

Abstract
A library of 10 Mn-containing complexes capable of switching reversibly between the Mn(II) and Mn(III) oxidation states was prepared and evaluated for potential usage as MRI reporters of tissue redox activity. We synthesized N-(2-hydroxybenzyl)-N,N′,N′-ethylenediaminetriacetic acid (HBET) and N-(2-hydroxybenzyl-N,N′,N′-trans-1,2-cyclohexylenediaminetriacetic acid (CyHBET) ligands functionalized (−H, −OMe, −NO2) at the 5-position of the aromatic ring. The Mn(II) complexes of all ligands and the Mn(III) complexes of the 5-H and 5-NO2 functionalized ligands were synthesized and isolated, but the Mn(III) complexes with the 5-OMe functionalized ligands were unstable. 1H relaxivity of the 10 isolable complexes was measured at pH 7.4 and 37 °C, 1.4 T. Thermodynamic stability, pH-dependent complex speciation, hydration state, water exchange kinetics of the Mn(II) complexes, and pseudo-first order reduction kinetics of the Mn(III) complexes were studied using a combination of pH-potentiometry, UV–vis spectroscopy, and 1H and 17O NMR measurements. The effects of ligand structural and electronic modifications on the Mn(II/III) redox couple were studied by cyclic voltammetry. The Mn(II) complexes are potent relaxation agents as compared to the corresponding Mn(III) species with [MnII(CyHBET)(H2O)]2– exhibiting a 7.5-fold higher relaxivity (3.3 mM–1 s–1) than the oxidized form (0.4 mM–1 s–1). At pH 7.4, Mn(II) exists as a mixture of fully deprotonated (ML) and monoprotonated (HML) complexes and Mn(II) complex stability decreases as the ligands become more electron-releasing (pMn for 10 μM [MnII(CyHBET–R′)(H2O)]2– decreases from 7.6 to 6.2 as R′ goes from −NO2 to −OMe, respectively). HML speciation increases as the electron-releasing nature of the phenolato-O donor increases. The presence of a water coligand is maintained upon conversion from HML to ML, but the water exchange rate of ML is faster by up to 2 orders of magnitude (kex310 for H[MnII(CyHBET)(H2O)]− and [MnII(CyHBET)(H2O)]2– are 1.2 × 108 and 1.0 × 1010 s–1, respectively). The Mn(II/III) redox potential can be tuned over a range of 0.30 V (E1/2 = 0.27–0.57 V) through electronic modifications to the 5-substituent of the aromatic ligand component. However, care must be taken in tuning the ligand electronics to avoid Mn(III)–ligand autoredox. Taken together, these results serve to establish criteria for optimizing Mn(III) versus Mn(II) relaxivity differentials, complex stability, and Mn(II/III) redox potential.
Short abstract
The Mn(II/III) redox couple represents a potentially powerful mechanism to generate MRI contrast in response to redox dynamics. Ten Mn complexes were synthesized, and the effects of ligand structure and electronics on Mn(II) versus Mn(III) relaxivity, solution structure, stability, solution dynamics, and redox potential were characterized by pH-potentiometry, UV−vis, 1H and 17O NMR, and cyclic voltammetry. A 7.5-fold relaxivity turn-on was observed upon reduction of Mn(III).
Introduction
Redox disregulation is a hallmark feature of numerous disease states, including cancers, ischemia, and chronic inflammation.1−6 Loss of the buffering mechanisms that regulate tissue redox activity can trigger biochemical cascades damaging to cellular or tissue components and exacerbate disease progression.7−9 Abnormal tissue redox status can have many causes. For example, tissue hypoxia leads to an aberrant, highly reducing microenvironment.10 Tissue redox status can also be depressed via remodeling of extracellular thiol/disulfide composition as a means to activate T-cells in immune response.11−13 Alternatively, reperfusion following periods of hypoxic ischemia results in oxidative stress through an uncontrolled spike in reactive oxygen species concentration.14,15 Abnormal concentrations of redox active cofactors and adventitious oxidation are associated with the onset and progression of neurological disorders such as Alzheimer’s, Parkinson’s, and Huntington’s diseases.16−19
Methods to monitor changes in redox activity in vivo could be highly useful for disease diagnosis, prognosis, or as a means to monitor response to therapy. Redox differentials between diseased and healthy tissues may also be exploited as a mechanism to control drug delivery in a specified manner.20−22 Indeed, the development of imaging techniques to monitor tissue redox represents a pressing challenge and is a highly sought goal in the field of biomedical imaging.23−28
Considerable effort has been placed toward the development of molecular probes capable of imaging redox activity. To date, some clinical success has been achieved using positron emission tomography (PET) probes that target hypoxic tissue. In some cases, imaging data acquired using radiotracers such as 64Cu(II)-diacetyl-bis(N4-methylthiosemi-carbazone) (64Cu(II)-ATSM) and 18F-fluoromisonidazole (18F-MISO) have been predictive of treatment outcome in patients undergoing curative radiotherapy.29−34 The hypoxia targeting mechanism of 18F-MISO uptake has also been extended to MRI contrast agents and fluorescent reporters.35,36
The hypoxia targeting PET probes operate through irreversible reaction and retention in oxygen-deprived tissue. Probes that respond to redox stimuli in a rapid and reversible manner could open the possibility of tracking tissue redox dynamics in real time.4,37,38 Magnetic resonance imaging (MRI) techniques could feasibly be utilized in this regard.39,40 The recent literature has seen numerous elegant examples of reversibly activated probes that provide MRI contrast using the quinolinium/1,4-dihydroquinoline,41 Co(II/III),42 and TEMPO-H/TEMPO4,37,38,43 redox couples. Redox triggered spiropyran/merocyanine isomerization has also been explored.44
Our group and others are interested in using the Mn(II/III) redox couple as a means to monitor redox imbalance.45,46 Mn can support more than one oxidation state within the physiological realm, and Mn(II) is a potent T1-relaxation agent.47
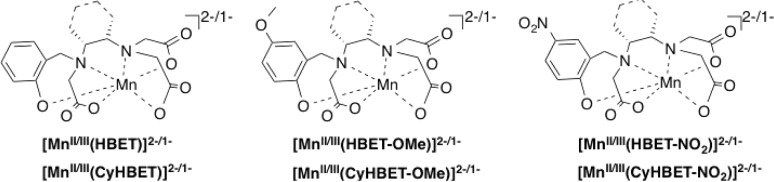
Previously, we demonstrated that the Mn(II) complex of N-(2-hydroxybenzyl)-N,N,′N′-ethylenediaminetriacetic acid (HBET) afforded 3.3-fold relaxivity enhancement as compared to the Mn(III) complex.45 This resulted in an increase of MR signal (turn-on effect) when the Mn(III) complex was reduced with glutathione and a decrease in signal (turn-off) when the Mn(II) complex was oxidized with hydrogen peroxide. HBET represents a promising functionalizable ligand scaffold for the optimization of a reversible, redox responsive MR relaxation agent. The N2O4 donor set with a single phenolato-O donor enables facile conversion between the Mn(II/III) couple, and both oxidation states are isolable and stable in solution. Inspired by the favorable redox and MRI signal enhancing properties of [MnII/III(HBET)]2–/1–, we aimed to optimize the Mn(II) versus Mn(III) relaxivity differential, maximize complex stability, and predictably control the redox potential. To this end, we prepared five new derivatives of the HBET ligand prototype featuring systematic structural and electronic modifications. The trans-1,2-cyclohexylenediamine (CyHBET series) provided backbone rigidification and preorganization (Chart 1). Electronic changes were introduced via substituent changes (R′ = −H, −OMe, −NO2) at the 5-position of the aromatic ring. For these six ligands, we prepared the corresponding Mn(II) and Mn(III) complexes and compared their relaxivities, thermodynamic stability, pH-dependent speciation, hydration state, water exchange kinetics, redox potential, and Mn(III) reduction kinetics in the presence of cysteine.
Chart 1. Mn(II/III) Complexes Considered in This Study.
Results
Synthesis
The ligands were prepared in a few simple steps (Scheme 1). Starting with mono N-BOC protected ethylenediamine or trans-1,2-diaminocylohexane, the 5-R′-2-hydroxybenzyl arm was appended to the backbone via reductive amination. The N-BOC protecting group was subsequently removed by stirring in TFA. The 5-nitro-2-hydoxybenzyl-appended diamines were then O-protected as tert-butyldimethyl silyl ethers. O-Protection of the hydroxybenzyl and 5-methoxy-2-hydroxybenzyl arms was unnecessary. Next, the diamine backbone was exhaustively alkylated using tert-butyl bromoacetate. The ligands were generated by TFA removal of the protecting groups. TFA is associated with the isolated ligand (determined through titration with NaOH as described below).
Scheme 1. Generalized Synthetic Scheme for Mn(II/III) Complexes of HBET–R′ and CyHBET–R′.

The Mn(II) complexes were generated by raising the pH of a 1:1 mixture of MnCl2 and ligand to pH 6.5. Alternatively, the complexes could be spontaneously generated via mixing in pH 7.4 buffered solution. The corresponding Zn(II) complexes, prepared for comparative study (see below), were generated by stoichiometric mixing in pH 7.4 buffer.
The Mn(III) complexes were prepared by addition of solid MnF3 to an aqueous solution of the ligand at pH 8. The pH was maintained during Mn(III) chelation by careful addition of 1 M NaOH. MnF3 is insoluble in water, and this ligand-aided dissolution strategy was chosen to minimize disproportionation of free aqueous Mn(III) to Mn(II) and Mn(IV). After MnF3 addition, the red-brown reaction mixtures contained a small amount of the Mn(II) complex, which was subsequently removed via RP-HPLC. This strategy afforded the Mn(III) complexes in higher yield than the previously reported aerial oxidation procedure employed to prepare [MnIII(HBET)]−.45 Upon purification, the Mn(III) form of the 5-H and 5-NO2 derivatives remained stable in solution for hours.
Reaction mixture analysis after addition of MnF3 to the ligands of the CyHBET–R′ series by LC–MS revealed two unique species of mass corresponding to the Mn(III) complex, which we attribute to diastereomers. The UV–vis profiles of these chromatographically unique species were monitored by a diode array detector coupled to the LC and were found to be indifferentiable. The species are separable by preparative HPLC (Figure 1, Supporting Information Figures S7–S9), but equilibrium mixtures were recovered from isolated product. It is noted that complexation of the CyHBET–R′ ligands with Zn(II) also afforded two chromatographically resolved species of identical mass corresponding to the Zn(II) complex (Supporting Information Figures S13–S15).
Figure 1.
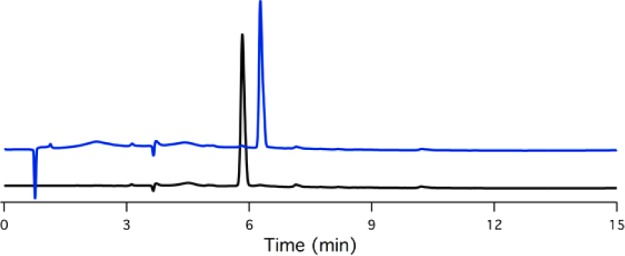
LC traces with UV detection (254 nm) of two chromatographically separable species corresponding to [MnIII(CyHBET–NO2)]−.
Synthesis of the Mn(III) complexes of HBET, CyHBET, HBET–NO2, and CyHBET–NO2 proceeded in a straightforward fashion. However, addition of MnF3 to HBET–OMe and CyHBET–OMe resulted in a complex product distribution (Supporting Information Figure S16). LC–MS analysis revealed the presence of desired product, free ligand, a m/z+= 847.4 species (best attributed to ligand dimerization via C–C bond formation, i.e., [(CyHBET–OMe)2 – 2H + H]+), and the Mn(II) and/or Mn(III) occupied forms of this dimer. We were unsuccessful in isolation of [MnIII(HBET–OMe)]− and [MnIII(CyHBET–OMe)]−.
Synthesis of pure, isolable [MnIII(HBET–OMe)]− was also attempted by stoichiometric oxidation using potassium ferricyanide, but this was also unsuccessful. To gain qualitative insight into the seemingly unstable nature of this complex, 0.8 mM of the [MnII(HBET)]2– or [MnII(HBET–OMe)]2– was combined with 1 mol equiv of ferricyanide in pH 9.0 Tris buffer, and the disappearance of Mn(III) was monitored by UV–vis spectroscopy (Figure 2). An absorbance at 496 nm, best attributed to a Mn(III) ligand field transition, was used as the spectroscopic handle;45 ferri- and ferrocyanide and the corresponding Mn(II) complex do not absorb in this region. After 2 min, the oxidized products afforded nearly identical UV–vis profiles. [MnIII(HBET)]− generated in this manner remained stable in solution for 2 h, but [MnIII(HBET–OMe)]− was 50% decomposed at ∼10 min. Product analysis by LC–MS confirmed the presence of the dimeric [(HBET–OMe)2 – 2H + H]+ species (m/z = 739.8) and corresponding Mn(II) complex.
