

Abstract
Arabidopsis thaliana is a model organism commonly used to understand and manipulate various cellular processes in plants, and it has been used extensively in the study of secondary cell wall formation. Secondary cell wall deposition occurs after the primary cell wall is laid down, a process carried out exclusively by specialized cells such as those forming vessel and fiber tissues. Most secondary cell walls are composed of cellulose (40–50%), hemicellulose (25–30%), and lignin (20–30%). Several mutations affecting secondary cell wall biosynthesis have been isolated, and the corresponding mutants may or may not exhibit obvious biochemical composition changes or visual phenotypes since these mutations could be masked by compensatory responses. Staining procedures have historically been used to show differences on a cellular basis. These methods are exclusively visual means of analysis; nevertheless their role in rapid and critical analysis is of great importance. Congo red and calcofluor white are stains used to detect polysaccharides, whereas Mäule and phloroglucinol are commonly used to determine differences in lignin, and toluidine blue O is used to differentially stain polysaccharides and lignin. The seemingly simple techniques of sectioning, staining, and imaging can be a challenge for beginners. Starting with sample preparation using the A. thaliana model, this study details the protocols of a variety of staining methodologies that can be easily implemented for observation of cell and tissue organization in secondary cell walls of plants.
Keywords: Cellular Biology, Issue 87, Xylem, Fibers, Lignin, polysaccharides, Plant cell wall, Mäule staining, Phloroglucinol, Congo red, Toluidine blue O, Calcofluor white, Cell wall staining methods
Introduction
The plant cell wall holds a plethora of information within its various components: lignin, cellulose, hemicelluloses (xylan, glucuronoxylan, xyloglucan, arabinoxylan, mixed linkage glucan, or glucomannan), and pectin. Histological techniques provide important visual cues in studying differences within the secondary cell walls at organizational and cellular levels. Various histological techniques were developed and can be found in the literature, but these techniques can be challenging and time-consuming for beginners because very detailed protocols with simple visual instructions are seldom if ever available. The goal of this study is to provide simple guidelines for histological staining techniques to obtain high-quality images.
Sectioning the stem tissue is the first step in visualization of cell walls and cell shapes. Although hand-cut sections are inexpensive and take less time to prepare, use of a vibratome offers consistency and yields high-quality images. Using a vibratome allows producing better data quality by generating even sections with the same thickness, which helps to produce sharp images and reduces significantly the risk of producing inaccurate differences between samples that would be simply caused by a bad sample preparation. Using resins to fix fresh specimens can be a challenge for beginners and may still be time-consuming even for experts when an analysis needs to be done quickly. In addition, it becomes impossible to measure any biological activity with the sample when it has been embedded in a resin. One simple technique that employs agarose and a homemade mold is helpful for embedding stem tissues, and it can also be utilized in other applications requiring dissection of soft tissues. Compared to embedding specimens in resin, this method has the advantage of keeping the tissues alive and reducing sample manipulation. Sectioning tissues via a vibratome is highly precise and generates homogenous sections, which, depending on the goal of the study, can then be used with several different staining techniques.
The simplest methods to visualize lignin and other aromatics employ ultraviolet (UV) light. Excitation of aromatic-based molecules by UV light is an old technique, but it is still one of the quickest approaches for visualization of lignin. However, UV visualization actually is not ideal for lignin detection because UV light will excite other aromatics. Lignin is made up primarily of three building blocks, the monolignols (hydroxycinnamyl alcohols: coniferyl alcohol, sinapyl alcohol, and p-coumaryl alcohol)1-3. Phloroglucinol stain can provide clues to the extent of cinnamaldehydes present in the xylem, fiber, and tracheal tissues4. Phloroglucinol is a good indicator of general cinnamaldehydes and can differentiate between cinnamaldehydes and other aromatics. The sinapyl alcohol monomers can be detected and differentiated by the use of Mäule stain. Toluidine blue O is a polychromatic dye and therefore has the ability to stain different elements of the cell wall in different colors5,6. The primary use of toluidine blue O is to detect pectin and lignin5,6. The advantage of using toluidine blue O is that many elements of the cell wall can be visualized in a single step. Both calcofluor white and congo red are easy to work with and can be used to visualize cellulose. Calcofluor white stains cellulose, callose, and other non-substituted or weakly substituted β-glucans6-9, whereas congo red stains directly to β-(1→4)-glucans and particularly to cellulose10,11. The goal of this study is to provide simple guidelines for the use of the aforementioned staining techniques to obtain high-quality images from A. thaliana stems.
Protocol
1. Stem Embedding
Make a 7% agarose solution in water (7 g of electrophoresis-grade agarose in 100 ml distilled water). Dissolve the agarose by autoclaving for 20 min or by microwaving for 20 min at lowest intensity (e.g., 10% intensity of a 1,250 watt microwave).
- Prepare a homemade mold for embedding the stems using plastic vials.
- Using a razor blade, cut off the conical bottom of a 2 ml screw-cap microcentrifuge tube (Part A). With a syringe needle, puncture a hole in the tube cap slightly larger than the diameter of the stem to be embedded. Next, cut 0.5 cm off the bottom of a 0.6 ml microcentrifuge tube (Part B).
- Add the cut-off 0.5 cm portion of the 0.6 ml microcentrifuge tube into the precut 2 ml microcentrifuge tube. Seal the two parts using Parafilm (Figure 1). NOTE: The bottom of the tube is cut to facilitate the removal of the embedded sample after the agarose has solidified. The hole in the cap will aid in holding the stem steady when the stem is embedded in agarose. The mold will serve to hold the agarose and hold the stem straight when embedding. The Parafilm will hold the two parts, A and B, until the agarose is set and the stem is embedded.
