

Abstract
Background
Recent studies indicate that α1‐adrenergic receptors (α1‐ARs) are cardioprotective by preventing cardiac myocyte death and augmenting contractility in heart failure. Although G‐protein‐coupled receptors are assumed to localize to and signal at the plasma membrane, we previously demonstrated that endogenous α1‐ARs localize to the nuclei in adult cardiac myocytes. However, the functional consequence of this nuclear localization remains unclear. Here, we attempted to reconcile nuclear localization of α1‐ARs with their physiologic function by examining α1‐AR‐induced contractility in adult cardiac myocytes.
Methods and Results
By measuring shortening in unloaded, cultured adult cardiac myocytes, we found that the α1A‐subtype regulated contractility through phosphorylation of cardiac troponin I (cTnI) at the protein kinase C (PKC) site, threonine 144. Reconstitution of an α1A‐subtype nuclear localization mutant in cardiac myocytes lacking α1‐ARs failed to rescue nuclear α1A‐mediated phosphorylation of cTnI and myocyte contractility. Leptomycin B, the nuclear export inhibitor, also blocked α1A‐mediated phosphorylation of cTnI. These data indicate that α1‐AR signaling originates in the nucleus. Consistent with these observations, we localized the α1A‐subtype to the inner nuclear membrane, identified PKCα, δ, and ε in the nucleus, and found that α1‐ARs activate PKCδ in nuclei isolated from adult cardiac myocytes. Finally, we found that a PKCδ nuclear localization mutant blunted α1‐induced phosphorylation of cTnI.
Conclusions
Together, our data identify a novel, “inside‐out” nuclear α1A‐subtype/PKCδ/cTnI‐signaling pathway that regulates contractile function in adult cardiac myocytes. Importantly, these data help resolve the discrepancy between nuclear localization of α1‐ARs and α1‐AR‐mediated physiologic function.
Keywords: cardiac myocyte, cardiac troponin I, nucleus, protein kinase C, α1‐adrenergic receptors
Introduction
In heart failure, elevated catecholamine levels leading to increased activation of adrenergic receptors (ARs) are believed to exacerbate pathologic ventricular remodeling. However, recent clinical and animal studies indicate that α1‐ARs have an important and previously unpredicted cardioprotective role in heart failure, despite accounting for only 11% of total ARs in the heart.1–2 In clinical trials, α1‐AR antagonists doubled the risk of heart failure in hypertensive patients3 and tended to reduce survival in heart failure patients.4 These findings are consistent with α1‐AR cardioprotection and are further supported by analogous studies in mice. In mice lacking cardiac α1‐ARs (α1A‐ and α1B‐AR double knockout mice; α1ABKO), we found that pathologic stress from pressure overload reduced survival and induced heart failure in surviving mice characterized by cardiac myocyte cell death, contractile dysfunction, fibrosis, and a failure of cardiac‐specific gene transcription.5–6 Furthermore, we identified an α1A subtype/extracellular signal‐regulated kinase (ERK) survival‐signaling pathway in adult cardiac myocytes, the absence of which might explain the maladaptive response to pathologic stress in α1ABKO mice.7–8 In addition, others have shown that transgenic overexpression of the α1A subtype prevented pathologic remodeling in response to pressure overload and ischemic injury.9–10 These studies contradict the widely held idea that all AR signaling in heart failure is detrimental, and suggest that activation of cardioprotective α1‐AR signaling could be a novel therapy for heart failure.1–2
There are 3 α1‐AR subtypes (α1A, α1B, and α1D), and whereas all 3 subtype mRNAs are expressed in the heart, only the α1A‐ and α1B‐ARs are detected at the protein level in cardiac myocytes.1 α1‐ARs are G‐protein‐coupled receptors (GPCRs) that signal through Gq. As with other Gq‐coupled receptors, including endothelin (ET) and angiotensin (AT) receptors, α1‐ARs, until recently, were thought to exacerbate pathologic remodeling in heart failure.11–12 Interestingly, ET‐Rs, AT‐Rs, and α1‐ARs were also recently shown to localize to the nucleus in adult cardiac myocytes.13–16 Specifically, we localized endogenous α1‐ARs to the nuclear membrane in adult cardiac myocytes, but failed to detect functional receptors at the plasma membrane.15 We identified nuclear localization sequences (NLS) in both the α1A and α1B subtypes, and found that mutation of the NLS in each subtype resulted in loss of nuclear localization and ability to induce phosphorylation of ERK.16 We also found that the α1‐AR‐signaling partners, Gαq and phospholipase Cβ1 (PLCβ1), colocalized with α1‐ARs only at the nuclear membrane.15 Furthermore, we demonstrated that nuclear α1‐AR signaling was facilitated by rapid catecholamine uptake mediated by the membrane transporter, organic cation transporter 3.15 However, whereas our data indicated that at least 80%, and possibly all, α1‐ARs localized to the nucleus in adult cardiac myocytes,15 previous reports suggested that only 5% of ET‐Rs localized to the nucleus (AT‐R levels are too low to make such measurements).13,16 Based on this differential localization, we previously suggested that Gq‐coupled receptor signaling might be compartmentalized, with nuclear Gq‐coupled receptors, such as α1‐ARs, being cardioprotective.16
However, mechanisms by which nuclear GPCRs signal and regulate physiologic function remain difficult to define.17 ET‐R‐induced calcium transients and AT‐R‐ and β‐AR‐induced gene transcription have been observed in nuclei isolated from adult cardiac myocytes.13–14,18 Yet, ascribing a physiological function to these nuclear receptors is difficult because the majority of these receptors localize to the plasma membrane. Conversely, α1‐ARs localize primarily to the nucleus,15–16 which would suggest that α1‐AR signaling must arise from the nucleus. Therefore, to reconcile nuclear localization of α1‐ARs with α1‐AR physiologic function, we examined α1‐AR‐mediated contractile function in adult cardiac myocytes. Our results define a novel “inside‐out” contractile signaling pathway in adult cardiac myocytes, where nuclear α1‐ARs activate protein kinase Cδ (PKCδ), leading to phosphorylation of cardiac troponin I (cTnI) at the sarcomere. Finally, our data help resolve the discrepancy between nuclear localization of α1‐ARs and α1‐AR‐mediated physiological function.
Methods
Experimental Animals
Generation of α1ABKO double knockout mice was previously described.5 Congenic C57BL/6J mice (12th to 15th generation, between 10 and 15 weeks of age) were used in all experiments. The use of all animals in this study conformed to the Public Health Service Guide for Care and Use of Laboratory Animals and was approved by The University of Minnesota and Sanford Research/University of South Dakota Institutional Animal Care and Use Committees.
Culture of Adult Mouse Cardiac Myocytes
Procedures for the isolation and culture of adult mouse cardiac myocytes were previously described.19
Chemicals
All reagents were prepared with chemicals purchased from Sigma‐Aldrich (St. Louis, MO), unless otherwise noted.
Adenoviruses
The α1A‐GFP (green fluorescent protein) and α1A‐NLSmut constructs were described previously.8,16 PKCδ constructs were made using a human cDNA encoding PKCδ (GenBank L07860), amplified by PCR with primers (IDT) containing Xho I (5′) and Kpn I (3′) restriction sites, and subcloned into the multicloning site of pDsRed monomer‐C1 (Clontech Laboratories, Mountain View, CA) in frame. Dominant negative PKCδ (PKCδ‐DN) was made by a K→A mutation at position 378 by site‐directed mutagenesis using the QuikChange kit (Stratagene, La Jolla, CA).20 The PKCδ nuclear localization mutant (PKCδ‐NLSmut) was made with a DNA minigene (IDT) containing the PKCδ NLS flanked by unique XcmI and AscI sites, but with the arginines (R) or lysines (K) replaced with alanine (A) at positions 613, 614, 615, 621, 623, 625, and 628 corresponding to the human sequence.20 All DsRed‐PKCδ constructs were subcloned into the AdEasy system (Stratagene) for adenovirus production. For all experiments, cardiac myocytes were infected at a multiplicity of infection of 1000 for the α1A‐AR constructs, resulting in 2.5‐fold overexpression for the α1A‐AR,8 and 5‐fold overexpression for the PKCδ constructs.
Measurement of ERK Activation in HeLa Cells
HeLa cells were plated at a density of 70 000 cells per 35‐mm plate overnight. The next day, cells were infected with an α1‐NLS mutant at a multiplicity of infection of 1000. Forty hours postinfection, cells were treated with 20 μmol/L of phenylephrine (PE) for 20 minutes at 37°C (5% CO2). For each treatment, 20 μg of total cell protein were resolved by SDS‐PAGE, transferred to PVDF, and probed with either antibodies to phospho‐ERK (pERK) or total ERK (tERK) (1:1000; Cell Signaling Technology, Danvers, MA).