Figure 2.

Left: UV–vis spectra acquired 2 min after 1 mol-equiv addition of potassium ferricyanide to [MnII(HBET)]2– (−) and [MnII(HBET–OMe)]2– (· · ·) at pH 9. Right: Absorbance at 496 nm as a function of time after oxidation of [MnII(HBET)]2– (●) and [MnII(HBET–OMe)]2– (▽).
Relaxivity at pH 7.4
The T1- and T2-relaxivities (r1, r2) of the 10 isolable complexes were measured at pH 7.4 (Tris buffer), 37 °C, 1.4 T. The results are summarized in Table 1 and Figure 3. The relaxivities of the Mn(II) complexes are all increased relative to the corresponding Mn(III) complexes. [MnII/III(CyHBET)]2–/1– showed the greatest increase in relaxivity upon reduction where a 7.5-fold r1 turn-on is observed. Large r2 differentials were also observed between the Mn(II) and Mn(III) oxidation states. For example, r2 of [MnII(CyHBET)]2– is over 5-fold greater than that of [MnIII(CyHBET)]−.
Table 1. T1- and T2-Relaxivity (mM–1 s–1) of Isolable Mn(II) and Mn(III) Complexes at pH 7.4, 37 °C, 1.4 T.
| Mn(II) |
Mn(III) |
|||
|---|---|---|---|---|
| r1 | r2 | r1 | r2 | |
| HBET | 2.8 | 9.4 | 1.1 | 2.7 |
| HBET–OMe | 3.1 | 11.1 | ||
| HBET–NO2 | 2.3 | 4.8 | 0.5 | 1.0 |
| CyHBET | 3.3 | 6.0 | 0.4 | 0.9 |
| CyHBET–OMe | 3.3 | 5.8 | ||
| CyHBET–NO2 | 2.3 | 3.7 | 0.5 | 0.9 |
Figure 3.

r1 values of the 10 isolable Mn complexes at pH 7.4 and 37 °C, 1.4 T; Mn(II) (black), Mn(III) (gray).
Across the separate HBET–R′ and CyHBET–R′ series, Mn(II) r1 at pH 7.4 appears to increase with the pKa of the phenolate donor (see below); r2 follows a similar trend. The r2 of the Mn(II) complexes of the HBET–R′ series is also markedly increased as compared to those of the CyHBET–R′ series. Little variance was observed across the relaxivity values of the 3 new Mn(III) complexes prepared for this study.
To highlight the differences in MRI signal generating efficacy between the Mn(II) and Mn(III) complexes, T1-weighted MRI images were also recorded on phantoms containing the four isolable Mn(III) complexes and their sister Mn(II) species. Figure 4 shows a T1-weighted image of pure water, 0.5 mM [MnIII(HBET–NO2)]−, and [MnII(HBET–NO2)]2– with accompanying signal intensities and relaxivities. As expected, the large r1 differential results in striking contrast in a standard T1-weighted image.
Figure 4.

Left: T1-weighted image recorded at room temperature at 4.7 T of samples containing water (top), 0.5 mM [MnIII(HBET–NO2)]− (middle), and [MnII(HBET–NO2)]2– (bottom). Right: r1 measured at room temperature at 4.7 T.
Mn(II) Stability and Speciation
pH-potentiometric measurements were performed to determine ligand pKa values, thermodynamic stability constants, and the pH-dependence on Mn(II) complex speciation. Measurements were not performed on the Mn(III) systems because the Mn(III) aqua ion is unstable in aqueous solutions and Mn(III) stabilization is contingent on coordination of the multidentate ligand. However, by analogy with the Fe(III)–HBET system,48 we expect Mn(III) to remain fully complexed across the pH range considered in this study. It is noted that we have generated isolable [MnIII(HBET)]− at pH 12.45 Also, isolated Mn(III) chelates can be characterized by LC–MS using a mobile phase buffered with 0.1% TFA without any sign of decomposition/dechelation. Measurements were performed on 1:1 mixtures of Mn(II) and ligand. The pH titration profiles of the free ligands and 1:1 Mn(II) ligand mixtures are shown in Supporting Information Figures S17,18. The protonation and formation constants for all ligand species and Mn(II) complexes, respectively, are found in Table 2. Distribution curves describing the pH-dependent speciation of [MnII(HBET)]2–, [MnII(HBET–OMe)]2–, and [MnII(HBET-NO2)]2– are shown in Figure 5 (remaining complexes in Supporting Information Figures S19–21). For all complexes, a mixture of fully deprotonated (ML) and protonated (HML) species exists at pH 7.4. There is no evidence of Mn–hydroxide formation up to pH 9.5. The pKa values of the HML species correlate with the electronic nature of the aromatic substituent R′. In this regard, the phenolate protonation was monitored using UV–vis spectroscopy (Figure 6, Supporting Information Table S2, Figures S22–32). The ligands and Mn(II) complexes are strongly absorbing in the near-UV region, and this spectral feature is pronouncedly red-shifted upon phenol deprotonation. The pKa values were estimated through spectrophotometric titrations by measuring absorbance at the λmax value of the phenolate as a function of pH and the data fit accordingly.50 pKa values determined by UV–vis report on the microscopic phenol/phenolate equilibrium, while the potentiometric approach yields macroscopic constants that are not specific to a site of protonation. For these ligands, the microscopic pKa values for phenol deprotonation are in good accord with pH-potentiometric data (Supporting Information Table S1). For the R′ = −H, −OMe ligands, the first protonation of the ligand is at the phenolate. The nitro group depresses the phenol pKa to such an extent that for these ligands the first protonation occurs at one of the tertiary amines. For all of the complexes, the HML species corresponds to protonation of the phenolato-O.
Table 2. Protonationa and Formationb Constants of Ligands and Their Corresponding Mn(II) Complexes.
| log KLH | log KLH2 | log KLH3 | log KLH4 | log KML | log KHML | pMn (pH 7.4)c | |
|---|---|---|---|---|---|---|---|
| HBET | 11.05 ± 0.04 | 8.83 ± 0.04 | 4.81 ± 0.04 | 2.22 ± 0.07 | 13.07 ± 0.02 | 7.29 ± 0.02 | 6.62 |
| HBET–OMe | 11.61 ± 0.02 | 9.10 ± 0.02 | 4.86 ± 0.02 | 2.46 ± 0.02 | 13.32 ± 0.03 | 7.61 ± 0.02 | 6.48 |
| HBET–NO2 | 9.32 ± 0.04 | 7.48 ± 0.04 | 4.26 ± 0.04 | 2.67 ± 0.07 | 11.29 ± 0.11 | 4.96 ± 0.12 | 7.01 |
| CyHBET | 11.36 ± 0.06 | 9.85 ± 0.06 | 3.94 ± 0.07 | 3.40 ± 0.07 | 14.16 ± 0.04 | 7.45 ± 0.03 | 6.68 |
| CyHBET–OMe | 12.58 ± 0.22 | 9.87 ± 0.22 | 3.99 ± 0.22 | 2.97 ± 0.22 | 14.61 ± 0.07 | 7.73 ± 0.07 | 6.24 |
| CyHBET–NO2 | 10.22 ± 0.05 | 8.05 ± 0.06 | 3.32 ± 0.08 | 2.43 ± 0.13 | 13.66 ± 0.09 | 4.49 ± 0.10 | 7.55 |
| EDTAd | 9.35 ± 0.01 | 5.98 ± 0.01 | 2.48 ± 0.03 | 2.23 ± 0.03 | 12.61 ± 0.15 | 2.90 ± 0.29 | 7.82 |
| CDTAd | 9.43 ± 0.02 | 6.01 ± 0.02 | 3.68 ± 0.02 | 2.51 ± 0.05 | 14.69 ± 0.17 | 2.42 ± 0.34 | 8.82 |
KLHn defined as [HnL]/([H+] × [Hn–1L]). Values were obtained by pH-potentiometry (25 °C, I = 0.1 M NaCl).
KML defined as [ML]/([M] × [L]); KHML defined as [HML]/([ML] × [H+]) (charges omitted for clarity).
pMn defined as −log[free Mn] when [M] = [L] = 10 μM.
Measurements performed independently by another group yielded nearly identical protonation and formation constants.49
Figure 5.

Distribution diagrams for 1:1 Mn(II):ligand mixtures of HBET (top), CyHBET (middle), and HBET–NO2 (bottom). ML, HML, and free M are depicted by solid, dashed, and dotted traces, respectively ([M] = [L] = 1 mM, 25 °C, I = 0.1 M NaCl); pH dependence of r1 (37 °C, 1.4 T) is overlaid in “●”.
Figure 6.
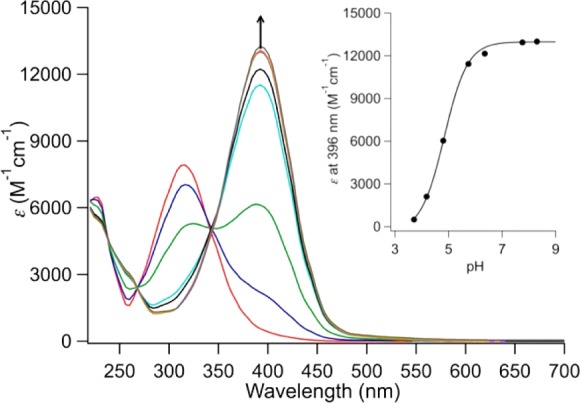
UV–vis spectrum of [MnII(HBET–NO2)]2– as a function of pH between pH 3 and 9. Arrow denotes increase in 396 nm absorbance with increasing pH. Inset: Absorbance at 396 nm as a function of pH. Solid line represents fit to data giving a pKa of 4.84.
The thermodynamic stability constants increase with the electron-releasing character of the R′ group. The formation constants determined for the CyHBET–R′ series were between 1 and 2 orders of magnitude greater than those for the corresponding HBET–R′ complexes. This parallels the formation constant increase observed going from [MnII(EDTA)]2– to [MnII(CDTA)]2–.49 pMn values were calculated for 10 μM complex at pH 7.4. At pH 7.4, the trend correlating thermodynamic stability and the electron-releasing character of the R′ group is reversed. Switching from HBET–R′ to CyHBET–R′ backbone does not significantly influence pMn under these conditions. In fact, when R′ = −OMe, the CyHBET–R′ backbone results in a reduced pMn at pH 7.4, which is a consequence of the increased basicity of this ligand.
Mn(II) Relaxivity as a Function of pH
T1-relaxivity of the Mn(II) complexes was also studied as a function of pH to glean insight into the effects of complex speciation. Measurements were performed on 1:1 mixtures of Mn(II) and ligand between pH 3 and 9.5 at 37 °C, 1.4 T (Figure 5, Supporting Information Figures S19–21). An r1 of 5.1 was observed for all systems at pH 3, corresponding to free Mn(II). The relaxivity rapidly decreases as the pH approaches 5, by which point the values are within 0.2 mM–1 s–1 of the values recorded at pH 7.4. Between pH 6.5 and 9.5, the relaxivity of the R′ = −H and −OMe functionalized complexes decreases slightly. The largest change is observed for [MnII(HBET)]2–, where r1 drops by 0.7 mM–1 s–1 (25%). The relaxivity of the R′ = −NO2 functionalized complexes remains unchanged between pH 5 and pH 9.5.
Mn(II) Hydration State and Water Exchange
We also sought to determine how the Mn(II) hydration state (q) is effected by complex speciation. H217O NMR data were acquired in the presence of the Mn(II) complexes between −10 and 60 °C in both pH 6 MES buffer or pH 9 Tris buffer. With the exception of the R′ = −NO2 functionalized complexes, pH 6 speciation is nearly entirely comprised of HML; at pH 9 the ML species is predominant. From the 17O NMR data, q can be determined through analysis of either the paramagnetically induced chemical shift (Δωp) or the line-width (full-width at half-height = Δν1/2 = 1/(πT2)).51,52 In a previous study, we demonstrated that q can be directly inferred through the line-width at the temperature where line-broadening is greatest (r2maxO, Supporting Information Appendix).52 The temperature at which r2max occurs depends on the mean residency time of the water coligand (τm; the inverse of the exchange rate, kex).