- Using a razor blade, cut a stem section from the area of interest, (e.g. middle of the first stem internode). NOTE: Ideally, the stem should be cut just before embedding to avoid destruction by dehydration. Alternatively, the stem can be temporarily stored in a plate containing a filter paper wetted with water. This plate can be placed on ice until ready to be embedded to prevent tissue dehydration and deformation. Storing the stem for periods longer than 30 min without high moisture will cause the stem to dry out.
Melt the agarose in the microwave at low intensity. CAUTION: The bottle containing agarose may be hot. Use a mitt to pick up the bottle. Let the melted agarose stand at room temperature (between 20 °C and 25 °C) until it cools to about 50 °C.
- Slowly pipette 5 ml of the agarose into the premade plastic mold to fill the vial. NOTE: Make sure there are no air bubbles in the molds. Air bubbles will cause gaps in the agarose mold and will not completely cover the stem sample, which will eventually lead to uneven cutting of the sample sections.
- Let the agarose cool down for 30 to 60 sec to become semisolid. Test with a small toothpick, making sure that it can penetrate and stand up, but not float, in the agarose. NOTE: If the stem is placed when the agarose is still hot it will damage the sample by shriveling the stem, destroying the cell morphology.
- Place the stem in this agarose-filled vial. Make sure that the stem stays straight. Place the perforated cap in such a way that the tip of the stem is held by the cap. Do not turn the screw-on cap. Sometime more than one stem (of a single kind) can be embedded in a single vial; but in that particular case, do not use the cap. NOTE: If the screw-on cap is turned, the stem will get twisted.
Leave the vial at room temperature or at 4 °C for 10-30 min, until it solidifies.
Remove the mold by gently sliding it out of the tube. Open the Parafilm. Push the bottom of Part B of the mold gently with thumb to release the solidified agarose from Part A onto a glass microscope slide.
- Cut the agarose embedded stem into three approximately equal pieces of 1.2 cm in length. Cut out the part of the stem that was not in the agarose and discard it.
- Store these pieces of agarose with the embedded stems in airtight 2 ml microcentrifuge tubes in a dark box at 4 °C until ready to section. Alternatively, store the tubes at 4 °C for a maximum of one week. NOTE: Storage is possible, but the experimenter needs to be aware that the embedded stems are still alive and, depending of the experiment, artifacts could potentially be introduced.
2. Stem Sectioning
CAUTION: Read the vibratome manual carefully and follow all safety instructions.
- Make sure the specimen disc is clean and completely dry (free of any solid or liquid). Clean the specimen disc with a razor blade to remove any residual glue from prior experiments and wash with water. Then dry with a paper towel. NOTE: This will ensure that the adhesive works well and binds to the agarose specimen. If this step is not done properly, it is possible that the specimen may not adhere to the disc and will later cause the specimen to fall off the disc during sectioning.
- Use soft paper wipes to remove any moisture from the agarose block containing the specimens.
- Place a small drop of tissue adhesive on the specimen disc. Streak out the adhesive to cover the area in the middle of the plate using the tip of the adhesive bottle. Quickly place the agarose block so that the specimens are either perpendicular to or in parallel with the plate for transversal or longitudinal cross-sections, respectively. Allow the adhesive to fix the sample to the specimen disc at room temperature or at 4 °C for 10-30 min. NOTE: This is a time-sensitive step.
Fix the specimen disc in place on the buffer tray or trough. The block/s of agarose with the sections should be in parallel with the razor blade. Fill the buffer tray with distilled water at room temperature until the samples are completely submerged.
- Cut a razor blade in half and then trim the ends with a sturdy scissors so that the blade stays completely flat. Attach one-half of the precut razor blade onto the knife holder.
- Set the angle on the knife holder to 84°, the speed to 0.90 mm/sec (position 8 on the vibratome) and frequency to 50 Hz (position 5 on the vibratome). Cut sections of 100 µm thickness. Use continuous mode. NOTE: This thickness was selected to assure that the tissue can withstand the various acid and base treatments used in some of the staining protocols. Set the window for sectioning and allow 15-20 min on a continuous mode for the vibratome to section an agarose block of approximately 1.2 cm.
Collect the sections using a disposable plastic pipette during sectioning in the buffer tray. Alternatively, the sections can be transferred from the buffer tray into a glass beaker and then to a clear Petri plate. Transfer few sections to the 2.0 ml microcentrifuge tubes. Note: The specimens are collected from a Petri plate because it is easier to see the sections on a clear background, and because the buffer tray can be primed for the next specimen.
The sections can be stored in a 50 ml tube or collected and stored in 1.5 to 2 ml microcentrifuge tubes at 4 °C for 30 to 60 min. NOTE: Storing the sections for longer periods will result in poor image quality.
3. Microscope and Camera Setting for Imaging
- Adjust the Koehler illumination on the microscope. Choose an objective. Focus on the specimen first. Bring the light intensity to one-half or lower. Close the field diaphragm (FD) and aperture (AP) or bring it to the lowest possible setting on that particular objective.
- The higher the magnification, the larger the ring will be. Use the condenser focusing knob to focus the field iris as sharply as possible.
- Slowly bring the image of the field iris (hexagon-shaped small ring) to the center using the condenser-centering knobs (pin-shaped screws surrounding the condenser). Slowly increase the diaphragm (FD) such that the field iris reaches the edge. Stop increasing the FD just after the field iris has passed the edge.
- Slowly increase the aperture (AP) to adjust contrast. Adjust the light intensity as required. NOTE: Koehler illumination needs to be adjusted for every objective, every experimental day. Note the numbers obtained after adjustment for the day. These numbers will be reused each time that objectives are switched back and forth for that particular day.
Adjust the aperture, intensity, exposure time, gain, and filters as described in Table 1. This information should be used only as a guide.