Measurement of Cardiac Myocyte Contractility
For measurement of α1‐AR‐mediated contractility, isolated cardiac myocytes were plated on coverslips, infected with adenovirus, and cultured for 40 hours. After 40 hours and 1 hour before measurement of contractile function, the culture medium was replaced with Tyrode's buffer (140 mmol/L of NaCl, 10 mmol/L of glucose, 10 mmol/L of HEPES, 4 mmol/L of KCl, 1 mmol/L of MgCl2, pH 7.45), supplemented with 1.2 mmol/L of Ca2+ at 37°C. Twenty minutes before the experiment, myocytes were loaded with 1 μmol/L of fura2‐AM (Invitrogen, Carlsbad, CA) in Tyrode's buffer at 37°C, washed with Tyrode's buffer, and placed into a stimulation chamber (Cell MicroControls, Norfolk, VA). Myocytes were paced at 1 Hz at 20 to 30 V and perfused continuously with Tyrode's buffer. For all experiments, myocytes were pretreated with the β‐antagonist, timolol (2 μmol/L), and, in some cases, also with the nuclear export inhibitor, leptomycin B (lepB; 18.5 nmol/L) for 30 minutes followed by the α1‐AR agonist, PE (10 μmol/L), for 5 minutes at room temperature. To measure sarcomere shortening and Ca2+ transients, changes in sarcomere length and fura2 ratio (340/380 nm), respectively, were recorded using the Fluorescence and Contractility System (IonOptix LLC, Milton, MA). In each experiment, data were collected from 4 to 8 myocytes per heart or treatment.
Measurement of the Phosphorylation of cTnI and Phospholamban
To measure the phosphorylation state of cTnI and phospholamban (PLB), isolated cardiac myocytes were plated in 35‐mm dishes, infected with adenovirus, and cultured for 40 hours. After 40 hours, myocytes were pretreated with timolol (2 μmol/L) and, in some cases, also with lepB (18.5 nmol/L) for 30 minutes followed by PE (20 μmol/L) for 5 minutes at 37°C. Whole‐cell lysates were prepared as described previously.16 Western blots were performed with primary antibodies to Thr144 phospho‐cTnI (1:1000; Abcam, Cambridge, MA), Ser23,24 phospho‐cTnI, total cTnI (1:1000; Cell Signaling), Thr17 phospho‐PLB (1:5000; Badrilla Ltd., Leeds, UK), Ser16 phospho‐PLB, and total PLB (1:2000; Millipore, Billerica, MA) followed by appropriate secondary antibodies conjugated to HRP (1:5000, Cell Signaling). All antibodies were prepared in 5% BSA in TBS‐T (20 mmol/L of Tris‐HCl, 150 mmol/L of NaCl, pH 7.6, 0.1% Tween‐20). Blots were developed using enhanced chemiluminescence (GE Healthcare, Fairfield, CT) and analyzed using ImageJ software (National Institutes of Health [NIH], Bethesda, MD).
Localization of α1A‐ARs by Differential Permeabilization and Immunocytochemistry
Isolated cardiac myocytes were plated on coverslips, infected with adenovirus expressing the α1A‐GFP, and cultured for 40 hours. After 40 hours, myocytes were fixed with 4% paraformaldehyde. For total membrane permeabilization, 0.2% Triton X‐100 in 1× PBS (137 mmol/L of NaCl, 27 mmol/L of KCl, 43 mmol/L of Na2HPO4, 14 mmol/L of KH2PO4) was used followed by 2 washes with 1× PBS at room temperature. For plasma membrane‐only permeabilization, fixed coverslips were placed in tissue culture dishes containing ice‐cold 0.01% digitonin in 1× PBS on ice for 5 minutes followed by 2 washes with ice‐cold 1× PBS on ice. Subsequent to permeabilization, myocytes were probed with an antibody against GFP to detect the α1A‐GFP (Santa Cruz Biotechnology, Santa Cruz, CA) followed by a cyanine (Cy)3‐conjugated secondary antibody (Invitrogen). Myocytes were also probed with antibodies against either Lamin A/C or cTnI (Cell Signaling) followed by Cy5‐conjugated secondary antibodies (Invitrogen). All primary antibodies were diluted at 1:50, and all secondary antibodies were diluted at 1:1000 in TBS (20 mmol/L of Tris‐HCl, 150 mmol/L of NaCl, pH 7.6). Myocytes were counterstained with the DNA marker, 4',6‐diamidino‐2‐phenylindole (DAPI). Coverslips were mounted on slides with Fluoromount G (Electron Microscopy Sciences, Hatfield, PA). All fluorescent images were captured by confocal microscopy using an Olympus inverted microscope (BX50) and Fluoview software (Olympus, Tokyo, Japan).
Biochemical Fractionation of Cardiac Myocytes
Procedures for the biochemical fractionation of freshly isolated cardiac myocytes were described elsewhere.15 The purity of each fraction was verified by Western blot using primary antibodies against Na+/Ca2+ exchanger (NCX; plasma membrane, 1:1000 dilution; Millipore), glyceraldehyde 3‐phosphate dehydrogenase (GAPDH; cytosol, 1:100 000 dilution; Fitzgerald Industries International, Inc., Acton, MA), and lamin‐associated protein 2 (LAP2; inner nuclear membrane marker, 1:1000 dilution; BD Biosciences, San Jose, CA).
Measurement of Nuclear PKC Expression and Signaling
To measure subcellular distribution of PKC isozymes, cardiac myocyte fractions were resolved as described above and then examined by Western blot. Western blots were probed with primary antibodies to PKC α, δ, and ε (Santa Cruz Biotechnology), and PKC distribution was quantified using ImageJ software (NIH). To measure nuclear PKC isozymes by immunocytochemistry, isolated myocyte nuclei were incubated with antibodies to PKC α, δ, and ε (1:1000; Santa Cruz Biotechnology) overnight at 4°C. Nuclei were washed with PBS and then incubated with a Texas Red–conjugated secondary antibody (Invitrogen) for 1 hour at 4°C. Nuclei were again washed with PBS and then mounted onto glass slides with Fluoromount G (Electron Microscopy Sciences) and counterstained with the DNA marker, DAPI. Fluorescent images were captured by confocal microscopy as described above.
To measure nuclear PKC activation and activity, 100 μg of isolated nuclei were treated with PE (20 μmol/L) for 20 minutes at 37°C and then nuclear proteins were resolved by SDS‐PAGE. To assess nuclear PKCδ activation by Western blot, an antibody against phospho‐PKCδ at Thr505 was used (1:1000; Cell Signaling). To determine nuclear PKC activity by Western blot, an antibody against phospho‐myristoylated alanine‐rich C kinase substrate (pMARCKS) was used (1:1000; Cell Signaling). LAP2 levels were measured as described above and used as loading controls and proteins were quantified as above.
Statistical Analysis
All values are expressed as mean±SEM. All contractility and Western blot data were analyzed for normality using both D'Agostino‐Pearson's and Shapiro‐Wilk's normality tests. In Figures 1A through 1C, 2B through 2D, 3A through 3C, and 6C, untransformed data were not normally distributed, but log transformation resulted in P>0.05 in these normality tests. Data in Figure 6B were normally distributed (P>0.05), so no further transformation was performed. Subsequently, data were analyzed by 2‐way ANOVA with repeated measures followed by Bonferroni's post‐test to compare groups, except for PKC activation and activity, where Student's t test was performed, and P<0.05 was considered significant. The specifics of each statistical analysis and P values are indicated in each figure legend. The number of measurements (n) is stated in each figure legend. In this case, the unit of measurement for the statistical analysis was the cardiac myocytes. However, both the number of different hearts used and the total number of myocytes are included to conform with convention. The exception was for nuclear fractionation studies, where 2 hearts were used in each preparation. A potential limitation of this study was the relatively small numbers used, resulting in a low power in the statistical analysis. Prism 6 software (GraphPad Software, Inc., La Jolla, CA) was used to conduct all statistical analyses.
Figure 1.