Line-width analysis could be used to determine q for all HML complexes (Figure 7, Supporting Information Figure S33). With the exception of [MnII(HBET–NO2)]2–, the ML adducts existed within the fast exchange regime throughout the entire temperature range studied; this obviated the determination of q via line-width analysis. For the ML complexes, we used chemical shift analysis to obtain q (Supporting Information Figures S34–36). Because water exchange was so fast for the ML complexes, the 17O line-width at higher temperatures was relatively narrow and allowed for accurate determination of the chemical shift.
Figure 7.

r2O plotted as a function of temperature for Mn(II) complexes of HBET (●), HBET–OMe (○), and HBET–NO2 (▲) at pH 6 (left) and pH 9 (right).
Hydration state discerned through chemical shift analysis was assigned to the nearest half-integer value affording a reasonable value for the Mn–17O hyperfine coupling constant (A0/ℏ, 3.3(±0.8) × 107 rad/s).53−67 For q obtained from r2maxO, temperature dependence on r2 was fit to a previously described three-parameter model yielding A0/ℏ, τm, and the activation enthalpy of water exchange (ΔH⧧).52 For q obtained through chemical shift data, A0/ℏ was estimated directly from the slope of the temperature dependence of Δωp and held constant as r2O was fit to the exchange parameters. The results are tabulated in Table 3.
Table 3. Hydration State, Mn–17O(Water) Hyperfine Coupling Constant, Mean Water Residency Time at 37 °C, and Enthalpy of Activation for Water Exchange Measured for Mn(II) Complexes in HML and ML Formsa.
| HML |
ML |
|||||||
|---|---|---|---|---|---|---|---|---|
| q | Ao/ℏ (×107 rad/s) | τm310 (ns) | ΔH⧧ (kJ/mol) | q | Ao/ℏ (×107 rad/s) | τm310 (ns) | ΔH⧧ (kJ/mol) | |
| HBET | 1 | 2.51 ± 0.04 | 22 ± 1 | 40.0 ± 0.9 | 1 | 3.54 ± 0.55 | 0.27 ± 0.02 | 33.8 ± 1.5 |
| HBET–OMe | 1 | 2.44 ± 0.04 | 23 ± 1 | 41.2 ± 0.9 | 1 | 4.15 ± 0.98 | 0.28 ± 0.01 | 40.7 ± 1.1 |
| HBET–NO2 | 0.5 | 3.02 ± 0.04 | 2.4 ± 0.1 | 39.9 ± 0.6 | 0.5 | 3.48 ± 0.09 | 2.1 ± 0.1 | 41.2 ± 1.1 |
| CyHBET | 1 | 2.53 ± 0.03 | 8.0 ± 0.2 | 38.4 ± 0.5 | 1 | 3.36 ± 0.99 | 0.13 ± 0.01 | 41.2 ± 3.4 |
| CyHBET–OMe | 1 | 2.46 ± 0.03 | 8.8 ± 0.7 | 38.6 ± 1.5 | 1 | 4.02 ± 0.63 | 0.33 ± 0.02 | 20.7 ± 1.6 |
| CyHBET–NO2 | 1 | 3.75 ± 0.58 | 0.52 ± 0.04 | 30.4 ± 2.3 | 1 | 3.97 ± 0.60 | 0.67 ± 0.02 | 31.3 ± 1.0 |
Charges omitted for clarity. Hydration state q = 1 is maintained for all measured HML species. Hydration state remains unchanged upon conversion to ML with the exception of [MnII(HBET–NO2)]2–, which was measured as q = 0.5, which implies a mixture of q = 0 and q = 1 species. Water exchange is accelerated about 3-fold in complexes of the CyHBET–R′ series as compared to HBET–R′.
Mn(III/II) Redox Behavior
Cyclic voltammetry (CV) measurements were performed to understand the influence that ligand electronic and structural changes exert over redox response. Measurements performed on isolable Mn(II) and Mn(III) sister complexes afforded identical voltammograms. Scanning between −0.20 and 0.75 V, the complexes displayed a reversible redox response between 0.45 and 0.57 V vs NHE (Table 4, Figure 8, Supporting Information Figures S37–40). The redox potentials vary little between Mn chelated by the R′ = −H and −OMe functionalized ligands (0.45–0.47 V), whereas the redox potentials of the −NO2 functionalized complexes occur at approximately 0.12 V more oxidizing potential. Scanning to 1.20 V reveals an additional oxidation event between 0.89 and 1.07 V for the R′ = −H and −OMe functionalized ligands (Supporting Information Figures S37–39, denoted Eox2 in Table 7). This second oxidation event is not observed for the −NO2 functionalized complexes. For [MnII/III(HBET–OMe)]2–/1– and [MnII/III(CyHBET–OMe)]2–/1–, the redox wave of the first event is no longer reversible upon scanning back in the reductive direction from this second oxidation event. Rather, a new reduction event emerges at 0.06 V.
Table 4. Redox Potential versus NHE of Mn(II/III) Couple, Irreversible Second Oxidation Event, Irreversible Oxidation Event of Corresponding Zn(II) Complexes, and Potential Difference between Eox of Zn(II) Complex and Mn(II/III) Couple (ΔEox Zn(II)–Mn(II)) at pH 7.4, 0.5 M KNO3.
| E1/2 Mn(III/II) (V) | Eox2 (V) | Eox Zn(II) (V) | ΔEox Zn(II)–Mn(II (V) | |
|---|---|---|---|---|
| HBET | 0.46 | 1.07 | 1.13 | 0.58 |
| HBET–OMe | 0.47 | 1.04 | 0.79 | 0.22 |
| HBET–NO2 | 0.56 | |||
| CyHBET | 0.45 | 0.98 | 1.01 | 0.45 |
| CyHBET–OMe | 0.46 | 0.94 | 0.76 | 0.23 |
| CyHBET–NO2 | 0.57 |
Figure 8.

CV of Mn(II) scanning from −0.20 to 0.75 V (−) and Zn(II) scanning from −0.30 to 1.25 V (- - -) complexes of HBET (left) and HBET–OMe (right), 5 mM complex, GC working electrode, Pt counter electrode, pH 7.4 with 0.5 M KNO3 as supporting electrolyte, scan rate: 100 mV/s. Arrows indicate position from which scans were initiated.
For [MnII/III(HBET–NO2)]2–/1– and [MnII/III(CyHBET–NO2)]2–/1–, scanning in the oxidative direction from −0.80 V affected the appearance of a new redox couple at 0.27 and 0.29 V, respectively (Supporting Information Figure S40). This is attributed to reduction of −NO2 to the more electron-releasing R′ = −NH2 functional group.68 Scanning from −0.80 V imparts no changes when R′ = −H, −OMe.
To confirm the participation of Mn in the reversible redox events, electrochemical characterization of the corresponding Zn(II) complexes was performed (Table 4, Figure 8, Supporting Information Figures S37–40). Zn(II) is redox innocent within the potential window analyzed and allows for unambiguous assignment of ligand-based activity.69−72 It should be noted that no electrochemical response was observed when the ligands were scanned in the absence of metals between −0.30 and 1.20 V at pH 7.4. The reversible events occurring between 0.45 and 0.57 V were absent in the CVs of the Zn(II) complexes. Scanning from −0.30 V, irreversible events attributed to ligand oxidation were found in all complexes except those featuring R′ −NO2 functionalization. Scanning in the oxidizing direction from −0.80 V brought upon the appearance of irreversible oxidation events at 0.42 and 0.39 V for [ZnII(HBET–NO2)]2– and [ZnII(CyHBET–NO2)]2–, respectively (Supporting Information Figure S40).
Reduction of Mn(III) by l-Cysteine
The reduction kinetics of the four isolable Mn(III) complexes by cysteine were measured under pseudo-first-order conditions (0.5 mM Mn(III), 10 mM cysteine). Conversion to Mn(II) was monitored by following disappearance of absorbances unique to Mn(III). For [MnIII(HBET)]− and [MnIII(CyHBET)]−, λmax 375 (ε = 1.88 × 103 and 1.06 × 103 M–1 s–1, respectively) was used as the spectroscopic handle; for [MnIII(HBET–NO2)]− and [MnIII(CyHBET–NO2)]−, λmax 496 (ε = 1.15 × 103 and 1.25 × 103 M–1 s–1, respectively) was used (Supporting Information Figures S41,42). The observed pseudo-first-order reaction rates (kobs) are depicted in Table 5. Separately analyzing complexes of R′ = −H and −NO2, kobs does not appear to be heavily influenced by the structural differences between the HBET–R′ and CyHBET–R′ ligand backbones. The reduction kinetics do however reflect the electronic nature of the R′ substituent. Reduction of the R′ = −H complexes occurs an order of magnitude more slowly than those featuring −NO2 functionalization.
Table 5. Observed Rate Constant for Conversion of 0.5 mM Mn(III) to Mn(II) in the Presence of 10 mM l-Cysteine at pH 7.4, 37 °C.
| kobs (s–1) | |
|---|---|
| [Mn(HBET)]− | 0.042 ± 0.001 |
| [Mn(HBET–NO2)]− | 0.732 ± 0.006 |
| [Mn(CyHBET)]− | 0.063 ± 0.000 |
| [Mn(CyHBET–NO2)]− | 0.563 ± 0.004 |
Discussion
Of the six ligands synthesized for this study, we were successful in isolating all six Mn(II) complexes and four Mn(III) complexes. The Mn(II) complexes are more potent relaxation agents at 1.4 T than sister Mn(III) complexes and afford greater MRI signal enhancement in T1-weighted images at 4.7 T.
At a given field strength, relaxation in the presence of a paramagnetic species is influenced by three dynamic parameters: τm, the rotational correlation time (τR), and longitudinal electronic relaxation time (T1e). Whichever process occurs on the fastest time scale will limit relaxivity.73−77 The determinants limiting nuclear relaxation in the presence of Mn(III) versus Mn(II) differ. The Mn(III) ion is characterized by very rapid T1e and is thus less sensitive to changes in τR and τm.46,78−80 For Mn(II), the influence of T1e is negligible at 1.4 T and above, and relaxivity is controlled by τm and τR.75 Given this mechanistic divergence, we anticipate that we can further amplify Mn(III) versus Mn(II) relaxivity differentials through fine-tuning the solution dynamics of the Mn-containing species.
The library of 12 Mn complexes studied here provides a platform for systematic evaluation of the effects of ligand structural and electronic modifications on complex stability, solution structure and water exchange parameters. These physical properties control relaxivity, as well as Mn(II/III) redox potential and Mn(III) reduction kinetics.
The pH-potentiometric measurements indicate that at pH 7.4, the complexes exist as mixtures of ML and HML. The fraction of HML composition at pH 7.4 increases with the pKa of the phenol moiety, and stability at pH 7.4 decreases as ligand pKa increases. Monitoring the UV–vis absorbance profile as a function of pH indicated that the HML species corresponds to protonation at the phenolato-O donor. Surprisingly, the preorganizing trans-1,2-cyclohexylenediamine backbone does not confer the increase in pH 7.4 stability that we anticipated through analogy with [Mn(EDTA)2– and [Mn(CDTA)]2–.
Defining the pH-dependence on complex speciation laid the framework to measure the hydration state and water exchange parameters of the HML and ML species using 17O NMR. Variable-temperature 17O measurements performed at pH 6 and 9, where Mn(II) speciation is comprised of predominantly HML or ML, respectively, reveal that the Mn(II) hydration state remains unchanged upon complex deprotonation. Mn(II) remains q = 1 for all species, except [MnII(HBET–NO2)]2–, which is q = 0.5. Mn(II) is 7-coordinate for the monoaqua ML complexes. Monoaqua MLH is either 6- or 7-coordinate, depending on whether the phenol remains coordinated upon protonation. The precise nature of this interaction cannot be conclusively determined from the available data.
Although complex speciation does not affect q, the water exchange rate is accelerated by 2 orders of magnitude upon deprotonation of HML. In fact, the water exchange rates exhibited by the R′ = −H and −OMe ML species are among the fastest reported.81 The R′ = −NO2 ML species exhibit slightly slower kinetics, but water exchange is still very rapid. It appears that the CyHBET–R′ ligands promote approximately 3-fold faster exchange then their HBET–R′ analogues in both the HML and the ML forms.
We note that the relaxivity of HML species is slightly higher than that of deprotonated ML. Because both HML and ML are the same size, the rotational correlation time should be very similar. They also each have a water coligand. One explanation for the slightly higher HML relaxivity could be prototropic exchange of the protonated phenol moiety. Another explanation could be the extremely rapid water exchange kinetics for the ML species. The dominant correlation time for these small Mn(II) complexes is expected to be rotation, but for some of the ML species where τR at 37 °C is on the order of 100 ps, this rapid exchange rate could also limit relaxivity.