Use a high-resolution digital camera with a large format interline CCD chip with no mechanical shutter to capture fluorescence images; and a high-resolution, high-speed camera for bright-field images.
Process the slides using standard software. Load the software. Click “Acquire” and “Set camera/board,” then select the camera to be used. Click “OK.” Click “Acquire” followed by “Color tab.” Under “Color tab,” choose “Bayer method” followed by “Average four.” Add the suggested gain (red, green, blue) from Table 1. Select the “Set save” tab and click on the directory where images are to be stored. Set “Exposure time” from the suggestions in Table 1. Set “Binning” at “1” and “Live binning” at “1.” Focus on the region of interest and then click on “Save live.”
Output images on the camera software are in the “TIF” format. Analyze the images for histochemistry of the cell types. Use standard computer software for downstream uses of presentation and publication.
4. Ultraviolet and Bright-field Imaging
Use a 1 ml pipette with the pipette tip cut so that the sections can be pipetted out easily. Gently pipette sections into the tip and on to a microscope slide. Cover the sections with a cover slip. Observe the sections under UV light or bright-field lighting.
5. Phloroglucinol-HCl (Wiesner) Staining12
Dissolve 0.3 g of phloroglucinol in 10 ml absolute ethanol to prepare a 3% phloroglucinol solution. Mix one volume of concentrated HCl (37 N) to two volumes of 3% phloroglucinol in ethanol; this solution is phloroglucinol-HCl (Ph-HCl) or Wiesner stain. NOTE: This solution is to be made fresh on the day of the staining and cannot be stored, because the solution will degrade over time and the staining will become ineffective. CAUTION: HCl is highly corrosive, so use extreme caution while handling the Ph-HCl stain.
- Transfer stem sections to a 2.0 ml microcentrifuge tube. Add 1 ml of the Ph-HCl solution to the tube containing the sections and cap it immediately because the HCl in the Ph-HCl stain is highly corrosive. Gently move the tube to assure that all the sections are stained. An alternative to this step is to keep the tube cap open so that the Ph-HCl solution can be pipetted up and down, preferably without disturbing the sections. NOTE: Be careful not to pipette the sections into the pipette tip, as this may damage those sections.
- Use a 1 ml pipette with the pipette tip cut in such a way that the sections can be pipetted out easily without damage. Gently pipette sections into the tip and onto a microscope slide. Cover the sections with cover slip. Observe the sections under bright-field lighting. NOTE: The Ph-HCl solution dries up in 5-10 min, causing a deterioration of the specimens. Therefore, the imaging has to be completed within that period of time.
6. Mäule Staining13,14
- Dissolve 0.2 g of potassium permanganate in 40 ml distilled water to prepare a 0.5% potassium permanganate solution. Store in a dark bottle at room temperature for a maximum of 7 days.
- Prepare a 3.7% HCl solution by adding 1 ml concentrated HCl (37 N) to 9 ml distilled water (1:10). Prepare a fresh 3.7% HCl solution on the day of the experiment. Make sure that the 37 N HCl stock solution being used is not too old. Concentrated ammonium hydroxide solution (14.8 M) should be stored at 4 °C to prevent the molarity of the saturated solution from decreasing.
- Transfer stem sections to a 2.0 ml microcentrifuge tube. Add 1 ml of the 0.5% potassium permanganate solution to the tube containing the sections.
- Pipette the 0.5% potassium permanganate solution up and down gently, preferably without disturbing the sections. Incubate for 2 min and repeat the pipetting. Let the microcentrifuge tube stand until all the sections settle down. NOTE: Be careful not to pipette the sections into the pipette tip as the sections may get damaged. It may be hard to see the stem sections as the solution is dark, so keep the tube with the sections on test tube rack and allow them to settle for 2 min.
- Using a 1 ml pipette, draw out 700 µl of 0.5% potassium permanganate solution. Add 700 µl of distilled water to rinse out the potassium permanganate solution. Repeat 3-4x or until the water solution stays clear.
- Discard the water. Quickly add 1 ml of 3% HCl until the deep brown color is discharged from the sections. This may happen within 3-5 min or may require 2 washes of 5 min each. Pipette out all the 3% HCl solution and immediately proceed to the next step.
- Add 1 ml of concentrated ammonium hydroxide solution. Use a 1 ml pipette with the tip cut in such a way that the sections can be pipetted out easily without damage. Gently pipette sections into the tip and onto a microscope slide. Cover the sections with a coverslip. Observe the sections under bright-field lighting. CAUTION: Because ammonium hydroxide is extremely corrosive, it requires extra care and attention to avoid causing corrosion to the stage of the microscope or lens. NOTE: The ammonium hydroxide fumes can fog the coverslip, making it difficult to observe the sample. Therefore be extra careful before adding the coverslip. The solution dries up in 5-10 min, so the imaging has to be completed within that period of time.
7. Congo Red Staining11,15
Dissolve 0.5 g congo red in 100 ml of distilled water to prepare a 0.5% congo red solution.
- Transfer stem sections to a 2.0 ml microcentrifuge tube. Add 1 ml of the 0.5% congo red solution to the tube. Gently pipette the 0.5% congo red solution up and down without disturbing the sections. NOTE: Be careful not to pipette the sections into the pipette tip as this may damage the sections.
- Incubate at room temperature for 10 min and repeat the pipetting. Follow with another 3 min of incubation at room temperature.
- Use a 1 ml pipette to draw out 700 µl of 0.5% congo red solution. Add 700 µl of distilled water to rinse out the congo red solution. Repeat the wash 3-4x until the washing solution is clear.
