

Abstract
The anaerobic degradation of cyclohexane carboxylic acid (CHC) has so far been studied only in Rhodopseudomonas palustris, in which CHC is activated to cyclohexanoyl coenzyme A (cyclohexanoyl-CoA [CHCoA]) and then dehydrogenated to cyclohex-1-ene-1-carboxyl-CoA (CHeneCoA). This intermediate is further degraded by reactions of the R. palustris-specific benzoyl-CoA degradation pathway of aromatic compounds. However, CHeneCoA is not an intermediate in the degradation of aromatic compounds in all other known anaerobic bacteria; consequently, degradation of CHC was mostly unknown in anaerobic bacteria. We identified a previously unknown CHC degradation pathway in the Fe(III)-reducing Geobacter metallireducens by determining the following CHC-induced in vitro activities: (i) the activation of CHC to CHCoA by a succinyl-CoA:CHC CoA transferase, (ii) the 1,2-dehydrogenation of CHCoA to CHeneCoA by CHCoA dehydrogenase, and (iii) the unusual 1,4-dehydrogenation of CHeneCoA to cyclohex-1,5-diene-1-carboxyl-CoA. This last represents a previously unknown joint intermediate of the CHC and aromatic compound degradation pathway in bacteria other than R. palustris. The enzymes catalyzing the three reactions were purified and characterized as specific enzymes after heterologous expression of the encoding genes. Quantitative reverse transcription-PCR revealed that expression of these genes was highly induced during growth with CHC but not with benzoate. The newly identified CHC degradation pathway is suggested to be present in nearly all CHC-degrading anaerobic bacteria, including denitrifying, Fe(III)-reducing, sulfate-reducing, and fermenting bacteria. Remarkably, all three CHC degradation pathways always link CHC catabolism to the catabolic pathways of aromatic compounds. We propose that the capacity to use CHC as a carbon source evolved from already-existing aromatic compound degradation pathways.
INTRODUCTION
Cyclohexane carboxylic acid (CHC) occurs in nature predominantly as a functional group in ubiquitous biological compounds. Examples are naphthenic acids in crude oil (1, 2) or polyketide antibiotics from Streptomyces species (3). The biosynthetic origin of CHC is the shikimate pathway, as shown for Alicyclobacillus acidocaldarius species and Streptomyces collinus (3, 4). Moreover, CHC has been reported to be a product during the fermentation of benzoate or crotonate (see Fig. 1C) (5, 6).
FIG 1.

Known degradation and formation pathways of CHC in anaerobic bacteria. Degradation in aerobic bacteria (A) or R. palustris (B) or formation as a fermentation product from benzoate in S. aciditrophicus (C) is shown.
Aerobic and anaerobic microorganisms have developed different strategies to use CHC as a carbon and/or energy source. The aerobic degradation of CHC was investigated in Alcaligenes and Arthrobacter strains and in Corynebacterium cyclohexanicum and involves monooxygenase- and oxidase-dependent steps yielding the aromatic 4-hydroxybenzoate (see Fig. 1A) (7, 8, 9, 10). Further degradation proceeds via the central intermediate protocatechuate that is channeled into the β-ketoadipate pathway.
The oxygen-independent degradation of CHC has so far been studied only in the facultatively anaerobic, photosynthetic alphaproteobacterium Rhodopseudomonas palustris. Here, CHC is first activated by an AMP-forming cyclohexanoyl coenzyme A (cyclohexanoyl-CoA [CHCoA]) synthetase (AliA) (see Fig. 1B) (11, 12). In a second step, CHCoA is oxidized to cyclohex-1-ene-1-carboxyl-CoA (CHeneCoA) by a specific acyl-CoA dehydrogenase (AliB) (13]). The product is subsequently hydrated to the corresponding alcohol by a specific enoyl-CoA hydratase and oxidized to 2-oxocyclohexanoyl-CoA; the ring structure is then cleaved hydrolytically, yielding the aliphatic pimeloyl-CoA (13, 14). The subsequent modified β-oxidation steps yield three acetyl-CoA molecules and one CO2 molecule. In R. palustris, CHeneCoA is the characteristic joint intermediate of the degradation of CHC and aromatic compounds, such as benzoate. During the degradation of the latter, the central benzoyl-CoA intermediate is dearomatized by a four-electron reduction to CHeneCoA, catalyzed by an ATP-dependent class I benzoyl-CoA reductase (15, 16). This so-called R. palustris type of benzoyl-CoA degradation pathway via the characteristic CHeneCoA intermediate has also been suggested to be present in the archaeon Ferroglobus placidus by genomic and transcriptomic analyses (17). However, it differs from the Thauera aromatica type of benzoyl-CoA degradation pathway present in all other known aromatic compound-degrading anaerobic bacteria. In denitrifying, sulfate-reducing, Fe(III)-reducing, and fermenting bacteria, the class I and II benzoyl-CoA reductases reduce their substrate only by two electrons, yielding cyclohex-1,5-diene-1-carboxyl-CoA (CHdieneCoA) (18, 19). As a consequence, a different set of enzymes is involved in CHdieneCoA degradation, including specific hydratases, dehydrogenases, and ring-cleaving hydrolases that form 3-hydroxypimeloyl-CoA and not pimeloyl-CoA. Notably, the enzymes involved in these reactions do not convert the corresponding intermediates of the R. palustris-type pathway (18, 20).
Only little is known about CHC degradation in anaerobic bacteria other than R. palustris. The complete degradation of CHC was reported for the sulfate-reducing Desulfococcus multivorans (21) and the fermenting Syntrophus aciditrophicus (22) (both are deltaproteobacteria) and in the denitrifying betaproteobacterium T. aromatica. These organisms degrade aromatic compounds via the T. aromatica type of benzoyl-CoA degradation pathway, where CHeneCoA is not a true intermediate. This finding suggests that CHC degradation in most anaerobic bacteria must be different from that reported for R. palustris.
In this work, we report on a novel CHC degradation pathway in Geobacter metallireducens that is likely to be present in all CHC-degrading anaerobic bacteria other than R. palustris. The specific enzymes involved were characterized as a CoA transferase and two acyl-CoA dehydrogenases that convert CHC to CHdieneCoA, a previously unknown joint intermediate of the anaerobic aromatic compound and CHC degradation pathways.
