

Abstract
FipB, an essential virulence factor of Francisella tularensis, is a lipoprotein with two conserved domains that have similarity to disulfide bond formation A (DsbA) proteins and the amino-terminal dimerization domain of macrophage infectivity potentiator (Mip) proteins, which are proteins with peptidyl-prolyl cis/trans isomerase activity. This combination of conserved domains is unusual, so we further characterized the enzymatic activity and the importance of the Mip domain and lipid modification in virulence. Unlike typical DsbA proteins, which are oxidases, FipB exhibited both oxidase and isomerase activities. FipA, which also shares similarity with Mip proteins, potentiated the isomerase activity of FipB in an in vitro assay and within the bacteria, as measured by increased copper sensitivity. To determine the importance of the Mip domain and lipid modification of FipB, mutants producing FipB proteins that lacked either the Mip domain or the critical cysteine necessary for lipid modification were constructed. Both strains replicated within host cells and retained virulence in mice, though there was some attenuation. FipB formed surface-exposed dimers that were sensitive to dithiothreitol (DTT), dependent on the Mip domain and on at least one cysteine in the active site of the DsbA-like domain. However, these dimers were not essential for virulence, because the Mip deletion mutant, which failed to form dimers, was still able to replicate intracellularly and retained virulence in mice. Thus, the Mip domains of FipB and FipA impart additional isomerase functionality to FipB, but only the DsbA-like domain and oxidase activity are essential for its critical virulence functions.
INTRODUCTION
Francisella tularensis is the causative agent of tularemia, a vector-borne disease that can also be contracted by inhalation of aerosolized material (1). There are several subspecies of F. tularensis that vary in virulence. Francisella tularensis subsp. tularensis is the most virulent subspecies and the only subspecies classified by the Centers for Disease Control and Prevention as a tier 1 select agent (http://www.selectagents.gov/select%20agents%20and%20toxins%20list.html), which is the designation given to those biological agents that have the greatest potential to be misused as biological weapons.
F. tularensis is an intracellular pathogen that escapes the phagosome and replicates in the cytoplasm. FipB (Francisella infectivity potentiator B; encoded by the FTT1103 locus) is an essential virulence factor of F. tularensis subsp. tularensis that is required for phagosomal escape and intracellular replication (2). F. tularensis subsp. tularensis fipB mutants are also completely avirulent in mice; mice survive and show no symptoms after challenge with as many as 1010 CFU (2). The FipB protein consists of a combination of two conserved domains (http://www.ncbi.nlm.nih.gov/cdd/), the amino-terminal dimerization domain (FKBP_N) found in macrophage infectivity potentiator (Mip) proteins (3) and a disulfide bond formation A (DsbA)-like domain (4, 5). So far, this combination of domains has been found only in Francisella and in the related species Fangia hongkongensis (6). Mip proteins are typically lipoproteins that form homodimers via their alpha-helical amino-terminal FKBP_N domain and also contain a peptidylprolyl cis/trans isomerase (FKBP_C) domain (7, 8). The fipB gene is transcribed in an operon with fipA, which encodes a short polypeptide of 96 amino acids that also has similarity to FKBP_N, though it has only ∼37% identity to the FKBP_N-related region of FipB (2, 9).
In Gram-negative bacteria, DsbA is a periplasmic protein that catalyzes disulfide bond formation by donating its disulfide bond to a nascent protein. DsbB, an inner membrane protein, reoxidizes DsbA so that it can fold another nascent protein (10). In a separate pathway, DsbC, a periplasmic disulfide bond isomerase that is a dimer with reduced cysteines in its active state, refolds proteins that have been misfolded by DsbA (11). DsbC is restored to its reduced state by an inner membrane protein, DsbD. FipB is an outer-membrane-anchored lipoprotein that, although not unique, is not typical of most DsbA-like proteins (12). In a number of other pathogens, DsbA is key for the folding, and thus the assembly and function, of a variety of critical virulence factors (12). Therefore, we hypothesized that FipB was a variant DsbA protein that acts as an oxidoreductase and is required for the folding of critical virulence factors. However, in this paper, we demonstrate that FipB has both oxidase and isomerase functions. We provide evidence that FipA and the Mip domain potentiate the isomerase activity of FipB. FipB represents an unusual model for a bacterial protein disulfide isomerase, because the active form does not appear to be a homodimer. Isomerase activity was not essential for FipB's role in virulence, but we suggest that isomerase activity is important for repairing misfolded proteins that result from oxidative stress induced by redox-active metals, such as copper.
MATERIALS AND METHODS
Bacterial strains and media.
All bacterial strains and plasmids used in this study are listed in Table S1 in the supplemental material. Francisella strains were grown on cysteine-supplemented Mueller-Hinton agar (MHA/c) plates or in Trypticase soy broth supplemented with cysteine (TSB/c). Studies involving Schu S4 and derivatives of that strain were carried out in an approved biosafety level 3 laboratory. Escherichia coli strains were grown in Luria-Bertani (LB) broth or on LB agar plates with kanamycin (50 μg/ml), ampicillin (100 μg/ml), tetracycline (10 μg/ml), or chloramphenicol (35 μg/ml) as required.
Construction of mutant strains.
Site-directed mutation, plasmid construction, and in cis complementation by homologous integration into the blaB locus were performed as described previously (9). Disruption of blaB, which encodes beta-lactamase resistance, is otherwise phenotypically neutral and provides a convenient screening mechanism (13).
Intracellular-replication assay.
Human lung epithelial A549 cells were propagated in high-glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum. Human monocyte THP-1 cells were maintained in RPMI containing 10% fetal bovine serum and 3 mg/ml l-glutamine and differentiated into macrophage-like cells in 24-well plates containing 100 ng/ml phorbol 12-myristate 13-acetate (PMA) over 48 h. The cells were then washed with phosphate-buffered saline (PBS) and recovered in RPMI without PMA for 24 h before infection. Bacteria were added to cells in tissue culture wells in 24-well plates, centrifuged at 1,000 × g for 8 min, and incubated at 37°C, 5% CO2. After 1 h, the cells were washed, and then the medium was replaced with fresh medium containing 50 μg/ml gentamicin. At the indicated time points, the cells were washed and then lysed with 0.1% sodium deoxycholate in PBS for 5 min or in water for 10 min. The number of bacteria was determined by plating serial dilutions on MHA/c plates. These experiments were repeated at least three times. The Student two-tailed t test was used for statistical analysis.
Mouse model of tularemia.
All mouse studies were approved by the University of Virginia Animal Care and Use Committee. For intranasal inoculation, 9- to 12-week-old C57BL/6 mice (Jackson Laboratory) were anesthetized with a ketamine-HCl–xylazine mixture. Twenty microliters of bacteria were inoculated into the nares. The actual challenge dose was confirmed by parallel viable plate counting. The mice were monitored twice daily. Mice showing signs of irreversible mortality were humanely euthanized, and death was considered to occur within 24 h.
Subcellular localization of FipB.
