

Abstract
Biofilm formation on central lines or peripheral catheters is a serious threat to patient well-being. Contaminated vascular devices can act as a nidus for bloodstream infection and systemic pathogen dissemination. Staphylococcal biofilms are the most common cause of central-line-associated bloodstream infections, and antibiotic resistance makes them difficult to treat. As an alternative to antibiotic intervention, we sought to identify anti-staphylococcal biofilm targets for the development of a vaccine or antibody prophylactic. A screening strategy was devised using a microfluidic system to test antibody-mediated biofilm inhibition under biologically relevant conditions of shear flow. Affinity-purified polyclonal antibodies to target antigen PhnD inhibited both Staphylococcus epidermidis and S. aureus biofilms. PhnD-specific antibodies blocked biofilm development at the initial attachment and aggregation stages, and deletion of phnD inhibited normal biofilm formation. We further adapted our microfluidic biofilm system to monitor the interaction of human neutrophils with staphylococcal biofilms and demonstrated that PhnD-specific antibodies also serve as opsonins to enhance neutrophil binding, motility, and biofilm engulfment. These data support the identification of PhnD as a lead target for biofilm intervention strategies performed either by vaccination or through passive administration of antibodies.
INTRODUCTION
The Centers for Disease Control and Prevention estimates that ∼41,000 cases of central-line associated bloodstream infection (CLABSI) occurred in United States hospitals in 2009, with a similar number in outpatient hemodialysis centers (∼37,000 cases in 2008) (1). In addition to the significant patient morbidity associated with these infections, direct medical costs are approximately $20,000 per occurrence (2, 3). As such, prevention of CLABSI has been declared a United States health care priority (4). The incidence of CLABSI in hospital intensive care units has gone down in recent years relative to its baseline measurement in 2001 (1), primarily due to implementation of evidence-based best practices for the insertion and maintenance of central lines (5). However, the combined burden of infection in intensive care units, inpatient wards, and outpatient hemodialysis centers is still unacceptably high. Given the continuing medical need, our goal is to develop a new vaccine or monoclonal antibody product to advance prevention efforts.
Data from the National Healthcare Safety Network show that coagulase-negative staphylococci are the leading cause of CLABSI (20.5%), followed by Staphylococcus aureus (12.3%) and then Enterococcus faecalis (8.8%) (6). S. epidermidis is the most clinically relevant species among the coagulase-negative staphylococci, accounting for >70% of catheter-related infections within the group (7, 8). Therefore, we focused on S. epidermidis for our studies but maintained an emphasis on targets conserved across the entire genus.
S. epidermidis is normally a harmless commensal found in abundance on skin and mucous membranes. Its success as an opportunistic pathogen causing CLABSI is due primarily to its ability to colonize and form biofilms on catheters, which subsequently act as a nidus for systemic dissemination (reviewed in references 9 and 10). Biofilms are communities of individual cells held together by a secreted matrix of proteins, polysaccharides, and extracellular DNA. The matrix protects ensconced bacteria from environmental stresses such as host defenses and antibiotics; as a result, biofilms are very difficult to treat, and clinical guidelines for CLABSI generally call for catheter removal in addition to antibiotic therapy (11). Since biofilms are integral to the establishment of staphylococcal CLABSI, we focused our intervention effort on this essential virulence factor.
In this report, we describe the identification and characterization of an anti-staphylococcal biofilm target for vaccination or passive antibody prophylaxis. Traditional biofilm assays using microtiter plates (12) are not suitable for modeling CLABSI because static conditions cannot reproduce the fluid dynamics of the in vivo milieu. For example, the circulation of blood around vascular catheters exerts shear flow forces on developing biofilms, replenishes nutrients, and removes bacterial waste products and signaling molecules—none of which are reproduced by the microtiter plate method. Therefore, we developed a biofilm model using a flow-based live cell imaging system (BioFlux 1000; Fluxion) (13, 14) and performed quantitative comparisons among new and previously described targets using time-lapse video microscopy for longitudinal monitoring of the complete biofilm development cycle. This flow-based assay was further adapted to assess potential host-pathogen interactions via the introduction of freshly isolated human neutrophils to the system. Based on the accumulated data, staphylococcal protein PhnD is presented as a leading candidate for antibody-mediated biofilm inhibition strategies for the prevention of CLABSI.
MATERIALS AND METHODS
Strains, media, and growth conditions.
See Table S1 in the supplemental material for a detailed list of strains and plasmids. S. epidermidis, S. aureus, S. haemolyticus, and S. hominis were grown at 30°C or 37°C in tryptic soy broth (TSB). Plasmids were transformed into staphylococci by the use of a published protocol (15). In brief, staphylococci were grown in basic medium to the mid-exponential phase, washed 4 times, and then resuspended in 10% (vol/vol) glycerol at a cell concentration of 1E10 CFU/ml. The resulting competent cells were incubated with 3 μg of plasmid DNA and then subjected to electroporation at 2.5 kV, 200 Ω, and 25 μF. After 2 h of incubation in TSB at 30°C, transformants were selected with the appropriate antibiotics (Sigma-Aldrich), which were used at the following concentrations: kanamycin at 50 μg/ml, trimethoprim at 10 μg/ml, erythromycin at 10 μg/ml, and ampicillin at 100 μg/ml.
To construct the 1457 ΔphnD strain, pHL008 was transformed into strain 1457 with selection on erythromycin at 30°C to select for the replication-competent plasmid. Transformants were then shifted to the restrictive temperature (42°C) to force chromosomal integration of pHL008 at the phnD locus. Single clones were screened for sensitivity to erythromycin and resistance to trimethoprim as a marker for the allele exchange. Positive clones were confirmed by PCR and Western analysis.
Plasmid construction.
The pHL007 fluorescent reporter plasmid was constructed by fusing 500 bp of the ftsA 5′ untranslated region (constitutive promoter) to the coding sequence for a red fluorescence protein that was codon optimized for expression in S. epidermidis (GenScript, Piscataway, NJ). This fragment was cloned into pUC19 via SalI-EcoRI. The resulting construct was then cut and ligated into the PstI site of pE194.
The pHL008 allele exchange vector was constructed by cloning dhfr (conferring trimethoprim resistance) between 800 bp of upstream DNA and 800 bp of downstream DNA flanking phnD. This cassette was cloned into pUC19 as an EcoRI-PstI fragment. The resulting construct was cloned into the PstI site of pROJ6448.
Expression plasmids were constructed for the designated amino acids of each protein as follows: PhnD/SERP2286 (amino acids 21 to 318), SesC/SERP2264 (amino acids 34 to 649), GapA/SERP0442 (amino acids 1 to 336), and EmbP/SERP1011 (amino acids 6599 to 7340). The appropriate nucleotide sequences were amplified by PCR from RP62A chromosomal DNA and cloned into pET-30 Ek/LIC according to the manufacturer's instructions for expression with an N-terminal His tag (Novagen). Complete plasmid sequences are available upon request.
Antigen expression and purification and generation of antibodies.
Proteins were expressed in Escherichia coli BL21(DE3) using Overnight Express Autoinduction System 1 (EMD Millipore), per the manufacturer's protocol. For Aap/SERP2398, a 20-amino-acid peptide (PGKPGVKNPDTGEVVTPPVD) was synthesized for immunization (GenScript), based on its published identification as an antibiofilm epitope (16). Expression cultures were harvested by centrifugation, and cell pellets were stored at −80°C.
