

Abstract
Pertussis disease, characterized by severe and prolonged coughing episodes, can progress to a critical stage with pulmonary inflammation and death in young infants. However, there are currently no effective treatments for pertussis. We previously studied the role of pertussis toxin (PT), an important Bordetella pertussis virulence factor, in lung transcriptional responses to B. pertussis infection in mouse models. One of the genes most highly upregulated in a PT-dependent manner encodes an epithelial transporter of bicarbonate, chloride, and thiocyanate, named pendrin, that contributes to asthma pathology. In this study, we found that pendrin expression is upregulated at both gene and protein levels in the lungs of B. pertussis-infected mice. Pendrin upregulation is associated with PT production by the bacteria and with interleukin-17A (IL-17A) production by the host. B. pertussis-infected pendrin knockout (KO) mice had higher lung bacterial loads than infected pendrin-expressing mice but had significantly reduced levels of lung inflammatory pathology. However, reduced pathology did not correlate with reduced inflammatory cytokine expression. Infected pendrin KO mice had higher levels of inflammatory cytokines and chemokines than infected pendrin-expressing mice, suggesting that these inflammatory mediators are less active in the airways in the absence of pendrin. In addition, treatment of B. pertussis-infected mice with the carbonic anhydrase inhibitor acetazolamide reduced lung inflammatory pathology without affecting pendrin synthesis or bacterial loads. Together these data suggest that PT contributes to pertussis pathology through the upregulation of pendrin, which promotes conditions favoring inflammatory pathology. Therefore, pendrin may represent a novel therapeutic target for treatment of pertussis disease.
INTRODUCTION
Bordetella pertussis, the etiologic agent of whooping cough, is a resurgent respiratory pathogen that is a major public health concern, especially for young infants (1). In 2012, almost 50,000 pertussis cases were reported in the United States, the highest number since the early 1950s, and new epidemics are emerging in various states in 2014 (http://www.cdc.gov/pertussis/outbreaks/about.html). Mechanisms proposed for the apparent reemergence of this disease include vaccine-driven evolution of the bacterium (2), a switch from whole-cell to acellular vaccine resulting in changes in the type and duration of immunity (3–7), and improved diagnosis (8). Severe disease, most often observed in infected infants, leads to hyperleukocytosis and can result in mortality (1, 9). However, no effective therapeutic is available to treat infants with severe pertussis.
B. pertussis modulates host innate and adaptive immune responses by releasing several toxins, including pertussis toxin (PT) (10). PT is an AB5 toxin that binds to glycosylated molecules on target cells (11, 12), enters the cell by endocytosis, and then undergoes retrograde transport to the Golgi complex and the endoplasmic reticulum (ER), from where it enters the cytosol, likely by using the ER-associated degradation (ERAD) pathway (13–15). In the cytosol, the active subunit of PT ADP-ribosylates the α subunit of heterotrimeric Gi/o proteins, leading to ablated G protein-coupled receptor (GPCR) signaling, loss of inhibition of adenylyl cyclase, and increased intracellular cyclic AMP (cAMP) (16, 17). Mouse models of B. pertussis infection have revealed a role for active PT in enhancing colonization of the airways (18), inhibiting the protective effect of resident airway macrophages (19), inhibiting early neutrophil influx to the lungs by suppressing chemokine upregulation (20, 21), upregulating proinflammatory cytokines after the initial stages of infection (22), suppressing serum antibody responses to B. pertussis (23), and exacerbating disease in various immunodeficient backgrounds (24, 25). PT is also critical for B. pertussis-induced infant mortality in mouse models (26), although it should be noted that the transposon insertion mutants used in that study were not complemented. However, despite our current knowledge, precisely how PT contributes to pathogenesis of pertussis remains unclear.
Through assessment of murine lung transcript levels following infection with B. pertussis wild-type (WT) or PT-deficient (ΔPT) strains, we previously identified a number of genes whose upregulation was associated with PT activity (27). One of the most highly upregulated genes was slc26a4, a gene that codes for pendrin (upregulated 9.4-fold versus phosphate-buffered saline [PBS]-treated mice and 5.4-fold versus ΔPT-infected mice). Pendrin is a transmembrane anion exchanger found on the apical surface of epithelial cells (28). This protein is highly expressed in the thyroid, kidney, and inner ear, with function-impairing mutations associated with a disease of prelingual deafness known as Pendred syndrome (28–30). However, low-level slc26a4 expression has also been described in several other organs, including the liver (31) and lungs (32). Lung expression was first identified when an allergy-driven and interleukin-13 (IL-13)-induced upregulation was observed in a murine model of asthma (32). Further studies have gone on to demonstrate that IL-13-stimulated enhanced airway slc26a4 expression is STAT6 dependent and may also be induced by IL-4 (33, 34). In a recent publication, slc26a4 was identified as the most highly upregulated gene in human asthmatic bronchi compared with control patients (35). In addition, levels of this anion transporter are enhanced in murine models of chronic obstructive pulmonary disease (COPD) (33) and in the sinonasal tissue of patients with allergic rhinitis and chronic rhinosinusitis (36).