- Use a 1 ml pipette with its tip cut in such a way that the sections can be pipetted out easily without damage. Gently pipette sections into the tip and onto a microscope slide, and cover them with coverslip. Observe the samples on the microscope slide under blue-light excitation using a filter with a bandpass of 560/40. NOTE: Do not store the sections in water for long prior to the microscopic analysis, as the water will wash out the congo red in 10-30 min.
8. Calcofluor White Staining9
Dissolve 0.02 g of fluorescent brightener 28 (calcofluor white M2R) in 10 ml distilled water to prepare a 0.2% stock solution. The 0.2% solution can be stored for six months in a dark bottle at 4 °C. Prepare a working solution of 0.02% by diluting the 0.2% stock solution 10x with distilled water.
- Transfer stem sections to a 2.0 ml microcentrifuge tube. Add to the tube 1 ml of the 0.02% calcofluor white solution. Gently pipette the 0.02% calcofluor white solution up and down. NOTE: Be careful not to pipette the sections into the pipette tip as this may damage the sections.
- Incubate at room temperature for 5 min and repeat the pipetting. Incubate the section for another 3 min at room temperature. NOTE: Allow the sections to settle onto the bottom of the tube to make it easier to pipette out the solution without damaging the sections.
- Use a 1 ml pipette to draw out 700 µl of 0.02% calcofluor white solution and add 700 µl of distilled water; wait for 5 min to rinse out the calcofluor white solution. Repeat the wash 3-4x. Use a 1 ml pipette with the pipette tip cut in such as way that the sections can be pipetted out easily without damage.
- Gently pipette sections into the tip and onto a microscope slide and cover them with a coverslip. Observe the sections under UV light.
9. Toluidine Blue O Staining16,17
Dissolve 0.02 g toluidine blue O in 100 ml distilled water to prepare a 0.02% solution. NOTE: The toluidine blue O solution can be stored for two weeks in a dark bottle at room temperature.
- Transfer stem sections to a 2.0 ml microcentrifuge tube. Add 1 ml of the 0.02% toluidine blue O solution to the tube. Gently pipette the 0.02% toluidine blue O solution up and down. Then incubate at room temperature for 5 min and repeat the pipetting once. NOTE: Be careful not to pipette the sections into the pipette tip as this may damage the sections. Let the microcentrifuge tube stand still until the sections settle down. This may be hard to see, as the solution is dark, so keep the tube containing the sections on test tube rack and allow them to settle for 2 min at room temperature.
- Use a 1 ml pipette to draw out 700 µl of 0.02% toluidine blue O solution. Add 700 µl of distilled water to rinse out the toluidine blue O solution. Repeat 3-4x or until the wash solution is clear. Use a 1 ml pipette with the pipette tip cut in such a way that the sections can be pipetted out easily without damaging them.
- Gently pipette sections into the tip and onto a microscope slide and cover them with a coverslip. Observe the sections under bright-field lighting.
Representative Results
Stem Embedding and Sectioning: The use of the homemade plastic mold to embed the stems in 7% agarose proved to be fast and easy (Figure 1). The two parts (A and B; Figure 1) of the embedded vial system make it simple to easily release the stem embedded in agarose as the agarose does not stick to the vial parts that are inert, keeping the system clean. The vials can be reused multiple times for years. The convenience of storing the embedded stem also makes the next steps easier. In order to save sometime, similar embedded stems can be grouped (up to 3) on a single sectioning plate for sectioning.
Observation of Aromatics Under UV: The stem sections were mounted in distilled water on to the glass slides and observed under UV light. The aromatic compounds, including the lignin in the cells, can be visualized by their autofluorescence under UV light. Xylem (Xy) and interfascicular fibers (Fi) showed autofluorescence under UV illumination but not in pith (Pi) and cortex or epidermis (Ep) (Figure 2), because lignin, an aromatic polymer, is deposited during secondary cell-wall biosynthesis. In some instances, when plants accumulate significant amounts of aromatics in their vacuoles (e.g., anthocyanin), fluorescence can be observed in cortical cells (data not shown). Stem cross-sections were analyzed under 5X, 10X, 20X, and 40X magnification (Figure 2 Panels A, B, C, and D-E, respectively) to better visualize cell wall aromatic distribution across the stem.
Observation of Lignin in the Stem Sections Stained with Phloroglucinol and Mäule Stains: The xylem composed of vessels and fibers and interfascicular fibers were the only elements of the stem that were stained by the lignin stains (phloroglucinol and Mäule) using a wild type stem section sample. Phloroglucinol stain reacts with cinnamaldehyde end-groups of lignin to give a pink or fuchsia color4 (Figure 3). As observed under UV illumination, a fuchsia coloration is observed in xylem and interfascicular fibers but is absent in pith and cortex or epidermis (Figure 3). Stem cross-sections were analyzed under 5X, 10X, 20X, and 40X magnification (Figure 3 Panels A, B, C, and D-E, respectively) to better visualize phloroglucinol stain distribution across the stem and differentiate the major tissues; xylem and interfascicular fibers; pith and cortex; and epidermis. Usually the intensity of the color correlates with the level of lignification in a qualitative manner — except when the analyzed mutant or transgenic is abnormally enriched in cinnamaldehyde-derived units (e.g. cad mutant)14. The Mäule stain is specific in detecting the syringyl lignin units in xylem and interfascicular fibers. Red coloration indicates the presence of syringyl lignin units in the lignin elements (Figure 4)18. However, a brighter red coloration is observed in the fibers when compared to the xylem tissues which suggests that the fibers contain a higher level of S lignin whereas the xylem is more enriched in G units. As observed after phloroglucinol staining, a red coloration is observed in xylem and interfascicular fibers but is absent in pith and cortex or epidermis (Figure 4). It also correlates with the lignin distribution observed under UV (Figure 2) and shows that syringyl units are present in every lignified tissue of this stem sample. Stem cross-sections were analyzed under 5X, 10X, 20X, and 40X magnification (Figure 2 Panels A, B, C, and D-E, respectively) to better visualize Mäule stain distribution across the stem and differentiate the major tissues: xylem and interfascicular fibers, pith and cortex, and epidermis. It is important to note that the amount and distribution of syringyl units between lignified tissues vary with the age of the stem and can be absent in a young stem (data not shown).