α1‐ARs regulate contractility through phosphorylation of cTnI at Thr144 in adult cardiac myocytes. A, Sarcomere dynamics and (B) Ca2+ transients were measured in WT and α1ABKO cardiac myocytes before and 5 minutes after phenylephrine (PE) treatment (10 μmol/L of PE, 2 μmol/L of timolol in all conditions). Single‐twitch contractions or calcium transients are shown at 5 minutes after the addition of PE. Averaged data for percent (%) sarcomere shortening and fold change in calcium (ΔCa2+) at 5 minutes after PE are presented as mean±SEM from 33 WT cardiac myocytes from 9 different cultures and 14 α1ABKO cardiac myocytes from 5 different cultures. C, cTnI phosphorylation at Thr144 and Ser23,24 and (D) PLB phosphorylation at Ser16 and Thr17 measured by Western blot from WT and α1ABKO cardiac myocytes treated with PE (20 μmol/L, 15 minutes). Quantitation of cTnI phosphorylation at Thr144 is presented as mean±SEM from 6 different cultures. All data were analyzed by 2‐way ANOVA with repeated measures and Bonferroni's post‐test. Percent sarcomere shortening, P=0.0123; ΔCa2+, P=NS; cTnI phosphorylation at Thr144, P=0.0384. Significant comparisons identified by Bonferroni's post‐test are indicated as P<0.05. ANOVA indicates analysis of variance; cTnI, cardiac troponin I; PLB, phospholamban; WT, wild type; α1ABKO, α1A‐ and α1B‐AR double knockout mice; α1‐ARs, α1‐adrenergic receptors.
Figure 2.

Nuclear localization of α1‐ARs is required for α1‐AR‐mediated contractility in adult cardiac myocytes. A, α1‐NLS mutants can activate ERK in HeLa cells. HeLa cells were infected with an α1‐NLS mutant at a multiplicity of infection of 1000. Forty hours postinfection, cells were treated with 20 μmol/L of phenylephrine (PE) for 20 minutes at 37°C (5% CO2). For each treatment, 20 μg of total cell protein were probed for phospho‐ERK (pERK) or total ERK (tERK). Representative results are from 3 separate experiments. B, Sarcomere dynamics and (C) Ca2+ transients were measured in α1ABKO cardiac myocytes expressing WT α1A‐AR (α1A‐WT) or the α1A‐ NLS mutant (α1A‐NLSmut) before and 5 minutes after PE treatment (10 μmol/L of PE, 2 μmol/L of timolol in all conditions). Single‐twitch contractions or calcium transients are shown at 5 minutes after the addition of PE. Averaged data for percent (%) sarcomere shortening and fold change in calcium (ΔCa2+) at 5 minutes after PE are presented as mean±SEM from 21 α1ABKO cardiac myocytes expressing the α1A‐WT or the α1A‐NLSmut from 7 different cultures for each. D, cTnI phosphorylation at Thr144 measured by Western blot from α1ABKO cardiac myocytes expressing α1A‐WT or α1A‐NLSmut treated with PE (20 μmol/L, 15 minutes). Quantitation of cTnI phosphorylation at Thr144 is presented as mean±SEM from 6 different cultures. All data were analyzed by 2‐way ANOVA with repeated measures and Bonferroni's post‐test. Percent sarcomere shortening, P=0.0066; ΔCa2+, P=NS; cTnI phosphorylation at Thr144, P=0.0032. Significant comparisons identified by Bonferroni's post‐test are indicated as P<0.05. ANOVA indicates analysis of variance; cTnI, cardiac troponin I; ERK, extracellular signal‐regulated kinase; NLS, nuclear localization sequences; WT, wild type; α1ABKO, α1A‐ and α1B‐AR double knockout mice; α1‐ARs, α1‐adrenergic receptors.
Figure 3.
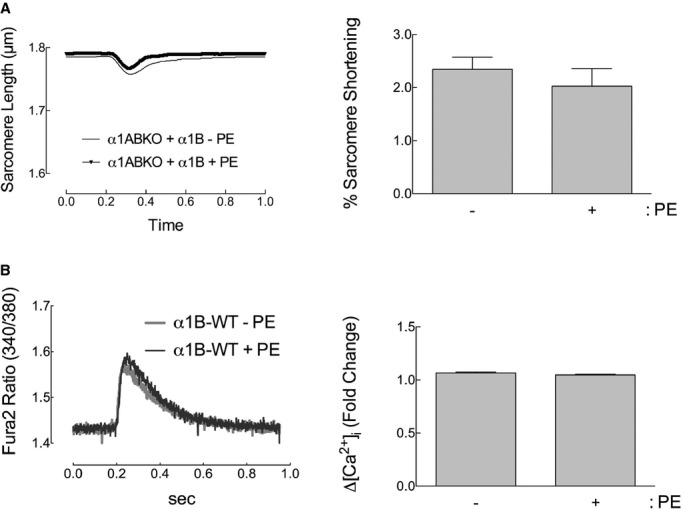
The α1B subtype does not affect adult cardiac myocyte contractility. A, Sarcomere dynamics and (B) Ca2+ transients in α1ABKO cardiac myocytes expressing WT α1B‐AR (α1B‐WT) before and 5 minutes after phenylephrine (PE) treatment (10 μmol/L of PE, 2 μmol/L of timolol in all conditions). Single‐twitch contractions or calcium transients are shown at 5 minutes after the addition of PE. Averaged data for percent (%) sarcomere shortening and fold change in calcium (ΔCa2+) at 5 minutes after PE are presented as mean±SEM from 33 WT cardiac myocytes from 8 different cultures. All data were analyzed by 2‐way ANOVA with repeated measures and Bonferroni's post‐test. Percent sarcomere shortening, P=NS; ΔCa2+, P=NS. ANOVA indicates analysis of variance; WT, wild type; α1ABKO, α1A‐ and α1B‐AR double knockout mice; α1‐ARs, α1‐adrenergic receptors.
Figure 6.
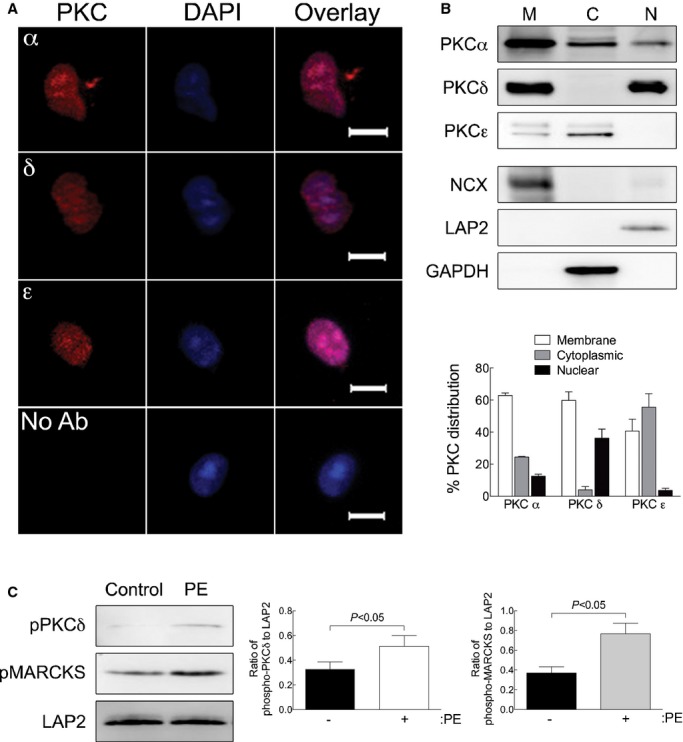
α1‐ARs activate PKC in nuclei isolated from adult cardiac myocytes. A, Nuclei were isolated from adult cardiac myocytes and stained with antibodies to the PKCα, δ, or ε isoforms followed by DAPI counterstain (scale bar=5 μm). B, PKCα, δ, or ε isoform distribution in membrane (M), cytosolic (C), and nuclear (N) was measured by Western blot analyses. Fraction purity was verified by Western blots for NCX (membrane), GAPDH (cytosolic), and LAP2 (nuclear). Relative distribution of the PKC isozymes is presented as mean±SEM for 3 separate nuclear preparations. C, PKC activation was measured in isolated nuclei treated with PE (20 μmol/L, 2 μmol/L of timolol) by phosphorylation of PKCδ at Thr505. PKC activity was measured by phosphorylation of the PKC substrate, MARCKS (pMARCKS). LAP2, a nuclear marker, was used as a loading control. Quantitation of PKCδ phosphorylation at Thr505 and MARCKS phosphorylation is presented as mean±SEM from 3 separate nuclear preparations. Data were analyzed by Student's t test, and significant comparisons are indicated as P<0.05. GAPDH indicates glyceraldehyde 3‐phosphate dehydrogenase; LAP2, lamin‐associated protein 2; NCX, Na+/Ca2+ exchanger; PE, phenylephrine; PKC, protein kinase C; pMARCKS, phospho‐myristoylated alanine‐rich C kinase substrate; α1‐ARs, α1‐adrenergic receptors.