MATERIALS AND METHODS
Materials.
Chemicals and biochemicals were obtained from Roche Diagnostics (Mannheim, Germany), Fluka (Neu-Ulm, Germany), Merck (Darmstadt, Germany), Roth (Karlsruhe, Germany), Sigma-Aldrich (Deisenhofen, Germany), Applichem (Darmstadt, Germany), and Bio-Rad (Munich, Germany). Coenzyme A was obtained from Applichem. CoA esters were synthesized as described earlier (23, 24). Materials and equipment for protein purification were obtained from GE Healthcare (Munich, Germany), Millipore (Bedford, MA), and Sigma-Aldrich (Deisenhofen, Germany). The Champion pET101 directional Topo expression kit and chemically competent Escherichia coli OneShotTOP10 and BL21Star (DE3) OneShot were purchased from Life Technologies (Grand Island, NY).
Computational analysis.
Plots were prepared and regression coefficients were calculated using the GraphPad Prism 6 software package. BLAST searches were performed using the NCBI BLAST server (http://www.ncbi.nlm.nih.gov/BLAST/). Phylogenetic alignments were performed using the Mega5 software package (www.megasoftware.net).
Cultivation of Geobacter metallireducens.
G. metallireducens was obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ 7210) and was cultivated at 25°C in 400-ml or 2-liter bottles under strictly anoxic conditions in a mineral salt medium (25) with nitrate (10 mM) as the electron acceptor. Either benzoate or CHC was added from sterile, pH-adjusted stock solutions (5 mM final concentrations) as a carbon and energy source. Cells were harvested at an optical density (578 nm) of approximately 0.5 under anaerobic conditions and stored at −20°C until further use.
Determination of CHC and nitrate via ion chromatography.
Medium samples were taken during growth of G. metallireducens. After centrifugation, the supernatants were analyzed by ion chromatography (system, Dionex ICS 2100; column, Dionex IonPac AG11-HC RFIC, 2 by 250 mm; conductivity monitor; Thermo Scientific). Concentrations of ions in the medium were calculated based on calibration series with 5 μM to 5 mM CHC, nitrite, and nitrate. The ions eluted after a 3-min isocratic start in a 24-min gradient from 1 to 60 mM potassium hydroxide.
Heterologous gene expression and enzyme production in Escherichia coli.
The genes encoding the putative CHCoA dehydrogenase (Gmet_3307), CHeneCoA dehydrogenase (Gmet_3306), and the CHC-activating CoA transferase (Gmet_3304) were amplified from G. metallireducens chromosomal DNA by PCR using primers suitable for the Champion pET101 directional Topo expression kit (Life Technologies) and for introducing a C-terminal 6×His tag according to the manufacturer's instructions. For the oligonucleotide sequences used, see the supplemental material (Table S1). The following PCR conditions were used: 35 cycles of 10 s of denaturation at 98°C, 30 s of primer annealing at 63°C, and 50 s of elongation at 72°C; Q5 DNA polymerase (New England BioLabs) was used. Gel elution, ligation, cloning, and expression were performed according to the manufacturer's protocol. Chemically competent Escherichia coli BL21 Star(DE3) OneShot cells were transformed with the purified vector and grown at 37°C in LB medium containing 100 μg ml−1 ampicillin and 50 mg liter−1 riboflavin. Cells were induced with 1.0 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at an optical density of 0.4 to 0.6. After growth overnight at 20°C, the cells were harvested by centrifugation and stored at −20°C until use.
Purification of recombinant proteins from G. metallireducens. (i) Preparation of cell extracts.
Frozen E. coli cells (Gmet_3306 and Gmet_3307, 2 to 3 g wet cell mass per 5 ml; Gmet_3304, 10 g per 10 ml) were suspended in 20 mM potassium phosphate, 200 mM sodium chloride, and 20 mM imidazole, pH 7.9 (buffer A), containing 0.1 mg of DNase I and 0.5 mg of lysozyme per ml. The cell suspension was passed through a French pressure cell at 138 MPa (4°C). The cell lysate was centrifuged at 100,000 × g for 60 min.
(ii) Affinity chromatography.
For nickel affinity chromatography, the supernatant obtained after ultracentrifugation was applied onto a 1-ml Ni-chelating affinity column (HisTrap HP; GE Healthcare) that had been equilibrated with buffer A. After washing with buffer A, the His-tagged proteins eluted in a gradient from 0.02 to 0.5 M imidazole in buffer A over 10 ml at a flow rate of 1.0 ml min−1. Active fractions (2.5 ml) were pooled. Imidazole was removed by passing the enzyme over a desalting PD10 column (GE Healthcare). It eluted in 3.5 ml of 20 mM potassium phosphate, pH 7.9 (buffer B). Fifty percent glycerol in buffer B was added to a final concentration of 25% (vol/vol) glycerol. The enzymes were stored at −20°C.
Acyl-CoA dehydrogenase assays.
CHCoA dehydrogenase and CHeneCoA dehydrogenase activities were measured in a discontinuous assay at 30°C via reverse-phase ultrahigh-performance liquid chromatography (RP-UPLC). The 200-μl assay volumes contained 200 mM morpholinepropanesulfonic acid (MOPS), 15 mM MgCl2, 0.2 mM ferrocenium hexafluorophosphate, and 1 to 10 μg enzyme, and the reaction was started by addition of 200 μM substrate. Twenty-microliter samples were taken at different time points and stopped by addition of 20 μl methanol. After centrifugation, the supernatants were applied to RP-UPLC (Waters) using an acetonitrile gradient (2% to 10% in 90 s, step to 20% in 1 s, and 20% to 30% in 139 s) in 10 mM phosphate buffer, pH 6.8. CHCoA, CHeneCoA, cyclohex-2-ene-1-carboxyl-CoA, cyclohex-3-ene-1-carboxyl-CoA, cyclohexa-1,5-diene-1-carboxyl-CoA (CHdieneCoA), 6-hydroxy-cyclohex-1-ene-1-carboxyl-CoA, butyryl-CoA, crotonyl-CoA, and isobutyryl-CoA were tested as substrates (0.2 mM [each]). For determination of Km values, substrate concentrations were varied from 6.25 to 200 μM. Spectrophotometric assays were discarded as cell extracts of G. metallireducens exhibited high unspecific activities with ferrocenium hexafluorophosphate.