Whole-cell lysates were prepared from an overnight bacterial culture by mixing equal amounts of an overnight culture and 2× SDS-PAGE loading buffer and then boiling for 5 min. Membrane fractions and culture supernatants were prepared from a 25-ml overnight culture. Bacteria were concentrated by centrifugation, and culture supernatants were filtered through a 0.22-μm syringe filter, followed by trichloroacetic acid (TCA) precipitation and resuspension in 1× SDS-PAGE sample buffer. Soluble and insoluble membrane fractions were prepared by suspending bacterial pellets in 1 volume of lysis buffer (0.75 M sucrose, 10 mM Tris, 20 mM EDTA), adding 300 μg/ml of lysozyme, and then incubating on ice for 2 h. After this incubation, 4 volumes of cold water was slowly added, with occasional rocking. The samples were incubated at room temperature (RT) for 30 min, and then protease inhibitor cocktail was added. The samples were subjected to 5 cycles of freezing and thawing in liquid nitrogen and at RT. Samples were then pelleted in a microcentrifuge at 12,000 × g for 30 min to remove unbroken bacteria. The supernatants were passed through a 0.22-μm syringe filter and then spun at 165,000 × g for 2 h at 4°C in an ultracentrifuge. Supernatants containing soluble proteins were TCA precipitated and then suspended in 1× SDS-PAGE buffer. Insoluble pellets containing outer and inner membrane proteins were washed with 10 mM HEPES and then suspended in 1× SDS-PAGE loading buffer. Samples were applied to SDS-PAGE for protein separation, and specific proteins were visualized by Western blotting. Antibodies included rabbit anti-FipB (2), rabbit anti-FupA (14), anti-IglB1 (BEI Resources), and anti-GroEL (Sigma-Aldrich).
Proteinase K treatment.
Bacteria grown on MHA/c plates were suspended in PBS containing 5 mM Mg2+ to an optical density at 595 nm (OD595) of 1 and incubated with 10 μg/ml proteinase K at 37°C for 30 min. Proteinase K digestion was quenched by the addition of 2 mM EGTA. The bacterial pellets were washed with protease inhibitor cocktail (Sigma-Aldrich), resuspended in 1× SDS-PAGE buffer, and then boiled for 5 min before loading onto SDS-PAGE for Western blot detection.
Motility assay.
Overnight cultures of each strain were adjusted to the same OD595. Bacteria were stabbed into a soft agar (0.2%) plate with LB medium containing 35 μg/ml chloramphenicol to maintain plasmid selection. After incubation at RT for 18 h, the motility from the inoculation site was measured. An E. coli (JCB571) dsbA mutant (kindly provided by J. C. Bardwell) was used as the parent strain (5). pACYC vectors containing the fipA, fipB, or fipAB gene were introduced into JCB571 by electroporation.
PDI detector assay.
To test for isomerase activity, PCR products corresponding to the fipA, fipB, or fipAB gene were inserted into the pACYC184 plasmid in the tetracycline resistance gene and then introduced into RGP665 (a generous gift from J. C. Bardwell) by electroporation. RGP665 is a ΔdsbC E. coli mutant that carries a protein disulfide isomerase (PDI) detector plasmid, which contains an engineered β-lactamase with two nonconsecutive cysteine substitutions (S81C and T108C) (15). Incorrect disulfide bond formation leads to increased sensitivity to ampicillin. In the presence of a protein disulfide isomerase, the β-lactamase can be refolded into a functional conformation and therefore confers increased ampicillin resistance on the host strain. Accordingly, bacterial cultures were grown in LB medium to an OD595 of 1 with appropriate antibiotics added. The bacteria were then diluted 1:100 and grown at 37°C in a shaker for 3 h with or without 2.5 mg/ml ampicillin. The cells were then serially diluted and plated on LB plates with appropriate antibiotics for plasmid maintenance, but without ampicillin.
Detection of free sulfhydryls.
Overnight cultures were pelleted by centrifugation and then suspended in 10% TCA at 4°C overnight. The precipitates were pelleted and washed in ice-cold acetone, dissolved in buffer (1 M Tris, pH 8.0, 0.1% SDS, 1 mM EDTA) containing 20 mM methoxypolyethylene glycol-maleimide (MAL-PEG) (Sigma-Aldrich), and incubated at RT for 30 min in the dark. After incubation, TCA was added to the samples at a 10% final concentration, and incubated on ice for 30 min. Protein pellets were washed twice with ice-cold acetone. The MAL-PEG-labeled samples were suspended in SDS-PAGE buffer containing dithiothreitol (DTT) for SDS-PAGE and Western blot analysis. Reduced controls were prepared by first incubating samples in buffer (1 M Tris, pH 8.0, 0.1% SDS, 1 mM EDTA) that contained 10 mM DTT at RT for 30 min, followed by TCA (10%) precipitation and acetone washing before MAL-PEG treatment. Untreated negative controls were also prepared by the same procedures but without DTT or MAL-PEG in the buffer. Western blot films were scanned, and bands were quantitatively analyzed using ImageQuant software. FipB bands on Western blots were visualized with polyclonal anti-FipB (2). Rabbit anti-GroEL serum (Sigma-Aldrich) or guinea pig anti-FupA (a gift from Girija Ramakrishnan) (14) served as the loading control.
FipB and FipA protein purification.
The fipB gene, minus the signal peptide and lipobox, was synthesized with an E. coli codon bias and ligated into the pHis-parallel 1 vector with a 6× His tag at the amino terminus (16). The fipA gene was amplified by PCR using Schu S4 DNA as the template and then inserted into plasmid pET-20b (Novagen) in frame with the vector-provided 6× His tag. Each clone was verified by DNA sequencing. Proteins were purified from lysates of induced cultures using Talon beads (Clontech). The beads were washed with wash buffer (50 mM Na2HPO4, 500 mM NaCl, containing 0.5 ml of 10-mg/ml stock aprotinin and 0.5 ml of 10-mg/ml stock leupeptin, pH 8.0), followed by a wash with 10 ml of the wash buffer, which contained 10 mM imidazole. The proteins were eluted with 500 mM imidazole in a 50 mM Na2HPO4, 500 mM NaCl buffer at pH 6.8 and then dialyzed with PBS. Purification was analyzed by SDS-PAGE, followed by silver stain (Bio-Rad) to assess purity. Proteins were aliquoted and stored at −20°C. The protein concentration was assessed using the BCA protein kit (Pierce) with bovine serum albumin (BSA) as the standard.
Assay for protein disulfide isomerase activity.
Isomerization activity was measured using the TEM1 β-lactamase assay described by Kpadeh et al. (17). Briefly, spent culture supernatants were collected from wild-type and ΔdsbC E. coli strains containing the engineered TEM1 β-lactamase and concentrated 24-fold on a Vivaspin 2 spin column with a molecular weight cutoff of 10,000 (Sartorius Stedium Biotech). Samples were examined by SDS-PAGE, followed by staining with Gelcode Blue (Thermo Scientific) to determine the relative concentration of β-lactamase in each fraction. For the enzyme reaction, ΔdsbC supernatants were added to 200 μM FipA and/or FipB. The tubes were incubated for 15 min at 37°C. After incubation, samples were transferred to a 96-well plate, and β-lactamase activity was measured upon the addition of 10 μl nitrocefin (500-μg/ml stock solution). Cleavage of the substrate was measured every minute at a wavelength of 490 nm for 30 min at 30°C on a Synergy H4 Hybrid reader (Biotek). Reaction mixtures were tested in triplicate.
Copper sensitivity assay.
Overnight cultures of F. tularensis or Francisella novicida were inoculated into 2 ml of TSB/c with or without CuCl2 and incubated at 37°C with 200 rpm shaking. Growth of the bacteria was monitored by measuring the culture OD595 at 24 h and 48 h.
RESULTS
FipB has both oxidoreductase and isomerase activities in E. coli.
