

Abstract
A unique decelerated hydrolytic procedure is developed and reported here for the preparation of ultrasmall nanoparticles (NPs) of PVP-coated BiOI with a narrow size distribution, i.e., 2.8 ± 0.5 nm. The crystal structure of this compound is determined by X-ray powder diffraction using the bulk materials. The stability, cytotoxicity, and potential use of the PVP-coated ultrasmall BiOI NPs as a CT contrast agent are investigated. Because of the combined X-ray attenuation effect of bismuth and iodine, such NPs exhibit a CT value that is among the best of those of the inorganic nanoparticle-based CT contrast agents reported in the literature.
Short abstract
The controlled hydrolysis reaction for producing BiO+ ions under acidic conditions in the presence of I− ions and a polymer coating agent leads to the formation of ultrasmall nanoparticles with a narrow size distribution (∼2.8 ± 0.5 nm). Such nanoparticles possess very high X-ray attenuation power and can be readily internalized by HeLa cells via endocytosis, suggesting their potential as a cellular X-ray CT contrast agent.
Introduction
In the search for suitable media to enhance X-ray contrast for medical imaging of the human body, the development of metal-based contrast agents predates the advent of the iodinated organics.1 Several compounds with metallic elements with high atomic numbers, including silver,2 cesium,3 bismuth,4 thorium,5 and tungsten,6 to name but a few, were investigated for potential contrast applications because of their high X-ray attenuation power. For example, almost 100 years ago, barium sulfate (BaSO4) was first introduced as an aqueous suspension for imaging the gastrointestinal (GI) tract.7 It has remained in clinical use with a little or no change in the formulation.8 On the other hand, the use of thorium oxide (ThO2) as an X-ray contrast agent, first used some 80 years ago, would later result in tragic consequences.9 Thorium (Z = 90) is the second heaviest naturally occurring element after uranium (Z = 92). A suspension of ThO2 showed superb image quality with virtually no acute toxicity or any immediate side effects.5b,5c This formulation was introduced into clinical use as an X-ray contrast agent (Thorotrast) in 1928 and quickly gained widespread applications for imaging cerebral arteries, liver, spleen, lymph nodes, and other organs.5 However, thorium has a naturally occurring radioactive isotope that is an alpha emitter with an extremely long radioactive half-life (4.08 MeV; t1/2 = 1.41 × 1010 years). This problem, compounded with the unusually long biological half-life of the ThO2 colloidal formulations (t1/2 = ∼22 years), would later cause liver cancer and leukemia in millions of patients who were injected with Thorotrast in Europe, North America, and Japan.10
Today, almost all the intravascular X-ray contrast agents are iodinated organic compounds.11 Despite their great water solubility, good safety profile, and high tolerance by the human body, the performance of these organic X-ray contrast agents is sometimes hampered by their non-organ-specific distribution, a short imaging time (i.e., ∼1.5 h), and occasionally acute renal toxicity.12 Consequently, the clinical need for organ-specific X-ray contrast agents with a longer blood circulation half-life has not been met.
The use of various nanoparticles as contrast agents has been considered as one possible solution to increasing imaging efficacy and blood circulation time to increase the time window for imaging.13−17 For example, a polymer-coated Bi2S3 nanoparticle preparation was found to have a high level of X-ray adsorption, long circulation times, and excellent imaging efficacy.18 However, the in vivo hydrolytic stability of such foul-smelling formulations will remain an unsolved problem. It should be noted that hydrolysis of Bi2S3 under endogenous acidic conditions may lead to the release of hydrogen sulfide (H2S), a gas that is known to be more toxic than hydrogen cyanide (HCN) to the neural and circulating system.19 We have set out to search for bismuth compounds that are hydrolytically stable and can readily be prepared as ultrasmall nanoparticles as particulate CT contrast agents that are biocompatible and renal clearable.17 As the U.S. Food and Drug Administration (FDA) currently requires that all injectable contrast agents be excreted from the body completely in a reasonable period of time, renal clearance provides the most viable pathway for achieving this goal.20−23 Bismuth (Z = 83) is the heaviest stable nonradioactive element with a strong X-ray attenuation power.4 In general, bismuth compounds are considered to be nontoxic compared to those of its heavy metal neighbors such as Hg, Tl, and Pb.24 Some bismuth compounds have a venerable history of applications in treating cephalitis and stomach ulcers.25 However, bismuth complexes usually have a low solubility in aqueous solutions.24 As a result, the small molecule platform has not yet afforded clinically useful bismuth-containing CT contrast agents.26 In this article, we describe a simple one-step aqueous solution route for preparing biocompatible and ultrasmall bismuth oxyiodide BiOI nanoparticles (NPs) and investigate their potential application as an efficient CT contrast agent. We prepared the PVP-coated BiOI NPs with an average size of 2.8 ± 0.5 nm and confirmed that the inorganic core of such NPs has the expected layered structure using X-ray powder diffraction. We have also shown that such NPs have no cytotoxicity and can be readily internalized by cells to act as intracellular CT contrast agent. To the best of our knowledge, the CT value measured for such NPs is the highest ever reported on a molarity basis because of the combined X-ray attenuation effect of both bismuth and iodine.
