

Abstract
Context:
Interruption of the renin-angiotensin-aldosterone system prevents incident diabetes in high-risk individuals, although the mechanism remains unclear.
Objective:
To test the hypothesis that activation of the endogenous renin-angiotensin-aldosterone system or exogenous aldosterone impairs insulin secretion in humans.
Design:
We conducted a randomized, blinded crossover study of aldosterone vs vehicle and compared the effects of a low-sodium versus a high-sodium diet.
Setting:
Academic clinical research center.
Participants:
Healthy, nondiabetic, normotensive volunteers.
Interventions:
Infusion of exogenous aldosterone (0.7 μg/kg/h for 12.5 h) or vehicle during low or high sodium intake. Low sodium (20 mmol/d; n = 12) vs high sodium (160 mmol/d; n = 17) intake for 5–7 days.
Main Outcome Measures:
Change in acute insulin secretory response assessed during hyperglycemic clamps while in sodium balance during a low-sodium vs high-sodium diet during aldosterone vs vehicle.
Results:
A low-sodium diet increased endogenous aldosterone and plasma renin activity, and acute glucose-stimulated insulin (−16.0 ± 5.6%; P = .007) and C-peptide responses (−21.8 ± 8.4%; P = .014) were decreased, whereas the insulin sensitivity index was unchanged (−1.0 ± 10.7%; P = .98). Aldosterone infusion did not affect the acute insulin response (+1.8 ± 4.8%; P = .72) or insulin sensitivity index (+2.0 ± 8.8%; P = .78). Systolic blood pressure and serum potassium were similar during low and high sodium intake and during aldosterone infusion.
Conclusions:
Low dietary sodium intake reduces insulin secretion in humans, independent of insulin sensitivity.
The renin-angiotensin-aldosterone system (RAAS) maintains sodium and fluid balance during periods of reduced intake. Inappropriate RAAS activation also contributes to hypertension and cardiovascular complications, however, and pharmacological blockade reduces blood pressure and cardiovascular risk. Angiotensin receptor blockers and angiotensin I converting enzyme inhibitors also improve glucose homeostasis and prevent diabetes progression in most studies, but the mechanism remains unclear (1–4).
Although obesity results in insulin resistance and impaired glucose tolerance, a compensatory increase in insulin secretion normally maintains glycemic control early in the course of disease. Angiotensin and aldosterone impair insulin sensitivity in animal studies by producing vascular remodeling and impairing skeletal muscle insulin receptor signaling and glucose transporter expression (5, 6). Insulin resistance alone is not sufficient to cause diabetes, however, due to the dynamic insulin response and ability to meet the increased insulin requirement. Therefore, overt hyperglycemia occurs only in individuals who develop a relative insulin secretory defect (7–9).
Recently, we determined that glucose-stimulated insulin secretion is increased in aldosterone-deficient mice in vivo and that aldosterone reduces insulin secretion in isolated islets ex vivo via reactive oxygen species (10). Aldosterone synthase deficiency further protects against high fat feeding-induced hyperglycemia and hepatic steatosis, but not against development of peripheral insulin resistance (11). Aldosterone production is increased in obesity (12, 13) and could contribute to diabetes progression by promoting β-cell dysfunction. Recent clinical studies also suggest that β-cell function is impaired in patients with aldosterone-producing adenomas compared to those with essential hypertension (14, 15). Furthermore, at least one group has reported that acute angiotensin II infusion, which stimulates aldosterone secretion, decreases glucose-stimulated insulin secretion in humans (16).
The effect of endogenous RAAS activation on insulin secretion has not been previously investigated in clinical studies. Acute dietary sodium reduction activates the RAAS, worsens glucose tolerance, and impairs insulin sensitivity as assessed by either the fasting glucose-insulin product (homeostasis model of assessment) or hyperinsulinemic clamps in most studies (17–21), but not all (22). The effect of dietary sodium reduction on insulin secretion has not been previously investigated, however. In the present study, we tested the hypothesis that activation of the RAAS by dietary sodium restriction decreases glucose-stimulated insulin secretion, as measured by hyperglycemic clamp. To further assess the effect of increasing aldosterone independently of angiotensin II, we compared the effect of overnight iv administration of exogenous aldosterone or vehicle during both low and high sodium intake.
Subjects and Methods
Screening
All studies were approved by the Vanderbilt University Institutional Review Board and conducted in accordance with the Declaration of Helsinki. We recruited healthy subjects from the Vanderbilt research participant volunteer registry. Informed consent was obtained, and subjects underwent a screening history and physical before study enrollment. Exclusion criteria included body mass index ≥ 30 kg/m2, fasting glucose > 110 mg/dL, or the use of anti-diabetes medication, serum triglycerides > 150 mg/dL, total cholesterol > 200 mg/dL, supine blood pressure > 130/85, recent glucocorticoid therapy, renal insufficiency, serum potassium < 3.5 mEq/L, or any serious medical condition. Pregnancy was excluded in women of childbearing potential by measurement of urine β-human chorionic gonadotropin at screening and on each study day.
Study protocol
Subjects were provided a 20-mmol/d sodium, calorie-controlled diet for 9 days (Figure 1A) or a 160-mmol/d sodium diet for 7 days (which represents the typical sodium intake in our region). All meals were prepared by the Vanderbilt Clinical Research Center (CRC) kitchen under the guidance of a registered dietician. Diets were similar in caloric content from carbohydrate (56%), fat (27%), and protein (17%) and were controlled for potassium (80 mEq/d) and calcium (10 mEq/d). The duration of diets was based on the time required to achieve sodium balance during respective low and high sodium intake. Compliance was assessed by measurement of 24-hour urine sodium excretion, plasma renin activity, and plasma aldosterone.