We also observed variability in the relaxivity of the Mn(III) complexes. The mechanism of high-spin Mn(III)-induced nuclear relaxation is less well understood. Presumably, the dominant correlation time is the electronic T1e. This relaxation time should be influenced in part by the ligand field, and it may not be surprising that modifying the ligand can change r1 for the Mn(III) complexes by up to 3-fold. More work on the Mn(III) complexes is required to better understand the relaxation mechanism and how the ligand alters relaxivity.
CV measurements taken on the 10 isolated complexes revealed reversible redox events occurring near the midpoint of the quasi-reversible [MnII/III(HBED)]2– and [MnII/III(EDTA)]2– redox couples and thus are attributed to Mn(II/III) activity.45 The Mn(II/III) events are influenced by the electron-releasing properties of the 5-R′ group. Within the series of isolated complexes, changing the R′ substituent caused a 0.12 V change in the Mn(II/III) couple. If we expand our analysis to include the electrochemically generated R′ = −NH2 complexes, redox tuning over 0.30 V is achieved via changing a single R′ substituent. Similarly, the potential of the second irreversible oxidation event is depressed between 0.30 and 0.40 V when R′ = −OMe as compared to −H, whereas this event is not observed up to 1.20 V when R′ = −NO2. This second oxidation event associated with the Mn complexes is of less concern, however. Within the extracellular spaces, there are few endogenous redox partners capable of achieving this oxidation.
With the exception of −NO2 reduction below −0.80 V, CV measurements performed on the analogous Zn(II) complexes exhibit only irreversible ligand-based oxidation and confirm Mn participation in the reversible events observed between 0.45 and 0.57 V. It is noted that changes to R′ effect more dramatic shifts to ligand oxidation potential than to the Mn(II/III) potential. For example, switching from R′ = −H to −OMe effects a 0.25 and 0.34 V depression in oxidation potential of the Zn(II) complexes of HBET–R′ and CyHBET–R′, respectively, whereas this modification leaves the reversible couple in the Mn complexes virtually unchanged.
The probability of ligand participation in the reversible redox event increases as the difference between ligand and Mn(II/III) oxidation potential (ΔEox Zn(II)–Mn(II)) decreases.82 This could potentially explain why reaction of the R′ = −H and −NO2 containing ligands with MnF3 cleanly afforded the corresponding Mn(III) complexes, whereas the −OMe functionalized ligands yielded a complex product mixture from which the target Mn(III) complex could not be isolated but products of oxidative ligand coupling could be identified. The smaller ΔEox Zn(II)–Mn(II) when R′ = −OMe (0.22–0.23 V) suggests a strong possibility of Mn(III)–ligand autoredox processes.82 Additionally, there is prior precedence of decomposition of nascent Mn(III) through oxidative C–C bond formation involving 2-hydroxybenzyl containing ligands.83 The rapid decomposition of [MnIII(HBET–OMe)]− generated in situ through stoichiometric oxidation of Mn(II) confirms the instability of Mn(III) within this ligand frame. The Mn(II/III) couple can be modulated through fine-tuning of phenol substituents, but Mn redox must be carefully balanced against ligand oxidation by Mn(III).
It is not only important to consider the Mn(II/III) redox potential but also the reduction kinetics in the presence of redox partners encountered in vivo. In this regard, the rate of conversion of Mn(III) to Mn(II) was measured in the presence of cysteine. Cysteine/cystine composition largely dictates the redox status of the extracellular spaces we aim to study by MRI. The reduction kinetics of the R′ = −H functionalized complexes proceed more slowly than those of R′ = −NO2 by an order of magnitude. Structural differences engendered by the ligand backbone are of lesser importance. Although a detailed kinetic and mechanistic analysis is beyond the scope of this Article, we have previously explored the reduction kinetics of [MnIII(HBET)]− in the presence of glutathione.45 Conversion to Mn(II) was found to exhibit first-order dependence on both [Mn(III)] and [thiol]. In human plasma, the cysteine concentration has been reported at 8–10 μM, and cysteine concentration between 40 and 50 μM.84 Assuming reduction in the presence of cysteine is a mechanistically analogous process, we can anticipate the Mn(III) complexes studied here could be expected to exhibit plasma half-lives on the order of 30–300 min. Importantly, these results demonstrate that it is possible to exercise control over Mn(III) reduction kinetics through tuning ligand electronics.
Conclusions
The development of imaging probes to monitor redox activity in vivo represents a difficult but important challenge in biomedical research. Mn complexed by the HBET–R′ ligands described in this study represents an excellent mechanism toward achieving this end through redox-stimulated MR signal enhancement. The experiments described above were performed to probe the influence of structural and electronic modifications on relaxivity turn-on, Mn(II) stability, speciation and solvation dynamics, Mn(II/III) redox response, and Mn(III) reduction kinetics.
Some relationships emerge from the series of experiments described above. (1) Mn(II) versus Mn(III) signal turn-on is influenced by the surrounding ligand environment. For the small molecules studied here, we observed between 2.5- and 7.5-fold change in r1. (2) Increasing Mn(II) HML speciation at pH 7.4 correlates to reduced thermodynamic stability. (3) Switching the ligand backbone from ethylenediamine to trans-1,2-cyclohexylenediamine does not confer the anticipated increase in stability at pH 7.4. (4) Mn(II) water exchange kinetics for ML are roughly 2 orders of magnitude faster than the corresponding HML species. (5) The reversible Mn(II/III) couple can be tuned through substitutions at the phenol aromatic ring. (6) The Mn(II/III) oxidation potential must be weighed against that of the ligand; Mn(III)–ligand autoredox presents a pathway for Mn(III) decomposition. (7) The rate of Mn(III) reduction in the presence of cysteine is influenced by the electron-releasing nature of the phenolato-O donor.
The structure–redox–relaxivity relationships outlined in this study serve to unveil rich and hitherto unexplored Mn coordination chemistry that can be exploited to overcome limitations in the available molecular imaging toolset. These relationships provide a chemical guide by which to optimize reversibly activated Mn(II/III) MR imaging probes for translational use. For example, decelerating rotational motion represents one possible strategy to amplify Mn(II) relaxivity.85−88 Understanding how ligand modifications influence ML versus HML composition, q, and τm provides a framework to predict the influence of changing τR on relaxivity differentials a priori.
We are presently pursuing strategies to incorporate the Mn-based probes into larger, more slowly tumbling entities. We are also working to establish molecular features key to translational success through experiments in animal models. The findings from this study provide a context by which to interpret results in this next phase of exploration.
Experimental Section
General
All chemicals and solvents were purchased commercially and used without further purification. NMR spectra were recorded on a 500 MHz Varian spectrometer. Chemical shifts are reported in δ (ppm). For 1H and 13C NMR spectra, the residual solvent peaks were used as internal reference except for the 13C NMR of the ligand where tert-BuOH was used as the internal reference. Liquid chromatography–mass spectrometry (LC–MS) was performed using an Agilent 1100 Series apparatus with an LC/MSD trap and Daly conversion dynode detector with UV detection at 220, 254, and 280 nm. The methods used on this system are as follows: (a) Luna C18 column (100 × 2 mm); eluent A, H2O/0.1% formic acid, B, MeCN/0.1% formic acid; gradient, 5% B to 95% B over 9 min; flow rate 0.8 mL/min (used for characterization of organic compounds); (b) Kromasil C18 column (250 × 4.6 mm); eluent C, 95% MeCN/5% 10 mM ammonium acetate; D, 10 mM ammonium acetate; gradient 5% C to 8% C over 14 min; flow rate 0.8 mL/min (used for characterization of manganese complexes); (c) Kromasil C4 column (250 × 4.6 mm); eluent C, 95% MeCN/5% 10 mM ammonium acetate; D, 10 mM ammonium acetate; gradient 5% C to 95% C over 10 min; flow rate 0.8 mL/min (used for characterization of manganese complexes). Reverse-phase semipreparative purification was performed on the Rainin Dynamax HPLC system with UV detection from 220 to 280 nm using a Polaris C18 column. Mobile phase A was 50 mM ammonium acetate buffer, pH 6.5, and mobile phase B was a mixture of 5% 50 mM ammonium acetate buffer, pH 6.5 and 95% MeCN. The methods used for purification are as follows: (a) starting from 5% B, the fraction of B increased to 8% over 23 min. The column was washed with 95% B for 2 min and then ramped to 5% B. The system was re-equilibrated at 5% B for 3 min. (b) Starting from 5% B, the fraction of B increased to 50% over 23 min. The column was washed with 95% B for 2 min and then ramped to 5% B. The system was re-equilibrated at 5% B for 3 min. Cyclic voltammetry measurements were performed using a Nuvant EZstat Pro potentiostat; the ferri/ferrocyanide couple was used as the internal standard. pH-potentiometric measurements were performed using an MPT 798 Titrino equipped with an Orion ROSS Ultra pH electrode and temperature-controlled reaction vessel held at 298 K. Samples were purged with Ar prior to measurement, and an inert atmosphere was maintained by constant Ar passage over the titration vessel. The data were analyzed using the Hyperquad2013 software package.89 All other pH measurements were performed using a ThermoOrion pH meter connected to a VWR Symphony glass electrode. UV–vis spectra were recorded on a SpectraMax M2 spectrophotometer using quartz cuvettes with a 1 cm path length. Manganese concentrations were determined using either an Agilent 7500a or 8800-QQQ ICP-MS system. All samples were diluted with 0.1% Triton X-100 in 5% nitric acid containing 20 ppb of Lu (as internal standard). The ratio of Mn (54.94) to Lu (174.97) was used to quantify the manganese concentration. A linear calibration curve ranging from 0.1 to 200 ppb was generated daily for the quantification.
1H/17O Relaxometry
Relaxivity measurements were performed on a Bruker mq60 minispec, 1.41 T and 37 °C. Longitudinal (T1) relaxation was acquired via an inversion recovery experiment using 10 inversion times of duration ranging between 0.05 × T1 and 5 × T1; tranverse (T2) relaxation was measured using a Carl–Purcell–Meiboom–Gill spin–echo experiment. Relaxivity (r1,2) was determined from the slope of a plot of 1/T1,2 versus [Mn] for at least four concentrations. The transverse (T2) relaxation times of 17O were acquired at 11.7 T from the full-width at half-height of the H217O signal.52 Previous work has shown that T2 times acquired through line-width data are nearly identical to those obtained using the CPMG pulse sequence. 17O T2 relaxivity (r2O) was calculated by dividing the Mn-imparted increase in 1/T2 relative to neat H2O at pH 3 by the Mn concentration in millimolar. Samples were enriched with a small amount of H217O. The 17O chemical shift measurements were performed in 10% D2O for frequency locking. The correction of the chemical shift to bulk magnetic susceptibility was taken from the 1H chemical shift of tert-BuOH in the presence and absence of Mn(II).
MR Imaging
Images were acquired using a Bruker Biospec 4.7 T system. Phantoms were positioned in a homemade sample holder and imaged using a volume coil. Acquisition matrix was 185 × 120 for 0.378 mm × 0.250 mm in-plane resolution; slice thickness = 3 mm. T1 images were obtained with a T1-weighted rapid acquisition refocused echo (RARE) sequence: TR/TE = 1000/25.9 ms. T1 times were determined using a 2D RARE inversion recovery sequence: TR = 3300 ms, TE = 9.7 ms. Inversion times (TI): 1, 38, 158, 225, 318, 450, 638, 850, 1200, and 3000 ms. T1 was obtained from a nonlinear least-square fit of the signal intensity (SI(t)) versus TI curve (eq 1), where T1, SI(0), and a are adjustable parameters.
| 1 |
Reduction Kinetics
To 400 μL of a 0.625 mM Mn(III) complex in pH 7.4 Tris buffer was added 100 μL of 50 mM l-cysteine. Final concentrations: 0.5 mM Mn, 10 mM l-cysteine. Conversion to Mn(II) was monitored by observing disappearance of a UV–vis absorbance (A) unique to Mn(III) (375 and 496 nm when R′ = −H, −NO2, respectively). The observed pseudo-first-order rate constant (kobs) was determined by fitting eq 2 to the data, where Ao and Af correspond to the absorbances at t = 0 and at the end of the measurement.
| 2 |
Synthesis
HBET, Na2[MnII(HBET)], and Na[MnIII–HBET)] were prepared as described previously. The syntheses of the CyHBET, Na2[MnII(CyHBET)], and Na[MnIII(CyHBET)] are described below. The other ligands and complexes were prepared analogously and are described in detail in the Supporting Information. The numerical naming system used for simplicity is described in Supporting Information Scheme S1.
tert-Butyl (2-(2-Hydroxy-5-methoxybenzyl)aminoethyl)carbamate (1)
To a solution of 2-hydroxy-5-methoxybenzaldehyde (12.0 mmol, 1.83 g) in 90 mL of MeOH was added a solution of tert-butyl N-(2-aminoethyl)carbamate (12.0 mmol, 1.92 g) in MeOH (30 mL), and the solution was stirred for 1 h. To this stirring solution was added solid NaBH4 (24.0 mmol, 0.908 g). Rapid evolution of gas was observed, and the solution turned colorless from pale yellow. After being stirred for 3 h, all volatiles were removed under reduced pressure, and a white solid was obtained. The residue was dissolved in 200 mL of CH2Cl2 extracted with 200 mL of saturated NaHCO3 solution. The aqueous layer was extracted with CH2Cl2 (2 × 100 mL). All of the organics were combined, washed with brine (200 mL), and dried over anhydrous MgSO4. The solvent was evaporated under reduced pressure to obtain 1 as a pale yellow solid (11.8 mmol, 3.49 g, 98.1%). 1H NMR (500 MHz, CDCl3) δ (ppm): 6.76 (m, 1H), 6.72 (m, 1H), 6.57 (d, 1H), 5.30 (s, 1H), 3.97 (s, 2H), 3.73 (s, 3H), 3.28 (m, 2H), 2.78 (t, 2H), 1.44 (s, 9H). 13C{1H} NMR (100 MHz, CDCl2) δ (ppm): 156.2, 152.3, 151.7, 123.1, 116.5, 114.2, 113.5, 79.3, 55.6, 52.2, 48.3, 39.9, 28.0. Molecular weight for C15H24N2O4: 296.36. MS (ESI) m/z: calcd, 297.37 (M + H)+; observed, 297.4.