Results
α1‐ARs Regulate Contractility Through Phosphorylation of cTnI at Thr144 in Adult Cardiac Myocytes
Here, we investigated the mechanisms regulating α1‐AR‐mediated contractility by measuring sarcomeric shortening and calcium transients in unloaded adult mouse cardiac myocytes.21 We found that the α1‐agonist, PE (10 μmol/L), in the presence of the β‐blocker, timolol (TML; 2 μmol/L), produced the standard triphasic response, although the third phase remained negative, as others have observed in isolated adult mouse cardiac myocytes.22–23 After 5 minutes, PE significantly decreased sarcomere shortening in WT adult mouse cardiac myocytes (Figure 1A, single‐twitch contraction shown at 5 minutes, and percent [%] sarcomere shortening at 5 minutes shown on right). As expected, PE had no effect on sarcomere shortening in α1ABKO adult cardiac myocytes, which lack endogenous α1‐ARs (Figure 1A). Interestingly, PE had no effect on calcium transients in WT or α1ABKO cardiac myocytes (Figure 1B, single calcium transient shown at 5 minutes and ΔCa2+ at 5 minutes shown on right).
Older studies proposed that α1‐ARs induce a positive inotropic response by increasing myofilament calcium sensitivity through phosphorylation of myosin light chain 224 or phosphorylation of cTnI at the protein kinase A (PKA) sites, serine 23,24 (Ser23,24).23 Here, we examined α1‐AR‐mediated phosphorylation of cTnI at multiple sites, including threonine 144 (Thr144), a PKC site identified as decreasing myofilament calcium sensitivity, and the PKA sites, Ser23,24, which are known to increase the rate of relaxation.25 In WT adult cardiac myocytes, PE increased the phosphorylation of cTnI at Thr144, but had no effect on the phosphorylation of cTnI at Ser23,24 (Figure 1C). As expected, PE had no effect on cTnI phosphorylation in α1ABKO adult cardiac myocytes (Figure 1C). To control for potential off‐target PE activation of β‐ARs, we measured phosphorylation of PLB at the PKA site Ser16 and the calcium/calmodulin‐dependent kinase site, Thr17.25 In WT and α1ABKO adult cardiac myocytes, PE had no effect on the phosphorylation of PLB at Thr17 (Figure 1D, no signal detected at Ser16), consistent with the lack of an effect on the calcium transient (Figure 1A and 1B). Together, our data suggest that α1‐ARs induce inotropic responses through phosphorylation of cTnI at Thr144 and hence a decrease in myofilament calcium sensitivity.
Nuclear Localization of α1‐ARs Is Required for α1‐AR‐Mediated Contractility in Adult Cardiac Myocytes
To test whether α1‐AR contractile signaling pathways originate in the nucleus in adult cardiac myocytes, we employed multiple approaches. First, we recently identified NLS in the cytoplasmic tail of both the α1A‐ and α1B‐AR subtypes and demonstrated that mutation of these NLS resulted in mislocalization of each receptor away from the nucleus, although, interestingly, neither receptor relocated to the plasma membrane.16 Furthermore, neither NLS overlapped with residues important for ligand binding or G‐protein binding/activation. Functionally, neither the α1A‐ nor α1B‐NLS mutants (α1‐NLSmut) mediated PE‐induced phosphorylation of ERK in adult cardiac myocytes, suggesting that nuclear localization is required for α1‐signaling.16 Importantly, both the α1A‐ and α1B‐NLSmuts mediated PE‐induced phosphorylation of ERK in HeLa cells, which do not express endogenous α1‐ARs and where exogenously expressed α1‐ARs do not localize to the nucleus (despite the NLS in each subtype), indicating that the NLS mutations did not simply inactivate the receptors (Figure 2A).
Here, we tested whether nuclear localization was required for α1‐AR signaling using the α1‐NLSmuts to reconstitute signaling in α1ABKO adult cardiac myocytes. Reconstitution of the WT α1A subtype, but not the α1B subtype, in α1ABKO adult cardiac myocytes restored the PE‐induced reduction of sarcomeric shortening and had no effect on the calcium transients, similar to WT adult cardiac myocytes (α1A‐WT shown in Figure 2B and 2C; α1B‐WT shown in Figure 3). This finding is consistent with previous studies indicating that the α1A subtype regulates contractility.26–27 In addition, reconstitution of the WT α1A subtype in α1ABKO adult cardiac myocytes restored the phosphorylation of cTnI at Thr144 (Figure 2D). More important, reconstitution with the α1A‐NLSmut in α1ABKO adult cardiac myocytes failed to restore PE‐mediated sarcomeric shortening or phosphorylation of cTnI at Thr144 (Figure 2B through 2D).
Blockade of Nuclear Export Inhibits α1‐AR‐Mediated Contractility in Adult Cardiac Myocytes
As an alternative approach to demonstrate that α1‐AR signaling originates at the nucleus in adult cardiac myocytes, we employed the nuclear export inhibitor, lepB, which blocks transport through the nuclear pore complex. We previously found that lepB inhibits α1‐AR‐mediated activation of ERK in adult cardiac myocytes,16 and others have used lepB to block histone deacetylase export from the nucleus in cardiac myocytes.28 In the presence of vehicle, PE reduced sarcomeric shortening (Figure 4A and 4B, single‐twitch contraction shown at 5 minutes) and increased the phosphorylation of cTnI at Thr144 (Figure 4C) in WT adult cardiac myocytes. In contrast, pretreatment of WT adult cardiac myocytes with lepB tended to reduce the effect of PE on sarcomeric shortening (Figure 4A), but significantly reduced PE‐induced cTnI phosphorylation (Figure 4C). The ability of lepB to inhibit α1‐mediated phosphorylation of cTnI agrees with our results with the α1A‐NLSmut (Figure 2), indicating that α1‐inotropic signaling originates in the nucleus.
Figure 4.

Blockade of nuclear export inhibits α1‐AR‐mediated contractility in adult cardiac myocytes. A, Sarcomere dynamics and (B) Ca2+ transients were measured in WT cardiac myocytes pretreated with vehicle or leptomycin B (lepB; 18.5 μmol/L, right) before and 5 minutes after phenylephrine (PE) treatment (10 μmol/L of PE, 2 μmol/L of timolol in all conditions). Single‐twitch contractions or calcium transients are shown at 5 minutes after the addition of PE. Averaged data for percent (%) sarcomere shortening and fold change in calcium (ΔCa2+) at 5 minutes after PE are presented as mean±SEM from 21 WT cardiac myocytes treated with vehicle and 29 WT cardiac myocytes treated with lepB from 10 different cultures. C, cTnI phosphorylation at Thr144 measured by Western blot from WT cardiac myocytes pretreated with vehicle or leptomycin B followed by PE (20 μmol/L, 15 minutes). Quantitation of cTnI phosphorylation at Thr144 is presented as mean±SEM from 8 different cultures. All data were analyzed by 2‐way ANOVA with repeated measures and Bonferroni's post‐test. Percent sarcomere shortening, P=NS; ΔCa2+, P=NS; cTnI phosphorylation at Thr144, P=0.0425. Significant comparisons identified by Bonferroni's post‐test are indicated as P<0.05. ANOVA indicates analysis of variance; cTnI, cardiac troponin I; WT, wild type; α1‐ARs, α1‐adrenergic receptors.
