

Abstract
SUMMARY
The aim of this review is to present the current state of knowledge on human latent tuberculosis infection (LTBI) based on clinical studies and observations, as well as experimental in vitro and animal models. Several key terms are defined, including “latency,” “persistence,” “dormancy,” and “antibiotic tolerance.” Dogmas prevalent in the field are critically examined based on available clinical and experimental data, including the long-held beliefs that infection is either latent or active, that LTBI represents a small population of nonreplicating, “dormant” bacilli, and that caseous granulomas are the haven for LTBI. The role of host factors, such as CD4+ and CD8+ T cells, T regulatory cells, tumor necrosis factor alpha (TNF-α), and gamma interferon (IFN-γ), in controlling TB infection is discussed. We also highlight microbial regulatory and metabolic pathways implicated in bacillary growth restriction and antibiotic tolerance under various physiologically relevant conditions. Finally, we pose several clinically important questions, which remain unanswered and will serve to stimulate future research on LTBI.
INTRODUCTION
Despite intensified efforts, tuberculosis (TB) remains a major global health problem. The World Health Organization (WHO) estimates that in 2007, there were ∼8.6 million new cases worldwide, up from 8 million in 1997 (1). TB is responsible for nearly 1.3 million deaths annually, second only to human immunodeficiency virus (HIV) as an infectious cause of death. In the great majority of immunocompetent persons, infection with Mycobacterium tuberculosis is initially contained by host defenses, resulting in latent TB infection (LTBI). However, persons with LTBI can progress to active TB at any time, often many years or even decades after initial infection (2), thereby serving as a source of new infections. Although identification and treatment of infectious persons are paramount, global TB eradication efforts must also focus on detecting and treating cases of LTBI. According to the Institute of Medicine, “to make significant progress toward the elimination of tuberculosis in the United States, efforts to prevent cases from occurring must be amplified” (3). Current diagnostic tests do not discriminate between LTBI and active TB, and treatment for LTBI requires prolonged administration of antibiotics (4). An improved understanding of the host and pathogen mechanisms underlying LTBI may yield novel assays which can identify persons at increased risk for progression to active disease (5), as well as new drugs to shorten the duration of LTBI treatment (6).
Latency, Persistence, and Dormancy: Definitions
Although “latency,” “persistence,” and “dormancy” often are used loosely and even interchangeably in the literature, these terms refer to distinct phenomena which may be phenotypically related. LTBI is defined clinically by a reactive tuberculin skin test (TST), indicating a delayed-type hypersensitivity (DTH) response to intradermal injection of M. tuberculosis-derived purified proteins or a T cell response to M. tuberculosis-specific antigens (Ag), in the absence of clinical and radiographic findings (7). In general, tubercle bacilli cannot be recovered from sputum or other sites in latently infected persons (8), indicating a low bacillary burden. Treatment of LTBI requires 9 months of treatment with isoniazid, which inhibits the mycolic acid synthesis pathway required for cell wall synthesis (9, 10), 4 months of treatment with the transcriptional inhibitor rifampin, or 2 months of rifampin and the sterilizing drug pyrazinamide, which is believed to target inactive bacilli residing within an acidic environment (11), although the last combination regimen is no longer recommended because of increased risk of hepatotoxicity among HIV-seronegative individuals (3). A recent clinical trial has shown that a once-weekly regimen of rifapentine and isoniazid for just 3 months is as effective as a standard self-administered 9-month daily regimen of isoniazid alone and that it has a significantly higher completion rate (12).
The term “persistence” has been used in the vernacular sense throughout the scientific literature to describe the ability of M. tuberculosis to persist or survive in host tissues or under various stress conditions. However, the term “persisters” was originally used by Bigger in 1944 to designate a small number of genetically drug-susceptible organisms among a growing population of Staphylococcus species, which could survive prolonged therapy with penicillin (13). Borrowing this terminology, McDermott defined M. tuberculosis persistence as the “capacity of drug-susceptible organisms to survive drug attack when subsisting in an animal body” (14). Therefore, in the classical sense, M. tuberculosis persistence is related to antibiotic pressure, while LTBI results from host immune defenses. Nevertheless, these two phenomena appear to be phenotypically related and may reflect similar physiological states of the organism. As in the case of LTBI, persistent bacilli appear to be relatively more susceptible to the sterilizing drugs rifampin and pyrazinamide than to the bactericidal drug isoniazid, as the introduction of rifampin into the anti-TB regimen reduced the duration of TB treatment from 18 months to 9 months, and the addition of pyrazinamide further reduced treatment to the current 6 months (15). “Bactericidal” drugs (e.g., isoniazid) are those that kill rapidly dividing bacilli. In contrast, “sterilizing” drugs (e.g., rifampin and pyrazinamide) more effectively kill persistent, nonreplicating organisms. Reduced susceptibility to killing by cell wall-active antibiotics, such as isoniazid, is referred to as “antibiotic tolerance” (16) and is a common feature of persistent bacilli and LTBI (17). This phenomenon, in which the rate of bacterial killing by cell wall synthesis inhibitors is directly proportional to the rate of bacterial replication and metabolic activity (18), is not unique to mycobacteria and has been described for various other organisms, including Streptococcus pneumoniae, Streptococcus pyogenes, Escherichia coli, and Treponema pallidum (19–21).
“Dormancy” is an anthropomorphic term derived from the Latin dormire, meaning “to sleep,” and has been used to designate multiple conditions in which the bacteria are viable but exhibit reduced metabolic activity. The term has been most commonly used in the context of an in vitro model of progressive hypoxia, in which M. tuberculosis shuts down metabolism and replication and becomes phenotypically tolerant to isoniazid (20) (see Modeling LTBI below).
Based on the inability to isolate the causative organisms and their relative refractoriness to cell wall synthesis inhibitors, as well as the lack of selection of resistance following monotherapy, LTBI traditionally has been thought to comprise a paucibacillary population of nonreplicating, metabolically quiescent organisms which have entered a “dormant” state as an adaptive response to immune-based containment mechanisms (22). However, the observation that isoniazid can prevent reactivation disease (23), albeit after prolonged treatment, indicates that some portion of bacilli are at least sporadically multiplying. Consistent with the hypothesis that bacillary replication continues during LTBI, studies using whole-genome sequencing showed the development of mutations in M. tuberculosis isolated from “latently” infected cynomolgus macaques (24). However, it is possible that such mutations could have accumulated as a result of oxidative damage in nonreplicating or sporadically dividing bacilli in the absence of any negative selective pressure. Furthermore, two studies by Lillebaek et al. provide compelling evidence from human LTBI that bacillary replication is minimal, as restriction fragment length polymorphism and insertion sequence patterns do not change over decades of latent infection (2, 25). Similarly, although the number of isolates studied was very small, Colangeli et al. used whole-genome sequencing to show that the mutation rates were not elevated in cases occurring more than 20 years after a TB outbreak relative to those representing recent transmission (361).
In order to reconcile these conflicting findings, a theory advanced in recent years speculates that LTBI does not refer to a single entity, but rather represents an array of microbiological and immunohistological findings giving rise to a clinical spectrum spanning LTBI and active disease (26–28). According to this school of thought, LTBI, like active TB, comprises heterogeneous bacillary populations, including those that are more rapidly or frequently replicating, which consequently are susceptible to killing by isoniazid, and those that are metabolically quiescent, which exhibit tolerance to isoniazid.
The classical paradigm that antibiotic tolerance reflects a small population of metabolically quiescent, nonreplicating “persister” organisms has been challenged by recent work highlighting the importance of efflux pumps in mediating this phenomenon (29). However, in the absence of direct data from the human condition, the precise physiological and metabolic status of the bacilli in LTBI, as well as the role of efflux pumps in mediating antibiotic tolerance, remains unknown (28).
Location of Bacilli in LTBI
It was commonly thought that after M. tuberculosis is inhaled into the lower respiratory tract and phagocytosed by alveolar macrophages, the inevitable outcome is chronic infection, leading to the phrase “once infected, always infected” (30). However, this long-held belief was challenged by a recent study which showed the disappearance of M. tuberculosis-specific Th1-type T cell responses in untreated TST-negative contacts of TB patients during a school outbreak (31), suggesting that early infection may be eradicated in a minority of individuals. Fewer than 10% of infected persons develop clinically evident primary TB, and the majority are able to contain the primary infection (32) with the induction of cell-mediated immune responses and the formation of granulomas, which consist primarily of lymphocytes and M. tuberculosis-infected macrophages (33, 34). At 6 to 8 weeks after infection in humans, and coincident with the development of DTH responses to M. tuberculosis antigens, these granulomas undergo necrosis, resulting in the death of the majority of tubercle bacilli and destruction of the surrounding host tissue. The small proportion of bacilli surviving in an altered physiological state have been postulated to result in LTBI (17). However, despite more than a century of investigation, the precise location(s) of the responsible organisms remains a mystery.
Soon after the discovery of the tubercle bacillus by Robert Koch (35), autopsy studies of persons who died of non-tuberculous-related causes revealed the presence of viable bacilli in various postmortem tissues. The findings of such studies gave rise to the still-prevalent belief that the bacilli responsible for LTBI are contained within caseous and necrotic granulomas. Rabinowitsch showed that the chalk-like contents of mesenteric and bronchial lymph node lesions from patients without active TB produced TB in rabbits in four out of five cases (36). These and similar observations in experimental animal models have led to the theory that spread of M. tuberculosis to the lymphatic system is essential to induce adaptive immunity, which is crucial for the generation of transmissible pathology (37). Examining 304 lesions from the lungs and lymph nodes of 169 persons, Opie and Aronson found living bacilli in more than a quarter of cases, including in 33% of partially fibrotic caseous lesions, 23% of caseous encapsulated nodules, 20% of calcified nodules, and only 4.4% of caseous calcified nodules (38). Interestingly, approximately one-quarter of apical fibrous scars showing no gross or histological evidence of TB grew M. tuberculosis, and calcified nodules from lungs with apical lesions produced TB in guinea pigs much more frequently (30.2% of cases) than did calcified nodules from lungs in which the apices appeared normal (8.1% of cases). Consistent with the hypothesis that LTBI may comprise a spectrum of microbiological and histopathological phenomena (26), Robertson found active TB lesions in 4% of 1,725 autopsy cases in whom the diagnosis was not suspected clinically (30).
In a study of 209 surgically removed lung tissues from 72 patients treated with antituberculous agents, M. tuberculosis growth was detected in 83% of open cavities, 24% of filled-in cavities, and 7% of solid necrotic lesions (39). Although acid-fast bacilli were found on smears in a high proportion of all types of lesions, the authors noted that bacillary growth was obtained consistently only from open sloughing lesions, leading them to speculate that some populations of bacilli could not be adequately revived using standard culturing techniques. Following up on these findings, Hobby et al. cultured a total of 85 necrotic lesions from 40 patients who had received TB treatment, incubating specimens in liquid cultures for up to 9 months (40). These investigators were able to culture viable tubercle bacilli from 78% of closed necrotic or healed lesions and noted that in 9 of the 25 lesions tested, viability was first detected only after prolonged incubation for more than 9 to 12 weeks. These observations led the authors to conclude that “although in many instances the surviving microbial cells appear to be in a suppressed metabolic state, if such a state exists, it is obviously reversible” (40). Although the surviving but not readily cultivable organisms from these lesions may be best described as persistent bacilli based on the definitions advanced above, the authors argued that “evidence is lacking to indicate the extent to which chemotherapy may be responsible for this effect,” suggesting that this phenomenon may also apply to LTBI (40).
Autopsy studies of patients who died from non-TB-related causes also have found evidence of viable bacilli in “normal” lung and lymphatic tissue. As early as 1890, Loomis reported that the bronchial lymph nodes from eight of 30 cases with no gross evidence of TB disease contained tubercle bacilli, as demonstrated by the development of TB in rabbits inoculated with homogenized material (41). Reviewing 357 cases from the literature which were determined to be free of TB at necropsy, Wang found that M. tuberculosis grew in 12% of cases from grossly and histologically normal cervical, bronchial, and mesenteric lymph nodes (42). In addition to providing some of the earliest pathological evidence for LTBI, these studies also demonstrated the importance of lymphatic spread of bacilli even in the absence of active pulmonary infection. Opie and Aronson injected guinea pigs with normal lung or mediastinal lymph node tissue obtained at a site distant from visible lesions and found that 45% of these tissues grew M. tuberculosis, with the highest incidence in cases containing apical fibrocaseous lesions or fibrous scars (38). In contrast to the relatively high rate of bacillary recovery from nontuberculous lung tissues reported in this study, Feldman and Baggenstoss were able to demonstrate tubercle bacilli in only two of 50 autopsy specimens from persons with presumed LTBI (43). Possible explanations for the discrepancy in the findings include the significantly lower prevalence of TB in the latter population (Rochester, MN), as well as the fact that the majority of cases in the latter study showed evidence only of the calcified Ghon complex, which typically does not contain viable bacilli (44). Lending support to these anatomic pathology studies, Hernández-Pando and colleagues applied molecular techniques to study sections of macroscopically normal lung tissue from 13 individuals from Ethiopia and 34 from Mexico who had died from non-TB-related causes, with sections of lung tissue from six Norwegian individuals and six Ethiopian TB cases serving as negative and positive controls, respectively (45). In situ PCR and conventional PCR revealed evidence of M. tuberculosis in five of 13 Ethiopian cases and 10 of 34 Mexican cases. Interestingly, PCR-positive cells also included type II pneumocytes, endothelial cells, and fibroblasts. More recently, the same group of investigators used similar techniques to show that M. tuberculosis DNA can be detected in various organs, including lungs, liver, and spleen, of latently infected individuals (46). In addition, 16S rRNA expression was detected in 10 positive tissue samples, confirming the viability of bacilli at these sites. Using in situ and conventional PCR to study autopsy samples of persons who died from non-TB-related causes, Neyrolles et al. detected M. tuberculosis DNA in adipose tissues surrounding the kidneys, stomach, lymph nodes, heart, and skin of 9/57 Mexican autopsy samples (6/19 individuals) and 8/26 French samples (6/20 individuals) (47), suggesting that adipocytes also may serve as a haven in which infecting bacilli can avoid killing by antimicrobials and recognition by the host immune system. These findings challenge the common belief that bacilli are confined to macrophages and other professional phagocytic cells during LTBI and reinforce the idea that M. tuberculosis can persist in tissues without histological evidence of TB infection, including at extrapulmonary sites.
MODELING LTBI
In Vitro Models of M. tuberculosis Growth Restriction and Antibiotic Tolerance
Clearly, in vitro models cannot duplicate all of the complex host-pathogen interactions underlying the clinical phenomenon of LTBI. However, several models exhibiting important features of LTBI, including bacillary growth restriction, reduced metabolic activity, and phenotypic tolerance to antituberculous drugs, have been proposed.
Nutrient starvation.
It has been known for some time that deprivation of essential nutrients, potentially simulating the microenvironment encountered by M. tuberculosis within necrotic granulomas, leads to bacillary growth arrest and reduced metabolism (48). Hobby and Lenert later showed that upon removal of carbon sources of energy from logarithmically growing cultures by washing and resuspending the bacilli in buffered saline, M. tuberculosis growth ceased and the organisms became refractory to killing by isoniazid and para-aminosalicylic acid (49). More recently, the antibiotic susceptibility profile of nutrient-starved M. tuberculosis was shown to mirror that of LTBI in that the minimum bactericidal concentration (MBC), defined as the minimum concentration required to kill 99% of the starting bacterial population, of rifampin increased from <0.625 mg/liter to 10 mg/liter, while the MBC of isoniazid increased from <0.625 mg/liter to 80 mg/liter against log-phase bacilli relative to 6-week-old nutrient-starved cultures, respectively (50). Consistent with earlier observations, gene and protein expression data derived from nutrient-starved M. tuberculosis revealed evidence of slowing down of the transcription apparatus, energy metabolism, lipid biosynthesis, and cell division, as well as induction of the stringent response (51).
Progressive hypoxia.
Perhaps the best-characterized model of M. tuberculosis dormancy, in which the bacilli are exposed to progressive hypoxia in vitro, which is intended to simulate the microaerobic conditions encountered by bacilli within host necrotic granulomas, was developed by Wayne and Hayes (20). In the eponymous Wayne model, M. tuberculosis cultures are slowly stirred within sealed containers with a 0.5 ratio of air to culture medium. When the dissolved oxygen content drops below 1%, the organisms enter the microaerobic nonreplicating persistence (NRP) stage 1, characterized by thickening of the outer cell wall and termination of replication and transcription, as evidenced by cessation of uptake of radiolabel from [3H]uracil into total RNA and DNA. As the dissolved oxygen drops to below about 0.06% saturation, the bacilli enter NRP stage 2 and show greater refractoriness to isoniazid than to rifampin (20). Several rounds of synchronous replication of M. tuberculosis result upon dilution of NRP stage 2 cultures into fresh, oxygen-rich medium, simulating reactivation when adverse, growth-restricting conditions are removed. Interestingly, progressive hypoxia renders M. tuberculosis susceptible to metronidazole (20, 52). Metronidazole was also found to have anti-TB activity in rabbits and nonhuman primates (53, 54). Although its efficacy in treating LTBI in humans has not been evaluated, a recent clinical trial found no difference in 2-month sputum culture conversion among patients with multidrug-resistant (MDR) TB receiving metronidazole or placebo in addition to optimized background regimens, but those receiving metronidazole had a more than 4-fold increased risk of developing peripheral neuropathy (55). It remains to be determined whether newer nitroimidazoles with a more favorable toxicity profile will have a role in the treatment of LTBI.
Stresses Simulating the Environment within the Macrophage Phagolysosome
Several in vitro conditions have been used to model the microenvironment encountered by M. tuberculosis residing within the macrophage phagolysosome. Although M. tuberculosis can inhibit maturation of the phagosome and limit its acidification to a pH of ∼6.2 by excluding vacuolar proton-ATPase in immunologically naive macrophages, this block is relieved and the phagosomal compartment acidifies to a pH of 4.5 to 5.0 in gamma interferon (IFN-γ)-activated macrophages (56). Importantly, exposure of M. tuberculosis to a pH of <5.5 in vitro leads to growth arrest (57, 58) and phenotypic tolerance to isoniazid (59). The in vivo relevance of the acidic pH model of M. tuberculosis dormancy is underscored by the fact that pyrazinamide, which exhibits antituberculous activity only under acidic conditions (60), is highly active in combination with rifampin for the treatment of LTBI (3).
Inorganic phosphate limitation may be another important microenvironmental condition within the macrophage phagolysosome (61). Phosphate limitation restricts M. tuberculosis growth in a dose-dependent manner, and the stringent response is induced in phosphate-starved cultures (62). Consistent with the hypothesis that phosphate-starved M. tuberculosis is in a nonreplicating state, the MBC of isoniazid was shown to increase from 0.06 μg/ml against logarithmically growing cultures to 20 μg/ml against 28-day-old phosphate-starved bacilli in the absence of genetic drug resistance (62).
Multiple-Stress Models
Although experimentally expedient, in vitro models using single-stress conditions cannot accurately represent the multiple stresses thought to be encountered by M. tuberculosis during human LTBI. To this end, a novel in vitro dormancy model involving multiple stresses, including hypoxia (5% O2 content), high CO2 (10%), nutrient deprivation (10% Dubos medium), and acidic pH (5.0), was developed (63). In this model, M. tuberculosis growth is arrested, and the organisms store lipids, lose their acid-fast staining properties, and become phenotypically tolerant to isoniazid to a greater degree than to rifampin. Global gene expression analysis of M. tuberculosis under these conditions revealed induction of stress response genes, accompanied by downregulation of biosynthetic pathways, as well as transcription and translation machineries (63).
An alternative in vitro model has been developed in which live tubercle bacilli or mycobacterial antigen-coated beads are used to generate granulomas characterized by recruitment of macrophages, their differentiation into multinucleated giant cells and epithelioid cells, and the final recruitment of a ring of activated lymphocytes (64). In this in vitro granuloma model, M. tuberculosis and other pathogenic mycobacteria that produce oxygenated mycolic acids are capable of forming foamy macrophages, which are unable to kill M. tuberculosis but restrict its growth as evidenced by induction of a genetic program associated with mycobacterial dormancy (65). However, it is not yet known if the bacilli in this model acquire phenotypic tolerance to bactericidal drugs.
In Vivo Models of M. tuberculosis Growth Restriction and Antibiotic Tolerance
Murine models.
Unlike in vitro models of M. tuberculosis growth restriction and antibiotic tolerance, animal models provide the entire repertoire of host immune defenses, which are critical to the establishment of LTBI in humans. The mouse is the best characterized and most extensively used model to study M. tuberculosis pathogenesis (Fig. 1 shows a histological comparison of tuberculosis in different experimental animals). However, unlike human LTBI, the classical murine model of TB infection is characterized by a high bacillary burden, with progressive lung pathology and early death of the animals (66). Traditionally, the stable bacillary census in the lungs of mice during the chronic phase of infection has been viewed as a static equilibrium based on evidence that the organisms are in a metabolically quiescent and slowly replicating state (67, 68). However, a recent study using M. tuberculosis containing an unstable plasmid that is lost at a steady rate from dividing bacilli in the absence of antibiotic selection found that the organisms continue to multiply in the lungs of chronically infected mice (69), and genome-wide transcriptional profiling of M. tuberculosis revealed that the organisms remain metabolically active during chronic TB in murine lungs (70). Nevertheless, M. tuberculosis during the chronic phase of infection in mice exhibits antibiotic tolerance in that the bactericidal activity of isoniazid is dramatically reduced when the drug is initiated 42 days after infection relative to initiation of the drug during the acute phase of infection (at day 0 or day 14 after infection) (71).
FIG 1.
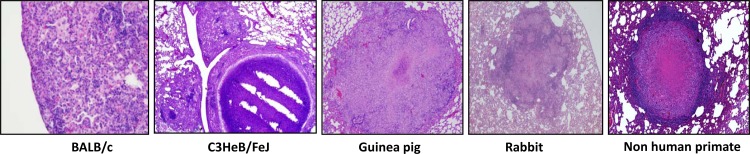
Histological comparison of TB lesions in different experimental animals.
McCune and McDermott at Cornell University developed a model of paucibacillary infection in which M. tuberculosis-infected mice are treated with isoniazid and pyrazinamide to induce a culture-negative state, after which bacteriological relapse of “persisters” can be achieved spontaneously following cessation of therapy or by administration of high-dose corticosteroids (72, 73). Although it is used to study immunological mechanisms involved in LTBI (74, 75), the so-called “Cornell model” is more accurately described as a model of M. tuberculosis persistence, since low-burden infection is achieved by antibiotic treatment rather than by immune control mechanisms.
A murine model of immune-based M. tuberculosis containment recently was developed, which overcomes the problem of the high lung bacillary load resulting in premature death of infected mice. In this model, mice are immunized with a recombinant BCG strain overexpressing the 30-kDa major secretory protein (76) via the aerosol route and challenged 6 weeks later with virulent M. tuberculosis (77, 78), leading to a stable lung census of ∼104 bacilli, which approximates the bacillary burden in human LTBI (38). Perhaps most importantly, this paucibacillary model accurately recapitulates the hierarchy of sterilizing activities of the various drug regimens used to treat human LTBI (78). Recently, this model has been enhanced by the use of C3Heb/FeJ mice, which develop necrotic lung granulomas, and infection may be reactivated by TNF-α neutralization (362). Of course, this model differs from human LTBI in that prior vaccination is required for immune-based control and establishment of a paucibacillary, asymptomatic infection.
A novel mouse hypoxic granuloma model involving the encapsulation of M. tuberculosis within semidiffusible hollow fibers and their subcutaneous implantation in mice has been proposed (79). Although the site of infection is not physiological, this model reliably establishes an immune-controlled, paucibacillary infection consisting of nonreplicating, metabolically quiescent organisms with reduced susceptibility to isoniazid relative to rifampin, while the infected animals remain healthy throughout. Interestingly, although the granulomatous tissues in this model stain with the hypoxia-specific probe pimonidazole, the anaerobic drug metronidazole is inactive (80), suggesting that the oxygen tension in this model is higher than that in NRP stage 2 of the Wayne model of progressive hypoxia (52). The extracellular localization of M. tuberculosis within hypoxic granulomas is a unique feature of this model (79, 80), which may simulate that of at least some M. tuberculosis populations during human LTBI (38).
Guinea pigs and rabbits.
Although mouse TB granulomas serve a function similar to that of human TB granulomas in that they contain infection and provide a localized environment for host immune defenses to kill M. tuberculosis, the histopathology of the standard mouse TB model differs significantly from human TB lung pathology. The cellular composition of murine TB granulomas is similar to that of human granulomas, with the exception of the absence of multinucleated giant cells; however, the former lesions are poorly organized and can be best described as a collection of activated and epithelioid macrophages and lymphocytic clusters (81). In addition to being excessively cellular, mouse TB lesions lack tissue necrosis, which is the pathological hallmark of human TB granulomas (81, 82), where at least some tubercle bacilli are believed to reside during human LTBI (38).
Relative to those in mice, granulomas in guinea pigs and rabbits more closely approximate their human counterparts with respect to cellular composition, granuloma architecture, and the presence of caseation necrosis (81). While TB granulomas in standard mouse models are not hypoxic (83, 84), tissue hypoxia is observed in those of guinea pigs (53, 85) and rabbits (53). In addition, the guinea pig model of TB allows for the differentiation between primary granulomas and secondary lesions, which appear to result from hematogenous dissemination (86, 87). However, like mice, guinea pigs infected with M. tuberculosis develop high-burden disease and succumb to infection (88), indicating that LTBI is not adequately modeled in this species. Also, despite recent advances (87, 89, 90), the study of TB immune control mechanisms in the guinea pig model has been complicated by the lack of appropriate reagents.
Mycobacterium bovis infection of rabbits leads to high-burden disease characterized by caseous granulomas, liquefaction of lesions, and cavitation (91). On the other hand, rabbits are naturally resistant to M. tuberculosis infection, resulting in a paucibacillary population in the lungs of animals at 10 weeks, which can be reactivated with immune-modulating agents (92, 93). Since prior sensitization can alter disease pathology in the rabbit and different strains of M. tuberculosis cause various spectrums of disease (94, 95), these parameters may be manipulated to establish a model of latency. Recently, Subbian et al. have described a highly relevant rabbit model of LTBI in which there is spontaneous and complete clearance of lung bacilli and pathological features by 12 weeks after aerosol infection of New Zealand White rabbits with M. tuberculosis CDC1551 (96). Importantly, these animals appear to establish LTBI rather than tissue sterilization, since reactivation can be achieved by administration of systemic corticosteroids. Validation of this model would require demonstration of the hierarchy of sterilizing activities of the various LTBI regimens in clinical use. As in the case of guinea pigs, immunological reagents for use in rabbits are limited.