CoA transferase assay.
CoA transferase activity was determined in 100-μl assay volumes with 10 to 100 μg enzyme in buffer (100 mM Tris and 10 mM MgCl2, pH 7.8) with 0.25 mM acyl-CoA as a CoA donor and 5 mM free acid as a CoA acceptor. For determination of Km values, the initial concentrations were varied from 0.125 mM to 5 mM for the free organic acid and from 7.8 μM to 250 μM for the CoA donor. The reaction was started by addition of the CoA acceptor. Twenty-microliter samples were taken at different time points and stopped by addition of 20 μl 1 M HCl. After centrifugation, the supernatants were applied to RP-UPLC using an acetonitrile gradient (2% to 10% in 90 s, step to 20% in 1 s, and 20% to 30% in 139 s) in 10 mM phosphate buffer, pH 6.8.
Determination of molecular mass.
Native masses were determined by analytical gel filtration via Superdex 200 (10/300 GL column [GE Healthcare], 25-ml volume, 0.5 ml min−1) using 20 mM triethanolamine-HCl, pH 7.8, 4 mM MgCl2, and 150 mM KCl. For calibration, ferritin (440 kDa), catalase (245 kDa), aldolase (158 kDa), bovine serum albumin (BSA) (67 kDa), ovalbumin (43 kDa), and carbonic anhydrase (29 kDa) were used.
Determination of cofactor content.
For flavin content determination, 100 μl of purified enzyme was precipitated with 200 μl methanol. The supernatants obtained after centrifugation were analyzed by RP-UPLC (see enzyme assays) and compared with a calibration curve from 0.1 to 10 μM flavin adenine dinucleotide (FAD). For reconstitution experiments, heterologously expressed and purified enzyme was incubated with 1 mM FAD for 60 min at 30°C. After desalting using a PD10 column (GE Healthcare), the protein was used for determination of activity and of protein and FAD content.
Quantitative RT-PCR.
For each tested growth substrate, cells from four independently grown cultures of Geobacter metallireducens GS-15 were used. Cells were anaerobically harvested in the mid-exponential phase. After removal of the supernatant, pellets were immediately stored at −80°C. Total RNA was isolated using a Direct-zol RNA MiniPrep kit (Zymo Research) and DNase I treated using a Turbo DNA-free kit (Life Technologies). RNA concentration, purity, and integrity were determined using a Nanodrop-1000 spectrophotometer (Peqlab) and sodium hypochlorite agarose gels (26). Reverse transcription (RT) of 300 ng DNase I-digested RNA was performed using a RevertAid First Strand cDNA synthesis kit (Thermo) with random hexamer primers. For quantitative PCR (qPCR) analysis, cDNA was diluted 20-fold (vol/vol) in nuclease-free H2O. The diluted cDNA was added to SSoAdvanced SYBR green supermix (Bio-Rad) containing 250 nM (each) gene-specific primers (see Table S2 in the supplemental material). Primers were designed using the web-based software Primer3 (27, 28), and specificities were validated using BLAST. qPCR was performed on a CFX96 touch real-time PCR cycler (Bio-Rad). Reference genes (Gmet_0899, encoding a Delta-1-pyrroline-5-carboxylate reductase, and Gmet_0918, encoding an MreB homolog) were identified using the software program qBase+ (Biogazelle), using the geNorm algorithm (29). Transcription was normalized (30) for both of these reference genes using qBase+. To rule out contamination with genomic DNA, non-reverse transcription controls were used.
Protein determinations.
SDS-PAGE (12.5%) was carried out according to the method of Laemmli. Proteins were visualized using the Simply-Blue SafeStain stain (Invitrogen). Protein was routinely determined by the method of Bradford using BSA as a standard.
RESULTS
Identification of putative genes involved in CHC degradation in deltaproteobacteria.
During axenic growth with benzoate (Fig. 1C) and/or crotonate, S. aciditrophicus forms the fermentation products CHC, CO2, and acetate (5). In both cases, CHdieneCoA is an intermediate that is formed either by a class II benzoyl-CoA reductase (benzoate fermentation) or by reversely operating enzymes of the T. aromatica-type benzoyl-CoA degradation pathway (crotonate fermentation). CHdieneCoA is then reduced to CHCoA by two consecutively operating CHeneCoA and CHCoA dehydrogenases (31) (Fig. 1C). Notably, S. aciditrophicus can also ferment CHC to acetate, although the enzymes involved have not been studied so far (22). This observation led us to the hypothesis that identical, reversibly operating CHeneCoA and CHCoA dehydrogenases are involved in both CHC formation and degradation. These dehydrogenases from S. aciditrophicus share high amino acid sequence similarities (>70% identities) with putative gene products from the Fe(III)-reducing, aromatic compound-degrading species Geobacter metallireducens, Geobacter bemidjiensis, and Geobacter daltonii and from the sulfate-reducing, CHC and aromatic compound-degrading Desulfococcus multivorans. Similar genes are also present in an uncultivated deltaproteobacterium (32) (contig ACD_75C01489). A phylogenetic tree of representative well-described acyl-CoA dehydrogenases is presented in the supplemental material (see Fig. S1).
