were purified from yeast and subjected to mass spectrometry analysis. Two adjacent tyrosine residues, Tyr-182 and Tyr-183, were identified as potential processing sites (Fig. 4A). Mutation of each single tyrosine to alanine (Ala) resulted in only a slight decrease of HA-Lsb1 protein processing, whereas substitution of both tyrosines (Y182A,Y183A), completely prevented it (Fig. 4, B and C). These mutations did not affect protein levels and stability of Lsb1 (Fig. 4D). We made a truncated form of Lsb1 (1–183 amino acids) corresponding to the processed form Lsb1′ and showed that it is accumulated at the levels similar to the wild type protein and has the same size as Lsb1′ as judged by mobility on SDS-PAGE (Fig. 4D).
FIGURE 4.

Lsb1 is processed at residues Tyr-182,Tyr-183. A, detection of the C-terminal peptide of processed Lsb1 protein by LC-MS/MS. Y.MQAPPPQQQQAPLPYPPPFTNY.Y was identified as the potential C-terminal peptide. B, Y182A,Y183A substitution blocks Lsb1 processing. HA-tagged wild type, single, and double mutants of Lsb1 were induced from PCUP1 for the indicated period of time and analyzed by immunoblotting with anti-HA Ab. C, thermal stress induces processing of wild type but not the Y182A,Y183A mutant. Lsb1 proteins are expressed from the endogenous promoter. Protein levels were analyzed at the indicated time points using anti-Lsb Ab. D, molecular weights of the unprocessed and processed forms of Lsb1 are equal to molecular weights of the full size Y182A,Y183A mutant Lsb1 protein and truncated Lsb1 protein (1–183 amino acids), respectively, as judged from the SDS-PAGE gel. Wild type, mutant, and truncated proteins, tagged with HA at N termini, were expressed from the copper-inducible promoter for the indicated periods of time. Protein extracts were run on SDS-PAGE gel and detected with the anti-HA antibody. Pgk1 protein detected by anti-Pgk1 Ab was used as a loading control (B–D). E, double substitution Y182A,Y183A in LSB1 (in combination with lsb2Δ) affects [PSI+] destabilization and recovery during HS the same way as lsb1Δ. Yeast were grown and treated by HS as in Fig. 1B.
Lsb1 Processing Is Essential for Its Function in the Maintenance of [PSI+] Prion
The Lsb1 protein with the Y182A,Y183A substitution, preventing Lsb1 processing, did not significantly affect the HS-induced prion curing destabilization (data not shown). However, the combination of the lsb1-Y182A,Y183A mutant allele with lsb2Δ increased [PSI+] destabilization by HS in a manner similar to the double deletion of LSB1 and LSB2 (Fig. 4E). The strain bearing both lsb2Δ and lsb1-Y182A,Y183A exhibited increased prion loss after a longer incubation at 39 °C. These data confirm the physiological relevance of Lsb1 processing, implicating it as a key event involved in Lsb1 function in prion maintenance during HS.
Lsb Proteins Co-localize with Each Other and with Sup35
Next, we employed fluorescence microscopy to determine whether Lsb1 and Lsb2 are co-localized in live cells. These experiments employed Lsb2-mCherry and both N-terminal and C-terminal GFP fusions to Lsb1. The N-terminal GFP fusion should allow visualization of both full size and processed Lsb1 isoforms, respectively, GFP-Lsb1 and GFP-Lsb1′ (Fig. 3B). In contrast, the C-terminal GFP fusion should allow only visualization of the full size Lsb1-GFP (Lsb1′ is not identifiable because GFP is cleaved off by processing) and GFP-tagged C-terminal fragment of Lsb1 (Lsb1CT-GFP) released during processing. We expected that the difference in localization of GFP-Lsb1 and Lsb1-GFP would permit a distinction between localization of full sized Lsb1 and cleaved Lsb1 (Lsb1′ and Lsb1CT). Surprisingly the pattern of cellular distribution of GFP-Lsb1 and Lsb1-GFP was the same: the GFP signal was distributed throughout the cytoplasm and accumulated in multiple punctate structures and in larger aggregates (Fig. 5A). This suggests that in both cases we are most likely observing localization of a full size Lsb1 (GFP-Lsb1 and Lsb1-GFP). The cleaved forms of Lsb1 (Lsb1′ and Lsb1CT) either are present at the same location as a full size protein or are not detectable. Indeed the amount of cleaved Lsb1 as detected by Western blot was smaller then the amount of the full size Lsb1 present in the same cells (Fig. 3, B and C). As it has been shown previously (26), Lsb2 was observed throughout the cytoplasm, accumulated in multiple punctate structures associated with actin patches, and in larger aggregates (Fig. 5A). In most (although not all) punctate structures and aggregates, Lsb1 and Lsb2 proteins were co-localized (Fig. 5A). N-terminal domains of Lsb1 and Lsb2 (1–53 and 1–52, respectively), tagged with fluorescent proteins did not form the punctate actin patch-associated structures (Fig. 5A), confirming our previous data (26) showing the requirement for the SH3 domains of Lsb proteins in the association with actin structures.
FIGURE 5.