Observation of Stem Sections Stained with Calcofluor White and Congo Red Stains: Calcofluor white stains cellulose, callose, and other nonsubstituted or weakly substituted β-glucans; whereas congo red stains directly to β-(1→4)-glucans and particularly to cellulose. Stem sections stained with calcofluor white were observed under UV light6-9. Congo red-stained sections were observed under blue-light excitation using a filter with a bandpass of 560/40. Both calcofluor white and congo red stained the epidermis, cortex, and pith; this is because all of these tissues contain cellulose as major polysaccharide polymer in their cell walls (Figure 5 and Figure 6, respectively). In contrast to calcofluor white, congo red stained polysaccharides better in the xylem and interfascicular fibers and seems less affected by the presence of lignin (Figure 5 and Figure 6)10,11. Stem cross-sections were analyzed under 5X, 10X, 20X, and 40X magnification (Panels A, B, C, and D-E, respectively of Figures 5 and 6) to better visualize calcofluor white and congo red stain distributions across the stem and the major tissues (xylem and interfascicular fibers, pith and cortex, and epidermis).
Observation of Stem Sections Stained with Toluidine Blue O: Toluidine blue O is classified as a polychromatic dye because it reacts with different chemical components of cells differently and results in a multi-colored specimen (Figure 7). The colors generated can provide information on the nature of the cell and its walls. Toluidine blue O is a cationic dye that binds to negatively charged groups5. An aqueous solution of this dye is blue, but different colors are generated when the dye binds with different anionic groups in the cell6,9. For example, a pinkish purple color will appear when the dye reacts with carboxylated polysaccharides such as pectic acid; green, greenish blue, or bright blue with poly-aromatic substances such as lignin and tannins; and purplish or greenish blue with nucleic acids. Toluidine blue O of stem cross-sections revealed that xylem and interfascicular fibers are lignified since they show a greenish blue or blue coloration (Figure 7, Panel C), which is in agreement with the UV and Ph-HCl staining observations (Figures 2 and 3). In contrast, the pith and cortex and epidermis tissues show greenish blue or blue coloration because, although they are not lignified, they contain some pectin polymers in their cell walls. Stem cross-sections were analyzed under 5X, 10X, 20X, and 40X magnification (Figure 7 Panels A, B, C, and D-E, respectively) to better visualize Toluidine blue O stain distribution across the stem and to differentiate the major tissues.
 Figure 1. Homemade mold for embedding the stems. On the left side; top of the 2 ml screw-cap microcentrifuge tube (Part A) and the bottom from the 0.6 ml microcentrifuge tube (Part B). On the right side; the Parafilm holding the two parts; A and B to form the final mold.
Figure 1. Homemade mold for embedding the stems. On the left side; top of the 2 ml screw-cap microcentrifuge tube (Part A) and the bottom from the 0.6 ml microcentrifuge tube (Part B). On the right side; the Parafilm holding the two parts; A and B to form the final mold.
 Figure 2. UV Fluorescence of A. thaliana stem cross-sections. Transverse sections of a wild type A. thaliana stem under UV light fluorescence showing the presence of aromatic compounds, including lignin, only in the walls of interfascicular fibers and xylem cells (A-E). The positions of the epidermis and cortex (Ep), interfascicular fibers (Fi), pith (Pi), and xylem (Xy) are indicated. Magnifications: Panel A: 5X; Panel B: 10X; Panel C: 20X; Panels D
and
E: 40X. Bar = 100 µm.
Figure 2. UV Fluorescence of A. thaliana stem cross-sections. Transverse sections of a wild type A. thaliana stem under UV light fluorescence showing the presence of aromatic compounds, including lignin, only in the walls of interfascicular fibers and xylem cells (A-E). The positions of the epidermis and cortex (Ep), interfascicular fibers (Fi), pith (Pi), and xylem (Xy) are indicated. Magnifications: Panel A: 5X; Panel B: 10X; Panel C: 20X; Panels D
and
E: 40X. Bar = 100 µm.
 Figure 3. Phloroglucinol-HCl staining of A. thaliana stem cross-sections. Phloroglucinol-HCl staining (pink or fuchsia color) of a wild type A. thaliana stem section showing the normal lignin deposition in the walls of interfascicular fibers and xylem cells (A-E). The positions of the epidermis and cortex (Ep), interfascicular fibers (Fi), pith (Pi), and xylem (Xy) are indicated. Magnifications: Panel A: 5X; Panel B: 10X; Panel C: 20X; Panels D
and
E: 40X. Bar = 100 µm.
Figure 3. Phloroglucinol-HCl staining of A. thaliana stem cross-sections. Phloroglucinol-HCl staining (pink or fuchsia color) of a wild type A. thaliana stem section showing the normal lignin deposition in the walls of interfascicular fibers and xylem cells (A-E). The positions of the epidermis and cortex (Ep), interfascicular fibers (Fi), pith (Pi), and xylem (Xy) are indicated. Magnifications: Panel A: 5X; Panel B: 10X; Panel C: 20X; Panels D
and
E: 40X. Bar = 100 µm.
 Figure 4. Mäule staining of A. thaliana stem cross-sections. Mäule staining (red color) of a wild type A. thaliana stem section showing the normal lignin deposition in the walls of interfascicular fibers and xylem cells (A-E). The positions of the epidermis and cortex (Ep), interfascicular fibers (Fi), pith (Pi), and xylem (Xy) are indicated. Magnifications: Panel A: 5X; Panel B: 10X; Panel C: 20X; Panels D
and
E: 40X. Bar = 100 µm.