In all cases, the verified or putative genes encoding CHeneCoA and CHC dehydrogenases are located adjacent to each other in a gene cluster along with other conserved genes (Fig. 2). In G. metallireducens, G. bemidjiensis, and G. daltonii, this cluster contains open reading frames that were annotated as putative CoA transferases (>78% amino acid sequence identity to each other) (Fig. 2). They share >75% amino acids identities with the previously described succinyl-CoA:benzoate CoA transferase from G. metallireducens (33). The putative CHC degradation gene clusters of G. daltonii, D. multivorans, and the uncultured deltaproteobacterium contain a gene encoding a putative AMP-forming acyl-CoA synthetase. Both, the CoA transferases and/or the acyl-CoA synthetases are likely candidates for the CHCoA-forming enzyme. Interestingly, the gene cluster in G. metallireducens includes a second copy of a bamA gene putatively encoding the ring cleaving 6-oxocyclohex-1-ene-1-carboxyl-CoA hydrolase. It has >90% amino acid sequence identity to the product of the experimentally verified bamA gene present in the benzoic acid metabolism gene cluster (20). Finally, a protein with high similarities to a transcriptional regulator of the DNA-binding FIS family (factor for inversion stimulation) is part of the gene cluster (Fig. 2).
FIG 2.

Gene clusters putatively involved in CHC degradation in deltaproteobacteria. Genes encoding proteins with significant sequence similarities are displayed in the same colors: CHCoA and CHeneCoA dehydrogenases (light and dark orange), proteins annotated as σ54-dependent regulators (pink), CoA transferases (light blue), carboxylic acid CoA ligases (dark blue), and similar hypothetical proteins (gray). The gene cluster in G. metallireducens contains an additional bamA gene encoding a ring-opening hydrolase (red). Abbreviations: σ54-trsc-reg, sigma54-dependent transcription regulator; acdh, acyl-CoA dehydrogenase; act, acyl-CoA transferase; hyp, hypothetical protein; kat, ketoacyl-CoA thiolase; Nact, N-acetyltransferase; kdac, zinc-dependent lysine deacetylase; acs, acyl-CoA synthetase; FeSOx, FeS oxidoreductase; etf, electron-transferring flavoprotein; M24pept, Met24 peptidase.
Growth of G. metallireducens with CHC.
Although growth with CHC has not been reported before, G. metallireducens was chosen as a well-characterized model organism to study the hypothesized alternative CHC degradation pathway. G. metallireducens indeed grew exponentially with CHC (5 mM) as the sole carbon source and nitrate (10 mM) as a terminal electron acceptor (ammonification conditions). Per mol CHC, 7.5 mol nitrate was consumed, suggesting a complete oxidation of CHC to CO2. The doubling time with CHC was similar to that with benzoate as a growth substrate (7.8 ± 0.5 h at 25°C). With 5 mM CHC, the optical density reached 0.47 (Fig. 3). At 30°C, the specific substrate consumption rate of exponentially growing cells was determined to be maximally 29 nmol min−1 [mg protein]−1.
FIG 3.

Growth of G. metallireducens with cyclohexane carboxylate (black circles) and benzoate (white circles). For each substrate, four independent cultures were measured. Error bars represent standard deviations of the means.
Enzyme activities in extracts from G. metallireducens.
Amino acid sequence similarities of known enzymes and the putative proteins involved in CHC degradation suggested the activation of CHC by a succinyl-CoA-dependent CoA transferase followed by the oxidation to CHdieneCoA by CHCoA and CHeneCoA dehydrogenase activities. To verify this assumption, in vitro enzyme activities were determined, in which substrate consumption and product formation were monitored by RP-UPLC analysis. The succinyl-CoA-dependent formation of CHCoA from CHC was observed with a specific activity of 16 nmol min−1 mg−1 in extracts from cells grown with CHC. This value is slightly below the specific substrate consumption rate of exponentially growing cells, which might be caused by unspecific thioesterases or suboptimal assay conditions. Such extracts also catalyzed the time-, protein-, and ferrocenium hexafluorophosphate-dependent oxidation of CHCoA to CHeneCoA (61 nmol min−1 mg−1) and CHeneCoA to CHdieneCoA (64 nmol min−1 mg−1). The latter was further oxidized to benzoyl-CoA, a side reaction that has also been observed for CHeneCoA dehydrogenase from S. aciditrophicus (31). The dehydrogenase activities were 4- to 6-fold higher than in cells grown on benzoate (CHCoA, 17 nmol min−1 mg−1; CH1eneCoA, 11 nmol min−1 mg−1).
Heterologous expression of putative genes involved in CHC degradation.
The genes Gmet_3304, Gmet_3307, and Gmet_3306, (Fig. 2), putatively coding for a CoA transferase and two acyl-CoA dehydrogenases, were individually expressed in E. coli BL21 Star with a C-terminal His tag. After purification by Ni-chelating affinity chromatography, in each case highly enriched protein bands were obtained, as evidenced by SDS-polyacrylamide gel analysis (Fig. 4). According to the presence of 6-fold His tags, each protein migrated at a slightly higher molecular mass than calculated from the amino acid sequence (47.8 kDa, 41.2 kDa, and 40.9 kDa for Gmet_3304, Gmet_3307, and Gmet_3306, respectively).
FIG 4.
