

Abstract
Background
Reactive oxygen species-mediated cysteine sulfenic acid modification has emerged as an important regulatory mechanism in cell signaling. The stability of sulfenic acid in proteins is dictated by the local microenvironment and ability of antioxidants to reduce this modification. Several techniques for detecting this cysteine modification have been developed, including direct and in situ methods.
Scope of review
This review presents a historical discussion of sulfenic acid chemistry and highlights key examples of this modification in proteins. A comprehensive survey of available detection techniques with advantages and limitations is discussed. Finally, issues pertaining to rates of sulfenic acid formation, reduction, and chemical trapping methods are also covered.
Major conclusions
Early chemical models of sulfenic acid yielded important insights into the unique reactivity of this species. Subsequent pioneering studies led to the characterization of sulfenic acid formation in proteins. In parallel, the discovery of oxidant-mediated cell signaling pathways and pathological oxidative stress has led to significant interest in methods to detect these modifications. Advanced methods allow for direct chemical trapping of protein sulfenic acids directly in cells and tissues. At the same time, many sulfenic acids are short-lived and the reactivity of current probes must be improved to sample these species, while at the same time, preserving their chemical selectivity. Inhibitors with binding scaffolds can be rationally designed to target sulfenic acid modifications in specific proteins.
General significance
Ever increasing roles for protein sulfenic acids have been uncovered in physiology and pathology. A more complete understanding of sulfenic acid-mediated regulatory mechanisms will continue to require rigorous and new chemical insights. This article is part of a Special Issue entitled Current methods to study reactive oxygen species - pros and cons and biophysics of membrane proteins. Guest Editor: Christine Winterbourn.
Keywords: Sulfenic acid, Sulfenic acid chemistry, Sulfenic acid detection method, Cellular lifetimes of sulfenic acid
1. Introduction
Cysteine (Cys, C) is a unique thiol containing amino acid coded by codon triplets UGU and UGC. Even though it is coded by two codons, Cys is mathematically underrepresented in the proteins of all organisms. It has been shown that Cys content of different species increases with the increase in complexity. For example, Cys content of the proteins in Archea is as low as 0.4%–0.5%, whereas mammalian protein may have as much as about 2.26%. The increase in protein-Cys content with increasing organism complexity may be attributed to its relative reactivity compared to other amino acids, particularly in the context of oxidation. One such predominant reactivity feature is disulfide bond formation which is a unique characteristic of nucleophilic thiol present on Cys-side chain [1,2]. Protein cysteine thiols may broadly be divided in four categories—(1) permanent structural disulfide bonds which are typically observed in oxidizing environments; (2) thiols that coordinate with metals, typically iron, zinc or copper; (3) those that are permanently in the reduced state and (4) those that are susceptible to reversible and irreversible oxidation [3]. The irreversible protein thiol oxidation has been shown to be a biomarker for the failure of protective mechanisms and indicative of diseases related to oxidative stress. Reversibly oxidized protein thiols are often required for enzyme catalysis and regulation of protein activity [4]. Cysteine sulfenic acid is one such oxoform formed by the action of reactive oxygen species (ROS) on protein thiols. Due to their inherently reactive nature, sulfenic acids have been viewed as transient species en route to other more stable oxidation states. Indeed, for a long time, sulfenic acid was speculated to be the transient oxidized species leading to the formation of disulfide bond. However, recent advances in the identification of sulfenic acids for their antioxidant activity and roles in signal transduction and transcription regulation events have placed them at the forefront of redox biology [5–7]. They have also been shown to play catalytic and structural roles in enzymes [7,8]. This review article will focus on principles which govern the sulfenic acid biochemistry. The foundation is laid by considering small molecule sulfenic acid chemistry and the observations from that are exported to the biological systems. We considered the kinetic factors responsible for the formation and metabolism of sulfenic acids to elaborate on their stability and reactivity. Finally, a general survey of current sulfenic acid detection techniques is presented.
2. The chemistry of sulfenic acid
Sulfenic acids have long been characterized as transient reaction intermediates of cysteine thiols formed under oxidative stress. A gap of almost half a century in the synthesis of first and second stable sulfenic acids is a testament of their unstable nature. Before considering the generation, stability and role of sulfenic acids in biological systems, it is imperative to go over the properties and fundamental reaction pathways of sulfenic acid, as deciphered by many decades of extensive and elegant chemical studies [9]. The purpose of this section is to touch upon the basic principles which govern the generation and reactions of stable as well as transient sulfenic acids in small molecules. A general survey of salient features of sulfenic acids in small molecules will serve as conceptual foundation toward an understanding of reaction properties in protein sulfenic acids.
2.1. Sulfur and sulfenic acid S – O bond
Although, the elemental components of living systems predominantly are carbon, nitrogen, oxygen and phosphorus, it is not easy to overlook the biological importance of another essential element, sulfur. It is no coincidence that sulfur is a neighbor of the elements that comprise most organic compounds thus sharing the electronic properties with them. Sulfur occurs in all major classes of biomolecules, including proteins, sugars, nucleic acids, vitamin cofactors and metabolites and is required by all living organisms. The flexibility of sulfur-containing molecules stems from the versatile chemistry shown by this element. As a member of same periodic group as oxygen, sulfur shares many similarities in chemical reactivity. However, sulfur’s position lower in the periodic table endows its compounds with many distinct properties, which are exploited in proteins and other biomolecules. For example, due to the combination of lower electronegativity and availability of lone pairs, thiols are superior nucleophiles compared to alcohols. Disulfide bonds (RS – SR) are more stable than peroxide bonds (RO – OR) and this enhanced stability is a key advantage due to which disulfide bonds are integral part of the structural features of proteins. Since sulfur is the second-row element of group VIa of the periodic table with an electronic configuration of [Ne]3s23p43d0, it has empty d-orbital available for bonding. This feature allows sulfur to attain valences beyond divalent compounds to valences of 4 and 6 at oxidation states ranging from −2 to +6.
In cysteine, the sulfur atom is fully reduced with an oxidation state of −2 and due to its low redox potential in proteins (E∘′ −0.27 to −0.125 V) the thiol side chain can readily undergo a range of oxidative post-translational modifications, some of which are illustrated in Fig. 1 [10]. Disulfide bond formation is the most commonly considered thiol oxidation reaction, which may proceed via two general pathways: (a) thiol–disulfide exchange and (b) condensation of a thiol with sulfenic acid [11]. Higher oxidation states of cysteine are energetically less stable and the nucleophilic character decreases as the oxidation number increases. This decrease in nucleophilic character is attributed to the acquisition of greater positive charge on sulfur. This overwhelming variety of oxidation states and their different chemical reactivity make the chemistry and biology of cysteine oxidation an important and fertile research area.
Fig. 1.
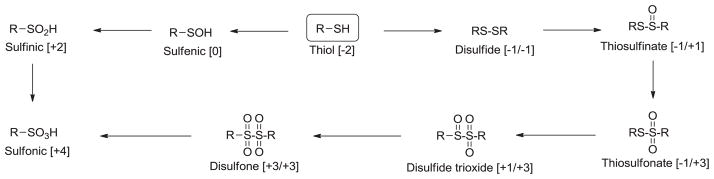
Biological cysteine oxidation states.
Sulfenic acids have been discovered in a variety of chemical and biological settings. Since sulfenic acids in small molecule systems are generally too unstable to isolate, a study of their physical and chemical properties and their reaction mechanism is not possible in normal fashion. This is highlighted by the fact that for many years, even the correct tautomeric structure of sulfenic acid was debatable. Several pieces of spectroscopic evidence indicated that sulfenic acid exists in the divalent sulfur tautomer (1) over the tetravalent sulfoxide structure (2) (Fig. 2). IR studies in solution on 1-anthraquinonesulfenic acid and 1,4-anthraquinonedisulfenic acid indicated that S – O bond length is distinctly longer than that of sulfoxide thus favoring the O-protonated form. This conclusion was supported by similar studies on other stable sulfenic acids [12–14].
Fig. 2.

Sulfenic acid tautomers.
2.2. Chemical reactivity of sulfenic acid
Sulfenic acids have been studied in many chemical reactions, which have been reviewed extensively [15,16]. The formal oxidation state of sulfur in sulfenic acid is 0 which results in its unique ability to function as both a nucleophile and an electrophile. Although the nucleophilic character of sulfenic acids is not very strong, it can still play a prominent role where the possibility of the formation of intermolecular hydrogen bonding is present. This dual nature is most clearly illustrated in Allicin chemistry which is propagated primarily by the formation of a hydrogen-bonded sulfenic acid dimer [17]. The condensation of two sulfenic acids results in the formation of thiosulfinates (Fig. 3, reaction 1). This is a unique feature among organic acids, in that favorable formation of thiosulfinate means that aqueous equilibrium favors the anhydride form of sulfenic acid. As a direct result of this feature, less is known about sulfenic acids than about corresponding higher sulfur oxyacids. The prevalence of thiosulfinates in biology is largely unknown; however, it must be acknowledged that due to the abundance of cellular thiols the interfacing of two sulfenic acids is likely to be a rare event [18]. This leads to another reaction of sulfenic acid where it may readily act as an electrophile to a nucleophilic thiol and form disulfide (Fig. 3, reaction 2).
Fig. 3.

Electrophilic reactions of sulfenic acid.
In 1974, Allison and coworkers reported a reaction of sulfenic acid which is highly selective and would prove to be a most useful reaction in detection. In this condensation reaction, sulfenic acid would act as an electrophile and react selectively with 1,3-diketones such as 5,5-dimethyl-1,3-cyclohexanedione (dimedone) under physiological conditions to give a corresponding thioether derivative (Fig. 3, reaction 4) [19]. The mechanism of this reaction has not been rigorously analyzed, however it is believed to proceed via a concerted mechanism with concomitant loss of water. Sulfenic acid may also react with an amine to give sulfenamide (Fig. 3, reaction 5) [19,20]. Though amide nitrogen atoms are considered poor nucleophiles, when fixed in close proximity, they act as nucleophiles and react with sulfur atom in a sulfenic acid to form a cyclic sulfenamide (Fig. 3, reaction 5) [21,22]. Finally, several inorganic and organic molecules, such as sodium arsenite, triphenylphosphine (Fig. 3, reaction 3), hydrazine and alkyl hydrazines (Fig. 3, reaction 6) and sodium azide can reduce RSOH back to the thiol [23,24]. Although the abovementioned reactions span across several classes, they can be categorized as and attributed to the electrophilic character of sulfur atom in sulfenic acid.
It has been emphasized above that sulfenic acids are generally unstable functional groups en route to other more stable oxidation states; it is only fair to discuss this over-oxidation in context with sulfenic acid’s nucleophilicity. Hyperoxidation of sulfenic acid to sulfinic acid requires nucleophilic attack on hydrogen peroxide (Fig. 4, reaction 1). A number of factors are likely to regulate this reaction, including the stability of sulfenic acid, nucleophilicity and oxidant concentration [15]. A kinetic study of 1-anthraquinonesulfenic acid oxidation indicated a pH independent rate constant (85 M−1 s−1). However, only two pH values were evaluated, so no specific conclusion could be drawn from those studies [25]. Kinetic studies have shown that the rate constant for oxidation of protein cysteine sulfenic acid of mammalian Prx1 is almost two to three orders of magnitude slower than the initial thiolate oxidation. Moreover, the pH profile of peroxide-mediated sulfenic acid oxidation indicated the sulfenate as the reacting species [26].
Fig. 4.

Nucleophilic reactions of sulfenic acid.
One particularly important and useful class of reactions of sulfenic acids is the concerted addition to both alkenes and alkynes. Given the transient nature of sulfenic acids, these addition reactions provide dual purpose. First, they act as effective trapping reagents for unstable sulfenic acids. Second, they are useful reactions for the synthesis of desired sulfoxides. Under organic conditions, sulfenic acids add to activated as well as unactivated alkenes to give sulfoxides (Fig. 4, reaction 2). The addition usually follows Markovnikov’s rule and the reaction has been extended to a wide variety of alkenes. The yields were much lower in case of addition reaction with cycloalkenes [15]. The addition is a cyclic concerted process which is the reverse of elimination of sulfenic acid from sulfoxides and thus proceeds through the same transition state. Sulfenic acids add to alkynes by a concerted mechanism similar to that described for additions to alkenes (Fig. 4, reaction 3). In case of alkynes as well, the concerted addition follows Markovnikov’s rule [15]. Although, these reactions are not facile in biological systems, their applications in organic synthesis cannot be underestimated. Aversa and coworkers have investigated the addition of enantiopure sulfenic acids to substituted alkynes in the synthesis of carbo- and hetero-sulfinyl dienes which were then used in many stereoselective transformations, such as Diels–Alder cycloaddition or nucleophilic conjugated additions. In most cases, the reaction of enantiopure sulfenic acids to enyne system is chemo-, regio- and stereoselective [16].
Sulfenic acids can also undergo nucleophilic substitution reaction on activated halogenated compounds such as 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole (NBD-Cl) (Fig. 4, reaction 4). This has served as a method to discriminate between thiols and sulfenic acids based on the unique spectral properties of respective products [27]. There have been few reports of nucleophilic sulfenic acid addition to different activated and unactivated alkyl halides as well (Fig. 4, reaction 5) [15,28–30]. In the end, it must be emphasized that even though sulfenic acids show greater variety of nucleophilic than electrophilic reactions, a major difficulty in capitalizing on the nucleophilic reactivity is that in biological systems, the thiolate is a much stronger nucleophile and can carry out much of the same chemistry as sulfenic acids, albeit yielding a different product.
2.3. Synthesis of sulfenic acid—chemical perspective
Due to their high reactivity, the majority of small molecule sulfenic acids cannot be isolated under usual laboratory conditions due to the tendency to undergo self-condensation to give thiosulfinate. Such unstable sulfenic acids have a very short half-life of <1 min. However, for a long time, various researchers devoted considerable efforts to synthesize stable sulfenic acids and use them as models to decipher the complex fate of evasive, but common species like Cys-SOH in biological systems. The general aim was to generate a sulfenic acid model system, which could be stored for hours if not days without appreciable self-condensation.
2.3.1. Generation of transient sulfenic acids in small molecules
The most common pathways toward the generation of transient sulfenic acids are displayed by nature itself during the generation of unstable Cys-SOH from Cys-SH during oxidative stress and in the chemistry of Allium species. Upon cutting garlic, alliin (4) is dissected by allinase and 2-propenesulfenic acid (5) is generated along with ammonium pyruvate (8). The transient sulfenic acid 5 undergoes self-condensation to form corresponding diallyl thiosulfinate allicin (7), which is responsible for the odor, flavor and antioxidant activity of garlic (Scheme 1, equation 1). Similarly, the lachrymatory effect of onion cut is due to the enzyme-catalyzed conversion of isoalliin (10) into 1-propenesulfenic acid (11), followed by its rearrangement in propanthial S-oxide 12 (Scheme 1, equation 2). In both cases, the sulfoxide precursors bear a migrant hydrogen beta- to the sulfur atom [17]. Taking inspiration from nature, organic chemists have devised similar pathways to generate transient sulfenic acids by syn-elimination of sulfoxide precursors through thermolysis or ionic stepwise β-elimination (Scheme 2, equation 1). Again, the characteristic feature here is the acidic hydrogen atom beta- to the sulfur due to the proximal presence of electron-withdrawing group (EWG) [15].
Scheme 1.
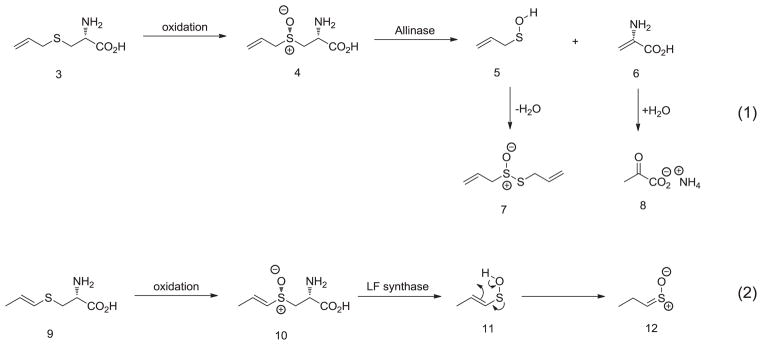
Generation of transient sulfenic acid—nature’s perspective.
Scheme 2.

(1) Fundamental synthetic strategy. (2) General syntheses of transient sulfenic acids.
Several applications of such scheme have been reported in the literature of which the work of Jones and coworkers is discussed below. Jones et al. first reported the synthesis of sulfenic acid from β-sulfinylpropionitriles under harsh thermal conditions [31]. However, in order to generate sulfenic acids under milder conditions, they reported new precursors 14, 16 and 18 which upon heating rearranged to benzenesulfenic acid (20) (Scheme 2, equation 2) [32]. Indeed, as hypothesized, the presence of two electron-withdrawing groups in the β-position, together with alkyl residue in α- to the sulfinyl group, enhanced the β-hydrogen mobility, thereby inducing the thermolysis of 19 even at 40 °C (Scheme 2, equation 2). This work by Jones was the inspiration behind the work of Aversa and coworkers. They used this chemistry and focused their attention on the generation of sulfenic acids bearing natural residues such as carbohydrate and amino acid side-chains. These were further used in the synthesis of target molecules containing sulfoxide or sulfone functionalities. In another application, recently Aversa et al. have reported easy two-step procedure starting from commercially available REM resin to prepare suitable solid-support based precursors of sulfenic acids [16].
Another fascinating application of this chemistry was displayed by Gates and coworkers. They used ortho substitution of β-sulfinyl propionic acid ester on a benzamide derivative (21) for in situ sulfenic acid generation. The sulfenic acid 23 was generated upon the incubation of 21 in aqueous buffered solution (250 mM sodium phosphate, pH 7.5, containing 30% MeCN) at 37 °C. It must be noted that the overall reaction is extremely slow (~36 h). Under organic conditions (DCM) the vicinal amide acted as a trap for sulfenic acid (23) to give a 3-isothiazolidinone system (24) which was used as a chemical model to better understand the redox-regulation of protein tyrosine phosphatase 1B (PTP1B) activity (Scheme 3) [33].
Scheme 3.
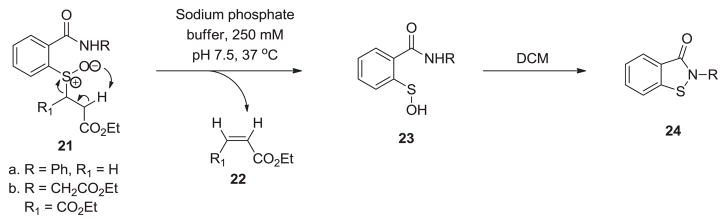
Sulfenic acid mediated generation of 3-isothiazolidinone heterocycle which serves as a chemical model to understand the redox-regulation of PTP1B activity.
2.3.2. Generation of stable sulfenic acids
The first stable sulfenic acid was reported by Fries in 1912—an anthraquinone derived sulfenic acid (27) (Scheme 4, equation 1) prepared by converting an antharaquinonesulfenyl bromide (25) to the less electrophilic corresponding methyl ester (26) followed by base-mediated hydrolysis [12]. The main feature that led to the enhanced stability of anthraquinone-1-sulfenic acid 27 is the formation of an intramolecular hydrogen bond with neighboring carbonyl group. Thus, generally speaking, circumstances hampering intermolecular hydrogen bonding (which promotes thiosulfinate formation) impart stability to corresponding sulfenic acid. The stability afforded by hydrogen bonding was demonstrated in two subsequent syntheses. Cohn et al. reported the isolation and characterization of a pyrimidine sulfenic acid (30) via alkali-mediated scission of disulfide in methyl analog of bis(4-thiouridine)disulfide (28) (Scheme 4, equation 2) [34]. In another report, stable lumazinesulfenates were prepared from corresponding disulfides (Scheme 4, equation 3). Again, the disulfide bond was cleaved under alkaline conditions to give the silver salt of 1,3,6-trimethyllumazine-7-thionate (31) which was alkylated with i-propyliodide followed by m-CPBA mediated oxidation to give corresponding sulfoxide (32). Thermal cleavage of sulfoxide 32 resulted in the cope elimination and gave analytically pure 1,3,6-trimethyllumazine-7-sulfenic acid (33) [35].
Scheme 4.