Recombinant His-tagged proteins were purified from the soluble fraction under native conditions using Ni Sepharose High Performance resin (GE Healthcare Life Sciences). Cells were thawed and resuspended in Ni Sepharose binding buffer (20 mM sodium phosphate, 0.3 to 0.5 M NaCl, 10% glycerol, 30 mM imidazole, pH 7 to 8) with EDTA-free protease inhibitor tablets (Roche). Cell disruption was performed by two passages through a microfluidizer at 18,000 lb/in2 on ice. The resulting lysate was clarified from insoluble material by centrifugation at 30,000 to 45,000 × g for 40 to 60 min at 4°C followed by filtration through a 0.22-μm-pore-size filter prior to column binding. Ni Sepharose binding buffer with 0.05% Tween 80 was used to wash bound protein to ensure endotoxin removal followed by extensive washing with binding buffer alone. Protein was eluted by a linear imidazole gradient (30 to 250 mM) in binding buffer. When purity was <95% or endotoxin levels were >1 endotoxin unit/μg, additional purification procedures—including the use of hydrophobic interaction chromatography (HiTrap Phenyl HP resin) and/or ion exchange chromatography (HiTrap Q HP resin) with buffer systems recommended by GE Healthcare Life Sciences—were employed. Purified proteins were dialyzed against phosphate-buffered saline (PBS)–10% glycerol and then sterilized by passage through a 0.22-μm-pore-size filter. The purity and stability of purified proteins were analyzed by SDS-PAGE and Western blot analyses. Endotoxin levels were estimated by the use of Endosafe PTS cartridges with an Endosafe portable test system.
Affinity-purified recombinant protein or keyhole limpet hemocyanin-conjugated Aap peptide was used to generate rabbit antisera using a 10-week immunization protocol (GenScript). Antigen-specific antibodies were affinity purified from the harvested sera using the purified proteins or peptide (GenScript). All antibodies were dialyzed into PBS and brought to a standard working concentration of 10 mg/ml. Affinity-purified rabbit anti-rat IgG antibody (R3756; Sigma-Aldrich) was used as an irrelevant control antibody in most experiments.
Flow-based biofilm inhibition assays.
A BioFlux 1000 system (Fluxion) with a Nikon Eclipse Ti-S microscope and temperature-controlled housing was used for all imaging experiments. Automated microscopy and image processing were performed with BioFlux Montage software.
For experiments performed in prophylactic mode, BioFlux 48-well plates (Fluxion) (catalog no. 910-0047), allowing up to 24 individual treatment conditions, were primed with TSB plus the indicated concentration of antibody from the outlet well at a shear setting of 2 dyn/cm2 for 10 min. Cultures of S. epidermidis or S. aureus were grown to the mid-log phase, briefly sonicated using a low power output to break up clumps, and normalized to an optical density at 600 nm (OD600) of 0.15. Bacteria were preincubated with the indicated concentration of antibody for 25 min at room temperature. For specificity of inhibition experiments, purified PhnD antigen or bovine serum albumin (BSA) (Sigma-Aldrich) was added to the priming medium and bacterial preincubation step at 20 μg/ml. Bacteria were seeded from the outlet well into the channel and viewing window at a shear setting of 2 dyn/cm2 for 3 s. After 1 h of incubation at 30°C or 37°C (as specified in the figure legend), TSB with the indicated concentration of antibody was flowed from the inlet well at a shear setting of 0.4 dyn/cm2 for the duration of the experiment. Images were automatically acquired every 20 min at multiple stage positions with bright-field illumination; images were also acquired in the red channel (86013v2 tetramethylrhodamine isothiocyanate [TRITC] filter set; Chroma, Bellows Falls, VT) using a 200-ms exposure time when strain 1457-FL was used. The background-corrected average pixel intensity per image was used to quantify biofilm formation for different antibody treatments.
For experiments performed in early therapeutic mode, the protocol was identical to that described above, except that BioFlux plates were primed using TSB without antibodies, and bacteria were not preincubated with antibodies prior to seeding. Under these conditions, bacteria were allowed to undergo initial attachment to the channel surface without antibody interference.
For experiments performed with human neutrophils, BioFlux 48-well plates were primed with TSB at a shear setting of 2 dyn/cm2 for 10 min. Bacterial cultures were grown to the late log phase, briefly sonicated at a low power output to break up clumps, and normalized to an OD600 of 0.8. S. epidermidis 1457 was seeded from the outlet well into the channel and viewing window at a shear setting of 2 dyn/cm2 for 3 s. After 30 min of incubation at 37°C, TSB was flowed from inlet well at a shear setting of 0.4 dyn/cm2 for 3.5 h to allow the establishment of biofilm. During the incubation period, polymorphonuclear leukocytes (PMNs [primarily neutrophils]) were purified from freshly drawn, EDTA-treated human blood (Research Blood Components, Brighton, MA). Density fractionation of PMNs was performed using Polymorphprep (Axis-Shield, Oslo, Norway) according to the manufacturer's instructions. Contaminating red blood cells were lysed with ACK lysis buffer (Lonza) for 45 s, and PMNs were then washed four times with Dulbecco's modified Eagle's medium (DMEM)-F12 medium (Invitrogen) and enumerated using a Vicell counter. The preparation was adjusted to 1E6 viable cells/ml. TSB from the inlet well (used for the establishment of biofilm) was then replaced with PMNs in DMEM-F12 suspension. PMNs were flowed at a shear setting of 0.2 dyn/cm2, and images were acquired at 2-min intervals under bright-field illumination conditions.
For experiments performed with human plasma, BioFlux 48-well plates were primed with Tris-buffered saline (TBS; 20 mM Tris [pH 6.8], 150 mM NaCl) plus 200 μg/ml of antibody from the outlet well at a shear setting of 2 dyn/cm2 for 10 min. Cultures of S. epidermidis strain 1457-FL were grown to the mid-log phase in TSB and then harvested by low-speed centrifugation, resuspended in an equal volume of filtered (0.45 μm pore size) 100% human plasma (Innovative Research, Novi, MI), and grown for an additional 3 h. The bacteria were harvested by low-speed centrifugation and then resuspended in TBS and briefly sonicated at a low power output to break up clumps and finally normalized to an OD600 of 0.5. Bacteria were preincubated with 200 μg/ml of anti-PhnD or control antibody for 20 min at room temperature. Seeding was performed in TBS at a shear setting of 2 dyn/cm2 for 3 s. After incubation for 1 h at 37°C, 100% human plasma with the indicated concentration of antibody was flowed from the inlet well at a shear setting of 0.1 dyn/cm2 for the duration of the experiment (using 1.4 cP as the viscosity of human plasma at 37°C). Images were automatically acquired every 20 min at multiple stage positions (86013v2 TRITC filter set; Chroma, Bellows Falls, VT) using a 200-ms exposure time. The background-corrected average pixel intensity per image was used to quantify biofilm formation for different antibody treatments.
Immunoblotting.
S. epidermidis, S. aureus, S. haemolyticus, and S. hominis strains were grown to the mid-log phase and normalized to an OD600 of 1.0 to ensure equal loading. Whole-cell lysates were separated by SDS-PAGE using NuPAGE Novex 4% to 12% Bis-Tris gels (Invitrogen). Proteins were transferred onto nitrocellulose using an iBlot apparatus according to the manufacturer's instructions (Invitrogen) and probed for PhnD using affinity-purified rabbit antibodies (1/2,000) and a donkey anti-rabbit secondary antibody conjugated to alkaline phosphatase.