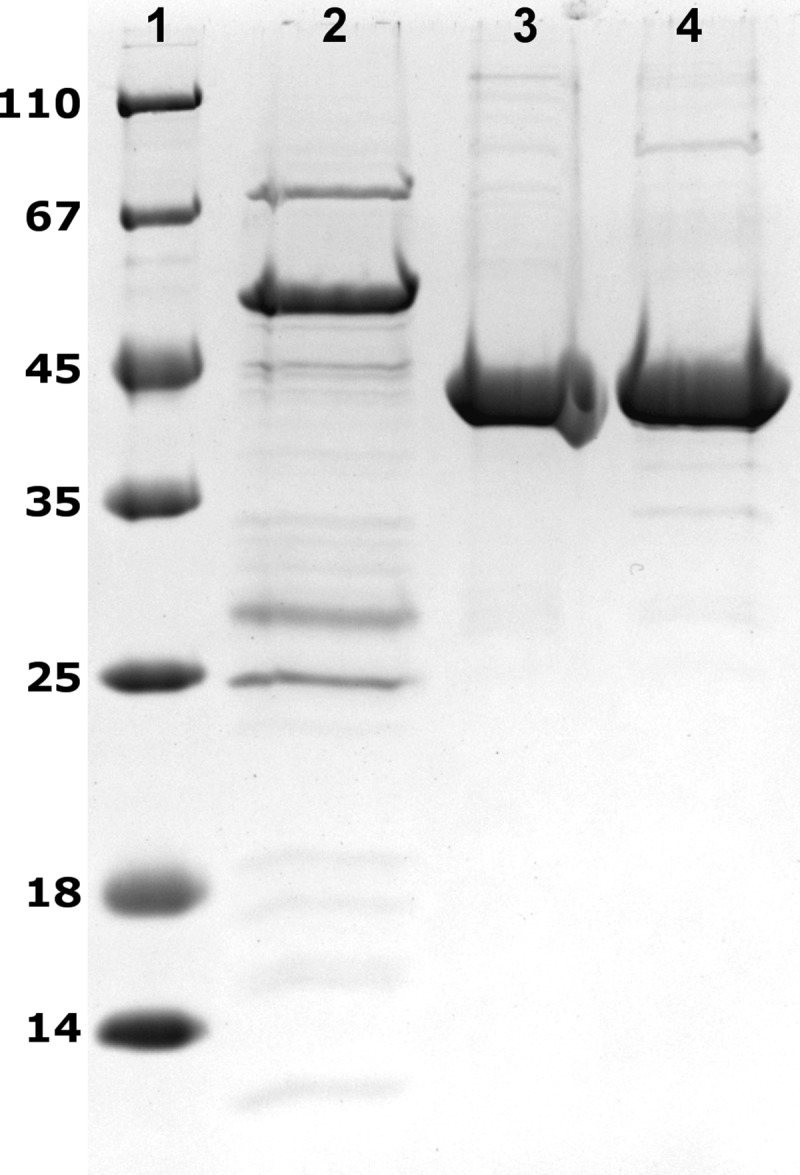
SDS-polyacrylamide gels of heterologously expressed and purified proteins involved in the degradation of CHC. Lane 1, molecular mass standard; lane 2, succinyl-CoA:CHC CoA transferase; lane 3, CHeneCoA dehydrogenase; lane 4, CHCoA dehydrogenase (7 μg each).
Characterization of the CHCoA-forming enzyme.
The gene Gmet_3304 was predicted to code for a class I CoA transferase that operates in a ping-pong mechanism (34). The heterologously produced product had a homodimeric composition with a native molecular mass of 95 ± 7 kDa as determined by gel filtration analysis. CoA transferase activity was tested by RP-UPLC analysis using various CoA donors (0.25 mM) and carboxylic acids (5 mM). Activation of CHC to CHCoA showed highest activities with succinyl-CoA (2.0 μmol min−1 mg−1) and butyryl-CoA (2.7 μmol min−1 mg−1) as a CoA donor; benzoate and cyclohex-1-ene-1-carboxylate were converted with lower specific activities (Table 1). The equilibrium constants (Keq) were extracted from the equilibrium substrate/product concentrations reached from the forward and reverse directions and gave highly similar numbers. The values obtained revealed that the formation of CHCoA from succinyl-CoA or butyryl-CoA as donors was favored over the reverse reaction, supporting that CHCoA formation is the in vivo role of the CoA transferase. Interestingly, the Km values and specific activities with CHC, benzoate, and cyclohex-1-ene-1-carboxylate were apparently affected by the choice of the CoA donor (Table 1) and were always lower with butyryl-CoA than with succinyl-CoA or other CoA donors. Such observations were not expected for a true ping-pong mechanism, and the mechanism of the CoA transferase needs to be studied in future detailed kinetic analyses. The results obtained suggest, on the first view, that butyryl-CoA serves as a natural CoA donor. However, a mass spectrometry-based CoA-ester pool analysis in exponentially grown cells revealed that succinyl-CoA is the most abundant CoA ester, whereas butyryl-CoA was hardly detectable, with a ratio of succinyl-CoA to butyryl-CoA of ≫1,000 (P. Kiefer, ETH Zürich, personal communication). For this reason, succinyl-CoA is considered a natural CoA donor for CHC activation.
TABLE 1.
Kinetic properties of the succinyl-CoA:CHC CoA transferasea
| Acceptor | CoA donor (0.25 mM) | Kmb (μM) | Activity (%) | Equilibrium Keq |
|---|---|---|---|---|
| Cyclohexane carboxylic acid | Succinyl-CoA | 56 ± 15 | 100 | 1.7 |
| Butyryl-CoA | 14 ± 6 | 135 | 5 | |
| Acetyl-CoA | >500 | 31 | NDc | |
| Propionyl-CoA | NDc | 9 | ND | |
| Acetoacetyl-CoA | ND | 5 | ND | |
| Crotonyl-CoA | ND | 50 | ND | |
| Glutaryl-CoA | ND | 28 | ND | |
| CH1eneCoA | >200 | 22 | ND | |
| Benzoic acid | Succinyl-CoA | 416 ± 50 | 15 | 0.001 |
| Butyryl-CoA | 125 ± 48 | 42 | 0.003 | |
| Cyclohex-1-ene-1-carboxylic acid | Succinyl-CoA | 158 ± 56 | 40 | 0.007 |
| Butyryl-CoA | 52 ± 12 | 140 | 0.03 |
“100%” refers to 2.0 μmol min−1 mg−1. Km values are given for the free acids.
Apparent Km values as calculated using the GraphPad software package Prism 6.
ND, not determined.
Characterization of CHCoA and CHeneCoA dehydrogenases.
Gel filtration analysis revealed a native molecular mass of 140 ± 10 kDa for both Gmet_3306 and Gmet_3307, suggesting a homotetrameric composition, respectively, that is typical for acyl-CoA dehydrogenases. The enzymes contained 0.80 (Gmet_3307) and 0.45 (Gmet_3306) FAD per subunit after incubation with free FAD followed by a desalting step.
The Gmet_3307 gene product catalyzed the ferrocenium hexafluorophosphate-dependent oxidation of CHCoA to CHeneCoA with a specific activity of 29 μmol min−1 mg−1; the product formed was not further dehydrogenated to CHdieneCoA. The reaction followed a Michaelis-Menten kinetic, with a Km value of <5 μM. As an alternative substrate, cyclohex-3-ene-1-carboxyl-CoA was converted to the corresponding cyclohexadiene-1-carboxyl-CoA isomers (30% rate compared to CHC). No activity was observed (<0.1%) with 6-hydroxycyclohex-1-ene-1-carboxyl-CoA, cyclohex-2-ene-1-carboxyl-CoA, butyryl-CoA, crotonyl-CoA, isobutyryl-CoA, glutaryl-CoA, or succinyl-CoA. Due to the low Km and high specificity, Gmet_3307 is referred to as a CHCoA dehydrogenase.