Synthesis of stable sulfenic acids.
Indeed, in past few decades, many researchers were successful in synthesizing new stable sulfenic acids in which the functionality is protected by a sterically or electronically demanding group to prevent formation of inter- or intramolecular disulfide bonds. In order to explore the actual influence of steric factors upon the stabilization of sulfenic acid system, Yoshimura and co workers explored the synthesis of trans-decalin-9-sulfenic acid 37 (Scheme 4, equation 4) [36]. Their reasoning was that trans-9-decalyl group with four axial protons would provide sufficient steric hindrance to protect the sulfenic acid functional group. The key step in the synthesis was acid hydrolysis of trans-decahydro-4a-[(methoxymethyl)sulfinyl]naphthalene (36) using the Okuyama method [37]. Although sulfenic acid 37 thus generated was somewhat stable, it disproportionated in benzene at room temperature within 30 min to give corresponding disulfide and sulfonic acid. This was the first example of a stable sulfenic acid having neither an unsaturated bond nor a heteroatom. The stability is attributed to the steric resistance caused by the four axial protons of trans-9-decalyl group when the SOH groups of two molecules approach each other.
Tripolt et al. reported unexpected stability of 4,6-dimethoxy-1,3,5-triazine-2-sulfenic acid. The oxidation of 4,6-dimethoxy-1,3,5-triazine-2(1H)-thione (38) with 2-benzenesulfonyl-3-p-nitrophenyloxaziridine in THF solution gave 4,6-dimethoxy-1,3,5-triazine-2-sulfenic acid (39) as a stable crystalline solid (Scheme 4, equation 5). According to their 13C NMR and X-ray analysis, 39 adopts the sulfenyl structure in condensed phase but in the crystal, the molecules form dimers linked by two strong intermolecular hydrogen bonds [38]. In another interesting study on the synthesis of unnaturally configured cephalo-sporins from 6-phthalimidopenicillanates, Baldwin et al. isolated an unusually stable azitidinone sulfenic acid (41) (Scheme 4, equation 6). They attributed the stability of these sulfenic acids to both steric bulk of azetidinone moiety which prevents the thiosulfinate synthesis and to the relative thermodynamic instability of the substrates 40 [39].
In addition to these six examples, there have been several successful synthetic endeavors based on different approaches viz. a stable sulfenic acid species bearing a tropylium cation which was the first instance of a stable sulfenic acid cation neither bearing a bulky residue nor forming a stabilizing hydrogen bond [40] or first synthesis of sulfenic acid by oxidation of sterically hindered thiophenetriptycene thiolate [41]. Although this sulfenic acid was stable toward intermolecular condensation with another sulfenic acid or thiol bearing same thiophenetriptycene group, it was susceptible toward photochemical and thermal decomposition. Finally, extensive work conducted by Okazaki and Goto toward the formation of stable sulfenic acids provides a readily accessible chemical system which simulates the environment of clefts present in protein sulfenic acids [42–44]. The stability of these molecules is attributed to the kinetic stabilization afforded by bulky bowl-type environment of cyclophanes. The cyclophane used in their system was abbreviated as Bmt and corresponding Bmt-SOH (43) was generated by the action of iodosobenzene, a mild oxidant, on Bmt-SH (42). Since the generation of symmetrical disulfide (Bmt-S-S-Bmt) is sterically hindered, they successfully recovered pure Bmt-SOH as a crystalline solid in 41% yield after silica gel column chromatography (Scheme 5, equation 1). Because sulfenic acid functionality is incorporated in a molecular cavity of Bmt, it was stable toward self-condensation and thiosulfinate formation was not observed even after weeks in the presence of air. In a demonstration of typical protein sulfenic acid redox process, Bmt-SOH readily reacted with an oxidant and reductant to give the corresponding sulfinic acid (44) and Bmt-SH (42), respectively (Scheme 5, equation 2). Bmt-SOH readily reacted with small molecule thiols to give unsymmetrical disulfides (45a–c) and underwent DTT mediated reduction to Bmt-SH (Scheme 5, equation 2). Moreover, Bmt-SOH underwent condensation reaction with benzylamine to give Bmt-sulfenamide (46) (Scheme 5, equation 2).
Scheme 5.

Synthesis and reactivity of sulfenic acid embedded in bowl-type cyclophane. (1) Synthesis of Bmt-SOH from Bmt-SH by a mild oxidant. (2) Reactions of Bmt-SOH.
It has been suggested that a major factor in the stabilization of sulfenic acid in proteins is the structural geometry where sulfenic acid is embedded in cavity and shielded from other reactive groups. Bmt- and analogs replicate a similar environment to impart stabilization to sulfenic acid and maybe used to better understand protein sulfenic acid [44]. In addition to the steric factors, some trends in the contribution of other factors can be concluded from the discussion above, as well as in literature. For aromatic sulfenic acids (like 27 and 39), presence of electron-withdrawing-substituents diminishes the nucleophilicity of sulfenic acid sulfur, thus stabilizing the aromatic sulfenic acids. Intramolecular hydrogen bonding between sulfenic acid and a nearby hydrogen bond acceptor also imparts substantial stability, as seen in 27, 30, 33, 39 and 41. Finally, the formation of thiosulfinates would be suppressed if the sulfenic acid is present in the form of sulfenate anion as the ionization reduces the hydrogen bond donating potential which is key to thiosulfinate formation. This is exemplified by the enhanced stability of 1-methyluracil-4-sulfenic acid 30 (pKa 6.3) and lumazine-sulfenic acid 33 (pKa 4.8) in alkaline solutions [34,35].
3. Sulfenic acids in proteins
Cysteine sulfenic acid (Cys-SOH) is a post-translational modification in proteins. Reversible oxidation of Cys-SH to this oxyacid has been implicated in several biological events. It has been shown that in the presence of molecular oxygen and trace metals, protein-SOH is spontaneously formed in unfolded proteins, even in the absence of exogenously added oxidants. The Cys-SOH could then serve as an intermediate in disulfide bond formation during nonenzymatic protein folding [45].
The antioxidant activity of many proteins is attributed to the oxidation of cysteine to sulfenic acid during the catalytic cycle. Peroxiredoxins (Prxs) which are highly expressed in all organisms constitute the largest family of peroxidases and have been reported to represent about 0.2–0.4% of the total soluble proteins in mammalian tissues. This makes Prxs the most abundant antioxidant enzymes in the cytosol. Prxs have very high specificity for hydrogen peroxide, peroxynitrite and other organic hydroperoxides. Their role as antioxidant as well as in signaling and the mechanism of action has been extensively studied and reported in many review articles [46–48]. They are broadly divided into six subfamilies (AhpC/Prx1, BCP/PrxQ, Prx5, Prx6, Tpx and AhpE) [49]. A related class of antioxidant enzymes is glutathione peroxidase (GPx) [50]. They also play an important role in cellular detoxification of H2O2 [51]. While mammalian GPxs in general are selenoproteins (SecGPxs), the vast majority of non-vertebrate GPxs like Drosophila melanogaster GPx are Cys homologues (2-CysGPxs) [52]. Families of both Prxs and GPxs share common enzyme-substitution mechanism. This implies that the mechanism does not involve any central enzyme complex where all the substrates are bound simultaneously. Instead the catalytic cycle comprises of a sequence of bimolecular reactions between the enzymes and their substrates. Concisely, a typical 2-CysPrx or 2-CysGPx constitutes two cysteines, one present in active site is ‘peroxidatic’ Cys and the other is ‘resolving’ Cys. In the first step, the peroxidatic Cys residue is oxidized to Cys-SOH and then rapidly forms a disulfide with resolving Cys. Then, this disulfide is reduced by thioredoxin (Trx) or related proteins at the expense of NADPH [53].
In addition to their occurrence as an intermediate in the catalytic cycle of many antioxidant enzymes, sulfenic acids have also been associated with cellular signaling and oxidative stress sensing [54]. Sulfenic acid formation is shown to be involved in H2O2-mediated inactivation of protein tyrosine phosphatases (PTPs). Tyrosine phosphorylation plays a central regulatory role in cell metabolism, growth, proliferation, differentiation, immune response, motility, tissue homeo-stasis and apoptosis. The phosphorylation status of the tyrosine residue is controlled by the opposing protein tyrosine kinases (PTKs) and PTPs, and their mechanisms of actions are well established [55,56]. This redox reaction of catalytic Cys-SH has been shown to be an important mechanism for the regulation of cellular PTP activity [56,57]. Human serum albumin, the most abundant protein in plasma, possesses a single free thiol which has been shown to form a stable sulfenic acid [58]. Recently, work done in our research group showed that sulfenic acid is involved in the redox regulation of epidermal growth factor receptor (EGFR) signaling where oxidation of active site Cys797 led to increased kinase activity [59,60]. Also, reversible sulfenic acid modification is shown to switch on or off the enzymatic activity of transcription and transduction factors like Escherichia coli OxyR [61], Bacillus subtilis OhrR [62], yeast Orp1-Yap1 [63] and RegB [64].
3.1. Historical perspective
As early as 1923, researchers were gathering the evidence for the existence of a peroxide-like derivative of cysteine. Based on the studies conducted on the oxidation of low molecular mass thiols by H2O2 and dithioformamidine, some even ventured ahead and speculated the formation of disulfide to involve two bimolecular reactions that involved sulfenic acid as an intermediate [65]. Although interesting, these studies on small molecule thiols were in no way indicative of sulfenic acid formation occurring in more complex biological systems. There was a general consensus in the scientific community that the systems with solvent-accessible cysteines are uniquely suited for oxidation to sulfenic acid by mild oxidants like H2O2 or iodosobenzoate. Moreover, when structural topology of the proteins hinders the formation of inter- and intra-molecular disulfide bond, it provides an ideal environment for the formation and stabilization of sulfenic acid [24].
Due to the absence of distinguishable spectroscopic features and direct detection techniques, initial evidence for the existence of protein-SOH was indirect, but compelling. In particular, the experimental studies conducted on the active site cysteine of glyceraldehydes 3-phosphate dehydrogenase (GAPDH) and papain provided convincing indirect evidence in favor of the formation of stable sulfenic acids in specific proteins. That specific protein thiols may oxidize to sulfenic acids was supported by the following evidence: (1) precedence for the formation of species of similar oxidation state as sulfenic acid, like sulfenyl iodide and sulfenyl thiosulfate; (2) the stoichiometry of oxidation in some protein thiols was consistent with the formation of sulfenic acids and not disulfides; (3) the oxidation of catalytically active thiols of papain and GAPDH by mild oxidants introduces electrophilic center in each of the enzymes with properties similar to sulfenyl halides. Fraenkel-Conrat published a study on the reaction of tobacco mosaic virus (TMV) coat protein with iodine which was the first to show that thiol sulfur may exist in an oxidation state similar to that of sulfenic acid [66]. Based on the evidence regarding reaction stoichiometry, radioisotope labeling and reversibility, it was shown that TMV coat protein thiol may be converted to a stable sulfenyl iodide (Fig. 5A). Upon denaturing, the sulfenyl iodide group regained its typical reactivity and instability. This is indicative that the unusual stability of sulfenyl halide is likely conferred by the surrounding protein environment.
Fig. 5.

Preliminary evidences for protein thiol modifications with sulfenic acid like oxidation state. (A) Generation of stable sulfenyl halide in TMV coat protein. (B) GAPDH reactivity with tetrathionate, o-iodosobenzoate and hydrogen peroxide.
With this evidence, a general hypothesis involving two-step mechanism for the disulfide formation was proposed—firstly, the conversion of protein thiol to corresponding sulfenyl iodide or sulfenic acid, followed by the attack from another thiolate to form disulfide. The take-home lesson from these studies was two-fold. First, that the local environment of the sulfenic acid may protect and preserve normally unstable reaction intermediates. Second, it was a testament of the involvement of sulfenic acid-like intermediates in disulfide formation in complex biological systems. A similar study which involved the treatment of GAPDH with tetrathionate reported the formation of a protein thiol derivative with same oxidation state as sulfenic acid [67]. Tetrathionate oxidizes simple thiols to the corresponding disulfides, presumably via formation of a sulfenyl thiosulfate intermediate (Fig. 5B, reaction 1). GAPDH, upon treatment with tetrathionate, was inactivated with the incorporation of thiosulfate and activity was restored by the addition of small thiols. GAPDH-sulfenyl thiosulfate was irreversibly inactivated by heat mediated denaturing presumable due to the formation of intramolecular disulfide (Fig. 5B, reaction 2). Moreover, reversible inactivation of GAPDH was demonstrated upon treatment with stoichiometric amount of o-iodosobenzoate. This inactivation was attributed to the formation of sulfenic acid as the enzyme was reactivated by sodium arsenite treatment (Fig. 5B, reaction 3) [68,69]. pH dependent inactivation of GAPDH with stoichiometric H2O2 provided another proof in favor of sulfenic acid formation [70]. Again, arsenite-mediated reversibility and lack of disulfide formation were the key features. Moreover, the increase in enzyme inactivation rate with increasing pH implicated the thiolate anion, and not thiol, as the substrate in the oxidation process. Excess H2O2 and longer incubation times led to the formation of further oxidized products characterized by their irreversibility to typical reducing agents.
Another study on iodine-mediated oxidation of creatine kinase enzyme points toward the formation of unstable sulfenyl iodide, which readily underwent hydrolysis to form sulfenic acid. In the presence of excess iodine, the consumption of 12 equivalents of iodine/mole of enzyme indicated toward the presence of six cysteine thiols in the protein. Addition of 4 equivalents of iodine/mole resulted in the oxidation of only two cysteine thiols, but complete loss of activity. This was indicative of the presence of two cysteine thiols at the active site. Radiolabeling with 125I2 resulted in the incorporation of minimal radioactivity in the enzyme. Thus, the oxidation may proceed through the formation of unstable sulfenyl halide which may hydrolyze to sulfenic acid. Finally, the inactive enzyme prepared by adding slightly less than 4 equivalents of iodine/mol was reactivated by the incubation with mercaptoethanol or dithiothreitol (DTT). This reactivation presumably occurs through the intermediate formation of a mixed disulfide with subsequent regeneration of the original cysteine thiol [71]. The examples discussed above, and many more, pointed toward the formation of sulfenic acid under oxidizing conditions, but the biological significance of this modification was unknown and speculative.
The discovery that the specific conversion of Cys-149 in GAPDH to corresponding sulfenic acid converts the enzyme from dehydrogenase to an acyl phosphatase was probably the first to hint at a biological role for protein sulfenic acid [68,72,73]. Another study demonstrated oxidation of the lone free thiol in albumin upon incubation with xanthine oxidase [74]. This modification, mediated by enzyme-generated oxidants, inferred that sulfenic acids may be generated in biological systems. Indeed, now with the development of several modern detection techniques like NMR, X-ray crystallography, mass spectrometry and chemical probes, many significant roles of protein sulfenic acids have been identified. To date, sulfenic acids have been implicated in the reversible inhibition or activation of enzymes, act as catalytically critical redox centers or transient intermediates during peroxide reduction, function in disulfide formation, as well as transcription activation/inactivation to direct the appropriate antioxidant response.
3.2. Protein sulfenic acid formation
As mentioned above, cysteine is peerless among all naturally occurring amino acids due to the high nucleophilic character of the side-chain thiol. Almost all physiological oxidants can react with thiols (or more likely thiolates), albeit they may differ substantially in their reaction rate and products. One-electron oxidants like superoxide ( ) may react with protein thiolates and give rise to thiyl radical (R – S•), which under aerobic conditions may react with another thiolate to eventually give a disulfide accompanied with superoxide formation [75,76]. Although, one must note that the relevance of thiols as biological targets of superoxide is debatable. Oxidation rates ranging from 10 to 103 M−1 s−1 have been reported but these values are considered imprecise due to the assumptions made in interpreting the complex kinetic mechanism [77]. On the other hand, similar reaction with two-electron oxidants gives sulfenic acid as the initial product [8,78,79]. H2O2 is most commonly implicated in conversion of protein thiolates to corresponding sulfenic acids. However, several other oxidants like alkyl hydroperoxides, peroxynitrite, hypochlorous acid and chloramines also generate sulfenic acid. Moreover, other pathways like alkaline hydrolysis of disulfides, hydrolysis of sulfenyl halides, sulfenyl esters, and nitrosothiols can generate sulfenic acids. Depending upon the protein microenvironment and oxidative stress, sulfenic acid may accumulate or undergo secondary reactions to form other, more stable products.
3.2.1. Hydrogen peroxide and organic hydroperoxide mediated sulfenic acid formation
Hydrogen peroxide (H2O2) is the simplest peroxide with oxidative properties (oxidation potential 1.8 V). It is mildly acidic with a pH of 6.2 in pure form. Under physiological conditions, it exists in predominantly unionized form (pKa = 11.75). H2O2 was once considered a toxic byproduct of aerobic life (for example, mitochondrial respiration) that must be swiftly eradicated to maintain cellular redox balance. However, studies conducted over past two decades showed that it may also be deliberately generated as a secondary product by the spontaneous dismutation of superoxide (~105 M−1 s−1), a reaction which is catalyzed by superoxide dismutase (SOD) (~109 M−1 s−1) [80,81]. It is also generated by peroxisomal enzymes and endoplasmic reticulum [82]. The levels of this diffusible oxidant are tightly regulated by peroxide-reducing antioxidant systems. The large majority of the biological activity of H2O2 is attributed to its ability to oxidize a cysteine thiolate to sulfenic acid (Fig. 6a). Although, H2O2 is a strong oxidant, its reaction with nucleophiles is often sluggish due to high activation barrier (46.6 kcal/mol) [76,83]. As shown in Table 1, at physiological pH, the second order rate constant for the oxidation of small molecule thiols decreases with increasing pKa but their thiolates have almost same rate constants with H2O2 (~20 M−1 s−1) [77]. This implies that the mechanism involves the nucleophilic attack of the cysteine thiolate on an oxygen atom of the peroxide molecule leading to the oxidation of thiol to corresponding sulfenic acid. But −OH is a poor leaving group which makes the SN2 mechanism for such reactions unfavorable. It has been shown that acid-catalyzed reaction of H2O2 with thiolates is much more facile due to the concomitant conversion of poor leaving group (−OH) to a good leaving group (H2O) [84]. Since the pKas of protein-thiols are variable and highly dependent on the native microenvironment, this would imply that thiol pKa would be the sole factor in determining their respective tendencies toward oxidation. But just low pKa is not a hallmark of superior tendency toward oxidation, as discussed later. Several studies have been conducted to evaluate the impact of H2O2–cysteine–SH interactions on cellular function and have resulted in the better understanding of the roles of protein sulfenic acid intermediates. For example, protein sulfenic acids have been established as key intermediates in cellular detoxification of H2O2 by active site thiol-containing peroxidase enzymes in which the reduction of peroxide is countered by the oxidation of the cysteine residue [85–87]. Also, H2O2-mediated sulfenic acid formation at catalytically active cysteine has been shown to inhibit PTPs and activate kinases, leading to a general enhancement of phosphorylation thus influencing signal transduction cascades [56,57].
Fig. 6.