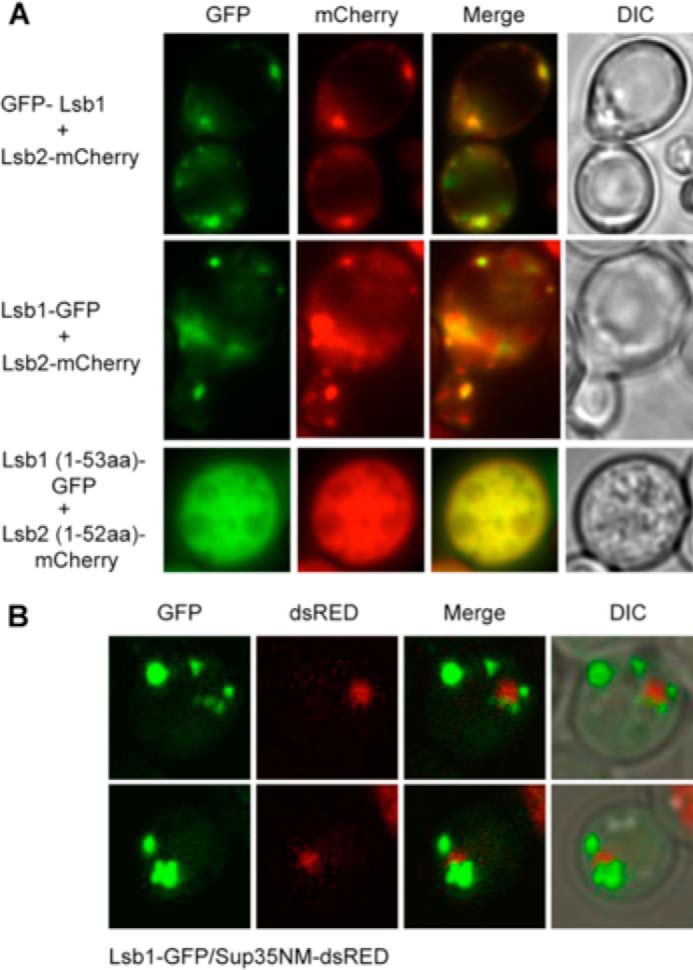
Colocalization of Lsb proteins with each other and Sup35. A, Lsb1 and Lsb2 wild type or truncated proteins tagged with GFP (Lsb1) and mCherry (Lsb2) at the N or C terminus as indicated on the figure were co-expressed from the PCUP1 promoter for 3 h at 30 °C in [psi−rnq−] strain. Fluorescent microscopy images of live cells are shown. B, aggregates of Sup35NM-dsRED are surrounded by puncta of Lsb1-GFP. Both proteins were co-expressed from the PCUP1 promoter for 24 h in the [psi−RNQ+] strain. Projection of 15 z-focal planes collected with 0.4-μm step size on Zeiss LSM510 confocal microscope (Carl Zeiss, Inc., Thornwood, NY) with a ×100 1.3 NA Zeiss oil immersion objective. DIC, differential interference contrast.
To determine whether Sup35 aggregates are associated with Lsb1 structures, Lsb1-GFP and PrD-containing fragment of Sup35 (Sup35NM) tagged with dsRed were co-expressed at high levels in the [psi−RNQ+] strain. Sup35 aggregates were partially overlapping with or fully encircled by Lsb1 puncta (Fig. 5B), as shown previously for Sup35 and Lsb2 (26).
Lsb1 Is Ubiquitinated at Lys-79 by the E3 Ligase Rsp5
Previous large scale mass spectrometry analysis identified Lsb1 as a protein ubiquitinated at Lys-41 and Lys-79 residues (39). To verify this, we expressed Lsb1 with the N-terminal HA tag at high levels under the PCUP1 promoter in the presence of Myc-Ub (29). HA-Lsb1 accumulated as higher Mr bands in addition to full-length and processed forms (Fig. 6A). IP of HA-Lsb1 from the strain expressing Myc-Ub confirmed that these high Mr bands indeed represent ubiquitinated Lsb1 (Fig. 6B). The truncated form of Lsb1 (1–183 amino acids), corresponding to Lsb1′, was also ubiquitinated (Fig. 4D). Similarly, the Y182A,Y183A substitution did not affect ubiquitination of Lsb1 (Fig. 4D). The amount of ubiquitinated Lsb1′ varied among experiments. Possibly, Lsb1′-Ub is less abundant or more efficiently degraded, compared with Lsb1-Ub. Also, as Lsb1′-Ub migrates just above Lsb1, high levels of Lsb1 might hinder detection of Lsb1′-Ub at higher exposures (Fig. 6D).
FIGURE 6.

Effects of ubiquitin-proteasome system on Lsb1 processing. A, substitution of a single lysine residue, K79R, in Lsb1 prevents accumulation of high molecular weight (Mr) conjugates. HA-Lsb1, its derivatives, and Myc-Ub were expressed from the copper-inducible promoter PCUP1. Lsb1 was detected with anti-HA Ab. B, high Mr conjugates represent ubiquinated Lsb1. HA-Lsb1 and Myc-Ub were expressed from PCUP1. Lsb1 was immunoprecipitated with anti-HA Ab. 10% of total protein used for immunoprecipitation was loaded as the “lysate.” High Mr conjugates of HA-Lsb1 react to anti-Myc Ab, confirming that they contain Myc-Ub. C, double P135A,P136A substitution in the potential Rsp5 binding site prevents Lsb1 ubiquitination. HA-Lsb1 wild type, mutant, and Myc-Ub were expressed from PCUP1. Lsb1 was detected with anti-HA Ab. D, E3 enzyme Rsp5 is involved in Lsb1 ubiquitination. HA-Lsb1 was expressed from PCUP1 in wild type and rsp5-1 temperature-sensitive mutant cells growing at 37 °C. HA-Lsb1 accumulates in mutant cells after 3 h of induction (left panel). Ubiquitinated Lsb1 can be detected in the wild type, but not in mutant cells after 24 h of induction (right panel). Lsb1 protein was identified by anti-HA Ab. E, partial stabilization of Lsb1 in cells deficient in Rsp5 activity. HA-tagged Lsb1 was expressed from the PCUP1 promoter in cells transformed either with vector (control) or plasmid expressing mutant Rsp5ΔC from PGAL promoter. Cells were grown in the presence of copper and galactose and protein levels of Lsb1 were analyzed at the indicated time points after the addition of cycloheximide (CHX). F, processing of Lsb1 is not affected in the mutant strain deficient in the trypsin-like (pre3-T20A) and postacidic/post-glutamic-like (pup1-T30A) activities of the proteasome. G, Lsb1 is processed in the mutant strain deficient in chymotrypsin-like activity of the proteasome (pre2-1/doa3-1). Cells were grown overnight, diluted in the morning, and incubated with shaking at 30 °C (F) or at the temperature indicated (G). Equal amounts of cells were collected, lysed by boiling, and protein was detected by Western blot with anti-Lsb Ab. H, the Lsb1 processing is inhibited by MG132. Processed Lsb1 is not detected after inhibition of chymotryptic peptidase activity of the proteasome by the addition of the proteasome inhibitor MG132 to the wild-type (WT) strain or to the mutant, pup1-T30A,pre3-T20A defective in the other peptidase activities of the proteasome. Cells incubated with MG132 for the indicated periods of time were collected, lysed by boiling, and analyzed by Western blot with anti-Lsb Ab. Pgk1 protein detected by anti-Pgk1 Ab (A, C, D, F, and G) and Rpt5 protein detected by anti-Rpt5 Ab (H) were used as a loading control.