Results and Discussion
Synthesis and Characterization of the Bulk Sample and Biocompatible NPs of BiOI
Bismuth is known to form stable oxyhalides BiOX (X = F, Cl, Br, and I) that crystallize in space group P4/nmm (No. 129).27 As an emerging class of semiconductor photocatalysts, the synthesis of BiOX materials with various nano- and microstructures has recently attracted an increasing level of attention.28 However, almost all of these studies are aimed at producing one-dimensional (1D) nanorods/wires, two-dimensional (2D) nanoplates/sheets, and three-dimensional (3D) hierarchical architectures as well as supported thin films. Thus far there has been only one article in the literature that reports on the synthesis of BiOI nanoparticles. Kaskel and co-workers successfully prepared BiOI nanoparticles with a relatively wide size range from 5 to 10 nm using the reverse microemulsion technique.29 We developed a simple aqueous solution-based synthetic procedure to prepare biocompatible and ultrasmall BiOI NPs for our intended applications. The essence of our new method is the decelerated hydrolysis of the hydrated Bi3+ ion under acidic conditions (i.e., pH 2.75) to produce the BiO+ ion that in turn reacts with the I– ion to form BiOI. It is known that the following reaction is the rate-limiting step in the formation of BiO+:30
In the presence of a polymer coating agent, such a hydrolysis reaction conducted in an acidic solution is so slow that it becomes the rate-limiting step for the nucleation of BiOI NPs, as well, thus leading to the formation of ultrasmall and monodispersed NPs. To impart water dispersibility and biocompatibility, we used polyvinylpyrrolidone (PVP; MW = 40000) as the coating agent. To the best of our knowledge, this approach to controlling the size and size distribution of BiOI NPs has been hitherto unknown.31,32 It should be noted that hydrolysis of the hydrated Bi3+ ions in acidic solutions often led to the formation of polynuclear clusters.33 To investigate the phase identity of our NP core produced under acidic conditions, we prepared the bulk samples of BiOI via this hydrolysis reaction under the same conditions by omitting the use of the polymer coating agent for phase determination.
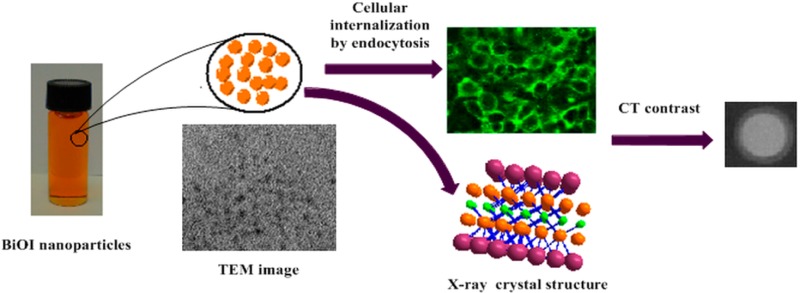
The X-ray powder diffraction (XRD) patterns of the bulk samples revealed that the product obtained from the procedure of slow aqueous hydrolysis reaction was phase-pure and could be readily indexed in space group P4/nmm (No. 129), indicating that BiOI is isostructural with BiOCl.27 Therefore, atomic parameters of the BiOCl structure were used as the initial parameters in the Rietveld refinement performed using TOPAS software from Bruker. The final refinement yielded a = 3.9940(1) Å, c = 9.1549(1) Å, V = 146.038(3) Å3, and ρcalc = 8.002 g/cm3 and converged at Rwp = 3.6% and Rp = 2.7% (see Figure S1 of the Supporting Information). The refinement included 54 atomic and isotropic displacement parameters (Table S1 of the Supporting Information) refined against 148 reflections within a 2θ angular range of 7–109°. The structure of BiOI can be viewed as consisting of layers of BiOI perpendicular to the c direction as shown in Figure 1. Within each BiOI layer, there are five alternating I–Bi–O–Bi–I sublayers. The O layer is situated at the center of the double sandwich. Each O atom has tetrahedral geometry. The Bi layer is cushioned by an O layer from the bottom and capped by an I layer from the top. Each Bi atom is eight-coordinated and bound by four O atoms from a tetragonal base and four I atoms from a tetragonal cap to form a tetragonal antiprism. The fractional atomic coordinates and isotropic displacement parameters are listed in Table S2 of the Supporting Information, while the selected interatomic distances and bond angles are summarized in Table S3 of the Supporting Information.
Figure 1.
X-ray structure of BiOI viewed along the b axis showing the layers stacked in the direction perpendicular to the c axis (top), the view of a single layer (bottom left), and the coordination geometry of the Bi atom (bottom right).