Direct two-electron oxidation and hydrolytic mechanisms of sulfenic acid formation.
Table 1.
Second order rate constants and conditions for H2O2 mediated oxidation of some biological small molecule thiols.
| Thiol | pKa | Rate constant (M−1 s−1) | Conditions | References |
|---|---|---|---|---|
| Penicillamine | 7.9 | 4.5 | 37 °C, pH 7.4 | [77] |
| Cysteine | 8.3 | 2.9 | 37 °C, pH 7.4 | [77] |
| 1.0 | 20 °C, pH 7.4 | [78] | ||
| Cysteamine | 8.3 | 2.9 | 37 °C, pH 7.4 | [77] |
| 1.1 | 20 °C, pH 7.4 | [78] | ||
| Glutathione | 8.8 | 0.9 | 37 °C, pH 7.4 | [77] |
| Dithiothreitol | 9.1 | 0.4 | 37 °C, pH 7.4 | [77] |
| 0.3 | 25 °C, pH 7.4 | [189] | ||
| N-Acetylcysteine | 9.5 | 0.2 | 37 °C, pH 7.4 | [77] |
| 0.9 | 25 °C, pH 7.4 | [190] |
In addition to H2O2, organic hydroperoxides (ROOH) have also been implicated in the oxidation of protein-SH to sulfenic acids. The modus operandi of organic hydroperoxides is a similar two-electron oxidation involving the attack of a thiolate on the oxygen of peroxide resulting in the oxidation of thiol to sulfenic acid and reduction of peroxide to corresponding alcohol (Fig. 6b). As with H2O2, early investigators observed that the oxidation products in many cases were not disulfides. In the 1960s, fatty acid hydroperoxides like linoleic acid hydroperoxide (LAHP) were found to inhibit active site thiol containing enzymes by oxidation, presumably to sulfenic acid [88]. Investigators have also observed that the oxidation of both proteins and small thiols with LAHP was faster than with H2O2 [89,90]. LAHP and many other organic hydroperoxides like ethyl hydroperoxide, benzoyl hydroperoxide, cumene hydroperoxide (CHP) and tert-butyl hydroperoxide (t-THP) have been recognized as Prx substrates and sulfenic acid generators. Wood and coworkers have shown sulfenic acid formation in response to both CHP and t-BHP treatment in E. coli [91]. Additionally, it was demonstrated that the OhrR of bacterial factor B. subtilis was reversibly inactivated by organic hydroperoxide, thereby repressing the transcription of the ohrA resistance gene [62].
3.2.2. Peroxynitrite mediated sulfenic acid formation
Peroxynitrite anion (ONOO−) is a potent oxidant formed by the radical reaction between superoxide anion ( ) and nitric oxide (•NO) [92]. Its conjugate acid is peroxynitrous acid (ONOOH) (pKa = 6.8 at 37 °C). Because NO is relatively long lived compared to , the sites of ONOOH formation are associated with the sites of formation in vivo. Peroxynitrite may oxidize the thiols directly or it can decompose to hydroxyl and nitrogen dioxide radicals which are also thiol reactive and act via one electron oxidation mechanism [93]. It has been shown that for the physiological concentrations of GSH, the direct reaction with ONOO− is fast enough to suppress the decomposition. Although, peroxynitrite is a stronger oxidant than H2O2, it also reacts with carbon dioxide to produce nitrosoperoxycarbonate ( ). The in vivo concentration of carbon dioxide is 1 mM and nucleophilic reaction with peroxynitrite is very facile making it an important ONOO− reaction pathway. Nitrosoperoxycarbonates readily homolyze to produce carbonate radical and nitrogen dioxide making GSH oxidation more radical mediated [94,95]. The oxidation is pH sensitive and pH dependence studies have proposed two models for the reactivity—a thiolate reacting with ONOOH or a thiol reacting with ONOO− (Fig. 6c and d) [93]. Second order rate constants for the peroxynitrite mediated oxidation of free cysteine and glutathione were reported to be 4500 M−1 s−1 and 1360 M−1 s−1 at 37 °C and pH 7.4 (compared to 2.9 M−1 s−1 and 0.87 M−1 s−1 with H2O2) respectively (Table 2). Nathan et al. detected the formation of protein-NBD adducts of Prx AhpC upon ONOO− treatment indicative of protein-SOH formation. Thus, they have provided indirect evidence that peroxiredoxins (Prx) rapidly reduce ONOO− to nitrite and act as detoxifying agents [96]. Later efforts focused on ONOO− mediated oxidation of parasite, yeast and human Prxs further provided evidence for the direct two-electron oxidation of their catalytic site Cys to sulfenic acid [97–99].
Table 2.
Second order rate constants and conditions for ONOO− mediated oxidation of some biological small molecule thiols.a
| Thiol | pKa | Rate constant (M−1 s−1) | Conditions |
|---|---|---|---|
| Penicillamine | 7.9 | 6420 | 37 °C, pH 7.4 |
| Cysteine | 8.3 | 4500 | 37 °C, pH 7.4 |
| Glutathione | 8.8 | 1360 | 37 °C, pH 7.4 |
| Homocysteine | 9.1 | 700 | 37 °C, pH 7.4 |
| N-Acetylcysteine | 9.5 | 415 | 37 °C, pH 7.4 |
Values taken from reference [99].
3.2.3. Oxidative action of hypohalous acids and chloramines on protein thiols
Another pathway to protein-SOH proceeds through the hypohalous (HOX) acid (Fig. 6h and e) and N-chloramine (R-NHCl) (Fig. 6i and e). Hypohalous acids (HOX; X = Cl, Br, I, SCN) are oxidants produced by neutrophils through H2O2-mediated oxidation of halides. This reaction is catalyzed by myeloperoxidase, which is a heme-dependent peroxidase [100–103]. The relative oxidant strength of HOX is HOCl > HOBr ≫ HOI ~ HOSCN. The pKa values are 4.85 for HOSCN, 7.59 for HOCl, 8.59 for HOBr and 10.4 for HOI [104]. These oxidants react preferentially with thiol compounds and methionine. Chloramines, which are generally weaker oxidants than HOCl, are generated by the action of HOCl upon biological amines. Thiols are among the most reactive substrates to HOCl and amines show almost 100-fold less reactivity, but the abundance of amines compared to thiols makes the reaction of HOCl with amines biologically relevant. The reaction of HOCl with thiols is extremely rapid and the rate constant for GSH is estimated to be >107 M−1 s−1 [104]. General reactivity comparisons with other small molecule thiols result in very similar rate constants indicating a lack of pKa dependence. On the other hand, the reaction rates of chloramines with various thiols varied by a factor of 20, showing inverse pKa dependence [105]. These findings highlight that the ionization of thiols is a major determinant of chloramine reactivity. The generation of sulfenic acid from thiols is indirect following the hydrolysis of sulfenyl chloride products formed by the reaction of HOCl or R-NHCl on thiols [105,106]. Sulfenic acid formation was shown in murine S100A8 (A8), a cytoplasmic protein in neutrophils. Upon incubation with HOCl, A8 was converted to four irreversible oxidation products corresponding to sulfinamide formation between reactive cysteine and different lysines (K6, K34/35, K87) and a sulfonic acid. They were able to trap the initially formed sulfenic acid by oxidizing A8 in the presence of sulfenic acid specific reagent dimedone [107]. Although, this is debatable because dimedone can ostensibly react with sulfenyl chlorides as well.
Although chloramines are less reactive than hypochlorous acid, it was shown that they have higher selectivity for thiols. Winterbourn and coworkers demonstrated that taurine chloramine preferentially oxidize the Cys-SH of creatinine kinase (CK) and GAPDH with concomitant loss of enzymatic activity. Although, sulfenic acid formation was not detected when GAPDH or CK was treated with taurine chloramine but intramolecular disulfide and some unidentified oxidant products were reported respectively. But, when reduced human serum albumin (HSA) was exposed to taurine chloramine and treated with NBD-Cl, the spectral shift to 356 nm did indicate the sulfenic acid formation [108].
3.2.4. Nitric oxide mediated sulfenic acid generation
Nitric oxide (•NO) is a highly diffusible, stable free radical gaseous molecule which is generated from L-arginine, oxygen and NADPH by a reaction catalyzed by nitric oxide synthase enzymes. The three generally accepted mechanisms of formation of nitrosothiols (RSNO) are: (a) by the action of nitrosating agent like dinitrogen trioxide (N2O3) which is generated from the reaction of •NO and oxygen; (b) the autooxidation of •NO generating nitrogen dioxide ( ) which, in turn, forms a thiyl radical from thiol and radical–radical combination of thiyl radical and •NO and (c) by the direct reaction of thiol with •NO resulting in a thionitroxide radical intermediate (RSNOH) which upon reduction of oxygen results in the formation of nitrosothiol and superoxide [109–111]. RSNO formation is one form of redox signaling and has been linked to various signaling pathway discussions of which is out of scope of this section. However, another reaction pathway, which is relevant here, is the conversion of nitrosothiols to sulfenic acids (Fig. 6f). First evidence was provided by the anaerobic oxidation of thiol group of human serum albumin (HSA) with •NO which resulted in the formation of sulfenic acid and nitrous oxide (N2O) [112]. Another interesting evidence for the sulfenic acid formation from nitrosothiols was demonstrated by the inhibition of a cysteine protease cathepsin K by an •NO donor. Upon cotreatment of cathepsin K with NOR-1 (a non-thiol •NO donor) and dimedone, a sulfenic acid specific reagent, resulted in a mass increase of 138 Da specific to the formation of dimedone adduct of sulfenic acid [113]. Although, a rational mechanism has not been proposed for such •NO mediated sulfenic acid formation, but hydrolysis of initially formed RSNO has been suggested.
3.2.5. Sulfenic acid generation by disulfide hydrolysis and other mechanisms
One of the criteria for the formation of a stable sulfenic acid is the absence of vicinal thiol. This is because there is sufficient evidence that cysteine sulfenic acid readily reacts with thiolate to form disulfide formation [45]. Formation of sulfenic acid by alkaline hydrolysis of disulfide is interesting because such a reaction would be physiologically inefficient due to other competing reactions. Hydrolysis of disulfides under alkaline conditions is known but the products are highly substrate dependent and do not result in the formation of stable sulfenic acids [114]. However, several such enzyme-catalyzed reactions have been reported in biological systems to result in the formation of sulfenic acid (Fig. 6g). The disulfide bond splitting and exchange have been previously observed in bovine serum albumin dimers [115]. The scrambling of sensitive disulfide bonds of plasmin which is a serine protease in alkaline solution is another such example [116]. Plasmin acts as an enzyme as well as substrate and undergoes cleavage at pH 11 leading to the formation of micro plasmin. Although, no proof for sulfenic acid formation was provided, the authors speculated that Cys511 of Cys511–Cys535 disulfide first reacted with hydroxide ion to expel the Cys535 thiolate, which in turn, reacted with another disulfide to give disulfide scrambling. Another study that proposed sulfenic acid formation upon disulfide hydrolysis, was conducted to elaborate on plasmin reduction activity of phosphoglycerate kinase (PGK). In this study, the researchers showed that the reduction of plasmin disulfide by PGK is a thiol independent process. Mutation of all seven Cys of PGK to Ala did not affect the plasmin reductase activity [117,118]. They proposed that PGK facilitates the cleavage of Cys512–Cys536 disulfide bond of plasmin by hydroxyl anion (−OH) and results in the formation of sulfenic acid at Cys512 along with a Cys536 thiolate thus presenting an opportunity for disulfide rearrangement.
Protein sulfenic acid generation by several other biological mechanisms has been proposed. Initial generation of thiyl radical offers many direct pathways to sulfenic acid. For example, a thiyl radical may combine with a hydroxyl radical to generate sulfenic acid directly [119,120]. The reaction of small molecule thiosulfinate allicin with papain resulted in the formation of mixed disulfide. The proposed mechanism for mixed disulfide formation consists of papain-SH condensing with allicin and expelling a small molecule sulfenic acid [121]. A similar mixed disulfide was obtained when glutathione disulfide-S-oxide was reacted with rat brain proteins generating a glutathione sulfenic acid in the process [122].
3.3. Detection of protein sulfenic acids
The discovery of oxidant-mediated cell signaling pathways and pathological oxidative stress has led to significant interest in methods to detect protein sulfenic acid modifications. At the outset, we note that the propensity of protein-SOH to undergo further reactions, absence of characteristic UV–visible spectral properties, and small size of this post-translational modification present a variety of inherent challenges to detection. Although many X-ray and NMR structures of protein sulfenic acids have been reported, a substantial amount of data collected in support of their existence has been based on their reactivity with chemically selective probes and the detection of higher, more stable oxidation states that imply RSOH intermediacy. This section presents a comprehensive survey of direct and indirect methods for detection protein sulfenic acids.
3.3.1. Detection by spectroscopic methods
X-ray crystallography, NMR and mass spectrometry (MS) approaches have offered significant insight into the structure and local environment of protein-SOH. For example, X-ray crystallography was used to characterize the Cys42 redox center in the flavoprotein NADH peroxidase in the sulfonic acid form, but the oxidation pathway was not clearly identified [123]. Subsequent innovations, which involved reduced exposure of the crystals to ambient oxygen and data collection at low temperature, resulted in structure refined to 2.8 Ǻ depicting the Cys42-sulfenic acid redox center, thus supporting the proposed catalytic role of this residue [124]. The existence of Cys42-SOH was supported by 13C NMR indicating a chemical shift of 13Cβ (carbon bearing thiol or sulfenic acid) to be 30.8 ppm (for Cys42-SH) and 41.3 ppm (for Cys42-SOH). These values were comparable to the predicted values [125]. In another example, Nakamura and coworkers used X-ray crystallography to decipher the oxidation mechanism of active site Cys50 and the stability of corresponding sulfenic acid form in an archaeal peroxiredoxin (Prx) from Aeropyrum pernix K1 (ApTPx) [126]. Since the stability of Cys50-sulfenic acid is crucial to the overall function of Prx, they attributed the stability to the formation of unique N – S (between His42 and Cys50-sulfenic acid) covalent bond. In a more recent example, the reduced, sulfenic acid, and mixed disulfide form of global transcriptional regulator SarZ in Staphylococcus aureus was resolved by crystallography [127]. Additionally, in many instances, protein-SOH formation has been detected by MS as an m/z shift equivalent to the addition of one oxygen atom (+16 Da). Other examples of MS detection are the transcription factor OhrR [62], methionine sulfoxide reductase (MsrA) from E. coli [128] and aldose reductase in the ischemic heart [129]. Although important, observations of protein sulfenic acids by spectroscopic methods are limited to in vitro analysis of purified or recombinant proteins.
3.3.2. Detection methods based on chemical reactivity
Methods based on the reaction of protein sulfenic acids with chemical probes are based on both the electrophilic and weak nucleophilic character of the sulfur atom. Fig. 7 summarizes reactions of sulfenic acid with nucleophiles, electrophiles and reducing agents that have inspired the majority of detection methods. Chemoselective reaction of dimedone with sulfenic acid is most often used for detection (Fig. 7, reaction 1), but the lack of an easily distinguishable product (e.g., by SDS-PAGE gel) initially required the use of radiolabeling or MS to monitor adduct formation. Recent development of fluorescent- and biotin-dimedone conjugates has enabled facile detection by in-gel fluorescence and Western blot techniques. Other nucleophiles have also been used as probes for sulfenic acid. For example, reaction of sulfenic acid and benzylamine to give the corresponding sulfenamide was used to detect GAPDH-SOH (Fig. 7, reaction 2). In this study, 14C benzylamine was reacted with oxidized GAPDH and the amount of incorporated radioactivity was equivalent to the degree of acyl phosphatase inactivation [19,20]. Nonetheless, the unstable nature of sulfenamides in the presence of biological amines, thiols and strong reducing agents was a major drawback and proved a major deterrent to the general use of this reaction. The facile reaction of sulfenic acid with thiols to produce disulfides has also been exploited to detect RSOH formation (Fig. 7, reaction 3). Aromatic thiol compound, TNB (5-mercapto-2-nitrobenzoic acid) ionizes to TNB2− in water at neutral and alkaline pH, and has a characteristic absorbance maximum at 412 nm. Using this, Poole and coworkers devised a spectrophotometric assay for protein sulfenic acid detection and successfully demonstrated the formation of sulfenic acid in the non-flavin redox center of the streptococcal NADPH peroxidase [130]. A few years later, formation of protein-SOH during peroxide reduction by bacterial Prx enzyme, AhpC was shown using a similar strategy [86]. A limitation of this method is that it requires the treatment of protein under anaerobic conditions, as TNB2− readily converts to DTNB (Ellman’s reagent) under aerobic conditions, leading to false positive identifications from free thiol labeling. A different strategy based on arsenite, and other mild reducing agents, mediated reduction of sulfenic acid and was incorporated into a variation of biotin switch technique (BST) (Fig. 7, reactions 4 and 5). Although known for its electrophilic nature, the sulfur atom of sulfenic acid is also a weak nucleophile, which inspired the detection by the electrophilic reagent 7-chloro-4-nitrobenz-2-oxa-1,3-diazole (NBD-Cl) (Fig. 7, reaction 6) [27]. The product thus formed is a sulfoxide with a unique absorbance maximum.
Fig. 7.

Biochemically relevant reactions of sulfenic acid upon which detection techniques are based.
3.3.2.1. Indirect detection methods
Indirect SOH-detection methods require ‘protection’ of free thiols using alkylating reagents like iodoacetamide (IAM) or N-ethylmaleimide (NEM). They diverge in subsequent steps, where different reagents/conditions are applied to convert sulfenic acid to a thiol or sulfonic acid. For instance, the BST was originally developed to identify protein S-nitrosylation [131]. This method consists of three major steps: (1) blocking of free cysteine thiols by S-methylthiolation with methyl methanethiosulfonate (MMTS); (2) reduction of S-nitrosothiols with ascorbate; and (3) labeling nascent thiols with biotin-HPDP. In subsequent work, Eaton et al. replaced ascorbate with the RSOH-reducing agent, sodium arsenite [132,133]. Their protocol involved: (1) blocking of free thiols with maleimide (S-alkylating agent) to prevent reactions of RSOH with thiols as well as indiscriminant labeling; (2) arsenite-mediated reduction of protein-SOH; and (3) labeling of nascent thiols with biotin-maleimide (Fig. 8A). Biotin thus incorporated at the sites of sulfenic acid formation enabled affinity isolation of labeled proteins. Subsequent SDS-PAGE, in-gel digestion and MALDI-TOF MS analysis led to the identification of several proteins. However, applications of this technique are limited because: 1) protein samples undergo extensive processing under denaturing conditions, and 2) the selectivity of the arsenite reduction step has not been rigorously investigated. In a variation on this theme, the extent of protein sulfenic acid modification was estimated by monitoring the change in enzyme activity, before and after arsenite treatment (Fig. 8B). Using this strategy, evidence for protein-SOH modification has been obtained for GAPDH [134], lipid and proteins within human plasma [74], and tissue treated with exogenous H2O2 [132]. Another strategy relies on S-hyperoxidation to detect sulfenic acid modification of protein tyrosine phosphatases (PTPs). Protein thiols are first blocked in lysates by S-alkylation using iodoacetic acid (IAA), followed by immunoprecipitation and oxidation of any surviving sulfenic acid to sulfonic acid using pervanadate. The sulfonic acid form of the highly conserved PTP active site is then detected by an antibody followed by immunoblot and/or enrichment and LC–MS/MS analysis (Fig. 8C) [135,136]. Major drawbacks of this approach include: 1) false positive signal from incomplete thiol blocking, and 2) lengthy chemical manipulations in cell lysate.
Fig. 8.

Indirect detection of sulfenic acid. (A) Indirect chemical detection using arsenite reduction. (B) Enzyme activity assay using arsenite mediated reduction of sulfenic acid. (C) Indirect chemical detection using pervanadate mediated hyperoxidation and selective immunoprecipitation.
3.3.2.2. Electrophilic probes
The reaction of NBD-Cl with sulfenic acid is facile, but it is not selective per se since it reacts with biological thiols and, under some circumstances, with amines and phenols [137]. The reaction product of NBD-Cl and sulfenic acid is sulfoxide, R-S(O)-NBD, with an absorbance maximum at 347 nm, whereas the thioether product formed by reaction with a thiol shows an absorption maxima at 420 nm and may have fluorescent properties (Fig. 9) [137]. Therefore, a shift in the absorbance upon reaction of NBD-Cl with oxidant-treated proteins has been used to confirm the presence sulfenic acid. Although sulfenic acid reaction products with NBD-Cl often show a distinctive absorption maximum, many exceptions exist and the only way that this reagent can be definitively used to identify sulfenic acid is through MS analysis [27]. Using this approach, Poole and coworkers observed the sulfenic acid intermediate that forms during the catalytic cycle of AhpC and NADH peroxidase [27]. Another study conducted by Denu et al. used NBD-Cl to demonstrate that the active site PTP cysteine underwent oxidation to sulfenic acid by H2O2 [57]. Using similar approach, sulfenic acid modification of OhrR [62], human serum albumin [138] and recombinant human alpha 1-antitrypsin [139] has also been identified.
Fig. 9.
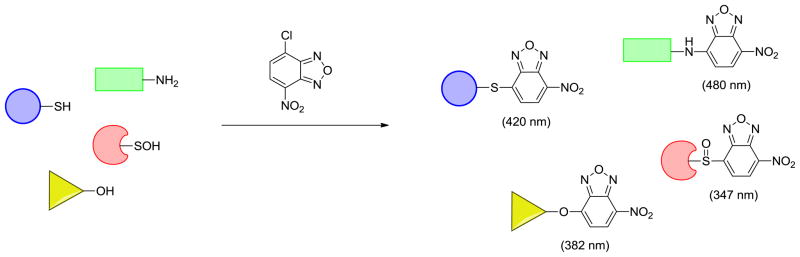
Reaction of NBD-Cl with protein nucleophiles. NBD-Cl may react with various biological nucleophiles with each conjugation product having different absorbance maximum.
3.3.2.3. Genetically-encoded probes
Wood and coworkers have reported a genetically-encoded probe to trap sulfenic acid-modified proteins in E. coli exposed to H2O2. Their strategy was to exploit the propensity of a cysteine residue (Cys598) in the Saccharomyces cerevisiae transcription factor, Yap1, to condense with the sulfenic acid intermediate formed at Cys36 of the redox sensing protein, Orp1 (also known as Gpx3). The probe used in their study consisted of 85 amino acids of the C-terminal cysteine rich domain of Yap1 along with a His-tag for affinity purification. Two of the three native cysteine residues in that domain were mutated (C620A and C629T), leaving a single cysteine C598 that could form a mixed disulfide at the sites of RSOH formation. E. coli cultures expressing the His-tagged Yap1 domain were treated with H2O2 and proteins that formed mixed disulfide with the probe were copurified on a Ni+ affinity column. Elution with DTT, separation by SDS-PAGE, tryptic digestion, and analysis by LC–MS/MS resulted in the identification of 32 proteins that may form sulfenic acid (Fig. 10) [91]. In a follow up study, 42 proteins that may form Cys-SOH were identified in S. cerevisiae exposed to exogenous H2O2 [140]. Although some interesting features are noted in this system, the Yap1-cCRD variant must be expressed in host cells and, since it is protein-based, it may exhibit substrate bias when compared to chemical-based probes.
Fig. 10.

Detection of in-cell protein sulfenylation with genetically encoded Yap1-cCRD probe. Cells expressing Yap1-cCRD are exposed to peroxide and protein conjugates are extracted with trichloroacetic acid (TCEP). Free thiols are capped by alkylation with iodoacetamide (IAA). Yap1-cCRD-captured proteins are affinity enriched and eluted with reducing agents (DTT or TCEP). After sample enrichment and protease digestion, the resulting peptides are analyzed and identified by LC–MS/MS.
3.3.2.4. Direct detection by selective small molecule nucleophilic probes
The nucleophilic reaction of dicoordinated sulfur compounds such as sulfenyl halides was first identified by Foss in 1947 (Fig. 11A) [141]. Further observations that nucleophiles with activated methylenes (like dimedone) form corresponding thioether bonds with sulfenyl halides, and the assumption that dicoordinated sulfenic acids in protein-SOH might also undergo similar reactivity, formed the foundation of a seminal article published by William S. Allison in 1974 [19]. This work utilized the mild carbon nucleophilicity of dimedone (a 1,3-dione) to detect GAPDH-SOH. The precise mechanism for this reaction has not been proven, but it is widely believed to proceed via direct substitution (Fig. 11B). To date, several analogs based upon the 1,3-dione core have been incorporated into various probes for detecting protein-SOH in vitro and in cells. Based on their chemical structures and detection mechanisms, these reagents and related strategies are classified and discussed in this section.
Fig. 11.

Detection of sulfenic acid by chemical reaction. (A) Nucleophilic reaction of dicoordinated sulfur. (B) Reaction of dimedone with sulfenic acid.
3.3.2.4.1. Detection by fluorescent and affinity based probes
In 2005, King and coworkers reported the first functionalized derivatives of dimedone analog, 1,3-cyclohexanedione [142]. These reagents are comprised of: 1) 1,3-cyclohexanedione “warhead”, 2) linker, and 3) isatoic acid (DCP-MAB) or 7-methoxycoumarin (DCP-MCC) fluorophores (Fig. 12A). The syntheses were straightforward and accomplished over 4 linear steps, but low yielding in both cases. These reagents were tested using mutant form of AhpC, a Prx from Salmonella typhimurium, in which the sulfenic acid intermediate is stabilized owing to the absence of the resolving thiol. These studies showed covalent labeling of AhpC-SOH with DCM-MAB and DCP-MCC and both probes were unreactive toward protein thiols, disulfides, nitrosothiols, sulfinic and sulfonic acids.
Fig. 12.