Nonhuman primates.
The nonhuman primate has been advanced as the model which most accurately reproduces the clinical, histological, and microbiological characteristics of human LTBI (97–99). Unlike mouse granulomas, and like those of guinea pigs and rabbits, necrotic granulomas in the nonhuman primate model are hypoxic (53). Although cynomolgus macaques infected with high doses of M. tuberculosis (104 to 105 bacilli) develop an acute, rapidly progressive, highly fatal multilobar pneumonia (99), approximately 40% of such monkeys infected with a low dose (∼25 bacilli) of virulent M. tuberculosis via bronchoscope develop a positive tuberculin skin test with no clinical evidence of disease for at least 6 months (97). Unlike monkeys with active disease, manifested by radiographic infiltrates or cavities and a wide spectrum of histological lesions harboring a high bacillary burden, monkeys with latent infection exhibit no abnormalities by chest roentgenogram and only a limited number of small granulomas in the lungs and hilar lymph nodes with central caseation, calcification, and peripheral fibrosis, from which few bacilli can be cultured (97, 100). Recent studies, however, have highlighted the overlap in histological, immunological, and microbiological features among animals with overt signs of disease (“active TB”) and those lacking clinical findings (“LTBI”) (101). Interestingly, M. tuberculosis could be readily cultured by bronchoalveolar lavage in a small proportion of asymptomatic monkeys termed “percolators” (98), perhaps representing subclinical disease and further reinforcing the concept that latency encompasses a range of different clinical, microbiological, and histopathological phenomena (26, 28).
Infection with simian immunodeficiency virus (SIV) (102) and simian-human immunodeficiency virus (SHIV) (103), an HIV-SIV chimera, may be used to model HIV infection and cause reactivation disease. Alternatively, LTBI can be reactivated in macaques using tumor necrosis factor alpha (TNF-α) neutralization (104). Although, as in mice, the availability of immunological reagents facilitates the study of latency and reactivation in nonhuman primates, the use of the latter model has been very limited due to its cost and resource-intensive nature.
HOST FACTORS INVOLVED IN LTBI
CD4 T Cells
Among people with latent TB infection, HIV infection is the strongest known risk factor for reactivation disease in humans and is associated with a number of immunological defects, including reductions in CD4 cell levels, impaired T cell proliferation, decreased cytolytic T cell responses, deranged intracellular killing, and reduced cytokine elaboration in response to mycobacterial antigen challenge (Table 1 lists a few important host genes/factors related to LTBI) (105). Attesting to the importance of CD4 T cells in host defenses during LTBI, the risk of TB reactivation increases dramatically in patients with HIV coinfection, from 5 to 10% per lifetime in immunocompetent hosts to as high as 10% annually in coinfected patients (106), and the risk of TB reactivation rises as the CD4 cell count declines (107–110).
TABLE 1.
Host genes implicated in establishment/maintenance of LTBI
| Gene/factor | Symbol | Importance and/or phenotype (reference[s]) |
|---|---|---|
| ATP-binding cassette, subfamily B (MDR/TAP1)a | Abcb2 | High susceptibility, severe granulomatous pneumonia (133) |
| β2-Microglobulina | B2m | Higher susceptibility, increased granuloma necrosis (127) |
| CD4+ T cellb | Th(1), Th(2) | Influences expression of disease in individuals with pulmonary TB (334) |
| CD8+ T cellb | CD8+ | T cells are crucial in immune surveillance against M. tuberculosis (335) |
| Chemokine (C-C motif) ligand 2b | CCL2 | Establishment of latency and maintenance of granuloma (336) |
| Cluster of differentiation 4a | CD4+ | Necessary to prevent reactivation, may have roles controlling persistent infection (121) |
| Inducible nitric oxide synthasea | Nos2 | High susceptibility, necrotic granulomatous pneumonitis (183) |
| Gamma interferona | IFN-γ | High susceptibility, increased granuloma necrosis (180, 181) |
| Gamma interferon receptora | IFN-γR | High susceptibility, elevated eosinophil infiltration of granulomas (337) |
| Interleukin-1a and interleukin-1ba | IL-1a and IL-1b | Moderate susceptibility, larger granulomas, but no necrosis (338) |
| Interleukin-10a,b | IL-10 | Role during the chronic/latent stage of pulmonary TB and in promoting reactivation (339, 340) |
| Interleukin-12 (p40)a | IL-12b | High susceptibility, defective granuloma formation (166) |
| Interleukin-18a | IL-18 | Moderate susceptibility, larger granulomas in the lung (341) |
| Neutrophil cytosolic factor1 NADPH oxidase subunit, 47 kDaa | Ncf1 | Increase of bacterial growth in the early phase of infection (342) |
| Solute carrier family 11 4 (NRAMP1)a,b | Slc11a1 | High susceptibility, phenocopy of the Nramp1D169/D169 susceptibility allele (343, 344) |
| Tumor necrosis factor alphaa,b | TNF-α | High susceptibility, increased granuloma necrosis, formation/maintenance of granulomas (151, 155, 345) |
| Tumor necrosis factor alpha p55 receptora,b | TNFRs1a | High susceptibility, increased granuloma necrosis (155) |
Studies were performed using in vivo (mouse) experimental models.
Studies were performed using in vitro (human macrophage and/or human lung biopsy/tissue/plasma samples) experimental models.
Whether HIV-infected persons are more susceptible to acquisition of M. tuberculosis infection following exposure is unknown; however, rapid progression to active TB is frequently observed in coinfected patients following exposure to TB contacts, which has resulted in numerous outbreaks in institutions such as hospitals, nursing homes, and prisons, as well as in community settings (105). Molecular analysis revealed that 38% of individuals residing in a long-term care facility for HIV-infected persons in San Francisco developed active pulmonary TB with the same strain of M. tuberculosis within 120 days of exposure to the index case (111). However, depletion of CD4 T cells is not the only immune defect predisposing HIV-infected individuals to TB disease. A retrospective cohort analysis of over 20,000 mineworkers in South Africa showed that the risk of TB doubled within the first year of HIV infection, prior to any significant reduction in CD4 cell counts (112). In addition, HIV-infected persons treated with antiretroviral drugs remain at increased risk for TB disease despite elevated CD4 counts and suppressed viral loads (113). Nevertheless, it is possible that predisposition to active TB in coinfected persons is determined not by total CD4 counts but by the number of M. tuberculosis-specific CD4 T cells. A recent study of 5 women with LTBI from a cohort of commercial sex workers in Tanzania, who were followed before and after HIV seroconversion, found that acute HIV infection was associated with a rapid depletion of M. tuberculosis-specific CD4 T cells (114).
The role of CD4 T cells in anti-TB immunity has been investigated most extensively in the mouse. Antibody-mediated depletion of CD4 T cells in thymectomized C57BL/6 mice led to a greater than 10-fold increase in bacillary numbers in the spleens of mice 3 weeks after infection (115). Antibody-mediated depletion of this lymphocyte subset in thymectomized CBA/Ca mice resulted in a significantly increased lung bacillary burden and accelerated animal death relative to those in nondepleted, thymectomized mice and nonthymectomized control mice (116). Similarly, significantly increased numbers of bacilli were detected in the lungs and spleens of thymectomized C57BL/6 mice treated with anti-CD4 monoclonal antibodies up to day 84 after infection with M. tuberculosis Erdman and Mycobacterium kansasii relative to those in thymectomized control mice (117). In elegant adoptive transfer experiments, Orme showed that CD4 T cells derived from M. tuberculosis-infected mice having undergone a protracted course of isoniazid chemotherapy were capable of restricting bacillary growth in the spleens of gamma-irradiated mice relative to control mice following intravenous challenge with M. tuberculosis (118). M. tuberculosis infection of transgenic mice defective in expression of either major histocompatibility complex (MHC) class II or CD4 molecules was associated with significantly increased bacterial numbers in the lungs, liver, and spleen and accelerated animal death relative to those in isogenic wild-type mice (119). Subsequent studies confirmed the importance of CD4-mediated immunity during subacute TB infection in mice, as wild-type mice were able to restrict bacillary growth and had a median survival time of 259 days, while MHC class II-deficient mice experienced progressive bacillary growth in the lungs and other organs beyond day 20, with a mean survival of 77 days (120).
In addition to their critical role in controlling M. tuberculosis growth during initial infection, CD4 T cells significantly contribute to restricting bacillary growth and preventing reactivation during the chronic phase of infection in mice. Antibody-mediated depletion of CD4 T cells at 6 months after M. tuberculosis infection in mice led to rapid reactivation of infection, characterized by a marked increase in the organ bacillary burden, increased lung pathology, and decreased survival of mice (121). CD4 T cell depletion did not result in diminished expression of IFN-γ and inducible nitric oxide synthase activity, suggesting that the ability of CD4 T cells to prevent reactivation during chronic TB infection may be independent of their roles in IFN-γ production and macrophage activation. Although infiltration of CD8 T cells into M. tuberculosis-infected lungs and their production of IFN-γ were similar in CD4−/− and wild-type mice, cytotoxic activity of CD8 T cells was impaired in the lungs of M. tuberculosis-infected CD4−/− mice, perhaps due to reduced lung expression of interleukin-2 (IL-2) and IL-15, which are critical for the development of cytotoxic effector cells (122).
The role of CD4 T cells in TB immunity has not been investigated directly in guinea pigs due to a lack of transgenic animals and immunological reagents; however, recent studies have used new flow cytometry and immunohistochemistry techniques to track these and other immune cells during the course of M. tuberculosis infection in guinea pigs (123, 124). Challenging the dogma that delayed-type hypersensitivity due to excessive cell-mediated immunity is responsible for necrosis of TB granulomas (91), Turner et al. found that primary lesion necrosis occurs by day 21 after M. tuberculosis infection of guinea pigs, prior to the peak influx of CD4 T cells, likely reflecting the effects of granulocyte degranulation (124). The number of CD4 T cells in the lungs of M. tuberculosis-infected guinea pigs drops precipitously after day 30 and is accompanied by the influx of B cells, neutrophils, and eosinophils and accelerated worsening of lung pathology (123).
HIV and TB coinfection has been modeled successfully in the nonhuman primate, allowing an assessment of the role of CD4 T cells in controlling M. tuberculosis infection in this model. In rhesus macaques coinfected with M. tuberculosis H37Rv and simian immunodeficiency virus (SIV), high viremia appeared to be a predictor of disseminated TB during 6 months of follow-up (125). Recently, Diedrich et al. showed that cynomolgus macaques latently infected with M. tuberculosis Erdman uniformly developed microbiological and histopathological evidence of reactivation following infection with the virus strain SIVmac251 and that reactivation was independent of viral load but correlated with depletion of peripheral T cells during acute SIV infection (102). Thus, monkeys reactivating within 17 weeks after SIV infection had fewer CD4 T cells in the periphery and in bronchoalveolar cells and did not recover from the initial depletion as well as monkeys that reactivated later.
CD4 T cell-based immunity is clearly an integral component of host defenses against M. tuberculosis infection. The precise molecular mechanisms by which these lymphocytes are able to prevent reactivation of LTBI, other than IFN-γ production and macrophage activation, require further elucidation. In addition, whether depletion of M. tuberculosis-specific CD4 T cells during acute HIV infection is responsible for the elevated risk of TB disease in coinfected individuals (114) and whether peripheral CD4 T cell depletion correlates with depletion of this T cell subset in TB granulomas, as suggested in the nonhuman primate model (102), remain to be determined.
CD8 T Cells
While it is generally accepted that class II MHC-restricted CD4 T cells are essential for immunity to tuberculosis, M. tuberculosis infection also elicits CD8 T cell responses in both humans and experimental animals (126). Relative to the case for CD4 T cells, clinical data are lacking to definitively implicate CD8 T cells in host control of human LTBI. However, the role of this T cell subset in TB immunity has been investigated in the mouse model. After intravenous infection with 106 M. tuberculosis Erdman organisms, mice deficient in the β2-microglobulin (β2m) component of the MHC class I molecule, which fail to develop functional CD8 T cells, exhibited caseous necrosis of lung granulomas, as well as greater numbers of tubercle bacilli in the lungs and markedly reduced survival relative to similarly infected control mice (127). However, β2m is also a component of several other antigen-presenting molecules, including the CD1 family of proteins (128), which have been shown to present lipid-based mycobacterial antigens to human T cells (129–131). In order to directly address this issue, Behar et al. studied the course of M. tuberculosis infection in mice deficient in transporter associated with antigen processing (TAP), which is required for peptide processing by the MHC class I pathway and for positive selection of CD8 T cells in the thymus (132), and in mice deficient in CD1D, whose expression and recognition by T cells are TAP independent (126, 133). Consistent with the data from the β2m-deficient mouse study (127), M. tuberculosis infection of TAP1-deficient mice was associated with greater organ bacillary burdens, more severe tissue pathology, and accelerated time to death, whereas CD1D-deficient mice were not more susceptible to infection than control mice (133). Furthermore, Kamath et al. showed that large numbers of culture filtrate protein 10 (CFP-10)-specific cytolytic CD8 T cells are recruited to the lungs of mice following M. tuberculosis infection (134). However, other studies have found little or no effect of antibody-mediated depletion of CD8 T cells on organ bacillary burden, tissue pathology, and survival in mice (117, 120).
The cytotoxic activity of CD8 T cells is dependent on different mechanisms, including apoptosis via the Fas-FasL pathway and killing via perforin and granulysin (135). The antimicrobial peptide granulysin released by human CD8 T cells has been shown to lead to killing of intracellular mycobacteria (135). It has been postulated that the relative unimportance of CD8 T cells in control of M. tuberculosis infection in mice (120) may be partially attributable to the absence of an analog of granulysin in this species (136). In addition to their cytolytic function, CD8 T cells also have cytokine-producing functions, which are predicted to play an important role in protective immune responses to M. tuberculosis infection by mathematical models based on published and unpublished data (136). Thus, M. tuberculosis-specific CD8 T cells are involved in the production of the proinflammatory cytokines IFN-γ and TNF-α (137, 138), which appear to be critical for control of M. tuberculosis infection and will be discussed further below.
TNF-α
Following the widespread use of TNF-α antagonists for the treatment of rheumatoid arthritis and other inflammatory conditions, a relationship was observed between the use of these agents and the development of granulomatous infections (139). Pulmonary and extrapulmonary TB was reported with significantly increased frequency among patients soon after initiation of treatment with the humanized mouse anti-TNF-α monoclonal antibody infliximab (140). Subsequently, an elevated risk of TB was found in patients treated with etanercept, which is a fusion of the extracellular portions of two human p75 TNF receptors linked to the Fc domains of human IgG1 (141). Since 44% of infliximab-related TB cases occurred within 90 days of treatment initiation, while only 10% of etanercept-related cases occurred during the same time period (142), it was suggested that the former cases may represent reactivation of LTBI, whereas the latter may have occurred as a result of the inability to control new infection (143). The rates of tuberculosis (TB) due to infliximab and etanercept use in the United States were reported to be 54 and 28 cases per 100,000 treated patients, respectively. A recent study controlling for potential differences in TB risk factors among patients showed that the risk of TB is higher for patients receiving the monoclonal antibodies infliximab and adalimumab than for patients receiving soluble-receptor anti-TNF-α therapy (144). In addition to helping to elucidate the basic biology of TNF-α, these clinical observations provide compelling evidence that TNF-α is critical for control of M. tuberculosis infection in humans.
TNF-α is produced by macrophages, dendritic cells, and T cells, as a transmembrane protein which is arranged in stable homotrimers that are cleaved to yield the soluble form (145). TNF-α binds to the cell surface receptors TNFR1 and TNFR2, which can signal through antiapoptotic and proinflammatory pathways (146, 147), and contributes to anti-TB immunity through a variety of mechanisms, including secretion of chemokines (148), upregulation of adhesion molecules (149), and induction of macrophage apoptosis (150). Treatment with anti-TNF-α monoclonal antibodies has been shown to inhibit IFN-γ-induced maturation of M. tuberculosis-containing phagosomes in human macrophages (151) and CD4 T cell activation and IFN-γ production in whole blood cultures (152), as well as to stimulate apoptosis of key immune cells, including monocytes, CD4 T helper cells, and M. tuberculosis-reactive CD8 T cells (145, 153). Mouse models of TB infection have confirmed the importance of TNF-α in macrophage activation and identified additional roles for this key proinflammatory cytokine, including immune cell recruitment to the site of infection and granuloma formation, as well as maintenance of the structural integrity of the granuloma (81). Mice deficient in TNF-α are unable to control M. tuberculosis multiplication, leading to poorly formed granulomas with necrosis, widespread dissemination, and death within 28 to 35 days after infection (154). Fulminant TB characterized by high bacillary loads and early death also was observed in M. tuberculosis-infected mice containing a disruption in the gene for the 55-kDa TNF-α receptor (155). Similarly, monoclonal antibody-mediated (155) or receptor-mediated (156) depletion of TNF-α in acutely infected mice yielded a high bacillary burden in the lungs, greater dissemination of disease, and accelerated animal death. Continued growth of M. tuberculosis in acutely infected mice lacking TNF-α appears to be due to delayed activation of macrophages (155) and inadequate granuloma formation as a result of altered chemokine expression by CD11b+ cells (157) and deficient immune cell recruitment (154). Both T cell-derived TNF-α and macrophage-derived TNF-α appear to play distinct roles in orchestrating the protective inflammatory response and enhancing mouse survival during acute infection with M. tuberculosis (158).
In addition to its role in granuloma formation, TNF-α is also important for maintenance of granuloma structure during chronic infection in mice. Monoclonal antibody-mediated neutralization of TNF-α beginning 6 months after intravenous infection with M. tuberculosis Erdman in a low-dose chronic TB murine model led to accelerated death of animals relative to control mice receiving rat IgG (159). Since mice depleted of TNF-α in this study showed a modest 10-fold increase of lung bacillary load at the time of death, their early demise was attributed to marked histopathological deterioration manifesting as increased cellularity, squamous metaplasia, and disorganization of granulomas. In a subsequent study, the same group of investigators used laser capture microdissection to study gene expression of lung lesions within 1 week after TNF-α neutralization, when lung bacterial burdens were equivalent in the experimental and control groups, and found enhanced expression of genes encoding the proinflammatory cytokines IFN-γ and IL-12p40, as well as those encoding the chemokines CCR6 and CCL20 in the experimental group (160). They speculated that altered chemokine expression may have led to B cell mistrafficking and the observed dissolution of B lymphocyte aggregates (160), which appear to be required for optimal control of M. tuberculosis infection (161). Additional studies have confirmed the importance of TNF-α in controlling paucibacillary infection and preventing reactivation. Following treatment with antituberculous drugs to reduce the lung bacillary load to undetectable levels, TNF-α-deficient mice developed reactivation of infection with high bacillary loads in the lungs, spleen, and liver, and animals died within 13 to 18 weeks (162). These findings were accompanied by diminished recruitment and activation of T cells and macrophages into the lung, with defective granuloma formation and reduced inducible NO synthase expression.
In order to investigate the differential propensity for TB disease among patients receiving various TNF-α antagonists, Plessner et al. studied the effects of a monoclonal anti-TNF-α antibody and the soluble TNFR fusion molecule etanercept in a mouse model of chronic TB (163). Although systemic TNF-α neutralization was equivalent between these two molecules and treatment with each beginning on the day prior to infection resulted in the death of mice within a month, administration of the receptor fusion molecule during the chronic phase of infection was associated with mycobacterial control, whereas chronically infected mice receiving the monoclonal antibody experienced a 10-fold increase in lung bacillary burden and succumbed to infection within a month. The discrepancy in phenotypes between mice treated with these molecules was attributed to decreased penetration into the granulomas by the receptor fusion molecule compared with the monoclonal antibody (163). The differential risk of TB among patients receiving different TNF-α antagonists has also been attributed to differential induction of target cell death, differential TNF-α receptor signaling, and differential net inhibition of TNF-α bioavailability (164). Studies in other animal models have further elucidated the role of TNF-α in controlling M. tuberculosis infection, in some cases corroborating and in other cases challenging observations in the mouse model. TNF-α was found to be important for control of M. tuberculosis replication in guinea pig alveolar and peritoneal macrophages, and exposure of resident macrophages to recombinant guinea pig TNF-α was associated with enhanced expression of the gene encoding IL-12p40 (165). The proinflammatory cytokine IL-12 has been shown to be crucial to the development of protective immunity against TB in the mouse model (166, 167) and has been used successfully as an adjuvant to standard antituberculous therapy to treat human TB (168). Further confirming the role of TNF-α in activation of a Th1-type response in guinea pigs, TNF-α neutralization of cultures of immune T cells and macrophages obtained from M. bovis BCG-vaccinated guinea pigs was associated with significant downregulation of IL-12p40 and IFN-γ mRNA expression, reduced antigen-induced lymphoproliferation, and enhanced intracellular viability of both attenuated and virulent M. tuberculosis strains (169). Using a guinea pig model of tuberculous pleuritis, Ly et al. showed that TNF-α neutralization resulted in increased cell-associated bacillary loads, which was accompanied by the absence of necrosis in pleural granulomas, a significant reduction in the proportion of macrophages in the effusions, and an accelerated transition to an anti-inflammatory cytokine response in pleural granulomas relative to the case for control animals (89).
Relative to more resistant outbred rabbits, susceptible inbred rabbits, which show reduced DTH responses and greater bacillary loads per pulmonary tubercle following M. tuberculosis infection, may be deficient in TNF-α, as purified thioglycolate-elicited peritoneal macrophages from these animals show reduced production of TNF-α in response to stimulation with lipopolysaccharide and M. tuberculosis infection (170).
Lin et al. showed that TNF-α neutralization of acutely infected cynomolgus macaques led to uncontrolled pulmonary and disseminated disease by 8 weeks after M. tuberculosis infection, and similar treatment of latently infected macaques resulted in a high rate of reactivation disease (104). Interestingly, although monkeys treated with TNF-α-neutralizing agents developed miliary disease characterized by increased bacillary burden and multiple organ involvement, the cellular composition of granulomas and their overall architecture were preserved when TNF-α was neutralized during both primary infection and latent infection. Relative to control monkeys with latent infection and control monkeys with active infection, anti-TNF-α-treated monkeys showed increased expression of IL-12p40 in hilar lymph nodes and significantly reduced expression of CCL4 in lung tissues (104). The finding that early granuloma formation is independent of TNF-α signaling is consistent with data from the zebrafish model of Mycobacterium marinum infection, in which normal granulomas formed in the absence of TNF-α despite increased bacterial growth (171).
The preponderance of evidence indicates that TNF-α is important for maintenance of LTBI and prevention of reactivation disease. Although production of TNF-α is generally considered beneficial to the host response, overexpression of this cytokine has been shown to yield severe inflammation in the lungs and spleen and earlier death of Mycobacterium bovis BCG-infected mice (172). In addition, TNF-α appears to modulate the immunopathological benefit conferred by M. bovis BCG vaccination following infection with virulent M. tuberculosis in guinea pigs (146). Thus, M. bovis BCG-vaccinated guinea pigs treated with anti-TNF-α antibodies developed splenomegaly following aerosol infection with M. tuberculosis, and antigen-specific lymphoproliferation was observed in splenocyte cultures from these animals upon TNF-α neutralization (173). Therefore, it appears that expression of this cytokine must be tightly regulated during M. tuberculosis infection in order to effect bacillary killing while minimizing tissue destruction (81). Consistent with this hypothesis, high levels of TNF-α were associated with clinical deterioration as evidenced by a statistically significant decrease in mean Karnofsky score shortly after initiation of antituberculous therapy in TB patients (174). However, the precise mechanisms by which TNF-α contributes to control of M. tuberculosis infection in humans remain unclear.
In contrast to the mouse data and consistent with the nonhuman primate data, human cases of extrapulmonary and disseminated TB following TNF-α blockade generally exhibit intact granulomas on biopsy of affected tissues (140, 175). However, it is still possible that TNF-α may have some role in the localization of immune cells and their interactions within the granuloma, rather than in the overall granuloma structure, and disseminated disease may result from TNF-α neutralization because of dysregulation of critical cytokines and chemotactic factors (104). A computational model simulating the pleiotropic functions attributed to TNF-α in the TB granuloma predicted that the key effector mechanisms for controlling mycobacterial growth are macrophage activation and stimulation of cytokine production (176). In this model, when bacterial numbers were held steady, the granuloma structure was found to be essentially unaffected in the absence of TNF-α, leading the authors to conclude that increased bacterial numbers resulting from loss of TNF-α are the main driver of aberrant granuloma formation. Recently, a novel two-compartment mathematical model predicted that TNF-α is a major mediator of phagocyte recruitment to the lungs but that it does not influence the total number of classically activated and alternatively activated macrophages in the lung (177). While useful, the predictions of such computational models require experimental verification. Improved characterization of the role of TNF-α in promoting LTBI is expected to accompany continued clinical experience with TNF-α antagonists, as well as ongoing research efforts using the nonhuman primate and other animal models of paucibacillary infection.
IFN-γ
Although robust data are lacking, the protective role of the IFN-γ pathway against mycobacterial infection in humans is supported by several studies. Children bearing mutations in the gene for IFN-γ receptor 1, leading to absence of receptors on cell surfaces and a functional defect in TNF-α upregulation by macrophages in response to IFN-γ, were found to develop severe infections by nonpathogenic mycobacteria (178). Humans latently infected with M. tuberculosis were shown to have up to 15-fold-higher proportions of IFN-γ+ IL-2+ and IFN-γ+ CD4 T cells with effector and central memory phenotype than subjects with active TB, in whom multifunctional (IFN-γ+ IL-2+ TNF-α+) CD4 T cells with primarily effector phenotype predominated (179), suggesting that IFN-γ plays an important role in human LTBI.