The α1A Subtype Is Localized in the Inner Nuclear Membrane in Adult Cardiac Myocytes
Our current (Figure 2) and previous data15–16 demonstrate that signaling occurs at the nucleus, and our results with lepB further suggest that α1‐AR signaling originates inside the nucleus (Figure 4), implying localization of α1‐ARs to the inner nuclear membrane. To determine α1A‐AR localization and orientation in the nuclear bilayer, we used differential permeabilization29 and immunocytochemistry in α1ABKO cardiac myocytes expressing the α1A‐AR tagged with GFP at the carboxyl tail. This alternative approach was necessitated by the lack of validated, subtype‐specific α1‐ARs antibodies,30 preventing conventional electron microscopy approaches. We previously demonstrated that the α1A‐GFP reproduces the nuclear localization of endogenous α1‐ARs in adult cardiac myocytes.8,15 Here, α1ABKO cardiac myocytes expressing the α1A‐GFP from the same culture were treated with either Triton X‐100, which completely permeabilizes all membranes, or with low‐percentage digitonin (0.01%), which permeabilizes only the plasma membrane. As controls for permeabilization, we used antibodies to cTnI, which should stain cTnI in both Triton X‐100 and digitonin‐treated cardiac myocytes in the cytoplasm, and antibodies to lamin A/C, an inner nuclear membrane protein,31 which should stain lamin A/C in the nucleus only in Triton X‐100–treated cardiac myocytes. To confirm α1A‐subtype localization independently, antibodies to GFP were used (in addition to GFP fluorescence). In Triton X‐100–treated cardiac myocytes, both lamin A/C (Figure 5A) and cTnI (Figure 5B) were detected, validating the Triton X‐100 permeabilization technique. Furthermore, the α1A‐GFP was detected by fluorescence and independently with a GFP antibody, as expected (Figure 5A and 5B). Conversely, in digitonin‐treated cardiac myocytes, cTnI (Figure 5D), but not lamin A/C (Figure 5C), staining was detected, which indicated that antibodies were not reaching the inside of the nucleus. Importantly, whereas α1A‐GFP fluorescence was detected, the GFP antibody failed to detect the α1A‐GFP (Figure 5C and 5D). The failure of the GFP antibody to detect the α1A‐GFP in digitonin‐treated myocytes indicated that the GFP tag, as with lamin A/C, was not accessible to the antibody. Based on the failure of the GFP antibody in this situation, we would suggest that α1A subtype, as with lamin A/C, is localized to the inner nuclear membrane (not necessarily colocalized, but in the same compartment), with the C‐terminus facing the nucleoplasm.
Figure 5.

The α1A‐subtype is localized in the inner nuclear membrane in adult cardiac myocytes. α1ABKO adult cardiac myocytes expressing α1A‐GFP were subjected to differential permeabilization with Triton X‐100 (permeabilized plasma and nuclear membranes, left) or with digitonin (permeabilize plasma membrane only, right) and then stained with an anti‐GFP antibody, detected by a Cy3‐labeled secondary antibody, shown in red (A through D). Fluorescence from the α1A‐GFP is shown in green. As controls for the differential permeabilization, cardiac myocytes were also counterstained with the inner nuclear membrane protein, lamin A/C (A and C) or cTnI (B and D) and detected by Cy5‐labeled secondary antibody, shown in white. Scale bar=20 μm. Results are representative from 3 separate cultures. cTnI indicates cardiac troponin I; DAPI, 4',6‐diamidino‐2‐phenylindole; GFP, green fluorescent protein; α1ABKO, α1A‐ and α1B‐AR double knockout mice.
α1‐ARs Activate PKC in Nuclei Isolated From Adult Cardiac Myocytes
Consistent with the idea that α1‐ARs signal at the nucleus, we previously demonstrated that the α1A and α1B subtypes, Gαq and PLCβ1, colocalize only at the nucleus in adult cardiac myocytes.15 Signaling through α1‐ARs also involves activation of PKC. Therefore, we screened for the expression of PKCα, δ, and ε, the primary PKC isoforms induced by α1‐AR signaling,32 in nuclei isolated from WT adult mouse cardiac myocytes. Immunocytochemistry with PKC isoform‐specific antibodies indicated that all 3 PKC isoforms were detected in nuclei isolated from WT cardiac myocytes (Figure 6A). Next, we determined the subcellular distribution of PKCs α, δ, and ε by isolating membrane, cytosolic, and nuclear fractions from WT adult cardiac myocytes. PKCα and δ were detected in the nuclear fraction, and among the nuclear PKCs, PKCδ expression was proportionally the highest (Figure 6B). Membrane, cytosolic, and nuclear fractions were validated by Western blots for NCX, GAPDH, and LAP2, respectively (Figure 6B).
Based on our identification of α1‐AR‐signaling molecules in the nucleus, we subsequently measured α1‐AR signaling directly in nuclei isolated from WT adult cardiac myocytes. In isolated nuclei, PE significantly increased the phosphorylation of PKCδ at Thr505, an autophosphorylation site required for activation, and the phosphorylation of the known PKC substrate, MARCKS (Figure 6C). In summary, our results clearly demonstrate that stimulation of nuclear α1‐ARs activates PKC, specifically PKCδ, and shows that cardiac myocyte nuclei contain machinery sufficient to initiate α1‐AR signaling.
Expression of a PKCδ Nuclear Localization Mutant Blunts α1A‐Receptor Signaling in Adult Cardiac Myocytes
To this point, our results suggest that α1‐AR signaling originates in the nucleus and is transduced from the nucleus to the sarcomere to induce phosphorylation of cTnI and thereby regulate contractility (Figures 1 through 5). Next, we examined PKCδ as a potential mediator of α1‐AR‐induced contractility. DeVries et al. have previously characterized an NLS in PKCδ.20 Because we detected a nuclear population of PKCδ that was activated by α1‐ARs in WT adult cardiac myocytes (Figure 5B and 5C), we tested whether nuclear localization of PKCδ would affect cardiac myocyte contractility. First, we compared localization of WT PKCδ (PKCδ‐WT) to a PKCδ NLS mutant (PKCδ‐NLSmut) in WT adult cardiac myocytes. As shown in Figure 7A, PKCδ‐WT was found throughout the cell, notably in the nucleus and Z‐disks. In contrast, disruption of the NLS in PKCδ prevented its nuclear localization. Next, we found that expression of the PKCδ‐NLSmut tended to reduce the effect of PE on sarcomeric shortening (Figure 7B), but significantly reduced PE‐induced cTnI phosphorylation (Figure 7C), similar to results obtained upon treatment with the nuclear export inhibitor, lepB (Figure 4). Together, our results further show that nuclear localization of α1‐ARs and PKCδ can regulate physiologic responses in adult cardiac myocytes.
Figure 7.

Expression of a PKCδ nuclear localization mutant blunts α1A‐receptor signaling in adult cardiac myocytes. A, Adult cardiac myocytes expressing WT PKCδ tagged with DsRed (PKCδ‐WT, left) or a PKCδ‐nuclear localization mutant tagged with DsRed (PKCδ‐NLSmut, right) were counterstained with antibodies to cTnI and the nuclear stain, DAPI, and examined by confocal microscopy (scale bar=20 μm). B, Sarcomere dynamics were measured in WT cardiac myocytes expressing PKCδ‐WT or PKCδ‐NLSmut before and 5 minutes after phenylephrine (PE; 10 μmol/L of PE, 2 μmol/L of timolol) treatment. Single‐twitch contractions are shown at 5 minutes after the addition of PE. Averaged data for percent (%) sarcomere shortening are presented as mean±SEM at 5 minutes after PE from 26 WT cardiac myocytes expressing PKCδ‐WT from 10 different cultures and 16 WT cardiac myocytes expressing PKCδ‐NLSmut from 8 different cultures. C, cTnI phosphorylation at Thr144 measured by Western blot from WT cardiac myocytes expressing PKCδ‐WT or PKCδ‐NLSmut treated with PE (20 μmol/L, 15 minutes). Quantitation of cTnI phosphorylation at Thr144 is presented as mean±SEM from 6 different cultures. All data were analyzed by 2‐way ANOVA with repeated measures and Bonferroni's post‐test. Percent sarcomere shortening, P=NS; cTnI phosphorylation at Thr144, P=0.0258. Significant comparisons identified by Bonferroni's post‐test are indicated as P<0.05. ANOVA indicates analysis of variance; cTnI, cardiac troponin I; PKC, protein kinase C; WT, wild type.
Discussion
Here, we sought to establish a physiologic function for nuclear α1‐AR signaling in adult cardiac myocytes. Based on our current findings and previous work,8,15–16 we propose a completely novel model for inside‐out α1‐AR signaling in adult cardiac myocytes (Figure 8), where α1‐ARs, localized to the inner nuclear membrane, induce signals in the nuclei that are transported to cytosolic (sarcomere) or membrane targets to regulate contractile function and survival signaling.
Figure 8.

Model of nuclear α1‐AR signaling in adult cardiac myocytes. The figure depicts α1‐ARs localized to the inner nuclear membrane (INM; Figure 5). Furthermore, α1‐ARs colocalize with Gαq, a phospholipase C isozyme (PLCβ1?), as well as PKCα, δ, and ε (Figure 6) only at the nucleus. Catecholamines, such as norepinephrine (NE), are actively transported into cardiac myocytes through organic cation transporter‐3 (OCT). NE binding to α1‐ARs leads to activation of PKC in the nucleus (Figure 6), and nuclear localization is required for signaling (Figure 2). Signals are transported out of the nucleus through the nuclear pore complex (NPC; inhibited by lepB; Figure 4). PKCδ induces phosphorylation of cTnI at the sarcomere to regulate contractility in adult cardiac myocytes (Figures 1 and 7). In addition, nuclear α1‐AR signaling induces activation of ERK in caveolae at the plasma membrane to regulate survival signaling in adult cardiac myocytes. cTnI indicates cardiac troponin I; DAG, diacylglycerol; ER/SR, endoplasmic reticulum/sarcoplasmic reticulum; ERK, extracellular signal‐regulated kinase; MEK, mitogen‐activated protein kinase kinase; NR, nucleoplasmic reticulum; PKC, protein kinase C; PTP, permeability transition pore; RYR, ryanodine receptor; α1‐ARs, α1‐adrenergic receptors.