Di-tert-butyl 2,2′-((2-((2-(tert-Butoxy)-2-oxoethyl)(2-((tert-butyl dimethylsilyl) oxy)-5-methoxybenzyl)amino)ethyl)azanediyl)diacetate (2)
1 (8.00 mmol, 2.37 g) was dissolved in CH2Cl2 (100 mL) followed by addition of 50 mL of trifluoroacetic acid (TFA). The reaction was stirred for 5 h, and then the volatiles were removed under reduced pressure. The reaction was taken up in 50 mL of water, washed with Et2O, and the water fraction was freeze-dried to produce the free amine quantitatively as a pale yellow solid, which was used in subsequent reaction without further purification.
The round-bottom flask containing the amine was charged with nitrogen, and dry CH2Cl2 (80 mL) was added and cooled in an ice bath. Under counter argon flow, N,N-diisopropylethylamine (40.0 mmol, 6.97 mL) was added, followed by addition of tert-butyldimethylsilyl chloride (8.80 mmol, 1.33 g) as a CH2Cl2 solution (10 mL). The solution was allowed to warm to room temperature and stirred for 5 h. The reaction was cooled back to 0 °C, and tert-butyl bromoacetate (24.8 mmol, 3.66 mL) was added dropwise. The reaction was stirred for 18 h under nitrogen atmosphere. The solution was diluted with CH2Cl2 (200 mL) and washed with saturated NaHCO3 (3 × 200 mL) and brine (1 × 200 mL). All of the organics were combined, dried over anhydrous MgSO4, and evaporated under reduced pressure to obtain a crude yellow oil. The product was purified as a colorless oil (1.46 g, 4.13 mmol, 51.7%) by using column chromatography; eluent: hexane/ethyl acetate, 9:1. 1H NMR (400 MHz, CDCl3) δ (ppm): 7.04 (d, 1H), 6.62 (m, 1H), 6.57 (m, 1H), 3.71 (s, 2H), 3.70 (s, 3H), 3.40 (s, 4H,), 3.27 (s, 2H), 2.79 (m, 4H), 1.40 (s, 9H), 1.37 (s, 18H), 0.94 (s, 9H), 0.13 (s, 6H). 13C{1H} NMR (100 MHz, CDCl2) δ (ppm): 171, 170.8, 154.1, 147.4, 130.8, 119.2, 114.7, 113.0, 80.8, 80.6, 56.3, 56.1, 55.6, 53.1, 52.8, 52.7, 28.3, 28.2, 26.0, 18.4. Molecular weight for C34H60N2O8Si: 652.93. MS (ESI) m/z: calcd, 653.94 (M + H)+; observed, 653.9.
2,2′-(2-((Carboxymethyl)((2-hydroxy-5-methoxybenzyl)amino)ethyl) azanediyl) Diacetic Acid (HBET–OMe) (3)
2 (2.24 mmol, 1.46 g) was dissolved in TFA (40 mL) followed by addition of triisopropylsilane (2.35 mL), 1-dodecanethiol (2.35 mL), and water (2.35 mL). The reaction was stirred for 5 h, and then the volatiles were removed under reduced pressure. The residue was dissolved in water (40 mL) and washed with Et2O (3 × 40 mL). The water fraction was freeze-dried to produce 3 quantitatively as a white solid. 1H NMR (500 MHz, D2O) δ (ppm): 7.02 (m, 2H) 6.95 (m, 1H), 4.52 (5, 2H), 4.09 (s, 2H), 3.80 (s, 3H), 3.58 (s, 4H), 3.48 (m, 2H), 3.26 (m, 2H). 13C{1H} NMR (125 MHz, D2O) δ (ppm): 173.6, 169.0, 152.5, 149.6, 117.9, 117.6, 117.0, 116.4, 55.9, 54.8, 51.5, 49.0. Molecular weight for C16H22N2O8: 370.35. MS (ESI) m/z: calcd, 371.36 (M + H)+; observed, 371.4.
Na2[MnII(HBET–OMe)] (4)
3 (0.23 mmol, 0.085 g) was dissolved in 5 mL of water. The pH was adjusted to 8 using 1 N NaOH solution. MnCl2·4H2O (0.23 mmol, 0.046 g) was then added to the solution, and the pH was carefully adjusted to 6.5. The reaction was stirred for 1 h, filtered, and freeze-dried to yield a white solid. The complex was injected onto a reverse phase C18 (Polaris) column and desalted using the method as described earlier. The fractions were collected and lyophilized to yield 4 as a white solid (0.19 mmol, 0.090 g, 81%). Molecular weight for C16H22MnN2O8: 421.26. MS (ESI) m/z: calcd, 424.28 (M + 3H)+; observed, 424.3.
tert-Butyl (trans-2-((2-Hydroxybenzyl)amino)cyclohexyl)carbamate (10)
To a solution of N-BOC-trans-1,2-diaminocyclohexane·HCl (3.99 mmol, 1.00 g) in 90 mL of MeOH was added NEt3 (4.39 mmol, 0.600 mL), and the reaction was stirred for 30 min. To the above mixture was added a solution of salicylaldehyde (3.99 mmol, 0.487 g) in MeOH (30.0 mL). After being stirred for 1 h, solid NaBH4 (8.38 mmol, 0.317 g) was added, and the reaction was stirred for 3 h. All of the volatiles were removed under reduced pressure to yield a pale yellow solid. The residue was dissolved in 200 mL of CH2Cl2 and extracted with 200 mL of saturated NaHCO3 solution. The aqueous layer was extracted with CH2Cl2 (2 × 100 mL). All of the organics were combined, washed with brine (200 mL), and dried over anhydrous MgSO4. The solvent was evaporated under reduced pressure to obtain 10 as a pale yellow solid (3.83 mmol, 1.23 g, 96.2%). 1H NMR (500 MHz, CDCl3) δ (ppm): 7.15 (m, 1H), 6.96 (m, 1H), 6.81 (m, 1H), 6.75 (m, 1H), 4.43 (s, 1H), 4.05 (d, 1 H), 3.93 (d, 1H), 3.41 (s, 1H), 2.31 (m, 1H), 2.17 (m, 1 H), 1.99 (m, 1H), 1.70 (m, 2H), 1.46 (s, 9H), 1.31 (m, 1H), 1.17 (m, 2H). 13C{1H} NMR (125 MHz, CDCl3) δ (ppm): 158.3, 156.0, 128.4, 128.0, 123.1, 118.7, 116.3, 79.4, 60.9, 53.9, 49.8, 33.0, 31.2, 28.3, 24.9, 24.5. Molecular weight for C18H28N2O3: 320.43. MS (ESI) m/z: calcd, 321.43 (M + H)+; observed, 321.5.
Di-tert-butyl 2,2′-((trans-2-((2-(tert-Butoxy)-2-oxoethyl)(2-hydroxybenzyl)amino) cyclohexyl)azanediyl)diacetate (11)
10 (3.15 mmol, 1.01 g) was dissolved in CH2Cl2 (100 mL) followed by addition of 50 mL of trifluoroacetic acid (TFA). The reaction was stirred for 5 h, and then the volatiles were removed under reduced pressure. The reaction was taken up in 50 mL of water, washed with Et2O, and the water fraction was freeze-dried to produce the diamine quantitatively as a pale yellow solid, which was used in subsequent reaction without further purification.
To the round-bottom flask containing the amine was added potassium iodide (6.30 mmol, 1.04 g), and the system was purged with nitrogen. Under counter nitrogen flow, dry dimethylformamide (2 mL) was added followed by the addition of N,N-diisopropylethylamine (15.80 mmol, 2.74 mL) and dropwise addition of tert-butyl bromoacetate (9.77 mmol, 1.90 g). The reaction was stirred for 18 h and then partitioned between saturated NaHCO3 solution and Et2O. The Et2O layer was separated and washed with several changes of H2O to remove DMF before drying over Na2SO4 and concentration to 1.00 g of yellow oil. Molecular weight for C31H50N2O7: 562.74. MS (ESI) m/z: calcd, 563.75 (M + H)+; observed, 563.8. The crude product was carried on in the next step without further purification.
2,2′-((trans-2-((Carboxymethyl)(2-hydroxybenzyl)amino) cyclohexyl)azanediyl) Diacetic Acid (CyHBET) (12)
The crude product (11) from the previous step was dissolved in TFA (40 mL) followed by addition of triisopropylsilane (2.35 mL), 1-dodecanethiol (2.35 mL), and water (2.35 mL). The reaction was stirred for 5 h, and then the volatiles were removed under reduced pressure. The residue was dissolved in water (40 mL) and washed with Et2O (3 × 40 mL). The water fraction was freeze-dried to produce crude 12. The product was then purified via preparative HPLC using method B. The fractions were collected and lyophilized to yield 12 as a white solid (1.0 mmol, 0.51 g, 32% from 10). 1H NMR (500 MHz, D2O) δ (ppm): 7.45 (m, 1H), 7.37 (m, 1H), 6.98 (m, 2H), 4.42 (s, 2H), 4.17 (d, 1H), 3.90 (d, 1H), 3.51 (br, 2H), 3.35 (br, 1H), 3.24 (br, 1H), 3.00 (br, 1H), 2.91 (br, 1H), 2.33–1.12 (8H).13C{1H} NMR (125 MHz, D2O) δ (ppm): 174.5, 170.4, 156.1, 133.3, 132.9, 121.9, 117.1, 116.6, 62.0, 59.6, 53.8, 52.3, 51.0, 48.3, 24.4. Molecular weight for C19H26N2O7: 394.42. MS (ESI) m/z: calcd, 395.43 (M + H)+; observed, 395.5.
Na2[MnII(CyHBET)] (13)
12 (0.260 mmol, 0.103 g) was dissolved in 5 mL of water. The pH was adjusted to 8 using 1 N NaOH solution. MnCl2·4H2O (0.260 mmol, 0.0510 g) was then added to the solution, and the pH was carefully adjusted to 6.5. The reaction was stirred for 1 h, filtered, and lyophilized to yield a white solid. The complex was injected onto a reverse phase C18 (Polaris) column and desalted using the method described above. Fractions were collected and lyophilized to yield 13 as a white solid (0.216 mmol, 0.106 g, 83.0%). Molecular weight for C19H21MnN2O7 445.34. MS (ESI) m/z: calcd, 448.35 (M + 3H)+; observed, 448.4.