IFN-γ appears to be the most important cytokine for control of M. tuberculosis infection in mice. Thus, IFN-γ gene knockout mice infected intravenously or via aerosol with a sublethal inoculum of M. tuberculosis Erdman developed severe tissue destruction and necrosis associated with widespread bacillary dissemination, very high bacillary burdens in the organs, and a fulminant clinical course (180). Similarly, Flynn et al. showed that although IFN-γ-deficient mice could develop granulomas, they were unable to restrict bacillary growth following intravenous infection with virulent M. tuberculosis, exhibiting severe tissue necrosis and accelerated mortality, which could be delayed but not prevented by treatment with exogenous recombinant IFN-γ (181). Using a variant of the Cornell model to first achieve a paucibacillary infection, Scanga et al. showed that subsequent neutralization of IFN-γ induced a slow but sustained increase in the number of bacilli in the lungs, supporting a role for this cytokine in the maintenance of latent infection (75).
Early studies revealed that IFN-γ knockout mice had undetectable or very low expression levels of the inducible nitric oxic synthase (iNOS/NOS2) gene in spleens, as well as greatly diminished serum levels of reactive nitrogen intermediates (RNI) relative to those in isogenic wild-type mice following M. tuberculosis infection (181). Confirming the importance of RNI in host resistance against M. tuberculosis, subsequent studies revealed that treatment of mice with the iNOS inhibitor aminoguanidine during the acute phase of infection resulted in increased tissue bacillary burdens and histopathology, as well as accelerated mortality (182). Furthermore, NOS2−/− mice behaved similarly to wild-type mice treated with systemic glucocorticoids during acute M. tuberculosis infection vis-à-vis organ bacillary burdens and time to death, and administration of a NOS2-specific inhibitor during the chronic phase of infection led to increased lung bacillary counts and reduced survival (183). In another study, mice which were infected with a relatively low inoculum of M. tuberculosis to mimic latent infection and then treated with aminoguanidine at 6 months postinfection were found to develop reactivation, as manifested by hepatosplenomegaly, intense tissue granulomatous reaction, and increased organ bacillary load (74). In the same study, mice receiving isoniazid and pyrazinamide for 4 weeks to yield a paucibacillary infection also had increasing bacillary counts in the lungs, liver, and spleen upon subsequent treatment with aminoguanidine. However, M. tuberculosis infection could not be reactivated in another modified Cornell model despite treatment with the NOS2-specific inhibitor l-N6-(1-iminoethyl)lysine for 210 days (75).
Interestingly, Flynn et al. found that iNOS expression in the livers of latently infected mice started to decrease 6 weeks after treatment with aminoguanidine despite regression of hepatic granulomatous lesions (74), suggesting that RNI-independent mechanisms may contribute to the prevention of tuberculous reactivation. Subsequent studies showed that control of virulent M. tuberculosis growth in the lungs and mouse survival were significantly greater in NOS2−/− mice than in those lacking IFN-γ, IFN-γR1, or the IFN-γR signaling protein STAT-1 (184), again indicating the existence of IFN-γ-dependent, NOS2-independent immunity against TB. In the same study, MacMicking et al. found that mice lacking the IFN-γ-responsive 47-kDa GTPase LRG-47 failed to control M. tuberculosis replication in the tissues, leading to significantly reduced survival of these mice relative to wild-type mice, despite intact NOS2 activity in the mutant mice. The enhanced TB susceptibility of Lrg-47−/− mice was attributed to defective bacillary killing in IFN-γ-activated macrophages as a result of reduced recruitment of H+ V-ATPase pumps to M. tuberculosis-containing phagosomes, resulting in impaired acidification and maturation of these organelles (184). More recently, it was shown that certain members of the 65-kDa guanylate-binding protein (Gbp) family, which is part of the IFN-γ-inducible GTPase superfamily, are required for control of M. bovis BCG infection in mice by inducing host defense proteins, including the phagocyte oxidase, antimicrobial peptides, and autophagy effectors, to kill intracellular bacteria (185).
IL-12
Since IL-12 is important in promoting the emergence of Th1, IFN-γ-producing T cells, the role of this cytokine in resistance to M. tuberculosis infection has been investigated. Early studies showed that administration of exogenous IL-12 enhanced resistance of mice to intravenous infection with virulent M. tuberculosis in a dose-dependent manner, which was associated with greater splenocyte production of IFN-γ upon stimulation with mycobacterial antigens than that of splenocytes from control infected animals, while treatment of mice with neutralizing monoclonal anti-IL-12 antibody led to increased bacillary numbers in the organs, as well as diffuse, poorly formed granulomas (186). Since IL-12 is a heterodimeric cytokine made up of two disulfide-linked subunits (p35 and p40) (187), subsequent studies focused on characterizing the role of each of these units in IL-12 protective responses. Mice deficient in the p35 subunit of IL-12 (IL-12p35−/−), which are able to produce endogenous IL-12p40, cleared M. bovis BCG infection and retained Ag-specific specific Th1 and cytotoxic T cell responses, as well as the ability to form protective granulomas, whereas IL-12p35−/−p40−/− mice were unable to control such infection and exhibited accelerated mortality following M. tuberculosis infection (188). Similarly, mice lacking the p40 subunit of IL-12 were unable to control M. tuberculosis growth, exhibiting reduced production of IFN-γ (166, 189) and increased mortality (189) relative to control mice, while mice lacking the p35 subunit showed only a modest defect in bacillary growth control with an intact ability to generate Ag-specific IFN-γ responses and prolonged survival relative of infected p40-deficient mice (189). This cytokine is composed of the IL-12 p40 subunit and a p19 subunit. More recently, IL-12p40-deficient mice were found to have impaired migration of dendritic cells from the lungs to the draining lymph nodes following M. tuberculosis infection, resulting in reduced activation of naive T cells, and treatment of IL-12p40-deficient dendritic cells with IL-12p40 homodimer restored M. tuberculosis-induced migration of these cells, as well as their ability to activate naive T cells (190).
Th17 Cells and T Regulatory Cells
Th17 cells have been reported to contribute to the adaptive immune response to M. tuberculosis in exposed persons and in patients with tuberculosis (191). Moreover, T regulatory (Treg) cells can modulate Th17 responses even in patients with latent infection (192). Superior Ag-specific responses and enhanced control of M. tuberculosis multiplication observed in p35 gene-disrupted mice relative to p40 gene-disrupted mice (189) have suggested that the p40 subunit may act other than as a component of IL-12. A candidate molecule capable of driving the protective responses in the p35 gene-disrupted mice is the novel cytokine interleukin-23 (IL-23). IL-23 is a heterodimeric cytokine consisting of p40 and p19 (IL-23 alpha subunit). In conjunction with IL-6 and transforming growth factor-β1 (TGF-β1), IL-23 stimulates naive CD4 T cells to differentiate into a novel subset of cells called Th17 cells, which produce IL-17, a proinflammatory cytokine that enhances T cell priming and stimulates the production of proinflammatory molecules such as IL-1, IL-6, TNF-α, NOS-2, and chemokines, resulting in inflammation (193) (194). This new pathway has been shown to play a critical role in both the development of autoimmune diseases and protection against infections (192). A study from India revealed an association between LTBI, as defined by asymptomatic tuberculin skin test positivity, and decreased levels of IL-23 and IL-17 (192). IL-23 was found to be essential for protection of ESAT-6 peptide-vaccinated mice against aerosol challenge with M. tuberculosis and for establishment of an IL-17-producing CD4+ T cell population in the lung (195). While Th17 cells, unlike Th1 cells, do not appear to be required for protection following primary M. tuberculosis infection in animal models, cross-regulation between these responses may be important for determining immunopathology (196).
Recently, T regulatory (Treg) populations, which suppress a variety of immune responses, have received significant attention (197). Treg cells mostly share the CD4+ CD25+ phenotype but can also be CD25− or CD8+. A more general feature is expression of the transcriptional regulator forkhead box P3 (FoxP3) (198). The number of CD4+ CD25+ FoxP3+ Treg cells was found to be increased in the blood or at the site of infection in patients with active TB relative to healthy control subjects, and FoxP3 expression was increased in patients with extrapulmonary TB compared with patients with purely pulmonary TB (199). Moreover, the frequency of CD4+ CD25+ FoxP3+ T lymphocytes was inversely correlated with local M. tuberculosis-specific immunity, and both blood and pleural Treg cells were able to suppress IFN-γ and IL-10 production in TB patients (200), thus possibly contributing to TB pathogenesis (199, 200). Expansion of the Treg cell population has been postulated to predispose to or be a marker of the progression of LTBI to active disease (201). Consistent with this hypothesis, ex vivo depletion of CD4+ CD25+ cells from peripheral blood mononuclear cells (PBMCs) resulted in increased numbers of M. tuberculosis antigen-specific IFN-γ-producing T cells in seven of eight patients with active TB (199). Depletion of CD25+ cells reduced lung and spleen bacillary loads and granuloma formation in M. tuberculosis-infected mice during acute infection but not during the later stage of infection (202).
The precise role of B cells in controlling TB infection remains to be determined. Ectopic follicle-like B cell aggregate formation, which is commonly observed in the lungs of patients with TB, has been suggested to be associated with containment of M. tuberculosis infection, perhaps through induction of antigen-specific IL-17 responses (203). However, B cell deficiency does not lead to enhanced susceptibility to M. tuberculosis infection in mice (204).
MICROBIAL FACTORS INVOLVED IN LTBI
Lipid and Energy Metabolism
Early studies demonstrated that tubercle bacilli grown in animals are metabolically distinct from those grown in vitro (Table 2 lists a few important M. tuberculosis genes related to LTBI) (205). Lipids have long been thought to play a key role in TB pathogenesis. M. tuberculosis preferentially utilizes fatty acids as a carbon and energy source for prolonged survival in the murine model of infection (205). The M. tuberculosis genome contains a large number of duplicated genes annotated as encoding β-oxidation enzymes, allowing the organism to catabolize a wide range of fatty acids through successive rounds of β-oxidation. Even-number-chain fatty acids are degraded to acetyl coenzyme A (AcCoA), and odd-number-chain fatty acids are degraded to both AcCoA and propionyl coenzyme A (PropCoA) (206).
TABLE 2.
M. tuberculosis genes implicated in LTBI
| Rv no. | Gene(s) | Importance and/or phenotypes (reference[s]) |
|---|---|---|
| Rv0126 | treS | Trehalose synthase, cells lacking this gene are as virulent as wild-type in mice (346) |
| Rv0350 | hsp70 | Heat shock protein 70, overexpression of heat shock proteins reduces survival of M. tuberculosis in the chronic phase of infection (347) |
| Rv0353 | hspR | HspR repressor, reduced long-term survival in mice (347) |
| Rv0467 | icl | Metabolism of fatty acids, reduced long-term survival in mice (213) |
| Rv0470c | pcaA | Cyclopropane synthase, reduced long-term survival in mice (208) |
| Rv0820, Rv0930 | phoT, pstA | Stringent response, phosphate transport protein, growth in alveolar macrophage (62) |
| Rv0981, Rv0982 | mprA, mprB | Two-component regulator, reduced long-term survival in mice (348, 349) |
| Rv1027/8c | kdpDE | Two-component regulator, increased lethality in mice (243) |
| Rv1032c | trcS | Two-component regulator, increased lethality in mice (243) |
| Rv1161 to -4 | narGHJI | Nitrate reductase, reduced long-term survival in mice (350, 351) |
| Rv1221 | sigE | Regulates fatty acid degradation by Icl1, cell wall biosynthesis, prolonged time to death in mice (257, 272) |
| Rv1736c | narX | Fused nitrate reductase, increased expression during hypoxia and in human granulomas (351) |
| Rv1737c | narK2 | Nitrate/nitrite transport, increased expression during hypoxia and in human granulomas (352) |
| Rv1832 | gcvB | Glycine decarboxylase, increased expression during hypoxia (353) |
| Rv1908c | katG | Catalase-peroxidase, reduced long-term survival in mice (354) |
| Rv2031c | hspX | α-Crystallin, increased expression during hypoxia and in mice (355) |
| Rv2109c, Rv2110c | prcBA | Proteosome activity, required for long-term survival (212) |
| Rv2583c | relA | Stringent response, (p)ppGpp synthase, reduced long-term survival in mice and guinea pigs (246, 255) |
| Rv2710 | sigB | Adaptation to stationary-phase growth in nutrient-poor environment, nonessential gene (263) |
| Rv2984, Rv0496 | ppk1, ppx1 | Poly(P) regulation, long-term survival in mice and guinea pigs (281, 283) |
| Rv3132c | dosR | Two-component regulator, hypoxic response, increased lethality in mice (79, 230, 245, 356) |
| Rv3139, Rv3140 | fad E23, fadE24 | Induced by INH, component of shunt pathway, detoxify accumulated fatty acids (357, 358) |
| Rv3223c | sigH | Regulates structural genes, DNA repair protein, enzymes in thiol metabolism, reduced pathology, prolonged time to death (265, 267, 270) |
| Rv3286c | sigF | Regulation of genes in early and late stationary phase, prolonged time to death in mice (359) |
| Rv3416 | whiB3 | Transcriptional regulator, reduced pathology, prolonged time to death (227) |
| Rv3583c | carD | rRNA transcription, essential for survival in mice (288, 360) |
| Rv3764/5c | tcrY | Two-component regulator, increased lethality in mice (243) |
Bacillary lipid accumulation has been linked to M. tuberculosis survival in the host and phenotypic tolerance to antibiotics. For example, a mutant deficient in triacylglycerol synthase1 (encoded by tgs1) showed impaired ability to accumulate triacylglycerol and reduced antibiotic tolerance, while complementation of the gene restored antibiotic tolerance (63). Pandey and Sassetti identified a mycobacterial gene cluster, mce4, which is specifically required for bacillary survival during chronic murine infection (207). Furthermore, they showed that mce4 encodes a cholesterol import system that enables M. tuberculosis to derive both carbon and energy from this ubiquitous component of host membranes (207). pcaA, a novel member of a family of cyclopropane synthetases, is necessary for chronic M. tuberculosis infection in mice (208), thus defining a role for cyclopropanated lipids in bacillary survival in vivo.
In addition to lipid metabolism, other metabolic pathways have been associated with long-term survival of M. tuberculosis in the host. These include dlaT (encoding dihydrolipoamide acyltransferase, a subunit of the pyruvate dehydrogenase complex) (209), menA, which is involved in menaquinone biosynthesis required for ATP (210), and cydC, which encodes a transporter for cytochrome bd assembly required for energy production during hypoxia and in vivo infection (211). Conditional depletion of the core proteasome subunits PrcB and PrcA impaired growth of M. tuberculosis in vitro, rendered bacilli more susceptible to nitric oxide, and impaired bacillary long-term survival during chronic infection in mice (212).
Glyoxylate shunt and gluconeogenesis.
M. tuberculosis utilizes two pathways, namely, the glyoxylate shunt and gluconeogenesis, for assimilating AcCoA and PropCoA from fatty acid and cholesterol catabolism. The glyoxylate shunt comprises the sequential activities of isocitrate lyase (Icl) and malate synthase (GlcB). Expression of the icl gene is significantly upregulated during the chronic phase of infection in mice (213), when bacillary growth appears to be restricted. Consistent with an important role for the glyoxylate shunt in the long-term survival of M. tuberculosis in host tissues, an icl-deficient mutant was shown to have a survival defect during the chronic phase of infection in the lungs of immunocompetent mice despite being dispensable for growth during the acute phase of infection (213). Interestingly, the virulence of the icl-deficient mutant was restored in IFN-γ knockout mice, and the mutant was markedly attenuated for survival in activated but not resting macrophages. Taken together, these data indicate that the glyoxylate shunt is critical for long-term bacillary survival in infected host tissues but that the requirement for this pathway is partially dependent on the immune status of the host.
An essential role for gluconeogenesis in mycobacterial metabolism in vivo has been confirmed by studies showing that disruption of pckA (214), which encodes phosphoenolpyruvate carboxykinase, in virulent Mycobacterium bovis (215) or the vaccine strain M. bovis bacillus Calmette-Guérin (216), resulted in reduced virulence in animal models of infection. Expression of the M. tuberculosis genes pckA and glpX, the latter of which encodes fructose 1,6-bisphosphatase, (215–218), was found to be upregulated in the lungs of chronically infected mice (216). Observations such as these have led to the concept that M. tuberculosis switches its carbon source from carbohydrates to fatty acids during the chronic phase of infection (219). Recent results have suggested the novel view that the metabolic adaptations of M. tuberculosis during in vivo infection reflect an adaptive response rather than a response to altered nutrient availability (206, 214–216, 220, 221).
FadE28 expression and hypervirulence.
M. tuberculosis fadE28 encodes acyl coenzyme A (acyl-CoA) dehydrogenase, which participates in the first step of β-oxidation (222). It has been postulated that the reported hypervirulence of certain strains may be attributable to upregulation of the fadE28 gene, as increased efficiency of β-oxidation may be important for survival within the macrophage phagolysosome, in which fatty acids may serve as the primary carbon source (223). The fadE28 gene, together with its adjacent genes, may also be critical in the process of lipid modification that could facilitate parasitism in human macrophages (223).
whiB-like genes.
The Wbl family represents an important group of M. tuberculosis proteins, of which WhiB3 represents the prevailing paradigm. WhiB3 is an intracellular redox sensor comprising a 4Fe-4S cluster, which specifically reacts with O2 and NO and other exogenous and endogenous metabolic signals to maintain redox balance (224). As a physiological regulator of lipid anabolism, WhiB3 differentially modulates the assimilation of propionate into complex polyketides under defined oxidizing and reducing conditions (225). WhiB3 interacts with the principal sigma factor SigA to affect host survival but is dispensable for in vivo growth of M. tuberculosis (226). In addition, WhiB3 may function as a transcriptional regulator, binding to the promoters of the genes pks2 and pks3 (225). Its expression is induced at 2 weeks postinfection in wild-type and immunodeficient mice and is inversely correlated with bacterial density (227).
Although nonessential for in vivo growth (226), WhiB3 may play an important role in sensing reductive stress generated by host lipid catabolism, controlling the biosynthesis of virulence polyketides and storage lipids necessary for achieving redox balance, and modulating host innate immunity (228). Interestingly, the latter study reported a link between WhiB3 modulation of lipid anabolism and the DosR-DosS-DosT signaling pathway, which is implicated in the M. tuberculosis dormancy response to progressive hypoxia (see below) (228).
A recent study reported that the transcriptional regulator WhiB5 is involved in metabolic adaptation during starvation and is required for normal growth and survival of M. tuberculosis in the lungs of BALB/c mice (229). Interestingly, mice infected with the whiB5-deficient mutant showed larger lung granulomas with more IFN-γ+ cells and fewer IL-10+ cells than in wild-type-infected mice, implicating WhiB5 in immunomodulation. In addition, reactivation of chronic TB infection in mice with systemic corticosteroids could not be achieved in the absence of WhiB5 (229). Global gene expression analysis identified 58 genes whose expression is WhiB5 dependent, including sigM, encoding an alternative sigma factor, and genes encoding the constituents of two type VII secretion systems, ESX-2 and ESX-4.
The Adaptive Response to Hypoxia
dosR-dosS-dosT.
The M. tuberculosis two-component system DosR-DosS (also called DevR-DevS) has received significant attention over the last decade as a key regulatory mechanism underlying M. tuberculosis dormancy. The histidine kinases DosS (DevS) and DosT phosphorylate and activate the cognate response regulator DosR in M. tuberculosis (230–232). Originally found to be induced by exposure of M. tuberculosis to progressive hypoxia, the dormancy survival regulator (DosR) regulon (233, 234) comprises 48 genes, including several believed to play an important role in M. tuberculosis adaptation to hypoxic stress, as well as energy acquisition from alternative carbon sources, such as fatty acid metabolism, and nitrate reductase genes. Thus, the DosR regulon includes the heat shock protein gene acr (hspX), narX, narK2, and fdxA (nitrate accumulation and alternative electron transport), nrdZ (deoxynucleoside triphosphate [dNTP] synthesis under microaerobic conditions), tgs1 (triglyceride synthase), and six M. tuberculosis orthologs of the universal stress protein family (resistance to DNA damage). Several DosR regulon genes are required for bacillary survival in animal models of TB infection containing tissue hypoxia (80). In support of a global regulatory role for DosR, the DosR-dependent regulon is also induced in response to other growth-limiting conditions, including nitric oxide (234), carbon monoxide (235), ascorbic acid (236), macrophage infection (237), and during the acute (79) and chronic (238) phases of infection in mice (234). Taneja et al. attributed ascorbic acid induction of DosR regulon to oxygen scavenging and creation of a hypoxic environment (236), while Honaker et al. demonstrated that it specifically induced the regulon via its reductive effect on the electron transport system, not in an artifactual manner through reaction with available oxygen (239). A second transcriptional program, named the enduring hypoxia response, was shown to be induced in long-term hypoxic cultures (231, 240). Expression of this set of ∼230 genes, which include the alternative sigma factor genes sigE and sigH (232), is DosR independent.
The DosR regulon appears to be essential for M. tuberculosis survival during progressive hypoxia by shifting metabolism away from aerobic respiration and maintaining energy levels and redox balance (241). Recently, Trauner et al. used microfluidic, proteomic, and ribosomal profiling techniques to show that the DosR-regulated protein ribosome-associated factor under hypoxia (RafH) promotes mycobacterial survival during hypoxia by stabilizing ribosomes in the associated form (70S) (242). Interestingly, phenotypes of dosS-dosR-deficient mutants have been varied in animal models. Thus, Parish et al. found that SCID mice infected intravenously with a dosR-dosS deletion mutant died more rapidly than mice infected with wild-type bacteria (243). Similarly, DBA2 mice infected with a dosR-dosS-deficient mutant had higher levels of bacteria in their organs at 15, 30, and 45 days after aerosol infection (231, 244). In a separate study, a dosR-dosS deletion mutant exhibited a growth defect in C57BL/6 mice and guinea pigs and did not replicate more than the wild type in the lungs of rabbits (245). In the latter study, the same mutant was found to have survival equivalent to that of the isogenic wild-type strain in an extracellular, hypoxic granuloma model in mice. Taken together, these data suggest that the dosR and dosS genes are required for wild-type growth and long-term survival in host tissues and that differences in phenotypes may be attributable to differences in the mutant strains and animal models used.
The Stringent Response
When microorganisms encounter an environment with limited nutrients or other stress-related stimuli, they enter a dramatically slowed growth state characterized by a decrease in rRNA, tRNA, and protein syntheses, modified RNA polymerase activities, diminished activity of many transport systems, and decreased carbohydrate, amino acid, and phospholipid metabolism. This is known as the stringent response, and it is mediated by the rapid accumulation of hyperphosphorylated guanine nucleotides, specifically the 3′-pyrophosphate derivative of GDP (ppGpp) and the 3′-pyrophosphate derivative of GTP (pppGpp), the mixture of both being referred to as (p)ppGpp (246). The alarmone (p)ppGpp interacts with RNA polymerase (247, 248), thus interfering with alternative sigma factor competition for RNA polymerase and modulating promoter recognition and activity (249, 250).
Role of RelA and the alarmone (p)ppGpp in M. tuberculosis growth restriction.
In M. tuberculosis, the enzyme RelA catalyzes both an ATP:GTP 3′-pyrophosphoryltransferase reaction and a (p)ppGpp 3′-pyrophosphohydrolase reaction [Fig. 2 illustrates the central hypothesis; i.e., a complex feedback regulatory loop involving poly(P)-dependent expression of M. tuberculosis rel and (p)ppGpp-mediated inhibition of poly(P) hydrolysis contributes to M. tuberculosis growth restriction and antibiotic tolerance] (251). The major physiological regulatory mechanism for RelA is its allosteric regulation by a complex of macromolecules consisting of the ribosome, tRNA, and mRNA, referred to as the RelA-activating complex (RAC). When uncharged tRNA binds nonenzymatically to the acceptor site of an elongating ribosome stalled at a codon on an mRNA, the ribosome-bound RelA is stimulated to synthesize (p)ppGpp, signaling that the environment is limited in amino acids. Conversely, when amino acid levels are sufficient, (p)ppGpp is degraded by the hydrolytic activity of RelA (252).
FIG 2.

A complex feedback regulatory loop involved in the M. tuberculosis stringent response. PPK, polyphosphate kinase; PPX, exopolyphosphatase; Pi, inorganic phosphate; rac, M. tuberculosis Rel-activating complex, consisting of the ribosome, tRNA, and mRNA. (Courtesy of Harvey Rubin, University of Pennsylvania School of Medicine.)
The ability to rapidly fine-tune cellular (p)ppGpp concentrations in response to available nutrients or other stresses is essential for the long-term survival of M. tuberculosis under growth-limiting conditions. Inactivation of the gene Rv2583c, encoding RelA, yields a strain (ΔrelA) unable to synthesize (p)ppGpp and defective in long-term survival upon nutrient starvation in vitro (246). Consistent with a broader role for (p)ppGpp as a global regulator of M. tuberculosis growth, drug-induced inhibition of mycobacterial respiration and mycolic acid synthesis is associated with upregulation of relA (253), and a ΔrelA mutant shows significantly impaired survival relative of the isogenic wild-type strain during extended anaerobic incubation (246) and during the chronic phase of infection in mouse lungs (254). In addition, relA-deficient M. tuberculosis strains showed significant survival defects compared to their respective isogenic wild-type strains within a hypoxic granuloma model of LTBI (79) and in the lungs of guinea pigs (255). Our recent studies suggest that (p)ppGpp may act as a metabolic “brake” upon exposure to growth-limiting conditions (unpublished data) (256), suggesting that impaired survival of a ΔrelA mutant relative to the isogenic wild-type strain in relevant models likely reflects the mutant strain's futile attempt to grow in the face of adverse conditions.
MprAB.
Like DosR-DosS, MprAB is a two-component system of Mycobacterium tuberculosis, which is associated with the regulation of several stress-responsive regulons and is induced in response to nutrient starvation and low concentrations of sodium dodecyl sulfate (SDS) (257). Compared to the parental strain, an M. tuberculosis ΔmprAB mutant was found to be more resistant to SDS and grew to a greater extent in human peripheral blood monocytes (257). In another study, an mprA-deficient M. tuberculosis mutant showed increased growth in resting murine macrophages relative to the isogenic wild-type strain but reduced long-term survival in a murine model (258).