We, and others have found that purified recombinant FipB has oxidoreductase activity in vitro using the insulin precipitation assay (18). To demonstrate that FipB is capable of oxidoreductase activity in vivo, we complemented an E. coli dsbA mutant with fipB and showed that it could restore motility, which is a DsbA-dependent activity in E. coli (Fig. 1). In this experiment, the fipB gene was introduced on the low-copy-number plasmid pACYC184 into E. coli ΔdsbA with or without the fipA gene. Motility was restored, though not to wild-type levels (Fig. 1). The presence of the fipA gene in addition to fipB did not influence the degree of motility. The growth of the strain expressing fipA alone had slight enhancement of growth compared to the vector control. We tested the growth of each of these strains in culture and found that the fipA strain grew slightly better than the vector control (see Fig. S1 in the supplemental material). Why this strain has a growth advantage is unclear.
FIG 1.

fipB complements E. coli dsbA. (A) ΔdsbA strains expressing fipA, fipB, or fipAB or carrying an empty vector, pACYC187, or wild-type (WT) bacteria with pACYC187 were stabbed into soft agar plates and grown for 18 h at RT. (B) Comparison of halo diameters of complemented strains, as well as wild-type E. coli carrying the empty vector. Strains expressing fipB or fipAB restored motility, while complementation with fipA was similar to the vector-only control. The error bars indicate standard deviations.
Homodimerization of DsbC, the E. coli protein disulfide isomerase, is a requirement for isomerase activity (19). Because FipB has a Mip-like dimerization domain, we hypothesized that FipB might have isomerase activity. To determine whether FipB has any isomerase activity, we used an E. coli-based PDI detector system developed by Ren and Bardwell (15). In this system, a plasmid carrying a bla gene that has been engineered to encode two extra cysteines is introduced into a dsbC mutant. In the absence of dsbC, a higher portion of the β-lactamase that is produced is misfolded, and thus, these strains have increased sensitivity to ampicillin. The introduction of a functional dsbC gene increases resistance to ampicillin. Using the PDI detector strain, we were able to demonstrate that FipB has isomerase activity in E. coli, but only when FipA was present (Fig. 2A). These results suggest that FipA influences the enzymatic activity of FipB.
FIG 2.
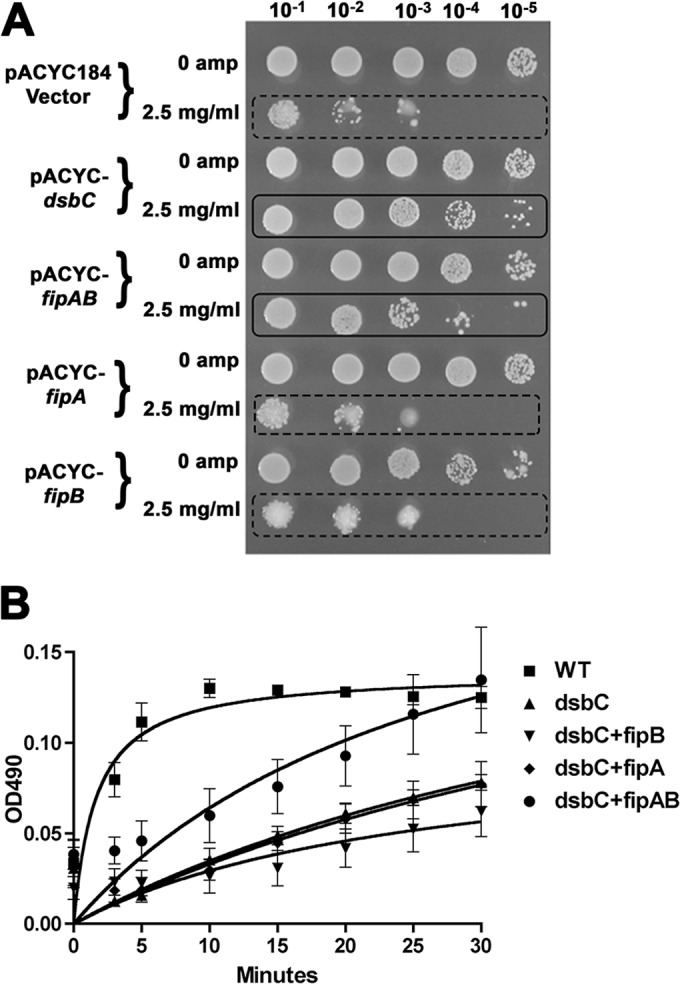
FipB demonstrates isomerase activity in E. coli that is dependent on fipA. (A) Cultures of a dsbC strain carrying the PDI detector plasmid and the indicated fip genes were grown in the presence of ampicillin (amp); the next day, they were serially diluted and spotted on LB plates without ampicillin. The solid boxes highlight cultures that were considered positive for isomerase activity, and the dashed boxes indicate cultures that were considered negative. (B) Recombinant FipB with or without FipA was incubated with supernatant from the dsbC strain used in panel A, which contains the PDI detector plasmid and produces the engineered, misfolded β-lactamase. β-Lactamase activity was measured upon the addition of nitrocefin. Cleavage of the substrate was measured every minute at a wavelength of 490 nm for 30 min at 30°C. Reaction mixtures were tested in triplicate. A supernatant from the wild-type PDI detector strain was used as a positive control. Using 2-way analysis of variance (ANOVA), FipAB was statistically significantly different from FipB, starting at 10 min; FipA and FipB were not statistically significantly different. The error bars indicate standard deviations.
An in vitro assay of isomerase activity was used to test for isomerase activity of recombinant FipB and the influence of the addition of FipA (17). Only FipB plus FipA demonstrated isomerase activity over background levels (Fig. 2B), further supporting isomerase activity of FipB and a FipA requirement.
FipA influences the oxidation state of FipB.
As demonstrated in an E. coli system, DsbC is a homodimer with two CXXC motifs that are in a reduced state (11). When DsbC encounters a misfolded protein, it first reduces the protein and then refolds it into its proper conformation. DsbC is returned to a reduced state by interaction with the inner membrane protein DsbD, which transfers reducing equivalents from the cytoplasm. The F. tularensis species lacks a recognizable DsbD or orthologs of other proteins that play similar roles (20). Since FipA was required for FipB isomerase activity in E. coli, we predicted that FipA would influence the oxidation state of FipB. We used MAL-PEG, a thioreactive compound that forms a covalent bond with free sulfhydryls and shifts the molecular mass of the protein by 5 kDa, to detect the oxidation states of FipB in the presence or absence of FipA. Cultures of wild-type and ΔfipA strains were treated with MAL-PEG in the presence or absence of DTT (Fig. 3). The percentage of FipB labeled by MAL-PEG for each strain was calculated by densitometry scans of the Western blots. In the samples treated with DTT and MAL-PEG, the migration of FipB was similarly increased to an apparent size of 75 to 100 kDa. Since FipB has two cysteines, one would have predicted only an increase of 10 kDa, but it appears that MAL-PEG significantly alters the migration of this protein on SDS-PAGE. Aberrant migration on SDS-PAGE after MAL-PEG modification has been observed by others (21). The addition of MAL-PEG without DTT produced three bands representing oxidized FipB, reduced FipB with one MAL-PEG molecule, and reduced FipB with two MAL-PEG molecules. These results indicated that in wild-type Schu S4, on average, 58% of FipB is in an oxidized form. Most of the reduced FipB was modified by only one MAL-PEG molecule. This was not too surprising, because based on the structure of E. coli DsbA, the second cysteine is buried in the molecule and not as accessible (22). As predicted, there was significantly more oxidized FipB in the ΔfipA strain than in the wild type (70%; P = 0.038) (Fig. 2B), supporting our premise that FipA helps to regulate the oxidation state of FipB.