Na[MnIII(CyHBET)] (14)
12 (0.11 mmol, 0.057 g) was dissolved in 8 mL of water. The pH was adjusted to 8 using 1 N NaOH solution. MnF3 (0.11 mmol, 0.013 g) was then added as a slurry in 2 mL of water, and pH 8 was maintained by addition of 1 M NaOH. The red-brown reaction mixture was filtered and immediately purified by RP-HPLC by method B. The fractions were collected and lyophilized to yield 14 as a brown solid (0.11 mmol, 0.050 g, 95%). Molecular weight for C19H21MnN2O7: 445.33. MS (ESI) m/z: calcd, 447.34 (M + 2H)+; observed, 447.4.
tert-Butyl (2-((2-Hydroxy-5-nitrobenzyl)amino)ethyl)carbamate (5)
To a solution of 2-hydroxy-5-nitrobenzaldehyde (3.51 mmol, 0.587 g) in 60 mL of MeOH was added a solution of tert-butyl N-(2-aminoethyl)carbamate (3.51 mmol, 0.562 g) in MeOH (30 mL), and the solution was stirred for 1 h. To this stirring solution was added solid NaBH4 (7.02 mmol, 0.266 g). Rapid evolution of gas was observed, and the solution turned colorless from pale yellow. After being stirred for 3 h, all volatiles were removed under reduced pressure, and a white solid was obtained. The residue was dissolved in a solvent mixture of 10 mL of MeOH and 200 mL of CH2Cl2 and extracted with 200 mL of saturated NaHCO3 solution. The aqueous layer was extracted with CH2Cl2 (2 × 100 mL). All of the organics were combined, washed with brine (200 mL), and dried over anhydrous MgSO4. The solvent was evaporated under reduced pressure to obtain 10 as a yellow solid (3.24 mmol, 1.01 g, 92.4%). 1H NMR (500 MHz, (CD3)2SO) δ (ppm): 8.01 (d, 1H), 7.92 (dd, 1H), 6.92 (t, 1H), 6.46 (d, 1H), 3.93 (s, 2H), 3.14 (m, 2H), 2.75 (t, 2H), 1.38 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3) δ (ppm): 172.1, 155.7, 133.6, 126.1, 126.0, 121.9, 117.3, 77.9, 48.7, 46.7, 37.9, 28.2. Molecular weight for C14H21N3O5: 311.33. MS (ESI) m/z: calcd, 312.34 (M + H)+; observed, 312.4.
Di-tert-butyl-2,2′-((2-((2-(tert-butoxy)-2-oxoethyl)(2-((tert-butyl diphenylsilyl) oxy)-5-nitrobenzyl)amino)ethyl)azanediyl)diacetate (6)
10 (3.24 mmol, 1.01 g) was dissolved in CH2Cl2 (100 mL) followed by addition of 50 mL of trifluoroacetic acid. The reaction was stirred for 5 h, and then the volatiles were removed under reduced pressure. The reaction was taken up in 50 mL of water, washed with Et2O, and the water fraction was freeze-dried to produce the free amine quantitatively as a pale yellow solid, which was used in subsequent reaction without further purification.
The round-bottom flask containing the amine was charged with nitrogen, and dry dimethylformamide (40 mL) was added and cooled in an ice bath. Under counter argon flow, N,N-diisopropylethylamine (16.2 mmol, 2.82 mL) was added, followed by addition of tert-butyldiphenylsilyl chloride (3.56 mmol, 0.979 g) as a dimethylformamide solution (5 mL). The solution was allowed to warm to room temperature and was stirred for 5 h. The reaction was cooled back to 0 °C, tert-butyl bromoacetate (10.0 mmol, 1.48 mL) was added dropwise, and the reaction was stirred for 18 h under nitrogen atmosphere. The solution was diluted with CH2Cl2 (200 mL) and washed with saturated NaHCO3 (3 × 200 mL) and brine (1 × 200 mL). All of the organics were combined, dried over anhydrous MgSO4, and evaporated under reduced pressure to obtain crude yellow oil. The product was purified as a colorless oil (0.777 mmol, 0.615 g, 23.9%) by using column chromatography; eluent: hexane/ethyl acetate, 9:1. 1H NMR (400 MHz, CDCI3) δ (ppm): 8.50 (d, 1H), 7.67 (m, 5H), 7.45 (m, 1H), 7.39 (m, 5H), 6.40 (d, 1 H), 4.08 (s, 2H), 3.48 (s, 4H), 3.44 (s, 2H), 2.93 (s, 4H), 1.48 (s, 9H), 1.43 (s, 18H), 1.1194 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3) δ (ppm): 170.8, 159.0, 142.1, 135.3, 131.4, 130.5, 128.2, 125.6, 123.3, 118.7, 81.1, 81.0, 56.5, 56.2, 53.0, 52.8, 52.7, 28.2, 26.5, 19.7. Molecular weight for C43H61N3O9Si: 792.04. MS (ESI) m/z: calcd, 793.05 (M + H)+; observed, 793.1.
2,2-((2-((Carboxymethyl)(2-hydroxy-5-nitrobenzyl)amino)ethyl)azanediyl) Diacetic Acid (HBET–NO2) (7)
6 (0.776 mmol, 0.615 g) was dissolved in trifluoroacetic acid (40 mL) followed by addition of triisopropylsilane (2.35 mL), 1-dodecanethiol (2.35 mL), and water (2.35 mL). The reaction was stirred for 5 h, and then the volatiles were removed under reduced pressure. The water fraction was freeze-dried to produce 7 quantitatively as a white solid. 1H NMR (500 MHz, D2O) δ (ppm): 8.33 (m, 1H), 8.22 (m, 1H), 7.06 (d, 1H), 4.57 (s, 2H), 4.07 (s, 2H), 3.60 (s, 4H), 3.49 (m, 2H), 3.27 (m, 2H). 13C{1H} NMR (125 MHz, D2O) δ (ppm): 173.5, 169.1, 162.2, 140.1, 129.1, 128.0, 116.7, 116.0, 54.8, 54.0, 51.9, 49.2. Molecular weight for C15H19N3O9: 385.33. MS (ESI) m/z: calcd, 386.33 (M + H)+; observed, 386.4.
Na2[MnII(HBET–NO2)] (8)
7 (0.25 mmol, 0.096 g) was dissolved in 5 mL of water. The pH was adjusted to 8 using 1 N NaOH solution. MnCl2·4H2O (0.25 mmol, 0.049 g) was then added to the solution, and the pH was carefully adjusted to 6.5. The reaction was stirred for 1 h, filtered, and freeze-dried to yield a white solid. The complex was injected onto a reverse phase C18 (Polaris) column and desalted using the method as described earlier. The fractions were collected and lyophilized to yield 8 as a light yellow solid (0.22 mmol, 0.10 g, 82%). Molecular weight for C15H17MnN3O9: 436.23. MS (ESI) m/z: calcd, 439.04 (M + 3H)+; observed, 439.4.
Na[MnIII(HBET–NO2)] (9)
MnF3 (0.054 mmol, 0.0060 g) was added to 7 (0.054 mmol, 0.021 g) with stirring in 5 mL of H2O at pH 8. The resultant red-orange solution was injected onto a reverse phase C18 (Polaris) column and purified using the method as described earlier. The fractions were collected and lyophilized to yield 9 as a brown solid (0.026 mmol, 0.012 g, 48%). Molecular weight for C19H21MnN3O9: 436.02. MS (ESI) m/z: calcd, 438.04 (M + 2H)+; observed, 438.0.
tert-Butyl (2-((2-Hydroxy-5-methoxybenzylidene)amino)cyclohexyl)carbamate (15)
trans-2-((tert-Butoxycarbonyl)amino)cyclohexane (1.30 mmol, 0.279 g) and 2-hydroxy-5-methoxybenzaldehyde (1.35 mmol, 0.206 g) were stirred together in 12 mL of MeOH at room temperature. Within minutes, copious precipitate fell from the bright yellow solution. After 90 min of stirring, 30 mL of H2O was added to the mixture to precipitate the Schiff base. The product was isolated by filtration and immediately carried through to the next step. 1H NMR (500 MHz, CDCl3) δ (ppm): 12.78 (s, 1H), 8.28 (s, 1H), 6.90 (d, 1H), 6.76 (d), 4.63 (s, 1H), 3.73 (s, 3H), 3.57 (s, 1H), 3.03 (s, 1H), 2.06 (m, 1H), 1.90 (m, 3H), 1.76 (t, 2H), 1.68 (q, 1H), 1.41 (m, 2H), 1.30 (s, 9H). 13C{1H} NMR (125.7 MHz, CDCl3) δ (ppm): 163.3, 155.4, 155.3, 151.9, 119.3, 118.5, 117.8, 115.0, 79.3, 72.7, 56.1, 54.2, 33.3, 31.6, 28.2, 24.8, 24.0. Molecular weight for C19H28N2O4: 348.44. MS (ESI) m/z: calcd, 349.21 (M + H)+; observed, 349.2
NaBH4 (1.35 mmol, 0.0510 g) was added portionwise to the isolated Schiff base (0.980 mmol, 0.342 g) with stirring in 20 mL of MeOH at room temperature. Within minutes, the yellow color of the solution bleached to pale beige. After 2 h, the solution was concentrated to dryness, taken up in CH2Cl2, and washed thoroughly with water and brine. The organic portion was washed, dried over MgSO4, and concentrated to the 2-hydroxy-5-methoxybenzyl appended amine, isolated as a beige solid (0.179 g, 0.00510 mmol, 52.2%). 1H NMR (500 MHz, CDCl3) δ (ppm): 6.72 (m, 2H), 6.54 (s, 1H), 4.53 (br s, 1H), 3.94 (dd, 2H), 3.38 (br s, 1H), 2.30 (m, 1H), 2.13 (m, 1H), 1.97 (m, 1H), 1.69 (m, 2H), 1.45 (s, 9H), 1.31–1.15 (m, 4H). Molecular weight for C19H30N2O4: 350.22. MS (ESI) m/z: calcd, 351.23 (M + H)+; observed, 351.3.
The N-BOC-protected product (0.510 mmol, 0.179 g) was then dissolved in 5 mL each CH2Cl2/TFA for 5 h. The solution was than concentrated to dryness, dissolved in 50 mL of CH2Cl2, and stirred over an excess of K2CO3(s) for 12 h. The K2CO3 was removed by filtration, and the mother liquor concentrated to 15 as a pale yellow oil in quantitative yield. 1H NMR (500 MHz, CDCl3) δ (ppm): 6.70 (m, 2H), 6.56 (s, 1H), 3.91 (dd, 2H), 3.72 (s, 3H), 2.39 (br t, 1H), 2.10 (m, 2H), 1.79 (m, 1H), 1.66 (m, 2H), 1.28–1.06 (m, 4H). 13C{1H} NMR (125.7 MHz, CDCl3) δ (ppm): 152.4, 152.0, 124.5, 116.7, 114.0, 113.3, 63.8, 55.9, 55.8, 50.3, 37.0, 30.9, 25.3, 24.9. Molecular weight for C14H22N2O2: 250.34. MS (ESI) m/z: calcd, 251.37 (M + H)+; observed, 251.1.
2,2′-((trans-2-((2-(tert-Butoxy)-2-oxoethyl)(2-hydroxy-5-methoxybenzyl)amino)cyclohexyl)azanediyl)diacetate (16)
To 24 (0.640 mmol, 0.161 g) with stirring in 3 mL of DMF with potassium iodide (0.470 mmol, 0.0780 g) and diisopropylethylamine (3.34 mmol, 0.432 g) was added tert-butyl bromoacetate (2.05 mmol, 0.399 g) at room temperature. The pale brown solution quickly developed a white precipitate. After 4 h of stirring, the solution was diluted with 100 mL of Et2O, and washed with Na2CO3(aq), copious water, and brine. The organic layer was concentrated to dryness and purified using a reverse phase C18 (Polaris) column; eluent A, H2O/0.1% TFA; B, MeCN/0.1%TFA; gradient 60% to 95% B over 25 min; flow rate, 20 mL/min. The fractions were lyophilized, than taken up in 50 mL of CH2Cl2, and stirred over solid K2CO3 for 6 h. The filtrate was concentrated to yield 16 (0.160 mmol, 0.0950 g, 25.0%). 1H NMR (500 MHz, CDCl3) δ (ppm): 9.53 (br s, 1H), 6.73 (m, 2H), 6.57 (d, 1H), 4.21 (d, 1H), 3.72 (s, 3H), 3.67 (d, 1H), 3.44 (m, 5H), 3.24 (d, 1H), 2.77 (t, 1H), 2.59 (t, 1H), 2.03 (m, 2H), 1.68 (m, 2H), 1.44 (2 s, 18H and 9H), 1.23 (m, 1H), 1.03 (m, 3H). 13C{1H} NMR (125.7 MHz, CDCl3) δ (ppm): 171.7, 171.3, 152.2, 151.9, 123.5, 116.7, 115.6, 113.7, 81.4, 80.8, 63.7, 59.6, 55.8, 55.5, 52.8, 28.2, 28.1, 25.8, 25.6 (one C could not be found in this spectrum; it is likely coincidental with another peak). Molecular weight for C32H52N2O8: 592.76. MS (ESI) m/z: calcd, 593.4 (M + H)+; observed, 593.5.