Disruption of mprAB has been shown to lead to upregulation of multiple stress-associated genes of the DosR, SigD, and IdeR regulons, including rpf (encoding resuscitation-promoting factor), narK2 (nitrate/nitrite transporter), pks4 (polyketide synthase), mmpL10 (fatty acid transport), and iron-responsive genes such as hisE (phosphoribosyl-ATP pyrophosphatase), mbtB and mbtD (mycobactin synthetase), fdxA (ferredoxin), and Rv2123 (PPE37) (259). In addition, global gene expression profiling revealed that many genes of the SigE regulon (257) were downregulated in the mprAB mutant, consistent with activation of sigE by MprA. Real-time PCR and primer extension analyses confirmed that MprAB activates the sigma factor genes sigE and sigB under SDS stress and during exponential growth (225). MprA regulates its own expression (260) and has complex interactions with the α-crystallin gene acr2 promoter and indirect effects on major housekeeping genes (261).
Sigma factor network, including SigB, SigE, and SigH.
Sigma (σ) factors are a class of transcription factors that control bacterial gene expression (262–267). Typically, most eubacteria encode a principal and a variable number of alternate σ factors, which control responses to specific environmental stimuli and adaptation to stress. The temporal expression of specific regulons controlled by one or more alternate sigma factors likely allows M. tuberculosis to survive during multiple phases of TB infection. Expression of the M. tuberculosis σ factors σH, σE, and σB can be triggered by either σH or σE (268, 269). A growing body of literature indicates that these sigma factors are induced in response to several different in vitro conditions mimicking in vivo stresses faced by the pathogen during the various stages of infection and disease (263–267, 270).
M. tuberculosis σE is induced upon uptake by macrophages and upon treatment with hydrogen peroxide and SDS, leading to activation of 23 genes, including sigB, mprA, mprB, and four other transcription factor genes (257). An M. tuberculosis sigH deletion (ΔsigH) mutant showed reduced survival and pathology in the lungs of rhesus macaques and reduced lung inflammation in mice (265, 267, 270). σB is the minor principal sigma factor of M. tuberculosis and appears to play a key role in defense against cell envelope damage (263, 266). σE is transcribed in either a σH- or MprB-dependent manner (225, 271).
An M. tuberculosis ΔsigE mutant was found to have defective growth within macrophages but equivalent growth and survival in the lungs of the C3H/HeJ mice relative to the isogenic wild-type strain. Time-to-death analysis revealed that 50% of C3H/HeJ mice and all BALB/c-SCID mice infected with the ΔsigE mutant had protracted survival relative to those infected with the wild type (272). Global transcriptional analysis revealed that 38 genes were downregulated in the ΔsigE mutant, including genes involved in protein translation, mycolic acid biosynthesis, transcriptional regulation, and electron transport. Conversely, the genes aceA (icl), Rv1130, and gltA1 (encoding citrate synthase 3) showed significantly increased expression in the ΔsigE mutant relative to the wild-type strain (206, 257, 273).
Inorganic polyphosphate.
Inorganic polyphosphate [poly(P)] has been postulated to play a regulatory role in the transition to bacterial persistence. Poly(P) is a linear polymer of many tens or hundreds of inorganic phosphate (Pi) residues linked by high-energy phosphoanhydride bonds (274). In bacteria, poly(P) is synthesized by polyphosphate kinase (PPK), which catalyzes the reversible transfer of the terminal (γ) phosphate of ATP to poly(P) (274), and is degraded by the exopolyphosphatase PPX, which maintains a dynamic balance of poly(P) in the cell (275). In many bacteria, the ppk and ppx genes comprise an operon, which is regulated by the two-component phosphate-response regulator PhoB (276). Accumulation of poly(P) occurs as a result of various growth-limiting conditions, including inorganic phosphate limitation, starvation for amino acids or nitrogen, nutrient downshifts, and osmotic stress (274).
Perhaps the best-characterized regulatory role for poly(P) is in the E. coli stringent response. Upon nutrient depletion, intracellular poly(P) levels increase up to 1,000-fold as a result of (p)ppGpp-mediated inhibition of PPX activity (275). Poly(P) induces expression of the rpoS gene (277, 278), which is also regulated by (p)ppGpp, and the encoded RNA polymerase sigma factor RpoS directs the transcription of >50 genes involved in cascades downshifting growth and metabolism, thereby adjusting the cell to a persistent state (278–280). Consistent with tight regulation of poly(P) levels in bacteria, overexpression of E. coli ppk and of ppx leads to poly(P) accumulation and depletion, respectively, and, in both cases, to loss of viability (274, 280).
Mycobacterium smegmatis deficient in ppk1 was found to be defective in poly(P) synthesis and more sensitive to surface stress, oxidative stress, and extended anaerobic incubation, confirming the importance of poly(P) in mycobacterial survival under various growth-limiting conditions (281). During stationary phase, ppk1 expression increases in M. smegmatis (281), suggesting that poly(P) accumulation in mycobacteria is attributable predominantly to an increase in its biosynthetic rate, unlike in the E. coli stringent response, in which poly(P) accumulates due to (p)ppGpp-mediated inhibition of PPX activity (275). Transcription of the relA gene and synthesis of the alarmone (p)ppGpp were impaired in the ppk1 mutant of M. smegmatis and restored after complementation with Rv2984 (ppk1) of M. tuberculosis, suggesting that PPK1 is required for initiating the stringent response (281). Furthermore, antisensing of Rv2984 in M. tuberculosis led to reduced intrabacillary poly(P) content and reduced survival in THP-1 macrophages (281). The ppk1 knockdown strain showed marked downregulation of mprA, mprB, sigE, and relA (281), supporting the presence of a poly(P)-mediated signaling pathway in M. tuberculosis (282).
Recently, the gene product of Rv0496/MT0516 was shown to be a PPX in M. tuberculosis (283, 284). An Rv0496-deficient M. tuberculosis mutant exhibited elevated intracellular levels of poly(P) and increased expression of the genes mprB, sigE, and relA relative to the isogenic wild-type strain, consistent with poly(P)-mediated signaling (283). Deficiency of Rv0496 resulted in decelerated bacillary growth during logarithmic phase in axenic cultures, as well as phenotypic tolerance to isoniazid, directly linking for the first time poly(P) accumulation and bacterial phenotypic tolerance to cell wall-active antibiotics. The Rv0496-deficient mutant also showed a significant survival defect in activated human macrophages and reduced long-term survival in the lungs of guinea pigs.
A recent study found that another putative PPX in M. tuberculosis, Rv1026, lacks detectable exopolyphosphatase activity against short-chain poly(P) substrates and that both Rv0496 and Rv1026 have modest ATPase activities (282, 284). As in E. coli, the exopolyphosphatase activity of Rv0496 was inhibited by pppGpp and, to a lesser extent, by ppGpp, linking poly(P) accumulation to induction of the stringent response.
Poly(P) appears to be essential for (p)ppGpp biosynthesis through the mprAB-sigE-relA signaling cascade during stationary phase in M. smegmatis (281, 285), indicating that poly(P) acts upstream of RelA in these bacteria and not downstream, as in E. coli (286, 287). Specifically, under growth-limiting conditions, poly(P) appears to act as a preferred phosphate donor for the two-component sensor histidine kinase MprB, which then phosphorylates the cognate response regulator MprA, facilitating transcription of the alternative RNA polymerase sigma factor sigE gene, thereby leading to increased transcription of relA and enhanced (p)ppGpp synthesis. In addition to being activated under various growth-limiting conditions, this stress response pathway also may be induced stochastically, leading to a small subpopulation of “persisters” with a competitive advantage in the eventuality of encountering adverse growth conditions (285). It remains to be determined if pppGpp-mediated inhibition of PPX activity leads to poly(P) accumulation in M. tuberculosis through a positive feedback loop (Fig. 2).
Control of rRNA transcription through CarD.
The deletion of an essential mycobacterial protein that controls rRNA transcription, CarD, leads to inability of the bacteria to grow in mice (288). CarD depletion leads to sensitivity to killing by oxidative stress, starvation, and DNA damage, accompanied by failure to reduce rRNA transcription. CarD can functionally replace DksA for stringent control of rRNA transcription, even though CarD associates with a different site on RNA polymerase. These findings highlight a distinct molecular mechanism for regulating rRNA transcription in mycobacteria that is critical for M. tuberculosis pathogenesis. RelA, in concert with CarD, mediates the downregulation of rRNA synthesis and ribosomal protein genes during starvation.
PhoY2.
Inactivation of E. coli PhoU, a negative regulator of the phosphate-specific transport (pst) operon involved in inorganic phosphate uptake, leads to reduced numbers of persistent bacteria and increased susceptibility to various antibiotics and stress conditions, including acid pH and nutrient starvation (289). Interestingly, the E. coli phoU-deficient mutant expressed high levels of genes involved in energy production and metabolism, efflux/transport, and flagellum and chemotaxis synthesis, suggesting that PhoU is a global repressor of cellular metabolism and that its inactivation leads to a hyperactive metabolic state, in which the bacteria become more susceptible to killing by antibiotics and other stresses. The homologous regulator PhoY2 (Rv0821c) appears to serve a similar function in M. tuberculosis (290). Thus, a phoY2-deficient mutant of M. tuberculosis showed increased susceptibility to rifampin and pyrazinamide by MIC testing and drug exposure assays in vitro, as well as a 10- to 30-fold decrease in the bacillary burden in lungs and spleens of mutant-infected mice compared with mice infected with the isogenic wild-type strain at 8 weeks after infection. Although PhoY2 is annotated as a transcriptional regulatory protein involved in phosphate transport, its precise role and regulatory mechanisms remain to be defined in M. tuberculosis.
Toxin-Antitoxin Modules
Toxin-antitoxin (TA) modules in M. tuberculosis may contribute to the establishment and maintenance of persistence and latent infection by regulating the synthesis of various macromolecules. There are 8 major families of TA modules: CcdBA, HigBA, HipBA, MazEF, ParDE, RelBE, VapBC, and Doc/PhD (291). A recent study revealed that 88 putative TA system candidates are present in M. tuberculosis, considerably more than in any other intracellular pathogen, potentially allowing bacilli to respond and adapt to the various stresses encountered during infection and rendering the bacilli refractory to antibiotics until conditions for growth are more favorable (292). Specifically, the toxins of the largest family of TA systems, VapBC, act by inhibiting translation via mRNA cleavage (293). Expression of these genes is differentially regulated in M. tuberculosis during stresses such as hypoxia and infection of macrophages and in vivo (294, 295). Furthermore, these systems are required for M. tuberculosis survival during chronic infection in mice and for the development of antibiotic tolerance in vitro (292).
In vitro transcriptome analysis of M. tuberculosis persisters surviving treatment with d-cycloserine showed overexpression of 10 TA modules, including Rv0549c/Rv0550c, Rv2021c/Rv2022c, Rv1989c/Rv1990c, Rv2865/Rv2866, Rv0918/Rv0919, Rv3180c/Rv3181c, Rv1955/Rv1956, Rv3188/Rv3189, and Rv2034/Rv2035 (296). Both the toxin and the antitoxin of Rv1989c/Rv1990c, Rv2865/Rv2866, and Rv2021c/Rv2022c were also upregulated in the enduring hypoxic response.
trans-Translation: a Mechanism of Action of Pyrazinamide
Pyrazinamide is a key component in successful first-line “short-course chemotherapy” of tuberculosis, with significant sterilizing activity against nonreplicating bacilli within an acidic environment in the host. In addition, the potent sterilizing activity of this drug, in combination with rifampin, is able to eradicate LTBI in humans in 2 months, although this regimen is no longer recommended due to concerns of hepatotoxicity in immunocompetent persons (3, 297). A recent study by Shi et al. elucidated a novel mechanism of action of pyrazinamide through inhibition of M. tuberculosis trans-translation (298). trans-Translation is essential for freeing scarce ribosomes in nonreplicating organisms, and its inhibition may explain the ability of pyrazinamide to eradicate dormant tubercle bacilli. A mycobacterial ribosomal protein called RpsA, which binds the dual-purpose bacterial transfer-mRNA (tmRNA), was identified by its ability to bind to the active form of pyrazinamide. Subsequently, a rare pyrazinamide-resistant isolate was isolated, which lacked an alanine at the C terminus of RpsA and consequently did not bind tmRNA. This binding activity is important for the survival of persisters, since tmRNA is required not only for protein synthesis but also for rescuing ribosomes that have become blocked in translation because of stress-induced damage, such as drug exposure. Elucidation of this mode of action may open a novel avenue for development of new TB drugs specifically targeting persistent bacilli (299, 300). The extent to which trans-translation contributes to bacillary survival during LTBI merits further study.
Mycobacterial Lipids and Biofilm Formation
The long-term survival of M. tuberculosis against host immune responses and antibiotics bears a striking resemblance to chronic infections involving biofilm-forming pathogens. Ojha et al. observed the in vitro formation of drug-tolerant M. tuberculosis biofilms containing a lipid-rich extracellular matrix rich in free methoxy mycolic acids (301). Development of such biofilms is dependent on the availability of certain nutrients, including zinc and iron, as well as the M. tuberculosis putative acyl-CoA synthase Rv1013 and GroEL1 (301), which modulates synthesis of mycolic acids specifically during mycobacterial biofilm formation (302).
Historically, M. tuberculosis was grown as a pellicle, a biofilm-like structure, at the liquid-air interface in a variety of synthetic media. A recent study by Sambandan et al. demonstrated that keto-mycolic acids are essential for mycobacterial organization into a pellicle and that growth within the pellicle confers drug tolerance to otherwise drug-susceptible bacilli (303). Intriguingly, an acellular rim surrounding the central necrosis of guinea pig primary lung granulomas was found to harbor drug-tolerant bacilli, raising the possibility of biofilms contributing to the in vivo phenomena of latency and persistence (85). However, it remains unclear whether tubercle bacilli form true biofilms in the human host and whether they have a role in LTBI.
Antimicrobial Efflux Pumps and Drug Tolerance
Challenging the dogma that antibiotic tolerance in LTBI reflects a population of nonreplicating or slowly replicating bacilli with reduced metabolism, recent work has focused on the role of mycobacterial efflux pumps. The M. tuberculosis genome contains many genes encoding putative drug efflux pumps, which may play a role in extruding antituberculous drugs during the course of treatment (304). Specifically, the M. tuberculosis isoniazid-inducible protein Rv0342/IniA, which functions through an MDR pump-like mechanism, confers drug tolerance to isoniazid and ethambutol, which is reversed by the pump inhibitor reserpine. Similarly, induction of the macrophage-induced efflux pump Rv1258c confers M. tuberculosis drug tolerance primarily to rifampin, which is reduced by the efflux pump verapamil. Interestingly, the addition of verapamil to the standard anti-TB regimen accelerates bacillary clearance in C3HeB/FeJ mice and significantly lowers relapse rates following 4 months of treatment compared with the case for mice receiving standard therapy alone (305). Recently, semisynthetic analogs of the protein synthesis inhibitor spectinomycin were found to have potent activity against drug-susceptible and drug-resistant M. tuberculosis, at least in part due to chemical modification, which allows the drugs to evade the Rv1258c efflux pump (306).
Certain metabolites, such as mannitol, generate a proton motive force, which facilitates uptake of aminoglycosides and enhances killing of Gram-negative and Gram-positive persisters in biofilms (307). The ability of such metabolites and efflux pump inhibitors to potentiate the activity of current LTBI regimens remains to be determined.
Reactivation of LTBI
Active TB can occur soon after initial exposure and infection (i.e., primary disease) or after a period of LTBI (reactivation disease). Although reactivation TB cannot be differentiated from primary disease on clinical or laboratory grounds, epidemiological studies have demonstrated that the majority of active TB cases in the United States and other countries with low TB prevalence occur as a result of reactivation of latent infection. The goal of LTBI treatment is to prevent reactivation, and this is especially recommended for persons who are at increased risk for progression from LTBI to disease (308). Persons at increased risk of reactivation of LTBI include those with HIV/AIDS, those receiving immunosuppressive treatment, including cancer chemotherapy, systemic steroids, and anti-TNF agents, and those with chronic systemic diseases, such as end-stage renal disease, rheumatic disorders, and diabetes mellitus (309).
Resuscitation-promoting factors.
Early studies observed that the addition of supernatant from “spent” (early-stationary-phase) cultures of M. tuberculosis H37Ra, containing an acid-labile and heat-stable “resuscitation factor,” enhanced the viability of aged cultures and aided the growth of small inocula in liquid culture (310). Resuscitation-promoting factors (Rpfs) are the first examples of mycobacterial proteins found to be associated with reactivation of chronic infection (311). The M. tuberculosis genome contains five Rpf-encoding genes, rpfA to -E. Rpfs have been shown to stimulate regrowth of nonreplicating cells in vitro (312) and to promote in vivo survival of bacilli in mice (313). The addition of Micrococcus luteus culture supernatant containing putative Rpfs significantly increased the speed with which M. tuberculosis was isolated from processed sputum samples (314). More recently, mycobacterial Rpf stimulation was found to enhance the cultivability of >80% of bacilli from 20 clinical sputum samples prior to the initiation of TB chemotherapy, and the proportion of Rpf-dependent organisms increased relative to the surviving colony-forming population during treatment (315). Site-directed mutagenesis studies have revealed that Rpf enzyme activity is indispensable for the resuscitation and growth-stimulatory effects of these proteins (316).
Deletion of rpfB (Rv1009) and various combinations of deletions in 3 of the 5 rpfA to -E genes yields M. tuberculosis strains that are unable to grow or divide under stressful conditions in vitro and in vivo (313, 317). RpfB and RpfE are lytic transglycosylases, which interact with Rpf-interacting protein (RipA), a peptidoglycan endopeptidase (318). RpfB and RipA appear to form a complex at the septa of dividing bacteria, and ripA depletion results in significantly reduced mycobacterial growth, formation of long, branched chains, and increased susceptibility to the cell wall-targeting beta-lactams (319).
Immunomodulation or vaccination in LTBI.
The M. bovis BCG vaccine has limited efficacy in protecting against reactivation of LTBI (320–322). DNA vaccines have shown promise in their ability to enhance the activity of antibiotic treatment and reduce reactivation of LTBI and transmission (323–325). Adjunctive immunotherapy with a DNA vaccine containing the LTBI marker α-crystallin (233, 328–330) reduces the duration of antibiotic treatment in chronically infected mice (331). Similarly, RUTI, a therapeutic vaccine comprising detoxified, fragmented M. tuberculosis cells delivered in liposomes, also has been shown to enhance the effect of short-course chemotherapy in animal models (326) and is currently being evaluated in clinical trials (327).
CONCLUSIONS AND UNANSWERED QUESTIONS
Progress in our understanding of LTBI has been impeded by the difficulty in obtaining relevant host tissue and microbiological samples from persons with LTBI, as well as the inadequacy of current imaging modalities. In turn, our poor understanding of human LTBI has made difficult the development and validation of clinically relevant research models. There is an urgent need for the establishment of well-characterized biobanks containing biospecimens from latently infected patients before and after reactivation and from those individuals who have had adequate follow-up to establish a long-term treatment outcome (332).
Based on human pathology data and emerging data from nonhuman primates, LTBI appears to represent a spectrum of microbiological and pathological states in which the organisms are viable but fail to produce respiratory or constitutional symptoms. The extremely low yield of acid-fast staining and culture of sputum and other tissue samples from latently infected individuals suggests that the infection is paucibacillary in nature. Further supporting this hypothesis, isoniazid preventive therapy generally does not lead to the emergence of isoniazid resistance, suggesting that the number of organisms is below the spontaneous mutation frequency for the drug (∼10−5). The antibiotic tolerance observed in LTBI, but also the differential susceptibility of bacilli to drugs with various mechanisms of action, suggests that at least some bacilli have slowed growth and/or metabolism. On the other hand, the eventual, albeit delayed, efficacy of isoniazid, which appears to target only actively replicating bacilli, suggests that these organisms sporadically multiply over the course of 6 to 9 months or that the drug is effective in killing at least those organisms responsible for clinical relapse. The detection of viable bacilli in normal-appearing lung tissue of latently infected individuals dispels the notion that necrotic granulomas are a sine qua non of LTBI, although such lesions may be sufficient for controlling TB infection. These observations have important implications for the development of novel experimental models which can further enhance our understanding of LTBI.
Abundant clinical and animal model data have underscored the importance of adaptive immunity, particularly CD4 T cells, for controlling TB infection. Likewise, the proinflammatory cytokines TNF-α and IFN-γ are clearly required for host control of M. tuberculosis growth. Many studies have highlighted the importance of various adaptive mechanisms in promoting the long-term survival of M. tuberculosis in host tissues, including the stringent response and a switch to utilization of fatty acids as a source of carbon and energy through the glyoxylate shunt.
Nevertheless, many unanswered questions remain. Clearly, the immunological mechanisms responsible for establishment and maintenance of LTBI require further investigation, as HIV-seropositive patients remain at increased risk for TB reactivation despite relatively high CD4 T cell counts, and a significant number of latently infected individuals who develop reactivation disease have no known immunological defect. In particular, the role of regulatory T cells in predisposing to reactivation of is an exciting area of current research.
From the perspective of the organism, it is still unknown whether antibiotic tolerance is due to host immune pressure or reflects a stochastic phenomenon in any given bacterial population, or perhaps both. An important scientific question is whether multiple external stimuli can be transduced through a common regulatory pathway to trigger bacterial growth arrest and antibiotic tolerance. Identification of such a “master regulator” or “dormancy switch” could have important implications for the development of new strategies to combat LTBI. Thus, inhibition of such pathways may “awaken” nonreplicating, metabolically quiescent bacilli, rendering them more susceptible to killing by currently available bactericidal drugs. Finally, the role of efflux pumps in promoting antibiotic tolerance during LTBI deserves further exploration. Animal and clinical studies in which efflux pump inhibitors are given together with standard LTBI regimens are being planned to determine if these combinations accelerate the eradication of LTBI.
It is hoped that improved imaging techniques and their greater accessibility in areas where TB is endemic will help identify the potential site(s) of LTBI and reactivation. Such approaches could address the question of whether reactivation occurs exclusively at necrotic granulomas in the lungs, with subsequent spread to other organs, or whether reactivation disease can also occur spontaneously at extrapulmonary sites.
It is readily apparent that no single host or mycobacterial pathway is responsible for LTBI and reactivation. Rather, these complex phenomena are attributable to multiple interdependent host and mycobacterial molecular networks, which cannot be deduced from any one particular model. We believe that further advances in the field will require innovative, multidisciplinary approaches involving genetic, transcriptional, proteomic, metabolomic, lipidomic, imaging, and computational techniques (333) in order to identify host cytokine networks responsible for immunological control of M. tuberculosis growth, as well as M. tuberculosis regulatory and metabolic pathways required for bacillary growth restriction and/or antibiotic tolerance and reactivation. Ultimately, findings from experimental models must be validated in clinical studies. Figure 3 shows a flowchart of the research strategy we have adopted to address these complex questions.
FIG 3.

Flowchart of a systems biology-based approach to studying latent TB infection and reactivation. Using an integrated, multidisciplinary approach, including the use of several novel animal models of LTBI in combination with transcriptional, proteomic, genetic, imaging, and computational techniques, researchers can begin to identify host cytokine networks responsible for immunological control of M. tuberculosis (Mtb) growth, as well as M. tuberculosis regulatory and metabolic pathways required for bacillary growth restriction and reactivation. Computational predictions of redundant host and M. tuberculosis molecular pathways required for LTBI and reactivation across experimental models can be tested directly by dual inhibition of host cytokine pathways and by generation of M. tuberculosis double knockouts. Finally, the biological relevance of the computational and experimental findings may be verified using plasma and peripheral blood mononuclear cell samples collected serially from latently infected persons before and after TB reactivation./
In addition to elucidating novel drug targets for LTBI treatment, an improved understanding of the molecular correlates of LTBI and reactivation disease could lead to the identification of biomarkers which can differentiate those latently infected individuals who are at highest risk of reactivation and, therefore, would benefit most from LTBI treatment. Identification and successful treatment of persons with LTBI at risk for reactivation are important components of global TB elimination efforts.
ACKNOWLEDGMENTS
The research reported in this publication was supported by the National Heart, Lung, and Blood Institute and the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award numbers R01 HL106786 and R01 AI083125, respectively.
The funding sources had no role in the study design, data collection, data analysis, data interpretation, or writing of the report. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors have no conflicts to report.
Biographies

Noton D. Dutta, Ph.D., is a senior postdoctoral researcher in the lab of Dr. Petros C. Karakousis at the Center for Tuberculosis Research, Johns Hopkins School of Medicine. He received his Ph.D. from Jadavpur University, India, and did his early postdoctoral research at the College of Veterinary Medicine, Seoul National University, Seoul, South Korea, and Tulane National Primate Research Center, Tulane University, New Orleans, LA. His research focuses on characterizing the efficacy of novel drugs and drug combinations against M. tuberculosis in murine and guinea pig models, with the goal of shortening the duration of TB chemotherapy. He is using multidisciplinary techniques, including computational modeling and high-throughput genetic screens in various animal models, including nonhuman primates, in order to elucidate M. tuberculosis essential pathways involved in latency and reactivation. He has published over 50 peer-reviewed papers on relevant topics.

Petros C. Karakousis, M.D., received his medical degree at the Washington University School of Medicine in St. Louis, MO, and completed internal medicine residency training at the Hospital of the University of Pennsylvania. After completing fellowship training in infectious diseases at Johns Hopkins University School of Medicine, he joined the Department of Medicine faculty in 2005. His primary research interest is the molecular basis of latent TB infection and persistence and identification of novel drug targets specific to the dormant state, with the ultimate goal of shortening the duration of TB chemotherapy. In addition, he has led the testing of novel anti-TB drugs and drug combinations in several animal models, including guinea pigs and mice. Over the last twelve years, he has developed several in vitro and in vivo models of TB latency and has used transcriptional and genetic-based techniques to study the regulatory and metabolic networks required for M. tuberculosis growth restriction and antibiotic tolerance.