Our proposed model for nuclear α1‐AR signaling and assertion that nuclear α1‐AR signaling is physiologically relevant are supported by the following observations. First, we found that the α1A subtype regulated contractility in adult cardiac myocytes, as reported by others,26–27 but we also identified a novel α1‐AR sarcomeric target: phosphorylation of cTnI at Thr144 (Figure 1). Second, by using an α1A‐NLSmut and the nuclear export inhibitor, lepB, we demonstrated that nuclear localization was required for α1‐AR signaling (Figure 2) and that α1‐AR signaling must have originated in the nucleus (Figure 4). Third, through differential permeabilization and immunocytochemistry, we found that α1‐ARs localized to the inner nuclear membrane with the C‐terminus of the receptor facing the nucleoplasm (Figure 5). Fourth, we identified PKCα, δ, and ε in nuclei isolated from adult cardiac myocytes. Along with our previous demonstration that α1‐ARs, Gαq, and phospholipase Cβ1 colocalize only at the nuclei in adult cardiac myocytes,15 this suggests that signaling could occur in the nucleus. In fact, we found that in nuclei isolated from adult cardiac myocytes, α1‐ARs induced PKC activation (Figure 6). More important, these findings are consistent with our results showing that nuclear localization was required for α1‐AR signaling and that α1‐AR signaling originated in the nucleus. Finally, we demonstrated that α1‐induced phosphorylation of cTnI at Thr144 was blunted by expression of a PKCδ nuclear localization mutant (Figure 7), further suggesting the signal arose in the nucleus.
Our identification of α1‐ARs localized to the inner nuclear membrane is provocative (Figure 5), because GPCRs are conventionally thought to localize to, and signal at, the plasma membrane. In fact, previous reports have suggested that α1‐ARs localize to the plasma membrane in cardiac myocytes, so how can our results be reconciled? Radioligand binding assays and antibodies are the most commonly used techniques to detect receptor expression and localization, but these techniques have limitations. Radioligand binding assays are typically performed on isolated membrane fractions, which typically do not distinguish membrane fractions (nuclear, for instance),5,26,33 and membrane‐impermeable ligands might miss intracellular receptors.34 Immunocytochemical approaches suffer from the lack of verified α1‐AR subtype‐specific antibodies,30 raising doubts about previous studies that relied upon antibodies subsequently shown to be nonspecific.27,35 Some previous studies employed membrane fractionation with binding assays to suggest that α1‐ARs localize to caveolae.35–36 However, using this technique, one study found that only 27% of α1‐AR binding was localized to the caveolar fraction in neonatal rat ventricular myocytes, suggesting that a majority of the receptor is localized elsewhere.36 Whether this undefined receptor population was at the nucleus was unclear in this study, as well as how this result might differ from adult cardiac myocytes. In contrast, using radioligand binding assays on fractionated adult cardiac myocytes or a fluorescent ligand to stain cultured adult cardiac myocytes, we previously demonstrated that at least 80% of α1‐ARs localize to the nucleus in adult cardiac myocytes.15
Our results indicated that nuclear localization was required to initiate signaling (Figure 2, α1A‐NLSmut fails to restore function), and that α1A‐subtype signaling arose in the nuclei (Figure 4C, lepB blocks α1‐cTnI phosphorylation). However, α1‐mediated contractile function was not entirely inhibited by lepB, suggesting that either not all α1 signaling arises in the nuclei or that not all intranuclear α1 signaling is exported by the nuclear pore complex. Our previous findings that α1 signaling does not originate at the plasma membrane,15 along with our results with the α1A‐NLSmut, are consistent with nuclear α1 signaling. In further support of our hypothesis that signals regulating α1‐mediated contractile function arise within the nucleus, previous studies indicated that the α1 agonist, PE, induced an increase in the frequency of inositol 1,4,5‐trisphosphate–mediated nuclear calcium transients and can trigger calcium‐induced calcium release in the cytosol in cardiac myocytes.37 Though we propose one mechanism whereby nuclear α1‐ARs regulate contractile function, namely, through activation of PKCδ and phosphorylation of cTnI, other mechanisms whereby intranuclear α1 signaling regulates contractile function are unclear. We cannot exclude the possibility of other contractile regulators that can be mediated by nuclear α1‐ARs. For example, although we previously detect PLCβ1 at the nucleus,15 recent studies indicate that the substrate for PLCβ1, phosphatidylinositol 4,5‐bisphosphate, is not in the nuclear membrane.38 Therefore, whether PLCβ1 is using a different substrate, or whether α1‐ARs signal through a different PLC isoform at the nucleus, remains to be determined.
As recently reviewed,1–2 α1‐ARs are cardioprotective and activating α1‐ARs might be a viable therapy in heart failure. This idea is contrary to previous assumptions that all Gq‐coupled receptors mediate pathologic remodeling in the heart.11–12 However, an important distinction between α1‐ARs and other Gq‐coupled receptors that do induce pathologic remodeling, such as ET‐Rs and AT‐Rs, is that α1‐ARs localize to the nucleus, whereas the majority of ET‐Rs and, possibly, the majority of AT‐Rs are expressed at the plasma membrane.13–15 This dichotomy in receptor localization (plasma membrane versus nucleus) and outcome (pathologic versus physiologic remodeling) suggests that distinct signaling pathways are activated at the nucleus. This holds true when comparing α1‐ARs to β1‐ARs as well. It could be argued that nuclear α1‐ARs, which mediate more chronic, protective effects,1–2 act in opposition to plasma membrane β1‐ARs, which acutely mediate contractile function, but are maladaptive with chronic stimulation in heart failure.39 In summary, the nuclear α1‐signaling pathway identified here emphasizes physiological distinctions between plasma membrane receptors, such as β1‐ARs, ET‐Rs, and AT‐Rs, versus α1‐ARs that might be defined by differential receptor localization.
In the basal state, α1‐AR‐mediated contractility is minor, compared to β‐AR‐mediated contractility, which provides the majority of inotropic response to catecholamine stimulation.32 However, evidence suggests that, in the failing heart, α1‐AR‐mediated contractility becomes proportionally more important. In human papillary muscle strips isolated from failing hearts, α1‐AR‐mediated inotropic responses were found to be proportionally greater, because β‐ARs were desensitized and down‐regulated, resulting in reduced β‐AR inotropic responses.40 This is supported by studies in both α1ABKO mice, where pathologic stress reduced contractile function,6 and α1A transgenic mice, where cardiac‐specific overexpression of the α1A subtype increased basal contractile function and protected the heart from pathological stress.9–10,26 However, our current data indicate that, in cultured, unloaded cardiac myocytes, α1‐ARs induced a net‐negative triphasic inotropic effect where the third phase fails to become positive (Figures 1 through 3), which agrees with other studies in isolated mouse cardiac myocytes and right ventricle trabeculae.22–23 In contrast, α1‐ARs mediated positive triphasic response in Langendorff whole mouse heart preparation41 and in rat and rabbit myocardial tissue.23,42–43 Therefore, we suspect that the negative inotropic response in the isolated mouse myocyte system simply reflects a unique aspect of this preparation.
cTnI is a key regulator of cardiac myocyte contractility. Its phosphorylation by β‐adrenergic activation of PKA at serines 23,24, which increases both the rate of relaxation and inotropy,44 is decreased in failing human hearts.45 In addition, activation of PKC has been shown to phosphorylate cTnI at serines 43,45 in addition to threonine 144 to decrease calcium sensitivity and contractility.46 More recently, it was suggested that the way PKCδ is activated could also affect the function of cTnI through differential phosphorylation effects.47 The aim of this study was to identify a novel nucleus to the myofilament pathway affecting cardiac myocyte contractility. Therefore, we focused on the phosphorylation of cTnI at Thr144, a known PKC site that decreases myofilament calcium sensitivity.48
Increased PKC activity has been reported in cardiac hypertrophy and heart failure.49–52 However, the development of therapies targeting abnormal PKC activity in heart failure have met with limited success, perhaps as a result of isoform‐specific activities. For example, the conventional (activated by calcium and lipid) PKC isoforms, α and β, have been shown to increase in heart failure.49–50 Recently, in a pig model of myocardial infarction, the inhibition of PKCα and β by ruboxistaurin was shown to improve cardiac function after injury.53 In contrast, for the novel PKC isoforms, δ and ε, which require only lipid for activation, the inhibition of PKCδ51–52 or the enhancement of PKCε51 activities have been shown to ameliorate pathologic hypertrophy. Here, we show that there is a significant amount of PKCδ in the nucleus of cardiac myocytes that can be activated by α1‐ARs (Figure 6). Because we have shown that α1‐ARs are protective against pathological stress,6,8 it is conceivable that increased PKCδ activity by α1‐ARs could instead be protective in addition to mediating α1‐AR myocyte contractility.