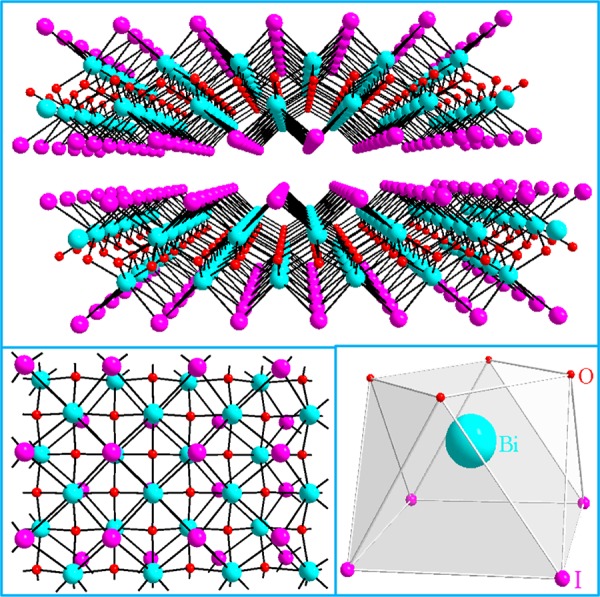
Transmission electronic microscopy (TEM) studies revealed ultrasmall BiOI NPs with a narrow size distribution of 2.8 ± 0.5 nm as shown in Figure 2. The size and size distribution were determined by measuring and averaging the size of 105 NPs from the TEM images. The latter also showed that the shape of these NPs is nearly spherical. The microstructure of individual NPs was examined in detail by high-resolution TEM (HRTEM) and selected area electron diffraction (SAED) on randomly selected NPs. The typical HRTEM image along with the SAED patterns of an NP, given as insets in Figure 2, revealed the crystalline nature of the NPs with a d spacing of 0.287 nm. This d spacing value matches well with the layer repeating distance found in BiOI from the X-ray structure determination, i.e., 2.876 Å. On the other hand, energy dispersive X-ray spectroscopy (EDX) of the PVP-coated BiOI NPs showed distinctive signals for Bi, O, and I with a peak intensity ratio of Bi to I that is essentially the same as that found in the bulk BiOI sample, suggesting that the Bi:I molar ratio in the PVP-coated BiOI NPs is also 1:1 (see Figure S2 of the Supporting Information). Furthermore, the XRD patterns of these PVP-coated BiOI NPs match well with those of the bulk BiOI sample, confirming that the inorganic core of such NPs has the BiOI structure (see Figure S3 of the Supporting Information). We attribute our successful control of the size and size distribution to the slow hydrolysis of the Bi3+ ion to form the BiO+ species in an acidic aqueous solution.
Figure 2.
Histogram of particle size (left), TEM image (top right) with HRTEM and SAED insets, and Z-contrast STEM image (bottom right) of BiOI NPs.
The PVP-coated BiOI NPs are highly dispersible in water and stable against aggregation for more than six months as monitored by the dynamic light scattering (DLS) technique. The latter also determined the hydrodynamic diameter of such NPs dispersed in water to be ∼16 nm (see Figure S4 of the Supporting Information). We also confirmed the existence of a robust PVP coating layer on the NPs that cannot be stripped off from the surfaces by prolonged dialysis against distilled water or sedimentation by centrifugation in a water/acetone mixture using Fourier transform infrared spectroscopy (see Figure S5 of the Supporting Information). Additionally, the results from thermal gravimetric analysis (TGA) showed the average surface loading of PVP to be 28 wt % (see Figure S6 of the Supporting Information).
Determination of the Solubility Product Constant of BiOX
The bulk BiOI solid is insoluble in water and hydrolytically stable at neutral pH. To quantitatively determine the concentrations of the dissociated [BiO+] and [I–] ions at equilibrium, the solubility product of the BiOI bulk phase was measured using both the spectrophotometric method and solution conductivity measurements. At 22 °C, the solubility product (Ksp) of the dissociation reaction BiOI(s) = BiO+(aq) + I–(aq) was found to be (3.34 ± 9) × 10–11 mol2 dm–6, thus giving saturated concentrations of the BiO+ and I– ions of ∼5.8 × 10–6 M each by the spectrophotometric method.34 At this temperature, the Ksp value obtained by solution conductivity measurements is (2.1 ± 2) × 10–11 mol2 dm–6, which gives saturated concentrations of the BiO+ and I– ions of ∼4.6 × 10–6 M each.35 We also monitored the leaching of the BiO+ ions from the PVP-coated BiOI NPs to aqueous solution by soaking ∼5 mg of NPs sealed in a dialysis bag [molecular weight cutoff (MWCO) of 3000] and periodically checking the solution concentrations of BiO+ ions outside the membrane bag using atomic adsorption (AA) spectrometry for 1 week. In all the analyzed solution samples, the bismuth concentration fell below the detection limit of this analytical technique for bismuth of 1 ppm, indicating that BiOI has sufficient thermodynamic stability and kinetic inertness to maintain its structural integrity for the intended application as a CT contrast agent.