Figure 1.
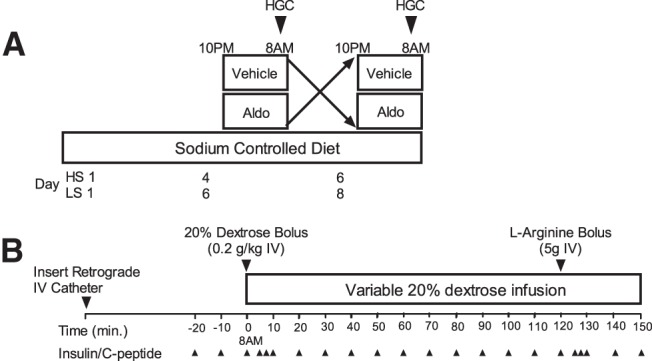
A, General study protocol. Subjects reported to the CRC in sodium balance on either a low-sodium (LS) or high-sodium-controlled diet (HS) and received either drug or vehicle infusion overnight and hyperglycemic clamps (HGC) the next morning. Subjects crossed over and received the remaining infusion on study day 2. B, Hyperglycemic clamp protocol. After insertion of catheters, baseline measurements were performed, and iv dextrose was infused to achieve hyperglycemia (∼200 mg/dL). Glucose was measured every 5 minutes, and dextrose infusion was adjusted to maintain hyperglycemia.
Subjects reported for admission to the CRC in the evenings of the sixth and eighth days of the low-sodium diet and of the fourth and sixth days of the high-sodium diet. Beginning at 10 pm, subjects were given either aldosterone (0.7 μg/kg/h in 5% dextrose water; Professional Compounding Corporation of America) or vehicle by iv infusion for 12.5 hours. Hyperglycemic clamps were performed as detailed in Figure 1B starting at 8–9 am, and drug infusion was continued until the completion of the clamp. Subjects continued the study diet and returned to the CRC on the following evening to complete the remaining infusion and second hyperglycemic clamp.
The order of drug administration was randomized and double-blinded by the Vanderbilt Investigational Drug Services. Blood pressure was monitored hourly during drug infusion (Dinamap; GE Medical). Serum potassium (I-Stat Analyzer; Abbott Labs) was monitored before, during, and after the drug infusion, and oral potassium chloride elixir was administered as needed before the clamp studies to maintain potassium > 3.7 mEq/L. The study was terminated if serum potassium fell below 3.2 mEq/L at any point. Hyperglycemic clamps were not initiated if serum potassium remained < 3.7 mEq/L after supplementation and repeat evaluation at 7–9 am. Potassium was not administered during the hyperglycemic clamp.
Hyperglycemic clamps
Subjects fasted after midnight, and clamp studies began at 8–9 am on each study day. During hyperglycemic clamp studies (Figure 1B), a catheter was inserted in a retrograde hand vein for blood sampling, and the hand was warmed throughout the study for blood arterialization. An antecubital catheter was inserted in the contralateral arm for glucose infusion. To account for pulsatile insulin secretion, baseline glucose, insulin, and C-peptide were measured at −20, −10, and −1 minute before glucose infusion, and the average value was used to calculate baseline values (t = 0). Blood for plasma glucose was drawn every 5 minutes and immediately centrifuged and analyzed using the glucose oxidase method (YSI 2300 STAT Plus Glucose Analyzer; YSI Life Sciences). A standardized priming 20% dextrose (Hospira) infusion was administered during the first 10 minutes (200 mg/kg body weight), and thereafter infusion rates were adjusted each 5 minutes to maintain plasma glucose at 200 mg/dL for 150 minutes, adjusted by the calculations by DeFronzo et al (23).
The acute glucose-stimulated insulin response (ΔInsulin0–10) was calculated as the maximum increase during the first 10 minutes above the averaged baseline value, and also as the area under the curve (AUC) for insulin above baseline value from time points 0 to 10 minutes (AUCΔInsulin0–10). The insulin sensitivity index was calculated by dividing the average glucose infusion rate (mg/kg body weight/min) by the average insulin concentration (μU/mL) during time 90–120 minutes multiplied by 100. This hyperglycemic clamp insulin sensitivity index estimate correlates well with insulin sensitivity measures obtained during hyperinsulinemic-euglycemic clamps (r = 0.84) (23, 24) and frequently sampled iv-glucose tolerance tests (r = 0.88) (25). The disposition index was calculated as the AUCΔInsulin0–10 multiplied by the insulin sensitivity index (26). Urinary glucose loss was assessed before and at the end of the study. No significant glucosuria was detected in the present studies.
Laboratory assays
Electrolytes and lipid panels were performed in the Vanderbilt Clinical Laboratory. Urine sodium and potassium concentrations were measured by flame photometry, and creatinine by the sodium picrate method. Blood was collected in EDTA, and plasma was separated and stored at −80°C until assayed. Aprotinin was also added to plasma for measurement of insulin and C-peptide by RIA (Millipore). Plasma aldosterone and plasma renin activity were measured by RIA as described previously (27). N-Terminal pro-brain natriuretic peptide (NT-pro-BNP) was measured by enzyme-linked immunoassay with colorimetric detection (BNP fragment EIA; Biochemica). Plasma catecholamines were measured using batch alumina extraction followed by HPLC with electrochemical detection and quantification.
Body composition measurements
Waist (horizontal umbilicus) circumference was measured with a spring-loaded tape measure (Gulick II; Country Technology). Body composition was determined by air displacement plethysmography with a BODPOD Composition System (Life Measurements Instruments).