RESULTS
Identifying antibodies with antibiofilm activity.
Since antibodies cannot penetrate a bacterial cell, their targets must be externally accessible. Therefore, publically available bioinformatic tools (expasy.org) were used to identify surface proteins of S. epidermidis as potential candidates for antibody-mediated biofilm inhibition. Predicted surface proteins were prioritized based on homology across the entire genus. The highest-ranking candidates were cloned and expressed in E. coli and then purified and used to generate hyperimmune sera in rabbits. Sera were affinity purified to obtain antigen-specific antibodies, and the resulting antibodies were screened for antibiofilm activity in vitro (Fig. 1A).
FIG 1.
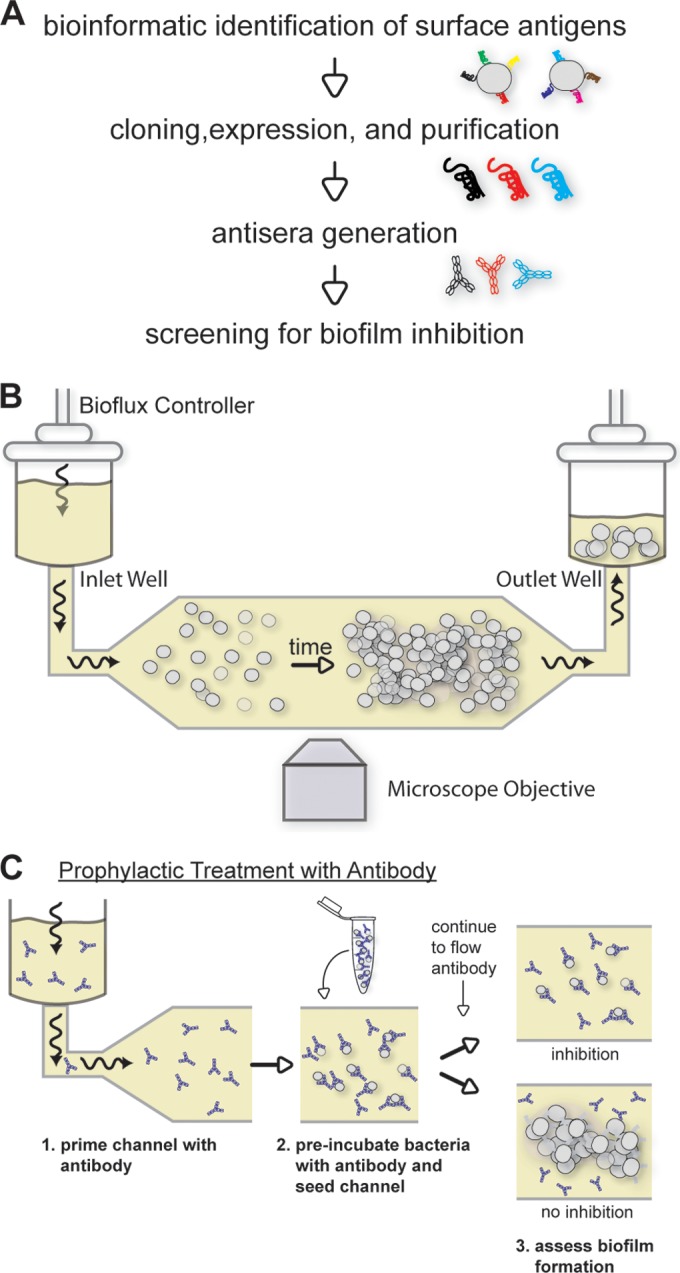
Overview of approach to identify biofilm-inhibiting antibodies. (A) High-level strategy for the identification of targets. (B) Schematic of the flow-based system for monitoring biofilm development. The BioFlux Controller applies pneumatic pressure to the inlet well which drives media at a defined flow rate over the attached bacteria in the microscope viewing window. (C) Diagram of the assay for biofilm inhibition, in which antibodies are present throughout (model for prophylactic intervention). Biofilm formation is monitored for the duration of the experiment. Wavy arrows indicate the direction of flow.
An important consideration for modeling nosocomial biofilm infections in vitro is to account for the effects of fluid dynamics in vivo; for example, blood flowing around an indwelling catheter exerts significant shear flow on a developing biofilm. Therefore, to mimic conditions that catheter-bound staphylococci encounter in the human circulatory system, biofilms were grown under conditions of flow and imaged by time-lapse microscopy using a Fluxion BioFlux 1000 system (Fig. 1B). This automated, fully integrated platform uses a multiwell plate format to allow sufficient throughput for screening (input and output wells are linked by microfluidic flow channels). A schematic of the assay modeling prophylactic administration of antibodies for biofilm inhibition is shown in Fig. 1C.
When grown in the presence of nonspecific control antibody, S. epidermidis 1457 attaches to the microfluidic flow channel and forms a robust biofilm (Fig. 2A; see also Movie S1 in the supplemental material). Test antibodies were screened for biofilm inhibition at a concentration of 200 μg/ml. Many candidates had no effect (data not shown), but antibodies to PhnD (SERP2286; annotated as a phosphonate ABC transporter substrate binding protein) strongly inhibited bacterial attachment and subsequent biofilm formation (Fig. 2A; see also Movie S1). The antibiofilm activity of the PhnD antibodies was dose dependent at from 5 to 100 μg/ml (Fig. 2B). To ensure that biofilm inhibition was specific to the PhnD target, the experiment was repeated in the presence of exogenous purified PhnD to bind and sequester PhnD-specific antibodies. Antibiofilm activity was lost in the presence of exogenous PhnD, whereas the addition of an irrelevant control protein (BSA) had no effect on inhibition (Fig. 2C). PhnD antibodies also showed antibiofilm activity in the microtiter plate assay based on endpoint staining with crystal violet (see Fig. S1). Of note, planktonic growth experiments were also performed in the presence or absence of PhnD antibody and showed no effect, suggesting that antibodies do not inhibit biofilm development through inhibition of S. epidermidis growth (data not shown).
FIG 2.

PhnD antibodies inhibit S. epidermidis biofilm formation. (A) Longitudinal monitoring of S. epidermidis 1457 biofilms grown at 37°C in the presence of PhnD or control (rabbit anti-rat IgG) antibodies (Ab) at 200 μg/ml. (B) Dose-dependent biofilm inhibition by PhnD antibodies at 37°C. Images for each antibody concentration were taken after 6 h and 10 h of biofilm development. (C) The antibiofilm activity of PhnD antibodies is target specific. Exogenous PhnD protein (but not BSA) is able to absorb specific antibodies and block the antibiofilm activity. A schematic illustration is provided below the microscope images. Scale bar, 15 μm.
Quantifying antibiofilm activity.
A fluorescent reporter strain of S. epidermidis (1457-FL) was constructed to enable a quantitative readout for antibiofilm activity. The reporter strain was comparable to its parent with respect to growth kinetics and biofilm development and showed a strong correlation between fluorescent signal intensity and cell number (data not shown). Treatment with PhnD antibodies at 100 μg/ml inhibited surface attachment and biofilm development of 1457-FL (Fig. 3A; see also Movie S2 in the supplemental material), and the endpoint reduction in fluorescent signal was 93% versus the control.
FIG 3.