FIG 3.
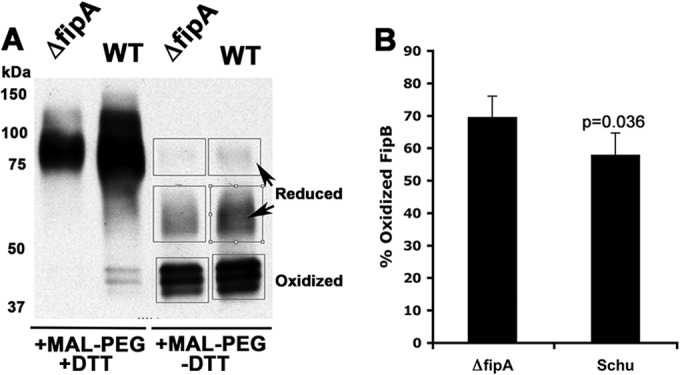
fipA influences the oxidation state of FipB. Overnight cultures of each strain were TCA precipitated and then treated with DTT and MAL-PEG, as indicated. (A) Western blot of labeled cultures with anti-FipB antibody. The boxed bands were subjected to densitometry scans. (B) Percentages of total FipB (the three boxed bands in panel A) that were present in the oxidized state as determined by densitometry scans. The data are combined from three separate labeling experiments. The P value was determined using Student's t test. The error bars indicate standard deviations.
FipB has isomerase activity in Francisella, as measured by copper sensitivity.
In E. coli, dsbC is required to refold proteins misfolded by copper oxidation (23). Therefore, to demonstrate that FipB has isomerase activity in Francisella, the copper sensitivities of fipB mutants of Schu S4 were examined. The growth of wild-type Schu S4 was unimpaired in 40 μM copper (Fig. 4). However, when fipB or fipAB was deleted, these strains were significantly more copper sensitive than wild-type bacteria (Fig. 4), supporting FipB's role as an isomerase in Francisella. The fipA mutant was also more sensitive than the wild type to copper, which is consistent with our in vitro and E. coli data that implied a role for FipA in potentiating isomerase activity (Fig. 4). Both wild-type and ΔfipA strains that overexpressed fipA were also more sensitive to copper, suggesting a dominant-negative effect of FipA. Deletion of the Mip domain led to increased copper sensitivity, indicating that the domain is also critical for isomerase activity. F. novicida strain U112 and a fipB mutant had similar copper sensitivity profiles (see Fig. S2 in the supplemental material).
FIG 4.
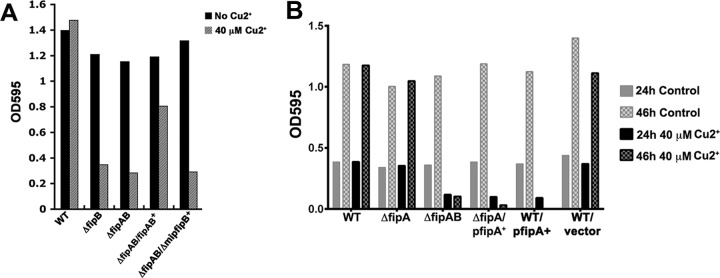
(A) A Schu S4 fipB mutant is more sensitive than the wild type to CuCl2. Bacteria were grown at 37°C for 21 to 24 h with shaking in TSB/c supplemented with 40 μM CuCl2. The ΔfipAB mutant was complemented in cis (indicated with a plus) with wild-type fipAB or Δmip fipB. (B) A Schu S4 fipA mutant is more sensitive to copper than the wild type. The indicated strains were grown at 37°C with shaking in TSB/c supplemented or not with 40 μM CuCl2 at the indicated time points. The WT and the ΔfipA mutants were complemented with fipA+ in trans, indicated by “p.”
The amino-terminal Mip domain and the lipobox motif are not essential for virulence.
As a potential DsbA-like protein, FipB has two atypical features: a lipobox motif, which specifies the addition of amino-terminal fatty acyl chains, and an FKBP_N domain characteristic of Mip proteins. To determine the importance of these domains in the essential virulence function of FipB, mutants that produced FipB with an altered lipobox (FipB-C22A) or deleted Mip domain (FipB::AS Δaa27–117, where FipB::AS indicates FipB with the insertion of two amino acids, alanine and serine, and Δaa27–117 indicates the deletion of amino acids 27 to 117) were constructed (Fig. 5) by integrating the engineered genes into the blaB locus in a ΔfipAB mutant of the Schu S4 strain of F. tularensis subsp. tularensis, along with wild-type fipA and the native promoter. To construct the Mip domain deletion mutant, two additional amino acids, alanine and serine, were introduced after the lipobox sequence. The addition of these two amino acids to an otherwise wild-type FipB did not negatively affect intracellular growth (Fig. 6A and B) but did, surprisingly, impact in vivo virulence (Table 1) (see Discussion). Expression of these genes was verified by Western blotting (Fig. 5B and C). Under nonreducing conditions, FipB formed higher-molecular-mass complexes. In Schu S4, FipB migrates as three isoforms, due at least in part to glycosylation differences (24). The lipobox mutant was poorly expressed, and protein was visible only after an extended exposure time (Fig. 5C and 6D).
FIG 5.

Construction and expression of fipB deletion and point mutants. (A) Diagram showing the domain structure of each mutant. Each construct contains the signal peptide (light-gray box), followed by the conserved lipobox motif (LVAC). In the FipB-C22A construct, the conserved cysteine (C) has been changed to an alanine (A). The black boxes indicate the insertion of 2 amino acids, alanine and serine (AS), after amino acid 26 as a result of the insertion of an NheI restriction site that was used to create the constructs. The endpoints of the deletions are indicated in the construct name; the numbering includes the amino-terminal signal peptide. Each mutant gene, along with wild-type fipA and the native promoter, was integrated into a ΔfipAB mutant of Schu S4 by homologous recombination at the blaB locus. (B) Western blot of a nonreducing SDS-PAGE showing the expression of the Mip deletion construct (FipB Δ27–117). Whole-cell lysates were separated by SDS-PAGE and then transferred to a nylon membrane. The membranes were incubated with both anti-FipB and anti-GroEL antibodies (loading control). Note that only the wild-type protein forms higher-molecular-mass complexes. (C) Western blot demonstrating the expression of FipB-C22A in a whole-cell lysate. Membranes were incubated with both anti-FipB and anti-FupA (used as a loading control) antibodies.
FIG 6.