Sample size calculation and randomization
The study was designed to achieve a sample size of 18 subjects, calculated to provide 80% power to detect a 30% (14 μU/mL based on published results) (23) mean difference in initial insulin response during the hyperglycemic clamp between aldosterone vs vehicle infusion, assuming α of 0.05 and standard deviation of 20 μU/mL using the paired t test. We anticipated that 30 subjects would need to enroll in order to complete 18 subjects after screening failure and dropout. The investigator and subjects were blinded to the treatment (aldosterone or vehicle), and treatment allocation was assigned by the Investigational Drug Pharmacy by randomized-block permutation generated by the study statistician (C.Y.). Dietary sodium was not blinded, but the order of sodium diet was varied.
Statistical analysis
Study data were collected and managed using REDCap electronic data capture tools hosted at Vanderbilt (28). Data are presented as mean ± SEM. Comparisons of baseline values were made using the Wilcoxon signed-rank test or Wilcoxon rank-sum test, as appropriate. Effects of aldosterone infusion and dietary sodium on outcome variables were assessed as fixed effects using linear mixed-effects models, with compound symmetry correlation structure for repeated measurements. Modeling was also performed using sex, race, and insulin sensitivity index as covariates, and results were not affected by these adjustments. All statistical analyses were performed using IBM SPSS for Windows (version 20, IBM SPSS) and SAS release 9. A two-tailed P value < .05 was considered statistically significant.
Results
Study participants
Thirty-four subjects consented, and after a screening physical, 23 enrolled in the study (five were excluded, and six declined participation, primarily due to scheduling difficulties). An additional three subjects were excluded or withdrew before the first clamp (due to loss of iv access, burning at the site of the iv catheter, and low potassium before overnight infusion). Twelve subjects were studied during low sodium intake, and 17 were studied under high sodium intake. This included nine subjects who completed both the low-sodium and high-sodium study periods. Demographic characteristics are presented in Table 1.
Table 1.
Study Participant Baseline Characteristics
| Low Sodium | High Sodium | |
|---|---|---|
| n | 12 | 17 |
| Sex, M:F | 4:8 | 7:10 |
| Race, Caucasian:AA | 9:3 | 13:4 |
| Age, y | 30.3 ± 2.0 | 27.9 ± 1.0 |
| Body mass index, kg/m2 | 25.7 ± 0.9 | 26.2 ± 0.9 |
| Waist circumference, cm | 87.9 ± 2.7 | 90.0 ± 2.9 |
| Body fat, % | 17.8 ± 3.9 | 18.5 ± 2.8 |
| Serum potassium, mEq/L | 3.96 ± 0.08 | 3.97 ± 0.07 |
| Creatinine, mg/dL | 0.94 ± 0.050 | 0.86 ± 0.041 |
| Glucose, mg/dL | 85.3 ± 2.0 | 85.2 ± 1.9 |
| Triglycerides, mg/dL | 56.4 ± 6.5 | 55.3 ± 7.6 |
| HDL cholesterol, mg/dL | 56.2 ± 3.7 | 58.9 ± 5.1 |
| LDL cholesterol, mg/dL | 92.9 ± 6.6 | 85.1 ± 5.7 |
Abbreviations: HDL, high-density lipoprotein; LDL, low-density lipoprotein, M, males; F, females; AA, African American.
Effect of dietary sodium intake on insulin secretion
Physiological measurements under low- and high-sodium diets are presented in Table 2. Plasma renin activity and aldosterone increased significantly during low sodium intake. Blood pressure and potassium were similar during low- and high-sodium diets. Urine sodium and potassium measurements indicate dietary compliance to achieve reduced sodium and constant potassium (Table 2). Baseline glucose and C-peptide were similar, but baseline insulin concentrations were lower during low sodium compared to high sodium intake (P = .004). During the clamp, equivalent hyperglycemia was achieved during low and high sodium (Figure 2A). The initial insulin response during the standardized priming glucose load was lower during the low-sodium diet compared to the high-sodium diet, as calculated by either maximal change in insulin (ΔInsulin0–10, −17.6 ± 6.7%; P = .012) or the AUC for insulin in the first 10 minutes (AUCΔInsulin0–10, −16.0 ± 5.6%; P = .007) (Table 2). The intraindividual correlation for the insulin response was high (linear R2 = 0.91 for intraindividual correlation during low and high sodium during vehicle infusion; P < .001). Glucose-stimulated insulin (Figure 2B) and C-peptide (Figure 2C) responses decreased similarly during the low-sodium diet (P < .01 for each). Results were significant after adjustment for age, sex, race, and insulin sensitivity index (effect of low-sodium diet β-coefficient, −9.6 ± 3.2; P = .005). Caucasian race (β-coefficient, −34.6 ± 11.2; P = .005) and insulin sensitivity index (β-coefficient, −0.75 ± 0.21; P < .001) correlated inversely with insulin response. C-peptide responses were also decreased by low-sodium diet (Table 2) during the initial 10 minutes (−21.8 ± 8.4%; P = .014) and at the end of the clamp (−33.8 ± 4.5%; P < .001), suggesting that altered insulin secretion rather than altered insulin clearance accounted for decreased insulin concentrations during hyperglycemia and low sodium intake. Insulin sensitivity was similar during low and high sodium intake (−1 ± 10.7%; P = .98), with a high intraindividual correlation (linear R2 = 0.81 for intraindividual correlation during low and high sodium during vehicle infusion; P < .001). The disposition index was not statistically different between dietary conditions (−0.7 ± 9.9%; P = .85).
Table 2.