REFERENCES
- 1.Lienhardt C, Glaziou P, Uplekar M, Lonnroth K, Getahun H, Raviglione M. 2012. Global tuberculosis control: lessons learnt and future prospects. Nat. Rev. Microbiol. 10:407–416. 10.1038/nrmicro2797 [DOI] [PubMed] [Google Scholar]
- 2.Lillebaek T, Dirksen A, Baess I, Strunge B, Thomsen VO, Andersen AB. 2002. Molecular evidence of endogenous reactivation of Mycobacterium tuberculosis after 33 years of latent infection. J. Infect. Dis. 185:401–404. 10.1086/338342 [DOI] [PubMed] [Google Scholar]
- 3.Jasmer RM, Nahid P, Hopewell PC. 2002. Latent tuberculosis infection. N. Engl. J. Med. 347:1860–1866. 10.1056/NEJMcp021045 [DOI] [PubMed] [Google Scholar]
- 4.Schluger NW. 2013. Advances in the diagnosis of latent tuberculosis infection. Semin. Respir. Crit. Care Med. 34:60–66. 10.1055/s-0032-1333545 [DOI] [PubMed] [Google Scholar]
- 5.Pai M. 2010. Spectrum of latent tuberculosis—existing tests cannot resolve the underlying phenotypes. Nat. Rev. Microbiol. 8:242 (Author reply, 8:242.) 10.1038/nrmicro2236-c2 [DOI] [PubMed] [Google Scholar]
- 6.Vernon A. 2013. Treatment of latent tuberculosis infection. Semin. Respir. Crit. Care Med. 34:67–86. 10.1055/s-0032-1333544 [DOI] [PubMed] [Google Scholar]
- 7.Horsburgh CR, Jr, Rubin EJ. 2011. Latent tuberculosis infection in the United States. N. Engl. J. Med. 364:1441–1448. 10.1056/NEJMcp1005750 [DOI] [PubMed] [Google Scholar]
- 8.Sia IG, Wieland ML. 2011. Current concepts in the management of tuberculosis. Mayo Clin. Proc. 86:348–361. 10.4065/mcp.2010.0820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Winder FG, Collins PB. 1970. Inhibition by isoniazid of synthesis of mycolic acids in Mycobacterium tuberculosis. J. Gen. Microbiol. 63:41–48. 10.1099/00221287-63-1-41 [DOI] [PubMed] [Google Scholar]
- 10.Takayama K, Wang L, David HL. 1972. Effect of isoniazid on the in vivo mycolic acid synthesis, cell growth, and viability of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2:29–35. 10.1128/AAC.2.1.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mitchison DA. 1985. The action of antituberculosis drugs in short-course chemotherapy. Tubercle 66:219–225. 10.1016/0041-3879(85)90040-6 [DOI] [PubMed] [Google Scholar]
- 12.Sterling TR, Villarino ME, Borisov AS, Shang N, Gordin F, Bliven-Sizemore E, Hackman J, Hamilton CD, Menzies D, Kerrigan A, Weis SE, Weiner M, Wing D, Conde MB, Bozeman L, Horsburgh CR, Jr, Chaisson RE, TB Trials Consortium PREVENT TB Study Team 2011. Three months of rifapentine and isoniazid for latent tuberculosis infection. N. Engl. J. Med. 365:2155–2166. 10.1056/NEJMoa1104875 [DOI] [PubMed] [Google Scholar]
- 13.Bigger JW. 1944. Treatment of staphylococcal infections with penicillin by intermittent sterilisation. Lancet i:497–500 [Google Scholar]
- 14.McDermott W. 1958. Microbial persistence. Yale J. Biol. Med. 30:257–291 [PMC free article] [PubMed] [Google Scholar]
- 15.Karakousis PC. 2009. Mechanisms of action and resistance of antimycobacterial agents, p 271–291 In Mayers DL. (ed), Antimicrobial Drug Resistance, vol 1 Humana Press, New York, NY [Google Scholar]
- 16.Tomasz A, Albino A, Zanati E. 1970. Multiple antibiotic resistance in a bacterium with suppressed autolytic system. Nature 227:138–140. 10.1038/227138a0 [DOI] [PubMed] [Google Scholar]
- 17.McDermott W. 1959. Inapparent infection: relation of latent and dormant infections to microbial persistence. Public Health Rep. 74:485–499. 10.2307/4590490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tuomanen E. 1986. Phenotypic tolerance: the search for beta-lactam antibiotics that kill nongrowing bacteria. Rev. Infect. Dis. 8(Suppl 3):S279–S291. 10.1093/clinids/8.Supplement_3.S279 [DOI] [PubMed] [Google Scholar]
- 19.Eagle H. 1952. Experimental approach to the problem of treatment failure with penicillin. I. Group A streptococcal infection in mice. Am. J. Med. 13:389–399 [DOI] [PubMed] [Google Scholar]
- 20.Wayne LG, Hayes LG. 1996. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect. Immun. 64:2062–2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. 2004. Bacterial persistence as a phenotypic switch. Science 305:1622–1625. 10.1126/science.1099390 [DOI] [PubMed] [Google Scholar]
- 22.Nuermberger E, Bishai WR, Grosset JH. 2004. Latent tuberculosis infection. Semin. Respir. Crit. Care Med. 25:317–336. 10.1055/s-2004-829504 [DOI] [PubMed] [Google Scholar]
- 23.Houk VN, Kent DC, Sorensen K, Baker JH. 1968. The eradication of tuberculosis infection by isoniazid chemoprophylaxis. Arch. Environ. Health 16:46–50. 10.1080/00039896.1968.10665013 [DOI] [PubMed] [Google Scholar]
- 24.Ford CB, Lin PL, Chase MR, Shah RR, Iartchouk O, Galagan J, Mohaideen N, Ioerger TR, Sacchettini JC, Lipsitch M, Flynn JL, Fortune SM. 2011. Use of whole genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection. Nat. Genet. 43:482–486. 10.1038/ng.811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lillebaek T, Dirksen A, Vynnycky E, Baess I, Thomsen VO, Andersen AB. 2003. Stability of DNA patterns and evidence of Mycobacterium tuberculosis reactivation occurring decades after the initial infection. J. Infect. Dis. 188:1032–1039. 10.1086/378240 [DOI] [PubMed] [Google Scholar]
- 26.Barry CE, III, Boshoff HI, Dartois V, Dick T, Ehrt S, Flynn J, Schnappinger D, Wilkinson RJ, Young D. 2009. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 7:845–855. 10.1038/nrmicro2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Young DB, Gideon HP, Wilkinson RJ. 2009. Eliminating latent tuberculosis. Trends Microbiol. 17:183–188. 10.1016/j.tim.2009.02.005 [DOI] [PubMed] [Google Scholar]
- 28.Gideon HP, Flynn JL. 2011. Latent tuberculosis: what the host “sees”? Immunol Res. 50:202–212. 10.1007/s12026-011-8229-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adams KN, Takaki K, Connolly LE, Wiedenhoft H, Winglee K, Humbert O, Edelstein PH, Cosma CL, Ramakrishnan L. 2011. Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell 145:39–53. 10.1016/j.cell.2011.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robertson HE. 1933. The persistence of tuberculous infections. Am. J. Pathol. 9:711–718 711 [PMC free article] [PubMed] [Google Scholar]
- 31.Ewer K, Millington KA, Deeks JJ, Alvarez L, Bryant G, Lalvani A. 2006. Dynamic antigen-specific T-cell responses after point-source exposure to Mycobacterium tuberculosis. Am. J. Respir. Crit. Care Med. 174:831–839. 10.1164/rccm.200511-1783OC [DOI] [PubMed] [Google Scholar]
- 32.Myers JA, Bearman JE, Dixon HG. 1963. The natural history of the tuberculosis in the human body. V. Prognosis among tuberculin-reactor children from birth to five years of age. Am. Rev. Respir. Dis. 87:354–369 [DOI] [PubMed] [Google Scholar]
- 33.Flynn JL, Chan J. 2001. Tuberculosis: latency and reactivation. Infect. Immun. 69:4195–4201. 10.1128/IAI.69.7.4195-4201.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaplan G, Post FA, Moreira AL, Wainwright H, Kreiswirth BN, Tanverdi M, Mathema B, Ramaswamy SV, Walther G, Steyn LM, Barry CE, III, Bekker LG. 2003. Mycobacterium tuberculosis growth at the cavity surface: a microenvironment with failed immunity. Infect. Immun. 71:7099–7108. 10.1128/IAI.71.12.7099-7108.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koch R. 1882. Aetiologie der Tuberculose. Berlin Klin. Wochenschr. 19:221–230 [Google Scholar]
- 36.Rabinowitsch L. 1907. Zur Frage latenter Tuberkelbacillen. Berlin Klin. Wochenschr. 44:35–39 [Google Scholar]
- 37.Behr MA, Waters WR. 2014. Is tuberculosis a lymphatic disease with a pulmonary portal? Lancet Infect. Dis. 14:250–255. 10.1016/S1473-3099(13)70253-6 [DOI] [PubMed] [Google Scholar]
- 38.Opie EL, Aronson JD. 1927. Tubercle bacilli in latent tuberculous lesions and in lung tissue without tuberculous lesions. Arch. Pathol. Lab. Med. 4:1–21 [Google Scholar]
- 39.Medlar EM, Bernstein S, Steward DM. 1952. A bacteriologic study of resected tuberculous lesions. Am. Rev. Tuberc. 66:36–43 [DOI] [PubMed] [Google Scholar]
- 40.Hobby GL, Auerbach O, Lenert TF, Small MJ, Comer JV. 1954. The late emergence of M. tuberculosis in liquid cultures of pulmonary lesions resected from humans. Am. Rev. Tuberc. 70:191–218 [DOI] [PubMed] [Google Scholar]
- 41.Loomis HP. 1890. Some facts in the etiology of tuberculosis, evidenced by thirty autopsies and experiments upon animals. M. Rec. 38:689–698 [Google Scholar]
- 42.Wang CY. 1916. An experimental study of latent tuberculosis. Lancet ii:417–419 [Google Scholar]
- 43.Feldman WH, Baggenstoss AH. 1939. The occurrence of virulent tubercle bacilli in presumably non-tuberculous lung tissue. Am. J. Pathol. 15:501–515 [PMC free article] [PubMed] [Google Scholar]
- 44.Ghon A. 1923. The primary complex in human tuberculosis and its significance. Am. Rev. Tuberc. 7:314–317 [Google Scholar]
- 45.Hernandez-Pando R, Jeyanathan M, Mengistu G, Aguilar D, Orozco H, Harboe M, Rook GA, Bjune G. 2000. Persistence of DNA from Mycobacterium tuberculosis in superficially normal lung tissue during latent infection. Lancet 356:2133–2138. 10.1016/S0140-6736(00)03493-0 [DOI] [PubMed] [Google Scholar]
- 46.Barrios-Payan J, Saqui-Salces M, Jeyanathan M, Alcantara-Vazquez A, Castanon-Arreola M, Rook G, Hernandez-Pando R. 2012. Extrapulmonary locations of mycobacterium tuberculosis DNA during latent infection. J. Infect. Dis. 206:1194–1205. 10.1093/infdis/jis381 [DOI] [PubMed] [Google Scholar]
- 47.Neyrolles O, Hernandez-Pando R, Pietri-Rouxel F, Fornes P, Tailleux L, Barrios Payan JA, Pivert E, Bordat Y, Aguilar D, Prevost MC, Petit C, Gicquel B. 2006. Is adipose tissue a place for Mycobacterium tuberculosis persistence? PLoS One 1:e43. 10.1371/journal.pone.0000043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Loebel RO, Shorr E, Richardson HB. 1933. The influence of foodstuffs upon the respiratory metabolism and growth of human tubercle bacilli. J. Bacteriol. 26:139–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hobby GL, Lenert TF. 1957. The in vitro action of antituberculous agents against multiplying and non-multiplying microbial cells. Am. Rev. Tuberc. 76:1031–1048 [DOI] [PubMed] [Google Scholar]
- 50.Xie Z, Siddiqi N, Rubin EJ. 2005. Differential antibiotic susceptibilities of starved Mycobacterium tuberculosis isolates. Antimicrob. Agents Chemother. 49:4778–4780. 10.1128/AAC.49.11.4778-4780.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Betts JC, Lukey PT, Robb LC, McAdam RA, Duncan K. 2002. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 43:717–731. 10.1046/j.1365-2958.2002.02779.x [DOI] [PubMed] [Google Scholar]
- 52.Wayne LG, Sramek HA. 1994. Metronidazole is bactericidal to dormant cells of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 38:2054–2058. 10.1128/AAC.38.9.2054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Via LE, Lin PL, Ray SM, Carrillo J, Allen SS, Eum SY, Taylor K, Klein E, Manjunatha U, Gonzales J, Lee EG, Park SK, Raleigh JA, Cho SN, McMurray DN, Flynn JL, Barry CE., III 2008. Tuberculous granulomas are hypoxic in guinea pigs, rabbits, and nonhuman primates. Infect. Immun. 76:2333–2340. 10.1128/IAI.01515-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin PL, Dartois V, Johnston PJ, Janssen C, Via L, Goodwin MB, Klein E, Barry CE, III, Flynn JL. 2012. Metronidazole prevents reactivation of latent Mycobacterium tuberculosis infection in macaques. Proc. Natl. Acad. Sci. U. S. A. 109:14188–14193. 10.1073/pnas.1121497109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carroll MW, Jeon D, Mountz JM, Lee JD, Jeong YJ, Zia N, Lee M, Lee J, Via LE, Lee S, Eum SY, Lee SJ, Goldfeder LC, Cai Y, Jin B, Kim Y, Oh T, Chen RY, Dodd LE, Gu W, Dartois V, Park SK, Kim CT, Barry CE, III, Cho SN. 2013. Efficacy and safety of metronidazole for pulmonary multidrug-resistant tuberculosis. Antimicrob. Agents Chemother. 57:3903–3909. 10.1128/AAC.00753-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vandal OH, Nathan CF, Ehrt S. 2009. Acid resistance in Mycobacterium tuberculosis. J. Bacteriol. 191:4714–4721. 10.1128/JB.00305-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chapman JS, Bernard JS. 1962. The tolerances of unclassified mycobacteria. I. Limits of pH tolerance. Am. Rev. Respir. Dis. 86:582–583 [DOI] [PubMed] [Google Scholar]
- 58.Portaels F, Pattyn SR. 1982. Growth of mycobacteria in relation to the pH of the medium. Ann. Microbiol. (Paris) 133:213–221 [PubMed] [Google Scholar]
- 59.Niki M, Tateishi Y, Ozeki Y, Kirikae T, Lewin A, Inoue Y, Matsumoto M, Dahl JL, Ogura H, Kobayashi K, Matsumoto S. 2012. A novel mechanism of growth phase-dependent tolerance to isoniazid in mycobacteria. J. Biol. Chem. 287:22743–22752. 10.1074/jbc.M111.333385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Y, Wade MM, Scorpio A, Zhang H, Sun Z. 2003. Mode of action of pyrazinamide: disruption of Mycobacterium tuberculosis membrane transport and energetics by pyrazinoic acid. J. Antimicrob. Chemother. 52:790–795. 10.1093/jac/dkg446 [DOI] [PubMed] [Google Scholar]
- 61.Rengarajan J, Bloom BR, Rubin EJ. 2005. Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc. Natl. Acad. Sci. U. S. A. 102:8327–8332. 10.1073/pnas.0503272102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rifat D, Bishai WR, Karakousis PC. 2009. Phosphate depletion: a novel trigger for Mycobacterium tuberculosis persistence. J. Infect. Dis. 200:1126–1135. 10.1086/605700 [DOI] [PubMed] [Google Scholar]
- 63.Deb C, Lee CM, Dubey VS, Daniel J, Abomoelak B, Sirakova TD, Pawar S, Rogers L, Kolattukudy PE. 2009. A novel in vitro multiple-stress dormancy model for Mycobacterium tuberculosis generates a lipid-loaded, drug-tolerant, dormant pathogen. PLoS One 4:e6077. 10.1371/journal.pone.0006077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Puissegur MP, Botanch C, Duteyrat JL, Delsol G, Caratero C, Altare F. 2004. An in vitro dual model of mycobacterial granulomas to investigate the molecular interactions between mycobacteria and human host cells. Cell. Microbiol. 6:423–433. 10.1111/j.1462-5822.2004.00371.x [DOI] [PubMed] [Google Scholar]
- 65.Peyron P, Vaubourgeix J, Poquet Y, Levillain F, Botanch C, Bardou F, Daffe M, Emile JF, Marchou B, Cardona PJ, de Chastellier C, Altare F. 2008. Foamy macrophages from tuberculous patients' granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog. 4:e1000204. 10.1371/journal.ppat.1000204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Manabe YC, Bishai WR. 2000. Latent Mycobacterium tuberculosis-persistence, patience, and winning by waiting. Nat. Med. 6:1327–1329. 10.1038/82139 [DOI] [PubMed] [Google Scholar]
- 67.Rees RJ, Hart PD. 1961. Analysis of the host-parasite equilibrium in chronic murine tuberculosis by total and viable bacillary counts. Br. J. Exp. Pathol. 42:83–88 [PMC free article] [PubMed] [Google Scholar]
- 68.Munoz-Elias EJ, Timm J, Botha T, Chan WT, Gomez JE, McKinney JD. 2005. Replication dynamics of Mycobacterium tuberculosis in chronically infected mice. Infect. Immun. 73:546–551. 10.1128/IAI.73.1.546-551.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gill WP, Harik NS, Whiddon MR, Liao RP, Mittler JE, Sherman DR. 2009. A replication clock for Mycobacterium tuberculosis. Nat. Med. 15:211–214. 10.1038/nm.1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Talaat AM, Ward SK, Wu CW, Rondon E, Tavano C, Bannantine JP, Lyons R, Johnston SA. 2007. Mycobacterial bacilli are metabolically active during chronic tuberculosis in murine lungs: insights from genome-wide transcriptional profiling. J. Bacteriol. 189:4265–4274. 10.1128/JB.00011-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Karakousis PC, Williams EP, Bishai WR. 2008. Altered expression of isoniazid-regulated genes in drug-treated dormant Mycobacterium tuberculosis. J. Antimicrob. Chemother. 61:323–331. 10.1093/jac/dkm485 [DOI] [PubMed] [Google Scholar]
- 72.McCune RM, Feldmann FM, Lambert HP, McDermott W. 1966. Microbial persistence. I. The capacity of tubercle bacilli to survive sterilization in mouse tissues. J. Exp. Med. 123:445–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McCune RM, Feldmann FM, McDermott W. 1966. Microbial persistence. II. Characteristics of the sterile state of tubercle bacilli. J. Exp. Med. 123:469–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Flynn JL, Scanga CA, Tanaka KE, Chan J. 1998. Effects of aminoguanidine on latent murine tuberculosis. J. Immunol. 160:1796–1803 [PubMed] [Google Scholar]
- 75.Scanga CA, Mohan VP, Joseph H, Yu K, Chan J, Flynn JL. 1999. Reactivation of latent tuberculosis: variations on the Cornell murine model. Infect. Immun. 67:4531–4538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Horwitz MA, Harth G. 2003. A new vaccine against tuberculosis affords greater survival after challenge than the current vaccine in the guinea pig model of pulmonary tuberculosis. Infect. Immun. 71:1672–1679. 10.1128/IAI.71.4.1672-1679.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nuermberger EL, Yoshimatsu T, Tyagi S, Bishai WR, Grosset JH. 2004. Paucibacillary tuberculosis in mice after prior aerosol immunization with Mycobacterium bovis BCG. Infect. Immun. 72:1065–1071. 10.1128/IAI.72.2.1065-1071.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang T, Zhang M, Rosenthal IM, Grosset JH, Nuermberger EL. 2009. Short-course therapy with daily rifapentine in a murine model of latent tuberculosis infection. Am. J. Respir. Crit. Care Med. 180:1151–1157. 10.1164/rccm.200905-0795OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Karakousis PC, Yoshimatsu T, Lamichhane G, Woolwine SC, Nuermberger EL, Grosset J, Bishai WR. 2004. Dormancy phenotype displayed by extracellular Mycobacterium tuberculosis within artificial granulomas in mice. J. Exp. Med. 200:647–657. 10.1084/jem.20040646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Klinkenberg LG, Sutherland LA, Bishai WR, Karakousis PC. 2008. Metronidazole lacks activity against Mycobacterium tuberculosis in an in vivo hypoxic granuloma model of latency. J. Infect. Dis. 198:275–283. 10.1086/589515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Flynn J, Chan J. 2004. Animal models of tuberculosis, p 237–250 In Rom W, Garay S. (ed), Tuberculosis, 2nd ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 82.McMurray DN. 2001. Disease model: pulmonary tuberculosis. Trends Mol. Med. 7:135–137. 10.1016/S1471-4914(00)01901-8 [DOI] [PubMed] [Google Scholar]
- 83.Tsai MC, Chakravarty S, Zhu G, Xu J, Tanaka K, Koch C, Tufariello J, Flynn J, Chan J. 2006. Characterization of the tuberculous granuloma in murine and human lungs: cellular composition and relative tissue oxygen tension. Cell. Microbiol. 8:218–232. 10.1111/j.1462-5822.2005.00612.x [DOI] [PubMed] [Google Scholar]
- 84.Aly S, Wagner K, Keller C, Malm S, Malzan A, Brandau S, Bange FC, Ehlers S. 2006. Oxygen status of lung granulomas in Mycobacterium tuberculosis-infected mice. J. Pathol. 210:298–305. 10.1002/path.2055 [DOI] [PubMed] [Google Scholar]
- 85.Lenaerts AJ, Hoff D, Aly S, Ehlers S, Andries K, Cantarero L, Orme IM, Basaraba RJ. 2007. Location of persisting mycobacteria in a guinea pig model of tuberculosis revealed by r207910. Antimicrob. Agents Chemother. 51:3338–3345. 10.1128/AAC.00276-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Smith DW, McMurray DN, Wiegeshaus EH, Grover AA, Harding GE. 1970. Host-parasite relationships in experimental airborne tuberculosis. IV. Early events in the course of infection in vaccinated and nonvaccinated guinea pigs. Am. Rev. Respir. Dis. 102:937–949 [DOI] [PubMed] [Google Scholar]
- 87.Ly LH, Russell MI, McMurray DN. 2008. Cytokine profiles in primary and secondary pulmonary granulomas of guinea pigs with tuberculosis. Am. J. Respir. Cell Mol. Biol. 38:455–462. 10.1165/rcmb.2007-0326OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ahmad Z, Klinkenberg LG, Pinn ML, Fraig MM, Peloquin CA, Bishai WR, Nuermberger EL, Grosset JH, Karakousis PC. 2009. Biphasic kill curve of isoniazid reveals the presence of drug-tolerant, not drug-resistant, Mycobacterium tuberculosis in the guinea pig. J. Infect. Dis. 200:1136–1143. 10.1086/605605 [DOI] [PubMed] [Google Scholar]
- 89.Ly LH, Jeevan A, McMurray DN. 2009. Neutralization of TNFalpha alters inflammation in guinea pig tuberculous pleuritis. Microbes Infect. 11:680–688. 10.1016/j.micinf.2009.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ly LH, Russell MI, McMurray DN. 2007. Microdissection of the cytokine milieu of pulmonary granulomas from tuberculous guinea pigs. Cell. Microbiol. 9:1127–1136. 10.1111/j.1462-5822.2006.00854.x [DOI] [PubMed] [Google Scholar]
- 91.Dannenberg AM., Jr 1994. Rabbit model of tuberculosis, p 149–156 In Bloom BR. (ed), Tuberculosis: pathogenesis, protection, and control. American Society for Microbiology, Washington, DC [Google Scholar]
- 92.Manabe YC, Kesavan AK, Lopez-Molina J, Hatem CL, Brooks M, Fujiwara R, Hochstein K, Pitt ML, Tufariello J, Chan J, McMurray DN, Bishai WR, Dannenberg AM, Jr, Mendez S. 2008. The aerosol rabbit model of TB latency, reactivation and immune reconstitution inflammatory syndrome. Tuberculosis (Edinb.) 88:187–196. 10.1016/j.tube.2007.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kesavan AK, Brooks M, Tufariello J, Chan J, Manabe YC. 2009. Tuberculosis genes expressed during persistence and reactivation in the resistant rabbit model. Tuberculosis (Edinb.) 89:17–21. 10.1016/j.tube.2008.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Manabe YC, Dannenberg AM, Jr, Tyagi SK, Hatem CL, Yoder M, Woolwine SC, Zook BC, Pitt ML, Bishai WR. 2003. Different strains of Mycobacterium tuberculosis cause various spectrums of disease in the rabbit model of tuberculosis. Infect. Immun. 71:6004–6011. 10.1128/IAI.71.10.6004-6011.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tsenova L, Ellison E, Harbacheuski R, Moreira AL, Kurepina N, Reed MB, Mathema B, Barry CE, III, Kaplan G. 2005. Virulence of selected Mycobacterium tuberculosis clinical isolates in the rabbit model of meningitis is dependent on phenolic glycolipid produced by the bacilli. J. Infect. Dis. 192:98–106. 10.1086/430614 [DOI] [PubMed] [Google Scholar]
- 96.Subbian S, O'Brien P, Kushner NL, Yang G, Tsenova L, Peixoto B, Bandyopadhyay N, Bader JS, Karakousis PC, Fallows D, Kaplan G. 2013. Molecular immunologic correlates of spontaneous latency in a rabbit model of pulmonary tuberculosis. Cell Commun. Signal. 11:16. 10.1186/1478-811X-11-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Capuano SV, III, Croix DA, Pawar S, Zinovik A, Myers A, Lin PL, Bissel S, Fuhrman C, Klein E, Flynn JL. 2003. Experimental Mycobacterium tuberculosis infection of cynomolgus macaques closely resembles the various manifestations of human M. tuberculosis infection. Infect. Immun. 71:5831–5844. 10.1128/IAI.71.10.5831-5844.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lin PL, Rodgers M, Smith L, Bigbee M, Myers A, Bigbee C, Chiosea I, Capuano SV, Fuhrman C, Klein E, Flynn JL. 2009. Quantitative comparison of active and latent tuberculosis in the cynomolgus macaque model. Infect. Immun. 77:4631–4642. 10.1128/IAI.00592-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Walsh GP, Tan EV, dela Cruz EC, Abalos RM, Villahermosa LG, Young LJ, Cellona RV, Nazareno JB, Horwitz MA. 1996. The Philippine cynomolgus monkey (Macaca fasicularis) provides a new nonhuman primate model of tuberculosis that resembles human disease. Nat. Med. 2:430–436. 10.1038/nm0496-430 [DOI] [PubMed] [Google Scholar]