The lipobox mutant (FipB-C22A) is competent for intracellular replication. (A) A549 cells were infected at a multiplicity of infection (MOI) of 100:1 and then lysed at the indicated times postinfection. The lysates were diluted and plated to enumerate intracellular bacteria. The data are representative of at least three independent experiments, each using an average of three independent wells. The strains used were Schu S4 (⧫) and mutants expressing ΔfipAB (□), FipB-C22A (△), or FipB::26AS (○). The error bars indicate standard deviations. (B) The lipobox mutant, FipB-C22A, does not associate with the membrane fraction. Bacteria were lysed and fractionated as described in Materials and Methods. Western blots of subcellular fractions of wild-type Schu S4 and strain BJM1100, which produces the lipobox mutant FipB-C22A, were prepared as described in Materials and Methods and incubated with the indicated antibodies. FupA is an outer membrane protein involved in ferrous iron transport, IglB is part of the F. tularensis subsp. tularensis type VI secretion system, and GroEL is a chaperone protein. FipB-C22A was faintly visible in total lysates and the soluble fraction, which was 25× concentrated. supe, supernatant. (C) PMA-differentiated THP-1 cells were infected with the indicated strains at an MOI of 50:1 and then lysed at 2 and 24 h postinfection. Negative controls included strains expressing FipB and FipB-C22A with mutated active sites (AMYA) and the ΔfipAB mutant. All mutated fipB genes were integrated into the bla site in the ΔfipAB mutant. The FipBc strain has the wild-type fipB gene integrated into the bla site of a ΔfipAB mutant. The data are representative of at least three independent experiments, each using an average of three independent wells. The strains indicated by asterisks were significantly different from wild-type Schu S4 (P < 0.0001). (D) Western blots depicting FipB in input bacteria and bacteria recovered from host cells at 24 h from the experiment depicted in panel C. Anti-FupA antibodies were used as a loading control.
TABLE 1.
Lipobox and Mip deletion mutants retain virulence
| Strain (genotype) | Intranasal infection dose(s)a (avg CFU/mouse) | Days to death postinfectionb | No. of survivors/total no. of mice (%) |
|---|---|---|---|
| Schu S4 | 12 and 24 | 5, 5, 5, 4, 4, 5 | 0/6 (0) |
| BJM1103 (fipB AMYA) | 24 and 45 | No deaths | 12/12 (100) |
| BJM1076 (fipB CMYC) | 10 and 100 | 6, 6, 7, 6, 6, 6 | 0/6 (0) |
| BJM1100 (fipB-C22A) | 31 | 5, 5, 5, 5 | 2/6 |
| 71 | 5, 5 | 4/6 | |
| 210 | 5, 5, 5 | 0/3 | |
| 21 | 6, 6, 8 | 0/3 | |
| 4 | 6, 9, 10 | 0/3 | |
| All tested doses | 6/21 (29) | ||
| BJM1099 (fipB::26AS) | 47 and 65 | 6, 6, 6, 6c | 8/12 (67) |
| BJM1111 (fipB::26AS Δaa27–117) | 37 | 5, 5, 5, 5, 5, 5 | 0/6 |
| 79 | 5, 5, 5, 5 | 2/6 | |
| All tested doses | 2/12 (17) |
Two values represent two independent trials with the indicated doses.
The number of days after challenge that mice showed the first signs of irreversible mortality and were euthanized. Survivors were all monitored for >21 days.
Two deaths occurred in each trial.
The intracellular-growth phenotypes of the lipobox mutant (FipB-C22A) and the Mip deletion mutant (FipB::AS Δaa27–117) in the human lung epithelial cell line A549 and in the human macrophage-like cell line THP-1 were determined using a gentamicin protection assay (Fig. 6 and 7). The lipobox mutant (FipB-C22A) replicated very similarly to wild-type bacteria in both cell lines, despite very low levels of detectable protein, indicating that the amino-terminal cysteine residue, and thus the fatty acyl modification, were not required for intracellular replication. The Mip deletion mutant was also found to be competent for intracellular replication (Fig. 7), though replication was reduced compared to wild-type levels. To rule out the possibility of contamination with wild-type bacteria restoring growth, the FipB proteins from input bacteria and from bacteria recovered from cells infected for 24 h were compared using Western blots (Fig. 6D and 7C). Using the FupA protein to normalize the values, the amounts of FipB-C22A protein in the input and recovered bacteria were 16% and 5% of the wild-type level, respectively. The FipB detected in cells infected with the Mip deletion mutant was the same size as the FipB with Mip deleted in the input bacteria. These Western blots indicated that contaminating bacteria were not responsible for the observed wild-type or near-wild-type levels of intracellular bacterial growth in the lipobox and Mip deletion mutant strains.
FIG 7.

The Mip domain deletion mutant is competent for intracellular replication. (A) A549 cells were infected with the indicated strains and then lysed at the indicated times postinfection. The lysates were diluted and plated to enumerate intracellular bacteria. The data represent an average of results from three independent wells and are representative of at least three independent experiments. The strains and MOIs used were as follows: Schu S4 (⧫), 130:1; ΔfipAB mutant (□), 133:1; FipB::AS Δaa27–117 mutant (Δmip) (●), 197:1; mutant with ΔfipAB in cis complemented with fipAB::AS26 (◊), 232:1. The MOIs were verified by plate counts. The error bars indicate standard deviations. (B) Differentiated THP-1 cells were infected with the indicated strains and then lysed at 2 or 24 h postinfection. The MOIs used were as follows: Schu S4, 39:1; ΔfipAB mutant, 112:1; FipB::AS Δaa27–117 mutant (Δmip), 104:1; mutant with ΔfipAB in cis complemented with fipAB::AS26, 69:1. The data represent an average of results from three independent wells and are representative of at least three independent experiments. (C) Western blots depicting FipB in input bacteria and bacteria recovered from host cells at 24 h from the experiment depicted in panel B. Anti-FupA antibodies were used as a loading control.
Both the lipobox and the Mip deletion mutants retained virulence in mice (Table 1). Although 6 out of 21 mice (29%) survived infection with the lipobox mutant (FipB-C22A), this level of survival was not statistically different from that of mice infected with the wild type or the in cis wild-type complement, where 100% of the mice died (Fisher's exact test). Only 2 out of 12 mice (17%) survived infection with the Mip deletion mutant (fipB::26AS Δaa27–117). However, infection with the strain that produced a FipB protein with two extra amino acids, AS, was not as virulent as infection with wild-type Schu S4 (67% survived). This result was surprising, because the FipB::AS strain did not exhibit any intracellular growth defects (Fig. 7). The fipB gene in this strain was resequenced to confirm that the AS insertion was still present. Western blots of bacteria recovered from the organs of infected mice ruled out the potential for contamination with wild-type bacteria in mice infected with FipB-C22A and FipB::AS ΔMip strains (see Fig. S3 in the supplemental material). All of the mice infected with a mutant that expressed a fipB gene with a mutation at the active site (AMYA) survived infection. A fipAB deletion mutant, as well as other CMYC mutants, have been previously shown to be avirulent at greater than 106 CFU (2, 9).
Cell fractionation demonstrated that the FipB-C22A protein was found in the soluble fraction, indicating that association of FipB with the membrane fraction was not essential for intracellular replication and virulence (Fig. 6B).
FipB forms a surface-exposed dimer that is sensitive to reduction.
In other bacteria, Mip proteins are homodimers mediated by the amino-terminal domain (FKBP_N) (3). Therefore, we predicted that FipB would also dimerize via this domain. The higher-molecular-mass complexes that were observed on Western blots with wild-type protein were not detectable when the Mip domain was deleted (Fig. 5B). However, these complexes were also sensitive to reducing conditions. FipB has three cysteine residues, one in the lipobox, which is acylated, and two in the active site, which in DsbA proteins is characterized by a CXXC motif; in FipB, this motif is CMYC. Mutations in the CMYC motif were constructed with alanine residues substituted for each cysteine. Wild-type FipB formed a higher-molecular-mass complex under nonreducing conditions (Fig. 8A). CMYA, and AMYC mutants of FipB also formed these complexes in enhanced portions, but an AMYA mutant did not. Since there is only one available Cys in the CMYA and AMYC mutants, these results also indicate that these ∼125-kDa complexes are dimers, though the apparent molecular mass is more than twice the monomer size of ∼47 kDa. The size of this complex is likely due to aberrant migration of the protein, because purified recombinant FipB also forms higher-molecular-mass complexes on nonreducing SDS-PAGE that migrate at a size that is larger than twice the monomer size (data not shown). The FipA protein appears to play a role in dimer formation, because although the deletion of fipA did not affect dimer formation, overexpression of fipA prevented the formation of these complexes (Fig. 8C). However, overexpression of fipA by episomal complementation of a ΔfipA mutant restored the intracellular-growth defect of the ΔfipA mutant, suggesting that this dimer form of FipB is not critical for intracellular growth (Fig. 9).