Figure 4. Mäule staining of A. thaliana stem cross-sections. Mäule staining (red color) of a wild type A. thaliana stem section showing the normal lignin deposition in the walls of interfascicular fibers and xylem cells (A-E). The positions of the epidermis and cortex (Ep), interfascicular fibers (Fi), pith (Pi), and xylem (Xy) are indicated. Magnifications: Panel A: 5X; Panel B: 10X; Panel C: 20X; Panels D
and
E: 40X. Bar = 100 µm.
 Figure 5. Calcofluor white staining of A. thaliana stem cross-sections. Calcofluor white staining of a wild type A. thaliana stem section showing cellulose deposition in the cell walls of cells of the epidermis, cortex, pith, interfascicular fibers, and xylem (A-E). The positions of the epidermis and cortex (Ep), interfascicular fibers (Fi), pith (Pi), and xylem (Xy) are indicated. Magnifications: Panel A: 5X; Panel B: 10X; Panel C: 20X; Panels D
and
E: 40X. Bar = 100 µm.
Figure 5. Calcofluor white staining of A. thaliana stem cross-sections. Calcofluor white staining of a wild type A. thaliana stem section showing cellulose deposition in the cell walls of cells of the epidermis, cortex, pith, interfascicular fibers, and xylem (A-E). The positions of the epidermis and cortex (Ep), interfascicular fibers (Fi), pith (Pi), and xylem (Xy) are indicated. Magnifications: Panel A: 5X; Panel B: 10X; Panel C: 20X; Panels D
and
E: 40X. Bar = 100 µm.
 Figure 6. Congo red staining of A. thaliana stem cross-sections. Congo red staining of a wild type A. thaliana stem section showing cellulose deposition in the cell walls of cells of the epidermis, cortex, pith, interfascicular fibers, and xylem (A-E). The positions of the epidermis and cortex (Ep), interfascicular fibers (Fi), pith (Pi), and xylem (Xy) are indicated. Magnifications: Panel A: 5X; Panel B: 10X; Panel C: 20X; Panels D
and
E: 40X. Bar = 100 µm.
Figure 6. Congo red staining of A. thaliana stem cross-sections. Congo red staining of a wild type A. thaliana stem section showing cellulose deposition in the cell walls of cells of the epidermis, cortex, pith, interfascicular fibers, and xylem (A-E). The positions of the epidermis and cortex (Ep), interfascicular fibers (Fi), pith (Pi), and xylem (Xy) are indicated. Magnifications: Panel A: 5X; Panel B: 10X; Panel C: 20X; Panels D
and
E: 40X. Bar = 100 µm.
 Figure 7. Toluidine blue O staining of A. thaliana stem cross-sections. Toluidine blue O staining of a wild type A. thaliana stem section showing the normal lignin deposition in the walls of interfascicular fibers and xylem cells (A-E). The positions of the epidermis and cortex (Ep), interfascicular fibers (Fi), pith (Pi), and xylem (Xy) are indicated. Magnifications: Panel A: 5X; Panel B: 10X; Panel C: 20X; Panels D and E: 40X. Bar = 100 µm.
Figure 7. Toluidine blue O staining of A. thaliana stem cross-sections. Toluidine blue O staining of a wild type A. thaliana stem section showing the normal lignin deposition in the walls of interfascicular fibers and xylem cells (A-E). The positions of the epidermis and cortex (Ep), interfascicular fibers (Fi), pith (Pi), and xylem (Xy) are indicated. Magnifications: Panel A: 5X; Panel B: 10X; Panel C: 20X; Panels D and E: 40X. Bar = 100 µm.
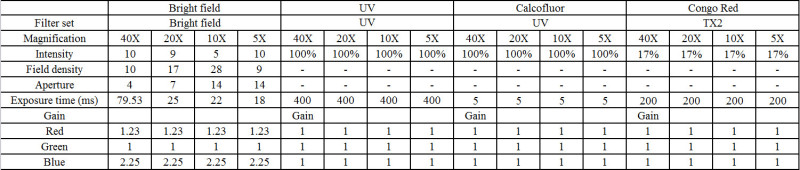 Table 1. Microscope and camera settings for imaging. Guidelines for working with fluorescence filters and bright-field imaging. Please click here to view a larger version of this figure.
Table 1. Microscope and camera settings for imaging. Guidelines for working with fluorescence filters and bright-field imaging. Please click here to view a larger version of this figure.
Discussion
A. thaliana stem sections are widely used to study the organization of the cells in the secondary cell wall and to qualitatively examine the differences between wild type and transgenic plants. The commonly used techniques for sectioning the specimens are direct hand cutting; or when specimens are embedded in agarose or fixative, sectioning can be done with a vibratome or microtome. In contrast to hand cutting, the last two allow reducing the risk of producing inaccurate differences between samples that would be simply caused by a bad sample preparation caused by thickness differences between samples and uneven surfaces. All of these methods have their pros and cons. We chose embedding in agarose, followed by sectioning using a vibratome, and did so for the following reasons: The ease of embedding and time spent in using agarose eliminates the need for elaborate processing of fresh tissue. Also, our homemade molds are easy and cheap to make, can hold the stem straight, can be used over many years, and can help in easy release of specimens without making a mess. The best way to dissolve high-percentage agarose and get a homogenous solution (full dissolution of 7 g of agarose in 100 ml without forming chunky granules) is to autoclave it or to microwave it at the lowest intensity. It is important to cut the stem tissue fresh, or alternatively to store it in a plate placed on ice containing a filter paper wetted with water to prevent tissue dehydration and deformation. Storing the stem for periods longer than 30 min without high moisture will cause the stem to dry out. The temperature of the agarose right before the stem is embedded should be around 50 °C. It is critical to check for the presence of air bubbles after the agarose has been poured into the vial, because air bubbles cause gaps in the mold and consequently the sectioning will be uneven. Air bubbles can be removed quickly by placing the solution under vacuum prior pouring it. The stem should be placed in the agarose when it is still soft and not warmer than 50 °C. The heat will damage the sample by shriveling the stem and will destroy the cell morphology. If the agarose is too cold the stem will not penetrate through the agarose and embedding will not be possible. After agarose embedding, stem samples can be stored at 4 °C up to one week.