Effect of Dietary Sodium on Hyperglycemic Clamp Measures
| Low Sodium | High Sodium | P Value | |
|---|---|---|---|
| n | 12 | 17 | |
| Serum potassium, mEq/L | 3.99 ± 0.04 | 3.92 ± 0.03 | .15 |
| Systolic blood pressure, mm Hg | 107.5 ± 2.7 | 112.5 ± 2.0 | .09 |
| Plasma renin activity, ng angiotensin I ·mL−1·h−1 | 2.93 ± 0.22 | 0.98 ± 0.17 | <.001 |
| Plasma aldosterone, ng/dL | 24.4 ± 1.8 | 7.2 ± 1.4 | <.001 |
| Plasma NT-pro-BNP, pmol/L | 3297 ± 2059 | 4983 ± 1195 | <.001 |
| Plasma norepinephrine, pg/mL | 83 ± 39 | 141 ± 15 | .22 |
| Plasma epinephrine, pg/mL | 4.80 ± 0.80 | 5.93 ± 0.91 | .49 |
| Urinary Na/Cr, mmol/g Cr | 10.0 ± 6.2 | 79.7 ± 4.8 | <.001 |
| Urinary K/Cr, mmol/g Cr | 50.3 ± 3.9 | 45.4 ± 3.1 | .25 |
| Urinary Na/K ratio | 0.20 ± 0.16 | 1.94 ± 0.12 | <.001 |
| Fasting | |||
| Glucose, mg/dL | 95.4 ± 1.9 | 95.0 ± 1.5 | .82 |
| Insulin, μU/mL | 9.87 ± 1.59 | 12.27 ± 1.49 | .004 |
| C-peptide, ng/mL | 1.88 ± 0.20 | 2.06 ± 0.18 | .31 |
| Glucose infusion rate60–90, mg·kg−1·min−1 | 7.7 ± 0.9 | 7.4 ± 0.7 | .72 |
| Glucose infusion rate90–120, mg·kg−1·min−1 | 10.1 ± 1.0 | 9.3 ± 0.9 | .29 |
| Insulin sensitivity index, mg·kg−1·min−1 per μU/mL·100 | 19.7 ± 3.3 | 19.9 ± 3.0 | .98 |
| Acute insulin response to glucose | |||
| ΔInsulin0–10, μU/mL | 40.7 ± 6.0 | 49.4 ± 5.5 | .012 |
| AUCΔInsulin 0–10, μU/mL·min | 349.2 ± 49.7 | 415.5 ± 47.2 | .007 |
| ΔC-peptide0–10, μU/mL | 3.40 ± 0.49 | 4.35 ± 0.45 | .014 |
| Late insulin response to glucose | |||
| ΔInsulin60–90, μU/mL | 44.2 ± 8.5 | 54.2 ± 8.2 | .006 |
| ΔInsulin90–120, μU/mL | 56.0 ± 10.2 | 64.5 ± 9.6 | .11 |
| ΔC-peptide90–120, μU/mL | 9.4 ± 1.1 | 14.2 ± 1.0 | <.001 |
| Disposition index | 6.89 ± 0.88 | 7.03 ± 0.76 | .85 |
Abbreviation: Ang, angiotensin. Data are expressed as least square mean ± SE, and P values are obtained from the linear mixed-effects model.
Figure 2.
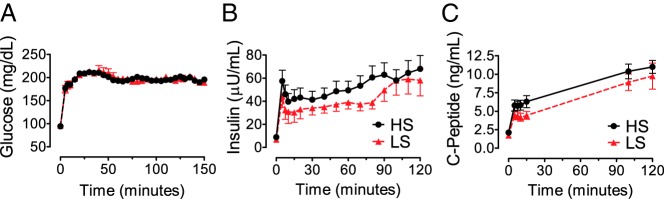
A, Plasma glucose during hyperglycemic clamps was “clamped” similarly during the study by standardized infusion of glucose, in order to measure glucose-stimulated insulin secretion. B, Plasma insulin increased in response to hyperglycemia, and this response was diminished during the low-sodium (LS, red triangle) compared to high-sodium (HS, black circle) diet. C, C-peptide concentrations paralleled those of insulin.
Because differences in menstrual cycle phase could affect insulin sensitivity, we explored the effect of diet on insulin secretion in males and found a significant effect of diet on the acute insulin response (P = .025). We also restricted analysis to the nine subjects who completed both the low- and high-sodium diets and obtained results that were qualitatively similar to those above, with a significant effect on acute insulin secretory response (P = .022; Supplemental Table 1). This effect also remained significant after adjusting for insulin sensitivity index (P = .0062 for diet effect; P = .0039 for association of insulin sensitivity index). Because circulating natriuretic peptides are decreased during a low-sodium diet and inversely associated with diabetes risk (29–31), we further analyzed plasma NT-pro-BNP. Plasma NT-pro-BNP correlated inversely with body mass index and positively with dietary sodium, but not with insulin secretion, insulin sensitivity, or disposition index. Adjustment for NT-pro-BNP did not affect the relationship between dietary sodium and initial insulin response (adjusted ΔInsulin0–10, −13.7 ± 3.8 μU/mL during a low-sodium vs a high-sodium diet; P = .001 for diet effect).
Effects of exogenous aldosterone
The effects of aldosterone are tabulated in Supplemental Table 2. Plasma aldosterone increased with aldosterone infusion during both low- and high-sodium diets (Figure 3A). Systolic blood pressure was not acutely altered by either aldosterone infusion or short-term dietary sodium modification (Figure 3B).
Figure 3.