Quantifying antibiofilm activity using a fluorescent reporter strain. (A) Longitudinal monitoring of S. epidermidis 1457-FL grown at 30°C in the presence of PhnD or control (rabbit anti-rat IgG) antibodies at 100 μg/ml. Images represent results of monitoring every 3 h at between 6 and 18 h of biofilm development. Scale bar, 20 μm. (B) Endpoint images at 20 h of 1457-FL biofilms grown at 30°C and treated with antibodies at 100 μg/ml. Scale bar, 30 μm. (C) Longitudinal quantification of the experiment described in the panel B legend using the average pixel intensity of images captured at 20-min intervals for 20 h. A.U., arbitrary units.
The fluorescent reporter was then used to compare PhnD antibodies to those for other targets, including several proteins with a known role in S. epidermidis biofilm formation: EmbP (SERP1011), Aap (SERP2398), and SesC (SERP2264) (17–20). Treatment with EmbP antibodies also led to a large decrease in biofilm formation (91% reduction versus the control); however, none of the other antibodies showed substantial effects at the endpoint (Fig. 3B). Longitudinal quantification showed that antibodies to SesC caused an initial delay in biofilm establishment, but this effect was eventually overwhelmed (Fig. 3C). Aap antibodies seemed to increase biofilm formation (Fig. 3B and C); given the repetitive structure of the Aap protein, a plausible explanation for this biofilm enhancement is that antibodies cross-linked neighboring cells and increased bacterial aggregation. Thus, antibody binding to the cell surface is not by itself sufficient to prevent biofilm formation, and inhibition of target function must also play a role.
PhnD antibodies block initial attachment and early aggregation.
To explore the kinetics of intervention with PhnD antibodies, we introduced antibodies at different stages of biofilm development (Fig. 4A). All previous experiments were performed in prophylactic mode, with antibodies present throughout (Fig. 1C). To test early therapeutic intervention, antibodies were not added until after bacterial attachment (Fig. 4B). In this format, the assay measures the ability of antibodies to inhibit at the aggregation stage. Under these conditions, PhnD antibody treatment still led to a significant decrease in biofilm development (85% endpoint reduction versus the control) (Fig. 4C and D; see also Movie S3 in the supplemental material). EmbP antibodies also reduced biofilm formation (65% versus the control), whereas the remaining antibodies either had no effect (anti-GapA and SERP0442) or led to increased biofilm levels (anti-SesC and -Aap; Fig. 4C and D). Intervention in the late therapeutic window was also tested by adding antibodies to a mature biofilm (Fig. 4A), but no effect was observed for any candidate (data not shown). Therefore, PhnD and EmbP antibodies inhibit biofilm formation at both the initial-attachment and early-aggregation stages of development but do not affect maturation or dispersal. Taken together, these quantitative data support the identification of PhnD as a lead target for antibody-mediated biofilm intervention strategies.
FIG 4.
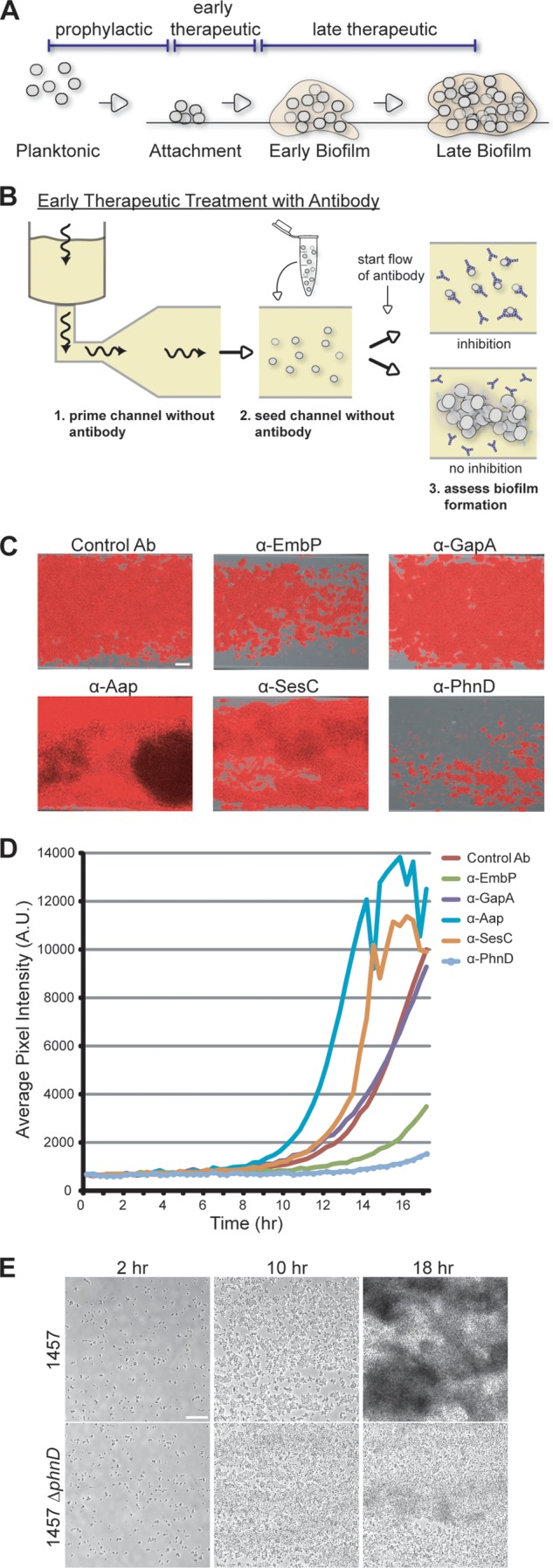
Assessing biofilm inhibition by antibodies added postattachment. (A) Biofilm development cycle. Possible antibody intervention points include the following: prophylactic (prior to bacterial attachment), early therapeutic (before intercellular aggregation), and late therapeutic (after biofilm establishment and maturation). (B) Diagram of the assay for biofilm inhibition, in which antibodies are introduced postattachment (model for early therapeutic intervention). Biofilm formation is monitored for the duration of the experiment. Wavy arrows indicate the direction of flow. (C) Endpoint images at 18 h of 1457-FL grown at 30°C and treated with antibodies postattachment at 100 μg/ml. Scale bar, 30 μm. (D) Longitudinal quantification of the experiment described in the panel C legend using average pixel intensity of images captured at 20-min intervals for 18 h. (E) Deletion of phnD results in defective biofilm formation. Biofilms of S. epidermidis 1457 and its isogenic ΔphnD mutant were grown under conditions of flow at 30°C. Images represent biofilm development after 2, 10, and 18 h. Scale bar, 25 μm.
Deletion of phnD results in defective biofilm formation.
To investigate the antibiofilm mechanism of PhnD antibodies, we constructed a phnD knockout strain in the S. epidermidis 1457 background. While planktonic growth levels were indistinguishable between the parental and mutant strains (data not shown), biofilm formation by the mutant strain was defective although not completely abolished (Fig. 4E; see also Movie S4 in the supplemental material). The ΔphnD strain formed biofilms that were less dense and only loosely attached to the channel surface. It is not intuitive that PhnD antibodies should be more inhibitory to biofilm formation than deletion of their target. One possibility is that antibiofilm activity is a cumulative effect of both functional target inhibition and steric interference of surface attachment by antibody binding.
PhnD antibodies promote phagocytosis of S. epidermidis biofilms.