In the respiratory system, pendrin functions to import Cl− from the airway apical surface and to export both HCO3− and thiocyanate (SCN−) into the airway surface liquid (ASL) (37, 38). Elevated lung pendrin levels also cause increased production of a major airway mucus protein, MUC5AC (33). In addition, SCN− secretion supports antimicrobial mechanisms in the lung, since SCN− is converted to the microbicidal OSCN− (hypothiocyanite) when oxidized by H2O2 in a reaction catalyzed by lactoperoxidase (39). Pendrin appears to contribute to airway inflammation and pathology, though the mechanism involved is unclear. In an OVA-induced model of airway hyperreactivity, pendrin knockout (KO) mice had reduced airway inflammation in response to challenge (40), while slc26a4 overexpression caused increased mucus production and elevated production of the neutrophil-attracting chemokines CXCL1 and CXCL2 (33). However, while upregulation of slc26a4 has been observed in response to rhinovirus infection in the presence of gamma interferon (IFN-γ) (40), to date no role for pendrin in a respiratory bacterial infection has been published.
We hypothesized that B. pertussis-induced elevated pendrin levels contribute to airway pathology associated with pertussis infection. In this study, we sought to examine B. pertussis-driven PT-dependent pendrin upregulation in the airways and to assess the contribution of this protein to respiratory inflammatory pathology in mouse models of B. pertussis infection.
MATERIALS AND METHODS
Bacterial strains.
The B. pertussis strain here designated wild type (WT), is a streptomycin- and nalidixic acid-resistant derivative of Tohama I (18). The ΔPT strain, generated from the WT, does not produce PT due to an in-frame deletion spanning all 5 PT genes (18). B1834 and B1831 are B. pertussis clinical isolates from a Netherlands outbreak that carry the PT promoters ptxP1 and ptxP3, respectively (41). All strains were grown on Bordet-Gengou (BG) agar plates supplemented with 10% defibrinated sheep blood. WT and ΔPT growth plates also contained 200 μg/ml streptomycin.
Mouse infections.
Six- to 10-week old BALB/c (Charles River Laboratories), C57BL/6 (Charles River Laboratories or in-house bred), Stat6−/− (on the BALB/c background [42], kindly donated by A. Keegan), Il-17a−/− (on the C57BL/6 background [43], kindly donated by M. Shirtliff), and Slc26a4−/− (on the C57BL/6 background [44], kindly donated by M. Soleimani) mice were all used in accordance with the University of Maryland, Baltimore (UMB), Institutional Animal Care and Use Committee. Bacterial inoculum was prepared from 48-h growth on appropriate BG agar plates in suspension with PBS. Mice were anesthetized with inhaled isoflurane, and the inoculum was administered intranasally in a 50-μl volume. Mice were inoculated with PBS, 2 × 106 CFU WT, B1831, or B1834, 1 × 107 CFU ΔPT, or 100 ng or 500 ng purified PT. Acetazolamide (100 mg/kg) was dissolved in dimethyl sulfoxide (DMSO), diluted 1:5 in sunflower oil, and administered daily from the day of infection by subcutaneous injection. A vehicle control was prepared with DMSO and sunflower oil alone. At time of harvest, mice were euthanized by carbon dioxide inhalation, followed by thoracotomy. The lungs and trachea were divided for bacterial counts, histology, and mRNA and protein analysis. Bacterial counts were determined by serial dilution of lung homogenates that were plated onto appropriate BG agar plates and counted after 4 to 5 days of growth at 37°C. All experiments were performed at least twice, with representative data from single experiments presented.
RNA isolation and processing.