1,3-Dione based probes directly conjugated to biotin or fluorescent tags.
Although this was an exciting advance forward in selective sulfenic acid detection, two limitations, poor detectability in polyacrylamide gels and the absence of affinity tag for isolation, led the same group to prepare a panel of seven new sulfenic acid probes (Fig. 12B) [143]. These probes can be divided into three categories: 1) 1,3-cyclohexanedione linked to biotin (DCP-bio series); 2) 1,3-cyclohexanedione linked to fluorescein (DCP-FL series), and 3) 1,3-cyclohexanedione linked to rhodamine (DCP-Rho series). Testing these reagents with C165S AhpC-SOH, verified reactivity and specificity similar to dimedone, DCP-MAB and DCP-MCC. A cysteine protease, papain was also used to validate the seven probes. Although, fluorescein- and rhodamine-linked probes are bright chromophores, their emissions were partially quenched after covalent attachment with protein-SOH. The emission intensities of DCP-FL1 conjugated to C165S AhpC and papain, were ~50% and ~75% quenched respectively. DCP-Rho1 and -Rho2 were similarly quenched and excitation/emission maxima shifted to longer wavelengths. Despite issues with emission quenching, both intact and tryptic MS analyses of probe-protein conjugates indicated selective probe incorporation at the expected cysteine. MS-based kinetic reactivity studies with DCP-Bio1 and C165S AhpC-SOH or papain-SOH were also performed. The initial rate studies indicated a modest reaction between C165S AhpC-SOH and dimedone. At 5 mM dimedone, the alkylated protein product was generated at an initial rate of 0.1 min−1. The rate constant corresponding to papain-SOH reaction with the DCP-Bio1 was found to increase with increasing probe concentration to a maximum value of 3 min−1 at 5 mM probe. The hyperbolic relationship of probe concentration and first order rate constant implied saturable or aggregation-based binding to the protein target.
Rates of DCP-Bio1 incorporation in recombinant protein-SOH were also characterized in a subsequent publication [144]. In this example, the protein targets were methionine sulfoxide reductase (fRMsr) of E. coli, C165S AhpC, and papain. For each rate study, 50 μM of reduced protein was incubated in a reaction mixture containing 1 mM DCP-Bio1, 25 mM potassium phosphate (pH 7.0), 100 μM DTPA (an aggressive chelating reagent) with one (AhpC) or two (papain) equivalents of H2O2 (fRMsr was oxidized with a 100-fold excess of methionine sulfoxide). Pseudo first order rate constants, as determined by MALDI-TOF MS, were 1.65 ± 0.22 min−1 for papain, 0.13 ± 0.014 min−1 for fRMsr, and 0.003 ± 0.0004 min−1 for C165S AhpC. The >500-fold differences among proteins in rate constants suggested that labeling might depend on protein-SOH accessibility and/or the presence of surrounding residues to facilitate covalent reaction. The effect of pH on labeling efficiency of DCP-Bio1 was evaluated using fRMsr. These data showed that labeling rates were essentially the same at pH 5.5, 7.0 and 8.0. The overall second order rate constant for the reaction of DCP-Bio1 with fRMsr was estimated at 2.0 ± 0.2 M−1 s−1.
The aforementioned fluorescent and biotin-linked probes have reported to tag sulfenic acid-modified (also termed sulfenylated) proteins in cell lysates and, depending on permeability, in cells. For example, DCP-Bio1 was used to detect sulfenylation of PTPs, SHP-1 and SHP-2 in cell lysates derived from activated CD8+ T cells [145]. Furdui et al. have also applied DCP-Bio1 to identify sulfenylation of the serine/threonine protein kinase, Akt2 at Cys124 [146]. Ushio-Fukai and coworkers have used DCP-Bio1 to investigate the role of VEGF-stimulated protein sulfenylation in human umbilical vein endothelial cells (HUVECs) [147]. Subsequently, Furdui group reported simplified synthesis of 4-alkylated 1,3-cyclopentanedione (1,3-CPD) probes [148]. A distinct advantage of this synthetic route over previously reported probes is the ease of synthesis and improved yields. Purified C165S AhpC treated with 5 mM probe, yielded first-order rate constants of 0.0024 min−1, 0.0018 min−1 and 0.0036 min−1 using dimedone, 1,3-CPD, and 4-(ethylthio)-1,3-cyclopentanedione (ethylthio-CPD), respectively. The two-fold rate enhancement afforded by the 4-thioalkyl substitution motivated its functionalization with biotin (BP1, Fig. 12C). Surprisingly, no rate studies were reported for the resulting biotinylated probe. However, analysis of pH dependent labeling by 4-(ethylthio)-1,3-cyclopentanedione showed that probe labeling increased at lower pH, in contrast to data obtained for DCP-Bio1. Lastly, although the 1,3-CPD-based probe was purported as kinetically superior to dimedone, the corresponding rate studies showed only marginal differences in rate constants. Moreover, rigorous kinetic analysis using model didpeptide and protein sulfenic acid shows that, by itself, 1,3-CPD reacts ≥20-fold slower than dimedone (ostensibly due to the stabilization of the carbanion in 1,3-CPD).
Along these lines, another biotinylated derivative of dimedone was prepared and used by Eaton and coworkers to probe oxidative stress in cardiac tissues (Fig. 12D) [149]. Biotin-dimedone incorporation was monitored by confocal fluorescence microscopy after staining permeabilized cells with ExtrAvidin-FITC. The resulting cells showed a broad diffuse staining pattern, which was elevated after H2O2 treatment. Affinity purification of tagged proteins via the biotin handle, SDS-PAGE separation, trypsin digestion, and LC–MS/MS analysis led to the identification of 24 proteins, largely consisting of abundant cytoskeletal elements. Our group reported a related biotinylated dimedone analog (DAz-1-biotin, Fig. 12E) in order to compare the effect of the biotin tag on the cell permeability of sulfenic acid probes [150].
3.3.2.4.2. Detection by azide- or alkyne-functionalized probes
Depending upon the application, one potential drawback for direct conjugation of any probe to biotin or a fluorophore is that the bulky chemical tags can reduce cell-permeability. Naturally, not all conjugated probes are entirely impermeant; however, comparative studies show time and again that tagged derivatives often suffer from diminished cell uptake and trafficking properties. Alternative mechanisms of uptake are possible, such as active transport, but may limit probe distribution to specific cellular compartments. A further consideration when functionalizing probes with large chemical tags is that increased steric bulk can lead to a significant bias in protein target labeling. The poor permeability of many biotin- and fluorophore-tagged probes typically necessitates labeling of proteins in lysates with associated limitations [151,152]. In this context, it is also important to bear in mind that labile and/or transient sulfenic acid modifications may be further oxidized or insufficiently trapped during the lysis procedure.
To address these core issues, we introduced the concept of azido-and alkyne-functionalized “chemical reporter” analogs of dimedone that enabled trapping and tagging of protein sulfenic acid modifications directly in living cells. The first probe to be developed in this series was DAz-1 (Fig. 13A). DAz-1 is a small azide-functionalized probe that, in later steps, can be coupled to biotin or a fluorophore through Staudinger ligation or Huisgen [3 + 2] cycloaddition also known as “click chemistry” (Fig. 13B–D) for detection, enrichment, and identification. Through a combination of biochemical, MS and Western blot analyses, we demonstrated that DAz-1 could selectively detect protein-SOH in purified proteins (papain and human serum albumin), complex protein mixtures, and in living mammalian cells. Notably, very distinct sulfenic acid labeling patterns were observed in live intact Jurkat cells as compared to lysates derived from the same line. At a minimum, these findings show that the profile of protein sulfenic acid modifications differs in cells, as compared cell lysates, thus highlighting the importance of in situ probing [150,153].
Fig. 13.

(A) Sulfenic acid specific probes with azide or alkyne handles. (B) Enrichment techniques. (C) Alkyne-biotin hapten. (D) Azide-biotin hapten. (E) General in vivo or in vitro protein-SOH labeling scheme.
Although direct detection of protein-SOH by DAz-1 in intact cells was unprecedented, early on we noted that the amide connection at the 5-position reduced its reactivity, as compared to dimedone. To address this issue, we developed a 4-substituted analog of 1,3-cyclohexanedione, 4-(3-azidopropyl)cyclohexane-1,3-dione or DAz-2 (Fig. 13A). Application of DAz-2 and the “reporter strategy” (Fig. 13E) to detect proteins that undergo sulfenic acid modifications in HeLa cells identified upwards of 200 candidates, including the majority of known sulfenic acid-modified proteins [154]. Cross-comparison of these data with those from disulfide and S-glutathionylation proteomes revealed modest overlap between these “redoxomes”, suggesting that a significant portion of sulfenic acid modifications may not be intermediates en route to S-thiolated forms and, instead, can be stabilized by the protein local environment. Alternatively, or in addition, it is also possible that: 1) lysate-based approaches employed in the S-thiolation proteomic studies resulted in fewer identifications and, therefore, lower overlap with the “sulfenome”, and/or 2) the modest rate constant for the reaction of many dimedone analogs with sulfenic acid (~103 M−1 min−1) may not be sufficient to trap especially transient modifications. Azido dimedone analogs have also been used to show that sulfenic acid modification of the thiol peroxidase, Gpx3 is essential for yeast to sense oxidative stress [155] and to identify a unique reducing system in the bacterial periplasm that protects single cysteine residues from oxidation [156].
Another study by our group examining protein sulfenylation during growth factor signaling led to the development of a new set of sulfenic acid probes, termed DYn-1 and DYn-2 (Fig. 13A) [59]. The underlying rationale for the development the new sulfenic acid probes was based on several studies demonstrating that alkynyl-chemical reporters, in combination with azido-detection tags, offer superior sensitivity relative to the reverse combination of azido-chemical reporter and alkynyl detection tags [157,158]. Another outstanding issue was the long and low yielding synthesis of DAz-2. Indeed, one synthetic route is characterized by three linear steps and ~10% overall yield, whereas the second route consisted of six linear steps with an overall yield of ~15% [154]. Optimization of the synthetic routes resulted in substantial improvement in the yields, as DYn-1 was synthesized in 2 steps (85% overall yield) and DYn-2 was synthesized in one step (96% yield). Furthermore, improvements in the synthesis of these compounds solved a longstanding issue in the functionalization of dimedone probes at the 4-position. In subsequent experiments, DYn-1 and DYn-2 were evaluated for their ability to detect protein sulfenic acid modifications in a recombinant protein, Gpx3. Analysis of these reactions by Western blot and MS revealed robust, H2O2-dependent protein-SOH labeling by DYn-2. In comparison to DYn-2, DYn-1 showed reduced sulfenic acid labeling (presumably due to steric hindrance during the click chemistry reaction) and, thus, this probe was not pursued further.
With DYn-2 in hand, we applied the probe to monitor changes in protein sulfenylation during cell signaling. As a brief introduction, binding of epidermal growth factor (EGF) to the extracellular domain of the EGF receptor (EGFR) leads to the assembly and activation of NADPH oxidase (Nox) that, in turn, generates endogenous H2O2 [60]. Employing our new reagents, we demonstrated that EGFR undergoes sulfenylation in response to EGF addition, even at low-nanomolar concentrations of growth factor. Interestingly, reciprocal immunoprecipitation analysis showed that EGFR and Nox2 associated in an EGF-dependent fashion. The intracellular kinase domain of EGFR contains six cysteine residues, one of which (Cys797) is located in the ATP-binding pocket and is conserved among nine additional receptor and non-receptor tyrosine kinases. Given its active site location and conservation, we hypothesized that Cys797 might be preferentially targeted by endogenous H2O2. Of particular relevance, this residue is selectively targeted by irreversible EGFR inhibitors, such as afatinib, extensively used in basic research and clinical trials for breast and non-small cell lung cancers [60]. Consistent with this proposal, pre-treatment of cells with inhibitors that irreversibly modify Cys797 prevented sulfenylation of EGFR. MS was subsequently used to verify Cys797 as the specific site of oxidation. Finally, in biochemical studies we showed that sulfenylation of Cys797 increased the EGFR kinase activity. To put these findings into context, a comparable degree of stimulation is observed for L858R and T790M oncogenic EGFR mutations [60]. Beyond the A431 cell line model, recent studies in our group also indicate that wild type and several activated mutant EGFR kinases undergo sulfenylation in both lung and breast tumors (T. Truong and K. Carroll, unpublished data). Hence, it appears that oxidation of specific residues in PTPs (catalytic Cys) and EGFR (Cys797) contributes to an increase in downstream signaling. Current work is directed toward understanding the molecular mechanism by which sulfenylation of EGFR Cys797 enhances kinase activity. The proximity of Cys797 to the ATP ligand as well as the C-helix and activation segment raises the possibility of transition state stabilization and/or destabilization of its auto inhibited conformation.
In this same study, three PTPs involved in the regulation of EGFR signaling, PTP1B, PTEN, and SHP2 were also demonstrated to undergo sulfenic acid modification in response to EGF stimulation. Interestingly, PTPs and EGFR displayed differential sensitivity to oxidation by EGF-induced endogenous H2O2 that correlated with the relative proximity of each enzyme to the oxidant source itself, Nox2. Lastly, we applied our chemical biology approach to monitor global changes in EGF-dependent protein sulfenylation [59]. Interestingly, growth factor addition led to widespread changes in protein sulfenylation within cells. EGF induced cysteine oxidation in a dose- and time-dependent manner and was accompanied by concomitant fluxes in intracellular H2O2. Additional experiments showed that treatment with cell-permeable PEGylated catalase attenuated EGF associated changes in protein sulfenylation, underscoring the importance of endogenous H2O2. Although sulfenylation of EGFR (and several PTPs) was the focus our recent study [59], many protein targets of EGF-induced endogenous H2O2 remain to be identified and will necessitate large-scale proteomic analysis. Nonetheless, our collective findings highlight sulfenylation as a signaling mechanism akin to phosphorylation, with regulatory implications for other receptor tyrosine kinases and irreversible inhibitors that target oxidant-sensitive cysteines in proteins.
3.3.2.4.3. Sulfenic acid probes with cleavable linkers
Affinity-based methods described above rely on biotin tags for enrichment and purification of tagged protein sulfenic acids. However, it is well known that fragmentation of the biotin handle can complicate MS2 analysis and subsequent database searching, especially for smaller peptides [157,159]. Thus, in many cases it is ideal to remove the biotin tag after enrichment of labeled proteins and/or peptides. Along these lines, a variety of cleavable linkers have been reported in order to facilitate proteomic analysis [160–163]. Taking cues from these approaches, our group designed a biotin linker consisting of three parts: 1) a biotin moiety, 2) a Boc-derivative cleavable by TFA, and 3) an alkyne group for bioorthogonal ligation with azide-functionalized sulfenic acid probes. The orthogonality of this linker with DAz-2 was first established using recombinant Gpx3. For enrichment, the biotinylated Gpx3 was captured on streptavidin-coated magnetic beads and on-resin digested with trypsin. Biotinylated peptides were then eluted from by the treatment with 8 M guanidine·HCl in water. In subsequent steps, the biotin moiety was efficiently cleaved by treatment with 5% TFA. The MS2 spectra of TFA-cleaved peptide showed complete mapping of the expected product [164]. This strategy was also successfully applied to quantify the relative amounts of oxidized Gpx3 in two different samples by reacting each sample with different concentrations of H2O2 and reacting them with either DAz-2 or heavy labeled d6-DAz-2 followed up by the biotinylation, enrichment and cleavage protocol [164]. In summary, this approach facilitates the mapping of specific site of cysteine oxidation. Moreover, this strategy may be used to monitor relative changes in protein oxidation (Fig. 14A).
Fig. 14.