In summary, we have uncovered a potentially significant physiological function for nuclear α1‐AR (GPCR) signaling in adult cardiac myocytes. Specifically, we identified a novel α1A subtype/PKCδ/cTnI inside‐out signaling pathway that regulates contractility in adult cardiac myocytes. This work has a broader implication in understanding α1‐AR‐mediated cardioprotection by identifying a novel nuclear signaling mechanism whereby α1‐ARs might protect the heart from pathological stress.
Sources of Funding
.This work was supported by the American Heart Association (SDG 0435338Z; to O'Connell), the NIH (P20 RR‐017662 [to O'Connell] and F32 HL085980‐01A1 [to Wright]), and the Pharmaceutical Research and Manufacturers of America (to Wright).
Disclosures
None.
Acknowledgments
The authors thank Yuan Huang, MD, and the Molecular Biology Core (funded by NIH P20 RR‐017662) for their assistance with construction of our adenoviral constructs.
References
- 1.Jensen BC, O'Connell TD, Simpson PC. Alpha‐1‐adrenergic receptors: targets for agonist drugs to treat heart failure. J Mol Cell Cardiol. 2011; 51:518-528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O'Connell TD, Jensen BC, Baker AJ, Simpson PC. Cardiac alpha1‐adrenergic receptors: novel aspects of expression, signaling mechanisms, physiologic function, and clinical importance. Pharmacol Rev. 2014; 66:308-333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Major cardiovascular events in hypertensive patients randomized to doxazosin vs chlorthalidone: the antihypertensive and lipid‐lowering treatment to prevent heart attack trial (ALLHAT). ALLHAT Collaborative Research Group. JAMA. 2000; 283:1967-1975 [PubMed] [Google Scholar]
- 4.Cohn JN. The vasodilator‐heart failure trials (V‐HeFT). Mechanistic data from the VA cooperative studies. Introduction. Circulation. 1993; 87:VI1-VI4 [PubMed] [Google Scholar]
- 5.O'Connell TD, Ishizaka S, Nakamura A, Swigart PM, Rodrigo MC, Simpson GL, Cotecchia S, Rokosh DG, Grossman W, Foster E, Simpson PC. The alpha(1A/C)‐ and alpha(1B)‐adrenergic receptors are required for physiological cardiac hypertrophy in the double‐knockout mouse. J Clin Invest. 2003; 111:1783-1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O'Connell TD, Swigart PM, Rodrigo MC, Ishizaka S, Joho S, Turnbull L, Tecott LH, Baker AJ, Foster E, Grossman W, Simpson PC. Alpha1‐adrenergic receptors prevent a maladaptive cardiac response to pressure overload. J Clin Invest. 2006; 116:1005-1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang Y, Wright CD, Kobayashi S, Healy CL, Elgethun M, Cypher A, Liang Q, O'Connell TD. Gata4 is a survival factor in adult cardiac myocytes but is not required for {alpha}1A‐adrenergic receptor survival signaling. Am J Physiol Heart Circ Physiol. 2008; 295:H699-H707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang Y, Wright CD, Merkwan CL, Baye NL, Liang Q, Simpson PC, O'Connell TD. An alpha1A‐adrenergic‐extracellular signal‐regulated kinase survival signaling pathway in cardiac myocytes. Circulation. 2007; 115:763-772 [DOI] [PubMed] [Google Scholar]
- 9.Du XJ, Fang L, Gao XM, Kiriazis H, Feng X, Hotchkin E, Finch AM, Chaulet H, Graham RM. Genetic enhancement of ventricular contractility protects against pressure‐overload‐induced cardiac dysfunction. J Mol Cell Cardiol. 2004; 37:979-987 [DOI] [PubMed] [Google Scholar]
- 10.Du XJ, Gao XM, Kiriazis H, Moore XL, Ming Z, Su Y, Finch AM, Hannan RA, Dart AM, Graham RM. Transgenic alpha1A‐adrenergic activation limits post‐infarct ventricular remodeling and dysfunction and improves survival. Cardiovasc Res. 2006; 71:735-743 [DOI] [PubMed] [Google Scholar]
- 11.Dorn GW., II Physiologic growth and pathologic genes in cardiac development and cardiomyopathy. Trends Cardiovasc Med. 2005; 15:185-189 [DOI] [PubMed] [Google Scholar]
- 12.Dorn GW, II, Brown JH. Gq signaling in cardiac adaptation and maladaptation. Trends Cardiovasc Med. 1999; 9:26-34 [DOI] [PubMed] [Google Scholar]
- 13.Boivin B, Chevalier D, Villeneuve LR, Rousseau E, Allen BG. Functional endothelin receptors are present on nuclei in cardiac ventricular myocytes. J Biol Chem. 2003; 278:29153-29163 [DOI] [PubMed] [Google Scholar]
- 14.Tadevosyan A, Maguy A, Villeneuve LR, Babin J, Bonnefoy A, Allen BG, Nattel S. Nuclear‐delimited angiotensin receptor‐mediated signaling regulates cardiomyocyte gene expression. J Biol Chem. 2010; 285:22338-22349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wright CD, Chen Q, Baye NL, Huang Y, Healy CL, Kasinathan S, O'Connell TD. Nuclear alpha1‐adrenergic receptors signal activated ERK localization to caveolae in adult cardiac myocytes. Circ Res. 2008; 103:992-1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wright CD, Wu SC, Dahl EF, Sazama AJ, O'Connell TD. Nuclear localization drives alpha1‐adrenergic receptor oligomerization and signaling in cardiac myocytes. Cell Signal. 2011; 24:794-802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tadevosyan A, Vaniotis G, Allen BG, Hebert TE, Nattel S. G protein‐coupled receptor signalling in the cardiac nuclear membrane: evidence and possible roles in physiological and pathophysiological function. J Physiol. 2012; 590:1313-1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vaniotis G, Del Duca D, Trieu P, Rohlicek CV, Hebert TE, Allen BG. Nuclear beta‐adrenergic receptors modulate gene expression in adult rat heart. Cell Signal. 2011; 23:89-98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Connell TD, Rodrigo MC, Simpson PC. Isolation and culture of adult mouse cardiac myocytes. Methods Mol Biol. 2007; 357:271-296 [DOI] [PubMed] [Google Scholar]
- 20.DeVries TA, Neville MC, Reyland ME. Nuclear import of PKCdelta is required for apoptosis: identification of a novel nuclear import sequence. EMBO J. 2002; 21:6050-6060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O'Connell TD, Ni Y, Lin KM, Han H, Yan Z. Isolation and culture of adult mouse cardiac myocytes for signaling studies. AfCS Res Rep. 2003; 1:1-9 [Google Scholar]
- 22.Sakurai K, Norota I, Tanaka H, Kubota I, Tomoike H, Endo M. Negative inotropic effects of angiotensin II, endothelin‐1 and phenylephrine in indo‐1 loaded adult mouse ventricular myocytes. Life Sci. 2002; 70:1173-1184 [DOI] [PubMed] [Google Scholar]
- 23.McCloskey DT, Rokosh DG, O'Connell TD, Keung EC, Simpson PC, Baker AJ. Alpha(1)‐adrenoceptor subtypes mediate negative inotropy in myocardium from alpha(1A/C)‐knockout and wild type mice. J Mol Cell Cardiol. 2002; 34:1007-1017 [DOI] [PubMed] [Google Scholar]
- 24.Andersen GO, Qvigstad E, Schiander I, Aass H, Osnes JB, Skomedal T. Alpha(1)‐AR‐induced positive inotropic response in heart is dependent on myosin light chain phosphorylation. Am J Physiol Heart Circ Physiol. 2002; 283:H1471-H1480 [DOI] [PubMed] [Google Scholar]
- 25.Solaro RJ. In: Page E, Fozzard H, Solaro RJ. (eds.). Modulation of cardiac myofilament activity by protein phosphorylation. Handbook of Physiology. 2001New York, NY: Oxford University Press; 264-300 [Google Scholar]