Lung tissue for RNA analysis was snap-frozen in a dry ice-ethanol bath. RNA was extracted with RNA Stat60 (TelTest, Inc.) as per the manufacturer's instructions. In brief, samples were homogenized in RNA Stat60 using an Omni TH mixer (Omni, Inc.), phase separated with the addition of chloroform, and precipitated with isopropanol. RNA was quantified, and 1 μg was reverse transcribed using a reverse transcription system (Promega). Quantitative real-time PCR (qRT-PCR) was performed with Maxima SYBR green/ROX quantitative PCR (qPCR) master mix (Thermo Scientific) in an Applied Biosystems 7500 Fast real-time PCR system. The hypoxanthine phosphoribosyltransferase (HPRT) gene was used as an internal housekeeping control gene (see primer details in Table S2 in the supplemental material), with all other genes normalized to the HPRT gene and expression calculated as fold change compared with PBS-inoculated control animal levels [calculated by the 2(−ΔΔCT) method]. For gene analyses by RT2 Profiler PCR array, RNA was isolated using a RNeasy microarray tissue minikit (Qiagen), reverse transcribed with the RT2 first-strand kit (Qiagen), and profiled by the mouse innate and adaptive immune responses array (Qiagen).
Western blotting.
Lung tissue for protein analysis was snap-frozen in a dry ice-ethanol bath. At the time of processing, tissue was weighed and suspended in 5 times volume HEENG buffer (20 mM HEPES [pH 7.6], 25 mM NaCl, 1 mM EDTA, 1 mM EGTA, and 10% [vol/vol] glycerol) containing 1% (vol/vol) Triton X-100, 0.05% (wt/vol) SDS, and protease inhibitor (Roche). Samples were cut, sonicated 3 times for 5-s pulses, and rotated at 4°C from 1 h. Insoluble proteins were pelleted by centrifugation at 13,000 rpm for 10 min at 4°C, and the supernatant was transferred into a new prechilled tube. Proteins were denatured in Laemmli buffer containing 15% (vol/vol) β-mercaptoethanol for 30 min at room temperature before being resolved on a 4 to 15% Mini-Protean TGX gel (Bio-Rad). Samples were transferred onto low-fluorescence polyvinylidene difluoride (PVDF) membranes (Bio-Rad) using a Trans-Blot Turbo system (Bio-Rad). The membrane was blocked in 5% (wt/vol) milk powder formulated in Tris-buffered saline (TBS) and probed using antibodies against actin (mouse monoclonal antibody from clone AC-15; Sigma) and pendrin (rabbit polyclonal antibody raised against the C-terminal 29 amino acids [aa] of rat pendrin, kindly donated by S. Wall) (45). Antibodies were detected using goat anti-mouse IgG DyLight 680 (Thermo Scientific) and donkey anti-rabbit IRDye 800CW (Li-Cor Biosciences) and visualized by an Odyssey infrared imaging system (Li-Cor Biosciences). Pendrin was normalized to actin and results expressed as fold change compared with the average PBS-inoculated mouse lung levels.
ELISArray.
Bronchoalveolar lavage (BAL) was performed in infected mice at 4 days postinoculation (dpi). In brief, an incision was made in the trachea, and a blunt-ended 21-gauge needle was inserted into the hole and sutured into place. The lungs were filled with 1 ml PBS containing 1 mM EDTA, and a volume of 700 to 800 μl was extracted. BAL fluid was kept at −80°C until processing. The ELISArray was performed as per the manufacturer's protocol (mouse inflammatory cytokine multianalyte ELISArray kit; Qiagen) using 50 μl undiluted BAL fluid per well.
Pathology.
Left lung samples were selected for immunopathology. When lungs were harvested for histology, the lungs were intracardially perfused with PBS, and the right lung (cranial, medial, caudal, and postcaval lobes) was tied off and removed for other analyses. The left lung was instilled with 10% (wt/vol) buffered formalin (Sigma) via an incision in the trachea, removed, and stored in 10% (wt/vol) buffered formalin. Slides were prepared and hematoxylin and eosin (H&E) stained by the Pathology EM and Histology Laboratory (UMB core facility). Histopathology was scored on a scale of 0 to 3, with 3 being the greatest degree of pathology, for each of (i) the degree of inflammation at the site of a bronchovascular bundle (BVB), (ii) the percentage of BVB involved, and (iii) the degree of pleuritis observed, with a total possible maximum score of 9.
Statistical analysis.
Graphs were plotted and data were analyzed using GraphPad Prism software. Fold changes were calculated per mouse compared with the average for the respective PBS-inoculated group. All plots represent the mean value ± the standard error of the mean (SEM). Significance was determined by Student's t test for two-group analyses and by analysis of variance (ANOVA) and the Kruskal-Wallis test for experiments containing three or more groups.