Cytotoxicity Assay and Cellular Uptake Studies of BiOI NPs
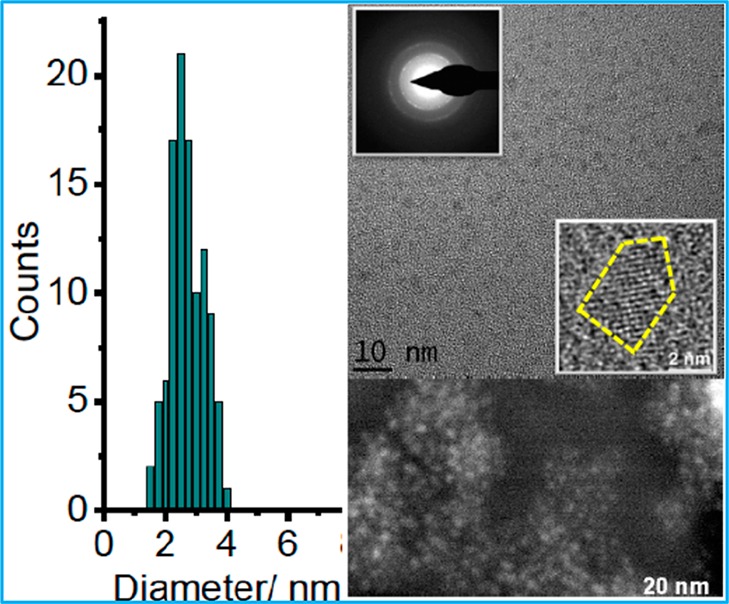
Although BiOCl has been widely used in the cosmetic industry as a nacreous pigment in fingernail polishes, lipsticks, and face powders since ancient times,24 the toxicity of BiOX (X = Br and I) has remained completely unknown. Additionally, compared to bulk materials, nanoparticles, especially ultrasmall nanoparticles, may exhibit unexpected or increased cytotoxicity because of their unusually large surface area relative to the total mass, which increases the chance of interacting with various endogenous biomolecules to trigger adverse intracellular responses.36,37 To explore the possibility of developing the BiOI NPs as an intravenous CT contrast agent, we examined the in vitro cytotoxicity of such NPs using the Trypan Blue exclusion method. HeLa cells were incubated for 24 h with varying concentrations of BiOI NPs. After a 24 h incubation with the highest dispersible concentration, i.e., 4.15 mg/mL BiOI NPs, the cell viability was found to be ∼92 ± 0.8%, indicating that the PVP-coated BiOI NPs exhibit no significant cytotoxicity (see Figure 3).
Figure 3.
Cell viability of HeLa cells after being incubated with PVP-coated BiOI NPs for 24 h.
On the other hand, the ability of the NPs to cross cell membranes provides a possibility to develop such NPs as a cellular CT contrasting agent. Currently, CT is not considered a cellular imaging modality, partly because of the lack of suitable contrast agents that are either cell-permeable or surface-modified with targeting agents that can selectively bind to certain receptors of the cell exterior.38 We explored the cellular uptake of PVP-coated BiOI NPs in HeLa cells using the confocal fluorescence microscopic technique. First, NPs were conjugated with molecules of the fluorescence dye carboxyfluorescein (CbF). The reason that the CbF-conjugated NPs were used as the fluorescent probe is that the CbF dye molecule itself is membrane-impermeable because of its low solubility in aqueous media and high anionic charge.39 Live HeLa cells were treated with the dye-labeled NPs, washed with phosphate-buffered saline (PBS), and directly imaged under a laser scanning confocal microscope without fixation. The images of confocal microscopy showed that the cells treated with the dye-labeled NPs, as compared to the control cells, exhibited strong and uniform green fluorescent signals, showing an even and untargeted distribution of NPs in the cytoplasm (see Figure 4). This finding is consistent with a cellular uptake mechanism of the NPs via endocytosis.
Figure 4.
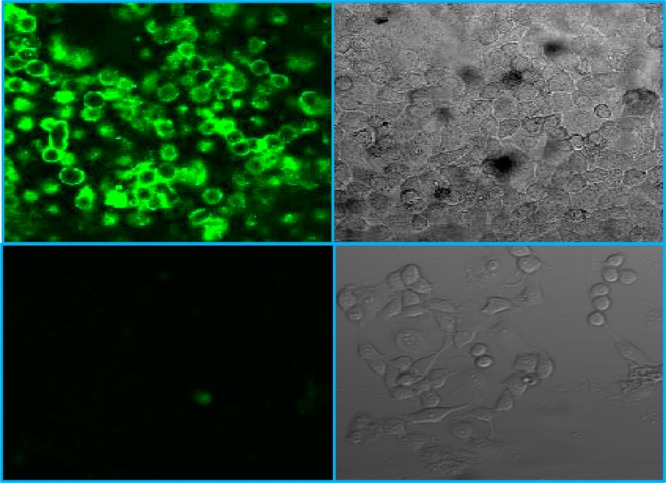
Confocal microscopic fluorescence image (top left) and bright field image (top right) of HeLa cells incubated with dye-conjugated BiOI NPs. Also shown are the fluorescence image (bottom left) and the bright field image (bottom right) of untreated HeLa cells as a negative control.
Measurements of X-ray CT Density Values of BiOI NPs
The X-ray attenuation of PVP-coated BiOI NPs was measured and reported as density values in Hounsfield units (HU). The latter is calculated by
where μ and μwater are the linear attenuation coefficients of the sample and air, respectively. By definition, the density value of water is assigned as zero. When the HU values of unknown materials are measured, the CT scanners are calibrated by measuring the density value of distilled water as the external reference. In this study, six different concentrations of NPs dispersed in aqueous media and a sample of distilled water were used in the measurement performed with a Gamma Medica Xspect microCT scanner operating at 75 kVp and 110 μA. As shown in Figure 5, the CT values expressed in HU exhibit a linear relationship with the Bi(III) concentration in NPs. The slope of this linear curve indicates the X-ray attenuation efficiency of the contrast agent in Hounsfield units per millimolar. In this regard, the X-ray attenuation efficiency of the current CT agent has a value of ∼20 HU/mM. In comparison, the PEGylated gold NPs gave a value of 5.3 HU/mM (i.e., 1.27 M PEG-coated Au NPs have a CT value of 6690 HU),15b while this value for PVP-coated Bi2S3 was reported to be 9.3 HU/mM in one article18 and 6.7 HU/mM in another.40 Similarly, the X-ray attenuation efficiency was measured and reported for a polymer-coated TaOx NP system to be 6.0 HU/mM.41 It is clear that our PVP-coated BiOI NPs appear to have the highest value on a molarity basis because of the combined X-ray attenuation effect of bismuth and iodine, as all the other reported inorganic NPs show a value of <20 HU/mM.42 It should be noted that the molar mass of BiOI of 351.88 g/mol is also the highest of those of the inorganic substances mentioned above, suggesting that when a given molar concentration of each contrast agent is to be injected into the body, the amount of mass required for BiOI NPs would turn out be the largest as well. However, even when the CT value is converted to the per milligram mass basis, the X-ray attenuation efficiency of BiOI NPs can still rival those of all of the inorganic nanoparticle-based CT contrast agents mentioned above.