General strategy for labeling and enrichment of sulfenic acid-modified protein and peptides. (A) Direct detection of sulfenylated proteins and peptides using modified TFA-cleavable biotin carbamate linker. (B) Direct detection of sulfenylated proteins with a β-ketoester probe amenable to cleavage by the reaction with hydroxylamine.
More recently, Furdui et al. reported the utilization of β-ketoesters as cleavable, sulfenic acid detection probes. In this example, the β-ketoester probe was functionalized with an alkyne handle using boric acid-mediated trans-esterification reaction (Fig. 14B). After testing of this probe with C165S AhpC, the alkyne-labeled protein was coupled to an azido-biotin hapten using Cu+-catalyzed click reaction. Removal of the biotin tag was achieved through NH2OH treatment, which gave a cyclic derivative amenable to MS analysis (Fig. 14B) [165]. The authors further demonstrated that the alkyne β-ketoester probe is cell permeable. However, the selectivity of the β-ketoester for protein sulfenic acids is not entirely certain as such linear 1,3-diketo derivatives have been reported to cross react with amines, such as lysine [166]. Moreover, our group has also documented spurious protein labeling with the β-ketoester functional group (P. Martinez and K. Carroll, unpublished data). Interestingly, despite the deactivating ester functional group adjacent to the carbon nucleophile, this study reports that the β-ketoester probe has enhanced reactivity, as compared to dimedone.
3.3.2.4.4. Quantification of protein sulfenic acid modifications
Probes designed for relative quantification of changes in protein sulfenic acid modification can be classified into two different methods. One strategy described above (Fig. 14A) employs DAz-2 and d6-DAz-2 probes to label protein-SOH in response to varying H2O2 concentrations. Labeled proteins are then conjugated to an acid-cleavable biotin tag via click reaction, followed by streptavidin-enrichment and LC–MS/MS analysis. This protocol allows for the fold-change in protein sulfenylation to be compared among samples [164]. Another method, shown in Fig. 15, uses a class of reagents termed isotope-coded d6-dimedone and iododimedone (ICDID) to label thiols and sulfenic acids, respectively [167]. The key feature of this strategy is that this pair of isotope mass probes generates chemically identical proteins, which differ only in the specific mass (6 Da). Overall, the ICDID method includes the following sequential steps: 1) Protein-SOHs are covalently modified by d6-dimedone; 2) excess reagent is removed and free thiols are labeled with iododimedone; 3) protein samples are proteolyzed, and 4) the resulting peptides are separated and analyzed by LC–MS. The extent of sulfenic acid modification at a particular cysteine residue can then be determined by dividing the intensity corresponding to heavy isotope labeled peak by a sum of the heavy- and light-isotope labeled peak intensities in mass spectrum. As proof-of-principle, this method was applied to quantify H2O2-dependent changes in sulfenic acid modification of Gpx3 and GAPDH. Importantly, this approach permits sulfenic acid modifications to be monitored at individual cysteines within a single protein and is amenable to peptide-based proteomic strategies. Another key feature of this approach is that it allows the occupancy of the site of modification to be determined. In some proteins, conversion of sulfenic acid to other cysteine oxoforms or incomplete chemical trapping of RSOH may preclude absolute quantification. However, either scenario still provides a lower limit for the extent of modification and our findings indicate that these phenomena are readily diagnosed in oxidant-dependence studies where the fraction of sulfenic acid modification levels off below one.
Fig. 15.
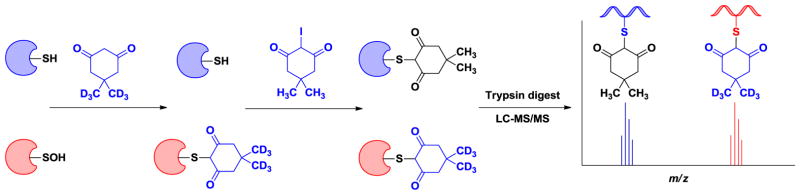
Quantification of protein sulfenylation using isotope-coded dimedone and iododimedone (ICDID).
3.3.2.4.5. Detection of protein sulfenic acid by immunochemical methods
In 2009, our group reported the first immunological method for detecting protein sulfenic acid modifications (Fig. 16A). Antibodies were elicited by a synthetic hapten mimicking dimedone-modified cysteine conjugated to KLH and are highly specific and sensitive for detecting protein-S-dimedone adducts by Western blot and immunofluorescence [168]. Application of this immunochemical approach to protein arrays and breast cancer cell lines revealed considerable differences in the level of protein sulfenic acid modifications among tumor subtypes. This method has also been used to demonstrate the colocalization of sulfenylated proteins with NOX2 during EGF signaling [59]. Subsequently, in 2011, Eaton and colleagues reported a similar antibody and used this reagent to study sulfenic acid modification of GAPDH in cardiac myocytes exposed to exogenous H2O2 [169].
Fig. 16.
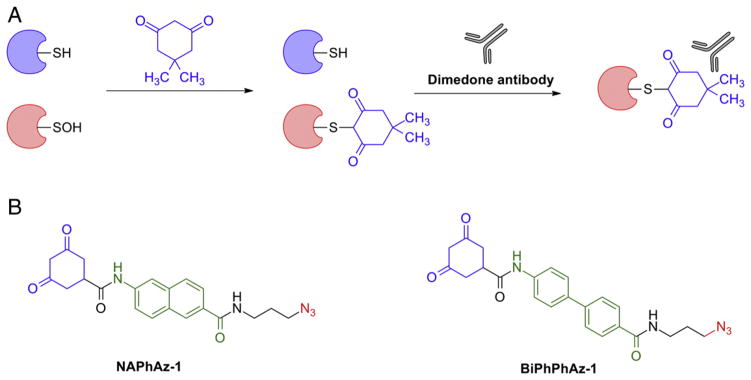
(A) Anti-dimedone antibody based detection of the cysteine-dimedone protein adducts. (B) Redox-based probes with affinity-based binding module for detecting reversible PTP oxidation.
3.3.2.4.6. Detection of sulfenic acids using protein-targeted probes
Analogous to irreversible electrophilic inhibitors that modify semi-conserved cysteines residues in protein tyrosine kinases currently in Phase II and III cancer clinical trials, we envision the development of nucleophile-functionalized small molecules that target a sulfenic acid-modified cysteine in a specific protein. Our design strategy is to conjugate the nucleophile “warhead” to a high affinity ligand that binds proximal to the target cysteine sulfenic acid. As proof of principle, we have developed small molecules that target PTPs, termed redox-based probes (RBPs, Fig. 16B), comprised of three parts: 1) a cyclohexanedione nucleophile, 2) a chemical scaffold that binds to the conserved PTP active site, and 3) an azide (or alkyne) chemical reporter to facilitate downstream detection and isolation of labeled PTPs (Fig. 16B) [170]. The RBPs exhibited enhanced binding and sensitivity for detecting sulfenic acid modification of the catalytic cysteine in the YopH and PTP1B, compared to the parent compound, DAz-1, which lacks the additional binding element. The RBP approach should facilitate cellular investigations of PTP redox regulation and other proteins that are amenable to such a targeted approach.
3.4. Cellular lifetime of sulfenic acid
3.4.1. Factors affecting the protein-thiol susceptibility to oxidation
Under oxidative stress, signaling or basal conditions, protein thiols may oxidize to protein sulfenic acid. Moreover, in biological systems, many types of ROS/RNS are generated that may oxidize a thiol to sulfenic acid. At this stage, further discussion of protein thiol oxidation must acknowledge that the cell milieu is not homogeneous and oxidant generation is often highly compartmentalized. Because of such compartmentalization, one needs to take into account the diffusion distances of various oxidants as well as the oxidative strength of different oxidants. Diffusion distance is defined as the distance for a 10-fold decrease in oxidant concentration [171]. A comparison of diffusion distances of various oxidants along with their oxidation rate constants with GSH is summarized in Table 3 [76,83].
Table 3.
Comparison of the rate constants and diffusion distances of common biological ROS.a
| GSH + oxidant | Diffusion distance (μm) | Rate constant (M−1 s−1) |
|---|---|---|
| GSH + H2O2 | 1600 | 0.9 |
| GSH + ONOO− | 60 | 700 |
| GSH + HOCl | 0.3 | 3 × 107 |
| GSH + NO2 | 0.4 | 3 × 107 |
| GSH + •OH | 0.02 | 1010 |
As detailed in the previous section, sulfenic acid formation shows a dependence on the pKa of small molecule thiol. This is highlighted by the fact that H2O2 mediated oxidation of Cys, GSH and N-acetylcysteine (NAC) thiolates has approximately same rate constants (20 M−1 s−1), whereas at pH 7.4 each compound is distinguished by a different rate constant [77]. However since low-molecular-weight thiols typically have pKas around 8–10, only a fraction of the thiol is available as the thiolate at physiological pH and, thus, only part of the reactivity is detectable. Hence, pH-independent rate constants are far better descriptors of inherent trends in reactivity. Using this as a principle, in case of small-molecule thiols, it has been demonstrated that higher pKa often implies higher reactivity [172]. In other words, a thiolate that is more basic will act as a stronger nucleophile. Based on this information, it is important to consider pKa in association with nucleophilicity of the thiol/thiolate in order to evaluate the tendency toward oxidation. The nucleophilicity depends upon several factors like basicity (pKa), polarizability, and presence of non-bonded electrons on adjacent atom. With respect to protein thiolates, such molecular and structural factors are discounted since in all cases the nucleophile is cysteine. The factors which affect the nucleophilicity of protein thiolates are environmental factors such as solvation, steric hindrance, hydrogen bonding, and the formation of cyclic transition states [172]. This means that same factors which affect the pKa of cysteine thiol in proteins also affect the nucleophilicity of the thiolate. The intrinsic pKa of free cysteine thiol in an aqueous solution is 8.3 [77]. Due to secondary and tertiary structures, cysteine pKas in proteins may vary over a large range. An often cited example of this environmental dependence is found in the thioredoxin (Trx) super-family. The pKa values of active-site cysteines in these proteins can vary from 3 to 14 [46]. When the nucleophilicity is considered, the extent of water solvation also becomes important. In an environment where the thiol/thiolate species are heavily solvated, their reactivities may be diminished because the solvation shell must be disrupted to arrive at the reaction-transition state.
Since the environment of thiolates in proteins is only partially aqueous and low dielectric factors like charge, dipole and hydrogen bonding from surrounding residues make the contribution of these factors quite prominent. Despite the fact that sulfur is less electronegative and larger in size compared to oxygen and nitrogen, it has been observed that thiols can act as modest hydrogen bond donors/acceptors [173]. Thus, backbone amide bonds could also play an important role in modulating the cysteine pKa. For example, such peptide linkages can donate a hydrogen bond and have a permanent dipole moment. In case of redox-active cysteines, the hydrogen-bonding parameters depend whether the accepting sulfur is neutral or in thiolate form. In the Trx superfamily, the number of hydrogen bonds formed by the Cys1 of conserved Cys1–X1–X2–Cys2 motif was found to directly affect pKa. Indeed, a structural analysis revealed that in reduced Trx (pKa = 6.3–7.1), Cys1 forms two hydrogen bonds, Grx (pKa = 4.0–5.0) form three and DsbA (pKa = 3.5) forms four [173].
Other factors, like the presence of charged side chains and long range electrostatic interactions play only a limited role in influencing the pKas [173]. Due to the presence of the macrodipole, secondary structures like α-helix can have a dramatic influence on cysteine pKas. Experimental evidence presented by Kortemme and Creighton has shown that the presence of macrodipole works in alliance with hydrogen-bonding to decrease the pKa of cysteines [174]. By and large, the effect of lowering the pKa of cysteine in proteins should be more important for thiolate leaving group tendencies than their function as a nucleophile. Moreover, in the context of cysteine oxidation, the effect of lowering the pKas on rate enhancement is more prominent when the pKa values are closer to the solution pH. Any further decrease in pKa will increase the thiolate concentration, but this effect will be undermined by the corresponding decrease in the nucleophilicity. Table 4 summarizes second-order rate constants for H2O2-sensitive cysteines in proteins.
Table 4.
Second order rate constants for the oxidation of selected protein thiols in comparison to cysteine.
| R-SH + H2O2 | Rate constant (M−1 s−1) | Conditions | References |
|---|---|---|---|
| Cysteine (pKa 8.3) | 20 | 37 °C, pH 7.4 | [77] |
| H. sapiens Prx 2 (2-Cys Prx) (pKa 5.3) | 1 × 108 | 25 °C, pH 7.4 | [191] |
| X. fastidiosa Prx Q | 4.5 × 107 | 25 °C, pH 7.4 | [192] |
| S. typhimurium AhpC (2-Cys Prx) | 4 × 107 | 25 °C, pH 7.0 | [193] |
| T. cruzi TXNPx | 3 × 107 | 25 °C, pH 7.4 | [97] |
| S. cerevisiae Tsa 1 (2-Cys Prx) | 2.2 × 107 | 25 °C, pH 7.4 | [99] |
| S. cerevisiae Tsa 2 (2-Cys Prx) | 1.3 × 107 | 25 °C, pH 7.4 | [99] |
| H. sapiens Prx 5 (2-Cys Prx) (pKa 5.1) | 3 × 105 | 25 °C, pH 7.4 | [194] |
| Glutathione peroxidases (GPxs) | 105–108 | pH 6.7–7.6 | [50] |
| M. tuberculosis AhpE (1-Cys Prx) (pKa 5.2) | 8.2 × 104 | 25 °C, pH 7.4 | [26] |
| OxyR | 2.3 × 105 | 37 °C, pH 7.4 | [9,61] |
| NADH peroxidase (H10Q mutant) | 1.94 × 104 | 5 °C, pH 7.0 | [195] |
| GAPDH (pKa 8.3) | 2.8 × 103 | 25 °C, pH 7.4 | [70] |
| Yeast GR EHR (Cys63) (pKa 3.7) | 0.005 | 5 °C, pH 7.0 | [178] |
| Cdc25A | 69 | 20 °C, pH 7.0 | [180] |
| Cdc25B (pKa 5.8) | 164 | 20 °C, pH 7.0 | [180] |
| 332 | 37 °C, pH 7.4 | [180] | |
| Cdc25C | 120 | 20 °C, pH 7.0 | [180] |
| Papain (Cys protease) (pKa 3.4) | 62 | 23 °C, pH 7.0 | [196] |
| PTP1B (PTPs) (pKa 5.5) | 9 | 30 °C, pH 7.0 | [57] |
| 42.8 | 25 °C, pH 7.0 | [197] | |
| LAR (PTPs) | 14 | 30 °C, pH 7.0 | [57] |
| VHR (PTPs) | 18 | 30 °C, pH 7.0 | [57] |
| SHP-1 | 2.0 | 25 °C, pH 7.5 | [198] |
| SHP-2 | 2.4 | 25 °C, pH 7.5 | [198] |
| ΔSHP-1 | 9.4 | 25 °C, pH 7.5 | [198] |
| ΔSHP-2 | 8.8 | 25 °C, pH 7.5 | [198] |
| MKP3 | 9.6 | 20 °C, pH 8.0 | [199] |
| LMV-PTP (isoform 1) | 13.1 | 25 °C, pH 7.5 | [200] |
| Human serum albumin (pKa 8.3–8.6) | 2.7 | 37 °C, pH 7.4 | [177] |
Although structural factors contribute to the increased reactivity of certain protein thiols (like Prxs), such features do not account for high susceptibility toward oxidation. For example, low abundance PTPs and high abundance Prxs, have similarly low pKa-thiols (pKa 5–6) but their rate constants for reaction with H2O2 vary substantially (PTPs = 10–40 M−1 s−1 compared with Prx = 105–108 M−1 s−1). Such low oxidation rate values for PTPs means that they would compete poorly with Prxs or even GSH. The higher rate of peroxide reduction in such cases is explained by the features in the structure of catalytic site [175]. Once the peroxide substrate is bound, features like the stabilization of an anionic transition state through a positive charge distribution in the binding pocket and a dynamic set of polar interactions play the key role in enhanced rates of oxidation. Moreover, the rates are kinetic parameters and are concentration dependent and the abundance of each protein-thiol will be a key factor to determine the oxidation tendencies and extent.
3.4.2. Factors affecting the stability of protein sulfenic acid
In 1976, Allison proposed that the ideal environment to stabilize a protein sulfenic acid is such where surface topology prevents the formation of intermolecular disulfides and proximal thiols that could produce intramolecular disulfides would also be absent [24]. Trends observed in small molecule sulfenic acids indicate that vicinal hydrogen bond acceptor moieties will also impart substantial stability. Moreover, limited solvent access and an apolar protein environment would contribute to the overall stabilization. Recently, a profiling study was conducted on sites of protein sulfenic acid modification that have been observed by crystallography or NMR [176]. Based on the sequence analysis where strongly and weekly interacting residues were analyzed, they observed that specific amino acid residues were overrepresented. They concluded that: 1) polar, but not necessarily charged residues, and 2) interactions with histidine and threonine residues are important. Such residues are generally hydrogen bond acceptors and could lower the pKa of modifiable cysteines. Thus, the intrinsic pKa of the cysteine in question may play an important role in stabilizing the related sulfenic acid. This would also mean that formation of sulfenate anion would impart stability to the sulfenic acid against self-condensation and disulfide formation. On the other hand, the sulfenate form is usually more prone to undergo sulfinic acid modification.
Although all these factors individually contribute to the stability of sulfenic acid, the absence of adjacent cysteines is still considered to be the major stabilizing factor. For example, human serum albumin, which is the most abundant protein in the vascular compartment, has 35 cysteines in its primary sequence. Of these, 34 are involved in disulfide bridges (17 bonds) and only one cysteine (Cys34) is free. In this case, Cys34 forms a sulfenic acid that can persist for hours and has been observed in several crystal structures [176,177]. Another study highlighting stability due to the absence of proximal thiols focused on a class of enzymes, called glutathione reductase (GR). The reduced form of GR (the EH2 form) has two functionally distinct cysteine thiols (Cys58 and Cys63). Cys58-SH can form a mixed disulfide with glutathione (GSH) or be alkylated by IAM to give an inactive species (the EHR form). Comparing the peroxide reactivity of GR EH2 and EHR states shows, quite clearly, the effect of a proximal thiol on the stability of sulfenic acid: Upon oxidation of EH2, only the intramolecular disulfide was detected. However, in the case of EHR, sulfenic acid modification of Cys63 was readily detected. Thus, elimination of neighboring Cys58 by alkylation or thiolation stabilizes Cys63-SOH [178].
Another example of a sulfenic acid whose the transient nature is attributed to the presence of a proximal cysteine is discussed below. Cdc25 phosphatase (Cdc25A, Cdc25B and Cdc25C in humans) belongs to the family of dual-specificity PTPs and they function as essential regulators of the cell cycle and as mediators of the checkpoint response in cells with DNA-damage [179]. As discussed above, PTPs are reversibly inactivated through the oxidation of active site cysteine to sulfenic acid. On the other hand, oxidation of the active site cysteine to sulfinic acid leads to irreversible enzyme inactivation. Interestingly, Cdc25 enzymes have a proximal cysteine also known as the “backdoor” cysteine that rapidly condenses with the active site Cys-SOH to give the intramolecular disulfide, serving as a protective mechanism against irreversible oxidation. X-ray structural analysis indicates that, in the reduced form, active site Cys473 and backdoor Cys426 are separated by 4.6 Ǻ (Fig. 17A). In the sulfenic acid form, Cdc25B has the same P-loop conformation but the structure is very different from the reduced form (Fig. 17B). In order to avoid steric clashes with oxidized Cys473, residues 475–477 (Phe475, Ser476 and Ser477) of the P-loop move above Cys473 capping part of the entrance to the active site. This results in major overall structural twist in oxidized Cdc25B active-site and oxygenated Cys473 orients differently in the pocket than expected. To form the disulfide, Cys473 shifts toward the back-door Cys426 and, in doing so, the beginning of the P-loop is distorted and the main chain direction is altered (Fig. 17C). The rate constant for intra-molecular disulfide formation in Cdc25B is 0.16 s−1, whereas oxidation of Cys473-SOH to Cys473-SO2H by H2O2 is 47 M−1 s−1. Assuming that these in vitro rate constants reflect the in vivo situation, they indicate that irreversible oxidation would not become a significant factor until very high H2O2 concentrations (≥1 mM). Thus, Cdc25B serves as an example of how disulfide bond formation protects a sulfenic acid-based redox switch from irreversible damage [180,181].
Fig. 17.
Active-site of Cdc25B showing sulfenic acid mediated disulfide formation between redox-active Cys473 and back-door Cys426. (A) The native Cdc25B (PDB ID: 1QB0) is shown with redox-active Cys473 and back-door Cys426. (B) Sulfenic acid formation at redox-active Cys473 (PDB ID: 1YML). (C) Disulfide formation between redox-active Cys473 and back-door Cys426 (PDB ID: 1CWR).
A notable example of sulfenic acid stabilization by hydrogen bonding is elaborated below. Human DJ-1, a protein associated with early-onset Parkinson’s disease, appears to guard against mitochondrial damage in cells [182,183]. The protective function of DJ-1 has been attributed to the facile “hyperoxidation” of active site Cys106 to Cys106-SO2H. Cys106 has an estimated pKa of 5.4, indicating that the thiolate form predominates at physiological pH [184]. At least in part, the low pKa of Cys106 is due to a highly conserved protonated glutamic acid (Glu18) residue, which is ideally situated to donate a hydrogen bond to the reactive thiolate. As shown in Fig. 18A, the hydrogen bond length is estimated at 3.2 Ǻ. After oxidation, the length of the hydrogen bond between Glu18 and Cys106-SO2H shortens to 2.47 Ǻ (Fig. 18B). To examine the relationship between hydrogen bonding and oxidation propensity, a DJ-1 mutant was generated in which Glu18 was changed to aspartic acid. Interestingly, the resulting Glu18Asp DJ-1 formed a stable sulfenic acid at Cys106, not sulfinic acid like wild-type protein. Moreover, Cys106-SOH was present as the sulfenate and was stabilized by a 2.88 Ǻ hydrogen bond to the protonated carboxylic side-chain of Asp18. This work, reported by Wilson and coworkers, is an excellent example of how the stability of sulfenic acid can be effectively modulated via proximal residues and hydrogen bonding (Fig. 18C) [184,185].
Fig. 18.
X-ray crystal structures of DJ-1. (A) Wild-type DJ-1 in reduced form (PDB ID: 1P5F); (B) wild type DJ-1 sulfinic acid showing the E18-Cys106 hydrogen bonding (PDB ID: 1SOA); (C) E18D DJ-1 mutant with stable Cys106 sulfenate, hydrogen bonded to D18 (PDB ID: 3CZA).
3.4.3. Protein sulfenic acid fates and metabolism
In this section, we discuss possible fates and metabolism of protein-SOH. As detailed above, protein-SOH is formed through thiolate oxidation (Fig. 19, reaction 1). In the presence of excess oxidant, protein-SOH may oxidize further to sulfinic acid or sulfonic acid (Fig. 19, reactions 2 and 4). Some rate constants for oxidation of sulfenic to sulfinic acid are summarized in Table 5. In contrast to sulfenic acid, these higher oxidation states are generally considered irreversible forms associated with protein misfolding, degradation, and pathology. In an interesting exception to this rule, sulfinic acid modification of Prxs was recently found to be reversible via an ATP-consuming reaction catalyzed by the enzyme, sulfiredoxin (Srx; Fig. 19, reaction 3) [186]. The proposed mechanism involves formation of an activated sulfinic acid phosphoryl ester. This species can undergo attack by Srx active site cysteine to give a thiosulfinate intermediate. Ensuing thiol–disulfide exchange reactions converts the thiosulfinate to sulfenic acid, mixed disulfide, and back to the thiol form (Fig. 19, reaction 9). Another pathway for thiosulfinate formation would be the condensation of two sulfenic acids (Fig. 19, reaction 8). This reaction is highly dependent on sulfenic acid pKa and, in a biological setting, would be most facile when pKa = 7.4 when equal amounts of sulfenic acid and sulfenate anion are present. Thiosulfinates can also undergo hydrolysis to revert back to the sulfinic acid and thiol (Fig. 19, reaction 10).
Fig. 19.