In a human infection, antibodies work in concert with other components of the immune system, often as opsonins to promote pathogen uptake by professional phagocytes. However, multiple studies have shown that staphylococcal biofilms impede the normal phagocytic activity of human neutrophils (reviewed in reference 21). Given the antibiofilm activity of PhnD antibodies, we hypothesized that they might circumvent the matrix defense and cooperate with neutrophils to promote biofilm clearance (Fig. 5A). To test this hypothesis, biofilms were formed and then freshly isolated human neutrophils were introduced in the presence of control or PhnD antibodies (all under conditions of flow). Addition of PhnD antibodies led to an immediate and marked increase in neutrophil binding and active engulfment of the S. epidermidis biofilm versus the control (Fig. 5B; see also Movie S5 in the supplemental material). In 4 independent experiments, treatment with PhnD antibodies increased neutrophil binding more than 8-fold versus the control. Tracking of individual neutrophils showed that a single cell was capable of moving toward and engulfing large numbers of biofilm bacteria (Fig. 5C; see also Movie S6). These results suggest that PhnD antibodies may enhance neutrophil phagocytosis of staphylococcal biofilms in vivo, which would act as a complement to their direct antibiofilm effects on the bacteria themselves.
FIG 5.

PhnD antibodies enhance neutrophil phagocytosis of S. epidermidis biofilms. (A) Diagram of the opsonophagocytosis assay on preformed biofilms under conditions of flow. (B) Neutrophils exhibit increased binding, motility, and engulfment of biofilms in the presence of PhnD antibody versus the control (rabbit anti-rat IgG; both antibodies at 5 μg/ml). Images were taken at 0, 20, 100, and 180 min after introduction of neutrophils. Scale bar, 20 μm. (C) Tracking the antibiofilm activity of an individual neutrophil in the presence of PhnD antibodies. (Top) A single neutrophil (arrow) is capable of moving toward and engulfing large numbers of biofilm bacteria. (Bottom) Schematic representation of top panels to highlight the individual neutrophil (colored purple) and its path (red line) as it engulfs biofilm. Other neutrophils in the field of view are depicted in green. Scale bar, 10 μm.
PhnD antibodies are active against biofilms with different matrix compositions.
Strains of S. epidermidis can differ significantly in biofilm phenotype. For example, the ica operon that codes for production of the matrix polysaccharide PNAG (poly-N-acetylglucosamine) is present only in a subset of clinical isolates (22–24). Since the properties of PNAG-positive and -negative biofilms are distinct, we wanted to test the antibiofilm activity of PhnD antibodies in both backgrounds. We identified a suitable PNAG-negative clinical isolate (S. epidermidis 7291) by screening for the presence of the ica locus. Strain 7291 was able to form biofilms under flow conditions (Fig. 6A, top), but its biofilms did not appear as dense or firmly attached as those formed by 1457. Once again, treatment with PhnD antibodies dramatically inhibited biofilm formation (Fig. 6A, bottom). This result suggests that the antibiofilm activity of PhnD antibodies is independent of the matrix, which implies coverage against a broad range of clinically relevant S. epidermidis strains.
FIG 6.
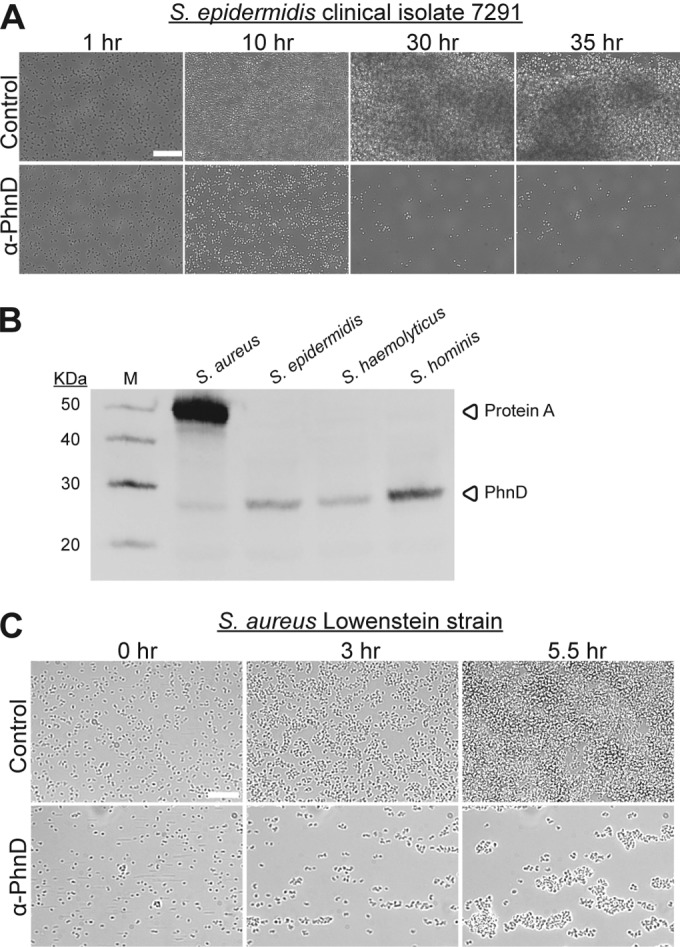
PhnD antibodies exhibit cross-reactivity and antibiofilm activity against multiple staphylococcal strains. (A) Longitudinal development of S. epidermidis 7291 biofilms grown at 37°C in the presence of PhnD or control antibodies (rabbit anti-rat IgG; each at 50 μg/ml). (B) Detection of PhnD homologs from multiple staphylococcal strains by Western immunoblotting with antibodies generated to PhnD from S. epidermidis. (C) PhnD antibodies inhibit S. aureus biofilm formation. Longitudinal development of S. aureus Lowenstein biofilms grown at 37°C in the presence of PhnD or control antibodies (rabbit anti-rat IgG; each at 100 μg/ml). Scale bar, 20 μm.
PhnD conservation and cross-species biofilm inhibition of S. aureus.
PhnD is a well-conserved (>75% identity) protein across all coagulase-negative staphylococci and S. aureus. Western blotting with polyclonal antibodies generated to PhnD from S. epidermidis showed cross-reactivity with its homologs from S. aureus (MRSA252), S. haemolyticus (SM 131), and S. hominis (R22) (Fig. 6B). To look at cross-species inhibition of biofilms, antibodies generated to PhnD from S. epidermidis were tested against S. aureus strain Lowenstein (a capsule-positive strain); strong antibiofilm activity was observed (Fig. 6C). These results suggest that PhnD antibody intervention strategies could be effective against many species of biofilm-forming staphylococci.
PhnD antibodies inhibit biofilms grown in plasma.
Biofilm formation is a regulated process that depends on environmental conditions. Likewise, bacterial gene expression patterns are subject to the medium in which cells are grown. In order to validate the biological relevance of PhnD as a target for the prevention of CLABSI, we tested the prophylactic effect of PhnD antibodies against biofilms grown in human plasma. Briefly, S. epidermidis 1457-FL grown in rich media was subcultured into 100% human plasma and grown for 3 h prior to seeding. In general, biofilms grown in human plasma developed more slowly and were less stable than those established using rich media. Although the dynamic range of the assay was reduced versus standard conditions, antibodies to PhnD still showed a clear antibiofilm effect versus the control (Fig. 7; see also Movie S7 in the supplemental material). These results demonstrate that PhnD antibodies are effective at inhibiting staphylococcal biofilms under conditions of growth and gene expression very similar to the in vivo milieu conditions.
FIG 7.