Figure 5.
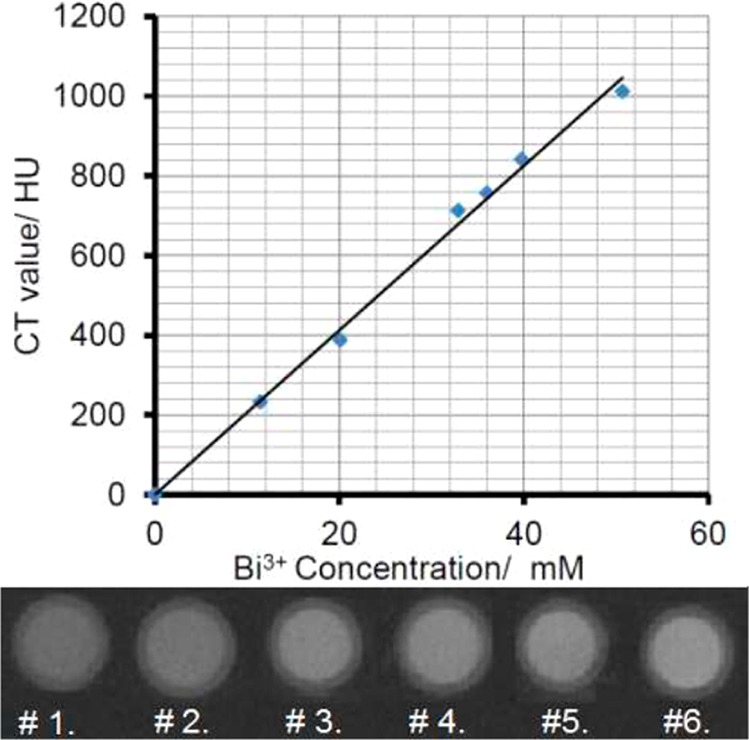
CT values and phantom images of the PVP-coated BiOI NPs as a function of Bi3+ concentration in aqueous solution for (1) 5.07, (2) 12.7, (3) 25.4, (4) 30.4, (5) 38.1, and (6) 50.7 mM.
Conclusion
In summary, the use of bismuth and iodine in a single compound gives rise to a favorable structural platform with an X-ray attenuation power that is among the best of those of all the known inorganic nanoparticulate systems thus far reported in the literature for CT contrast applications. Conceptually, BiOI can be viewed as a compound consisting of the BiO+ and I– ions. This notion has led to the development of a controlled hydrolytic procedure in this work for the preparation of biocompatible ultrasmall NPs of BiOI. On the other hand, BiOI is a covalent solid-state compound with a layered structure and a very low solubility product constant. The latter is a desirable feature for ensuring low osmolality if these NPs are to be developed as an intravenously injectable CT contrast agent. Our preliminary studies of the cytotoxicity and cellular internalization suggest that such NPs may have potential for CT cellular imaging and image-guided drug delivery applications.
Materials and Methods
Synthesis of the PVP-Coated BiOI Nanoparticles
A solution of 1.0 mM Bi(NO3)3 (0.1940 g of bismuth nitrate pentahydrate in 400 mL at pH ∼2.7) containing 2.0 g of PVP (average MW = 40000) was added slowly with a solution of 20 mM NaI (0.0600 g of sodium iodide in 20 mL) at room temperature to give a clear colorless to pale yellow solution. After the mixture had been stirred for 30 min, 10 mL aliquots of the solution were each added with an equal volume of acetone and centrifuged at 10000 rpm for ∼15 min, resulting in the formation of a pellet of nanoparticles in each centrifuge tube. The nanoparticles were redispersed in ∼10 mL of distilled water by sonication and separated again by the addition of an equal volume of acetone and centrifugation. The purification process was repeated twice more.
Synthesis of the Bulk BiOI Materials
Bulk BiOI materials were prepared using solutions of Bi(NO3)3 and NaI in the absence of a coating agent. Specifically, an aqueous solution of 400 mL of Bi(NO3)3 (1.0 mM) was added to an aqueous solution of 20 mL of NaI (20 mM) while being vigorously stirred at room temperature. This reaction resulted in a pale yellow precipitate in 1 h. After the mixture had been stirred for an additional 8 h at room temperature, the product was filtered and washed with water twice and with acetone twice. The product was dried in air at room temperature for 24 h.