Sulfenic acid metabolism pathways.
Table 5.
Rate constants for the oxidation of protein sulfenic acids to sulfinic acids.
| R-SOH + H2O2 → R-SO2H | Rate constant (M−1 s−1) | Conditions | References |
|---|---|---|---|
| Cdc25B-SOH | 60 | 20 °C, pH 7.0 | [180] |
| Cdc25C-SOH | 110 | 20 °C, pH 7.0 | [180] |
| C426S Cdc25B-SOH | 47 | 20 °C, pH 7.0 | [180] |
| C330S Cdc25C-SOH | 110 | 20 °C, pH 7.0 | [180] |
| ΔC218S MKP3-SOH | 101 | 20 °C, pH 8.0 | [199] |
| Mammalian Prx1-SOH | 57 | 25 °C, pH 7.4 | [26] |
| M. tuberculosis Prx-SOH | 40 | 25 °C, pH 7.4 | [26] |
| H10Q NADH Prx-SOH | 1.8 × 104 | 25 °C, pH 7.0 | [195] |
| HSA-SOH | 0.4 | 37 °C, pH 7.4 | [177] |
Disulfide bond formation is often named as the most biologically significant reaction of protein sulfenic acids. Oxidation of low-molecular-weight thiols, like Cys and GSH, results in the formation of disulfides and a 2:1 (thiol:oxidant) stoichiometry indicates the participation of a sulfenic acid intermediate. In proteins, disulfide formation can be intra- or intermolecular. A prominent example of sulfenic acid mediated intermolecular disulfide formation can be observed in 2Cys-Prxs (Fig. 19, reaction 5). The intramolecular disulfide bond generated during the catalytic cycle is then reduced by a class of small redox enzymes, called thioredoxins (Trxs) through thiol–disulfide exchange reactions (Fig. 19, reaction 6). Reversible formation of sulfenic acid mediated intramolecular disulfides is implicated as the mechanism of activation of the OxyR transcription factor and antioxidant defense against peroxides. Generation of mixed disulfides between proteins and low-molecular-weight thiols is referred to as S-thiolation. In general (but not always) this reaction proceeds via a protein-SOH intermediate. For example, GSH condensation with a protein sulfenic acid (known as S-glutathionylation) is promoted by oxidative/nitrosative stress, but can also occur in unstressed cells as an effective route of RSOH metabolism. One role of protein S-glutathionylation may be to prevent irreversible oxidation of protein thiols [187]. Protein-SSG mixed disulfides can be reduced by thiol–disulfide exchange with another molecule of GSH. Rate constants for the reaction between sulfenic acid and thiols are summarized in Table 6.
Table 6.
Rate constants for sulfenic acid-mediated disulfide formation and thiol-mediated reactivation of enzymes.
| Reaction | Rate Constant | Conditions | References |
|---|---|---|---|
| R-SOH + R′-SH → R-S-S-R′ | |||
| Cys-SOH + Cys | >105 M−1 s−1 | 18 °C, pH 7.0 | [79] |
| Cdc25B-SOH + back-door Cys | 0.16 s−1 | 20 °C, pH 7.0 | [180] |
| Cdc25C-SOH + back-door Cys | 0.012 s−1 | 20 °C, pH 7.0 | [180] |
| HSA-SOH + Cys | 21.6 M−1 s−1 | 25 °C, pH 7.4 | [177] |
| HSA-SOH + GSH | 2.9 M−1 s−1 | 25 °C, pH 7.4 | [177] |
| Reactivation of enzyme | |||
| MKP3ox + GSH/Trx-TrxR | 1–2/316 M−1 s−1 | 20 °C, pH 8.0 | [199] |
| Cdc25Box + DTT/Trx-TrxR | 0.5/1007 M−1 s−1 | 20 °C, pH 7.0 | [180] |
| PTP-SSG + Grx | 483 M−1 s−1 | 30 °C, pH 7.0 | [201] |
| SHP-1 + DTT/GSH | 0.64/0.026 | 25 °C, pH 7.5 | [198] |
| SHP-2 + DTT/GSH | 0.46/0.16 | 25 °C, pH 7.5 | [198] |
| ΔSHP-1 + DTT/GSH | 0.33/0.077 | 25 °C, pH 7.5 | [198] |
| ΔSHP-2 + DTT/GSH | 0.84/0.07 | 25 °C, pH 7.5 | [198] |
As mentioned above, protein sulfenic acids can also react with an amine functional group in small-molecules and peptide-backbone amides. Indeed, the sulfenic acid derivative of GAPDH has been detected by reaction of the oxidized protein with radiolabeled benzylamine to form a sulfenamide [19,20]. Moreover, the sulfenic acid in PTP1B undergoes intramolecular attack by the adjacent amide nitrogen atom which results in the formation of an isothiazolidinone species also referred to as a sulfenyl amide (Fig. 19, reaction 7) [21,22].
3.5. Considerations for chemical trapping of sulfenic acids
In a cellular context, rates of sulfenic acid metabolism by low-molecular weight or protein thiols are extremely important considerations in the chemical trapping (and detection) of this species. For this purpose, known rate constants for chemical reactions of sulfenic acid (oxidation, reduction, alkylation) are summarized in Table 7. Of particular interest, we note the significant difference in rate constants for the reaction between dimedone and various sulfenic acid models as well as between an individual sulfenic acid and chemical probes that differ in size and other physical characteristics. Collectively, these data suggest that: 1) the accessibility of sulfenic acid (or bulk of the chemical probe) affects labeling efficiency, and/or 2) some environments may facilitate the chemical reaction between the probe and RSOH.
Table 7.
Rate constants for reactions of model RSOH with oxidants, reductants, and probes.
| R-SOH + conditions → product | Rate constant (M−1 s−1) | Conditions | References |
|---|---|---|---|
| HSA-SOH + H2O2 | 0.4 | 37 °C, pH 7.4 | [177] |
| HSA-SOH + dimedone | 0.027 | 37 °C, pH 7.4 | [177] |
| HSA-SOH + TNB | 105 | 25 °C, pH 7.4 | [177] |
| HSA-SOH + Cys/GSH | 21.6/2.9 | 25 °C, pH 7.4 | [177] |
| HSA-SOH + arsenite | 0.036 | 37 °C, pH 7.4 | [177] |
| fRMsr + DCP-Bio1 | 2.0 | 37 °C, pH 5–8 | [144] |
| C165S AhpC-SOH + Dimedone | 1.7a | 37 °C, pH 7.0 | [143] |
| Papain-SOH + DCP-Bio1 | 27.5a | 37 °C, pH 7.0 | [144] |
| C165S AhpC-SOH + DCP-Bio1 | 0.05a | 37 °C, pH 7.0 | [144] |
| C165S AhpC-SOH + dimedone | 0.008a | 25 °C, pH 5.5 | [148] |
| C165S AhpC-SOH + 1,3-CPD | 0.006a | 25 °C, pH 5.5 | [148] |
| C165S AhpC-SOH + ethylthio-CPD | 0.012a | 25 °C, pH 5.5 | [148] |
| Dipeptide-SOH + dimedone | 25.5 | 25 °C, pH 7.4 | b |
| Dipeptide-SOH + DAz-1 | 3a | 25 °C, pH 7.4 | b |
| Dipeptide-SOH + DAz-2 | 13a | 25 °C, pH 7.4 | b |
| Dipeptide-SOH + DYn-2 | 11a | 25 °C, pH 7.4 | b |
| Papain-SOH + DAz-1 | 0.8 | 25 °C, pH 7.4 | c |
Calculated from pseudo-first order rate constant and probe concentration.
V. Gupta and K. Carroll, unpublished results.
S. Leonard and K. Carroll, unpublished results.
A comparison of rate constants for sulfenic acid trapping by chemical probes relative to other competing biological reactions indicates that current reagents sample only a fraction of the protein-SOH population in cells (note also that competing reactions can exist in vitro, but can be addressed more readily by adjusting probe, oxidant, and protein concentrations). Considerations of what makes an ideal chemical trap must take into account the ultimate downstream application and goal of the study. In general, however, the probe should be sufficiently reactive and selective under physiological conditions, compatible with an aqueous environment, and have minimal effect on the redox balance and cell viability. Dimedone-based probes satisfy the former criteria, but do so with moderate chemical reactivity. Using HSA as an example, we consider the reaction of HSA-SOH with dimedone in competition with mixed disulfide formation with GSH (Fig. 20). For HSA-SOH, the rate constant (k2) for dimedone-trapping (reaction 2) is 0.027 M−1 s−1 and the rate constant (k3) for S-glutathionylation (reaction 3) is 2.9 M−1 s−1. Estimated cellular GSH concentration is ~1 mM and generally, dimedone concentration between 5 and 10 mM (typical conditions, which are not toxic to cells) is used for labeling. Using these values in equation 3, we estimate that the proportion of HSA-SOH trapped by dimedone would be ~4–8%, while the rest would react with GSH. If the goal is to qualitatively profile changes in cellular protein sulfenylation, this level of reactivity and trapping is sufficient, given the high sensitivity afforded by subsequent chemiluminescent and fluorescent signals. Although probes with higher chemical reactivity may sample a higher proportion of biological sulfenic acids, this may also translate into a reagent that has reduced selectivity and/or toxicity. Thus, it is important to balance reactivity with selectivity in the design and application of a chemical probe. Extending these calculations, we estimate that for the chemical probe to sample at least 50% of HSA-SOH, k2 should be ~0.3 M−1 s−1 or 10-fold more reactive compared to dimedone. Similarly, to trap ~90% HSA-SOH, k2 would need to be ~2.6 M−1 s−1 or roughly 100-fold more reactive than dimedone. For obvious reasons, a chemical trap which would sample 100% of protein-SOH is mathematically improbable. Work under progress in our laboratory has already identified several potential probes with reactivity that are 10–30 fold higher than dimedone, while maintaining chemoselectivity (V. Gupta and K. Carroll, unpublished data).
Fig. 20.
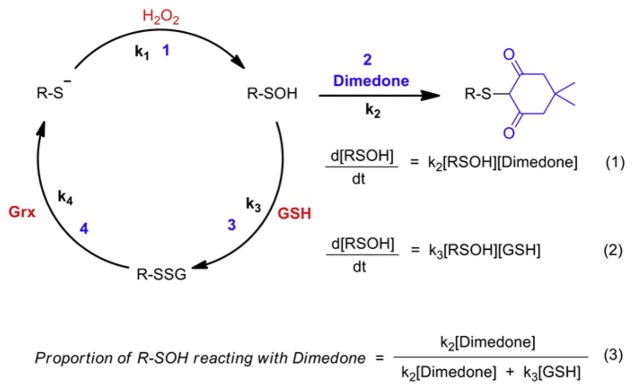
Kinetic representation of competitive in vivo trapping of sulfenic acid with dimedone in the presence of glutathione.
3.6. Cellular lifetime of sulfenic acid—kinetic aspects
Fig. 21 shows the three common sulfenic acid redox cycles. Cellular lifetimes of protein-SOH may be estimated on the basis of an estimate of steady-state concentration of protein-SOH (although this would not be applicable to signaling situations where steady-state is perturbed). For example, MAP kinase phosphatase 3 (MKP3) is redox regulated as shown by Fig. 21A. Assuming that: 1) MKP3 (0.1 μM) is inactivated under basal H2O2 concentrations (0.2 μM; the second order rate constant for this inactivation is 9.6 M−1 s−1), and 2) reactivation of MKP3 by Trx (~5 μM) with a second order rate constant of 316 M−1 s−1, using equation 3 the steady-state concentration of MKP3-SOH under these conditions is estimated to be 3 nM. Similar calculations for MKP3 under oxidative stress conditions (i.e., H2O2 ~ 20 μM) would result in a steady-state concentration of 0.3 μM for MKP3-SOH. Additional scenarios are available for protein-SOHs following the pathways shown in Fig. 21B and C, but the same general idea holds.
Fig. 21.