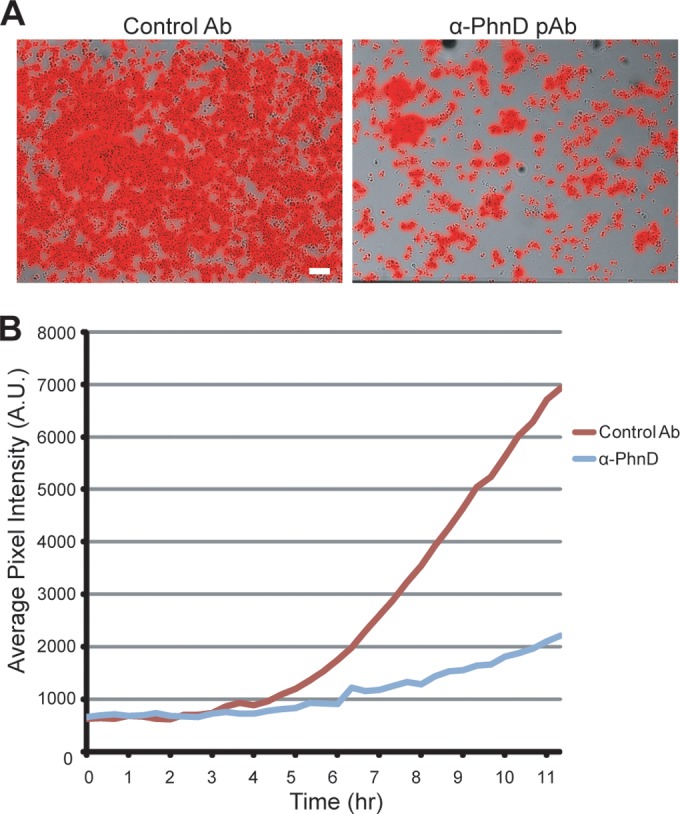
PhnD antibodies inhibit S. epidermidis biofilms grown in human plasma. (A) Endpoint images of 1457-FL grown for 12 h at 37°C with antibodies as indicated. All antibodies were administered in prophylactic mode at 200 μg/ml. Scale bar, 20 μm. (B) Longitudinal quantification of the experiment described in the panel A legend using average pixel intensity of images captured at 20-min intervals for 12 h.
DISCUSSION
Our goal was to identify a vaccine or antibody candidate to prevent CLABSI caused by staphylococcal biofilms (6, 25). We established a flow-based in vitro model using a BioFlux 1000 system (Fluxion) to screen antibodies for biofilm inhibition under conditions that simulate the fluid dynamics of circulating blood (Fig. 1B and C and 3B). This model represents a substantial improvement over standard static assays performed in microtiter plates, in which the wells are closed environments that rapidly exhaust nutrients, accumulate waste products, and do not subject the bacteria to shear flow forces. We further improved this model by developing a fluorescent reporter strain to allow quantitative comparisons among antibody candidates.
In our screen, antibodies to PhnD (SERP2286, annotated as a phosphonate ABC transporter substrate binding protein) inhibited S. epidermidis biofilms in a dose-dependent manner (Fig. 2), achieving nearly complete prevention of growth at biologically relevant concentrations. PhnD-specific antibodies outperformed those targeting other known surface proteins, including those with a predicted role in biofilm formation such as SesC and Aap (Fig. 3 and 4). Given that all targets in Fig. 3 and 4 are surface exposed (18, 19, 26, 27), it is clear that biofilm inhibition requires more than just antibody binding to the cell surface. In fact, abundant antibody binding to highly expressed surface antigens may induce cell aggregation and increase biofilm formation, as observed for Aap (Fig. 3 and 4) and SesC (Fig. 4); this phenomenon has been documented previously for Aap (16). Antibodies to EmbP performed well in the prophylactic assay (Fig. 3), but PhnD-specific antibodies were clearly superior when administered in early therapeutic mode (Fig. 4). Of note, subdomains of Aap and EmbP were used to generate the antibodies in our studies; therefore, we cannot rule out the possibility that alternate regions of the same antigens could result in antibodies with a different activity profile.
During our time-lapse monitoring, we observed for all antibody candidates with antibiofilm activity that small clumps of cells would sometimes form and then dissociate under conditions of shear flow (see Movies S1, S2, S3, and S7 in the supplemental material). Could the release of these clumped cells have biological consequences? We can only speculate, but biofilm clumps dissociated by antibody treatment are not expected to be equivalent to microcolonies or emboli released by the natural dispersal mechanisms that are implicated in the metastatic spread of infection (28). Such clumps would already be coated with antibodies to prevent their subsequent attachment at distal sites. Alternatively, one might expect that clumped cells released by active antibody would get filtered through the liver and trapped and destroyed by Kupffer cells (29). Indeed, we have performed intravenous challenge of healthy mice with S. epidermidis scraped from biofilms that had been minimally disrupted (i.e., that were still likely to exist in clumps), and the mice very rapidly cleared all signs of infection (data not shown).
PhnD was selected as our lead antigen for further study because it elicited the antibodies with the strongest antibiofilm activity and was the best-conserved (>75% identity) target among all species in the genus. Phosphonate-binding and -uptake proteins have not previously been described as playing a role in biofilm formation. In E. coli, phosphonates are taken into the cell and converted to phosphates, likely to meet nutritional requirements (30–33). It is not known if PhnD has an analogous role in S. epidermidis. But since phosphate-limiting conditions have been shown to suppress biofilm development in some bacteria (34), a plausible mechanistic hypothesis is that antibody binding to PhnD could lead to phosphate starvation and trigger a dispersal mechanism for individual bacteria to seek out a more nutritive environment. Consistent with the involvement of PhnD in biofilm development, deletion of phnD resulted in a strain unable to form normal biofilms (Fig. 4E). Future mechanistic studies could focus on defining the substrate specificity of PhnD and assessing the impact of this substrate on intracellular signaling pathways and biofilm formation.
Antibodies for the prevention of CLABSI could be elicited in a patient by either vaccination or passive administration. Vaccination requires a fully functioning immune system and sufficient lead time to mount a response (e.g., as in planned surgeries). However, many patients with central lines are critically ill or are admitted to intensive care units with no advance warning; passive antibody administration would be preferable in these circumstances. Since timing is a critical parameter for defining the clinical indication, we tested our candidate antibodies at different stages of biofilm development to establish viable intervention points. PhnD-specific antibodies were active in both the prophylactic and early therapeutic settings (Fig. 3 and 4) but could not disrupt late-stage biofilms—at least, they could not do so in isolation (data not shown). When considered in the broader context of the patient immune system, however, our studies with human neutrophils predict that PhnD antibodies would also have a therapeutic benefit on preformed biofilms.
Multiple studies have shown that staphylococcal biofilms are recalcitrant to phagocytosis (reviewed in reference 21), but we observed clearance of S. epidermidis biofilms by neutrophils in the presence of opsonizing PhnD antibodies (Fig. 5; see also Movies S5 and S6 in the supplemental material). Günther and coworkers have shown phagocytosis of S. aureus biofilms under static conditions (35), but to our knowledge, this is the first demonstration of biofilm opsonophagocytosis under conditions of shear flow. Since staphylococcal biofilms leading to CLABSI are bathed in flowing blood, the recruitment of neutrophils in our model is highly relevant to the in vivo infection: circulating neutrophils must adhere strongly to avoid being washed away by shear flow forces. Notably, the neutrophils recruited by PhnD antibodies were highly motile and actively engulfed sections of preformed biofilm. While we observed that antibodies to targets other than PhnD (e.g., SERP1011 and SERP2398) could mediate biofilm opsonophagocytosis (data not shown), it does not necessary follow that all surface-binding antibodies possess this quality. For example, Cerca and coworkers demonstrated marked differences in the opsonophagocytic killing of different S. epidermidis stains grown in either the planktonic or biofilm mode using antibodies to the PNAG surface polysaccharide (36). As such, we suggest that biofilm-specific opsonophagocytosis is an important feature to test empirically.