RESULTS
B. pertussis infection in mice upregulates pendrin production in a pertussis toxin-dependent manner.
In a previous microarray study, we identified PT-dependent upregulation of the pendrin gene slc26a4 in the lungs of B. pertussis-infected BALB/c mice (27). Further to this, we sought to confirm and examine this upregulation. Enhanced expression of the slc26a4 gene and production of pendrin in lungs of infected BALB/c mice were confirmed at the mRNA (by qRT-PCR) and protein (by Western blotting) levels, respectively, compared with PBS-inoculated control animals (Fig. 1A and B). Pendrin levels were significantly enhanced at 4 days postinoculation (dpi) compared with those in PBS-treated control animals, and they further increased at 7 dpi. These results indicate that the B. pertussis infection causes upregulation of slc26a4; however, whether this is through a direct or indirect mechanism is not clear. Similar slc26a4 gene and pendrin protein upregulations were also observed in lungs of B. pertussis-infected C57BL/6 mice, though transcription peaked at 4 dpi (Fig. 1C and D), indicating that this function is not mouse strain specific. Enhanced slc26a4 expression in the tracheae of infected C57BL/6 mice was not observed (data not shown), suggesting that this gene is upregulated in the bronchial and other lung tissue.
FIG 1.

Pendrin production is increased upon B. pertussis infection. (A and B) Infected BALB/c mice (n ≥ 4 per group) displayed upregulated Slc26a4 expression at the mRNA level by qRT-PCR (A) and pendrin protein production as assessed by Western blotting (B) at 4 and 7 days postinfection (dpi). In a representative Western blot, the upper panel shows pendrin upregulation, the lower panel shows actin as a loading control, and M indicates the protein standard marker. (C and D) C57BL/6 mice (n ≥ 4 per group) infected with B. pertussis were also evaluated at 4 and 7 dpi for Slc26a4 transcript (C) and pendrin protein (D). Expression levels were all normalized to control PBS-treated mice and displayed as mean ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (compared with control mice).
To confirm our previous findings on the role of PT in enhanced pendrin synthesis (27), we compared pendrin levels in C57BL/6 mice infected with the “wild-type” (WT) strain of B. pertussis to those in mice infected with a high dose of the PT-deficient strain (ΔPT). No significant difference was observed in bacterial loads between groups at 4 dpi (Fig. 2A). However, WT infection induced a significantly higher level of pendrin upregulation than ΔPT infection, supporting a role for PT in enhanced Slc26a4 expression (Fig. 2B) (P = 0.03). Given that B. pertussis required PT for maximal pendrin expression, we tested whether treatment of mice with the toxin alone could increase expression of this protein. Mice were administered a single intranasal dose of 100 or 500 ng purified PT and probed for pendrin production in lungs by Western blotting at 4 and 7 dpi. No enhancement of pendrin protein production was observed at either time point (data not shown), indicating that PT alone is not sufficient for pendrin upregulation.