The Lsb1 K79R mutation increased the level of non-ubiquitinated Lsb1 and eliminated high Mr bands, whereas the K41R mutation had no effect (Fig. 6, A and B), indicating that under our conditions, Lys-79 is the major Lsb1 ubiquitination site. We constructed a strain bearing both lsb2Δ and lsb1-K79R and demonstrated that the lsb1-K79R mutation did not affect prion loss. These data demonstrate that ubiquitination of Lsb1 is not critical for prion maintenance during HS.
We have confirmed previous data suggesting that the E3 Ub ligase Rsp5 is responsible for ubiquitination of Lsb1 (40, 41) by demonstrating that the P135A,P136A substitution within the Rsp5 recognition site (PY motif) of Lsb1, identified as PPSY138, abolishes Lsb1 ubiquitination (Fig. 6C). In agreement with these data, the levels of Lsb1 and Lsb1′ are dramatically increased in a strain harboring the temperature-sensitive mutation rsp5-1 (42) grown at non-permissive temperature, possibly due to blocked ubiquitination, leading to Lsb1 and Lsb1′ accumulation (Fig. 6D). It was difficult to use the rsp5-1 ts mutant for checking if Rsp5 regulates Lsb1 stability, because incubation at non-permissive temperature (37 °C) also affects levels and processing of Lsb proteins. Therefore, we have simultaneously expressed HA-Lsb1 and the dominant-negative mutant Rsp5ΔC from the PCUP and PGAL promoters, respectively, and used anti-HA Ab for Lsb1 detection. By using cycloheximide chase, we have observed partial stabilization of Lsb1 in the presence of Rsp5ΔC, compared with cells expressing endogenous wild type Rsp5 only (Fig. 6E).
Processing of Lsb1 Is Inhibited by the Proteasome Inhibitor MG132
Several key cellular factors, including NF-κB and Spt23, are activated via limited proteolytic processing by the proteasome, following protein ubiquitination (43). We thus asked whether Lsb1 processing depends on proteasome function and/or on ubiquitination. There are three eukaryotic 20 S proteasomal β-type proteolytically active subunits: Pre3, Pup1, and Pre2/Doa3, which provide the postacidic/post-glutamic-like, the trypsin-like, and the chymotrypsin-like activities, respectively (44). To analyze the role of proteasome in the Lsb1 processing, we analyzed processing of Lsb1 in the double mutant pup1-T30,pre3-T20 defective in both postacidic/post-glutamic-like and the trypsin-like activities and in temperature-sensitive mutant pre2-1/doa3-1 with defective chymotrypsin-like activity of the proteasome (Fig. 6, F and G). We have found that Lsb1 is processed in both mutant strains. In the pup1-T30,pre3-T20 mutant the amount of processed and full size Lsb1 was not different from the amount in wild type cells (Fig. 6F). Alternatively the amount of Lsb1 was increased in the doa3-1 mutant compared with a corresponding wild type strain. Therefore, there was more of processed Lsb1 (Lsb1′) present in the mutant then in a wild type cells grown at 25 °C (Fig. 6G). These differences could be explained by our previous finding that Lsb1 is a short-lived protein and its half-life is increased in the doa3-1 mutant (26). The fact that Lsb1 is processed in both mutant strains manifests a possibility that all three proteasome activities can be involved in Lsb1 processing and compensate for each other in mutant strains. To completely inhibit all three proteasome proteolytic activities we decided to use a combination of MG132 (chemical inhibitor of chymotrypsin-like activity) and the pup1-T30,pre3-T20 mutant as described (45). This also should eliminate an effect of higher temperature (37 °C) on Lsb1 processing, which could affect the results of the experiment involving the temperature-sensitive mutant pre2-1/doa3-1. We have treated wild type and pup1-T30,pre3-T20 mutant cells with the proteasome inhibitor MG132. This treatment entirely abolished the Lsb1 processing, suggesting that, the chymotrypsin-like activity of the proteasome inhibited by MG132 (36), could be responsible for processing at least in the absence of two other activities (Fig. 6H). An alternative explanation is that processing might be dependent on some other yet unknown MG132-sensitive protease, because MG132 is known to inhibit other proteases at higher concentrations (46).
Full-length Lsb1 Is a Membrane-associated Protein
Processing of Lsb1 removes the C-terminal hydrophobic tail, which could potentially act as a transmembrane anchor. To determine whether processing influences the intracellular localization of Lsb1, we studied distribution of HA-Lsb1 between the soluble and pelleted (membrane-bound) fractions separated by centrifugation. Processed Lsb1 was found exclusively in the soluble fraction, whereas full-length protein was detected in similar amounts in both the soluble and pellet fractions (Fig. 7A). Likewise, analysis of untagged Lsb1 protein expressed from the endogenous promoter demonstrates that only full-length Lsb1 is associated with the membrane fraction (data not shown). Distribution of HA-Lsb1 and untagged Lsb1 was the same in [PSI+] strain OT55 and [psi−] strain MHY501. Lsb1 was extracted from the pellet fraction by detergents such as Triton X-100 or SDS, but not by high salt nor in a high pH buffer (Fig. 7B), consistent with Lsb1 being a membrane-associated protein. Fractionation of the pellet fraction on a sucrose gradient confirmed co-sedimentation of Lsb1 exclusively with the fractions containing markers of endoplasmic reticulum (ER) or plasma membranes (Fig. 7C). The processing-defective HA-Lsb1 protein with the Y182A,Y183A mutation was present in both soluble and pellet fractions, indicating that this mutation does not affect Lsb1 association with membranes (Fig. 7A).
FIGURE 7.