Redox regulation of sulfenic acid—steady state approximation to estimate sulfenic acid concentration.
We can also estimate the half-life of MKP3-SOH based on the reaction with GSH (note that not all protein-SOHs are accessible to GSH). Using the known second order rate constant for the reaction (~1 M−1 s−1) and estimated GSH concentration (~1 mM), the first order rate constant for this reaction would be 0.001 s−1. If this value is substituted into equation 4 (Fig. 21), the estimated half-life of MKP3-SOH would be ~12 min. Similar calculations for the reaction between HSA-SOH and GSH give an estimated HSA-SOH half-life of 4 min. Indeed, these estimated protein-SOH half-lives correlate well to global sulfenylation studies published by our group, in which we have observed that protein sulfenylation in A431 cells peaks around 5 min after EGF addition, with a subsequent decay over 30 min [59]. These estimated half-lives for protein-SOHs are on par with the cellular lifetimes for many other post-translational modifications, including phosphorylation and O-GlcNAc (the half-lives for some of which are estimated to be less than 1 min) [188].
4. Conclusions and future perspectives
Over the years, several reviews have appeared explaining the generation and chemistry of small molecule sulfenic acids and biological roles of sulfenic acids separately. The review here presented a general survey of sulfenic acid formation from small molecules to proteins. In small molecule systems, sterics and hydrogen bonding with neighboring heteroatoms emerged as primary factors which stabilize the sulfenic acids. Moreover, electronic factors responsible for decreasing the pKa of sulfenic acid and resulting in formation of sulfenates were found to impart a stabilizing effect by suppressing the thiosulfinate formation. The knowledge gained from small molecule sulfenic acid chemistry was used as foundation to explain the stable/transient nature of sulfenic acid in proteins. Since disulfide formation is biologically most facile reaction for protein sulfenic acids, absence of nearby thiolates emerged as the primary stabilizing parameter. Moreover, modulation in hydrogen bonding is shown to stabilize/destabilize the sulfenic acids in proteins. Kinetic parameters involved in the oxidation of protein thiolates to protein sulfenic acids and eventual metabolism to more stable products were looked at in order to gain an insight into the lifetime of sulfenic acids. Moreover, a general survey of the sulfenic acid detection techniques used by various research groups is presented. Based on all the information reviewed through the process of writing this review, it is observed that with technological advances several new probes have been developed to recognize protein sulfenic acid. Their reactivity is based on the 1,3-dicarbonyl chemistry originally demonstrated with dimedone. Some were cell-permeable whereas others had fluorescent or biotin tags for identification and visualization. Even with many successful examples of sulfenic acid detection, it is still challenging to trap short-lived sulfenic acid modifications in cellular systems. This is because reaction rates of many biological reactions outpace the reaction rates shown by dimedone analogs. Although no chemoproteomic method for detecting any post-translational modification detection is absolutely quantitative, using protein-SOH probes to monitor qualitative changes in cellular sulfenome has allowed for a significant insight. At the same time, further improvement in detection rates will certainly permit for a more comprehensive analysis. Progress in the field of thiol redox proteomics should advance our knowledge of regulatory mechanisms that involve oxidation of cysteine residues and lead to a better understanding of oxidative biochemistry in health and disease.
Acknowledgments
This work was supported by NIH grant R01 GM102187.
Footnotes
This article is part of a Special Issue entitled Current methods to study reactive oxygen species - pros and cons and biophysics of membrane proteins. Guest Editor: Christine Winterbourn.
References
- 1.Miseta A, Csutora P. Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol Biol Evol. 2000;17:1232–1239. doi: 10.1093/oxfordjournals.molbev.a026406. [DOI] [PubMed] [Google Scholar]
- 2.Pe’er I, Felder CE, Man O, Silman I, Sussman JL, Beckmann JS. Proteomic signatures: amino acid and oligopeptide compositions differentiate among phyla. Proteins Struct Funct Bioinform. 2004;54:20–40. doi: 10.1002/prot.10559. [DOI] [PubMed] [Google Scholar]
- 3.Sanchez R, Riddle M, Woo J, Momand J. Prediction of reversibly oxidized protein cysteine thiols using protein structure properties. Protein Sci. 2008;17:473–481. doi: 10.1110/ps.073252408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15:247–254. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- 5.Poole LB, Nelson KJ. Discovering mechanisms of signaling-mediated cysteine oxidation. Curr Opin Chem Biol. 2008;12:18–24. doi: 10.1016/j.cbpa.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paulsen CE, Carroll KS. Orchestrating redox signaling networks through regulatory cysteine switches. ACS Chem Biol. 2009;5:47–62. doi: 10.1021/cb900258z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hall A, Karplus PA, Poole LB. Typical 2-Cys peroxiredoxins—structures, mechanisms and functions. FEBS J. 2009;276:2469–2477. doi: 10.1111/j.1742-4658.2009.06985.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poole LB, Karplus PA, Claiborne A. Protein sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol. 2004;44:325–347. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 9.Claiborne A, Mallett TC, Yeh JI, Luba J, Parsonage D. Structural, redox, and mechanistic parameters for cysteine-sulfenic acid function in catalysis and regulation. Adv Protein Chem. 2001;58:215–276. doi: 10.1016/s0065-3233(01)58006-7. [DOI] [PubMed] [Google Scholar]
- 10.Jacob C, Giles GI, Giles NM, Sies H. Sulfur and selenium: the role of oxidation state in protein structure and function. Angew Chem Int Ed. 2003;42:4742–4758. doi: 10.1002/anie.200300573. [DOI] [PubMed] [Google Scholar]
- 11.Nagy P. Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation-mediated pathways. Antioxid Redox Signal. 2013;18:1623–1641. doi: 10.1089/ars.2012.4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fries K. Über α-Anthrachinon-sulfensäure. Ber Dtsch Chem Ges. 1912;45:2965–2973. [Google Scholar]
- 13.Bruice TC, Sayigh AB. The structure of anthraquinone-1-sulfenic acid (Fries’ acid) and related compounds. J Am Chem Soc. 1959;81:3416–3420. [Google Scholar]
- 14.Penn RE, Block E, Revelle LK. Flash vacuum pyrolysis studies. 5. Methanesulfenic acid. J Am Chem Soc. 1978;100:3622–3623. [Google Scholar]
- 15.Hogg DR. Sulfenic Acids and Derivatives (1990) John Wiley & Sons, Ltd; 2010. Chemistry of sulphenic acids and esters; pp. 361–402. [Google Scholar]
- 16.Aversa MC, Bonaccorsi P, Madec D, Prestat G, Poli G. Innovative Catalysis in Organic Synthesis. Wiley-VCH Verlag GmbH & Co. KGaA; 2012. The fabulous destiny of sulfenic acids; pp. 47–76. [Google Scholar]
- 17.Vaidya V, Ingold KU, Pratt DA. Garlic: source of the ultimate antioxidants—sulfenic acids. Angew Chem. 2009;121:163–166. doi: 10.1002/anie.200804560. [DOI] [PubMed] [Google Scholar]
- 18.Jeong J, Jung Y, Na S, Jeong J, Lee E, Kim M-S, Choi S, Shin D-H, Paek E, Lee H-Y, Lee K-J. Novel Oxidative Modifications in Redox-Active Cysteine Residues. Mol Cell Proteomics. 2011;10:M110 000513. doi: 10.1074/mcp.M110.000513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benitez LV, Allison WS. The inactivation of the acyl phosphatase activity catalyzed by the sulfenic acid form of glyceraldehyde 3-phosphate dehydrogenase by dimedone and olefins. J Biol Chem. 1974;249:6234–6243. [PubMed] [Google Scholar]
- 20.Allison WS, Benitez LV, Johnson CL. The formation of a protein sulfenamide during the inactivation of the acyl phosphatase activity of oxidized glyceraldehyde-3-phosphate dehydrogenase by benzylamine. Biochem Biophys Res Commun. 1973;52:1403–1409. doi: 10.1016/0006-291x(73)90657-8. [DOI] [PubMed] [Google Scholar]
- 21.Salmeen A, Andersen JN, Myers MP, Meng T-C, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 22.van Montfort RLM, Congreve M, Tisi D, Carr R, Jhoti H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature. 2003;423:773–777. doi: 10.1038/nature01681. [DOI] [PubMed] [Google Scholar]
- 23.Kharasch N, Potempa SJ, Wehrmeister HL. The sulfenic acids and their derivatives. Chem Rev. 1946;39:269–332. doi: 10.1021/cr60123a004. [DOI] [PubMed] [Google Scholar]
- 24.Allison WS. Formation and reactions of sulfenic acids in proteins. Acc Chem Res. 1976;9:293–299. [Google Scholar]
- 25.Kice JL, Weclas-Henderson L, Kewan A. Equilibrium and kinetic studies of some reactions of 1-anthraquinonesulfenic acid and its methyl ester. J Org Chem. 1989;54:4198–4203. [Google Scholar]
- 26.Hugo MN, Turell LA, Manta B, Botti H, Monteiro G, Netto LES, Alvarez B, Radi R, Trujillo M. Thiol and sulfenic acid oxidation of AhpE, the one-cysteine peroxiredoxin from mycobacterium tuberculosis: kinetics: acidity constants, and conformational dynamics. Biochemistry. 2009;48:9416–9426. doi: 10.1021/bi901221s. [DOI] [PubMed] [Google Scholar]
- 27.Ellis HR, Poole LB. Novel application of 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole to identify cysteine sulfenic acid in the AhpC component of alkyl hydroperoxide reductase†. Biochemistry. 1997;36:15013–15018. doi: 10.1021/bi972191x. [DOI] [PubMed] [Google Scholar]
- 28.O’Donnell JS, Schwan AL. β-Sulfinyl acrylate esters as a convenient source of alkane- and arenesulfenate anions. Tetrahedron Lett. 2003;44:6293–6296. [Google Scholar]
- 29.Singh SP, O’Donnell JS, Schwan AL. Nucleophilic attack of 2-sulfinyl acrylates: a mild and general approach to sulfenic acid anions. Org Biomol Chem. 2010;8:1712–1717. doi: 10.1039/b917217c. [DOI] [PubMed] [Google Scholar]
- 30.Schwan AL, Verdu MJ, Singh SP, O’Donnell JS, Ahmadi AN. Diastereoselective alkylations of a protected cysteinesulfenate. J Org Chem. 2009;74:6851–6854. doi: 10.1021/jo901021r. [DOI] [PubMed] [Google Scholar]
- 31.Neville Jones D, Cottam PD, Davies J. Addition of sulphenic acids to unactivated alkynes to give alkenyl sulphoxides. Tetrahedron Lett. 1979;20:4977–4980. [Google Scholar]
- 32.Adams H, Anderson JC, Bell R, Neville Jones D, Peel MR, Tomkinson NCO. The synthesis and Diels–Alder reactions of (E)- and (Z)-1-methoxy-3-(phenylsulfinyl)buta-1,3-dienes. J Chem Soc Perkin Trans. 1998;1:3967–3974. [Google Scholar]
- 33.Sivaramakrishnan S, Keerthi K, Gates KS. A chemical model for redox regulation of protein tyrosine phosphatase 1B (PTP1B) activity. J Am Chem Soc. 2005;127:10830–10831. doi: 10.1021/ja052599e. [DOI] [PubMed] [Google Scholar]
- 34.Pal BC, Uziel M, Doherty DG, Cohn WE. Isolation and characterization of a pyrimidine sulfenic acid via scission of the sulfur–sulfur bond in the methyl analog of bis(4-thiouridine) disulfide. J Am Chem Soc. 1969;91:3634–3638. doi: 10.1021/ja01041a036. [DOI] [PubMed] [Google Scholar]
- 35.Heckel A, Pfieiderer W. Lumazinesulfenates—a new class of stable sulfenic acids. Tetrahedron Lett. 1983;24:5047–5050. [Google Scholar]
- 36.Yoshimura T, Tsukurimichi E, Yamazaki S, Soga S, Shimasaki C, Hasegawa K. Synthesis of a stable sulfenic acid, trans-decalin-9-sulfenic acid. J Chem Soc, Chem Commun. 1992:1337–1338. [Google Scholar]
- 37.Okuyama T, Toyoda M, Fueno T. Acid-catalyzed hydrolysis of methoxymethyl phenyl sulfoxide without concomitant racemization. Bull Chem Soc Jpn. 1990;63:1316–1321. [Google Scholar]
- 38.Tripolt R, Belaj F, Nachbaur E. Unexpectedly stable sulfenic acid—4,6-dimethoxy-1,3,5-triazine-2-sulfenic acid—synthesis, properties, molecular and crystal-structure. Z Naturforsch, B: Chem Sci. 1993;48:1212–1222. [Google Scholar]
- 39.Fekner T, Baldwin JE, Adlington RM, Schofield CJ. Unusually stable azetidinone sulfenic acids. Tetrahedron Lett. 1998;39:6983–6986. [Google Scholar]
- 40.Machiguchi T, Hasegawa T, Otani H, Yamabe S, Mizuno H. Tropothione S-oxide: the first example of a sulfine charge reversion (umpolung) J Am Chem Soc. 1994;116:407–408. [Google Scholar]
- 41.Ishii A, Komiya K, Nakayama J. Synthesis of a stable sulfenic acid by oxidation of a sterically hindered thiol (thiophenetriptycene-8-thiol)1 and its characterization. J Am Chem Soc. 1996;118:12836–12837. [Google Scholar]
- 42.Goto K, Tokitoh N, Okazaki R. Synthesis of a stable arenesulfenic acid bearing a bowl-shaped macrobicyclic cyclophane skeleton. Angew Chem Int Ed Engl. 1995;34:1124–1126. [Google Scholar]
- 43.Saiki T, Goto K, Tokitoh N, Okazaki R. Synthesis and structure of a bridged calix [6]arene with a sulfenic acid functionality in the cavity. J Org Chem. 1996;61:2924–2925. doi: 10.1021/jo960068q. [DOI] [PubMed] [Google Scholar]
- 44.Goto K, Holler M, Okazaki R. Synthesis, structure, and reactions of a sulfenic acid bearing a novel bowl-type substituent: the first synthesis of a stable sulfenic acid by direct oxidation of a thiol. J Am Chem Soc. 1997;119:1460–1461. [Google Scholar]
- 45.Rehder DS, Borges CR. Cysteine sulfenic acid as an intermediate in disulfide bond formation and nonenzymatic protein folding. Biochemistry. 2010;49:7748–7755. doi: 10.1021/bi1008694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hofmann B, Hecht HJ, Flohé L. Peroxiredoxins. Biol Chem. 2002;383:347. doi: 10.1515/BC.2002.040. [DOI] [PubMed] [Google Scholar]
- 47.Hall A, Nelson K, Poole LB, Karplus PA. Structure-based insights into the catalytic power and conformational dexterity of peroxiredoxins. Antioxid Redox Signal. 2011;15:795–815. doi: 10.1089/ars.2010.3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 49.Soito L, Williamson C, Knutson ST, Fetrow JS, Poole LB, Nelson KJ. PREX: peroxiredoxin classification indEX, a database of subfamily assignments across the diverse peroxiredoxin family. Nucleic Acids Res. 2011;39:D332–D337. doi: 10.1093/nar/gkq1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Toppo S, Flohé L, Ursini F, Vanin S, Maiorino M. Catalytic mechanisms and specificities of glutathione peroxidases: variations of a basic scheme. Biochim Biophys Acta, Gen Subj. 2009;1790:1486–1500. doi: 10.1016/j.bbagen.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 51.Brigelius-Flohé R. Tissue-specific functions of individual glutathione peroxidases. Free Radic Biol Med. 1999;27:951–965. doi: 10.1016/s0891-5849(99)00173-2. [DOI] [PubMed] [Google Scholar]
- 52.Tosatto SC, Bosello V, Fogolari F, Mauri P, Roveri A, Toppo S, Flohe L, Ursini F, Maiorino M. The catalytic site of glutathione peroxidases. Antioxid Redox Signal. 2008;10:1515–1526. doi: 10.1089/ars.2008.2055. [DOI] [PubMed] [Google Scholar]
- 53.Flohe L, Toppo S, Cozza G, Ursini F. A comparison of thiol peroxidase mechanisms. Antioxid Redox Signal. 2011;15:763–780. doi: 10.1089/ars.2010.3397. [DOI] [PubMed] [Google Scholar]
- 54.Zheng M, Wang X, Templeton LJ, Smulski DR, LaRossa RA, Storz G. DNA microarray-mediated transcriptional profiling of the Escherichia coli response to hydrogen peroxide. J Bacteriol. 2001;183:4562–4570. doi: 10.1128/JB.183.15.4562-4570.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hubbard SR, Till JH. Protein tyrosine kinase structure and function. Annu Rev Biochem. 2000;69:373–398. doi: 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- 56.Tanner JJ, Parsons ZD, Cummings AH, Zhou H, Gates KS. Redox regulation of protein tyrosine phosphatases: structural and chemical aspects. Antioxid Redox Signal. 2011;15:77–97. doi: 10.1089/ars.2010.3611. [DOI] [PubMed] [Google Scholar]
- 57.Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- 58.Alvarez B, Carballal S, Turell L, Radi R. Formation and reactions of sulfenic acid in human serum albumin. Methods Enzymol. 2010;473:117–136. doi: 10.1016/S0076-6879(10)73005-6. [DOI] [PubMed] [Google Scholar]
- 59.Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, Carroll KS. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat Chem Biol. 2012;8:57–64. doi: 10.1038/nchembio.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Truong TH, Carroll KS. Redox regulation of epidermal growth factor receptor signaling through cysteine oxidation. Biochemistry. 2012;51:9954–9965. doi: 10.1021/bi301441e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zheng M, Aslund F, Storz G. Activation of the OxyR transcription factor by reversible disulfide bond formation. Science. 1998;279:1718–1721. doi: 10.1126/science.279.5357.1718. [DOI] [PubMed] [Google Scholar]
- 62.Fuangthong M, Helmann JD. The OhrR repressor senses organic hydroperoxides by reversible formation of a cysteine-sulfenic acid derivative. Proc Natl Acad Sci U S A. 2002;99:6690–6695. doi: 10.1073/pnas.102483199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ma LH, Takanishi CL, Wood MJ. Molecular mechanism of oxidative stress perception by the Orp1 protein. J Biol Chem. 2007;282:31429–31436. doi: 10.1074/jbc.M705953200. [DOI] [PubMed] [Google Scholar]
- 64.Wu J, Cheng Z, Reddie K, Carroll K, Hammad LA, Karty JA, Bauer CE. RegB kinase activity is repressed by oxidative formation of cysteine sulfenic acid. J Biol Chem. 2013;288:4755–4762. doi: 10.1074/jbc.M112.413492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pirie NW. The oxidation of sulphydryl compounds by hydrogen peroxide: catalysis of oxidation of cysteine by thiocarbamides and thiolglyoxalines. Biochem J. 1933;27:1181–1188. doi: 10.1042/bj0271181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fraenkel-Conrat H. The reaction of tobacco mosaic virus with iodine. J Biol Chem. 1955;217:373–381. [PubMed] [Google Scholar]
- 67.Pihl A, Lange R. The interaction of oxidized glutathione, cystamine monosulfoxide, and tetrathionate with the-SH groups of rabbit muscle D-glyceraldehyde 3-phosphate dehydrogenase. J Biol Chem. 1962;237:1356–1362. [PubMed] [Google Scholar]
- 68.Ehring R, Colowick SP. The two-step formation and inactivation of acylphosphatase by agents acting on glyceraldehyde phosphate dehydrogenase. J Biol Chem. 1969;244:4589–4599. [PubMed] [Google Scholar]
- 69.Parker DJ, Allison WS. The mechanism of inactivation of glyceraldehyde 3-phosphate dehydrogenase by tetrathionate, o-iodosobenzoate, and iodine monochloride. J Biol Chem. 1969;244:180–189. [PubMed] [Google Scholar]
- 70.Little C, O’Brien PJ. Mechanism of peroxide-inactivation of the sulphydryl enzyme glyceraldehyde-3-phosphate dehydrogenase. Eur J Biochem. 1969;10:533–538. doi: 10.1111/j.1432-1033.1969.tb00721.x. [DOI] [PubMed] [Google Scholar]
- 71.Trundle D, Cunningham LW. Iodine oxidation of the sulfhydryl groups of creatine kinase. Biochemistry. 1969;8:1919–1925. doi: 10.1021/bi00833a023. [DOI] [PubMed] [Google Scholar]
- 72.Allison WS, Connors MJ. The activation and inactivation of the acyl phosphatase activity of glyceraldehyde-3-phosphate dehydrogenase. Arch Biochem Biophys. 1970;136:383–391. doi: 10.1016/0003-9861(70)90209-2. [DOI] [PubMed] [Google Scholar]
- 73.Allison WS, Benitez LV. An adenosine triphosphate–phosphate exchange catalyzed by a soluble enzyme couple inhibited by uncouplers of oxidative phosphorylation. Proc Natl Acad Sci. 1972;69:3004–3008. doi: 10.1073/pnas.69.10.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Radi R, Bush KM, Cosgrove TP, Freeman BA. Reaction of xanthine oxidase-derived oxidants with lipid and protein of human plasma. Arch Biochem Biophys. 1991;286:117–125. doi: 10.1016/0003-9861(91)90016-c. [DOI] [PubMed] [Google Scholar]
- 75.Wardman P, von Sonntag C. [3] Kinetic factors that control the fate of thiyl radicals in cells. In: Lester P, editor. Methods Enzymol. Vol. 251. Academic Press; 1995. pp. 31–45. [DOI] [PubMed] [Google Scholar]
- 76.Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45:549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 77.Winterbourn CC, Metodiewa D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic Biol Med. 1999;27:322–328. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 78.Barton JP, Packer JE, Sims RJ. Kinetics of the reaction of hydrogen peroxide with cysteine and cysteamine. J Chem Soc Perkin Trans. 1973;2:1547–1549. [Google Scholar]
- 79.Nagy P, Ashby MT. Reactive sulfur species: kinetics and mechanisms of the oxidation of cysteine by hypohalous acid to give cysteine sulfenic acid. J Am Chem Soc. 2007;129:14082–14091. doi: 10.1021/ja0737218. [DOI] [PubMed] [Google Scholar]
- 80.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 81.Bedard K, Krause K-H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 82.Yeldandi AV, Rao MS, Reddy JK. Hydrogen peroxide generation in peroxisome proliferator-induced oncogenesis. Mutat Res Fundam Mol Mech Mutagen. 2000;448:159–177. doi: 10.1016/s0027-5107(99)00234-1. [DOI] [PubMed] [Google Scholar]
- 83.Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 84.Choi H-J, Kang SW, Yang C-H, Rhee SG, Ryu S-E. Crystal structure of a novel human peroxidase enzyme at 2.0 A resolution. Nat Struct Mol Biol. 1998;5:400–406. doi: 10.1038/nsb0598-400. [DOI] [PubMed] [Google Scholar]
- 85.Chae HZ, Uhm TB, Rhee SG. Dimerization of thiol-specific antioxidant and the essential role of cysteine 47. Proc Natl Acad Sci U S A. 1994;91:7022–7026. doi: 10.1073/pnas.91.15.7022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ellis HR, Poole LB. Roles for the two cysteine residues of AhpC in catalysis of peroxide reduction by alkyl hydroperoxide reductase from Salmonella typhimurium. Biochemistry. 1997;36:13349–13356. doi: 10.1021/bi9713658. [DOI] [PubMed] [Google Scholar]
- 87.Baker LM, Poole LB. Catalytic mechanism of thiol peroxidase from Escherichia coli. Sulfenic acid formation and overoxidation of essential CYS61. J Biol Chem. 2003;278:9203–9211. doi: 10.1074/jbc.M209888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wills ED. Effect of unsaturated fatty acids and their peroxides on enzymes. Biochem Pharmacol. 1961;7:7–16. doi: 10.1016/0006-2952(61)90119-8. [DOI] [PubMed] [Google Scholar]
- 89.Little C, O’Brien PJ. Products of oxidation of a protein thiol group after reaction with various oxidizing agents. Arch Biochem Biophys. 1967;122:406–410. doi: 10.1016/0003-9861(67)90212-3. [DOI] [PubMed] [Google Scholar]
- 90.Little C, O’Brien PJ. The effectiveness of a lipid peroxide in oxidizing protein and non-protein thiols. Biochem J. 1968;106:419–423. doi: 10.1042/bj1060419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Takanishi CL, Ma LH, Wood MJ. A genetically encoded probe for cysteine sulfenic acid protein modification in vivo. Biochemistry. 2007;46:14725–14732. doi: 10.1021/bi701625s. [DOI] [PubMed] [Google Scholar]
- 92.Marla SS, Lee J, Groves JT. Peroxynitrite rapidly permeates phospholipid membranes. Proc Natl Acad Sci U S A. 1997;94:14243–14248. doi: 10.1073/pnas.94.26.14243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Quijano C, Alvarez B, Gatti RM, Augusto O, Radi R. Pathways of peroxynitrite oxidation of thiol groups. Biochem J. 1997;322(Pt 1):167–173. doi: 10.1042/bj3220167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bonini MG, Augusto O. Carbon dioxide stimulates the production of thiyl, sulfinyl, and disulfide radical anion from thiol oxidation by peroxynitrite. J Biol Chem. 2001;276:9749–9754. doi: 10.1074/jbc.M008456200. [DOI] [PubMed] [Google Scholar]
- 95.Denicola A, Freeman BA, Trujillo M, Radi R. Peroxynitrite reaction with carbon dioxide/bicarbonate: kinetics and influence on peroxynitrite-mediated oxidations. Arch Biochem Biophys. 1996;333:49–58. doi: 10.1006/abbi.1996.0363. [DOI] [PubMed] [Google Scholar]
- 96.Bryk R, Griffin P, Nathan C. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature. 2000;407:211–215. doi: 10.1038/35025109. [DOI] [PubMed] [Google Scholar]
- 97.Trujillo M, Budde H, Pineyro MD, Stehr M, Robello C, Flohe L, Radi R. Trypanosoma brucei and Trypanosoma cruzi tryparedoxin peroxidases catalytically detoxify peroxynitrite via oxidation of fast reacting thiols. J Biol Chem. 2004;279:34175–34182. doi: 10.1074/jbc.M404317200. [DOI] [PubMed] [Google Scholar]
- 98.Dubuisson M, Stricht DV, Clippe A, Etienne F, Nauser T, Kissner R, Koppenol WH, Rees JF, Knoops B. Human peroxiredoxin 5 is a peroxynitrite reductase. FEBS Lett. 2004;571:161–165. doi: 10.1016/j.febslet.2004.06.080. [DOI] [PubMed] [Google Scholar]
- 99.Ogusucu R, Rettori D, Munhoz DC, Netto LE, Augusto O. Reactions of yeast thioredoxin peroxidases I and II with hydrogen peroxide and peroxynitrite: rate constants by competitive kinetics. Free Radic Biol Med. 2007;42:326–334. doi: 10.1016/j.freeradbiomed.2006.10.042. [DOI] [PubMed] [Google Scholar]