Finally, to confirm the in vivo relevance of PhnD as an antibiofilm target, we tested the activity of PhnD antibodies against biofilms grown in human plasma to mimic the environment during CLABSI (Fig. 7; see also Movie S7 in the supplemental material). These experiments were critical because biofilm formation is a tightly regulated process that is heavily influenced by the bacterial growth medium. Biofilms formed under shear flow conditions in 100% human plasma were less stable than those established using standard laboratory growth media, but antibodies to PhnD still showed a clear antibiofilm effect (Fig. 7; see also Movie S7). Efficacy testing of PhnD antibodies in animals would be a logical next step, but the existing rat central venous catheter models for CLABSI (37–39) have significant limitations for evaluating vaccines or passive antibody administration. In particular, the route of bacterial challenge is a key consideration. Delivery of the challenge inoculum directly into the catheter lumen (37, 38) is not suitable for vaccine or passive antibody studies because the bacteria are protected from host humoral and cell-mediated effectors. Likewise, intravascular injection of bacteria to achieve hematogenous colonization of the external catheter surface is highly problematic because megadoses (>109 cells) of bacteria are required (39) and it is impossible to distinguish whether the intervention is targeting the planktonic form or the biofilm form. Further, given that the ideal strategy is to inhibit biofilm formation at its earliest stages, it is not practical to surgically implant catheters with preformed biofilms to circumvent the catheter colonization issue. Our conclusion is that existing animal models for CLABSI offer limited prognostic value for assessing vaccine or antibody interventions. Further studies are needed to develop new animal models that better reflect the natural human acquisition of CLABSI. In the interim, we suggest that a suite of flow-based studies similar to what we have presented should be seriously considered as an alternative to animal efficacy testing for this indication. Our assessment of various intervention points for biofilm inhibition and the interplay of antibodies with human neutrophils to clear existing biofilm together offer a holistic view of how a PhnD vaccine or antibody prophylactic might work in the context of a human infection.
In conclusion, PhnD is a promising target for a vaccine or passive antibody strategy against staphylococcal biofilms. Since it is highly conserved, a single vaccine or monoclonal antibody could have antibiofilm activity against the entire genus. Interestingly, a phnD homolog also exists in E. faecalis, the third leading cause of CLABSI. It is tempting to speculate that antibiofilm activity against the top three CLABSI pathogens could be achieved via the same target. Importantly, the tailored antibody strategies we envision would leave a patient's normal gastrointestinal flora intact. Also, by targeting a virulence factor rather than bacterial viability, we potentially avoid selective pressure that could lead to the development of resistance (40). Given the medical need to reduce the burden of CLABSI, a new vaccine or antibody prophylactic would be an important contribution to prevention efforts.
Supplementary Material
ACKNOWLEDGMENTS
We thank P. D. Fey for strains and plasmids, J. Switzer and P. Mott for cloning and expression experiments, and R. Charlebois and R. Oomen for assistance with Bioinformatics.
This work was funded by Sanofi Pasteur. All of us are employees of Sanofi Pasteur and may own stock in the company.
Footnotes
Published ahead of print 23 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02168-14.
REFERENCES
- 1.Centers for Disease Control and Prevention (CDC). 2011. Vital signs: central line-associated blood stream infections–United States, 2001, 2008, and 2009. MMWR Morb. Mortal. Wkly. Rep. 60:243–248 [PubMed] [Google Scholar]
- 2.Kilgore M, Brossette S. 2008. Cost of bloodstream infections. Am. J. Infect. Control 36:S172.e1-S172.e3. 10.1016/j.ajic.2008.10.004 [DOI] [PubMed] [Google Scholar]
- 3.Boyce JM. 2012. Prevention of central line-associated bloodstream infections in hemodialysis patients. Infect. Control Hosp. Epidemiol. 33:936–944. 10.1086/667369 [DOI] [PubMed] [Google Scholar]
- 4.Srinivasan A, Craig M, Cardo D. 2012. The power of policy change, federal collaboration, and state coordination in healthcare-associated infection prevention. Clin. Infect. Dis. 55:426–431. 10.1093/cid/cis407 [DOI] [PubMed] [Google Scholar]
- 5.Miller SE, Maragakis LL. 2012. Central line-associated bloodstream infection prevention. Curr. Opin. Infect. Dis. 25:412–422. 10.1097/QCO.0b013e328355e4da [DOI] [PubMed] [Google Scholar]
- 6.Sievert DM, Ricks P, Edwards JR, Schneider A, Patel J, Srinivasan A, Kallen A, Limbago B, Fridkin S. 2013. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009–2010. Infect. Control Hosp. Epidemiol. 34:1–14. 10.1086/668770 [DOI] [PubMed] [Google Scholar]
- 7.Aldea-Mansilla C, Garcia de Viedma D, Cercenado E, Martin-Rabadan P, Marin M, Bouza E. 2006. Comparison of phenotypic with genotypic procedures for confirmation of coagulase-negative Staphylococcus catheter-related bloodstream infections. J. Clin. Microbiol. 44:3529–3532. 10.1128/JCM.00839-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haslett TM, Isenberg HD, Hilton E, Tucci V, Kay BG, Vellozzi EM. 1988. Microbiology of indwelling central intravascular catheters. J. Clin. Microbiol. 26:696–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fey PD, Olson ME. 2010. Current concepts in biofilm formation of Staphylococcus epidermidis. Future Microbiol. 5:917–933. 10.2217/fmb.10.56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Mellaert L, Shahrooei M, Hofmans D, Eldere JV. 2012. Immunoprophylaxis and immunotherapy of Staphylococcus epidermidis infections: challenges and prospects. Expert Rev. Vaccines 11:319–334. 10.1586/erv.11.190 [DOI] [PubMed] [Google Scholar]
- 11.Mermel LA, Allon M, Bouza E, Craven DE, Flynn P, O'Grady NP, Raad II, Rijnders BJ, Sherertz RJ, Warren DK. 2009. Clinical practice guidelines for the diagnosis and management of intravascular catheter-related infection: 2009 Update by the Infectious Diseases Society of America. Clin. Infect. Dis. 49:1–45. 10.1086/599376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kwasny SM, Opperman TJ. 2010. Static biofilm cultures of Gram-positive pathogens grown in a microtiter format used for anti-biofilm drug discovery. Curr. Protoc. Pharmacol. 50:13A.18.1–13A.18.23. 10.1002/0471141755.ph13a08s50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benoit MR, Conant CG, Ionescu-Zanetti C, Schwartz M, Matin A. 2010. New device for high-throughput viability screening of flow biofilms. Appl. Environ. Microbiol. 76:4136–4142. 10.1128/AEM.03065-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moormeier DE, Endres JL, Mann EE, Sadykov MR, Horswill AR, Rice KC, Fey PD, Bayles KW. 2013. Use of microfluidic technology to analyze gene expression during Staphylococcus aureus biofilm formation reveals distinct physiological niches. Appl. Environ. Microbiol. 79:3413–3424. 10.1128/AEM.00395-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Augustin J, Gotz F. 1990. Transformation of Staphylococcus epidermidis and other staphylococcal species with plasmid DNA by electroporation. FEMS Microbiol. Lett. 54:203–207 [DOI] [PubMed] [Google Scholar]