Association of Lsb1 with membranes. A, total extracts (T) of cells expressing HA-tagged Lsb1 (WT or Y182A,Y183A mutant) from PCUP1 were separated into soluble (S) and pellet (P) fractions by centrifugation, separated by SDS-gel electrophoresis, and probed with anti-HA Ab. Unprocessed (Lsb1) and processed (Lsb1′) forms are indicated. B, the pellet fraction obtained from HA-Lsb1 expressing cells (A) was washed with detergents, salts, and buffer as indicated, separated into pellet and soluble fractions as in A, and probed with anti-HA Ab. C, sucrose gradient of pelleted Lsb1 protein. Left panel, total extracts of cells expressing HA-tagged Lsb1 were separated into soluble and pellet fractions and analyzed as in A. Right panel, the pellet fraction was subjected to sucrose gradient and fractions were analyzed by SDS-PAGE and anti-HA immunoblotting to detect Lsb1. A–C, protein markers of plasma membrane (Pma1), ER (Dpm1), and cytoplasm (Pgk1) were detected with protein-specific antibodies.
Fluorescence microscopy demonstrated that full-length Lsb1, tagged with fluorescent protein (GFP or mCherry) at the N terminus, is found in the cytosol, in multiple punctate structures associated with actin patches near the plasma membrane, and in the nuclear-ER rim when cells grow at 25 °C (Fig. 8A, supplemental Movie S1). This interpretation is confirmed in cells with the position of the nucleus indicated by Hoechst staining (Fig. 8B) and by co-localization with ER marker protein Sbh1 (31) and nuclear envelope protein Mlp1-RFP (47) (Fig. 8, C and D). At a higher temperature (30 °C), localization of GFP-Lsb1 to the nuclear-ER rim was lost (Fig. 8A), consistent with the increased abundance of the processed form, which is confined to cytosol (see above). GFP-Lsb1 (1–182), corresponding to the processed form of Lsb1, was detected only in the cytosol and not in the nuclear-ER rim (Fig. 8E). In contrast, the processing defective GFP-Lsb1Y182A,Y183A was localized to the nuclear-ER rim (Fig. 8, C and E).
FIGURE 8.

Association of Lsb1 with nuclear-ER rim. A–G, representative images of live cells are shown. At least 100 cells were analyzed for each experiment. Wild type GFP-Lsb1 and its mutant derivatives were expressed from the copper-inducible promoter, PCUP1. A, GFP-Lsb1 forms a rim and small dots at the cell periphery in 40–50% of cells growing at 25 °C. At 30 °C, GFP-Lsb1 is not present in the rim but is distributed over the cytoplasm and forms small dots in 96% of cells. B, GFP-Lsb1 rim, formed at 25 °C, is colocalized with the nuclear-ER rim. Live cells expressing GFP-Lsb1 from PCUP1 were stained with Hoechst to indicate the positions of the nuclei. C, Lsb1 colocalizes with ER marker protein Sbh1. Proteins were expressed from plasmid promoters PCUP1- mCherry-Lsb1 and PGAL-GFP-Sbh1 for 18 h. Colocalization was detected in all cells. D, GFP-Lsb1 expressed from the PCUP1 promoter colocalizes with nuclear envelope protein Mlp1-RFP expressed from endogenous promoter. Colocalization was detected in all cells. E, GFP-Lsb1 mutant deficient in processing (Y182A,Y183A) is present in the nuclear nuclear-ER rim in 40–50% of cells, whereas the truncated version (1–183 amino acids) corresponding to the processed form of Lsb1 is observed only in the cytoplasm in all cells growing at 25 °C. F, ubiquitination affects localization of Lsb1 to the nuclear-ER rim. GFP-Lsb1 WT and mutants not deficient in ubiquitination (K41R and W90S) do not associate with the nuclear-ER rim in almost all cells growing at 30 °C. However, Lsb1 mutants deficient in ubiquitination (K79R, and K41R,K79R, and P135A,P136A) associate with the nuclear-ER rim in 60–80% of cells growing at 30 °C. The W90S mutant, defective in interaction with Las17, does not form small dots as has been reported previously. G, deletion of get2Δ abolishes localization of wild type and ubiquitination-deficient (P135A,P136A) mutant GFP-Lsb1 to nuclear-ER rim at 25 °C.
Ubiquitination Regulates Localization of Lsb1 to the Nuclear-ER Rim
Mutants deficient in ubiquitination (K79R, double K41R,K79R, or double P135A,P136A) were accumulated in the ER-nuclear rim at 25 °C (supplemental Movie S2) as confirmed by co-localization with the nuclear envelope protein Mlp1 (Fig. 8D). At 30 °C mutants abolishing ubiquitination (K79R, double K41R,K79R, or double P135A,P136A) were still accumulated in the nuclear-ER rim to a high extent, whereas wild type and mutants with no defect in Lsb1 ubiquitination (K41R or W90S) were almost undetectable (Fig. 8F). These data indicate that although ubiquitination is not obligatory for Lsb1 processing (see above, Fig. 6, A–D), it may affect cellular localization of Lsb1 and specifically its accumulation in the nuclear-ER rim.
The GET Pathway Is Responsible for Targeting of Lsb1 to the ER Membrane
Membrane localization of proteins with C-terminal hydrophobic tails (tail-anchored, or TA proteins) depends on the GET-anchored proteins trafficking pathway. Interaction with the GET complex components prevents aggregation of cytosolic TA proteins until these proteins reach their target destination (48). The abundance of hydrophobic amino acid residues in the C-terminal region of Lsb1 suggests that this region might function as a hydrophobic tail anchor. Indeed, GFP-Lsb1 localization to the nuclear-ER rim was abolished in cells with deletion of GET2, one of the key components of the GET pathway (Fig. 8G). Moreover, the get2Δ cells accumulated large aggregates of Lsb1, as typical for mislocalized TA proteins. These data demonstrate that Lsb1 is indeed a TA protein that is targeted to the ER-nuclear rim via the GET pathway.