FIG 2.
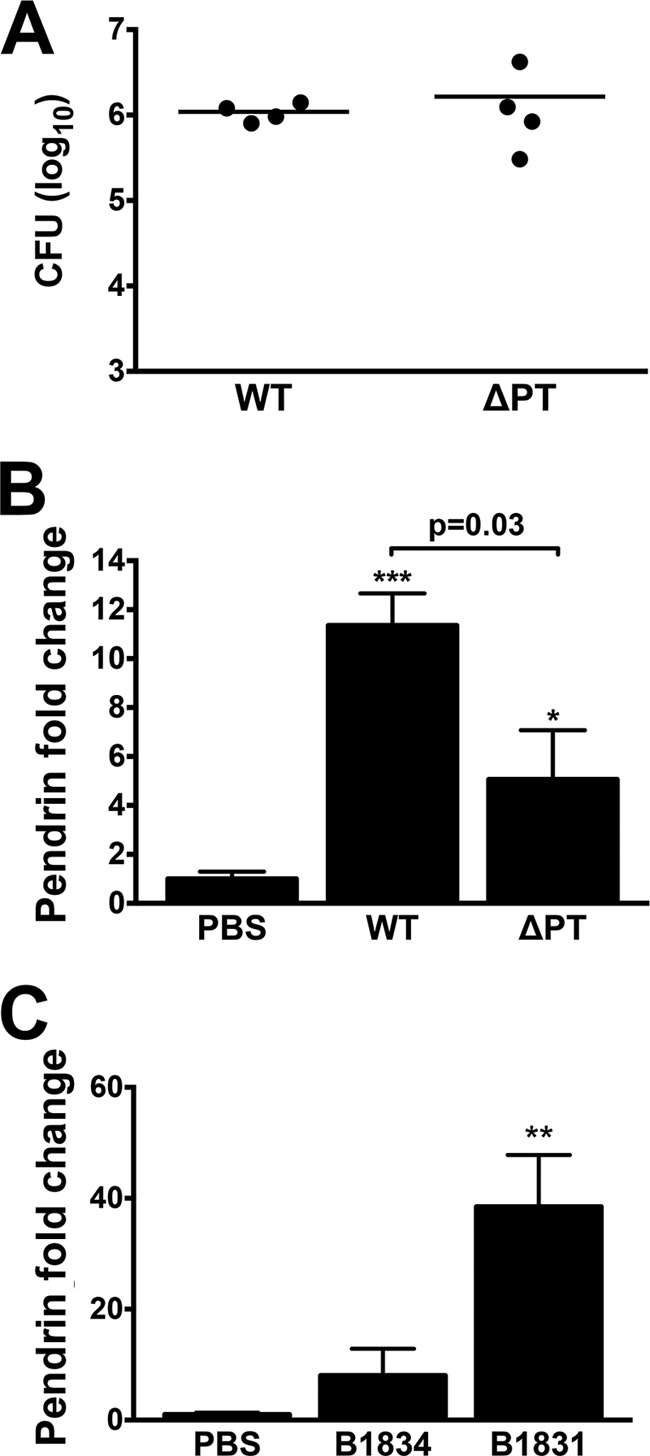
B. pertussis-induced pendrin upregulation is reduced in the absence of pertussis toxin (PT) and observed with recent clinical isolates. C57BL/6 mice (n = 4 per group) were infected with WT B. pertussis or a high dose of PT knockout B. pertussis (ΔPT). Lung loads were similar for both strains at 4 dpi (A). However, ΔPT-infected mice had significantly lower levels of pendrin upregulation (B). BALB/c mice (n ≥ 3 per group) were inoculated with recent B. pertussis ptxP1 (B1834) or ptxP3 (B1831) clinical isolates and assessed for pendrin protein levels by Western blotting (C). Pendrin production was normalized to the control PBS-treated mice. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (compared with control samples). Bars represent mean ± SEM.
The WT strain is derived from the prototype strain Tohama, isolated in the 1950s (46). More recent B. pertussis strains have several genetic differences from Tohama, including a shift from the PT promoter P1 allele (ptxP1) to the P3 allele (ptxP3) (41). To determine whether pendrin upregulation occurs after infection with a more recent clinical B. pertussis isolate, we assessed pendrin protein levels in BALB/c mice infected with a ptxP1 (B1834) or ptxP3 (B1831) strain isolated in 1999 (41). Colonizations with B1834 and B1831 were at a level similar to that for the WT (data not shown); however, B1831 induced an even higher level of pendrin upregulation than that of the WT in BALB/c mice at 4 dpi (see Fig. 1B), while pendrin upregulation in B1834-infected mice was similar to that in WT-infected mice (Fig. 2C). These data show that pendrin overproduction can be induced by multiple strains of B. pertussis and that this is independent of the PT promoter allele.
B. pertussis-induced pendrin upregulation is STAT6 independent but IL-17A dependent.
Since slc26a4 expression is upregulated by the TH2 cytokines IL-4 and IL-13 acting through the transcription factor STAT6 in mouse models of asthma (34), we assessed the role of STAT6 in our model. BALB/c and STAT6 KO (Stat6−/− on the BALB/c background) mice were inoculated with WT B. pertussis. At 4 dpi there was no significant STAT6-dependent difference in lung bacterial load (Fig. 3A) or in slc26a4 gene upregulation (Fig. 3B), consistent with the lack of IL-4 and IL-13 expression during B. pertussis infection in BALB/c mice (27). Therefore, STAT6 is not the crucial regulator of Slc26a4 in our infection model.
FIG 3.