- 100.Lin PL, Pawar S, Myers A, Pegu A, Fuhrman C, Reinhart TA, Capuano SV, Klein E, Flynn JL. 2006. Early events in Mycobacterium tuberculosis infection in cynomolgus macaques. Infect. Immun. 74:3790–3803. 10.1128/IAI.00064-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lin PL, Ford CB, Coleman MT, Myers AJ, Gawande R, Ioerger T, Sacchettini J, Fortune SM, Flynn JL. 2014. Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nat. Med. 20:75–79. 10.1038/nm.3412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Diedrich CR, Mattila JT, Klein E, Janssen C, Phuah J, Sturgeon TJ, Montelaro RC, Lin PL, Flynn JL. 2010. Reactivation of latent tuberculosis in cynomolgus macaques infected with SIV is associated with early peripheral T cell depletion and not virus load. PLoS One 5:e9611. 10.1371/journal.pone.0009611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pawar SN, Mattila JT, Sturgeon TJ, Lin PL, Narayan O, Montelaro RC, Flynn JL. 2008. Comparison of the effects of pathogenic simian human immunodeficiency virus strains SHIV-89.6P and SHIV-KU2 in cynomolgus macaques. AIDS Res. Hum. Retroviruses 24:643–654. 10.1089/aid.2007.0238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lin PL, Myers A, Smith L, Bigbee C, Bigbee M, Fuhrman C, Grieser H, Chiosea I, Voitenek NN, Capuano SV, Klein E, Flynn JL. 2010. Tumor necrosis factor neutralization results in disseminated disease in acute and latent Mycobacterium tuberculosis infection with normal granuloma structure in a cynomolgus macaque model. Arthritis Rheum. 62:340–350. 10.1002/art.27271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Karakousis PC, Chaisson RE. 2008. Mycobacterial infections and HIV infection, p 2487–2497 In Fishman AP, Elias JA, Fishman JA, Grippi MA, Senior RM, Pack AI. (ed), Fishman's pulmonary diseases and disorders, vol 2 McGraw-Hill, New York, NY [Google Scholar]
- 106.Selwyn PA, Hartel D, Lewis VA, Schoenbaum EE, Vermund SH, Klein RS, Walker AT, Friedland GH. 1989. A prospective study of the risk of tuberculosis among intravenous drug users with human immunodeficiency virus infection. N. Engl. J. Med. 320:545–550. 10.1056/NEJM198903023200901 [DOI] [PubMed] [Google Scholar]
- 107.Selwyn PA, Sckell BM, Alcabes P, Friedland GH, Klein RS, Schoenbaum EE. 1992. High risk of active tuberculosis in HIV-infected drug users with cutaneous anergy. JAMA 268:504–509. 10.1001/jama.268.4.504 [DOI] [PubMed] [Google Scholar]
- 108.Moreno S, Baraia-Etxaburu J, Bouza E, Parras F, Perez-Tascon M, Miralles P, Vicente T, Alberdi JC, Cosin J, Lopez-Gay D. 1993. Risk for developing tuberculosis among anergic patients infected with HIV. Ann. Intern. Med. 119:194–198. 10.7326/0003-4819-119-3-199308010-00003 [DOI] [PubMed] [Google Scholar]
- 109.Antonucci G, Girardi E, Raviglione MC, Ippolito G. 1995. Risk factors for tuberculosis in HIV-infected persons. A prospective cohort study. Gruppo Italiano di Studio Tubercolosi e AIDS (GISTA). JAMA 274:143–148 [DOI] [PubMed] [Google Scholar]
- 110.Markowitz N, Hansen NI, Hopewell PC, Glassroth J, Kvale PA, Mangura BT, Wilcosky TC, Wallace JM, Rosen MJ, Reichman LB. 1997. Incidence of tuberculosis in the United States among HIV-infected persons. The Pulmonary Complications of HIV Infection Study Group. Ann. Intern. Med. 126:123–132 [DOI] [PubMed] [Google Scholar]
- 111.Daley CL, Small PM, Schecter GF, Schoolnik GK, McAdam RA, Jacobs WR, Jr, Hopewell PC. 1992. An outbreak of tuberculosis with accelerated progression among persons infected with the human immunodeficiency virus. An analysis using restriction-fragment-length polymorphisms. N. Engl. J. Med. 326:231–235 [DOI] [PubMed] [Google Scholar]
- 112.Sonnenberg P, Glynn JR, Fielding K, Murray J, Godfrey-Faussett P, Shearer S. 2005. How soon after infection with HIV does the risk of tuberculosis start to increase? A retrospective cohort study in South African gold miners. J. Infect. Dis. 191:150–158. 10.1086/426827 [DOI] [PubMed] [Google Scholar]
- 113.Lawn SD, Myer L, Bekker LG, Wood R. 2006. Burden of tuberculosis in an antiretroviral treatment programme in sub-Saharan Africa: impact on treatment outcomes and implications for tuberculosis control. AIDS 20:1605–1612. 10.1097/01.aids.0000238406.93249.cd [DOI] [PubMed] [Google Scholar]
- 114.Geldmacher C, Schuetz A, Ngwenyama N, Casazza JP, Sanga E, Saathoff E, Boehme C, Geis S, Maboko L, Singh M, Minja F, Meyerhans A, Koup RA, Hoelscher M. 2008. Early depletion of Mycobacterium tuberculosis-specific T helper 1 cell responses after HIV-1 infection. J. Infect. Dis. 198:1590–1598. 10.1086/593017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Muller I, Cobbold SP, Waldmann H, Kaufmann SH. 1987. Impaired resistance to Mycobacterium tuberculosis infection after selective in vivo depletion of L3T4+ and Lyt-2+ T cells. Infect. Immun. 55:2037–2041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Leveton C, Barnass S, Champion B, Lucas S, De Souza B, Nicol M, Banerjee D, Rook G. 1989. T-cell-mediated protection of mice against virulent Mycobacterium tuberculosis. Infect. Immun. 57:390–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Flory CM, Hubbard RD, Collins FM. 1992. Effects of in vivo T lymphocyte subset depletion on mycobacterial infections in mice. J. Leukoc. Biol. 51:225–229 [DOI] [PubMed] [Google Scholar]
- 118.Orme IM. 1988. Characteristics and specificity of acquired immunologic memory to Mycobacterium tuberculosis infection. J. Immunol. 140:3589–3593 [PubMed] [Google Scholar]
- 119.Caruso AM, Serbina N, Klein E, Triebold K, Bloom BR, Flynn JL. 1999. Mice deficient in CD4 T cells have only transiently diminished levels of IFN-gamma, yet succumb to tuberculosis. J. Immunol. 162:5407–5416 [PubMed] [Google Scholar]
- 120.Mogues T, Goodrich ME, Ryan L, LaCourse R, North RJ. 2001. The relative importance of T cell subsets in immunity and immunopathology of airborne Mycobacterium tuberculosis infection in mice. J. Exp. Med. 193:271–280. 10.1084/jem.193.3.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Scanga CA, Mohan VP, Yu K, Joseph H, Tanaka K, Chan J, Flynn JL. 2000. Depletion of CD4(+) T cells causes reactivation of murine persistent tuberculosis despite continued expression of interferon gamma and nitric oxide synthase 2. J. Exp. Med. 192:347–358. 10.1084/jem.192.3.347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Serbina NV, Lazarevic V, Flynn JL. 2001. CD4(+) T cells are required for the development of cytotoxic CD8(+) T cells during Mycobacterium tuberculosis infection. J. Immunol. 167:6991–7000. 10.4049/jimmunol.167.12.6991 [DOI] [PubMed] [Google Scholar]
- 123.Ordway D, Palanisamy G, Henao-Tamayo M, Smith EE, Shanley C, Orme IM, Basaraba RJ. 2007. The cellular immune response to Mycobacterium tuberculosis infection in the guinea pig. J. Immunol. 179:2532–2541. 10.4049/jimmunol.179.4.2532 [DOI] [PubMed] [Google Scholar]
- 124.Turner OC, Basaraba RJ, Orme IM. 2003. Immunopathogenesis of pulmonary granulomas in the guinea pig after infection with Mycobacterium tuberculosis. Infect. Immun. 71:864–871. 10.1128/IAI.71.2.864-871.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Safi H, Gormus BJ, Didier PJ, Blanchard JL, Lakey DL, Martin LN, Murphey-Corb M, Vankayalapati R, Barnes PF. 2003. Spectrum of manifestations of Mycobacterium tuberculosis infection in primates infected with SIV. AIDS Res. Hum. Retroviruses 19:585–595. 10.1089/088922203322230950 [DOI] [PubMed] [Google Scholar]
- 126.Behar SM. 2013. Antigen-specific CD8(+) T cells and protective immunity to tuberculosis. Adv. Exp. Med. Biol. 783:141–163. 10.1007/978-1-4614-6111-1_8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Flynn JL, Goldstein MM, Triebold KJ, Koller B, Bloom BR. 1992. Major histocompatibility complex class I-restricted T cells are required for resistance to Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. U. S. A. 89:12013–12017. 10.1073/pnas.89.24.12013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Arora P, Foster EL, Porcelli SA. 2013. CD1d and natural killer T cells in immunity to Mycobacterium tuberculosis. Adv. Exp. Med. Biol. 783:199–223. 10.1007/978-1-4614-6111-1_11 [DOI] [PubMed] [Google Scholar]
- 129.Sieling PA, Chatterjee D, Porcelli SA, Prigozy TI, Mazzaccaro RJ, Soriano T, Bloom BR, Brenner MB, Kronenberg M, Brennan PJ, et al. 1995. CD1-restricted T cell recognition of microbial lipoglycan antigens. Science 269:227–230. 10.1126/science.7542404 [DOI] [PubMed] [Google Scholar]
- 130.Moody DB, Reinhold BB, Guy MR, Beckman EM, Frederique DE, Furlong ST, Ye S, Reinhold VN, Sieling PA, Modlin RL, Besra GS, Porcelli SA. 1997. Structural requirements for glycolipid antigen recognition by CD1b-restricted T cells. Science 278:283–286. 10.1126/science.278.5336.283 [DOI] [PubMed] [Google Scholar]
- 131.Beckman EM, Porcelli SA, Morita CT, Behar SM, Furlong ST, Brenner MB. 1994. Recognition of a lipid antigen by CD1-restricted alpha beta+ T cells. Nature 372:691–694. 10.1038/372691a0 [DOI] [PubMed] [Google Scholar]
- 132.Aldrich CJ, Ljunggren HG, Van Kaer L, Ashton-Rickardt PG, Tonegawa S, Forman J. 1994. Positive selection of self- and alloreactive CD8+ T cells in Tap-1 mutant mice. Proc. Natl. Acad. Sci. U. S. A. 91:6525–6528. 10.1073/pnas.91.14.6525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Behar SM, Dascher CC, Grusby MJ, Wang CR, Brenner MB. 1999. Susceptibility of mice deficient in CD1D or TAP1 to infection with Mycobacterium tuberculosis. J. Exp. Med. 189:1973–1980. 10.1084/jem.189.12.1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kamath AB, Woodworth J, Xiong X, Taylor C, Weng Y, Behar SM. 2004. Cytolytic CD8+ T cells recognizing CFP10 are recruited to the lung after Mycobacterium tuberculosis infection. J. Exp. Med. 200:1479–1489. 10.1084/jem.20041690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Stenger S, Hanson DA, Teitelbaum R, Dewan P, Niazi KR, Froelich CJ, Ganz T, Thoma-Uszynski S, Melian A, Bogdan C, Porcelli SA, Bloom BR, Krensky AM, Modlin RL. 1998. An antimicrobial activity of cytolytic T cells mediated by granulysin. Science 282:121–125. 10.1126/science.282.5386.121 [DOI] [PubMed] [Google Scholar]
- 136.Sud D, Bigbee C, Flynn JL, Kirschner DE. 2006. Contribution of CD8+ T cells to control of Mycobacterium tuberculosis infection. J. Immunol. 176:4296–4314. 10.4049/jimmunol.176.7.4296 [DOI] [PubMed] [Google Scholar]
- 137.Lalvani A, Brookes R, Wilkinson RJ, Malin AS, Pathan AA, Andersen P, Dockrell H, Pasvol G, Hill AV. 1998. Human cytolytic and interferon gamma-secreting CD8+ T lymphocytes specific for Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 95:270–275. 10.1073/pnas.95.1.270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Li L, Sad S, Kagi D, Mosmann TR. 1997. CD8Tc1 and Tc2 cells secrete distinct cytokine patterns in vitro and in vivo but induce similar inflammatory reactions. J. Immunol. 158:4152–4161 [PubMed] [Google Scholar]
- 139.Wallis RS, Broder MS, Wong JY, Hanson ME, Beenhouwer DO. 2004. Granulomatous infectious diseases associated with tumor necrosis factor antagonists. Clin. Infect. Dis. 38:1261–1265. 10.1086/383317 [DOI] [PubMed] [Google Scholar]
- 140.Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, Siegel JN, Braun MM. 2001. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N. Engl. J. Med. 345:1098–1104. 10.1056/NEJMoa011110 [DOI] [PubMed] [Google Scholar]
- 141.Mohan AK, Cote TR, Block JA, Manadan AM, Siegel JN, Braun MM. 2004. Tuberculosis following the use of etanercept, a tumor necrosis factor inhibitor. Clin. Infect. Dis. 39:295–299. 10.1086/421494 [DOI] [PubMed] [Google Scholar]
- 142.Wallis RS, Broder M, Wong J, Beenhouwer D. 2004. Granulomatous infections due to tumor necrosis factor blockade: correction. Clin. Infect. Dis. 39:1254–1255. 10.1086/424455 [DOI] [PubMed] [Google Scholar]
- 143.Wallis RS, Broder M, Wong J, Lee A, Hoq L. 2005. Reactivation of latent granulomatous infections by infliximab. Clin. Infect. Dis. 41(Suppl 3):S194–S198. 10.1086/429996 [DOI] [PubMed] [Google Scholar]
- 144.Tubach F, Salmon D, Ravaud P, Allanore Y, Goupille P, Breban M, Pallot-Prades B, Pouplin S, Sacchi A, Chichemanian RM, Bretagne S, Emilie D, Lemann M, Lortholary O, Mariette X. 2009. Risk of tuberculosis is higher with anti-tumor necrosis factor monoclonal antibody therapy than with soluble tumor necrosis factor receptor therapy: the three-year prospective French Research Axed on Tolerance of Biotherapies registry. Arthritis Rheum. 60:1884–1894. 10.1002/art.24632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Harris J, Keane J. 2010. How tumour necrosis factor blockers interfere with tuberculosis immunity. Clin. Exp. Immunol. 161:1–9. 10.1111/j.1365-2249.2010.04146.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ly LH, McMurray DN. 2009. The yin-yang of TNFalpha in the guinea pig model of tuberculosis. Indian J. Exp. Biol. 47:432–439 [PubMed] [Google Scholar]
- 147.Wallis RS. 2008. Tumour necrosis factor antagonists: structure, function, and tuberculosis risks. Lancet Infect. Dis. 8:601–611. 10.1016/S1473-3099(08)70227-5 [DOI] [PubMed] [Google Scholar]
- 148.Roach DR, Bean AG, Demangel C, France MP, Briscoe H, Britton WJ. 2002. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J. Immunol. 168:4620–4627. 10.4049/jimmunol.168.9.4620 [DOI] [PubMed] [Google Scholar]
- 149.Lopez Ramirez GM, Rom WN, Ciotoli C, Talbot A, Martiniuk F, Cronstein B, Reibman J. 1994. Mycobacterium tuberculosis alters expression of adhesion molecules on monocytic cells. Infect. Immun. 62:2515–2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Keane J, Balcewicz-Sablinska MK, Remold HG, Chupp GL, Meek BB, Fenton MJ, Kornfeld H. 1997. Infection by Mycobacterium tuberculosis promotes human alveolar macrophage apoptosis. Infect. Immun. 65:298–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Harris J, Hope JC, Keane J. 2008. Tumor necrosis factor blockers influence macrophage responses to Mycobacterium tuberculosis. J. Infect. Dis. 198:1842–1850. 10.1086/593174 [DOI] [PubMed] [Google Scholar]
- 152.Saliu OY, Sofer C, Stein DS, Schwander SK, Wallis RS. 2006. Tumor-necrosis-factor blockers: differential effects on mycobacterial immunity. J. Infect. Dis. 194:486–492. 10.1086/505430 [DOI] [PubMed] [Google Scholar]
- 153.Bruns H, Meinken C, Schauenberg P, Harter G, Kern P, Modlin RL, Antoni C, Stenger S. 2009. Anti-TNF immunotherapy reduces CD8+ T cell-mediated antimicrobial activity against Mycobacterium tuberculosis in humans. J. Clin. Invest. 119:1167–1177. 10.1172/JCI38482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bean AG, Roach DR, Briscoe H, France MP, Korner H, Sedgwick JD, Britton WJ. 1999. Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. J. Immunol. 162:3504–3511 [PubMed] [Google Scholar]
- 155.Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, Lowenstein CJ, Schreiber R, Mak TW, Bloom BR. 1995. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 2:561–572. 10.1016/1074-7613(95)90001-2 [DOI] [PubMed] [Google Scholar]
- 156.Adams LB, Mason CM, Kolls JK, Scollard D, Krahenbuhl JL, Nelson S. 1995. Exacerbation of acute and chronic murine tuberculosis by administration of a tumor necrosis factor receptor-expressing adenovirus. J. Infect. Dis. 171:400–405. 10.1093/infdis/171.2.400 [DOI] [PubMed] [Google Scholar]
- 157.Algood HM, Lin PL, Yankura D, Jones A, Chan J, Flynn JL. 2004. TNF influences chemokine expression of macrophages in vitro and that of CD11b+ cells in vivo during Mycobacterium tuberculosis infection. J. Immunol. 172:6846–6857. 10.4049/jimmunol.172.11.6846 [DOI] [PubMed] [Google Scholar]
- 158.Saunders BM, Briscoe H, Britton WJ. 2004. T cell-derived tumour necrosis factor is essential, but not sufficient, for protection against Mycobacterium tuberculosis infection. Clin. Exp. Immunol. 137:279–287. 10.1111/j.1365-2249.2004.02518.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Mohan VP, Scanga CA, Yu K, Scott HM, Tanaka KE, Tsang E, Tsai MM, Flynn JL, Chan J. 2001. Effects of tumor necrosis factor alpha on host immune response in chronic persistent tuberculosis: possible role for limiting pathology. Infect. Immun. 69:1847–1855. 10.1128/IAI.69.3.1847-1855.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chakravarty SD, Zhu G, Tsai MC, Mohan VP, Marino S, Kirschner DE, Huang L, Flynn J, Chan J. 2008. Tumor necrosis factor blockade in chronic murine tuberculosis enhances granulomatous inflammation and disorganizes granulomas in the lungs. Infect. Immun. 76:916–926. 10.1128/IAI.01011-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Maglione PJ, Xu J, Chan J. 2007. B cells moderate inflammatory progression and enhance bacterial containment upon pulmonary challenge with Mycobacterium tuberculosis. J. Immunol. 178:7222–7234. 10.4049/jimmunol.178.11.7222 [DOI] [PubMed] [Google Scholar]
- 162.Botha T, Ryffel B. 2003. Reactivation of latent tuberculosis infection in TNF-deficient mice. J. Immunol. 171:3110–3118. 10.4049/jimmunol.171.6.3110 [DOI] [PubMed] [Google Scholar]
- 163.Plessner HL, Lin PL, Kohno T, Louie JS, Kirschner D, Chan J, Flynn JL. 2007. Neutralization of tumor necrosis factor (TNF) by antibody but not TNF receptor fusion molecule exacerbates chronic murine tuberculosis. J. Infect. Dis. 195:1643–1650. 10.1086/517519 [DOI] [PubMed] [Google Scholar]
- 164.Ehlers S. 2005. Why does tumor necrosis factor targeted therapy reactivate tuberculosis? J. Rheumatol. Suppl. 74:35–39 [PubMed] [Google Scholar]
- 165.Cho H, Lasco TM, Allen SS, Yoshimura T, McMurray DN. 2005. Recombinant guinea pig tumor necrosis factor alpha stimulates the expression of interleukin-12 and the inhibition of Mycobacterium tuberculosis growth in macrophages. Infect. Immun. 73:1367–1376. 10.1128/IAI.73.3.1367-1376.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Cooper AM, Magram J, Ferrante J, Orme IM. 1997. Interleukin 12 (IL-12) is crucial to the development of protective immunity in mice intravenously infected with mycobacterium tuberculosis. J. Exp. Med. 186:39–45. 10.1084/jem.186.1.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Nolt D, Flynn JL. 2004. Interleukin-12 therapy reduces the number of immune cells and pathology in lungs of mice infected with Mycobacterium tuberculosis. Infect. Immun. 72:2976–2988. 10.1128/IAI.72.5.2976-2988.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Greinert U, Ernst M, Schlaak M, Entzian P. 2001. Interleukin-12 as successful adjuvant in tuberculosis treatment. Eur. Respir. J. 17:1049–1051. 10.1183/09031936.01.17510490 [DOI] [PubMed] [Google Scholar]
- 169.Cho H, McMurray DN. 2005. Neutralization of tumor necrosis factor alpha suppresses antigen-specific type 1 cytokine responses and reverses the inhibition of mycobacterial survival in cocultures of immune guinea pig T lymphocytes and infected macrophages. Infect. Immun. 73:8437–8441. 10.1128/IAI.73.12.8437-8441.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Dorman SE, Hatem CL, Tyagi S, Aird K, Lopez-Molina J, Pitt ML, Zook BC, Dannenberg AM, Jr, Bishai WR, Manabe YC. 2004. Susceptibility to tuberculosis: clues from studies with inbred and outbred New Zealand White rabbits. Infect. Immun. 72:1700–1705. 10.1128/IAI.72.3.1700-1705.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Clay H, Volkman HE, Ramakrishnan L. 2008. Tumor necrosis factor signaling mediates resistance to mycobacteria by inhibiting bacterial growth and macrophage death. Immunity 29:283–294. 10.1016/j.immuni.2008.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bekker LG, Moreira AL, Bergtold A, Freeman S, Ryffel B, Kaplan G. 2000. Immunopathologic effects of tumor necrosis factor alpha in murine mycobacterial infection are dose dependent. Infect. Immun. 68:6954–6961. 10.1128/IAI.68.12.6954-6961.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lasco TM, Cassone L, Kamohara H, Yoshimura T, McMurray DN. 2005. Evaluating the role of tumor necrosis factor-alpha in experimental pulmonary tuberculosis in the guinea pig. Tuberculosis (Edinb.) 85:245–258. 10.1016/j.tube.2005.01.001 [DOI] [PubMed] [Google Scholar]
- 174.Bekker LG, Maartens G, Steyn L, Kaplan G. 1998. Selective increase in plasma tumor necrosis factor-alpha and concomitant clinical deterioration after initiating therapy in patients with severe tuberculosis. J. Infect. Dis. 178:580–584. 10.1086/517479 [DOI] [PubMed] [Google Scholar]
- 175.Iliopoulos A, Psathakis K, Aslanidis S, Skagias L, Sfikakis PP. 2006. Tuberculosis and granuloma formation in patients receiving anti-TNF therapy. Int. J. Tuberc. Lung Dis. 10:588–590 [PubMed] [Google Scholar]
- 176.Ray JC, Flynn JL, Kirschner DE. 2009. Synergy between individual TNF-dependent functions determines granuloma performance for controlling Mycobacterium tuberculosis infection. J. Immunol. 182:3706–3717. 10.4049/jimmunol.0802297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Marino S, Myers A, Flynn JL, Kirschner DE. 2010. TNF and IL-10 are major factors in modulation of the phagocytic cell environment in lung and lymph node in tuberculosis: a next-generation two-compartmental model. J. Theor. Biol. 265:586–598. 10.1016/j.jtbi.2010.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Newport MJ, Huxley CM, Huston S, Hawrylowicz CM, Oostra BA, Williamson R, Levin M. 1996. A mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infection. N. Engl. J. Med. 335:1941–1949. 10.1056/NEJM199612263352602 [DOI] [PubMed] [Google Scholar]
- 179.Caccamo N, Guggino G, Joosten SA, Gelsomino G, Di Carlo P, Titone L, Galati D, Bocchino M, Matarese A, Salerno A, Sanduzzi A, Franken WP, Ottenhoff TH, Dieli F. 2010. Multifunctional CD4(+) T cells correlate with active Mycobacterium tuberculosis infection. Eur. J. Immunol. 40:2211–2220. 10.1002/eji.201040455 [DOI] [PubMed] [Google Scholar]
- 180.Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. 1993. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J. Exp. Med. 178:2243–2247. 10.1084/jem.178.6.2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. 1993. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J. Exp. Med. 178:2249–2254. 10.1084/jem.178.6.2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Chan J, Tanaka K, Carroll D, Flynn J, Bloom BR. 1995. Effects of nitric oxide synthase inhibitors on murine infection with Mycobacterium tuberculosis. Infect. Immun. 63:736–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, Nathan CF. 1997. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 94:5243–5248. 10.1073/pnas.94.10.5243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.MacMicking JD, Taylor GA, McKinney JD. 2003. Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science 302:654–659. 10.1126/science.1088063 [DOI] [PubMed] [Google Scholar]
- 185.Kim BH, Shenoy AR, Kumar P, Das R, Tiwari S, MacMicking JD. 2011. A family of IFN-gamma-inducible 65-kD GTPases protects against bacterial infection. Science 332:717–721. 10.1126/science.1201711 [DOI] [PubMed] [Google Scholar]
- 186.Cooper AM, Roberts AD, Rhoades ER, Callahan JE, Getzy DM, Orme IM. 1995. The role of interleukin-12 in acquired immunity to Mycobacterium tuberculosis infection. Immunology 84:423–432 [PMC free article] [PubMed] [Google Scholar]
- 187.Kobayashi M, Fitz L, Ryan M, Hewick RM, Clark SC, Chan S, Loudon R, Sherman F, Perussia B, Trinchieri G. 1989. Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J. Exp. Med. 170:827–845. 10.1084/jem.170.3.827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Holscher C, Atkinson RA, Arendse B, Brown N, Myburgh E, Alber G, Brombacher F. 2001. A protective and agonistic function of IL-12p40 in mycobacterial infection. J. Immunol. 167:6957–6966. 10.4049/jimmunol.167.12.6957 [DOI] [PubMed] [Google Scholar]