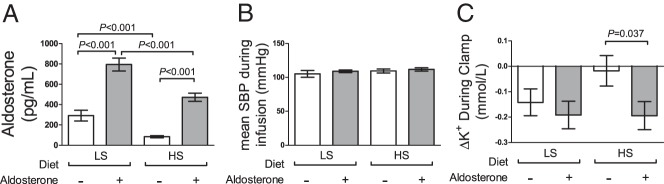
A, Plasma aldosterone increased during the low-sodium (LS) diet compared to the high sodium (HS) diet and during aldosterone infusion. B, Mean systolic blood pressure (SBP) was not altered by diet or drug. C, Change in serum potassium from beginning to end of the hyperglycemic clamp was altered by acute aldosterone infusion or short-term dietary sodium restriction.
During aldosterone/vehicle infusions, serum potassium was maintained similarly > 3.7 mEq/L by oral KCl administration, but required 8.3 ± 4.4 mEq more KCl supplementation during the high-sodium compared to the low-sodium diet (P = .027 for low- vs high-sodium diet; P = .67 for aldosterone; P = .67 for drug × diet interaction). During hyperglycemic clamps on the low-sodium diet, serum potassium then decreased from t = 0 to end of clamp, and this was not affected by aldosterone (−0.14 ± 0.05 vs −0.19 ± 0.05 mEq/L for vehicle and aldosterone; P = .50). During the high-sodium diet, however, serum potassium decreased minimally during vehicle infusion, and aldosterone infusion produced a significantly greater reduction in potassium (−0.03 ± 0.05 vs −0.19 ± 0.06 mEq/L for vehicle and aldosterone; P = .045; Figure 3C), similar to that observed during the low-sodium diet. The potassium decline was not associated with ΔGlucose during the clamp, but correlated negatively with ΔInsulin0–10 (P = .032; β-coefficient, −0.0022) and ΔInsulin90–120 (P = .009; β-coefficient, −0.0017). This correction did not attenuate the significance of aldosterone administration.
There was no effect of overnight aldosterone infusion on baseline fasting glucose (95.5 ± 1.6 vs 94.8 ± 1.7 mg/dL during vehicle vs aldosterone; P = .69) or insulin (11.2 ± 1.5 vs 10.9 ± 1.5 μU/mL during vehicle vs aldosterone; P = .63). At the end of the hyperglycemic clamps (time, 90–120 min), plasma glucose was clamped near 200 mg/dL, similarly during all study periods (overall mean, 195.4 ± 0.6 mg/dL; P = .92 for diet; P = .74 for aldosterone). Overnight aldosterone infusion did not affect the insulin response to glucose (ΔInsulin0–10 = 44.9 ± 5.6 vs 45.2 ± 5.8 μU/mL during vehicle vs aldosterone; P = .91), insulin sensitivity index (20.0 ± 3.0 vs 19.6 ± 3.1 mg·kg−1·min−1 per μU/mL during vehicle vs aldosterone; P = .78), or disposition index (6.97 ± 0.78 vs 6.95 ± 0.82 during vehicle vs aldosterone; P = .96) during either the low- or high-sodium diet. Adjustment for age, sex, race, or insulin sensitivity did not alter the significance of these results.
Discussion
Dietary sodium restriction and activation of the RAAS can impair insulin sensitivity and glucose tolerance (17–21, 32), but the effects on β-cell function have not been previously investigated. We found that dietary sodium restriction, which activates the endogenous RAAS, reduces glucose-stimulated insulin secretion in humans without affecting insulin sensitivity. Short-term administration of exogenous aldosterone did not alter insulin sensitivity or insulin secretion, but augmented the potassium response to hyperglycemic hyperinsulinemia.
Low sodium intake increases endogenous renin, angiotensin II, and aldosterone in parallel and does not allow discrimination between the effect of angiotensin II and effects of aldosterone on insulin secretion. Exogenous angiotensin II infusion decreases insulin secretion and glucose tolerance in humans, but the contribution of aldosterone has not been investigated (16). Studies in rodents and isolated perifused islets indicate that aldosterone decreases insulin secretion. For example, selective aldosterone deficiency in mice results in increased glucose- stimulated insulin secretion without affecting insulin sensitivity, and aldosterone impairs insulin secretion in isolated islets in vitro via reactive oxygen species (10). During high-fat feeding, aldosterone-deficient mice are protected against obesity-related complications such as hepatic fat accumulation and adipokine dysfunction, but not against the development of insulin resistance (11). In humans, the incidence of diabetes appears to be increased in patients with primary aldosteronism (33). However, other studies based on fasting measures did not reveal any significant effects of adrenalectomy on glucose or lipid abnormalities (34). Using clamp techniques, Fischer et al (15) demonstrated that first-phase glucose-stimulated, but not arginine-stimulated, insulin secretion is impaired in subjects with aldosterone-producing adenomas and is improved after adrenalectomy. Although an overnight infusion of exogenous aldosterone did not affect insulin secretion in our study, this does not exclude the possibility that a more prolonged increase in endogenous aldosterone contributes to the effect of low-sodium intake on insulin secretion. Further mechanistic studies are needed to define more clearly the role of endogenous aldosterone, potassium, and the mineralocorticoid receptor on insulin secretion.