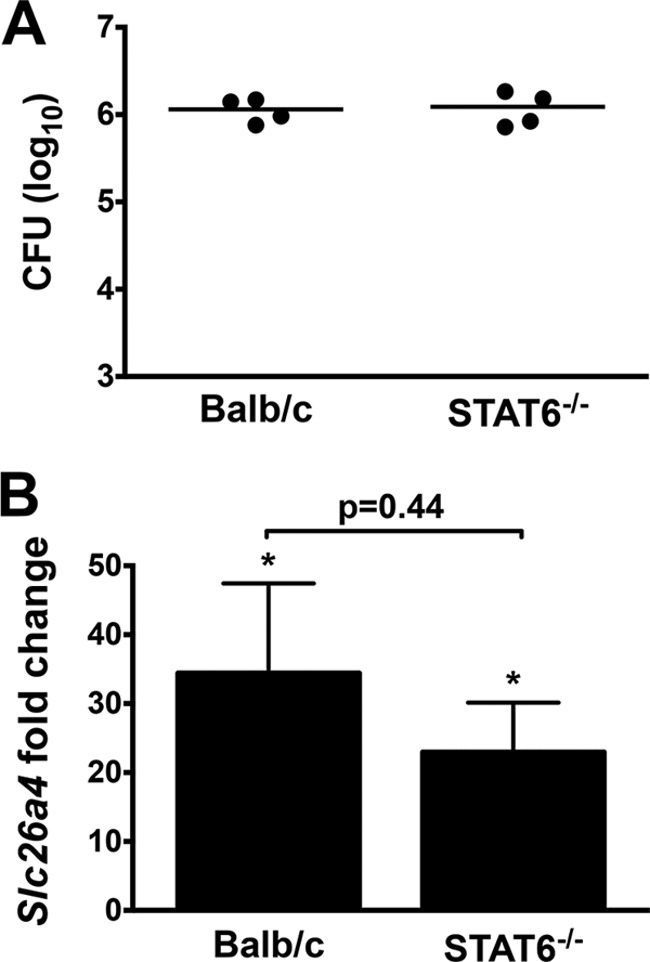
B. pertussis-induced enhanced Slc26a4 levels are STAT6 independent. Lung loads at 4 dpi with B. pertussis WT were similar in BALB/c and Stat6−/− mice (n = 4) (A). qPCR revealed no significant difference in infection-induced Slc26a4 upregulation (B). Expression levels were normalized to control PBS-treated BALB/c mice and displayed as mean ± SEM. *, P < 0.05 compared with control PBS-treated mice.
In a previous study, bicarbonate secretion by human bronchial epithelial cells was significantly increased by IL-17A treatment, and it was postulated that this was due to an effect on a member of the SLC26 family of proteins (47). Since B. pertussis infection is associated with elevated levels of IL-17A (22, 48), we evaluated pendrin expression in infected IL-17A KO mice (on the C57BL/6 background). Il-17a−/− mice and control C57BL/6 mice were inoculated with WT B. pertussis and assessed for lung bacterial load and pendrin expression at 4 dpi. No significant difference in bacterial loads was observed between the infected mouse strains (Fig. 4A). However, the level of slc26a4/pendrin upregulation in infected Il-17a−/− mice was 3-fold lower than that observed in C57BL/6 mice at both the mRNA and protein levels (Fig. 4B and C), indicating that IL-17A contributes to the enhancement of slc26a4 expression and pendrin synthesis in our model.
FIG 4.
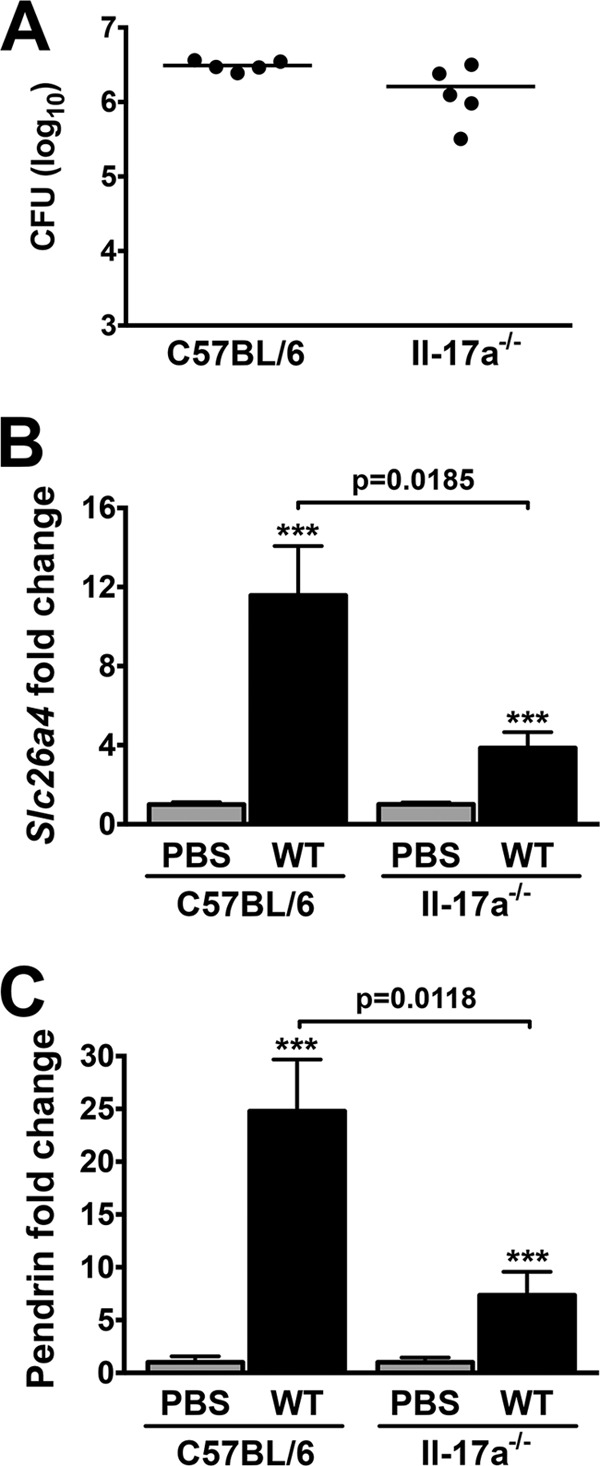
B. pertussis-induced pendrin upregulation is IL-17A dependent. Lung loads 4 dpi with WT B. pertussis were not significantly lower in Il-17a−/− mice (n = 5) (A). However, a significant reduction in infection-induced Slc26a4 mRNA (B) and pendrin protein (C) was observed in Il-17a−/− mice compared with C57BL/6 mice. Expression levels were normalized to control PBS-treated samples. ***, P < 0.001 (compared with control strain matched mice). Bars indicate mean ± SEM.