DISCUSSION
Lsb1 and Lsb2 Are Components of the Cytoskeletal Machinery, Protecting the Cell from the Damaging Effect of Stress-induced Protein Misfolding
Molecular chaperones and the ubiquitin proteasome system are major components of the protein quality control machinery that repairs or eliminates misfolded proteins during stress, as well as under normal conditions. Eukaryotic cells antagonize the toxicity of misfolded proteins not only by solubilizing them, but also by sequestering them into benign, amyloid-like species and/or depositing them into the quality control protein deposits (5–7, 49, 50). Many proteins can form amyloids in vitro suggesting that the amyloid conformation is an ancient protein-fold (51). In vivo, amyloids are formed in response to various perturbations in protein homeostasis, including aging-associated cell stress (52). It has been suggested that at least in some cases, amyloids might be cytoprotective, whereas difficult to characterize soluble oligomers of misfolded proteins are the toxic species (20, 49). Indeed, filamentous aggregates (“rings”) of overproduced Sup35 protein are toxic, whereas formation of self-perpetuating amyloids (prions) ameliorates this toxicity, becoming a “lesser of two evils” (6). Actin-rich complexes appear to provide initial sites for the assembly of protein aggregates (6). Similar events may occur when misfolded proteins are accumulated during stress.
It was proposed that amyloid-like aggregates formed during unfavorable conditions could also be beneficial because they protect important proteins from degradation during stress (1). Indeed, stress granules, assemblies that protect pre-initiation mRNA complexes during stress, are formed with participation of TIA-1 protein containing a prion-like Q/N-rich domain (53).
Asymmetric partitioning of stress-damaged and potentially toxic protein species between mother and daughter cells, known as spatial quality control, also serves as a mechanism of protection of “newborn” daughter cells from misfolded proteins. This partitioning depends on chaperones and the actin cytoskeleton (9, 15, 16, 54). Mechanisms of mother-specific aggregate accumulation remain a matter of debate. Some researchers argue that slower diffusion of large structures through the bud neck is sufficient to explain aggregate retention in mother cells (13). However, other data point to the involvement of targeted cytoskeleton-dependent aggregate retention and/or retrograde transport (10, 14) Notably, the yeast prion [PSI+] is also asymmetrically distributed in the cell divisions after stress, and this asymmetric distribution is chaperone-dependent (21). The actin cytoskeleton is also involved in [PSI+] maintenance (4, 22). This makes [PSI+] a powerful system for investigation of the mechanisms dealing with aggregated proteins during and after stresses.
Our previous studies have shown that Las17-binding protein Lsb2 plays a role in prion stabilization during HS (26). Here we demonstrate that the Lsb2 paralog, Lsb1 has a similar effect (Fig. 1, A and B). Moreover, double deletion of Lsb1 and Lsb2 exhibits a cumulative effect, resulting in prion loss even after prolonged incubation under stress conditions. The death of the mother cell after the first post-stress division is increased in lsb1Δ lsb2Δ cells compared with wild type cells, suggesting that cell death could be at least in part related to aggregated proteins that show the same pattern of asymmetry (Table 2).
LSB2 expression is regulated by the stress-inducible transcription factor Hsf1 (55) and HS dramatically increases Lsb2 levels (26). Surprisingly, this is not a case for full-length Lsb1 (Fig. 3C). However, we found that Lsb1 is processed at the C terminus and that HS increases this processing (Fig. 3). Processing-defective mutant Lsb1 is unable to prevent prion loss after long-term incubation (240 min) at high temperatures, similar to lsb1Δ lsb2Δ (Fig. 4E). This confirms that Lsb1 processing is biologically significant. The processed form of Lsb1 is important for prion maintenance during stress and/or proper segregation after stress.
Mechanism of Lsb1 Processing
We have shown that the full-length form Lsb1 is distributed through the cytoplasm, and associated with the plasma membrane and nuclear-ER rim, whereas the processed form is almost exclusively observed in the cytosol (Fig. 7). Our data does not distinguish whether full-length membrane-associated Lsb1 (located in the nuclear-ER rim) is a precursor, which gives rise to processed Lsb1, or if processing occurs by some other mechanism and the processed form is accumulated in the cytosol during stress (Figs. 3C and 7A). Lsb1 possesses a hydrophobic C-terminal tail, and its association with the membrane depends on proper functioning of the GET pathway (Fig. 8G), similar to known tail-anchored (TA) proteins. These proteins, whereas tethered to the ER membrane via hydrophobic tails, present their functional N-terminal domains to the cytosol (56). Hydrophobic tails must be protected from uncontrolled aggregation until the proteins reach their target destination. Therefore, failure of the GET pathway, due to genetic alterations or physiological conditions, results in the cytosolic aggregation of TA proteins, which is also observed for Lsb1 in the GET-deficient strains (Fig. 8G). Notably, some components of the GET pathway (e.g. Sgt2) also interact with amyloid-like aggregates (including yeast prions) and modulate chaperone effects on aggregates (31). It is an attractive possibility that Lsb1 may also be involved in the assembly and detoxification of aggregates of TA proteins in the case of the GET pathway failure. Elevated temperatures (and possibly other stresses) induce processing of Lsb1. Processed Lsb1 apparently plays a role in the assembly of aggregate deposits during stress and then is degraded. Indeed, we observed that the processed form of Lsb1 is quickly eliminated after processing is inhibited (Fig. 6H).
Processing of Lsb1 is abolished in the presence of the proteasome inhibitor MG132 (Fig. 6H). One known proteasome-dependent processing pathway is a regulated ubiquitin/proteasome-dependent processing, which is essential for regulation of certain transcription factors, including those that are tethered to the ER membrane by a C-terminal transmembrane anchor (57). Proteins must first be ubiquitinated to become subjects of regulated ubiquitin/proteasome-dependent processing. However, Lsb1 mutants with a defect in the major site of ubiquitination, or in the E3 ligase-binding site, still undergo efficient processing (Fig. 6, A-C). It is possible that Lsb1 mutants are transiently ubiquitinated by another E3 ligase through other yet unidentified sites to induce processing, or that proteasomal processing of Lsb1 is ubiquitination-independent. It is also not excluded that the proteasome may stimulate Lsb1 processing indirectly. Because MG132 is known to inhibit other proteases at higher concentrations (46), it is also possible that processing might be dependent on some other MG132-sensitive yet unknown protease. Either way, environmental stresses apparently trigger Lsb1 processing via stimulation of proteolytic activity.