Most previous studies examining the effect of RAAS activation on glucose metabolism have demonstrated an effect on insulin sensitivity (5, 6). Angiotensin II could impair insulin sensitivity by reducing tissue perfusion, increasing extracellular matrix deposition, or impairing cellular insulin responsiveness (5). Angiotensin II and angiotensin receptor activation impair insulin sensitivity within vascular smooth muscle and myocytes by increasing reactive oxygen species and increased insulin receptor phosphorylation, resulting in an impaired downstream signaling response to insulin (35–37). Angiotensin receptor blockade improves insulin signaling in animal models and reduces progression to diabetes in clinical trials (1, 2, 5, 35). Dietary sodium reduction in healthy subjects impaired insulin sensitivity in some studies, as measured during hyperinsulinemic clamps (18–20). On the other hand, some have observed either no difference or improvement in insulin sensitivity during low sodium intake (22, 38). We did not observe a difference in insulin sensitivity during dietary stimulation of the RAAS or during exogenous aldosterone infusion. Although we used the hyperglycemic clamp rather than hyperinsulinemic clamp estimate of insulin sensitivity, this measurement correlates well with established standards (23–25).
The increased potassium response to hyperglycemic hyperinsulinemia during RAAS stimulation or exogenous aldosterone administration stands in contrast to the lack of any glucose response. Although some have suggested that hypokalemia induces insulin resistance, insulin-stimulated potassium uptake occurs independently of glucose uptake. The potassium decrease during hyperglycemic clamps is due to hepatic and skeletal muscle uptake in response to insulin (39). The potassium response to insulin is more sensitive and longer lasting than the glucose uptake response and occurs even in the absence of glucose (40, 41). Acute mineralocorticoid administration increases potassium tolerance in anephric dialysis subjects without altering fecal potassium excretion, whereas spironolactone decreases potassium tolerance, suggesting that mineralocorticoids contribute to acute cellular potassium redistribution (42). Likewise, potassium tolerance after acute administration is increased by endogenous or exogenous mineralocorticoids in nephrectomized rats (43, 44). Our findings suggest that mineralocorticoids could increase the effectiveness of insulin during treatment of hyperkalemia, although further studies are needed to test this hypothesis. This study may also explain why some patients with hyperaldosteronism do not become hypokalemic until they are challenged with a thiazide diuretic (45), which worsens insulin sensitivity and increases circulating insulin concentrations (32).
We studied a healthy population to examine the effects of sodium intake on normal physiology. Although we attempted to control for potential confounding variables such as body mass index, results in our study will need to be validated in more insulin-resistant populations. We were unable to blind the dietary intervention to the participants in this study. Because we primarily hypothesized that aldosterone impairs insulin secretion, we used the hyperglycemic clamp technique, which is arguably the preferred method for determination of insulin secretion (46). Although these studies are labor-intensive, they have proven useful in smaller population-based genetic studies and also provide a measure of insulin sensitivity that correlates strongly with hyperinsulinemic-euglycemic clamps (23, 26, 47). We observed no effect of dietary sodium or aldosterone on insulin sensitivity as assessed by the hyperglycemic clamp (23), but we cannot exclude the possibility that such an effect would be seen using a more targeted technique such as a hyperinsulinemic-euglycemic clamp. The effect of a low-sodium diet remained significant when analysis was restricted to males only, demonstrating that altered insulin sensitivity due to menstrual phase does not explain these findings. Furthermore, infusion of aldosterone and vehicle was conducted within 2 days of each other during the same phase of the menstrual cycle in females.
Our study has potentially important clinical implications. Severe dietary sodium restriction may adversely affect glucose metabolism by impairing insulin secretion. The dietary sodium restriction used in this study was intentionally restrictive in order to maximally stimulate the endogenous RAAS. It is unlikely that individuals would achieve this degree of sodium reduction on standard available diets. Indeed, the sodium content of the study diet was 10–20% of the recommended daily sodium intake for patients with hypertension, chronic kidney disease, or congestive heart failure (<2300 mg, or 100 mEq sodium). On the other hand, diuretics such as hydrochlorothiazide are known to impair glucose tolerance, although the mechanism is not entirely understood (48, 49). Our findings suggest that these drugs may impair insulin secretion by affecting sodium balance and RAAS activation, independent of potassium. Conversely, interventions that block the RAAS improve glucose homeostasis in clinical trials. Valsartan reduces the risk of progression to diabetes in patients with impaired glucose tolerance (1), and ramipril improves glucose tolerance (2). These beneficial effects have been attributed to improved insulin sensitivity, but our study highlights the importance of improved insulin secretion also reported to occur in subjects during valsartan treatment (4, 50). The use of RAAS blockade in conjunction with thiazide diuretics may therefore improve glucose homeostasis by improving insulin secretion.
Acknowledgments
The authors thank Yan Ru Su and Dillon O'Neill in the Vanderbilt Cardiology Core Lab for Translational and Clinical Research for their technical expertise.
This work was supported by National Institutes of Health (NIH) Grants DK081662, DK096994, HL060906, and DK20593 (Vanderbilt Diabetes Research and Training Center Hormone Assay Core), and Grants UL1 RR024975 and UL1 TR000445 from National Center for Advancing Translational Sciences/NIH (Vanderbilt Institute for Clinical and Translational Research).
This study was registered on ClinicalTrials.gov NCT00732160.
J.M.L. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. J.M.L. designed, researched data, and wrote the manuscript. L.M.B., C.Y., T.J.W., and N.J.B. contributed to the design, researched data, and reviewed/edited the manuscript.
Disclosure Summary: The authors have nothing to disclose
Footnotes
- AUC
- area under the curve
- NT-pro-BNP
- N-terminal brain natriuretic peptide
- RAAS
- renin-angiotensin-aldosterone system.