- 100.Winterbourn CC. Comparative reactivities of various biological compounds with myeloperoxidase-hydrogen peroxide-chloride, and similarity of oxidant to hypochlorite. Biochim Biophys Acta, Gen Subj. 1985;840:204–210. doi: 10.1016/0304-4165(85)90120-5. [DOI] [PubMed] [Google Scholar]
- 101.Hawkins CL, Pattison DI, Davies MJ. Hypochlorite-induced oxidation of amino acids, peptides and proteins. Amino Acids. 2003;25:259–274. doi: 10.1007/s00726-003-0016-x. [DOI] [PubMed] [Google Scholar]
- 102.Pattison DI, Davies MJ. Reactions of myeloperoxidase-derived oxidants with biological substrates: gaining chemical insight into human inflammatory diseases. Curr Med Chem. 2006;13:3271–3290. doi: 10.2174/092986706778773095. [DOI] [PubMed] [Google Scholar]
- 103.Prütz WA. Hypochlorous acid interactions with thiols, nucleotides, DNA, and other biological substrates. Arch Biochem Biophys. 1996;332:110–120. doi: 10.1006/abbi.1996.0322. [DOI] [PubMed] [Google Scholar]
- 104.Nagy P, Winterbourn CC. Chapter 6—redox chemistry of biological thiols. In: James CF, editor. Advances in Molecular Toxicology. Vol. 4. Elsevier; 2010. pp. 183–222. [Google Scholar]
- 105.Peskin AV, Winterbourn CC. Kinetics of the reactions of hypochlorous acid and amino acid chloramines with thiols, methionine, and ascorbate. Free Radic Biol Med. 2001;30:572–579. doi: 10.1016/s0891-5849(00)00506-2. [DOI] [PubMed] [Google Scholar]
- 106.Folkes LK, Candeias LP, Wardman P. Kinetics and mechanisms of hypochlorous acid reactions. Arch Biochem Biophys. 1995;323:120–126. doi: 10.1006/abbi.1995.0017. [DOI] [PubMed] [Google Scholar]
- 107.Raftery MJ, Yang Z, Valenzuela SM, Geczy CL. Novel intra- and intermolecular sulfinamide bonds in S100A8 produced by hypochlorite oxidation. J Biol Chem. 2001;276:33393–33401. doi: 10.1074/jbc.M101566200. [DOI] [PubMed] [Google Scholar]
- 108.Peskin AV, Winterbourn CC. Taurine chloramine is more selective than hypochlorous acid at targeting critical cysteines and inactivating creatine kinase and glyceraldehyde-3-phosphate dehydrogenase. Free Radic Biol Med. 2006;40:45–53. doi: 10.1016/j.freeradbiomed.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 109.Tannenbaum SR, White FM. Regulation and specificity of S-nitrosylation and denitrosylation. ACS Chem Biol. 2006;1:615–618. doi: 10.1021/cb600439h. [DOI] [PubMed] [Google Scholar]
- 110.Foster MW, McMahon TJ, Stamler JS. S-Nitrosylation in health and disease. Trends Mol Med. 2003;9:160–168. doi: 10.1016/s1471-4914(03)00028-5. [DOI] [PubMed] [Google Scholar]
- 111.Foster MW, Hess DT, Stamler JS. Protein S-nitrosylation in health and disease: a current perspective. Trends Mol Med. 2009;15:391–404. doi: 10.1016/j.molmed.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Demaster EG, Quast BJ, Redfern B, Nagasawa HT. Reaction of nitric-oxide with the free sulfhydryl-group of human serum-albumin yields a sulfenic acid and nitrous-oxide. Biochemistry. 1995;34:11494–11499. doi: 10.1021/bi00036a023. [DOI] [PubMed] [Google Scholar]
- 113.Percival MD, Ouellet M, Campagnolo C, Claveau D, Li C. Inhibition of cathepsin K by nitric oxide donors: evidence for the formation of mixed disulfides and a sulfenic acid. Biochemistry. 1999;38:13574–13583. doi: 10.1021/bi991028u. [DOI] [PubMed] [Google Scholar]
- 114.Danehy JP, Hunter WE. Alkaline decomposition of organic disulfides. II. Alternative pathways as determined by structure. J Org Chem. 1967;32:2047–2053. [Google Scholar]
- 115.Andersson L-O. Hydrolysis of disulfide bonds in weakly alkaline media II. Bovine serum albumin dimer. Biochim Biophys Acta, Protein Struct. 1970;200:363–369. doi: 10.1016/0005-2795(70)90178-9. [DOI] [PubMed] [Google Scholar]
- 116.Wu HL, Shi GY, Wohl RC, Bender ML. Structure and formation of microplasmin. Proc Natl Acad Sci U S A. 1987;84:8793–8795. doi: 10.1073/pnas.84.24.8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lay AJ, Jiang XM, Kisker O, Flynn E, Underwood A, Condron R, Hogg PJ. Phosphoglycerate kinase acts in tumour angiogenesis as a disulphide reductase. Nature. 2000;408:869–873. doi: 10.1038/35048596. [DOI] [PubMed] [Google Scholar]
- 118.Lay AJ, Jiang XM, Daly E, Sun L, Hogg PJ. Plasmin reduction by phosphoglycerate kinase is a thiol-independent process. J Biol Chem. 2002;277:9062–9068. doi: 10.1074/jbc.M111531200. [DOI] [PubMed] [Google Scholar]
- 119.Wardman P. Evaluation of the “radical sink” hypothesis from a chemical-kinetic viewpoint. J Radioanal Nucl Chem. 1998;232:23–27. [Google Scholar]
- 120.Carballal S, Radi R, Kirk MC, Barnes S, Freeman BA, Alvarez B. Sulfenic acid formation in human serum albumin by hydrogen peroxide and peroxynitrite. Biochemistry. 2003;42:9906–9914. doi: 10.1021/bi027434m. [DOI] [PubMed] [Google Scholar]
- 121.Rabinkov A, Miron T, Konstantinovski L, Wilchek M, Mirelman D, Weiner L. The mode of action of allicin: trapping of radicals and interaction with thiol containing proteins. Biochim Biophys Acta, Gen Subj. 1998;1379:233–244. doi: 10.1016/s0304-4165(97)00104-9. [DOI] [PubMed] [Google Scholar]
- 122.Li J, Huang FL, Huang KP. Glutathiolation of proteins by glutathione disulfide S-oxide derived from S-nitrosoglutathione. Modifications of rat brain neurogranin/RC3 and neuromodulin/GAP-43. J Biol Chem. 2001;276:3098–3105. doi: 10.1074/jbc.M008260200. [DOI] [PubMed] [Google Scholar]
- 123.Stehle T, Claiborne A, Schulz GE. NADH binding site and catalysis of NADH peroxidase. Eur J Biochem. 1993;211:221–226. doi: 10.1111/j.1432-1033.1993.tb19889.x. [DOI] [PubMed] [Google Scholar]
- 124.Yeh JI, Claiborne A, Hol WGJ. Structure of the native cysteine-sulfenic acid redox center of enterococcal NADH peroxidase refined at 2.8 Å resolution†,‡. Biochemistry. 1996;35:9951–9957. doi: 10.1021/bi961037s. [DOI] [PubMed] [Google Scholar]
- 125.Crane EJ, Vervoort J, Claiborne A. 13C NMR analysis of the cysteine-sulfenic acid redox center of enterococcal NADH peroxidase†. Biochemistry. 1997;36:8611–8618. doi: 10.1021/bi9707990. [DOI] [PubMed] [Google Scholar]
- 126.Nakamura T, Yamamoto T, Abe M, Matsumura H, Hagihara Y, Goto T, Yamaguchi T, Inoue T. Oxidation of archaeal peroxiredoxin involves a hypervalent sulfur intermediate. Proc Natl Acad Sci. 2008;105:6238–6242. doi: 10.1073/pnas.0709822105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Poor CB, Chen PR, Duguid E, Rice PA, He C. Crystal structures of the reduced, sulfenic acid, and mixed disulfide forms of SarZ, a redox active global regulator in Staphylococcus aureus. J Biol Chem. 2009;284:23517–23524. doi: 10.1074/jbc.M109.015826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Boschi-Muller S, Azza S, Sanglier-Cianferani S, Talfournier F, Van Dorsselear A, Branlant G. A sulfenic acid enzyme intermediate is involved in the catalytic mechanism of peptide methionine sulfoxide reductase from Escherichia coli. J Biol Chem. 2000;275:35908–35913. doi: 10.1074/jbc.M006137200. [DOI] [PubMed] [Google Scholar]
- 129.Kaiserova K, Srivastava S, Hoetker JD, Awe SO, Tang X-L, Cai J, Bhatnagar A. Redox activation of aldose reductase in the ischemic heart. J Biol Chem. 2006;281:15110–15120. doi: 10.1074/jbc.M600837200. [DOI] [PubMed] [Google Scholar]
- 130.Poole LB, Claiborne A. The non-flavin redox center of the streptococcal NADH peroxidase. II. Evidence for a stabilized cysteine-sulfenic acid. J Biol Chem. 1989;264:12330–12338. [PubMed] [Google Scholar]
- 131.Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE. 2001;2001:pl1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- 132.Saurin AT, Neubert H, Brennan JP, Eaton P. Widespread sulfenic acid formation in tissues in response to hydrogen peroxide. Proc Natl Acad Sci U S A. 2004;101:17982–17987. doi: 10.1073/pnas.0404762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Eaton P. Protein thiol oxidation in health and disease: techniques for measuring disulfides and related modifications in complex protein mixtures. Free Radic Biol Med. 2006;40:1889–1899. doi: 10.1016/j.freeradbiomed.2005.12.037. [DOI] [PubMed] [Google Scholar]
- 134.Ishii T, Sunami O, Nakajima H, Nishio H, Takeuchi T, Hata F. Critical role of sulfenic acid formation of thiols in the inactivation of glyceraldehyde-3-phosphate dehydrogenase by nitric oxide. Biochem Pharmacol. 1999;58:133–143. doi: 10.1016/s0006-2952(99)00060-x. [DOI] [PubMed] [Google Scholar]
- 135.Persson C, Sjöblom T, Groen A, Kappert K, Engström U, Hellman U, Heldin C-H, den Hertog J, Östman A. Preferential oxidation of the second phosphatase domain of receptor-like PTP-α revealed by an antibody against oxidized protein tyrosine phosphatases. Proc Natl Acad Sci U S A. 2004;101:1886–1891. doi: 10.1073/pnas.0304403101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Karisch R, Neel BG. Methods to monitor classical protein-tyrosine phosphatase oxidation. FEBS J. 2013;280:459–475. doi: 10.1111/j.1742-4658.2012.08626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Birkett DJ, Price NC, Radda GK, Salmon AG. The reactivity of SH groups with a fluorogenic reagent. FEBS Lett. 1970;6:346–348. doi: 10.1016/0014-5793(70)80095-3. [DOI] [PubMed] [Google Scholar]
- 138.Carballal S, Alvarez B, Turell L, Botti H, Freeman BA, Radi R. Sulfenic acid in human serum albumin. Amino Acids. 2007;32:543–551. doi: 10.1007/s00726-006-0430-y. [DOI] [PubMed] [Google Scholar]
- 139.Griffiths SW, King J, Cooney CL. The reactivity and oxidation pathway of cysteine 232 in recombinant human alpha 1-antitrypsin. J Biol Chem. 2002;277:25486–25492. doi: 10.1074/jbc.M203089200. [DOI] [PubMed] [Google Scholar]
- 140.Takanishi CL, Wood MJ. A genetically encoded probe for the identification of proteins that form sulfenic acid in response to H2O2 in Saccharomyces cerevisiae. J Proteome Res. 2011;10:2715–2724. doi: 10.1021/pr1009542. [DOI] [PubMed] [Google Scholar]
- 141.Foss O. Kationoid reactivity of sulphur—sulphenyl compounds. Acta Chem Scand. 1947;1:307–327. [Google Scholar]
- 142.Poole LB, Zeng B-B, Knaggs SA, Yakubu M, King SB. Synthesis of chemical probes to map sulfenic acid modifications on proteins. Bioconjug Chem. 2005;16:1624–1628. doi: 10.1021/bc050257s. [DOI] [PubMed] [Google Scholar]
- 143.Poole LB, Klomsiri C, Knaggs SA, Furdui CM, Nelson KJ, Thomas MJ, Fetrow JS, Daniel LW, King SB. Fluorescent and affinity-based tools to detect cysteine sulfenic acid formation in proteins. Bioconjug Chem. 2007;18:2004–2017. doi: 10.1021/bc700257a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Klomsiri C, Nelson KJ, Bechtold E, Soito L, Johnson LC, Lowther WT, Ryu S-E, King SB, Furdui CM, Poole LB. Chapter 3—use of dimedone-based chemical probes for sulfenic acid detection: evaluation of conditions affecting probe incorporation into redox-sensitive proteins. In: Enrique C, Lester P, editors. Methods Enzymol. Vol. 473. Academic Press; 2010. pp. 77–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Michalek RD, Nelson KJ, Holbrook BC, Yi JS, Stridiron D, Daniel LW, Fetrow JS, King SB, Poole LB, Grayson JM. The requirement of reversible cysteine sulfenic acid formation for T cell activation and function. J Immunol. 2007;179:6456–6467. doi: 10.4049/jimmunol.179.10.6456. [DOI] [PubMed] [Google Scholar]
- 146.Wani R, Qian J, Yin L, Bechtold E, King SB, Poole LB, Paek E, Tsang AW, Furdui CM. Isoform-specific regulation of Akt by PDGF-induced reactive oxygen species. Proc Natl Acad Sci U S A. 2011;108:10550–10555. doi: 10.1073/pnas.1011665108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kaplan N, Urao N, Furuta E, Kim SJ, Razvi M, Nakamura Y, McKinney RD, Poole LB, Fukai T, Ushio-Fukai M. Localized cysteine sulfenic acid formation by vascular endothelial growth factor: role in endothelial cell migration and angiogenesis. Free Radic Res. 2011;45:1124–1135. doi: 10.3109/10715762.2011.602073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Qian J, Klomsiri C, Wright MW, King SB, Tsang AW, Poole LB, Furdui CM. Simple synthesis of 1,3-cyclopentanedione derived probes for labeling sulfenic acid proteins. Chem Commun (Camb) 2011;47:9203–9205. doi: 10.1039/c1cc12127h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Charles RL, Schroder E, May G, Free P, Gaffney PRJ, Wait R, Begum S, Heads RJ, Eaton P. Protein sulfenation as a redox sensor—proteomics studies using a novel biotinylated dimedone analogue. Mol Cell Proteomics. 2007;6:1473–1484. doi: 10.1074/mcp.M700065-MCP200. [DOI] [PubMed] [Google Scholar]
- 150.Seo YH, Carroll KS. Facile synthesis and biological evaluation of a cell-permeable probe to detect redox-regulated proteins. Bioorg Med Chem Lett. 2009;19:356–359. doi: 10.1016/j.bmcl.2008.11.073. [DOI] [PubMed] [Google Scholar]
- 151.Go YM, Jones DP. Redox compartmentalization in eukaryotic cells. Biochim Biophys Acta. 2008;1780:1273–1290. doi: 10.1016/j.bbagen.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lopez-Mirabal HR, Winther JR. Redox characteristics of the eukaryotic cytosol. Biochim Biophys Acta. 2008;1783:629–640. doi: 10.1016/j.bbamcr.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 153.Reddie KG, Seo YH, Muse WB, III, Leonard SE, Carroll KS. A chemical approach for detecting sulfenic acid-modified proteins in living cells. Mol Biosyst. 2008;4:521–531. doi: 10.1039/b719986d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Leonard SE, Reddie KG, Carroll KS. Mining the thiol proteome for sulfenic acid modifications reveals new targets for oxidation in cells. ACS Chem Biol. 2009;4:783–799. doi: 10.1021/cb900105q. [DOI] [PubMed] [Google Scholar]
- 155.Paulsen CE, Carroll KS. Chemical dissection of an essential redox switch in yeast. Chem Biol. 2009;16:217–225. doi: 10.1016/j.chembiol.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 156.Depuydt M, Leonard SE, Vertommen D, Denoncin K, Morsomme P, Wahni K, Messens J, Carroll KS, Collet J-F. A periplasmic reducing system protects single cysteine residues from oxidation. Science. 2009;326:1109–1111. doi: 10.1126/science.1179557. [DOI] [PubMed] [Google Scholar]
- 157.Charron G, Zhang MZM, Yount JS, Wilson J, Raghavan AS, Shamir E, Hang HC. Robust fluorescent detection of protein fatty-acylation with chemical reporters. J Am Chem Soc. 2009;131:4967–4975. doi: 10.1021/ja810122f. [DOI] [PubMed] [Google Scholar]
- 158.Speers AE, Cravatt BF. Profiling enzyme activities in vivo using click chemistry methods. Chem Biol. 2004;11:535–546. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 159.Borisov OV, Goshe MB, Conrads TP, Rakov VS, Veenstra TD, Smith RD. Low-energy collision-induced dissociation fragmentation analysis of cysteinyl-modified peptides. Anal Chem. 2002;74:2284–2292. doi: 10.1021/ac010974p. [DOI] [PubMed] [Google Scholar]
- 160.Fauq AH, Kache R, Khan MA, Vega IE. Synthesis of acid-cleavable light isotope-coded affinity tags (ICAT-L) for potential use in proteomic expression profiling analysis. Bioconjug Chem. 2006;17:248–254. doi: 10.1021/bc0503059. [DOI] [PubMed] [Google Scholar]
- 161.Szychowski J, Mahdavi A, Hodas JJ, Bagert JD, Ngo JT, Landgraf P, Dieterich DC, Schuman EM, Tirrell DA. Cleavable biotin probes for labeling of biomolecules via azide-alkyne cycloaddition. J Am Chem Soc. 2010;132:18351–18360. doi: 10.1021/ja1083909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Yang YY, Grammel M, Raghavan AS, Charron G, Hang HC. Comparative analysis of cleavable azobenzene-based affinity tags for bioorthogonal chemical proteomics. Chem Biol. 2010;17:1212–1222. doi: 10.1016/j.chembiol.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Park KD, Liu R, Kohn H. Useful tools for biomolecule isolation, detection, and identification: acylhydrazone-based cleavable linkers. Chem Biol. 2009;16:763–772. doi: 10.1016/j.chembiol.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 164.Truong TH, Garcia FJ, Seo YH, Carroll KS. Isotope-coded chemical reporter and acid-cleavable affinity reagents for monitoring protein sulfenic acids. Bioorg Med Chem Lett. 2011;21:5015–5020. doi: 10.1016/j.bmcl.2011.04.115. [DOI] [PubMed] [Google Scholar]
- 165.Qian J, Wani R, Klomsiri C, Poole LB, Tsang AW, Furdui CM. A simple and effective strategy for labeling cysteine sulfenic acid in proteins by utilization of [small beta]-ketoesters as cleavable probes. Chem Commun. 2012;48:4091–4093. doi: 10.1039/c2cc17868k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zhu X, Tanaka F, Lerner RA, Barbas CF, III, Wilson IA. Direct observation of an enamine intermediate in amine catalysis. J Am Chem Soc. 2009;131:18206–18207. doi: 10.1021/ja907271a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Seo YH, Carroll KS. Quantification of protein sulfenic acid modifications using isotope-coded dimedone and iododimedone. Angew Chem Int Ed. 2011;50:1342–1345. doi: 10.1002/anie.201007175. [DOI] [PubMed] [Google Scholar]
- 168.Seo YH, Carroll KS. Profiling protein thiol oxidation in tumor cells using sulfenic acid-specific antibodies. Proc Natl Acad Sci U S A. 2009;106:16163–16168. doi: 10.1073/pnas.0903015106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Maller C, Schroder E, Eaton P. Glyceraldehyde 3-phosphate dehydrogenase is unlikely to mediate hydrogen peroxide signaling: studies with a novel anti-dimedone sulfenic acid antibody. Antioxid Redox Signal. 2011;14:49–60. doi: 10.1089/ars.2010.3149. [DOI] [PubMed] [Google Scholar]
- 170.Leonard SE, Garcia FJ, Goodsell DS, Carroll KS. Redox-based probes for protein tyrosine phosphatases. Angew Chem Int Ed Engl. 2011;50:4423–4427. doi: 10.1002/anie.201007871. [DOI] [PubMed] [Google Scholar]
- 171.Lancaster JR. Diffusion of free nitric oxide. Methods Enzymol. 1996;268:31–50. doi: 10.1016/s0076-6879(96)68007-0. [DOI] [PubMed] [Google Scholar]
- 172.Ferrer-Sueta G, Manta B, Botti H, Radi R, Trujillo M, Denicola A. Factors affecting protein thiol reactivity and specificity in peroxide reduction. Chem Res Toxicol. 2011;24:434–450. doi: 10.1021/tx100413v. [DOI] [PubMed] [Google Scholar]
- 173.Roos G, Foloppe N, Messens J. Understanding the pK(a) of redox cysteines: the key role of hydrogen bonding. Antioxid Redox Signal. 2013;18:94–127. doi: 10.1089/ars.2012.4521. [DOI] [PubMed] [Google Scholar]
- 174.Kortemme T, Creighton TE. Ionisation of cysteine residues at the termini of model α-helical peptides. Relevance to unusual thiol pKa values in proteins of the thioredoxin family. J Mol Biol. 1995;253:799–812. doi: 10.1006/jmbi.1995.0592. [DOI] [PubMed] [Google Scholar]
- 175.Hall A, Parsonage D, Poole LB, Karplus PA. Structural evidence that peroxiredoxin catalytic power is based on transition-state stabilization. J Mol Biol. 2010;402:194–209. doi: 10.1016/j.jmb.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Salsbury FR, Jr, Knutson ST, Poole LB, Fetrow JS. Functional site profiling and electrostatic analysis of cysteines modifiable to cysteine sulfenic acid. Protein Sci. 2008;17:299–312. doi: 10.1110/ps.073096508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Turell L, Botti H, Carballal S, Ferrer-Sueta G, Souza JM, Duran R, Freeman BA, Radi R, Alvarez B. Reactivity of sulfenic acid in human serum albumin. Biochemistry. 2008;47:358–367. doi: 10.1021/bi701520y. [DOI] [PubMed] [Google Scholar]
- 178.Miller H, Claiborne A. Peroxide modification of monoalkylated glutathione reductase. Stabilization of an active-site cysteine-sulfenic acid. J Biol Chem. 1991;266:19342–19350. [PubMed] [Google Scholar]
- 179.Strausfeld U, Labbe JC, Fesquet D, Cavadore JC, Picard A, Sadhu K, Russell P, Doree M. Dephosphorylation and activation of a p34cdc2/cyclin B complex in vitro by human CDC25 protein. Nature. 1991;351:242–245. doi: 10.1038/351242a0. [DOI] [PubMed] [Google Scholar]
- 180.Sohn J, Rudolph J. Catalytic and chemical competence of regulation of Cdc25 phosphatase by oxidation/reduction†. Biochemistry. 2003;42:10060–10070. doi: 10.1021/bi0345081. [DOI] [PubMed] [Google Scholar]
- 181.Buhrman G, Parker B, Sohn J, Rudolph J, Mattos C. Structural mechanism of oxidative regulation of the phosphatase Cdc25B via an intramolecular disulfide bond. Biochemistry. 2005;44:5307–5316. doi: 10.1021/bi047449f. [DOI] [PubMed] [Google Scholar]
- 182.Canet-Aviles RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S, Baptista MJ, Ringe D, Petsko GA, Cookson MR. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci U S A. 2004;101:9103–9108. doi: 10.1073/pnas.0402959101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Meulener MC, Xu K, Thomson L, Ischiropoulos H, Bonini NM. Mutational analysis of DJ-1 in Drosophila implicates functional inactivation by oxidative damage and aging. Proc Natl Acad Sci U S A. 2006;103:12517–12522. doi: 10.1073/pnas.0601891103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Witt AC, Lakshminarasimhan M, Remington BC, Hasim S, Pozharski E, Wilson MA. Cysteine pKa depression by a protonated glutamic acid in human DJ-1. Biochemistry. 2008;47:7430–7440. doi: 10.1021/bi800282d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Blackinton J, Lakshminarasimhan M, Thomas KJ, Ahmad R, Greggio E, Raza AS, Cookson MR, Wilson MA. Formation of a stabilized cysteine sulfinic acid is critical for the mitochondrial function of the parkinsonism protein DJ-1. J Biol Chem. 2009;284:6476–6485. doi: 10.1074/jbc.M806599200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 187.Dalle-Donne I, Rossi R, Colombo G, Giustarini D, Milzani A. Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends Biochem Sci. 2009;34:85–96. doi: 10.1016/j.tibs.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 188.Hart GW, Akimoto Y. The O-GlcNAc modification. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Essentials of Glycobiology. Cold Spring Harbor (NY): 2009. [PubMed] [Google Scholar]
- 189.Zhang N, Schuchmann HP, Von Sonntag C. The reaction of superoxide radical anion with dithiothreitol: a chain process. J Phys Chem. 1991;95:4718–4722. [Google Scholar]
- 190.Aruoma OI, Halliwell B, Hoey BM, Butler J. The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, super-oxide, and hypochlorous acid. Free Radic Biol Med. 1989;6:593–597. doi: 10.1016/0891-5849(89)90066-x. [DOI] [PubMed] [Google Scholar]
- 191.Manta B, Hugo M, Ortiz C, Ferrer-Sueta G, Trujillo M, Denicola A. The peroxidase and peroxynitrite reductase activity of human erythrocyte peroxiredoxin 2. Arch Biochem Biophys. 2009;484:146–154. doi: 10.1016/j.abb.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 192.Horta BB, de Oliveira MA, Discola KF, Cussiol JR, Netto LE. Structural and biochemical characterization of peroxiredoxin Qbeta from Xylella fastidiosa: catalytic mechanism and high reactivity. J Biol Chem. 2010;285:16051–16065. doi: 10.1074/jbc.M109.094839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Poole LB. Bacterial defenses against oxidants: mechanistic features of cysteine-based peroxidases and their flavoprotein reductases. Arch Biochem Biophys. 2005;433:240–254. doi: 10.1016/j.abb.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 194.Trujillo M, Clippe A, Manta B, Ferrer-Sueta G, Smeets A, Declercq JP, Knoops B, Radi R. Pre-steady state kinetic characterization of human peroxiredoxin 5: taking advantage of Trp84 fluorescence increase upon oxidation. Arch Biochem Biophys. 2007;467:95–106. doi: 10.1016/j.abb.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 195.Crane EJ, Parsonage D, Claiborne A. The active-site histidine-10 of enterococcal NADH peroxidase is not essential for catalytic activity†. Biochemistry. 1996;35:2380–2387. doi: 10.1021/bi952347y. [DOI] [PubMed] [Google Scholar]
- 196.Lin WS, Armstrong DA, Gaucher GM. Formation and repair of papain sulfenic acid. Can J Biochem. 1975;53:298–307. doi: 10.1139/o75-042. [DOI] [PubMed] [Google Scholar]
- 197.Barrett WC, DeGnore JP, Keng Y-F, Zhang Z-Y, Yim MB, Chock PB. Roles of superoxide radical anion in signal transduction mediated by reversible regulation of protein-tyrosine phosphatase 1B. J Biol Chem. 1999;274:34543–34546. doi: 10.1074/jbc.274.49.34543. [DOI] [PubMed] [Google Scholar]
- 198.Chen CY, Willard D, Rudolph J. Redox regulation of SH2-domain-containing protein tyrosine phosphatases by two backdoor cysteines. Biochemistry. 2009;48:1399–1409. doi: 10.1021/bi801973z. [DOI] [PubMed] [Google Scholar]
- 199.Seth D, Rudolph J. Redox regulation of MAP kinase phosphatase 3. Biochemistry. 2006;45:8476–8487. doi: 10.1021/bi060157p. [DOI] [PubMed] [Google Scholar]
- 200.Caselli A, Marzocchini R, Camici G, Manao G, Moneti G, Pieraccini G, Ramponi G. The inactivation mechanism of low molecular weight phosphotyrosine-protein phosphatase by H2O2. J Biol Chem. 1998;273:32554–32560. doi: 10.1074/jbc.273.49.32554. [DOI] [PubMed] [Google Scholar]
- 201.Barrett WC, DeGnore JP, Konig S, Fales HM, Keng YF, Zhang ZY, Yim MB, Chock PB. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry. 1999;38:6699–6705. doi: 10.1021/bi990240v. [DOI] [PubMed] [Google Scholar]