Significance of Lsb1 Ubiquitination
The ubiquitin proteasome system plays a major role during environmental stress and also modulates prion behavior (58–61). As we have indicated previously, the effects of stress on prions can be mediated, in part, by the action of the ubiquitin proteasome system on Lsb2 abundance and/or function (26). In the case of Lsb1, both unprocessed and processed forms are ubiquitinated at the same residue, Lys-79, by the E3 ligase Rsp5 (Fig. 4D). Appearance of a ladder of ubiquitinated Lsb1 species is suggestive of Lsb1 polyubiquitination (Fig. 6B). This is in agreement with our previous finding that Lsb1 has a half-life of 15–20 min, and that both the half-life and steady-state levels of Lsb1 are dramatically increased in a proteasome mutant (26). Ubiquitination of Lsb1 is not required for its processing (Fig. 6, A–D) and proper localization (Fig. 8D). However, ubiquitination influences intracellular distribution of Lsb1 during stress. At high temperatures, localization of Lsb1 to the ER-nuclear rim is essentially lost in the wild type strain; however, it is still detected in the ubiquitination-deficient mutants (Fig. 8F). Therefore, Lsb1 ubiquitination interferes with its localization to membranes in stress conditions.
Role of Lsb Proteins in Aggregate Maintenance during Heat Shock
We propose that several pathways are involved in detoxification of misfolded aggregated proteins during and after stress. One of the pathways operates via assembling misfolded proteins into the large quality control deposits, showing asymmetric segregation in cell divisions. This pathway protects daughter cells at the expense of aggregate-accumulating mothers. Another pathway, involving Lsb1 and Lsb2, sequesters toxic misfolded proteins to the actin assembly sites during HS. This sequestration, either occurring via direct interaction of Lsb1/2 with aggregates or mediated by other components, decreases the toxic effects of protein aggregation on both mother and daughter cells and prevents aggregated proteins from forming huge assemblies exhibiting asymmetric distribution in the cell divisions. This explains why mortality of mother cells in the post-HS divisions is increased in the absence of Lsb proteins. In the case of self-perpetuating protein aggregates (prions), Lsb proteins protect them from stress-induced destabilization via antagonizing their sequestration into the large quality control deposits.
It is possible that in addition to generating an aggregate-interacting derivative of Lsb1, its processing is also involved in HS signaling. Lsb2 and both full size and processed forms of Lsb1 are short lived and quickly removed from cells via ubiquitination and degradation, which would be expected of signaling molecules. Moreover, after a quick surge at the beginning of stress treatment, Lsb2 levels go down if stress conditions persist (26). These properties restrict the roles of Lsb proteins in non-stressed cells (or in the cells adapted to new conditions), so that Lsb “pro-aggregation” properties become confined to the periods of environmental (or physiological) changes. Taken together, our results suggest that dynamic induction, processing, and degradation of actin cytoskeleton components serve as a regulatory mechanism, minimizing the toxic effects of protein misfolding and maximizing the protective effects of controlled protein aggregation in changing environment.
Supplementary Material
Acknowledgments
We thank W. P. Tansey, D. Bedwell, A. Corbett and M. Fasken for the reagents, D. H. Wolf for helpful discussion, and J. F. Reger for help with some experiments.
This work was supported, in whole or in part, by National Institutes of Health Grant GM093294 (to K. D. W., T. A. C., and Y. O. C.) and Project 1.50.2218.2013 from St. Petersburg State University (to Y. O. C.).

This article contains supplemental Movies 1 and 2.
- SH
- Src homology domain
- ER
- endoplasmic reticulum
- GET
- guided entry of tail-anchored proteins
- HS
- heat shock
- Ub
- ubiquitin
- IP
- immunoprecipitation
- TA
- tail-anchored.
REFERENCES
- 1. Chernoff Y. O. (2007) Stress and prions: lessons from the yeast model. FEBS Lett. 581, 3695–3701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li L., Kowal A. S. (2012) Environmental regulation of prions in yeast. PLoS Pathog. 8, e1002973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Verghese J., Abrams J., Wang Y., Morano K. A. (2012) Biology of the heat shock response and protein chaperones: budding yeast (Saccharomyces cerevisiae) as a model system. Microbiol. Mol. Biol. Rev. 76, 115–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chernova T. A., Wilkinson K. D., Chernoff Y. O. (2014) Physiological and environmental control of yeast prions. FEMS Microbiol. Rev. 38, 326–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kopito R. R. (2000) Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10, 524–530 [DOI] [PubMed] [Google Scholar]
- 6. Ganusova E. E., Ozolins L. N., Bhagat S., Newnam G. P., Wegrzyn R. D., Sherman M. Y., Chernoff Y. O. (2006) Modulation of prion formation, aggregation, and toxicity by the actin cytoskeleton in yeast. Mol. Cell. Biol. 26, 617–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang Y., Meriin A. B., Zaarur N., Romanova N. V., Chernoff Y. O., Costello C. E., Sherman M. Y. (2009) Abnormal proteins can form aggresome in yeast: aggresome-targeting signals and components of the machinery. FASEB J. 23, 451–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sontag E. M., Vonk W. I., Frydman J. (2014) Sorting out the trash: the spatial nature of eukaryotic protein quality control. Curr. Opin. Cell Biol. 26, 139–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aguilaniu H., Gustafsson L., Rigoulet M., Nyström T. (2003) Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science 299, 1751–1753 [DOI] [PubMed] [Google Scholar]