- 189.Cooper AM, Kipnis A, Turner J, Magram J, Ferrante J, Orme IM. 2002. Mice lacking bioactive IL-12 can generate protective, antigen-specific cellular responses to mycobacterial infection only if the IL-12 p40 subunit is present. J. Immunol. 168:1322–1327. 10.4049/jimmunol.168.3.1322 [DOI] [PubMed] [Google Scholar]
- 190.Khader SA, Partida-Sanchez S, Bell G, Jelley-Gibbs DM, Swain S, Pearl JE, Ghilardi N, Desauvage FJ, Lund FE, Cooper AM. 2006. Interleukin 12p40 is required for dendritic cell migration and T cell priming after Mycobacterium tuberculosis infection. J. Exp. Med. 203:1805–1815. 10.1084/jem.20052545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Scriba TJ, Kalsdorf B, Abrahams DA, Isaacs F, Hofmeister J, Black G, Hassan HY, Wilkinson RJ, Walzl G, Gelderbloem SJ, Mahomed H, Hussey GD, Hanekom WA. 2008. Distinct, specific IL-17- and IL-22-producing CD4+ T cell subsets contribute to the human anti-mycobacterial immune response. J. Immunol. 180:1962–1970. 10.4049/jimmunol.180.3.1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Babu S, Bhat SQ, Kumar NP, Kumaraswami V, Nutman TB. 2010. Regulatory T cells modulate Th17 responses in patients with positive tuberculin skin test results. J. Infect. Dis. 201:20–31. 10.1086/648735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, McClanahan TK, O'Shea JJ, Cua DJ. 2009. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat. Immunol. 10:314–324. 10.1038/ni.1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.van de Wetering D, de Paus RA, van Dissel JT, van de Vosse E. 2009. Salmonella induced IL-23 and IL-1beta allow for IL-12 production by monocytes and Mphi1 through induction of IFN-gamma in CD56 NK/NK-like T cells. PLoS One 4:e8396. 10.1371/journal.pone.0008396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, Cilley GE, Shen F, Eaton SM, Gaffen SL, Swain SL, Locksley RM, Haynes L, Randall TD, Cooper AM. 2007. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat. Immunol. 8:369–377. 10.1038/ni1449 [DOI] [PubMed] [Google Scholar]
- 196.Khader SA, Cooper AM. 2008. IL-23 and IL-17 in tuberculosis. Cytokine 41:79–83. 10.1016/j.cyto.2007.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Larson RP, Shafiani S, Urdahl KB. 2013. Foxp3(+) regulatory T cells in tuberculosis. Adv. Exp. Med. Biol. 783:165–180. 10.1007/978-1-4614-6111-1_9 [DOI] [PubMed] [Google Scholar]
- 198.Fontenot JD, Gavin MA, Rudensky AY. 2003. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 4:330–336. 10.1038/ni904 [DOI] [PubMed] [Google Scholar]
- 199.Guyot-Revol V, Innes JA, Hackforth S, Hinks T, Lalvani A. 2006. Regulatory T cells are expanded in blood and disease sites in patients with tuberculosis. Am. J. Respir. Crit. Care Med. 173:803–810. 10.1164/rccm.200508-1294OC [DOI] [PubMed] [Google Scholar]
- 200.Chen X, Zhou B, Li M, Deng Q, Wu X, Le X, Wu C, Larmonier N, Zhang W, Zhang H, Wang H, Katsanis E. 2007. CD4(+)CD25(+)FoxP3(+) regulatory T cells suppress Mycobacterium tuberculosis immunity in patients with active disease. Clin. Immunol. 123:50–59. 10.1016/j.clim.2006.11.009 [DOI] [PubMed] [Google Scholar]
- 201.Hougardy JM, Schepers K, Place S, Drowart A, Lechevin V, Verscheure V, Debrie AS, Doherty TM, Van Vooren JP, Locht C, Mascart F. 2007. Heparin-binding-hemagglutinin-induced IFN-gamma release as a diagnostic tool for latent tuberculosis. PLoS One 2:e926. 10.1371/journal.pone.0000926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ozeki Y, Sugawara I, Udagawa T, Aoki T, Osada-Oka M, Tateishi Y, Hisaeda H, Nishiuchi Y, Harada N, Kobayashi K, Matsumoto S. 2010. Transient role of CD4+CD25+ regulatory T cells in mycobacterial infection in mice. Int. Immunol. 22:179–189. 10.1093/intimm/dxp126 [DOI] [PubMed] [Google Scholar]
- 203.Zhang M, Wang Z, Graner MW, Yang L, Liao M, Yang Q, Gou J, Zhu Y, Wu C, Liu H, Zhou B, Chen X. 2011. B cell infiltration is associated with the increased IL-17 and IL-22 expression in the lungs of patients with tuberculosis. Cell. Immunol. 270:217–223. 10.1016/j.cellimm.2011.05.009 [DOI] [PubMed] [Google Scholar]
- 204.Turner J, Frank AA, Brooks JV, Gonzalez-Juarrero M, Orme IM. 2001. The progression of chronic tuberculosis in the mouse does not require the participation of B lymphocytes or interleukin-4. Exp. Gerontol. 36:537–545. 10.1016/S0531-5565(00)00257-6 [DOI] [PubMed] [Google Scholar]
- 205.Bloch H, Segal W. 1956. Biochemical differentiation of Mycobacterium tuberculosis grown in vivo and in vitro. J. Bacteriol. 72:132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Shi L, Sohaskey CD, Pfeiffer C, Datta P, Parks M, McFadden J, North RJ, Gennaro ML. 2010. Carbon flux rerouting during Mycobacterium tuberculosis growth arrest. Mol. Microbiol. 78:1199–1215. 10.1111/j.1365-2958.2010.07399.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Pandey AK, Sassetti CM. 2008. Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl. Acad. Sci. U. S. A. 105:4376–4380. 10.1073/pnas.0711159105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Glickman MS, Cox JS, Jacobs WR., Jr 2000. A novel mycolic acid cyclopropane synthetase is required for cording, persistence, and virulence of Mycobacterium tuberculosis. Mol. Cell 5:717–727. 10.1016/S1097-2765(00)80250-6 [DOI] [PubMed] [Google Scholar]
- 209.Bryk R, Gold B, Venugopal A, Singh J, Samy R, Pupek K, Cao H, Popescu C, Gurney M, Hotha S, Cherian J, Rhee K, Ly L, Converse PJ, Ehrt S, Vandal O, Jiang X, Schneider J, Lin G, Nathan C. 2008. Selective killing of nonreplicating mycobacteria. Cell Host Microbe 3:137–145. 10.1016/j.chom.2008.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Dhiman RK, Mahapatra S, Slayden RA, Boyne ME, Lenaerts A, Hinshaw JC, Angala SK, Chatterjee D, Biswas K, Narayanasamy P, Kurosu M, Crick DC. 2009. Menaquinone synthesis is critical for maintaining mycobacterial viability during exponential growth and recovery from non-replicating persistence. Mol. Microbiol. 72:85–97. 10.1111/j.1365-2958.2009.06625.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Dhar N, McKinney JD. 2010. Mycobacterium tuberculosis persistence mutants identified by screening in isoniazid-treated mice. Proc. Natl. Acad. Sci. U. S. A. 107:12275–12280. 10.1073/pnas.1003219107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Gandotra S, Lebron MB, Ehrt S. 2010. The Mycobacterium tuberculosis proteasome active site threonine is essential for persistence yet dispensable for replication and resistance to nitric oxide. PLoS Pathog. 6:e1001040. 10.1371/journal.ppat.1001040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.McKinney JD, Honer zu Bentrup K, Munoz-Elias EJ, Miczak A, Chen B, Chan WT, Swenson D, Sacchettini JC, Jacobs WR, Jr, Russell DG. 2000. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature 406:735–738. 10.1038/35021074 [DOI] [PubMed] [Google Scholar]
- 214.Marrero J, Rhee KY, Schnappinger D, Pethe K, Ehrt S. 2010. Gluconeogenic carbon flow of tricarboxylic acid cycle intermediates is critical for Mycobacterium tuberculosis to establish and maintain infection. Proc. Natl. Acad. Sci. U. S. A. 107:9819–9824. 10.1073/pnas.1000715107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Collins DM, Wilson T, Campbell S, Buddle BM, Wards BJ, Hotter G, De Lisle GW. 2002. Production of avirulent mutants of Mycobacterium bovis with vaccine properties by the use of illegitimate recombination and screening of stationary-phase cultures. Microbiology 148:3019–3027 [DOI] [PubMed] [Google Scholar]
- 216.Liu K, Yu J, Russell DG. 2003. pckA-deficient Mycobacterium bovis BCG shows attenuated virulence in mice and in macrophages. Microbiology 149:1829–1835. 10.1099/mic.0.26234-0 [DOI] [PubMed] [Google Scholar]
- 217.Gutka HJ, Rukseree K, Wheeler PR, Franzblau SG, Movahedzadeh F. 2011. glpX gene of Mycobacterium tuberculosis: heterologous expression, purification, and enzymatic characterization of the encoded fructose 1,6-bisphosphatase II. Appl. Biochem. Biotechnol. 164:1376–1389. 10.1007/s12010-011-9219-x [DOI] [PubMed] [Google Scholar]
- 218.Movahedzadeh F, Rison SC, Wheeler PR, Kendall SL, Larson TJ, Stoker NG. 2004. The Mycobacterium tuberculosis Rv1099c gene encodes a GlpX-like class II fructose 1,6-bisphosphatase. Microbiology 150:3499–3505. 10.1099/mic.0.27204-0 [DOI] [PubMed] [Google Scholar]
- 219.Bishai W. 2000. Lipid lunch for persistent pathogen. Nature 406:683–685. 10.1038/35021159 [DOI] [PubMed] [Google Scholar]
- 220.Munoz-Elias EJ, McKinney JD. 2005. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat. Med. 11:638–644. 10.1038/nm1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Sassetti CM, Rubin EJ. 2003. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. U. S. A. 100:12989–12994. 10.1073/pnas.2134250100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Munoz-Elias EJ, Upton AM, Cherian J, McKinney JD. 2006. Role of the methylcitrate cycle in Mycobacterium tuberculosis metabolism, intracellular growth, and virulence. Mol. Microbiol. 60:1109–1122. 10.1111/j.1365-2958.2006.05155.x [DOI] [PubMed] [Google Scholar]
- 223.Lam TH, Yuen KY, Ho PL, Wong KC, Leong WM, Law HK, Weng XH, Zhang WH, Chen S, Yam WC. 2008. Differential fadE28 expression associated with phenotypic virulence of Mycobacterium tuberculosis. Microb. Pathog. 45:12–17. 10.1016/j.micpath.2008.01.006 [DOI] [PubMed] [Google Scholar]
- 224.Singh A, Guidry L, Narasimhulu KV, Mai D, Trombley J, Redding KE, Giles GI, Lancaster JR, Jr, Steyn AJ. 2007. Mycobacterium tuberculosis WhiB3 responds to O2 and nitric oxide via its [4Fe-4S] cluster and is essential for nutrient starvation survival. Proc. Natl. Acad. Sci. U. S. A. 104:11562–11567. 10.1073/pnas.0700490104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Pang X, Vu P, Byrd TF, Ghanny S, Soteropoulos P, Mukamolova GV, Wu S, Samten B, Howard ST. 2007. Evidence for complex interactions of stress-associated regulons in an mprAB deletion mutant of Mycobacterium tuberculosis. Microbiology 153:1229–1242. 10.1099/mic.0.29281-0 [DOI] [PubMed] [Google Scholar]
- 226.Steyn AJ, Collins DM, Hondalus MK, Jacobs WR, Jr, Kawakami RP, Bloom BR. 2002. Mycobacterium tuberculosis WhiB3 interacts with RpoV to affect host survival but is dispensable for in vivo growth. Proc. Natl. Acad. Sci. U. S. A. 99:3147–3152. 10.1073/pnas.052705399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Banaiee N, Jacobs WR, Jr, Ernst JD. 2006. Regulation of Mycobacterium tuberculosis whiB3 in the mouse lung and macrophages. Infect. Immun. 74:6449–6457. 10.1128/IAI.00190-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Singh A, Crossman DK, Mai D, Guidry L, Voskuil MI, Renfrow MB, Steyn AJ. 2009. Mycobacterium tuberculosis WhiB3 maintains redox homeostasis by regulating virulence lipid anabolism to modulate macrophage response. PLoS Pathog. 5:e1000545. 10.1371/journal.ppat.1000545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Casonato S, Cervantes Sanchez A, Haruki H, Rengifo Gonzalez M, Provvedi R, Dainese E, Jaouen T, Gola S, Bini E, Vicente M, Johnsson K, Ghisotti D, Palu G, Hernandez-Pando R, Manganelli R. 2012. WhiB5, a transcriptional regulator that contributes to Mycobacterium tuberculosis virulence and reactivation. Infect. Immun. 80:3132–3144. 10.1128/IAI.06328-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Park HD, Guinn KM, Harrell MI, Liao R, Voskuil MI, Tompa M, Schoolnik GK, Sherman DR. 2003. Rv3133c/dosR is a transcription factor that mediates the hypoxic response of Mycobacterium tuberculosis. Mol. Microbiol. 48:833–843. 10.1046/j.1365-2958.2003.03474.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Rustad TR, Harrell MI, Liao R, Sherman DR. 2008. The enduring hypoxic response of Mycobacterium tuberculosis. PLoS One 3:e1502. 10.1371/journal.pone.0001502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Rustad TR, Sherrid AM, Minch KJ, Sherman DR. 2009. Hypoxia: a window into Mycobacterium tuberculosis latency. Cell Microbiol. 11:1151–1159. 10.1111/j.1462-5822.2009.01325.x [DOI] [PubMed] [Google Scholar]
- 233.Sherman DR, Voskuil M, Schnappinger D, Liao R, Harrell MI, Schoolnik GK. 2001. Regulation of the Mycobacterium tuberculosis hypoxic response gene encoding alpha-crystallin. Proc. Natl. Acad. Sci. U. S. A. 98:7534–7539. 10.1073/pnas.121172498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Voskuil MI, Schnappinger D, Visconti KC, Harrell MI, Dolganov GM, Sherman DR, Schoolnik GK. 2003. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J. Exp. Med. 198:705–713. 10.1084/jem.20030205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Kumar A, Deshane JS, Crossman DK, Bolisetty S, Yan BS, Kramnik I, Agarwal A, Steyn AJ. 2008. Heme oxygenase-1-derived carbon monoxide induces the Mycobacterium tuberculosis dormancy regulon. J. Biol. Chem. 283:18032–18039. 10.1074/jbc.M802274200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Taneja NK, Dhingra S, Mittal A, Naresh M, Tyagi JS. 2010. Mycobacterium tuberculosis transcriptional adaptation, growth arrest and dormancy phenotype development is triggered by vitamin C. PLoS One 5:e10860. 10.1371/journal.pone.0010860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, Dolganov G, Efron B, Butcher PD, Nathan C, Schoolnik GK. 2003. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J. Exp. Med. 198:693–704. 10.1084/jem.20030846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Shi L, Jung YJ, Tyagi S, Gennaro ML, North RJ. 2003. Expression of Th1-mediated immunity in mouse lungs induces a Mycobacterium tuberculosis transcription pattern characteristic of nonreplicating persistence. Proc. Natl. Acad. Sci. U. S. A. 100:241–246. 10.1073/pnas.0136863100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Honaker RW, Dhiman RK, Narayanasamy P, Crick DC, Voskuil MI. 2010. DosS responds to a reduced electron transport system to induce the Mycobacterium tuberculosis DosR regulon. J. Bacteriol. 192:6447–6455. 10.1128/JB.00978-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Chao MC, Rubin EJ. 2010. Letting sleeping dos lie: does dormancy play a role in tuberculosis? Annu. Rev. Microbiology 64:293–311. 10.1146/annurev.micro.112408.134043 [DOI] [PubMed] [Google Scholar]
- 241.Leistikow RL, Morton RA, Bartek IL, Frimpong I, Wagner K, Voskuil MI. 2010. The Mycobacterium tuberculosis DosR regulon assists in metabolic homeostasis and enables rapid recovery from nonrespiring dormancy. J. Bacteriol. 192:1662–1670. 10.1128/JB.00926-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Trauner A, Lougheed KE, Bennett MH, Hingley-Wilson SM, Williams HD. 2012. The dormancy regulator DosR controls ribosome stability in hypoxic mycobacteria. J. Biol. Chem. 287:24053–24063. 10.1074/jbc.M112.364851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Parish T, Smith DA, Kendall S, Casali N, Bancroft GJ, Stoker NG. 2003. Deletion of two-component regulatory systems increases the virulence of Mycobacterium tuberculosis. Infect. Immun. 71:1134–1140. 10.1128/IAI.71.3.1134-1140.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Malhotra V, Sharma D, Ramanathan VD, Shakila H, Saini DK, Chakravorty S, Das TK, Li Q, Silver RF, Narayanan PR, Tyagi JS. 2004. Disruption of response regulator gene, devR, leads to attenuation in virulence of Mycobacterium tuberculosis. FEMS Microbiol. Lett. 231:237–245. 10.1016/S0378-1097(04)00002-3 [DOI] [PubMed] [Google Scholar]
- 245.Converse PJ, Karakousis PC, Klinkenberg LG, Kesavan AK, Ly LH, Allen SS, Grosset JH, Jain SK, Lamichhane G, Manabe YC, McMurray DN, Nuermberger EL, Bishai WR. 2009. Role of the dosR-dosS two-component regulatory system in Mycobacterium tuberculosis virulence in three animal models. Infect. Immun. 77:1230–1237. 10.1128/IAI.01117-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Primm TP, Andersen SJ, Mizrahi V, Avarbock D, Rubin H, Barry CE., III 2000. The stringent response of Mycobacterium tuberculosis is required for long-term survival. J. Bacteriol. 182:4889–4898. 10.1128/JB.182.17.4889-4898.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Artsimovitch I, Patlan V, Sekine S, Vassylyeva MN, Hosaka T, Ochi K, Yokoyama S, Vassylyev DG. 2004. Structural basis for transcription regulation by alarmone ppGpp. Cell 117:299–310. 10.1016/S0092-8674(04)00401-5 [DOI] [PubMed] [Google Scholar]
- 248.Perederina A, Svetlov V, Vassylyeva MN, Tahirov TH, Yokoyama S, Artsimovitch I, Vassylyev DG. 2004. Regulation through the secondary channel-structural framework for ppGpp-DksA synergism during transcription. Cell 118:297–309. 10.1016/j.cell.2004.06.030 [DOI] [PubMed] [Google Scholar]
- 249.Braeken K, Moris M, Daniels R, Vanderleyden J, Michiels J. 2006. New horizons for (p)ppGpp in bacterial and plant physiology. Trends Microbiol. 14:45–54. 10.1016/j.tim.2005.11.006 [DOI] [PubMed] [Google Scholar]
- 250.Manganelli R. 2007. Polyphosphate and stress response in mycobacteria. Mol. Microbiol. 65:258–260. 10.1111/j.1365-2958.2007.05819.x [DOI] [PubMed] [Google Scholar]
- 251.Avarbock D, Avarbock A, Rubin H. 2000. Differential regulation of opposing RelMtb activities by the aminoacylation state of a tRNA.ribosome.mRNA.RelMtb complex. Biochemistry 39:11640–11648. 10.1021/bi001256k [DOI] [PubMed] [Google Scholar]
- 252.Sajish M, Tiwari D, Rananaware D, Nandicoori VK, Prakash B. 2007. A charge reversal differentiates (p)ppGpp synthesis by monofunctional and bifunctional Rel proteins. J. Biol. Chem. 282:34977–34983. 10.1074/jbc.M704828200 [DOI] [PubMed] [Google Scholar]
- 253.Boshoff HI, Myers TG, Copp BR, McNeil MR, Wilson MA, Barry CE., III 2004. The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: novel insights into drug mechanisms of action. J. Biol. Chem. 279:40174–40184. 10.1074/jbc.M406796200 [DOI] [PubMed] [Google Scholar]
- 254.Dahl JL, Kraus CN, Boshoff HI, Doan B, Foley K, Avarbock D, Kaplan G, Mizrahi V, Rubin H, Barry CE., III 2003. The role of RelMtb-mediated adaptation to stationary phase in long-term persistence of Mycobacterium tuberculosis in mice. Proc. Natl. Acad. Sci. U. S. A. 100:10026–10031. 10.1073/pnas.1631248100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Klinkenberg LG, Lee JH, Bishai WR, Karakousis PC. 2010. The stringent response is required for full virulence of Mycobacterium tuberculosis in guinea pigs. J. Infect. Dis. 202:1397–1404. 10.1086/656524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Klinkenberg LG, Karakousis PC. 2013. Preventing Mycobacterium tuberculosis persister formation to shorten TB chemotherapy. Keystone Symposia X8, abstr X8 2039 [Google Scholar]
- 257.Manganelli R, Voskuil MI, Schoolnik GK, Smith I. 2001. The Mycobacterium tuberculosis ECF sigma factor sigmaE: role in global gene expression and survival in macrophages. Mol. Microbiol. 41:423–437. 10.1046/j.1365-2958.2001.02525.x [DOI] [PubMed] [Google Scholar]
- 258.He H, Hovey R, Kane J, Singh V, Zahrt TC. 2006. MprAB is a stress-responsive two-component system that directly regulates expression of sigma factors SigB and SigE in Mycobacterium tuberculosis. J. Bacteriol. 188:2134–2143. 10.1128/JB.188.6.2134-2143.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Pang X, Samten B, Cao G, Wang X, Tvinnereim AR, Chen XL, Howard ST. 2013. MprAB regulates the espA operon in Mycobacterium tuberculosis and modulates ESX-1 function and host cytokine response. J. Bacteriol. 195:66–75. 10.1128/JB.01067-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.White MJ, Savaryn JP, Bretl DJ, He H, Penoske RM, Terhune SS, Zahrt TC. 2011. The HtrA-like serine protease PepD interacts with and modulates the Mycobacterium tuberculosis 35-kDa antigen outer envelope protein. PLoS One 6:e18175. 10.1371/journal.pone.0018175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Pang X, Howard ST. 2007. Regulation of the alpha-crystallin gene acr2 by the MprAB two-component system of Mycobacterium tuberculosis. J. Bacteriol. 189:6213–6221. 10.1128/JB.00492-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Manganelli R, Provvedi R, Rodrigue S, Beaucher J, Gaudreau L, Smith I. 2004. Sigma factors and global gene regulation in Mycobacterium tuberculosis. J. Bacteriol. 186:895–902. 10.1128/JB.186.4.895-902.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Lee JH, Karakousis PC, Bishai WR. 2008. Roles of SigB and SigF in the Mycobacterium tuberculosis sigma factor network. J. Bacteriol. 190:699–707. 10.1128/JB.01273-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Fontan PA, Voskuil MI, Gomez M, Tan D, Pardini M, Manganelli R, Fattorini L, Schoolnik GK, Smith I. 2009. The Mycobacterium tuberculosis sigma factor sigmaB is required for full response to cell envelope stress and hypoxia in vitro, but it is dispensable for in vivo growth. J. Bacteriol. 191:5628–5633. 10.1128/JB.00510-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Kaushal D, Schroeder BG, Tyagi S, Yoshimatsu T, Scott C, Ko C, Carpenter L, Mehrotra J, Manabe YC, Fleischmann RD, Bishai WR. 2002. Reduced immunopathology and mortality despite tissue persistence in a Mycobacterium tuberculosis mutant lacking alternative sigma factor, SigH. Proc. Natl. Acad. Sci. U. S. A. 99:8330–8335. 10.1073/pnas.102055799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Dutta NK, Mehra S, Kaushal D. 2010. A Mycobacterium tuberculosis sigma factor network responds to cell-envelope damage by the promising anti-mycobacterial thioridazine. PLoS One 5:e10069. 10.1371/journal.pone.0010069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Dutta NK, Mehra S, Martinez AN, Alvarez X, Renner NA, Morici LA, Pahar B, Maclean AG, Lackner AA, Kaushal D. 2012. The stress-response factor SigH modulates the interaction between Mycobacterium tuberculosis and host phagocytes. PLoS One 7:e28958. 10.1371/journal.pone.0028958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Sanyal S, Banerjee SK, Banerjee R, Mukhopadhyay J, Kundu M. 2013. Polyphosphate kinase 1, a central node in the stress response network of Mycobacterium tuberculosis, connects the two-component systems MprAB and SenX3-RegX3 and the extracytoplasmic function sigma factor, sigma E. Microbiology 159:2074–2086. 10.1099/mic.0.068452-0 [DOI] [PubMed] [Google Scholar]
- 269.Sachdeva P, Misra R, Tyagi AK, Singh Y. 2010. The sigma factors of Mycobacterium tuberculosis: regulation of the regulators. The FEBS journal. 277:605–626. 10.1111/j.1742-4658.2009.07479.x [DOI] [PubMed] [Google Scholar]
- 270.Mehra S, Golden NA, Stuckey K, Didier PJ, Doyle LA, Russell-Lodrigue KE, Sugimoto C, Hasegawa A, Sivasubramani SK, Roy CJ, Alvarez X, Kuroda MJ, Blanchard JL, Lackner AA, Kaushal D. 2012. The Mycobacterium tuberculosis stress response factor SigH is required for bacterial burden as well as immunopathology in primate lungs. J. Infect. Dis. 205:1203–1213. 10.1093/infdis/jis102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Wang X, Wang H, Xie J. 2011. Genes and regulatory networks involved in persistence of Mycobacterium tuberculosis. Sci. China Life Sci. 54:300–310. 10.1007/s11427-011-4134-5 [DOI] [PubMed] [Google Scholar]
- 272.Ando M, Yoshimatsu T, Ko C, Converse PJ, Bishai WR. 2003. Deletion of Mycobacterium tuberculosis sigma factor E results in delayed time to death with bacterial persistence in the lungs of aerosol-infected mice. Infect. Immun. 71:7170–7172. 10.1128/IAI.71.12.7170-7172.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Datta P, Shi L, Bibi N, Balazsi G, Gennaro ML. 2011. Regulation of central metabolism genes of Mycobacterium tuberculosis by parallel feed-forward loops controlled by sigma factor E (sigma(E)). J. Bacteriol. 193:1154–1160. 10.1128/JB.00459-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Kornberg A, Rao NN, Ault-Riche D. 1999. Inorganic polyphosphate: a molecule of many functions. Annu. Rev. Biochem. 68:89–125. 10.1146/annurev.biochem.68.1.89 [DOI] [PubMed] [Google Scholar]