Pendrin KO mice exhibit reduced pertussis-driven lung pathology.
Further to our finding that pendrin is upregulated in the lungs of B. pertussis-infected mice, we sought to determine the contribution of pendrin to pertussis infection and disease. To address this question, disease progression was assessed in infected pendrin-expressing and pendrin KO littermate mice (Slc26a4+/+ and Slc26a4−/−, respectively). We first analyzed pendrin production in the lungs of these infected mice by Western blotting (Fig. 5A) and confirmed a lack of expression in the knockout mice. There was no significant difference in lung bacterial loads between mouse strains at 4 and 7 dpi, but the bacterial burden was significantly greater in Slc26a4−/− mice at 14 and 21 dpi (Fig. 5B), demonstrating a potential antibacterial role for pendrin. However, bacterial numbers were low in both strains of mice at 28 dpi, indicating that there is no long-term deleterious effect on bacterial clearance in pendrin KO mice.
FIG 5.
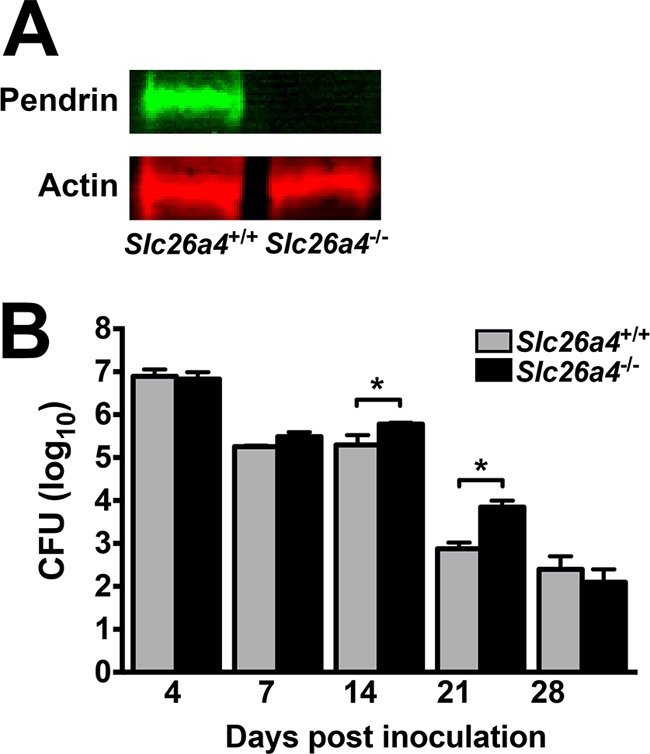
B. pertussis colonization is increased in Slc26a4−/− mice. (A) Lack of pendrin production (upper panel) in B. pertussis-infected Slc26a4−/− mice was confirmed by Western blotting, while actin levels (lower panel) remained normal. (B) Pendrin-expressing and pendrin KO littermate mice (n ≥ 5 per group) displayed a similar course of B. pertussis infection. However, at later time points, lung CFU were elevated in KO mice (black bars) compared with pendrin-expressing mice (gray bars). Bars represent the average per group ± SEM, *, P < 0.05.