- 10. Liu B., Larsson L., Caballero A., Hao X., Oling D., Grantham J., Nyström T. (2010) The polarisome is required for segregation and retrograde transport of protein aggregates. Cell 140, 257–267 [DOI] [PubMed] [Google Scholar]
- 11. Nyström T., Liu B. (2014) The mystery of aging and rejuvenation: a budding topic. Curr. Opin. Microbiol. 18, 61–67 [DOI] [PubMed] [Google Scholar]
- 12. Hernebring M., Brolén G., Aguilaniu H., Semb H., Nyström T. (2006) Elimination of damaged proteins during differentiation of embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 103, 7700–7705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhou C., Slaughter B. D., Unruh J. R., Eldakak A., Rubinstein B., Li R. (2011) Motility and segregation of Hsp104-associated protein aggregates in budding yeast. Cell 147, 1186–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu B., Larsson L., Franssens V., Hao X., Hill S. M., Andersson V., Höglund D., Song J., Yang X., Öling D., Grantham J., Winderickx J., Nyström T. (2011) Segregation of protein aggregates involves actin and the polarity machinery. Cell 147, 959–961 [DOI] [PubMed] [Google Scholar]
- 15. Erjavec N., Larsson L., Grantham J., Nyström T. (2007) Accelerated aging and failure to segregate damaged proteins in Sir2 mutants can be suppressed by overproducing the protein aggregation-remodeling factor Hsp104p. Genes Dev. 21, 2410–2421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tessarz P., Schwarz M., Mogk A., Bukau B. (2009) The yeast AAA+ chaperone Hsp104 is part of a network that links the actin cytoskeleton with the inheritance of damaged proteins. Mol. Cell. Biol. 29, 3738–3745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smethurst D. G., Dawes I. W., Gourlay C. W. (2011) Actin: a biosensor that determines cell fate in yeasts. FEMS Yeast Res. 408, 432–448 [DOI] [PubMed] [Google Scholar]
- 18. Prusiner S. B. (2012) Cell biology: a unifying role for prions in neurodegenerative diseases. Science 336, 1511–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Prusiner S. B. (1998) Prions. Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liebman S. W., Chernoff Y. O. (2012) Prions in yeast. Genetics 191, 1041–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Newnam G. P., Birchmore J. L., Chernoff Y. O. (2011) Destabilization and recovery of a yeast prion after mild heat shock. J. Mol. Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bailleul-Winslett P. A., Newnam G. P., Wegrzyn R. D., Chernoff Y. O. (2000) An antiprion effect of the anticytoskeletal drug latrunculin A in yeast. Gene Exp. 9, 145–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Weinberg J., Drubin D. G. (2012) Clathrin-mediated endocytosis in budding yeast. Trends Cell Biol. 22, 1–1322018597 [Google Scholar]
- 24. Lauwers E., Erpapazoglou Z., Haguenauer-Tsapis R., André B. (2010) The ubiquitin code of yeast permease trafficking. Trends Cell Biol. 20, 196–204 [DOI] [PubMed] [Google Scholar]
- 25. Spiess M., de Craene J. O., Michelot A., Rinaldi B., Huber A., Drubin D. G., Winsor B., Friant S. (2013) Lsb1 is a negative regulator of las17 dependent actin polymerization involved in endocytosis. PloS One 8, e61147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chernova T. A., Romanyuk A. V., Karpova T. S., Shanks J. R., Ali M., Moffatt N., Howie R. L., O'Dell A., McNally J. G., Liebman S. W., Chernoff Y. O., Wilkinson K. D. (2011) Prion induction by the short-lived, stress-induced protein Lsb2 is regulated by ubiquitination and association with the actin cytoskeleton. Mol. Cell 43, 242–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Longtine M. S., McKenzie A., 3rd, Demarini D. J., Shah N. G., Wach A., Brachat A., Philippsen P., Pringle J. R. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961 [DOI] [PubMed] [Google Scholar]
- 28. Storici F., Resnick M. A. (2006) The delitto perfetto approach to in vivo site-directed mutagenesis and chromosome rearrangements with synthetic oligonucleotides in yeast. Methods Enzymol. 409, 329–345 [DOI] [PubMed] [Google Scholar]
- 29. Hochstrasser M., Ellison M. J., Chau V., Varshavsky A. (1991) The short-lived MATα2 transcriptional regulator is ubiquitinated in vivo. Proc. Natl. Acad. Sci. U.S.A. 88, 4606–4610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shcherbik N., Zoladek T., Nickels J. T., Haines D. S. (2003) Rsp5p is required for ER bound Mga2p120 polyubiquitination and release of the processed/tethered transactivator Mga2p90. Curr. Biol. 13, 1227–1233 [DOI] [PubMed] [Google Scholar]
- 31. Kiktev D. A., Patterson J. C., Müller S., Bariar B., Pan T., Chernoff Y. O. (2012) Regulation of chaperone effects on a yeast prion by cochaperone Sgt2. Mol. Cell. Biol. 32, 4960–4970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sherman F. (2002) Getting started with yeast. Methods Enzymol. 350, 3–41 [DOI] [PubMed] [Google Scholar]
- 33. Chernoff Y. O., Uptain S. M., Lindquist S. L. (2002) Analysis of prion factors in yeast. Methods Enzymol. 351, 499–538 [DOI] [PubMed] [Google Scholar]
- 34. Ghahramani S. (2000) Fundamentals of Probability, Prentice Hall, UpperSaddle River, NJ [Google Scholar]
- 35. Rieder S. E., Emr S. D. (2001) Isolation of subcellular fractions from the yeast Saccharomyces cerevisiae. (Bonifacino J. S., et al. eds) Current Protocols in Cell Biology, Chapter 3, Unit 3 8 [DOI] [PubMed] [Google Scholar]
- 36. Liu C., Apodaca J., Davis L. E., Rao H. (2007) Proteasome inhibition in wild-type yeast Saccharomyces cerevisiae cells. BioTechniques 42, 158. [DOI] [PubMed] [Google Scholar]
- 37. James P., Halladay J., Craig E. A. (1996) Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 144, 1425–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Evangelista M., Klebl B. M., Tong A. H., Webb B. A., Leeuw T., Leberer E., Whiteway M., Thomas D. Y., Boone C. (2000) A role for myosin-I in actin assembly through interactions with Vrp1p, Bee1p, and the Arp2/3 complex. J. Cell Biol. 148, 353–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Peng J., Schwartz D., Elias J. E., Thoreen C. C., Cheng D., Marsischky G., Roelofs J., Finley D., Gygi S. P. (2003) A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 21, 921–926 [DOI] [PubMed] [Google Scholar]