References
- 1. NAVIGATOR Study Group, McMurray JJ, Holman RR, Haffner SM, et al. Effect of valsartan on the incidence of diabetes and cardiovascular events. N Engl J Med. 2010;362:1477–1490 [DOI] [PubMed] [Google Scholar]
- 2. DREAM Trial Investigators, Bosch J, Yusuf S, Gerstein HC, et al. Effect of ramipril on the incidence of diabetes. N Engl J Med. 2006;355:1551–1562 [DOI] [PubMed] [Google Scholar]
- 3. Scheen AJ. Prevention of type 2 diabetes mellitus through inhibition of the renin-angiotensin system. Drugs. 2004;64:2537–2565 [DOI] [PubMed] [Google Scholar]
- 4. Sowers JR, Raij L, Jialal I, et al. Angiotensin receptor blocker/diuretic combination preserves insulin responses in obese hypertensives. J Hypertens. 2010;28:1761–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Luther JM, Brown NJ. The renin-angiotensin-aldosterone system and glucose homeostasis. Trends Pharmacol Sci. 2011;32:734–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sowers JR, Whaley-Connell A, Epstein M. Narrative review: the emerging clinical implications of the role of aldosterone in the metabolic syndrome and resistant hypertension. Ann Intern Med. 2009;150:776–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846 [DOI] [PubMed] [Google Scholar]
- 8. Seino S. Cell signalling in insulin secretion: the molecular targets of ATP, cAMP and sulfonylurea. Diabetologia. 2012;55:2096–2108 [DOI] [PubMed] [Google Scholar]
- 9. Weyer C, Bogardus C, Mott DM, Pratley RE. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest. 1999;104:787–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Luther JM, Luo P, Kreger MT, et al. Aldosterone decreases glucose-stimulated insulin secretion in vivo in mice and in murine islets. Diabetologia. 2011;54:2152–2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Luo P, Dematteo A, Wang Z, et al. Aldosterone deficiency prevents high-fat-feeding-induced hyperglycaemia and adipocyte dysfunction in mice. Diabetologia. 2013;56:901–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goodfriend TL, Kelley DE, Goodpaster BH, Winters SJ. Visceral obesity and insulin resistance are associated with plasma aldosterone levels in women. Obes Res. 1999;7:355–362 [DOI] [PubMed] [Google Scholar]
- 13. Bentley-Lewis R, Adler GK, Perlstein T, et al. Body mass index predicts aldosterone production in normotensive adults on a high-salt diet. J Clin Endocrinol Metab. 2007;92:4472–4475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mosso LM, Carvajal CA, Maiz A, et al. A possible association between primary aldosteronism and a lower β-cell function. J Hypertens. 2007;25:2125–2130 [DOI] [PubMed] [Google Scholar]
- 15. Fischer E, Adolf C, Pallauf A, et al. Aldosterone excess impairs first phase insulin secretion in primary aldosteronism. J Clin Endocrinol Metab. 2013;98:2513–2520 [DOI] [PubMed] [Google Scholar]
- 16. Fliser D, Schaefer F, Schmid D, Veldhuis JD, Ritz E. Angiotensin II affects basal, pulsatile, and glucose-stimulated insulin secretion in humans. Hypertension. 1997;30:1156–1161 [DOI] [PubMed] [Google Scholar]
- 17. Ames RP. The effect of sodium supplementation on glucose tolerance and insulin concentrations in patients with hypertension and diabetes mellitus. Am J Hypertens. 2001;14:653–659 [DOI] [PubMed] [Google Scholar]
- 18. Townsend RR, Kapoor S, McFadden CB. Salt intake and insulin sensitivity in healthy human volunteers. Clin Sci Lond. 2007;113:141–148 [DOI] [PubMed] [Google Scholar]
- 19. Fliser D, Fode P, Arnold U, Nowicki M, Kohl B, Ritz E. The effect of dietary salt on insulin sensitivity. Eur J Clin Invest. 1995;25:39–43 [DOI] [PubMed] [Google Scholar]
- 20. Perry CG, Palmer T, Cleland SJ, et al. Decreased insulin sensitivity during dietary sodium restriction is not mediated by effects of angiotensin II on insulin action. Clin Sci Lond. 2003;105:187–194 [DOI] [PubMed] [Google Scholar]
- 21. Garg R, Williams GH, Hurwitz S, Brown NJ, Hopkins PN, Adler GK. Low-salt diet increases insulin resistance in healthy subjects. Metabolism. 2011;60:965–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Donovan DS, Solomon CG, Seely EW, Williams GH, Simonson DC. Effect of sodium intake on insulin sensitivity. Am J Physiol. 1993;264:E730–E734 [DOI] [PubMed] [Google Scholar]
- 23. DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. 1979;237:E214–E223 [DOI] [PubMed] [Google Scholar]
- 24. Mitrakou A, Vuorinen-Markkola H, Raptis G, et al. Simultaneous assessment of insulin secretion and insulin sensitivity using a hyperglycemia clamp. J Clin Endocrinol Metab. 1992;75:379–382 [DOI] [PubMed] [Google Scholar]
- 25. Korytkowski MT, Berga SL, Horwitz MJ. Comparison of the minimal model and the hyperglycemic clamp for measuring insulin sensitivity and acute insulin response to glucose. Metabolism. 1995;44:1121–1125 [DOI] [PubMed] [Google Scholar]
- 26. 't Hart LM, Simonis-Bik AM, Nijpels G, et al. Combined risk allele score of eight type 2 diabetes genes is associated with reduced first-phase glucose-stimulated insulin secretion during hyperglycemic clamps. Diabetes. 2010;59:287–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Luther JM, Gainer JV, Murphey LJ, et al. Angiotensin II induces interleukin-6 in humans through a mineralocorticoid receptor-dependent mechanism. Hypertension. 2006;48:1050–1057 [DOI] [PubMed] [Google Scholar]