Determination of the Structure of BiOI from the Bulk Sample
XRD patterns were recorded using a Bruker D8 Advance X-ray powder diffractometer using Cu Kα radiation with a Ni β-filter and a LynxEye PSD detector. Powder patterns were measured from 7° to 109° 2θ with a step size of 0.01446° and an exposition time of 800 s/step. The Rietveld refinement was conducted using TOPAS software from Bruker. The refinement included 54 atomic coordinate and isotropic displacement parameters (Tables S1–S3 of the Supporting Information) refined against 148 reflections within this angular range, which yielded a = 3.99399(4) Å, c = 9.15486(8) Å, V = 146.038(2) Å3, space group P4/nmm (No. 129), and ρcalc = 8.002 g/cm3 and converged at Rwp = 3.6% and Rp = 2.7%.
TEM Imaging and EDX Measurements
Nanoparticles were first suspended in water, and then a droplet of the suspension was placed on a carbon-coated copper TEM grid (400 mesh). The specimens were allowed to air-dry and analyzed at 200 kV using a FEI Tecnai F20 field emission transmission electron microscope equipped with an integrated scanning TEM (STEM) unit. The energy dispersive X-ray spectroscopy (EDX) results were obtained with an EDAX spectrometer in STEM mode. The spatial resolution is <1 nm through the acquisition of high-resolution high-angle angular dark field (HAADF) images, in which the contrast is sensitive to the atomic number (Z).
Thermogravimetric Analysis
Thermal analysis was conducted on a PVP-coated NP sample using a TA Instruments 2950 high-resolution thermogravimetric analyzer (Universal V3.9A) in air from room temperature to 600 °C with a heating rate of 10 °C/min.
Determination of the BiOI Solubility Product Constant
A spectrophotometric method was used to determine the I– concentration for the calculation of the BiOI solubility product constant. A bulk BiOI sample was allowed to equilibrate with deionized water at 22 °C for 48 h. Samples for spectroscopic analysis of the I– concentration at equilibrium were individually prepared by mixing 1.00 mL of the solution described above with 1.00 mL of KNO2 (0.1 M) and 1.00 mL of HNO3 (0.1 M) before the mixture was transferred to a cuvette. A solution containing the same amount of KNO2 and HNO3 was used as the reference blank. Absorbance at 525 nm was measured for the production of iodine based on the following reaction:
Standard solution series (5.0, 10.0, 25.0, 50.0, 100.0, and 300.0 μM) of KI were prepared and mixed with KNO2 and HNO3 using the same procedure to obtain a calibration curve (see Figure S7 of the Supporting Information). The Ksp value for BiOI was calculated to be (3.34 ± 9) × 10–11 mol2 dm–6, based on the measured I– concentration by assuming the following equilibrium:
Further, the solubility product constant was separately measured using solution conductivity measurements. First, the electric conductivity (κ) of the BiOI solution at equilibrium and that of deionized water were measured separately at 22 °C. The conductivity of deionized water was then subtracted from the solution conductivity to obtain the net conductivity of the dissolved BiOI solute. Before the conductivity measurements were taken, the conductivity probe was calibrated using a standard electrolyte solution with a known conductivity. The equilibrium concentration of the dissolved BiOI was calculated using the equation [BiOl]eq ≅ (κ/Λmo), where κ is the net conductivity of the dissolved BiOI and Λm is the limiting molar conductivity of BiOI at infinite dilution and is calculated from the contributions of individual ions using the tabulated values. The solubility product constant of BiOI was found to be (2.1 ± 2) × 10–11 mol2 dm–6.
Cell Viability Assay
The effect of PVP-coated BiOI NPs on cell viability was assessed using a Trypan Blue exclusion viability assay. HeLa cells were seeded in a 96-well plate at a density of 1 × 104 cells/well with the DMEM (Dulbecco’s modified Eagle’s medium) low-glucose medium containing 10% FBS (fatal bovine serum) with penicillin/streptomycin and incubated for 5 h at 37 °C in an atmosphere of 5% CO2 and 95% air to allow cells to attach to the surface. Cells in each well were then treated with 100 μL of fresh medium containing varying concentrations of the NPs and then incubated for 24 h. Control wells contained the same medium without NPs. After the cells had been stained with Trypan Blue, viable and nonviable cells were counted using a hemocytometer.
Conjugation of Fluorescence Dye Molecules to the Surfaces of Nanoparticles
To covalently attach the fluorescence dye molecules to the surfaces of NPs, the synthetic procedure used to prepare PVP-coated BiOI NPs was modified slightly. Specifically, an aqueous Bi3+ (1 mM, 50 mL) solution containing PVP (average MW = 40000, ∼500 mg) and 1.0 mL of 1 mM ethylenediamine was slowly added to the aqueous solution of KI (1 mM, 50 mL) while being vigorously stirred. In the dye coupling reaction, the 6-carboxyfluorescene dye (CbF, 0.012 g) was first reacted with N-[3-(dimethylamino)propyl]-N-ethylcarbodiimide hydrochloride (EDC, 0.004 g) in a water/ethanol mixture (3.2 mM, 10 mL). The primary -NH2 on BiOI (10 mL, ∼5 mM) NPs was then reacted with ∼1 mL of the EDC-coupled carboxyfluorescence dye (CbF) and stirred overnight. The reaction mixture was dialyzed against distilled water using a regenerated cellulose tubular membrane (MWCO of 12000) for 2 days to remove unreacted dye (see Figure S8 of the Supporting Information).