FIG 8.

FipB forms a dimer via cysteines in the enzymatic active site that is sensitive to proteinase K treatment and inhibited by overexpression of fipA. (A) Immunoblots of strains complemented in cis with fipB genes with mutations in the active-site cysteines. Bacteria were lysed in the presence or absence of DTT. The blots were incubated with anti-FipB and anti-GroEL antibodies. (B) Bacterial pellets were resuspended in PBS (top) or 10 μg/ml of proteinase K (bottom) and incubated for 30 min at 37°C. After incubation, samples were boiled in 2× nonreducing sample buffer, separated by SDS-PAGE, transferred to a nylon membrane, and then incubated with both anti-FipB and anti-FupA antibodies. (C) Immunoblotting was first performed with anti-FipB antibody, and immunoblots were then stripped and incubated with both anti-FupA and anti-FipA antibodies.
FIG 9.
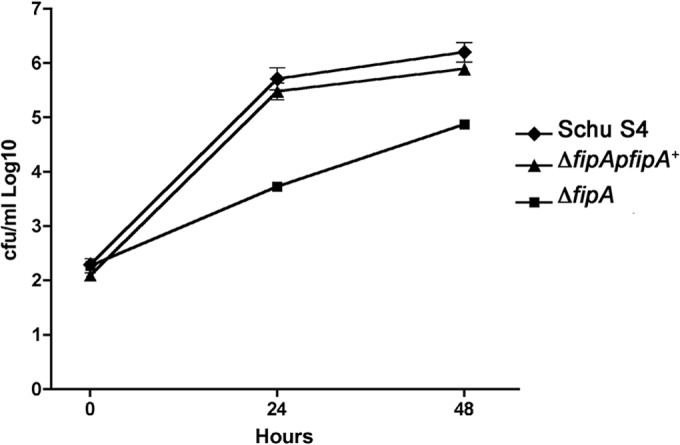
Complementation of the ΔfipA mutant restores the intracellular-growth defect. A549 cells were infected at an MOI of 100:1, as described in Materials and Methods, and then lysed at the indicated times postinfection to determine CFU/ml. The error bars indicate standard deviations.
Several members of the Mip family have been reported to be surface exposed (7, 8). We observed, using several different techniques (immunofluorescence, biotin labeling, and immunoprecipitation [data not shown]), that at least some portion of FipB was surface exposed. We tested the surface exposure of FipB using proteinase K digestion and found that only the higher-molecular-mass complexes of FipB were sensitive to digestion (Fig. 8B). Whether surface-exposed FipB has any function is unclear, but since FipB dimers were not detected in the Mip deletion mutant or the fipA overexpression strain and these strains were still able to replicate intracellularly, it appears that this dimer complex and surface exposure do not play essential roles in FipB-mediated virulence functions.
DISCUSSION
In this paper, we have demonstrated that the DsbA-related protein FipB is a bifunctional protein that acts as both an oxidoreductase and a protein disulfide isomerase. In many bacteria, these functions are carried out by separate pathways (10). The DsbA/DsbB pathway forms disulfide bonds in nascent proteins, and the DsbC/DsbD pathway refolds misfolded proteins by first reducing incorrect disulfide bonds and then reforming the correct bonds. For DsbC to function as an isomerase, it needs to be in a reduced state. Dimerization of DsbC protects it from being oxidized by DsbB. The isomerase activity was somewhat unexpected, because FipB does not appear to form a functional dimer. Using protease sensitivity, we were able to detect only surface-exposed dimers of FipB that formed through the cysteines in the active site. The periplasmic exposed forms of FipB appear to be monomers, though we cannot rule out the possibility that transient dimers form. In E. coli, DsbC is returned to its reduced state via an inner membrane protein, DsbD, that provides reducing equivalents it receives from cytoplasmic thioredoxin (25). Bioinformatic searches of F. tularensis have not revealed any DsbD ortholog. There are other related families of proteins that can also provide reducing equivalents, such as CcdA and ScsB, but there are no orthologs of these protein families either (20). Interestingly, there is a strain of a Francisella sp., Tx077308, which contains a DsbD ortholog (F7308_1227) that is lacking in other sequenced strains and species, suggesting that this gene may have been a later addition to the strain's genome. Strain Tx077308 belongs to a clade that is quite distant from the F. tularensis subspecies (26). However, the strain encodes both FipA and FipB proteins that are 79% and 73% identical, respectively, to the Schu S4 FipA and FipB proteins. Regardless, Francisella must have some other mechanism for maintaining FipB in a reduced state. We found that the short peptide FipA, which is cotranscribed with fipB, influenced the proportion of FipB in the oxidized state. When fipA was deleted, there was an increased amount of oxidized FipB. In E. coli, one function of the dimerization domain of DsbC is to prevent interaction with and oxidation by DsbB; when the dimerization domain of DsbC is mutated, DsbC can complement a dsbA mutant (27). These results suggest that F. tularensis subsp. tularensis FipA and FipB directly interact and prevent oxidation by DsbB. Our model is that FipA helps to maintain FipB in a reduced state through interaction with the Mip domain of FipB. The in vitro isomerase assay suggests that there is a direct FipA-FipB interaction (Fig. 2B), but we have been unable to demonstrate this by coimmunoprecipitation, pulldowns, or bacterial two-hybrid interaction (28). We were able to detect FipA only when it was overexpressed (Fig. 7B), so the protein is either present at levels below our limits of detection or unstable, which may limit our ability to detect a direct interaction. The dominant-negative effects of fipA overexpression on copper sensitivity suggest that FipA may need to dissociate for isomerase activity and interaction with substrates to occur. Overexpression of fipA, however, did not affect intracellular growth (Fig. 9). One potential explanation for this paradox is that in the reducing environment of the host cytoplasm, the dominant-negative effects of FipA have less impact or that only a low level of isomerase activity is required for late-stage bacterial growth. Thus far, Francisella is one of only a few bacteria that lack a recognizable member of the DsbD superfamily, which provides the reducing equivalents to periplasmic protein disulfide isomerase. The chimeric nature of FipB and its potential interaction with FipA may be an adaptation to the absence of DsbD. Regardless, F. tularensis is an unusual paradigm for disulfide bond formation and isomerase activity. FipB is not the only bifunctional DsbA-related protein. The DsbA2 protein of Legionella pneumophila also has both oxidase and protein disulfide isomerase activities (17). DsbA2 has an amino-terminal domain that functions in dimer formation, but the domain is not related to the FKBP_N domain of Mip proteins. A bifunctional DsbA has also been engineered in E. coli by fusing the FKBP_N domain to DsbA (19). This chimeric protein formed dimers, had both oxidase and isomerase activities, and conferred modest copper resistance.