- 40. Gupta R., Kus B., Fladd C., Wasmuth J., Tonikian R., Sidhu S., Krogan N. J., Parkinson J., Rotin D. (2007) Ubiquitination screen using protein microarrays for comprehensive identification of Rsp5 substrates in yeast. Mol. Syst. Biol. 3, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kaminska J., Spiess M., Stawiecka-Mirota M., Monkaityte R., Haguenauer-Tsapis R., Urban-Grimal D., Winsor B., Zoladek T. (2011) Yeast Rsp5 ubiquitin ligase affects the actin cytoskeleton in vivo and in vitro. Eur. J. Cell Biol. 90, 1016–1028 [DOI] [PubMed] [Google Scholar]
- 42. Huibregtse J. M., Yang J. C., Beaudenon S. L. (1997) The large subunit of RNA polymerase II is a substrate of the Rsp5 ubiquitin-protein ligase. Proc. Natl. Acad. Sci. U.S.A. 94, 3656–3661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hoppe T., Matuschewski K., Rape M., Schlenker S., Ulrich H. D., Jentsch S. (2000) Activation of a membrane-bound transcription factor by regulated ubiquitin/proteasome-dependent processing. Cell 102, 577–586 [DOI] [PubMed] [Google Scholar]
- 44. Heinemeyer W., Fischer M., Krimmer T., Stachon U., Wolf D. H. (1997) The active sites of the eukaryotic 20 S proteasome and their involvement in subunit precursor processing. J. Biol. Chem. 272, 25200–25209 [DOI] [PubMed] [Google Scholar]
- 45. Howard G. C., Collins G. A., Tansey W. P. (2012) Letter to the Editor. Yeast. 29, 93–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tawa N. E., Jr., Odessey R., Goldberg A. L. (1997) Inhibitors of the proteasome reduce the accelerated proteolysis in atrophying rat skeletal muscles. J. Clin. Investig. 100, 197–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fasken M. B., Stewart M., Corbett A. H. (2008) Functional significance of the interaction between the mRNA-binding protein, Nab2, and the nuclear pore-associated protein, Mlp1, in mRNA export. J. Biol. Chem. 283, 27130–27143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schuldiner M., Metz J., Schmid V., Denic V., Rakwalska M., Schmitt H. D., Schwappach B., Weissman J. S. (2008) The GET complex mediates insertion of tail-anchored proteins into the ER membrane. Cell 134, 634–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Douglas P. M., Treusch S., Ren H. Y., Halfmann R., Duennwald M. L., Lindquist S., Cyr D. M. (2008) Chaperone-dependent amyloid assembly protects cells from prion toxicity. Proc. Natl. Acad. Sci. U.S.A. 105, 7206–7211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kaganovich D., Kopito R., Frydman J. (2008) Misfolded proteins partition between two distinct quality control compartments. Nature 454, 1088–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chiti F., Dobson C. M. (2009) Amyloid formation by globular proteins under native conditions. Nat. Chem. Biol. 5, 15–22 [DOI] [PubMed] [Google Scholar]
- 52. Chiti F., Dobson C. M. (2006) Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 [DOI] [PubMed] [Google Scholar]
- 53. Gilks N., Kedersha N., Ayodele M., Shen L., Stoecklin G., Dember L. M., Anderson P. (2004) Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 15, 5383–5398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Guarente L. (2010) Forever young. Cell 140, 176–178 [DOI] [PubMed] [Google Scholar]
- 55. Harbison C. T., Gordon D. B., Lee T. I., Rinaldi N. J., Macisaac K. D., Danford T. W., Hannett N. M., Tagne J. B., Reynolds D. B., Yoo J., Jennings E. G., Zeitlinger J., Pokholok D. K., Kellis M., Rolfe P. A., Takusagawa K. T., Lander E. S., Gifford D. K., Fraenkel E., Young R. A. (2004) Transcriptional regulatory code of a eukaryotic genome. Nature 431, 99–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Borgese N., Brambillasca S., Colombo S. (2007) How tails guide tail-anchored proteins to their destinations. Curr. Opin. Cell Biol. 19, 368–375 [DOI] [PubMed] [Google Scholar]
- 57. Rape M., Jentsch S. (2004) Productive RUPture: activation of transcription factors by proteasomal processing. Biochim. Biophys. Acta 1695, 209–213 [DOI] [PubMed] [Google Scholar]
- 58. Chernova T. A., Allen K. D., Wesoloski L. M., Shanks J. R., Chernoff Y. O., Wilkinson K. D. (2003) Pleiotropic effects of Ubp6 loss on drug sensitivities and yeast prion are due to depletion of the free ubiquitin pool. J. Biol. Chem. 278, 52102–52115 [DOI] [PubMed] [Google Scholar]
- 59. Allen K. D., Chernova T. A., Tennant E. P., Wilkinson K. D., Chernoff Y. O. (2007) Effects of ubiquitin system alterations on the formation and loss of a yeast prion. J. Biol. Chem. 282, 3004–3013 [DOI] [PubMed] [Google Scholar]
- 60. Tyedmers J., Madariaga M. L., Lindquist S. (2008) Prion switching in response to environmental stress. PLoS Biol. 6, e294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kabani M., Redeker V., Melki R. (2014) A role for the proteasome in the turnover of Sup35p and in [PSI(+)] prion propagation. Mol. Microbiol. 92, 507–528 [DOI] [PubMed] [Google Scholar]
- 62. Chernoff Y. O., Galkin A. P., Lewitin E., Chernova T. A., Newnam G. P., Belenkiy S. M. (2000) Evolutionary conservation of prion-forming abilities of the yeast Sup35 protein. Mol. Microbiol. 35, 865–876 [DOI] [PubMed] [Google Scholar]
- 63. Chen P., Johnson P., Sommer T., Jentsch S., Hochstrasser M. (1993) Multiple ubiquitin-conjugating enzymes participate in the in vivo degradation of the yeast MATα2 repressor. Cell 74, 357–369 [DOI] [PubMed] [Google Scholar]
- 64. Newnam G. P., Wegrzyn R. D., Lindquist S. L., Chernoff Y. O. (1999) Antagonistic interactions between yeast chaperones Hsp104 and Hsp70 in prion curing. Mol. Cell. Biol. 19, 1325–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.