- 16.Hu J, Xu T, Zhu T, Lou Q, Wang X, Wu Y, Huang R, Liu J, Liu H, Yu F, Ding B, Huang Y, Tong W, Qu D. 2011. Monoclonal antibodies against accumulation-associated protein affect EPS biosynthesis and enhance bacterial accumulation of Staphylococcus epidermidis. PLoS One 6:e20918. 10.1371/journal.pone.0020918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Christner M, Franke GC, Schommer NN, Wendt U, Wegert K, Pehle P, Kroll G, Schulze C, Buck F, Mack D, Aepfelbacher M, Rohde H. 2010. The giant extracellular matrix-binding protein of Staphylococcus epidermidis mediates biofilm accumulation and attachment to fibronectin. Mol. Microbiol. 75:187–207. 10.1111/j.1365-2958.2009.06981.x [DOI] [PubMed] [Google Scholar]
- 18.Williams RJ, Henderson B, Sharp LJ, Nair SP. 2002. Identification of a fibronectin-binding protein from Staphylococcus epidermidis. Infect. Immun. 70:6805–6810. 10.1128/IAI.70.12.6805-6810.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shahrooei M, Hira V, Khodaparast L, Khodaparast L, Stijlemans B, Kucharikova S, Burghout P, Hermans PW, Van Eldere J. 2012. Vaccination with SesC decreases Staphylococcus epidermidis biofilm formation. Infect. Immun. 80:3660–3668. 10.1128/IAI.00104-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shahrooei M, Hira V, Stijlemans B, Merckx R, Hermans PW, Van Eldere J. 2009. Inhibition of Staphylococcus epidermidis biofilm formation by rabbit polyclonal antibodies against the SesC protein. Infect. Immun. 77:3670–3678. 10.1128/IAI.01464-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Otto M. 2012. Molecular basis of Staphylococcus epidermidis infections. Semin. Immunopathol. 34:201–214. 10.1007/s00281-011-0296-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arciola CR, Baldassarri L, Montanaro L. 2001. Presence of icaA and icaD genes and slime production in a collection of staphylococcal strains from catheter-associated infections. J. Clin. Microbiol. 39:2151–2156. 10.1128/JCM.39.6.2151-2156.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arciola CR, Gamberini S, Campoccia D, Visai L, Speziale P, Baldassarri L, Montanaro L. 2005. A multiplex PCR method for the detection of all five individual genes of ica locus in Staphylococcus epidermidis. A survey on 400 clinical isolates from prosthesis-associated infections. J. Biomed. Mater. Res. A 75:408–413. 10.1002/jbm.a.30445 [DOI] [PubMed] [Google Scholar]
- 24.Galdbart JO, Allignet J, Tung HS, Ryden C, El Solh N. 2000. Screening for Staphylococcus epidermidis markers discriminating between skin-flora strains and those responsible for infections of joint prostheses. J. Infect. Dis. 182:351–355. 10.1086/315660 [DOI] [PubMed] [Google Scholar]
- 25.Hidron AI, Edwards JR, Patel J, Horan TC, Sievert DM, Pollock DA, Fridkin SK. 2008. NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infect. Control Hosp. Epidemiol. 29:996–1011. 10.1086/591861 [DOI] [PubMed] [Google Scholar]
- 26.Sellman BR, Howell AP, Kelly-Boyd C, Baker SM. 2005. Identification of immunogenic and serum binding proteins of Staphylococcus epidermidis. Infect. Immun. 73:6591–6600. 10.1128/IAI.73.10.6591-6600.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hussain M, Herrmann M, von Eiff C, Perdreau-Remington F, Peters G. 1997. A 140-kilodalton extracellular protein is essential for the accumulation of Staphylococcus epidermidis strains on surfaces. Infect. Immun. 65:519–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boles BR, Horswill AR. 2011. Staphylococcal biofilm disassembly. Trends Microbiol. 19:449–455. 10.1016/j.tim.2011.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong CH, Jenne CN, Petri B, Chrobok NL, Kubes P. 2013. Nucleation of platelets with blood-borne pathogens on Kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nat. Immunol. 14:785–792. 10.1038/ni.2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alicea I, Marvin JS, Miklos AE, Ellington AD, Looger LL, Schreiter ER. 2011. Structure of the Escherichia coli phosphonate binding protein PhnD and rationally optimized phosphonate biosensors. J. Mol. Biol. 414:356–369. 10.1016/j.jmb.2011.09.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jochimsen B, Lolle S, McSorley FR, Nabi M, Stougaard J, Zechel DL, Hove-Jensen B. 2011. Five phosphonate operon gene products as components of a multi-subunit complex of the carbon-phosphorus lyase pathway. Proc. Natl. Acad. Sci. U. S. A. 108:11393–11398. 10.1073/pnas.1104922108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kamat SS, Williams HJ, Raushel FM. 2011. Intermediates in the transformation of phosphonates to phosphate by bacteria. Nature 480:570–573. 10.1038/nature10622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Metcalf WW, Wanner BL. 1993. Mutational analysis of an Escherichia coli fourteen-gene operon for phosphonate degradation, using TnphoA' elements. J. Bacteriol. 175:3430–3442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Monds RD, Newell PD, Gross RH, O'Toole GA. 2007. Phosphate-dependent modulation of c-di-GMP levels regulates Pseudomonas fluorescens Pf0–1 biofilm formation by controlling secretion of the adhesin LapA. Mol. Microbiol. 63:656–679 [DOI] [PubMed] [Google Scholar]
- 35.Günther F, Wabnitz GH, Stroh P, Prior B, Obst U, Samstag Y, Wagner C, Hänsch GM. 2009. Host defence against Staphylococcus aureus biofilms infection: phagocytosis of biofilms by polymorphonuclear neutrophils (PMN). Mol. Immunol. 46:1805–1813. 10.1016/j.molimm.2009.01.020 [DOI] [PubMed] [Google Scholar]
- 36.Cerca N, Jefferson KK, Oliveira R, Pier GB, Azeredo J. 2006. Comparative antibody-mediated phagocytosis of Staphylococcus epidermidis cells grown in a biofilm or in the planktonic state. Infect. Immun. 74:4849–4855. 10.1128/IAI.00230-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rupp ME, Fey PD, Heilmann C, Gotz F. 2001. Characterization of the importance of Staphylococcus epidermidis autolysin and polysaccharide intercellular adhesin in the pathogenesis of intravascular catheter-associated infection in a rat model. J. Infect. Dis. 183:1038–1042. 10.1086/319279 [DOI] [PubMed] [Google Scholar]
- 38.Rupp ME, Ulphani JS, Fey PD, Mack D. 1999. Characterization of Staphylococcus epidermidis polysaccharide intercellular adhesin/hemagglutinin in the pathogenesis of intravascular catheter-associated infection in a rat model. Infect. Immun. 67:2656–2659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ebert T, Smith S, Pancari G, Wu X, Zorman J, Clark D, Cook J, Burns C, Antonello JM, Cope L, Nagy E, Meinke A, McNeely T. 2011. Development of a rat central venous catheter model for evaluation of vaccines to prevent Staphylococcus epidermidis and Staphylococcus aureus early biofilms. Hum. Vaccin. 7:630–638. 10.4161/hv.7.6.15407 [DOI] [PubMed] [Google Scholar]
- 40.Rasko DA, Sperandio V. 2010. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 9:117–128. 10.1038/nrd3013 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.