Cellular Uptake Studies
Confocal scanning microscopy was used to study the cellular uptake of the fluorescence dye-labeled NPs in HeLa cells. The cells were first seeded in an eight-well chamber slide at a density of approximately 1.5 × 105 cells/well and incubated at 37 °C for 24 h in complete medium without antibiotics. The culture medium was then replaced with a medium containing dye-labeled NPs (500 μM). The cells were then incubated with fluorescence dye-labeled NPs for 4 h. After the cells had been washed three times with the PBS buffer solution to remove free NPs, fresh medium was added to the cells and the living cells without fixing were directly imaged under the confocal microscope with the 488 nm excitation wavelength.
X-ray Attenuation Measurements and CT Phantom Imaging
The linear X-ray attenuation coefficients of nanoparticle solutions at different concentrations, distilled water, and air were measured using a Gamma Medica Xspect instrument. The CT phantom imaging studies were conducted with the following parameters: 512 slices/360° rotation; 75 kVp, 110 μA; field of view, 39.47; resolution, 150 μm. The linear transformation of the raw data was performed to obtain the Hounsfield units for the various NP concentrations.
Acknowledgments
We thank the National Cancer Institute for financial support (1R21CA143408-01A1). We also thank Dr. Peter Y. Zavalij (Department of Chemistry and Biochemistry, University of Maryland, College Park, MD) for his assistance with the powder X-ray structure determination work. The TEM data were obtained using the Cryo TEM Facility at the Liquid Crystal Institute (Kent State University), supported by the Ohio Research Scholars Program.
Supporting Information Available
X-ray crystallographic data in CIF format and other data of spectroscopic characterization of BiOI materials, including EDX, DLS, IR, TGA, UV–vis, and fluorescence emission spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Yu S. B.; Watson A. D. Chem. Rev. 1999, 99, 2353. [DOI] [PubMed] [Google Scholar]; b Krause W. Adv. Drug Delivery Rev. 1999, 37, 159. [DOI] [PubMed] [Google Scholar]
- a Voelcker F.; von Lichtenberg A. Muench. Med. Wochenschr. 1906, 53, 105. [Google Scholar]; b Uhle A. A.; Pfahler G. E.; MacKinney W. H.; Miller A. G. Ann. Surg. 1910, 51, 546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe I. Japanese Patent Application 90-104206, 1990; Chem. Abstr.1990, 116, 262525
- a Rumpel T. Muench. Med. Wochenschr. 1897, 44, 420. [Google Scholar]; b Cannon W. B. Am. J. Physiol. 1898, 1, 359. [Google Scholar]
- a Burns J. E. Bull. Johns Hopkins Hosp. 1916, 27, 157. [PubMed] [Google Scholar]; b Moniz E.; Pinto A.; Lima A. Rev. Neurol. 1931, 32, 646. [Google Scholar]; c Dickson W. H. Can. Med. Assoc. J. 1932, 27, 125. [PMC free article] [PubMed] [Google Scholar]; d Macdonald I. G. Can. Med. Assoc. J. 1932, 27, 136. [PMC free article] [PubMed] [Google Scholar]
- Von Sailer R.; Kissler B.; Stauch G.; Franken T.; Huth F. Fortschr Geb Rontgenstr Nuklearmed 1973, 119, 727. [PubMed] [Google Scholar]
- Ikehata A.; Nakano Y.; Sakuma T. J. Gastroenterol. 1999, 28, 280. [DOI] [PubMed] [Google Scholar]
- Baert A. L., Ed. Encyclopedia of Imaging; Springer-Verlag: Berlin, 2007; Vol. 2. [Google Scholar]
- J. Am. Med. Assoc. 1932, 99, 2193. [Google Scholar]
- a van Kaick G.; Dalheimer A.; Hornik S.; Kaul A.; Liebermann D.; Lührs H.; Spiethoff A.; Wegener K.; Wesch H. Radiat. Res. 1999, 152, S64. [PubMed] [Google Scholar]; b Becker N.; Liebermann D.; Wesch H.; Van Kaick G. Eur. J. Cancer 2008, 44, 1259. [DOI] [PubMed] [Google Scholar]
- Idée J.-M.; Nachman I.; Port M.; Petta M.; Le Lem G.; Le Greneur S.; Dencausse A.; Meyer D.; Corot C. Top. Curr. Chem. 2002, 222, 151–171. [Google Scholar]