In Francisella tularensis subsp. holarctica, using the thermal aggregation of citrate synthase assay, Schmidt et al. have shown that FipB, which they refer to as DsbA, has chaperone activity in vitro (29). This result suggests a third function for FipB. However, not all of their data were consistent with ours. They found that deletion of the Mip domain in FipB rendered the strain avirulent and that a recombinant protein lacking the Mip domain was not able to reduce insulin. In contrast, our FipB strain with Mip deleted exhibited only partial attenuation in intracellular growth. The deletion of these genes in both subspecies results in a highly attenuated or avirulent phenotype (2, 29), but there are sequence differences between the F. tularensis subsp. tularensis FipB protein and the F. tularensis subsp. holarctica ortholog, DsbA (30), including a Thr-to-Ala change in a tripeptide that has been found to be critical for the resolution of substrate binding in E. coli DsbA (31). However, the most likely explanation is the difference in the extent of the deletion; our mutant lacked amino acids 27 to 117, while Schmidt et al. deleted amino acids 46 to 143. A BLAST search with known DsbA and Mip proteins indicates that DsbA similarity begins around amino acid 124 and that Mip similarity ends around amino acid 127. These authors have shown that their Mip deletion protein lacks oxidoreductase activity, so we suggest that the extent of their deletion disrupted the functions of both domains. When constructing our Mip deletion mutant, 2 amino acids (AS) were inserted as the result of the insertion of a restriction site. We tested the phenotype of a strain that expressed full-length FipB that, except for the insertion of AS, was otherwise wild type. Intracellular replication of this strain was similar to that of wild-type strains (Fig. 7); however, in vivo, the strain exhibited a higher level of attenuation than the Mip deletion strain (Table 1). This result indicated that the insertion of AS was not neutral, but the deletion of the Mip domain somewhat mitigated this effect. It also indicated that in Francisella, the intracellular-replication phenotype might not always indicate the level of in vivo virulence. The lack of correlation between intracellular growth and in vivo virulence has been observed for several other Francisella gene mutants, in particular amnK, pdpD, iglG, and iglI (32, 33).
In a number of bacterial pathogens, DsbA proteins have been shown to be required for the function of critical virulence factors, including the assembly of type III secretion machinery, flagella, pili, and toxins (12). We speculate that FipB is required for the folding or function of key virulence factors; however, specific substrates that reflect FipB's essential role in virulence have yet to be characterized. In the live vaccine strain (LVS), several proteins that increased in abundance in a fipB (dsbA) mutant were identified using a proteomic-labeling technique, but their link to virulence has not been established (18). In E. coli, where DsbA and DsbC have been the best studied, a clear phenotype for DsbA is evident, including loss of motility and decreased virulence (34–36), and multiple substrates have been identified (5, 31). Initially, although DsbC was characterized as having protein disulfide isomerase activity, few substrates could be identified, and dsbC mutants did not have a clear phenotype (37). However, Hiniker et al. have shown that DsbC is required for copper resistance (23). They found that copper catalyzed the formation of incorrect disulfide bonds that then could be repaired to the correct state by DsbC and proposed that the primary role of DsbC is to refold misfolded proteins formed during oxidative stress. Consistent with our isomerase assay results, we found that FipB, the Mip domain of FipB, and FipA were all required for copper resistance. These results, along with our in vitro data, support the role of FipB as a protein disulfide isomerase in F. tularensis subsp. tularensis and suggest that the isomerase activity of FipB may be more critical during oxidative stress and perhaps more critical for survival in the vector or environmental reservoirs. Combining its oxidase and isomerase activities in one protein, and possibly eliminating the need for DsbD, may contribute to its success as a low-dose pathogen that can survive in many hosts.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Jim Bardwell, Zegbeh Kpadeh, Karl Klose, Paul Hoffman, Carol Gilchrist, and Girija Ramakrishnan for strains and helpful discussions.
This work was supported by NIH/NIAID R56AI091746 to B.J.M.
Footnotes
Published ahead of print 4 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01359-13.
REFERENCES
- 1.Oyston PC, Sjostedt A, Titball RW. 2004. Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat. Rev. Microbiol. 2:967–978. 10.1038/nrmicro1045 [DOI] [PubMed] [Google Scholar]
- 2.Qin A, Scott DW, Thompson JA, Mann BJ. 2009. Identification of an essential Francisella tularensis subsp. tularensis virulence factor. Infect. Immun. 77:152–161. 10.1128/IAI.01113-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kohler R, Fanghanel J, Konig B, Luneberg E, Frosch M, Rahfeld JU, Hilgenfeld R, Fischer G, Hacker J, Steinert M. 2003. Biochemical and functional analyses of the Mip protein: influence of the N-terminal half and of peptidylprolyl isomerase activity on the virulence of Legionella pneumophila. Infect. Immun. 71:4389–4397. 10.1128/IAI.71.8.4389-4397.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bessette PH, Qiu J, Bardwell JC, Swartz JR, Georgiou G. 2001. Effect of sequences of the active-site dipeptides of DsbA and DsbC on in vivo folding of multidisulfide proteins in Escherichia coli. J. Bacteriol. 183:980–988. 10.1128/JB.183.3.980-988.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bardwell JC, McGovern K, Beckwith J. 1991. Identification of a protein required for disulfide bond formation in vivo. Cell 67:581–589. 10.1016/0092-8674(91)90532-4 [DOI] [PubMed] [Google Scholar]
- 6.Lau KW, Ren J, Fung MC, Woo PC, Yuen KY, Chan KK, Qian PY, Wong PK, Wu M. 2007. Fangia hongkongensis gen. nov., sp. nov., a novel gammaproteobacterium of the order Thiotrichales isolated from coastal seawater of Hong Kong. Int. J. Syst. Evol. Microbiol. 57:2665–2669. 10.1099/ijs.0.65156-0 [DOI] [PubMed] [Google Scholar]
- 7.Neff L, Daher S, Muzzin P, Spenato U, Gulacar F, Gabay C, Bas S. 2007. Molecular characterization and subcellular localization of macrophage infectivity potentiator, a Chlamydia trachomatis lipoprotein. J. Bacteriol. 189:4739–4748. 10.1128/JB.01889-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leuzzi R, Serino L, Scarselli M, Savino S, Fontana MR, Monaci E, Taddei A, Fischer G, Rappuoli R, Pizza M. 2005. Ng-MIP, a surface-exposed lipoprotein of Neisseria gonorrhoeae, has a peptidyl-prolyl cis/trans isomerase (PPIase) activity and is involved in persistence in macrophages. Mol. Microbiol. 58:669–681. 10.1111/j.1365-2958.2005.04859.x [DOI] [PubMed] [Google Scholar]