Use of a vibratome instead of hand-sectioning yields precisely cut specimens and better image quality. All safety instructions should be carefully read and followed while using a vibratome. It is important to ensure that the specimen disc is clean and dry to allow the tissue adhesive to hold the specimen strongly in place so it does not fall off during sectioning. The thickness of the specimens is specified to withstand the harsh acid and base treatments during staining procedures. The razor blade used in the vibratome should be completely flat to ensure that the section is cut evenly. Sectioning takes approximately 20 min per sample; in order to save time, similar specimens can be sectioned in groups on a single specimen plate. During sectioning, the sections are transferred from the buffer tray into a small Petri dish to make the sections easier to see on a clear background and to liberate the buffer tray for the next specimen. Sections can be immediately used for downstream staining procedures or stored for later use. Aliquoting the stem sections to 2.0 ml microcentrifuge tubes helps in the priming for staining procedures. Making correct adjustments and settings on the camera and microscope are very critical for achieving high quality images. Adjusting the Kohler illumination is one of the most important steps in the use of the microscope. The adjustments for aperture and light intensity, as described in the methods section, are meant only as a guideline. They may need to be modified depending on the microscope and camera utilized. We used a high-resolution digital camera with a large format interline CCD chip and no mechanical shutter for UV imaging, calcofluor white, and congo red staining; and a high-resolution and high-speed color camera for bright-field images of Mäule, phloroglucinol and toluidine blue O staining. Images were analyzed, processed and assembled using standard imaging software.
Phloroglucinol is the most commonly used stain for lignin determination; it is not a true lignin stain as it stains only cinnamaldehyde end-groups. The phloroglucinol stain, also known as Weisner stain, yields a characteristic cherry pink or fuchsia color in the xylem and interfascicular fibers where these aldehyde groups are present4. The intensity of the pink color varies with the extent of lignification. Although subtle differences are harder to observe, it is easy to spot differences between wild type and mutant stem sections that have variations in their lignin content19. It is also easy to observe differences between younger (towards the stem apex) and older (stem base) tissues, because lignification levels between such tissues vary. Usually in wild type stem, epidermis cortex and pith do not contain cinnamaldehydes (thus do not get colored) unless there is ectopic lignification20. Mäule stain, on the other hand, is specific toward syringyl lignin units and can be used to differentiate different lignin enrichments in S or G units (e.g. fiber versus xylem tissues)14,18. The potassium permanganate and hydrochloric acid act on the syringyl nucleus of the S unit in the lignin to form a methoxycatechol and then to methoxy-o-quinone following reaction with ammonium hydroxide. The Mäule stain is a good indicator for differentiating mutants that lack or are extensively depleted in S units of lignin (e.g. in A. thaliana f5h mutants and general gymnosperm plants), Mäule gives yellow-brown coloration instead of red in both the xylem and the sclerenchyma21. For the same reason, Mäule staining can be used to differentiate angiosperms, which primarily contain S lignin from gymnosperms that typically lack the S unit18. Because strong acid and base are used during phloroglucinol and Mäule staining procedures respectively, some experimental steps need to be performed with caution. Both concentrated HCl and ammonium hydroxide are strong corrosives; they can deteriorate the microscope stage and lens if the slides containing the Ph-HCl and Mäule stained samples are not handled properly. They can also dry up quickly, within 5-10 min, causing specimens to deteriorate, so the imaging has to be done rapidly. Toluidine blue O is a polychromatic stain and stains different components of the cell wall in different colors as it reacts with different chemical components of cell walls differently, producing a multi-colored specimen. The colors generated can provide information on the nature of the cell. Toluidine blue O is also a cationic dye that binds to negatively charged groups5,6. An aqueous solution of this dye is blue, but different colors are generated when the dye binds with different anionic groups in the cell. For example, the colors are a pinkish purple when the dye reacts with carboxylated polysaccharides such as pectin; green, greenish blue, or bright blue with aromatic substances such as lignin and tannins; and purplish or greenish blue with nucleic acids. The staining is easy and can last for two days.
Calcofluor white stains cellulose, callose, and other nonsubstituted or weakly substituted β-glucans6-9, and congo red stains directly to β-(1→4)-glucans and particularly to cellulose8,10,11. Calcofluor white stains brilliantly the epidermis, cortex, and pith but that brilliance is significantly reduced in xylem and interfascicular fibers. There are two possible explanations for the reduced fluorescence of calcofluor white in heavily lignified tissues; one attributes it to the presence of lignin, which would, in turn reduce the ability of calcofluor white to diffuse properly through to the polysaccharides. A second explanation points to a partial quenching of the fluorescence emitted by the calcofluor white excited under UV. In contrast to calcofluor white, congo red staining seems less affected by the presence of lignin. It could be related to the difference in fluorescence properties between calcofluor white and congo red; a better diffusion capability of congo red through the lignified tissues than calcofluor white; or a slight difference in polysaccharide binding properties22. It is important to note that the sections stained with congo red should not be stored too long before imaging, because congo red tends to get washed away in water.