The protein derived from Gmet_3306 converted CHeneCoA to CHdieneCoA at a rate of 2.3 μmol min−1 mg−1 and CHdieneCoA to benzoyl-CoA at a rate of 1.0 μmol min−1 mg−1 with a Km of 31 ± 16 μM for CHeneCoA and a Km of 85 ± 9 μM for CHdieneCoA. Neither CHCoA, cyclohex-2-ene-1-carboxyl-CoA, cyclohex-3-ene-1-carboxyl-CoA, butyryl-CoA, nor crotonyl-CoA was converted (<0.1% compared to result for CHeneCoA). The kinetic properties strongly suggest that Gmet_3306 encodes a specific CHdieneCoA-forming CHeneCoA dehydrogenase catalyzing a 1,4-dehydrogenation reaction with the capacity to oxidize its product further to benzoyl-CoA.
Transcriptional regulation of genes involved in benzoate and cyclohexane carboxylate degradation.
The differential transcription of selected genes during growth with benzoate and CHC was investigated by quantitative reversed transcription-PCR. The targeted genes included the two genes of the newly identified putative CHC degradation-related gene cluster (Gmet_3305 [encoding a putative 6-oxocyclohex-1-ene-1-carboxyl-CoA hydrolase] and Gmet_3306 [encoding CHeneCoA dehydrogenase]) and genes involved in the benzoyl-CoA degradation pathway (previously shown to be differentially translated during growth on benzoate [35]): bamA (encoding a 6-oxocyclohex-1-ene-1-carbonyl-CoA hydratase), bamB (encoding a class II benzoyl-CoA reductase active site-containing subunit), bamM (encoding glutaryl-CoA dehydrogenase), and bamQ (encoding 6-hydroxycyclohex-1-ene-1-carbonyl-CoA dehydrogenase). Analysis of transcripts revealed a 3-orders-of-magnitude-higher transcription of Gmet_3305 and Gmet_3306 during growth with CHC than during growth with benzoate. Genes involved in the benzoyl-CoA degradation pathways were not differentially transcribed (Fig. 5).
FIG 5.

Transcriptional analysis of genes involved in the anaerobic degradation of CHC and benzoate. Genes involved in the benzoyl-CoA degradation pathway (bamA, bamB-1, bamM, and bamQ) and genes of the CHC degradation cluster (Gmet_3305 and Gmet_3306) are shown. For each gene, relative transcription in cells grown on benzoate (white) was compared to that in cells grown on CHC (gray). Gmet_3305 and Gmet_3306 (P ≤ 0.001) but none of the other tested genes (P > 0.05) exhibited a statistically significant differential transcription. Error bars represent standard errors of the means.
DISCUSSION
In this work, we identified a third CHC degradation pathway via the characteristic intermediate CHdieneCoA, formed from CHeneCoA by CHeneCoA dehydrogenase, the key enzyme of the novel CHC degradation pathway. Notably, this reaction proceeds by an unusual 1,4-dehydrogenation and not by the 1,2-dehydrogenation catalyzed by conventional acyl-CoA dehydrogenases (36). The observed further dehydrogenation of CHdieneCoA to benzoyl-CoA by CHeneCoA dehydrogenase is not considered to play a physiological role, since the CHdieneCoA intermediate is readily converted to 6-hydroxycyclohex1-ene-1-carboxyl-CoA by a highly active CHdieneCoA hydratase (18) and other enzymes of the benzoyl-CoA degradation pathway.
It is striking that all three CHC degradation pathways convert CHC to intermediates of aromatic compound degradation pathways: (i) the oxygenase- and oxidase-dependent aromatization to 4-hydroxybenzoate in aerobic bacteria (8), (ii) the oxidation to CHeneCoA in R. palustris (14), and (iii) the oxidation to CHdieneCoA in G. metallireducens (Fig. 6). We propose that with the exception of R. palustris and probably F. placidus (17), the pathway identified in G. metallireducens is present in all CHC-degrading anaerobic microorganisms. This proposal is based on the finding that except for R. palustris (and F. placidus), all aromatic compound-degrading anaerobes employ the T. aromatica type of benzoyl-CoA degradation pathway, where CHdieneCoA but not CHeneCoA is the intermediate formed by class I or II benzoyl-CoA reductases (37). The former was identified as a joint intermediate involved in anaerobic aromatic compound and CHC degradation. For the initial activation of CHC to CHCoA, two alternatives appear to exist: while a CoA transferase is present in Geobacter species, other deltaproteobacteria, such as D. multivorans, lack such a transferase in the predicted CHC degradation gene cluster. In these organisms, a gene putatively coding for a CoA ligase is proposed to be involved in CHCoA formation. G. daltonii seems to contain both alternatives for CHC activation (Fig. 2).
FIG 6.

The newly identified cyclohexane carboxylic acid degradation pathway in G. metallireducens. The involved gene products are depicted in the colors used in Fig. 3. The intermediate CHdieneCoA interfaces the CHC and aromatics degradation pathways (shown in gray). The cyclohexa-1,5-diene-1-carboxyl-CoA dehydrogenase (Gmet_3306) represents a key enzyme that catalyzes a 1,4-dehydrogenation and is uniquely employed in the new CHC degradation pathway.
The genes encoding the CoA transferase and the two acyl-CoA dehydrogenases involved in the G. metallireducens type of CHC degradation pathway are always located in a gene cluster that are likely to form a transcriptional unit. The upregulation of gene transcription and the induction of the corresponding enzyme activities during growth on CHC support this assumption. The presence of the cluster in the genomes of the CHC-degrading D. multivorans and S. aciditrophicus confirms the predicted abundance, and its presence in the genomes of G. bemidijensis and G. daltonii strongly suggests that these organisms are able to utilize CHC as carbon source.