- 275.Kuroda A, Murphy H, Cashel M, Kornberg A. 1997. Guanosine tetra- and pentaphosphate promote accumulation of inorganic polyphosphate in Escherichia coli. J. Biol. Chem. 272:21240–21243. 10.1074/jbc.272.34.21240 [DOI] [PubMed] [Google Scholar]
- 276.Rao NN, Liu S, Kornberg A. 1998. Inorganic polyphosphate in Escherichia coli: the phosphate regulon and the stringent response. J. Bacteriol. 180:2186–2193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Rao NN, Kornberg A. 1996. Inorganic polyphosphate supports resistance and survival of stationary-phase Escherichia coli. J. Bacteriol. 178:1394–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Shiba T, Tsutsumi K, Yano H, Ihara Y, Kameda A, Tanaka K, Takahashi H, Munekata M, Rao NN, Kornberg A. 1997. Inorganic polyphosphate and the induction of rpoS expression. Proc. Natl. Acad. Sci. U. S. A. 94:11210–11215. 10.1073/pnas.94.21.11210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Hengge-Aronis R. 2002. Signal transduction and regulatory mechanisms involved in control of the sigma(S) (RpoS) subunit of RNA polymerase. Microbiol. Mol. Biol. Rev. 66:373–395. 10.1128/MMBR.66.3.373-395.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Ault-Riche D, Fraley CD, Tzeng CM, Kornberg A. 1998. Novel assay reveals multiple pathways regulating stress-induced accumulations of inorganic polyphosphate in Escherichia coli. J. Bacteriol. 180:1841–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Sureka K, Dey S, Datta P, Singh AK, Dasgupta A, Rodrigue S, Basu J, Kundu M. 2007. Polyphosphate kinase is involved in stress-induced mprAB-sigE-rel signalling in mycobacteria. Mol. Microbiol. 65:261–276. 10.1111/j.1365-2958.2007.05814.x [DOI] [PubMed] [Google Scholar]
- 282.Chuang YM, Belchis DA, Karakousis PC. 2013. The polyphosphate kinase gene ppk2 is required for Mycobacterium tuberculosis inorganic polyphosphate regulation and virulence. mBio 4(3):e00039-00013. 10.1128/mBio.00039-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Thayil SM, Morrison N, Schechter N, Rubin H, Karakousis PC. 2011. The role of the novel exopolyphosphatase MT0516 in Mycobacterium tuberculosis drug tolerance and persistence. PLoS One 6:e28076. 10.1371/journal.pone.0028076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Choi MY, Wang Y, Wong LL, Lu BT, Chen WY, Huang JD, Tanner JA, Watt RM. 2012. The two PPX-GppA homologues from Mycobacterium tuberculosis have distinct biochemical activities. PLoS One 7:e42561. 10.1371/journal.pone.0042561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Sureka K, Ghosh B, Dasgupta A, Basu J, Kundu M, Bose I. 2008. Positive feedback and noise activate the stringent response regulator rel in mycobacteria. PLoS One 3:e1771. 10.1371/journal.pone.0001771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Chouayekh H, Virolle MJ. 2002. The polyphosphate kinase plays a negative role in the control of antibiotic production in Streptomyces lividans. Mol. Microbiol. 43:919–930. 10.1046/j.1365-2958.2002.02557.x [DOI] [PubMed] [Google Scholar]
- 287.Magnusson LU, Farewell A, Nystrom T. 2005. ppGpp: a global regulator in Escherichia coli. Trends Microbiol. 13:236–242. 10.1016/j.tim.2005.03.008 [DOI] [PubMed] [Google Scholar]
- 288.Stallings CL, Stephanou NC, Chu L, Hochschild A, Nickels BE, Glickman MS. 2009. CarD is an essential regulator of rRNA transcription required for Mycobacterium tuberculosis persistence. Cell 138:146–159. 10.1016/j.cell.2009.04.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Li Y, Zhang Y. 2007. PhoU is a persistence switch involved in persister formation and tolerance to multiple antibiotics and stresses in Escherichia coli. Antimicrob. Agents Chemother. 51:2092–2099. 10.1128/AAC.00052-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Shi W, Zhang Y. 2010. PhoY2 but not PhoY1 is the PhoU homologue involved in persisters in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 65:1237–1242. 10.1093/jac/dkq103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Zhu L, Sharp JD, Kobayashi H, Woychik NA, Inouye M. 2010. Noncognate Mycobacterium tuberculosis toxin-antitoxins can physically and functionally interact. J. Biol. Chem. 285:39732–39738. 10.1074/jbc.M110.163105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Ahidjo BA, Kuhnert D, McKenzie JL, Machowski EE, Gordhan BG, Arcus V, Abrahams GL, Mizrahi V. 2011. VapC toxins from Mycobacterium tuberculosis are ribonucleases that differentially inhibit growth and are neutralized by cognate VapB antitoxins. PLoS One 6:e21738. 10.1371/journal.pone.0021738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Sharp JD, Cruz JW, Raman S, Inouye M, Husson RN, Woychik NA. 2012. Growth and translation inhibition through sequence-specific RNA binding by Mycobacterium tuberculosis VapC toxin. J. Biol. Chem. 287:12835–12847. 10.1074/jbc.M112.340109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Korch SB, Contreras H, Clark-Curtiss JE. 2009. Three Mycobacterium tuberculosis Rel toxin-antitoxin modules inhibit mycobacterial growth and are expressed in infected human macrophages. J. Bacteriol. 191:1618–1630. 10.1128/JB.01318-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Ramage HR, Connolly LE, Cox JS. 2009. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: implications for pathogenesis, stress responses, and evolution. PLoS Genet. 5:e1000767. 10.1371/journal.pgen.1000767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Keren I, Minami S, Rubin E, Lewis K. 2011. Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. mBio 2(3):e00100-00111. 10.1128/mBio.00100-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Stout JE, Engemann JJ, Cheng AC, Fortenberry ER, Hamilton CD. 2003. Safety of 2 months of rifampin and pyrazinamide for treatment of latent tuberculosis. Am. J. Respir. Crit. Care Med. 167:824–827. 10.1164/rccm.200209-998OC [DOI] [PubMed] [Google Scholar]
- 298.Shi W, Zhang X, Jiang X, Yuan H, Lee JS, Barry CE, III, Wang H, Zhang W, Zhang Y. 2011. Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science 333:1630–1632. 10.1126/science.1208813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Cole ST. 2011. Pyrazinamide—old TB drug finds new target. Science 333:1583–1584. 10.1126/science.1212450 [DOI] [PubMed] [Google Scholar]
- 300.Zhang Y, Yew WW, Barer MR. 2012. Targeting persisters for tuberculosis control. Antimicrob. Agents Chemother. 56:2223–2230. 10.1128/AAC.06288-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Ojha AK, Baughn AD, Sambandan D, Hsu T, Trivelli X, Guerardel Y, Alahari A, Kremer L, Jacobs WR, Jr, Hatfull GF. 2008. Growth of Mycobacterium tuberculosis biofilms containing free mycolic acids and harbouring drug-tolerant bacteria. Mol. Microbiol. 69:164–174. 10.1111/j.1365-2958.2008.06274.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Ojha A, Anand M, Bhatt A, Kremer L, Jacobs WR, Jr, Hatfull GF. 2005. GroEL1: a dedicated chaperone involved in mycolic acid biosynthesis during biofilm formation in mycobacteria. Cell 123:861–873. 10.1016/j.cell.2005.09.012 [DOI] [PubMed] [Google Scholar]
- 303.Sambandan D, Dao DN, Weinrick BC, Vilcheze C, Gurcha SS, Ojha A, Kremer L, Besra GS, Hatfull GF, Jacobs WR., Jr 2013. Keto-mycolic acid-dependent pellicle formation confers tolerance to drug-sensitive Mycobacterium tuberculosis. mBio 4(3):e00222-13. 10.1128/mBio.00222-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Schmalstieg AM, Srivastava S, Belkaya S, Deshpande D, Meek C, Leff R, van Oers NS, Gumbo T. 2012. The antibiotic resistance arrow of time: efflux pump induction is a general first step in the evolution of mycobacterial drug resistance. Antimicrob. Agents Chemother. 56:4806–4815. 10.1128/AAC.05546-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Gupta S, Tyagi S, Almeida DV, Maiga MC, Ammerman NC, Bishai WR. 2013. Acceleration of tuberculosis treatment by adjunctive therapy with verapamil as an efflux inhibitor. Am. J. Respir. Crit. Care Med. 188:600–607. 10.1164/rccm.201304-0650OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Lee RE, Hurdle JG, Liu J, Bruhn DF, Matt T, Scherman MS, Vaddady PK, Zheng Z, Qi J, Akbergenov R, Das S, Madhura DB, Rathi C, Trivedi A, Villellas C, Lee RB, Rakesh Waidyarachchi SL, Sun D, McNeil MR, Ainsa JA, Boshoff HI, Gonzalez-Juarrero M, Meibohm B, Bottger EC, Lenaerts AJ. 2014. Spectinamides: a new class of semisynthetic antituberculosis agents that overcome native drug efflux. Nat. Med. 20:152–158. 10.1038/nm.3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Allison KR, Brynildsen MP, Collins JJ. 2011. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature 473:216–220. 10.1038/nature10069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.American Thoracic Society, Centers for Disease Control and Prevention, Infectious Diseases Society of America. 2005. American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: controlling tuberculosis in the United States. Am. J. Respir. Crit. Care Med. 172:1169–1227. 10.1164/rccm.2508001 [DOI] [PubMed] [Google Scholar]
- 309.Herrera V, Perry S, Parsonnet J, Banaei N. 2011. Clinical application and limitations of interferon-gamma release assays for the diagnosis of latent tuberculosis infection. Clin. Infect. Dis. 52:1031–1037. 10.1093/cid/cir068 [DOI] [PubMed] [Google Scholar]
- 310.Sun Z, Zhang Y. 1999. Spent culture supernatant of Mycobacterium tuberculosis H37Ra improves viability of aged cultures of this strain and allows small inocula to initiate growth. J. Bacteriol. 181:7626–7628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Russell-Goldman E, Xu J, Wang X, Chan J, Tufariello JM. 2008. A Mycobacterium tuberculosis Rpf double-knockout strain exhibits profound defects in reactivation from chronic tuberculosis and innate immunity phenotypes. Infect. Immun. 76:4269–4281. 10.1128/IAI.01735-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Mukamolova GV, Turapov OA, Young DI, Kaprelyants AS, Kell DB, Young M. 2002. A family of autocrine growth factors in Mycobacterium tuberculosis. Mol. Microbiol. 46:623–635. 10.1046/j.1365-2958.2002.03184.x [DOI] [PubMed] [Google Scholar]
- 313.Downing KJ, Mischenko VV, Shleeva MO, Young DI, Young M, Kaprelyants AS, Apt AS, Mizrahi V. 2005. Mutants of Mycobacterium tuberculosis lacking three of the five rpf-like genes are defective for growth in vivo and for resuscitation in vitro. Infect. Immun. 73:3038–3043. 10.1128/IAI.73.5.3038-3043.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Freeman R, Dunn J, Magee J, Barrett A. 2002. The enhancement of isolation of mycobacteria from a rapid liquid culture system by broth culture supernate of Micrococcus luteus. J. Med. Microbiol. 51:92–93 [DOI] [PubMed] [Google Scholar]
- 315.Mukamolova GV, Turapov O, Malkin J, Woltmann G, Barer MR. 2010. Resuscitation-promoting factors reveal an occult population of tubercle bacilli in sputum. Am. J. Respir. Crit. Care Med. 181:174–180. 10.1164/rccm.200905-0661OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Mukamolova GV, Murzin AG, Salina EG, Demina GR, Kell DB, Kaprelyants AS, Young M. 2006. Muralytic activity of Micrococcus luteus Rpf and its relationship to physiological activity in promoting bacterial growth and resuscitation. Mol. Microbiol. 59:84–98. 10.1111/j.1365-2958.2005.04930.x [DOI] [PubMed] [Google Scholar]
- 317.Tufariello JM, Mi K, Xu J, Manabe YC, Kesavan AK, Drumm J, Tanaka K, Jacobs WR, Jr, Chan J. 2006. Deletion of the Mycobacterium tuberculosis resuscitation-promoting factor Rv1009 gene results in delayed reactivation from chronic tuberculosis. Infect. Immun. 74:2985–2995. 10.1128/IAI.74.5.2985-2995.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Hett EC, Chao MC, Steyn AJ, Fortune SM, Deng LL, Rubin EJ. 2007. A partner for the resuscitation-promoting factors of Mycobacterium tuberculosis. Mol. Microbiol. 66:658–668. 10.1111/j.1365-2958.2007.05945.x [DOI] [PubMed] [Google Scholar]
- 319.Hett EC, Chao MC, Deng LL, Rubin EJ. 2008. A mycobacterial enzyme essential for cell division synergizes with resuscitation-promoting factor. PLoS Pathog. 4:e1000001. 10.1371/journal.ppat.1000001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Andersen P. 2001. TB vaccines: progress and problems. Trends Immunol. 22:160–168. 10.1016/S1471-4906(01)01865-8 [DOI] [PubMed] [Google Scholar]
- 321.Orme IM. 2013. Vaccine development for tuberculosis: current progress. Drugs 73:1015–1024. 10.1007/s40265-013-0081-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Dutta NK, McLachlan J, Mehra S, Kaushal D. 2014. Humoral and lung immune responses to Mycobacterium tuberculosis infection in a primate model of protection. Trials Vaccinol. 3:47–51. 10.1016/j.trivac.2014.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Nuermberger E, Tyagi S, Williams KN, Rosenthal I, Bishai WR, Grosset JH. 2005. Rifapentine, moxifloxacin, or DNA vaccine improves treatment of latent tuberculosis in a mouse model. Am. J. Respir. Crit. Care Med. 172:1452–1456. 10.1164/rccm.200507-1047OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Andersen P. 2007. Vaccine strategies against latent tuberculosis infection. Trends Microbiol. 15:7–13. 10.1016/j.tim.2006.11.008 [DOI] [PubMed] [Google Scholar]
- 325.Repique CJ, Li A, Collins FM, Morris SL. 2002. DNA immunization in a mouse model of latent tuberculosis: effect of DNA vaccination on reactivation of disease and on reinfection with a secondary challenge. Infect. Immun. 70:3318–3323. 10.1128/IAI.70.7.3318-3323.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Cardona PJ. 2006. RUTI: a new chance to shorten the treatment of latent tuberculosis infection. Tuberculosis (Edinb.) 86:273–289. 10.1016/j.tube.2006.01.024 [DOI] [PubMed] [Google Scholar]
- 327.Lambert PH, Hawkridge T, Hanekom WA. 2009. New vaccines against tuberculosis. Clin. Chest Med. 30:811–826. 10.1016/j.ccm.2009.08.014 [DOI] [PubMed] [Google Scholar]
- 328.Wilkinson RJ, Wilkinson KA, De Smet KA, Haslov K, Pasvol G, Singh M, Svarcova I, Ivanyi J. 1998. Human T- and B-cell reactivity to the 16kDa alpha-crystallin protein of Mycobacterium tuberculosis. Scand. J. Immunol. 48:403–409. 10.1046/j.1365-3083.1998.00420.x [DOI] [PubMed] [Google Scholar]
- 329.Cunningham AF, Spreadbury CL. 1998. Mycobacterial stationary phase induced by low oxygen tension: cell wall thickening and localization of the 16-kilodalton alpha-crystallin homolog. J. Bacteriol. 180:801–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Stewart JN, Rivera HN, Karls R, Quinn FD, Roman J, Rivera-Marrero CA. 2006. Increased pathology in lungs of mice after infection with an alpha-crystallin mutant of Mycobacterium tuberculosis: changes in cathepsin proteases and certain cytokines. Microbiology 152:233–244. 10.1099/mic.0.28275-0 [DOI] [PubMed] [Google Scholar]
- 331.Chauhan P, Jain R, Dey B, Tyagi AK. 2013. Adjunctive immunotherapy with alpha-crystallin based DNA vaccination reduces tuberculosis chemotherapy period in chronically infected mice. Sci. Rep. 3:1821. 10.1038/srep01821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Wallis RS, Kim P, Cole S, Hanna D, Andrade BB, Maeurer M, Schito M, Zumla A. 2013. Tuberculosis biomarkers discovery: developments, needs, and challenges. Lancet Infect. Dis. 13:362–372. 10.1016/S1473-3099(13)70034-3 [DOI] [PubMed] [Google Scholar]
- 333.Dutta NK, Bandyopadhyay N, Veeramani B, Lamichhane G, Karakousis PC, Bader JS. 2014. Systems biology-based identification of Mycobacterium tuberculosis persistence genes in mouse lungs. mBio 5(1):e01066-13. 10.1128/mBio.01066-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Bai X, Wilson SE, Chmura K, Feldman NE, Chan ED. 2004. Morphometric analysis of Th(1) and Th(2) cytokine expression in human pulmonary tuberculosis. Tuberculosis (Edinb.) 84:375–385. 10.1016/j.tube.2004.05.001 [DOI] [PubMed] [Google Scholar]
- 335.Tully G, Kortsik C, Hohn H, Zehbe I, Hitzler WE, Neukirch C, Freitag K, Kayser K, Maeurer MJ. 2005. Highly focused T cell responses in latent human pulmonary Mycobacterium tuberculosis infection. J. Immunol. 174:2174–2184. 10.4049/jimmunol.174.4.2174 [DOI] [PubMed] [Google Scholar]
- 336.Hussain R, Ansari A, Talat N, Hasan Z, Dawood G. 2011. CCL2/MCP-I genotype-phenotype relationship in latent tuberculosis infection. PLoS One 6:e25803. 10.1371/journal.pone.0025803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Kirman J, Zakaria Z, McCoy K, Delahunt B, Le Gros G. 2000. Role of eosinophils in the pathogenesis of Mycobacterium bovis BCG infection in gamma interferon receptor-deficient mice. Infect. Immun. 68:2976–2978. 10.1128/IAI.68.5.2976-2978.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Yamada H, Mizumo S, Horai R, Iwakura Y, Sugawara I. 2000. Protective role of interleukin-1 in mycobacterial infection in IL-1 alpha/beta double-knockout mice. Lab. Invest. 80:759–767. 10.1038/labinvest.3780079 [DOI] [PubMed] [Google Scholar]
- 339.Turner J, Gonzalez-Juarrero M, Ellis DL, Basaraba RJ, Kipnis A, Orme IM, Cooper AM. 2002. In vivo IL-10 production reactivates chronic pulmonary tuberculosis in C57BL/6 mice. J. Immunol. 169:6343–6351. 10.4049/jimmunol.169.11.6343 [DOI] [PubMed] [Google Scholar]
- 340.O'Leary S, O'Sullivan MP, Keane J. 2011. IL-10 blocks phagosome maturation in mycobacterium tuberculosis-infected human macrophages. Am. J. Respir. Cell Mol. Biol. 45:172–180. 10.1165/rcmb.2010-0319OC [DOI] [PubMed] [Google Scholar]
- 341.Sugawara I, Yamada H, Kaneko H, Mizuno S, Takeda K, Akira S. 1999. Role of interleukin-18 (IL-18) in mycobacterial infection in IL-18-gene-disrupted mice. Infect. Immun. 67:2585–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Cooper AM, Segal BH, Frank AA, Holland SM, Orme IM. 2000. Transient loss of resistance to pulmonary tuberculosis in p47(phox−/−) mice. Infect. Immun. 68:1231–1234. 10.1128/IAI.68.3.1231-1234.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Stagas MK, Papaetis GS, Orphanidou D, Kostopoulos C, Syriou S, Reczko M, Drakoulis N. 2011. Polymorphisms of the NRAMP1 gene: distribution and susceptibility to the development of pulmonary tuberculosis in the Greek population. Med. Sci. Monitor 17:PH1–PH6. 10.12659/MSM.881312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Vidal S, Tremblay ML, Govoni G, Gauthier S, Sebastiani G, Malo D, Skamene E, Olivier M, Jothy S, Gros P. 1995. The Ity/Lsh/Bcg locus: natural resistance to infection with intracellular parasites is abrogated by disruption of the Nramp1 gene. J. Exp. Med. 182:655–666. 10.1084/jem.182.3.655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Quesniaux VF, Jacobs M, Allie N, Grivennikov S, Nedospasov SA, Garcia I, Olleros ML, Shebzukhov Y, Kuprash D, Vasseur V, Rose S, Court N, Vacher R, Ryffel B. 2010. TNF in host resistance to tuberculosis infection. Curr. Direct. Autoimmun. 11:157–179. 10.1159/000289204 [DOI] [PubMed] [Google Scholar]
- 346.Kalscheuer R, Syson K, Veeraraghavan U, Weinrick B, Biermann KE, Liu Z, Sacchettini JC, Besra G, Bornemann S, Jacobs WR., Jr 2010. Self-poisoning of Mycobacterium tuberculosis by targeting GlgE in an alpha-glucan pathway. Nat. Chem. Biol. 6:376–384. 10.1038/nchembio.340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Stewart GR, Snewin VA, Walzl G, Hussell T, Tormay P, O'Gaora P, Goyal M, Betts J, Brown IN, Young DB. 2001. Overexpression of heat-shock proteins reduces survival of Mycobacterium tuberculosis in the chronic phase of infection. Nat. Med. 7:732–737. 10.1038/89113 [DOI] [PubMed] [Google Scholar]
- 348.Zahrt TC, Deretic V. 2001. Mycobacterium tuberculosis signal transduction system required for persistent infections. Proc. Natl. Acad. Sci. U. S. A. 98:12706–12711. 10.1073/pnas.221272198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Zahrt TC, Wozniak C, Jones D, Trevett A. 2003. Functional analysis of the Mycobacterium tuberculosis MprAB two-component signal transduction system. Infect. Immun. 71:6962–6970. 10.1128/IAI.71.12.6962-6970.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Weber I, Fritz C, Ruttkowski S, Kreft A, Bange FC. 2000. Anaerobic nitrate reductase (narGHJI) activity of Mycobacterium bovis BCG in vitro and its contribution to virulence in immunodeficient mice. Mol. Microbiol. 35:1017–1025. 10.1046/j.1365-2958.2000.01794.x [DOI] [PubMed] [Google Scholar]
- 351.Fritz C, Maass S, Kreft A, Bange FC. 2002. Dependence of Mycobacterium bovis BCG on anaerobic nitrate reductase for persistence is tissue specific. Infect. Immun. 70:286–291. 10.1128/IAI.70.1.286-291.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Hutter B, Dick T. 2000. Analysis of the dormancy-inducible narK2 promoter in Mycobacterium bovis BCG. FEMS Microbiol. Lett. 188:141–146. 10.1111/j.1574-6968.2000.tb09185.x [DOI] [PubMed] [Google Scholar]
- 353.Wayne LG, Lin KY. 1982. Glyoxylate metabolism and adaptation of Mycobacterium tuberculosis to survival under anaerobic conditions. Infect. Immun. 37:1042–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Li Z, Kelley C, Collins F, Rouse D, Morris S. 1998. Expression of katG in Mycobacterium tuberculosis is associated with its growth and persistence in mice and guinea pigs. J. Infect. Dis. 177:1030–1035. 10.1086/515254 [DOI] [PubMed] [Google Scholar]
- 355.Hu Y, Movahedzadeh F, Stoker NG, Coates AR. 2006. Deletion of the Mycobacterium tuberculosis alpha-crystallin-like hspX gene causes increased bacterial growth in vivo. Infect. Immun. 74:861–868. 10.1128/IAI.74.2.861-868.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Majumdar SD, Vashist A, Dhingra S, Gupta R, Singh A, Challu VK, Ramanathan VD, Kumar P, Tyagi JS. 2012. Appropriate DevR (DosR)-mediated signaling determines transcriptional response, hypoxic viability and virulence of Mycobacterium tuberculosis. PLoS One 7:e35847. 10.1371/journal.pone.0035847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Wilson M, DeRisi J, Kristensen HH, Imboden P, Rane S, Brown PO, Schoolnik GK. 1999. Exploring drug-induced alterations in gene expression in Mycobacterium tuberculosis by microarray hybridization. Proc. Natl. Acad. Sci. U. S. A. 96:12833–12838. 10.1073/pnas.96.22.12833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Manganelli R, Voskuil MI, Schoolnik GK, Dubnau E, Gomez M, Smith I. 2002. Role of the extracytoplasmic-function sigma factor sigma(H) in Mycobacterium tuberculosis global gene expression. Mol. Microbiol. 45:365–374. 10.1046/j.1365-2958.2002.03005.x [DOI] [PubMed] [Google Scholar]
- 359.Williams EP, Lee JH, Bishai WR, Colantuoni C, Karakousis PC. 2007. Mycobacterium tuberculosis SigF regulates genes encoding cell wall-associated proteins and directly regulates the transcriptional regulatory gene phoY1. J. Bacteriol. 189:4234–4242. 10.1128/JB.00201-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Connolly LE, Cox JS. 2009. CarD tricks and magic spots: mechanisms of stringent control in mycobacteria. Cell Host Microbe 6:1–2. 10.1016/j.chom.2009.07.001 [DOI] [PubMed] [Google Scholar]
- 361.Colangeli R, Arcus VL, Cursons RT, Ruthe A, Karalus N, Coley K, Manning SD, Kim S, Marchiano E, Alland D. 2014. Whole genome sequencing of Mycobacterium tuberculosis reveals slow growth and low mutation rates during latent infections in humans. PLoS One 9:e91024. 10.1371/journal.pone.0091024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Dutta NK, Illei PB, Jain SK, Karakousis PC. 2014. Characterization of a novel necrotic granuloma model of latent tuberculosis infection and reactivation in mice. Am. J. Pathol. 184:2045–2055. 10.1016/j.ajpath.2014.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]