- a Idée J.-M.; Lancelot E.; Pines E.; Corot C. Invest. Radiol. 2004, 39, 155. [DOI] [PubMed] [Google Scholar]; b Morcos S. K.; Thomsen H. S.; Webb J. A. W. Eur. J. Radiol. 1999, 9, 1602. [DOI] [PubMed] [Google Scholar]; c Shigenaga A.; Tsuji D.; Nishioka N.; Tsuda S.; Itoh K.; Otaka A. ChemBioChem 2007, 8, 1929. [DOI] [PubMed] [Google Scholar]; d Jost G.; Pietsch H.; Lengsfeld P.; Hutter J.; Sieber M. A. Invest. Radiol. 2010, 45, 255. [DOI] [PubMed] [Google Scholar]
- Lewin M.; Carlesso N.; Tung C. H.; Tang X. W.; Cory D.; Scadden D. T.; Weissleder R. Nat. Biotechnol. 2000, 18, 410. [DOI] [PubMed] [Google Scholar]
- Rosi N. L.; Mirkin C. A. Chem. Rev. 2005, 105, 1547. [DOI] [PubMed] [Google Scholar]
- a Hainfeld J. F.; Slatkin D. N.; Focella T. M.; Smilowitz H. M. Br. J. Radiol. 2006, 79, 248. [DOI] [PubMed] [Google Scholar]; b Kim D.; Park S.; Lee J. H.; Jeong Y. Y.; Jon S. J. Am. Chem. Soc. 2007, 129, 7661. [DOI] [PubMed] [Google Scholar]
- deKrafft K. E.; Xie Z. G.; Cao G. H.; Tran S.; Ma L. Q.; Zhou O. Z.; Lin W. B. Angew. Chem., Int. Ed. 2009, 48, 9901. [DOI] [PubMed] [Google Scholar]
- Perera V. S.; Hao J.; Gao M.; Gough M.; Zavalij P. Y.; Flask C.; Basilion J. P.; Huang S. D. Inorg. Chem. 2011, 50, 7910. [DOI] [PubMed] [Google Scholar]
- Rabin O.; Perez J. M.; Grimm J.; Wojtkiewicz G.; Weissleder R. Nat. Mater. 2006, 5, 118. [DOI] [PubMed] [Google Scholar]
- Beauchamp R. O.; Bus J. S.; Popp J. A.; Boreiko C. J.; Andjelkovich D. A.; Leber P. Crit. Rev. Toxicol. 1984, 13, 25. [DOI] [PubMed] [Google Scholar]
- Liu J.; Yu M.; Zhou C.; Zheng J. Mater. Today 2013, 16, 477. [Google Scholar]
- Kim B. H.; Hackett M. J.; Park J.; Hyeon T. Chem. Mater. 2014, 26, 59. [Google Scholar]
- Bonitatibus P. J.; Torres A. S.; Kandapallil B.; Lee B. D.; Goddard G. D.; Colborn R. E.; Marino M. E. ACS Nano 2012, 6, 6650. [DOI] [PubMed] [Google Scholar]
- Torres A. S.; Bonitatibus P. J.; Colborn R. E.; Goddard G. D.; FitzGerald P. F.; Lee B. D.; Marino M. E. Invest. Radiol. 2012, 47, 578. [DOI] [PubMed] [Google Scholar]
- Briand G. G.; Burford N. Chem. Rev. 1999, 99, 2601. [DOI] [PubMed] [Google Scholar]
- Sadler P. J.; Li H.; Sun H. Coord. Chem. Rev. 1999, 185–186, 689. [Google Scholar]
- Lusic H.; Grinstaff M. W. Chem. Rev. 2013, 113, 1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sillén L. G. Sven. Kem. Tidskr. 1941, 53, 39. [Google Scholar]; b Keller E.; Kramer V. Z. Naturforsch. 2005, 60b, 1255. [Google Scholar]
- See, for example:Zhang X.; Ai Z.; Jia F.; Zhang L. J. Phys. Chem. C 2008, 112, 747. [Google Scholar]
- Henle J.; Simon A.; Frenzel A.; Scholz S.; Kaskel S. Chem. Mater. 2007, 19, 367. [Google Scholar]
- Näslund J.; Persson I.; Sandström M. Inorg. Chem. 2000, 39, 4012. [DOI] [PubMed] [Google Scholar]
- Uemura T.; Kitagawa S. J. Am. Chem. Soc. 2003, 125, 7814. [DOI] [PubMed] [Google Scholar]
- Uemura T.; Ohba M.; Kitagawa S. Inorg. Chem. 2004, 43, 7339. [DOI] [PubMed] [Google Scholar]
- Miersch L.; Rüffer T.; Lang H.; Schulze S.; Hietschold M.; Zahn D.; Mehring M. Eur. J. Inorg. Chem. 2010, 4763. [Google Scholar]
- Green D. B.; Rechtsteiner G.; Honodel A. J. Chem. Educ. 1996, 73, 787. [Google Scholar]
- Coury L. Curr. Sep. 1999, 18, 91. [Google Scholar]
- Nel A.; Xia T.; Madler L.; Li N. Science 2006, 311, 622. [DOI] [PubMed] [Google Scholar]
- Pan Y.; Neuss S.; Leifert A.; Fischler M.; Wen F.; Simon U.; Schmid G.; Brandau W.; Jahnen-Dechent W. Small 2007, 3, 1941. [DOI] [PubMed] [Google Scholar]
- Popovtzer R.; Agrawal A.; Kotov N. A.; Popovtzer A.; Balter J.; Carey T. E.; Kopelman R. Nano Lett. 2008, 8, 4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangburn T. O.; Georgiou K.; Bates F. S.; Kokkoli E. Langmuir 2012, 28, 12816. [DOI] [PubMed] [Google Scholar]
- Ai K.; Liu Y.; Liu J.; Yuan Q.; He Y.; Lu L. Adv. Mater. 2011, 23, 4886. [DOI] [PubMed] [Google Scholar]
- Oh M. H.; Lee N.; Kim H.; Park S. P.; Piao Y.; Lee J.; Jun S. W.; Moon W. K.; Choi S. H.; Hyeon T. J. Am. Chem. Soc. 2011, 133, 5508. [DOI] [PubMed] [Google Scholar]
- Jakhmola A.; Anton N.; Vandamme T. F. Adv. Healthcare Mater. 2012, 1, 413. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.