- 9.Qin A, Scott DW, Rabideau MM, Moore EA, Mann BJ. 2011. Requirement of the CXXC motif of novel Francisella Infectivity Potentiator Protein B FipB, and FipA in virulence of F. tularensis subsp. tularensis. PLoS One 6:e24611. 10.1371/journal.pone.0024611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kadokura H, Beckwith J. 2010. Mechanisms of oxidative protein folding in the bacterial cell envelope. Antioxid. Redox Signal. 13:1231–1246. 10.1089/ars.2010.3187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zapun A, Missiakas D, Raina S, Creighton TE. 1995. Structural and functional characterization of DsbC, a protein involved in disulfide bond formation in Escherichia coli. Biochemistry 34:5075–5089. 10.1021/bi00015a019 [DOI] [PubMed] [Google Scholar]
- 12.Heras B, Shouldice SR, Totsika M, Scanlon MJ, Schembri MA, Martin JL. 2009. DSB proteins and bacterial pathogenicity. Nat. Rev. Microbiol. 7:215–225. 10.1038/nrmicro2087 [DOI] [PubMed] [Google Scholar]
- 13.LoVullo ED, Molins-Schneekloth CR, Schweizer HP, Pavelka MS., Jr 2009. Single-copy chromosomal integration systems for Francisella tularensis. Microbiology 155:1152–1163. 10.1099/mic.0.022491-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramakrishnan G, Sen B, Johnson R. 2012. Paralogous outer membrane proteins mediate uptake of different forms of iron and synergistically govern virulence in Francisella tularensis tularensis. J. Biol. Chem. 287:25191–25202. 10.1074/jbc.M112.371856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ren G, Bardwell JC. 2011. Engineered pathways for correct disulfide bond oxidation. Antioxid. Redox Signal. 14:2399–2412. 10.1089/ars.2010.3782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheffield P, Garrard S, Derewenda Z. 1999. Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expr. Purif. 15:34–39. 10.1006/prep.1998.1003 [DOI] [PubMed] [Google Scholar]
- 17.Kpadeh ZZ, Jameson-Lee M, Yeh AJ, Chertihin O, Shumilin IA, Dey R, Day SR, Hoffman PS. 2013. Disulfide bond oxidoreductase DsbA2 of Legionella pneumophila exhibits protein disulfide isomerase activity. J. Bacteriol. 195:1825–1833. 10.1128/JB.01949-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Straskova A, Pavkova I, Link M, Forslund AL, Kuoppa K, Noppa L, Kroca M, Fucikova A, Klimentova J, Krocova Z, Forsberg A, Stulik J. 2009. Proteome analysis of an attenuated Francisella tularensis dsbA mutant: identification of potential DsbA substrate proteins. J. Proteome Res. 8:5336–5346. 10.1021/pr900570b [DOI] [PubMed] [Google Scholar]
- 19.Arredondo S, Segatori L, Gilbert HF, Georgiou G. 2008. De novo design and evolution of artificial disulfide isomerase enzymes analogous to the bacterial DsbC. J. Biol. Chem. 283:31469–31476. 10.1074/jbc.M803346200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cho SH, Parsonage D, Thurston C, Dutton RJ, Poole LB, Collet JF, Beckwith J. 2012. A new family of membrane electron transporters and its substrates, including a new cell envelope peroxiredoxin, reveal a broadened reductive capacity of the oxidative bacterial cell envelope. mBio. 3:e00291–00211. 10.1128/mBio.00291-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crow A, Liu Y, Moller MC, Le Brun NE, Hederstedt L. 2009. Structure and functional properties of Bacillus subtilis endospore biogenesis factor StoA. J. Biol. Chem. 284:10056–10066. 10.1074/jbc.M809566200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin JL, Waksman G, Bardwell JC, Beckwith J, Kuriyan J. 1993. Crystallization of DsbA, an Escherichia coli protein required for disulphide bond formation in vivo. J. Mol. Biol. 230:1097–1100. 10.1006/jmbi.1993.1226 [DOI] [PubMed] [Google Scholar]
- 23.Hiniker A, Collet JF, Bardwell JC. 2005. Copper stress causes an in vivo requirement for the Escherichia coli disulfide isomerase DsbC. J. Biol. Chem. 280:33785–33791. 10.1074/jbc.M505742200 [DOI] [PubMed] [Google Scholar]
- 24.Thomas RM, Twine SM, Fulton KM, Tessier L, Kilmury SL, Ding W, Harmer N, Michell SL, Oyston PC, Titball RW, Prior JL. 2011. Glycosylation of DsbA in Francisella tularensis subspecies tularensis. J. Bacteriol. 193:5498–5509. 10.1128/JB.00438-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kouwen TR, van Dijl JM. 2009. Interchangeable modules in bacterial thiol-disulfide exchange pathways. Trends Microbiol. 17:6–12. 10.1016/j.tim.2008.10.003 [DOI] [PubMed] [Google Scholar]
- 26.Watarai M, Tobe T, Yoshikawa M, Sasakawa C. 1995. Contact of Shigella with host cells triggers release of Ipa invasins and is an essential function of invasiveness. EMBO J. 14:2461–2470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bader MW, Hiniker A, Regeimbal J, Goldstone D, Haebel PW, Riemer J, Metcalf P, Bardwell JC. 2001. Turning a disulfide isomerase into an oxidase: DsbC mutants that imitate DsbA. EMBO J. 20:1555–1562. 10.1093/emboj/20.7.1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karna SL, Zogaj X, Barker JR, Seshu J, Dove SL, Klose KE. 2010. A bacterial two-hybrid system that utilizes Gateway cloning for rapid screening of protein-protein interactions. Biotechniques 49:831–833. 10.2144/000113539 [DOI] [PubMed] [Google Scholar]
- 29.Schmidt M, Klimentova J, Rehulka P, Straskova A, Spidlova P, Szotakova B, Stulik J, Pavkova I. 2013. Francisella tularensis subsp. holarctica DsbA homologue: thioredoxin-like protein with chaperone function. Microbiology 159:2364–2374. 10.1099/mic.0.070516-0 [DOI] [PubMed] [Google Scholar]
- 30.Balonova L, Hernychova L, Mann BF, Link M, Bilkova Z, Novotny MV, Stulik J. 2010. A multimethodological approach to identification of glycoproteins from the proteome of Francisella tularensis, an intracellular microorganism. J. Proteome Res. 9:1995–2005. 10.1021/pr9011602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kadokura H, Tian H, Zander T, Bardwell JC, Beckwith J. 2004. Snapshots of DsbA in action: detection of proteins in the process of oxidative folding. Science 303:534–537. 10.1126/science.1091724 [DOI] [PubMed] [Google Scholar]
- 32.Broms JE, Lavander M, Meyer L, Sjostedt A. 2011. IglG and IglI of the Francisella pathogenicity island are important virulence determinants of Francisella tularensis LVS. Infect. Immun. 79:3683–3696. 10.1128/IAI.01344-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ludu JS, de Bruin OM, Duplantis BN, Schmerk CL, Chou AY, Elkins KL, Nano FE. 2008. The Francisella pathogenicity island protein PdpD is required for full virulence and associates with homologues of the type VI secretion system. J. Bacteriol. 190:4584–4595. 10.1128/JB.00198-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miki T, Okada N, Kim Y, Abe A, Danbara H. 2008. DsbA directs efficient expression of outer membrane secretin EscC of the enteropathogenic Escherichia coli type III secretion apparatus. Microb. Pathog. 44:151–158. 10.1016/j.micpath.2007.09.001 [DOI] [PubMed] [Google Scholar]
- 35.Missiakas D, Georgopoulos C, Raina S. 1993. Identification and characterization of the Escherichia coli gene dsbB, whose product is involved in the formation of disulfide bonds in vivo. Proc. Natl. Acad. Sci. U. S. A. 90:7084–7088. 10.1073/pnas.90.15.7084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watarai M, Tobe T, Yoshikawa M, Sasakawa C. 1995. Disulfide oxidoreductase activity of Shigella flexneri is required for release of Ipa proteins and invasion of epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 92:4927–4931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berkmen M, Boyd D, Beckwith J. 2005. The nonconsecutive disulfide bond of Escherichia coli phytase (AppA) renders it dependent on the protein-disulfide isomerase, DsbC. J. Biol. Chem. 280:11387–11394. 10.1074/jbc.M411774200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.