2,2′-((2-((Carboxymethyl)(2-hydroxy-5-methoxybenzyl)amino)cyclohexyl)azanediyl) Diacetic Acid (CyHBET–OMe) (17)
16 (0.224 mmol, 0.0950 g) was dissolved in 3 mL each CH2Cl2/TFA. After 6 h of stirring, the reaction mixture was concentrated to quantitatively yield 17 as a white solid. 1H NMR (500 MHz, D2O) δ (D2O): 7.12 (s, 1H), 7.03–6.97 (m, 2H), 4.41 (s, 2H), 4.11 (m, 1H), 3.84 (s, 3H), 3.41 (br s, 2H), 3.27, (t, 1H), 3.12 (br s, 1H), 2.99 (t, 1H), 2.37 (m, 1H), 2.06 (m, 1H), 1.93 (m, 1H), 1.81 (m, 1H), 1.57 (m, 1H), 1.37–1.14 (m, 4H). 13C{H} NMR (125.7 MHz, D2O) δ (tert-BuOH): 174.6, 173.8, 170.4, 153.4, 150.0, 118.8, 118.0, 117.9, 117.5, 62.2, 59.6, 56.3, 53.7, 52.3, 50.9, 48.2, 30.2, 24.5, 24.4 (one C could not be found in this spectra; it is likely coincidental with another peak). Molecular weight for C20H29N2O8: 424.45. MS (ESI) m/z: calcd, 425.2 (M + H)+; observed, 425.2.
Na2[Mn(CyHBET–OMe)] (18)
17 (0.14 mmol, 0.074 g) was dissolved in 5 mL of water. The pH was adjusted to 6.5. MnCl2·4H2O (0.14 mmol, 0.028 g) was added, and the pH was readjusted to 6.5. The reaction mixture was purified using a reverse phase C18 (Polaris) column; eluent A, 50 mM NH4OAc; B, MeCN; gradient 5% to 6% B over 25 min; flow rate, 20 mL/min. The fractions were lyophilized to yield 18 as a white solid (0.052 g, 0.089 mmol, 62%). Molecular weight for C20H27MnN2O8: 475.36. MS (ESI) m/z: calcd, 478.1 (M + H)+; observed, 478.1.
tert-Butyl (trans-2-((2-Nitrobenzyl)amino)cyclohexyl)carbamate (19)
To a solution of N-BOC-trans-1,2-diaminocyclohexane·HCl (3.99 mmol, 1.00 g) in 90 mL of MeOH was added NEt3 (4.39 mmol, 0.600 mL), and the reaction was stirred for 30 min. To the above mixture was added a solution of 2-hydroxy-5-nitrobenzaldehyde (3.99 mmol, 0.667 g) in MeOH (30 mL). After being stirred for 1 h, solid NaBH4 (8.38 mmol, 0.317 g) was added, and the reaction was stirred for 3 h. All of the volatiles were removed under reduced pressure to yield a pale yellow solid. The residue was dissolved in 200 mL of CH2Cl2 extracted with 200 mL of saturated NaHCO3 solution. The aqueous layer was extracted with CH2Cl2 (2 × 100 mL). All of the organics were combined, washed with brine (200 mL), and dried over anhydrous MgSO4. The solvent was evaporated under reduced pressure to obtain 19 as a pale yellow solid (3.37 mmol, 1.23 g, 84.4%). 1H NMR (500 MHz, CD3OD) δ (ppm): 8.05 (m, 1H), 7.91 (m, 1H), 6.81 (m, 1H), 4.48 (d, 1H), 4.08 (m, 2H), 3.42 (d, 1H), 2.31 (m, 1H), 2.13 (m, 1H), 1.98 (m, 1H), 1.75 (m, 2H), 1.45 (s, 9H), 1.17 (m, 3H). Molecular weight for C18H27N3O5: 365.42. MS (ESI) m/z: calcd, 366.42 (M + H)+; observed, 366.5.
Di-tert-butyl 2,2′-((trans-2-((2-(tert-Butoxy)-2-oxoethyl)(2-hydroxy-5-nitrobenzyl)amino)cyclohexyl)azanediyl)diacetate (20)
19 (3.15 mmol, 1.15 g) was dissolved in CH2Cl2 (100 mL) followed by addition of 50 mL of trifluoroacetic acid. The reaction was stirred for 5 h, and then the volatiles were removed under reduced pressure. The reaction was taken up in 50 mL of water, washed with Et2O, and the water fraction was freeze-dried to produce the free amine quantitatively as a pale yellow solid, which was used in subsequent reaction without further purification.
To the round-bottom flask containing the amine was added potassium iodide (6.30 mmol, 1.04 g), and the system was purged with nitrogen. Under counter nitrogen flow, dry dimethylformamide (2 mL) was added followed by the addition of N,N-diisopropylethylamine (15.8 mmol, 2.74 mL) and dropwise addition of tert-butyl bromoacetate (9.77 mmol, 1.90 g). The reaction was stirred for 18 h and then partitioned between saturated NaHCO3 solution and Et2O. The Et2O layer was separated and washed with several changes of water to remove DMF before drying over Na2SO4 and concentration to 0.730 g of yellow oil. The crude product was carried immediately through to the next step without further purification. Molecular weight for C31H49N3O9: 607.74. MS (ESI) m/z: calcd, 608.74 (M + H)+; observed, 608.9.
2,2′-((trans-2-((Carboxymethyl)(2-hydroxy-5-nitrobenzyl)amino)cyclohexyl)azane-diyl) Diacetic Acid (CyHBET–NO2) (21)
The crude product (20) from the previous step was dissolved in trifluoroacetic acid (40 mL) followed by addition of triisopropylsilane (2.35 mL), 1-dodecanethiol (2.35 mL), and water (2.35 mL). The reaction was stirred for 5 h, and then the volatiles were removed under reduced pressure. The residue was dissolved in water (40 mL) and washed with Et2O (3 × 40 mL). The water fraction was freeze-dried to produce crude 21. The product was then purified via preparative HPLC using a Polaris C18 column; eluent A, H2O/0.1% TFA; B, MeCN/0.1% TFA; gradient 5% to 50% B over 25 min; flow rate, 15 mL/min. The fractions were collected and lyophilized to yield 21 as a white solid (1.13 mmol, 0.497 g, 35.6% from 19). 1H NMR (500 MHz, D2O) δ (ppm): 8.37 (d, J = 2.58 Hz, 1H), 8.20 (m, 1H), 7.06 (d, J = 9.10 Hz, 1H), 4.40 (s, 2H), 4.10 (d, 1H), 3.84 (d, 1H), 3.56 (br, 1H), 3.44 (br, 1H), 3.19 (br, 2H), 3.04 (br, 2H), 2.35 (m, 1H), 1.89 (m, 1H), 1.78 (m, 1H), 1.53 (m, 1H), 1.24 (br, 4H). 13C{1H} NMR (125 MHz, D2O) δ (ppm): 173.8, 171.4, 162.8, 141.1, 129.8, 128.5, 119.0, 117.1, 72.1, 71.7, 63.3, 60.0, 55.0, 43.2, 24.7, 24.5, 24.3. Molecular weight for C19H25N3O9: 439.42. MS (ESI) m/z: calcd, 440.42 (M + H)+; observed, 440.5.
Na2[MnII(CyHBET–NO2)] (22)
21 (0.260 mmol, 0.114 g) was dissolved in 5 mL of water. The pH was adjusted to 8 using 1 N NaOH solution. MnCl2·4H2O (0.260 mmol, 0.0510 g) was then added to the solution, and the pH was carefully adjusted to 6.5. The reaction was stirred for 1 h, filtered, and lyophilized to yield a white solid. The complex was injected onto a reverse phase C18 (Polaris) column and desalted using the method described above. Fractions were collected and lyophilized to yield 22 as a white solid (0.190 mmol, 0.102 g, 73.2%). Molecular weight for C19H23MnN3O9: 490.32. MS (ESI) m/z: calcd, 493.09 (M + 3H)+; observed, 494.1.
Na[MnIII(CyHBET–NO2)] (23)
MnF3 (0.080 mmol, 0.011 g) was added to 21 (0.080 mmol, 0.043 g) with stirring in 5 mL of H2O at pH 8. The resultant red-orange solution was purified using a reverse phase C18 (Polaris) column; eluent A, H2O (10 mM ammonium acetate), B, MeCN; gradient 5% to 60% B over 25 min; flow rate, 20 mL/min. The fractions were collected and lyophilized to yield 23 as a brown solid (0.050 mmol, 0.024 g, 63%). Molecular weight for C19H21MnN3O9: 490.32. MS (ESI) m/z: calcd, 492.09 (M + 2H)+; observed, 492.1.
Acknowledgments
This work was supported by grants from the National Cancer Institute (CA161221 to P.C. and a T32 postdoctoral fellowship to E.M.G., CA009502) and instrumentation funded by the National Center for Research Resources and the Office of the Director (RR14075, OD010650). We thank Anna Moore for access to a UV–vis spectrophotometer. We thank Nick Rotile and Iliyana Atanasova for assistance with MRI scans.