Although histochemical staining is neither quantitative nor absolutely conclusive, it can reveal through visual cues a wealth of information about important traits such as the integrity of tissues or abnormal cell wall deposition and lignification. The staining techniques described above are easy to perform and are applicable for critical elements in a plant cell wall, such as lignin and cellulose. By contrast, immunofluorescence using specific antisera and monoclonal antibodies is required for detection of various components of hemicelluloses and pectin. This report is meant to serve as a primer for beginners in the field of secondary cell wall staining who use A. thaliana as their model system.
Disclosures
The authors have no financial conflicts to disclose.
Acknowledgments
We are thankful to Sabin Russell for editing assistance. This work was part of the DOE Joint BioEnergy Institute (http://www.jbei.org) supported by the U. S. Department of Energy, Office of Science, Office of Biological and Environmental Research, through contract DE-AC02-05CH11231 between Lawrence Berkeley National Laboratory and the U.S. Department of Energy.
References
- Boerjan W, Ralph J, Baucher M. Lignin biosynthesis. Annual review of plant biology. 2003;54:519–546. doi: 10.1146/annurev.arplant.54.031902.134938. [DOI] [PubMed] [Google Scholar]
- Vanholme R, Demedts B, Morreel K, Ralph J, Boerjan W. Lignin biosynthesis and structure. Plant physiology. 2010;153:895–905. doi: 10.1104/pp.110.155119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys JM, Chapple C. Rewriting the lignin roadmap. Current opinion in plant biology. 2002;5:224–229. doi: 10.1016/s1369-5266(02)00257-1. [DOI] [PubMed] [Google Scholar]
- Adler E. Lignin chemistry—past, present and future. Wood Sci. Technol. 1977;11:169–218. [Google Scholar]
- Brien TP, Feder N, McCully ME. Polychromatic Staining of Plant Cell Walls by Toluidine Blue. 1964;59:368–373. [Google Scholar]
- Mori B, Bellani LM. Differential staining for cellulosic and modified plant cell walls. Biotechnic & histochemistry: official publication of the Biological Stain Commission. 1996;71:71–72. doi: 10.3109/10520299609117136. [DOI] [PubMed] [Google Scholar]
- Maeda H, Ishida N. Specificity of binding of hexopyranosyl polysaccharides with fluorescent brightener. Journal of biochemistry. 1967;62:276–278. doi: 10.1093/oxfordjournals.jbchem.a128660. [DOI] [PubMed] [Google Scholar]
- Wood PJ. Specificity in the interaction of direct dyes with polysaccharides. Carbohydrate Research. 1980;85:271–287. [Google Scholar]
- Hughes J, McCully ME. The use of an optical brightener in the study of plant structure. Stain technology. 1975;50:319–329. doi: 10.3109/10520297509117082. [DOI] [PubMed] [Google Scholar]
- Verbelen JP, Kerstens S. Polarization confocal microscopy and congo red fluorescence: a simple and rapid method to determine the mean cellulose fibril orientation in plants. Journal of microscopy. 2000;198:101–107. doi: 10.1046/j.1365-2818.2000.00691.x. [DOI] [PubMed] [Google Scholar]
- Anderson CT, Carroll A, Akhmetova L, Somerville C. Real-time imaging of cellulose reorientation during cell wall expansion in Arabidopsis roots. Plant physiology. 2010;152:787–796. doi: 10.1104/pp.109.150128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liljegren S. Phloroglucinol Stain for Lignin. Cold Spring Harbor Protocols. 2010. [DOI] [PubMed]
- Nakano J, Meshitsuka G, lin S, Dence C. The Detection of Lignin Methods in Lignin Chemistry. Berlin: Springer-Verlag; 1992. [Google Scholar]
- Sibout R, et al. CINNAMYL ALCOHOL DEHYDROGENASE-C and -D are the primary genes involved in lignin biosynthesis in the floral stem of Arabidopsis. The Plant cell. 2005;17:2059–2076. doi: 10.1105/tpc.105.030767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teather RM, Wood PJ. Use of Congo red-polysaccharide interactions in enumeration and characterization of cellulolytic bacteria from the bovine rumen. Applied and environmental microbiology. 1982;43:777–780. doi: 10.1128/aem.43.4.777-780.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung E. A beginner's guide to the study of plant structure. Proceedings of the 19th Workshop/Conference of the Association for Biology Laboratory Education (ABLE. 1998;19:365–36. [Google Scholar]
- Turner SR, Somerville CR. Collapsed xylem phenotype of Arabidopsis identifies mutants deficient in cellulose deposition in the secondary cell wall) The Plant Cell Online. 1997;9:689–701. doi: 10.1105/tpc.9.5.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iiyama K, Pant R. The mechanism of the Mäule colour reaction. Introduction of methylated syringyl nuclei into softwood lignin. Wood Sci. Technol. 1988;22:167–175. [Google Scholar]
- Yang F, et al. Engineering secondary cell wall deposition in plants. Plant biotechnology journal. 2013;11:325–335. doi: 10.1111/pbi.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong R, Ripperger A, Ye ZH. Ectopic deposition of lignin in the pith of stems of two Arabidopsis mutants. Plant physiology. 2000;123:59–70. doi: 10.1104/pp.123.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapple CC, Vogt T, Ellis BE, Somerville CR. An Arabidopsis mutant defective in the general phenylpropanoid pathway. The Plant cell. 1992;4:1413–1424. doi: 10.1105/tpc.4.11.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagard M, et al. PROCUSTE1 Encodes a Cellulose Synthase Required for Normal Cell Elongation Specifically in Roots and Dark-Grown Hypocotyls of Arabidopsis. The Plant Cell Online. 2000;12:2409–2423. doi: 10.1105/tpc.12.12.2409. [DOI] [PMC free article] [PubMed] [Google Scholar]