- 28. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)–a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lazo M, Young JH, Brancati FL, et al. NH2-terminal pro-brain natriuretic peptide and risk of diabetes. Diabetes. 2013;62:3189–3193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chang HR, Hsieh JC, Hsu BG, Wang LY, Chen MY, Wang JH. Inverse association of N-terminal pro-B-type natriuretic peptide with metabolic syndrome in patients with congestive heart failure. PLoS One. 2013;8:e79096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Heinisch BB, Vila G, Resl M, et al. B-type natriuretic peptide (BNP) affects the initial response to intravenous glucose: a randomised placebo-controlled cross-over study in healthy men. Diabetologia. 2012;55:1400–1405 [DOI] [PubMed] [Google Scholar]
- 32. Eriksson JW, Jansson PA, Carlberg B, et al. Hydrochlorothiazide, but not Candesartan, aggravates insulin resistance and causes visceral and hepatic fat accumulation: the mechanisms for the diabetes preventing effect of Candesartan (MEDICA) Study. Hypertension. 2008;52:1030–1037 [DOI] [PubMed] [Google Scholar]
- 33. Reincke M, Meisinger C, Holle R, et al. Is primary aldosteronism associated with diabetes mellitus? Results of the German Conn's Registry. Horm Metab Res. 2010;42:435–439 [DOI] [PubMed] [Google Scholar]
- 34. Matrozova J, Steichen O, Amar L, Zacharieva S, Jeunemaitre X, Plouin PF. Fasting plasma glucose and serum lipids in patients with primary aldosteronism: a controlled cross-sectional study. Hypertension. 2009;53:605–610 [DOI] [PubMed] [Google Scholar]
- 35. Velloso LA, Folli F, Perego L, Saad MJ. The multi-faceted cross-talk between the insulin and angiotensin II signaling systems. Diabetes Metab Res Rev. 2006;22:98–107 [DOI] [PubMed] [Google Scholar]
- 36. Folli F, Kahn CR, Hansen H, Bouchie JL, Feener EP. Angiotensin II inhibits insulin signaling in aortic smooth muscle cells at multiple levels. A potential role for serine phosphorylation in insulin/angiotensin II crosstalk. J Clin Invest. 1997;100:2158–2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wei Y, Whaley-Connell AT, Chen K, et al. NADPH oxidase contributes to vascular inflammation, insulin resistance, and remodeling in the transgenic (mRen2) rat. Hypertension. 2007;50:384–391 [DOI] [PubMed] [Google Scholar]
- 38. Grey A, Braatvedt G, Holdaway I. Moderate dietary salt restriction does not alter insulin resistance or serum lipids in normal men. Am J Hypertens. 1996;9:317–322 [DOI] [PubMed] [Google Scholar]
- 39. DeFronzo RA, Felig P, Ferrannini E, Wahren J. Effect of graded doses of insulin on splanchnic and peripheral potassium metabolism in man. Am J Physiol. 1980;238:E421–E427 [DOI] [PubMed] [Google Scholar]
- 40. Zierler KL, Rabinowitz D. Effect of very small concentrations of insulin on forearm metabolism. Persistence of its action on potassium and free fatty acids without its effect on glucose. J Clin Invest. 1964;43:950–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zierler KL. Effect of insulin on potassium efflux from rat muscle in the presence and absence of glucose. Am J Physiol. 1960;198:1066–1070 [DOI] [PubMed] [Google Scholar]
- 42. Sugarman A, Brown RS. The role of aldosterone in potassium tolerance: studies in anephric humans. Kidney Int. 1988;34:397–403 [DOI] [PubMed] [Google Scholar]
- 43. Alexander EA, Levinsky NG. An extrarenal mechanism of potassium adaptation. J Clin Invest. 1968;47:740–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bia MJ, Tyler KA, DeFronzo RA. Regulation of extrarenal potassium homeostasis by adrenal hormones in rats. Am J Physiol. 1982;242:F641–F644 [DOI] [PubMed] [Google Scholar]
- 45. Calhoun DA. Is there an unrecognized epidemic of primary aldosteronism? Pro. Hypertension. 2007;50:447–453; discussion 447–453 [DOI] [PubMed] [Google Scholar]
- 46. Stumvoll M, Fritsche A, Haring HU. Clinical characterization of insulin secretion as the basis for genetic analyses. Diabetes. 2002;51(suppl 1):S122–S129 [DOI] [PubMed] [Google Scholar]
- 47. Simonis-Bik AM, Nijpels G, van Haeften TW, et al. Gene variants in the novel type 2 diabetes loci CDC123/CAMK1D, THADA, ADAMTS9, BCL11A, and MTNR1B affect different aspects of pancreatic β-cell function. Diabetes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Carter BL, Einhorn PT, Brands M, et al. Thiazide-induced dysglycemia: call for research from a working group from the National Heart, Lung, and Blood Institute. Hypertension. 2008;52:30–36 [DOI] [PubMed] [Google Scholar]
- 49. Stears AJ, Woods SH, Watts MM, et al. A double-blind, placebo-controlled, crossover trial comparing the effects of amiloride and hydrochlorothiazide on glucose tolerance in patients with essential hypertension. Hypertension. 2012;59:934–942 [DOI] [PubMed] [Google Scholar]
- 50. van der Zijl NJ, Moors CC, Goossens GH, Hermans MM, Blaak EE, Diamant M. Valsartan improves β-cell function and insulin sensitivity in subjects with impaired glucose metabolism: a randomized controlled trial. Diabetes Care. 2011;34:845–851 [DOI] [PMC free article] [PubMed] [Google Scholar]