- 26.Lin F, Owens WA, Chen S, Stevens ME, Kesteven S, Arthur JF, Woodcock EA, Feneley MP, Graham RM. Targeted alpha(1A)‐adrenergic receptor overexpression induces enhanced cardiac contractility but not hypertrophy. Circ Res. 2001; 89:343-350 [DOI] [PubMed] [Google Scholar]
- 27.O‐Uchi J, Sasaki H, Morimoto S, Kusakari Y, Shinji H, Obata T, Hongo K, Komukai K, Kurihara S. Interaction of {alpha}1‐adrenoceptor subtypes with different G proteins induces opposite effects on cardiac L‐type Ca2+ channel. Circ Res. 2008; 102:1378-1388 [DOI] [PubMed] [Google Scholar]
- 28.Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, McKinsey TA. Protein kinases C and D mediate agonist‐dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol. 2004; 24:8374-8385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adam SA, Marr RS, Gerace L. Nuclear protein import in permeabilized mammalian cells requires soluble cytoplasmic factors. J Cell Biol. 1990; 111:807-816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jensen BC, Swigart PM, Simpson PC. Ten commercial antibodies for alpha‐1‐adrenergic receptor subtypes are nonspecific. Naunyn Schmiedebergs Arch Pharmacol. 2009; 379:409-412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RT. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest. 2004; 113:370-378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brodde OE, Michel MC. Adrenergic and muscarinic receptors in the human heart. Pharmacol Rev. 1999; 51:651-690 [PubMed] [Google Scholar]
- 33.Rokosh DG, Simpson PC. Knockout of the alpha 1A/C‐adrenergic receptor subtype: the alpha 1A/C is expressed in resistance arteries and is required to maintain arterial blood pressure. Proc Natl Acad Sci USA. 2002; 99:9474-9479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Filipeanu CM, Zhou F, Fugetta EK, Wu G. Differential regulation of the cell‐surface targeting and function of beta‐ and alpha1‐adrenergic receptors by Rab1 GTPase in cardiac myocytes. Mol Pharmacol. 2006; 69:1571-1578 [DOI] [PubMed] [Google Scholar]
- 35.Fujita T, Toya Y, Iwatsubo K, Onda T, Kimura K, Umemura S, Ishikawa Y. Accumulation of molecules involved in alpha1‐adrenergic signal within caveolae: caveolin expression and the development of cardiac hypertrophy. Cardiovasc Res. 2001; 51:709-716 [DOI] [PubMed] [Google Scholar]
- 36.Lanzafame AA, Turnbull L, Amiramahdi F, Arthur JF, Huynh H, Woodcock EA. Inositol phospholipids localized to caveolae in rat heart are regulated by alpha1‐adrenergic receptors and by ischemia‐reperfusion. Am J Physiol Heart Circ Physiol. 2006; 290:H2059-H2065 [DOI] [PubMed] [Google Scholar]
- 37.Luo D, Yang D, Lan X, Li K, Li X, Chen J, Zhang Y, Xiao RP, Han Q, Cheng H. Nuclear Ca2+ sparks and waves mediated by inositol 1,4,5‐trisphosphate receptors in neonatal rat cardiomyocytes. Cell Calcium. 2008; 43:165-174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang L, Malik S, Pang J, Wang H, Park KM, Yule DI, Blaxall BC, Smrcka AV. Phospholipase cepsilon hydrolyzes perinuclear phosphatidylinositol 4‐phosphate to regulate cardiac hypertrophy. Cell. 2013; 153:216-227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J Am Coll Cardiol. 2009; 54:1747-1762 [DOI] [PubMed] [Google Scholar]
- 40.Skomedal T, Borthne K, Aass H, Geiran O, Osnes JB. Comparison between alpha‐1 adrenoceptor‐mediated and beta adrenoceptor‐ mediated inotropic components elicited by norepinephrine in failing human ventricular muscle. J Pharmacol Exp Ther. 1997; 280:721-729 [PubMed] [Google Scholar]
- 41.Turnbull L, McCloskey DT, O'Connell TD, Simpson PC, Baker AJ. Alpha 1‐adrenergic receptor responses in alpha 1AB‐AR knockout mouse hearts suggest the presence of alpha 1D‐AR. Am J Physiol Heart Circ Physiol. 2003; 284:H1104-H1109 [DOI] [PubMed] [Google Scholar]
- 42.Endoh M, Blinks JR. Actions of sympathomimetic amines on the Ca2+ transients and contractions of rabbit myocardium: reciprocal changes in myofibrillar responsiveness to Ca2+ mediated through alpha‐ and beta‐adrenoceptors. Circ Res. 1988; 62:247-265 [DOI] [PubMed] [Google Scholar]
- 43.Terzic A, Puceat M, Clement O, Scamps F, Vassort G. Alpha 1‐adrenergic effects on intracellular pH and calcium and on myofilaments in single rat cardiac cells. J Physiol. 1992; 447:275-292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Solaro RJ, Moir AJ, Perry SV. Phosphorylation of troponin i and the inotropic effect of adrenaline in the perfused rabbit heart. Nature. 1976; 262:615-617 [DOI] [PubMed] [Google Scholar]
- 45.Bodor GS, Oakeley AE, Allen PD, Crimmins DL, Ladenson JH, Anderson PA. Troponin I phosphorylation in the normal and failing adult human heart. Circulation. 1997; 96:1495-1500 [DOI] [PubMed] [Google Scholar]
- 46.Goldspink PH, Montgomery DE, Walker LA, Urboniene D, McKinney RD, Geenen DL, Solaro RJ, Buttrick PM. Protein kinase Cepsilon overexpression alters myofilament properties and composition during the progression of heart failure. Circ Res. 2004; 95:424-432 [DOI] [PubMed] [Google Scholar]
- 47.Sumandea MP, Rybin VO, Hinken AC, Wang C, Kobayashi T, Harleton E, Sievert G, Balke CW, Feinmark SJ, Solaro RJ, Steinberg SF. Tyrosine phosphorylation modifies protein kinase C delta‐dependent phosphorylation of cardiac troponin I. J Biol Chem. 2008; 283:22680-22689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burkart EM, Sumandea MP, Kobayashi T, Nili M, Martin AF, Homsher E, Solaro RJ. Phosphorylation or glutamic acid substitution at protein kinase C sites on cardiac troponin I differentially depress myofilament tension and shortening velocity. J Biol Chem. 2003; 278:11265-11272 [DOI] [PubMed] [Google Scholar]
- 49.Bowling N, Walsh RA, Song G, Estridge T, Sandusky GE, Fouts RL, Mintze K, Pickard T, Roden R, Bristow MR, Sabbah HN, Mizrahi JL, Gromo G, King GL, Vlahos CJ. Increased protein kinase C activity and expression of Ca2+‐sensitive isoforms in the failing human heart. Circulation. 1999; 99:384-391 [DOI] [PubMed] [Google Scholar]
- 50.Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, Lorenz JN, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, DePaoli‐Roach AA, Robbins J, Hewett TE, Bibb JA, Westfall MV, Kranias EG, Molkentin JD. PKC‐alpha regulates cardiac contractility and propensity toward heart failure. Nat Med. 2004; 10:248-254 [DOI] [PubMed] [Google Scholar]
- 51.Chen L, Hahn H, Wu G, Chen CH, Liron T, Schechtman D, Cavallaro G, Banci L, Guo Y, Bolli R, Dorn GW, II, Mochly‐Rosen D. Opposing cardioprotective actions and parallel hypertrophic effects of delta PKC and epsilon PKC. Proc Natl Acad Sci USA. 2001; 98:11114-11119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hahn HS, Yussman MG, Toyokawa T, Marreez Y, Barrett TJ, Hilty KC, Osinska H, Robbins J, Dorn GW., II Ischemic protection and myofibrillar cardiomyopathy: dose‐dependent effects of in vivo deltaPKC inhibition. Circ Res. 2002; 91:741-748 [DOI] [PubMed] [Google Scholar]
- 53.Ladage D, Turnbull IC, Ishikawa K, Takewa Y, Rapti K, Morel C, Karakikes I, Hadri L, Muller‐Ehmsen J, Costa KD, Hajjar RJ, Kawase Y. Delivery of gelfoam‐enabled cells and vectors into the pericardial space using a percutaneous approach in a porcine model. Gene Ther. 2011; 18:979-985 [DOI] [PMC free article] [PubMed] [Google Scholar]