All studied and predicted CHC-degrading anaerobes have the capacity to degrade aromatic compounds, but not necessarily vice versa. In agreement, at least in G. metallireducens, CHC induces the expression of genes involved in both CHC and aromatic compound degradation, whereas benzoate did not induce the genes involved in CHC degradation. Among the 26 known aromatic compound-degrading anaerobes with available genomes (37), only a few contain genes similar to that encoding CHeneCoA dehydrogenase. This observation suggests that the enzymatic equipment for anaerobic CHC degradation expanded the existing carbon source utilization capacity among certain aromatic compound-degrading anaerobes. This suggestion is in line with the much higher abundance of aromatic functionalities in nature than of the CHC moiety. A likely scenario for the evolution of the CHC degradation pathway discovered in this work comprises a duplication of a gene encoding an acyl-CoA dehydrogenase (e.g., butyryl-CoA dehydrogenase). Notably, CHCoA and CHeneCoA dehydrogenases are always located adjacent to each other in the known/predicted CHC degradation gene clusters. The succinyl-CoA:CHC CoA transferase might have evolved from the succinyl-CoA:benzoate CoA transferase that has been described for G. metallireducens (Gmet_2054) (33). Based on these considerations, we postulate that organisms with the capability of degrading CHC very likely also utilize aromatic compounds.
Supplementary Material
ACKNOWLEDGMENTS
We thank Patrick Kiefer and Julia Vorholt (ETH Zurich) for mass spectrometry-based CoA-ester pool analyses.
This work was funded by the Deutsche Forschungsgemeinschaft (BO 1565/9-2) within SPP1319.
Footnotes
Published ahead of print 11 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02071-14.
REFERENCES
- 1.Brient JA, Wessner PJ, Doyle MN. 1995. Naphthenic acids. In Kroschwitz JI. (ed), Encyclopedia of chemical technology, 4th ed, vol 16, p 1017–1029 John Wiley & Sons, New York, NY [Google Scholar]
- 2.Holowenko FM, MacKinnon MD, Fedorak PM. 2001. Naphthenic acids and surrogate naphthenic acids in methanogenic microcosms. Water Res. 35:2595–2606. 10.1016/S0043-1354(00)00558-3 [DOI] [PubMed] [Google Scholar]
- 3.Handa S, Floss HG. 1997. Biosynthesis of ω-cyclohexyl fatty acids in Alicyclobacillus acidocaldarius: the stereochemistry of the initial 1,4-conjugate elimination. Chem. Comm. 1997:153–154. 10.1039/A607122H [DOI] [Google Scholar]
- 4.Cropp TA, Wilson DJ, Reynolds KA. 2000. Identification of a cyclohexylcarbonyl CoA biosynthetic gene cluster and application in the production of doramectin. Nat. Biotechnol. 18:980–983. 10.1038/79479 [DOI] [PubMed] [Google Scholar]
- 5.Mouttaki H, Nanny MA, McInerney MJ. 2007. Cyclohexane carboxylate and benzoate formation from crotonate in Syntrophus aciditrophicus. Appl. Environ. Microbiol. 73:930–938. 10.1128/AEM.02227-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mouttaki H, Nanny MA, McInerney MJ. 2008. Use of benzoate as an electron acceptor by Syntrophus aciditrophicus grown in pure culture with crotonate. Environ. Microbiol. 10:3265–3274. 10.1111/j.1462-2920.2008.01716.x [DOI] [PubMed] [Google Scholar]
- 7.Blakley ER. 1974. The microbial degradation of cyclohexane carboxylic acid: a pathway involving aromatization to form p-hydroxybenzoic acid. Can. J. Microbiol. 20:1297–1306. 10.1139/m74-202 [DOI] [Google Scholar]
- 8.Kaneda T. 1974. Enzymatic aromatization of 4-ketocyclohexanecarboxylic acid to p-hydroxybenzoic acid. Biochem. Biophys. Res. Commun. 58:140–144. 10.1016/0006-291X(74)90902-4 [DOI] [PubMed] [Google Scholar]
- 9.Smith DI, Callely AG. 1975. The microbial degradation of cyclohexane carboxylic acid. J. Gen. Microbiol. 91:210–212. 10.1099/00221287-91-1-210 [DOI] [PubMed] [Google Scholar]
- 10.Taylor DG, Trudgill PW. 1978. Metabolism of cyclohexane carboxylic acid by Alcaligenes strain W1. J. Bacteriol. 134:401–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Küver J, Xu Y, Gibson J. 1995. Metabolism of cyclohexane carboxylic acid by the photosynthetic bacterium Rhodopseudomonas palustris. Arch. Microbiol. 164:337–345. 10.1007/BF02529980 [DOI] [PubMed] [Google Scholar]
- 12.Samanta SK, Harwood CS. 2005. Use of the Rhodopseudomonas palustris genome sequence to identify a single amino acid that contributes to the activity of a coenzyme A ligase with chlorinated substrates. Mol. Microbiol. 55:1151–1159. 10.1111/j.1365-2958.2004.04452.x [DOI] [PubMed] [Google Scholar]
- 13.Pelletier DA, Harwood CS. 2000. 2-Hydroxycyclohexanecarboxyl coenzyme A dehydrogenase, an enzyme characteristic of the anaerobic benzoate degradation pathway used by Rhodopseudomonas palustris. J. Bacteriol. 182:2753–2760. 10.1128/JB.182.10.2753-2760.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perrotta J, Harwood CS. 1994. Anaerobic metabolism of cyclohex-1-ene carboxylate, a proposed intermediate of benzoate degradation by Rhodopseudomonas palustris. Appl. Environ. Microbiol. 60:1775–1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koch J, Eisenreich W, Bacher A, Fuchs G. 1993. Products of enzymatic reduction of benzoyl-CoA, a key reaction in anaerobic aromatic metabolism. Eur. J. Biochem. 211:649–661. 10.1111/j.1432-1033.1993.tb17593.x [DOI] [PubMed] [Google Scholar]