Supporting Information Available
Synthesis procedures, pH titration profiles, Mn distribution diagrams, UV–vis spectra, 17O NMR data, and CV data discussed but not depicted here. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
† E.M.G. and S.M. contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Mammoto T.; Jiang A.; Jiang E.; Panigrahy D.; Kieran M. W.; Mammoto A. Am. J. Pathol. 2013, 183, 1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail D. F.; Joyce J. A. Nat. Med. 2013, 19, 1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz N. W.; Le Fur Y.; Chiche J.; Pouysségur J.; Cozzone P. J. Cancer Res. 2013, 73, 4616. [DOI] [PubMed] [Google Scholar]
- Bakalova R.; Zhelev Z.; Aoki I.; Saga T. Clin. Cancer Res. 2013, 19, 2503. [DOI] [PubMed] [Google Scholar]
- Zhu X.; Cardounel A. J.; Zweier J. L.; He G. Antioxid. Redox Signaling 2007, 9, 447. [DOI] [PubMed] [Google Scholar]
- Galinier A.; Carrière A.; Fernandez Y.; Carpéné C.; André M.; Caspar-Bauguil S.; Thouvenot J.-P.; Périquet B.; Pénicaud L.; Casteilla L. J. Biol. Chem. 2006, 281, 12682. [DOI] [PubMed] [Google Scholar]
- Jorgenson T. C.; Zhong W.; Oberley T. D. Cancer Res. 2013, 73, 6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R. J. Biol. Chem. 2012, 287, 4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaiswing L.; Oberley T. D. Antioxid. Redox Signaling 2010, 13, 449. [DOI] [PubMed] [Google Scholar]
- Cannito S.; Novo E.; Compagnone A.; di Bonzo L. V.; Busletta C.; Zamara E.; Paternostro C.; Povero D.; Bandino A.; Bozzo F.; Cravanzola C.; Bravoco A.; Colombatto S.; Parola M. Carcinogenesis 2008, 29, 2267. [DOI] [PubMed] [Google Scholar]
- Yan Z.; Garg S. J.; Kipnis J.; Banerjee R. Nat. Chem. Biol. 2009, 5, 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg S. J.; Yan Z.; Vitvitsky V.; Banerjee R. Antioxid. Redox Signaling 2011, 15, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelini G.; Gardella S.; Ardy M.; Ciriolo M. R.; Filomeni G.; Di Trapani G.; Clarke F.; Sitia R.; Rubartelli A. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausenloy D. J.; Yellon D. M. J. Clin. Invest. 2013, 123, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweier J. L.; Flaherty J. T.; Weisfeldt M. L. Proc. Natl. Acad. Sci. U.S.A. 1987, 84, 1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D.; Shi S.; Zhang L.; Liu L.; Ding B.; Zhao B.; Yagnik G.; Zhou F. ACS Chem. Neurosci. 2013, 4, 1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskici G.; Axelsen P. H. Biochemistry 2012, 51, 6289. [DOI] [PubMed] [Google Scholar]
- Rosini M.; Simoni E.; Milelli A.; Minarini A.; Melchiorre M. J. Med. Chem. 2014, 57, 2821. [DOI] [PubMed] [Google Scholar]
- Sayre L. M.; Perry G.; Smith M. A. Chem. Res. Toxicol. 2008, 21, 172. [DOI] [PubMed] [Google Scholar]
- Lee M. H.; Yang Z.; Lim C. W.; Lee Y. H.; Dongbang S.; Kang C.; Kim J. S. Chem. Rev. 2013, 113, 5071. [DOI] [PubMed] [Google Scholar]
- Page S. M.; Martorella M.; Parelkar S.; Kosif I.; Emrick T. Mol. Pharmaceutics 2013, 10, 2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen H.; Marzenell P.; Jentzsch E.; Wenz F.; Veldwijk M. R.; Mokhir A. J. Med. Chem. 2012, 55, 924. [DOI] [PubMed] [Google Scholar]
- Pacheco-Torres J.; López-Larrubia P.; Ballesteros P.; Cerdán S. NMR Biomed. 2011, 24, 1. [DOI] [PubMed] [Google Scholar]
- De Leon-Rodriguez L. M.; Lubag A. J. M.; Malloy C. R.; Martinez G. V.; Gillies R. J.; Sherry A. D. Acc. Chem. Res. 2009, 42, 948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.; Wu K.; Sherry A. D. Angew. Chem., Int. Ed. 1999, 38, 3192. [PubMed] [Google Scholar]
- Raghunand N.; Howison C.; Sherry A. D.; Zhang S.; Gillies R. J. Magn. Reson. Med. 2003, 49, 249. [DOI] [PubMed] [Google Scholar]
- Gillies R. J.; Raghunand N.; Karczmar G. S.; Bhujwalla Z. M. J. Magn. Reson. Imaging 2002, 16, 430. [DOI] [PubMed] [Google Scholar]
- Garcia-Martin M. L.; Martinez G. V.; Raghunand N.; Sherry A. D.; Zhang S.; Gillies R. J. Magn. Reson. Med. 2006, 55, 309. [DOI] [PubMed] [Google Scholar]
- Eschmann S.-M.; Paulsen F.; Reimold M.; Dittmann H.; Welz S.; Reischl G.; Machulla H.-J.; Bares R. J. Nucl. Med. 2005, 46, 253. [PubMed] [Google Scholar]
- Alawneh J. A.; Moustafa R. R.; Marrapu S. T.; Jensen-Kondering U.; Morris R. S.; Jones P. S.; Aigbirhio F. I.; Fryer T. D.; Carpenter T. A.; Warburton E. A.; Baron J.-C. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 736. [DOI] [PubMed] [Google Scholar]
- Wood K. A.; Wong W. L.; Saunders M. I. Nucl. Med. Biol. 2008, 35, 393. [DOI] [PubMed] [Google Scholar]
- Bourgeois M.; Rajerison H.; Guerard F.; Mougin-Degraef M.; Barbet J.; Michel N.; Cherel M.; Faivre-Chauvet A.; Gestin J.-F. Nucl. Med. Rev. 2011, 14, 90. [DOI] [PubMed] [Google Scholar]
- Yoshii Y.; Yoneda M.; Ikawa M.; Furukawa T.; Kiyono Y.; Mori T.; Yoshii H.; Oyama N.; Okazawa H.; Saga T.; Fujibayashi Y. Nucl. Med. Biol. 2012, 39, 177. [DOI] [PubMed] [Google Scholar]
- Takasawa M.; Moustafa R. R.; Baron J.-C. Stroke 2008, 39, 1629. [DOI] [PubMed] [Google Scholar]
- Rojas-Quijano F. A.; Tircsó G.; Benyó E. T.; Baranyai Z.; Hoang H. T.; Kálmán F. K.; Gulaka P. K.; Kodibagkar V. D.; Aime S.; Kovács Z.; Sherry A. D. Chem.—Eur. J. 2012, 18, 9669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda K.; Okabe Y.; Kadonosono T.; Ueno T.; Youssif B. G. M.; Kizaka-Kondoh S.; Nagasawa H. Bioconjugate Chem. 2012, 23, 324. [DOI] [PubMed] [Google Scholar]
- Zhelev Z.; Aoki I.; Gadjeva V.; Nikolova B.; Bakalova R.; Saga T. Eur. J. Cancer 2013, 49, 1467. [DOI] [PubMed] [Google Scholar]
- Zhelev Z.; Bakalova R.; Aoki I.; Lazarova D.; Saga T. ACS Chem. Neurosci. 2013, 4, 1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsitovich P. B.; Burns P. J.; McKay A. M.; Morrow J. R. J. Inorg. Biochem. 2014, 133, 143. [DOI] [PubMed] [Google Scholar]
- Do Q. N.; Ratnakar S. J.; Kovács Z.; Sherry A. D. ChemMedChem 2014, 9, 1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnakar S. J.; Viswanathan S.; Kovacs Z.; Jindal A. K.; Green K. N.; Sherry A. D. J. Am. Chem. Soc. 2012, 134, 5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsitovich P. B.; Spernyak J. A.; Morrow J. R. Angew. Chem., Int. Ed. 2013, 52, 13997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnakar S. J.; Soesbe T. C.; Lumata L. L.; Do Q. N.; Viswanathan S.; Lin C.-Y.; Sherry A. D.; Kovacs Z. J. Am. Chem. Soc. 2013, 135, 14904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu C.; Nagao R.; Louie A. Y. Angew. Chem., Int. Ed. 2009, 48, 6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loving G. S.; Mukherjee S.; Caravan P. J. Am. Chem. Soc. 2013, 135, 4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aime S.; Botta M.; Gianolio E.; Terreno E. Angew. Chem., Int. Ed. 2000, 39, 747. [DOI] [PubMed] [Google Scholar]
- Lauffer R. B. Chem. Rev. 1987, 87, 901. [Google Scholar]
- Ma R.; Motekaitis R. J.; Martell A. E. Inorg. Chim. Acta 1995, 233, 137. [Google Scholar]
- Kálmán F. K.; Tircsó G. Inorg. Chem. 2012, 51, 10065. [DOI] [PubMed] [Google Scholar]
- Billo J.Excel for Chemists: A Comprehensive Guide; Wiley: New York, 2001. [Google Scholar]
- Dynamics of Solutions and Fluid Mixtures by NMR; Delpuech J.-J., Ed.; Wiley: West Sussex, England, 1995. [Google Scholar]
- Gale E. M.; Zhu J.; Caravan P. J. Am. Chem. Soc. 2013, 135, 18600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetter M. S.; Grant M. W.; Wood E. J.; Dodgen H. W.; Hunt J. P. Inorg. Chem. 1972, 11, 2701. [Google Scholar]
- Aime S.; Anelli P. L.; Botta M.; Brocchetta M.; Canton S.; Fedeli F.; Gianolio E.; Terreno E. J. Biol. Inorg. Chem. 2002, 7, 58. [DOI] [PubMed] [Google Scholar]
- Drahoš B.; Kotek J.; Hermann P.; Lukeš I.; Tóth E. Inorg. Chem. 2010, 49, 3224. [DOI] [PubMed] [Google Scholar]
- Drahoš B.; Pniok M.; Havlíčková J.; Kotek J.; Císařová I.; Hermann P.; Lukeš I.; Tóth E. Dalton Trans. 2011, 40, 10131. [DOI] [PubMed] [Google Scholar]
- Drahoš B.; Kotek J.; Císařová I.; Hermann P.; Helm L.; Lukeš I.; Tóth E. Inorg. Chem. 2011, 50, 12785. [DOI] [PubMed] [Google Scholar]
- Dees A.; Zahl A.; Puchta R.; van Eikama Hommes N. J. R.; Heinemann F. W.; Ivanović-Burmazović I. Inorg. Chem. 2007, 46, 2459. [DOI] [PubMed] [Google Scholar]
- Lieb D.; Friedel F. C.; Yawer M.; Zahl A.; Khusniyarov M. M.; Heinemann F. W.; Ivanovíc-Burmazovíc I. Inorg. Chem. 2013, 52, 222. [DOI] [PubMed] [Google Scholar]
- Maigut J.; Meier R.; Zahl A.; van Eldik R. Inorg. Chem. 2008, 47, 5702. [DOI] [PubMed] [Google Scholar]
- Maigut J.; Meier R.; Zahl A.; van Eldik R. J. Am. Chem. Soc. 2008, 130, 14556. [DOI] [PubMed] [Google Scholar]
- de Sá A.; Bonnet C. S.; Geraldes C. F. G. C.; Tóth É.; Ferreira P. M. T.; André J. P. Dalton Trans. 2013, 42, 4522. [DOI] [PubMed] [Google Scholar]
- Tei L.; Gugliotta G.; Fekete M.; Kálmán F. K.; Botta M. Dalton Trans. 2011, 40, 2025. [DOI] [PubMed] [Google Scholar]
- Rolla G. A.; Platas-Iglesias C.; Botta M.; Tei L.; Helm L. Inorg. Chem. 2013, 52, 3268. [DOI] [PubMed] [Google Scholar]
- Drahoš B.; Lukeš I.; Tóth E. Eur. J. Inorg. Chem. 2012, 1974. [Google Scholar]
- Patinec V.; Rolla G. A.; Botta M.; Tripier R.; Esteban-Gomez D.; Platas-Iglesias C. Inorg. Chem. 2013, 52, 11173. [DOI] [PubMed] [Google Scholar]
- Esteban-Gómez D.; Cassino C.; Botta M.; Platas-Iglesias C. RSC Adv. 2014, 4, 7094. [Google Scholar]
- Yang Y.-L.; Unnikrishnan B.; Chen S.-M. Int. J. Electrochem. Sci. 2011, 6, 3902. [Google Scholar]
- Sokolowski A.; Müller J.; Weyhermüller T.; Schnepf R.; Hildebrandt P.; Hildenbrand K.; Bothe E.; Wieghardt K. J. Am. Chem. Soc. 1997, 119, 8889. [Google Scholar]
- Bill E.; Müller J.; Weyhermüller T.; Wieghardt K. Inorg. Chem. 1999, 38, 5795. [Google Scholar]
- Lu C. C.; Bill E.; Weyhermüller T.; Bothe E.; Wieghardt K. J. Am. Chem. Soc. 2008, 130, 3181. [DOI] [PubMed] [Google Scholar]
- Kal S.; Filatov A. S.; Dinolfo P. H. Inorg. Chem. 2013, 52, 13963. [DOI] [PubMed] [Google Scholar]
- Caravan P.; Ellison J. J.; McMurry T. J.; Lauffer R. B. Chem. Rev. 1999, 99, 2293. [DOI] [PubMed] [Google Scholar]
- Caravan P. Chem. Soc. Rev. 2006, 35, 512. [DOI] [PubMed] [Google Scholar]
- Caravan P.; Farrar C. T.; Frullano L.; Uppal R. Contrast Media Mol. Imag 2009, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas S.; Jacques V.; Sun W.-C.; Troughton J. S.; Welch J. T.; Chasse J. M.; Willich-Schmitt H.; Caravan P. Invest. Radiol. 2010, 45, 600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacques V.; Dumas S.; Wei-Chuan S.; Troughton J. S.; Greenfield M. T.; Caravan P. Invest. Radiol. 2010, 45, 613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayburt T.; Sharp R. R. J. Chem. Phys. 1990, 92, 5892. [Google Scholar]
- Koenig S. H.; Brown R. D. III; Spiller M. Magn. Reson. Med. 1987, 4, 252. [DOI] [PubMed] [Google Scholar]
- Cheng W.; Haedicke I. E.; Nofiele J.; Martinez F.; Beera K.; Scholl T. J.; Cheng H.-L. M.; Zhang X.-a. J. Med. Chem. 2014, 57, 516. [DOI] [PubMed] [Google Scholar]
- Helm L.; Merbach A. E. Chem. Rev. 2005, 105, 1923. [DOI] [PubMed] [Google Scholar]
- D’Alessandro D. M.; Keene F. R. Chem. Soc. Rev. 2006, 35, 424. [DOI] [PubMed] [Google Scholar]
- Yu M.; Beyers R. J.; Gorden J. D.; Cross J. N.; Goldsmith C. R. Inorg. Chem. 2012, 51, 9153. [DOI] [PubMed] [Google Scholar]
- Jones D. P.; Mody V. C. Jr.; Carlson J. L.; Lynn M. J.; Sternberg P. Jr. Free Radical Biol. Med. 2002, 33, 1290. [DOI] [PubMed] [Google Scholar]
- Koenig S. H.; Brown R. D.; Brewer C. F. III. Proc. Natl. Acad. Sci. U.S.A. 1973, 70, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troughton J. S.; Greenfield M. T.; Greenwood J. M.; Dumas S.; Wiethoff A. J.; Wang J.; Spiller M.; McMurry T. J.; Caravan P. Inorg. Chem. 2004, 43, 6313. [DOI] [PubMed] [Google Scholar]
- Aime S.; Canton C.; Crich S. G.; Terreno E. Magn. Reson. Chem. 2002, 40, 41. [Google Scholar]
- Fanali G.; Cao Y.; Ascenzi P.; Fasano M. J. Inorg. Biochem. 2012, 117, 198. [DOI] [PubMed] [Google Scholar]
- Gans P.; Sabatini A.; Vacca A. Talanta 1996, 1996, 1739. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.