- 16.Egland PG, Pelletier DA, Dispensa M, Gibson J, Harwood CS. 1997. A cluster of bacterial genes for anaerobic benzene ring biodegradation. Proc. Natl. Acad. Sci. U. S. A. 94:6484–6489. 10.1073/pnas.94.12.6484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holmes DE, Risso C, Smith JA, Lovley DR. 2012. Genome-scale analysis of anaerobic benzoate and phenol metabolism in the hyperthermophilic archaeon Ferroglobus placidus. ISME J. 6:146–157. 10.1038/ismej.2011.88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peters F, Shinoda Y, McInerney MJ, Boll M. 2007. Cyclohexa-1,5-diene-1-carbonyl-coenzyme A (CoA) hydratases of Geobacter metallireducens and Syntrophus aciditrophicus: evidence for a common benzoyl-CoA degradation pathway in facultative and strict anaerobes. J. Bacteriol. 189:1055–1060. 10.1128/JB.01467-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Löffler C, Kuntze K, Vazquez JR, Rugor A, Kung JW, Böttcher A, Boll M. 2011. Occurrence, genes and expression of the W/Se-containing class II benzoyl-coenzyme A reductases in anaerobic bacteria. Environ. Microbiol. 13:696–709. 10.1111/j.1462-2920.2010.02374.x [DOI] [PubMed] [Google Scholar]
- 20.Kuntze K, Shinoda Y, Mouttaki H, McInerney MJ, Vogt C, Richnow HH, Boll M. 2008. 6-Oxocyclohex-1-ene-1-carbonyl-coenzyme A hydrolases from obligately anaerobic bacteria: characterization and identification of its gene as a functional marker for aromatic compounds degrading anaerobes. Environ. Microbiol. 10:1547–1556. 10.1111/j.1462-2920.2008.01570.x [DOI] [PubMed] [Google Scholar]
- 21.Peters F, Rother M, Boll M. 2004. Selenocysteine-containing proteins in anaerobic benzoate metabolism of Desulfococcus multivorans. J. Bacteriol. 186:2156–2163. 10.1128/JB.186.7.2156-2163.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elshahed MS, Bhupathirju VH, Wofford NQ, Nanny MA, McInerney MJ. 2001. Metabolism of benzoate, cyclohex-1-ene carboxylate, and cyclohexane carboxylate by “Syntrophus aciditrophicus” strain SB in syntrophic association with H2-using microorganisms. Appl. Environ. Microbiol. 67:1728–1738. 10.1128/AEM.67.4.1728-1738.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thiele B, Rieder O, Jehmlich N, von Bergen M, Müller M, Boll M. 2008. Aromatizing cyclohexa-1,5-diene-1-carboxyl-coenzyme A oxidase. Characterization and its role in anaerobic aromatic metabolism. J. Biol. Chem. 283:20713–20721. 10.1074/jbc.M802841200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gross GG, Zenk MH. 1966. Darstellung und Eigenschaften von Coenzym A-Thiolestern substituierter Zimtsauren. Z. Naturforschg. 21b:683–690 [Google Scholar]
- 25.Lovley DR, Phillips EJ. 1988. Novel mode of microbial energy metabolism: organic carbon oxidation coupled to dissimilatory reduction of iron or manganese. Appl. Environ. Microbiol. 54:1472–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aranda PS, LaJoie DM, Jorcyk CL. 2012. Bleach gel: a simple agarose gel for analyzing RNA quality. Electrophoresis 33:366–369. 10.1002/elps.201100335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koressaar T, Remm M. 2007. Enhancements and modifications of primer design program Primer3. Bioinformatics 23:1289–1291. 10.1093/bioinformatics/btm091 [DOI] [PubMed] [Google Scholar]
- 28.Untergasser A, Cutcutache I, Koresaar T, Ye J, Faircloth BC, Remm M, Rozen SG. 2012. Primer3—new capabilities and interfaces. Nucleic Acids Res. 40:e115. 10.1093/nar/gks596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Spelemann F. 2002. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3:RESEARCH0034.1–RESEARCH0034.12. 10.1186/gb-2002-3-7-research0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kung JW, Seifert J, von Bergen M, Boll M. 2013. Cyclohexanecarboxyl-coenzyme A (CoA) and cyclohex-1-ene-1-carboxyl-CoA dehydrogenases, two enzymes involved in the fermentation of benzoate and crotonate in Syntrophus aciditrophicus. J. Bacteriol. 195:3193–3200. 10.1128/JB.00322-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wrighton KC, Thomas BC, Sharon I, Miller CS, Castelle CJ, VerBerkmoes NC, Wilkins MJ, Hettich RL, Lipton MS, Williams KH, Long PE, Banfield JF. 2012. Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337:1661–1665. 10.1126/science.1224041 [DOI] [PubMed] [Google Scholar]
- 33.Oberender J, Kung JW, Seifert J, von Bergen M, Boll M. 2012. Identification and characterization of a succinyl-coenzyme A (CoA):benzoate CoA transferase in Geobacter metallireducens. J. Bacteriol. 194:2501–2508. 10.1128/JB.00306-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heider J. 2001. A new family of CoA-transferases. FEBS Lett. 509:345–349. 10.1016/S0014-5793(01)03178-7 [DOI] [PubMed] [Google Scholar]
- 35.Wischgoll S, Heintz D, Peters F, Erxleben A, Sarnighausen E, Reski R, Van Dorsselaer A, Boll M. 2005. Gene clusters involved in anaerobic benzoate degradation of Geobacter metallireducens. Mol. Microbiol. 58:1238–1252. 10.1111/j.1365-2958.2005.04909.x [DOI] [PubMed] [Google Scholar]
- 36.Thorpe C, Kim JJ. 1995. Structure and mechanism of action of the acyl-CoA dehydrogenases. FASEB J. 9:718–725 [DOI] [PubMed] [Google Scholar]
- 37.Boll M, Löffler C, Morris BE, Kung JW. 2014. Anaerobic degradation of homocyclic aromatic compounds via arylcarboxyl-coenzyme A esters: organisms, strategies and key enzymes. Environ. Microbiol. 16:612–627. 10.1111/1462-2920.12328 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.