The most striking difference between infected Slc26a4+/+ and Slc26a4−/− mice was observed in lung inflammatory pathology. B. pertussis infection in mice is typically characterized by peribronchial and perivascular inflammation (49). At 4 dpi, both mouse strains displayed low levels of pathology (Fig. 6E). At 7 dpi, littermates expressing pendrin exhibited peribronchiolar and perivascular lymphoid cuffing, in addition to alveolitis, slight edema, and intra-alveolar hemorrhage (Fig. 6A). In contrast, infected pendrin KO mice exhibited relatively minor bronchus-associated lymphoid tissue (BALT) hyperplasia and perivascular inflammation, in addition to a near absence of alveolitis, edema, and hemorrhage (Fig. 6B and E). At 14 dpi, infected pendrin KO mice continued to exhibit significantly reduced pathology compared with infected pendrin-expressing littermates (Fig. 6C to E). At 21 and 28 dpi, there was continued low-level pathology in both strains that was not significantly different (Fig. 6E). We conclude that the absence of pendrin is protective against the lung inflammatory pathology induced by B. pertussis infection.
FIG 6.
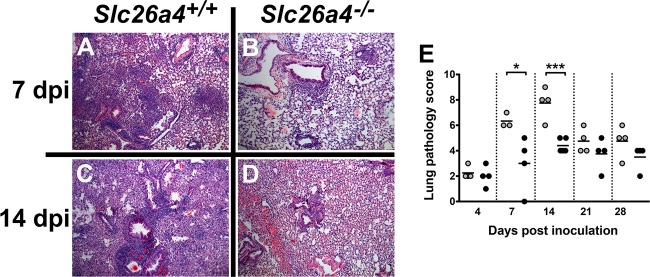
Lung inflammatory pathology is reduced in pendrin KO mice. A time course of B. pertussis infection-induced pathology was performed in pendrin-expressing (Slc26a4+/+) and pendrin KO (Slc26a4−/−) mice (n ≥ 3), and pathology was scored based on the percentage of bronchovascular bundles involved, degree of involvement, and pleuritis. (A to D) Representative H&E-stained images at 7 dpi and 14 dpi. (E) Pathology score was lower in Slc26a4−/− mice at all time points, and this was significant at 7 dpi and 14 dpi. *, P < 0.05; ***, P < 0.001.
B. pertussis-infected pendrin KO mice have higher levels of inflammatory cytokines and chemokines.
B. pertussis infection in mice yields a TH1/TH17-type response associated with elevated levels of IL-17A, IFN-γ, IL-6, and tumor necrosis factor alpha (TNF-α) (22), in addition to the neutrophil- and macrophage-recruiting chemokines CXCL1, CXCL2, CXCL5, CXCL10, and CCL2 (22, 27). To determine whether the reduced lung pathology in infected pendrin KO mice was due to lower levels of proinflammatory cytokines than in infected pendrin-expressing mice, we first examined gene expression at 4 dpi with WT B. pertussis in Slc26a4+/+ and Slc26a4−/− mice by PCR array. Infection in both strains of mice resulted in elevated lung expression of various proinflammatory cytokines and chemokines compared with those in PBS-treated mice (see Table S1 in the supplemental material). However, infected pendrin KO mice displayed increased levels of a number of other inflammatory mediators that were not observed in infected Slc26a4+/+ mice (see Table S1 in the supplemental material). Surprisingly, transcript levels of most of the cytokines and chemokines assayed were higher in infected Slc26a4−/− mice than in Slc26a4+/+ mice (Fig. 7A; see Table S1 in the supplemental material). Expression of only one cytokine, IL-5, was lower in Slc26a4−/− mice. This cytokine is typically associated with a TH2 response and is not known to play a role in pertussis disease. To support the array data and to elaborate on the course of cytokine and chemokine expression, we assessed transcript levels at 4 and 7 dpi by qPCR for Il-1β, Il-6, Il-10, Il-17a, Ifn-γ, Cxcl1, Cxcl2, Cxcl10, and Ccl5. In line with the array results, at 4 dpi most genes tested were upregulated to a significantly higher extent in Slc26a4−/− than in Slc26a4+/+ mice (see Fig. S1 in the supplemental material). The only gene tested whose expression was not significantly higher in pendrin KO infected tissue was Il-10 (see Fig. S1 in the supplemental material), indicating that pathology is not reduced due to an increase in anti-inflammatory responses. At 7 dpi, Il-17a expression was still higher in pendrin KO mice, but there was little difference between mouse strains for the other cytokines and chemokines (see Fig. S1 in the supplemental material). Protein levels in bronchoalveolar lavage (BAL) fluid from infected pendrin-expressing and pendrin KO mice were assessed at 7 dpi by ELISArray. Consistent with the mRNA data, higher cytokine and chemokine protein levels were observed in Slc26a4−/− mice than in Slc26a4+/+ mice (Fig. 7B). Taken together these data indicate that the reduced pathology in pendrin KO mice is not due to reduced production or secretion of inflammatory mediators.
FIG 7.
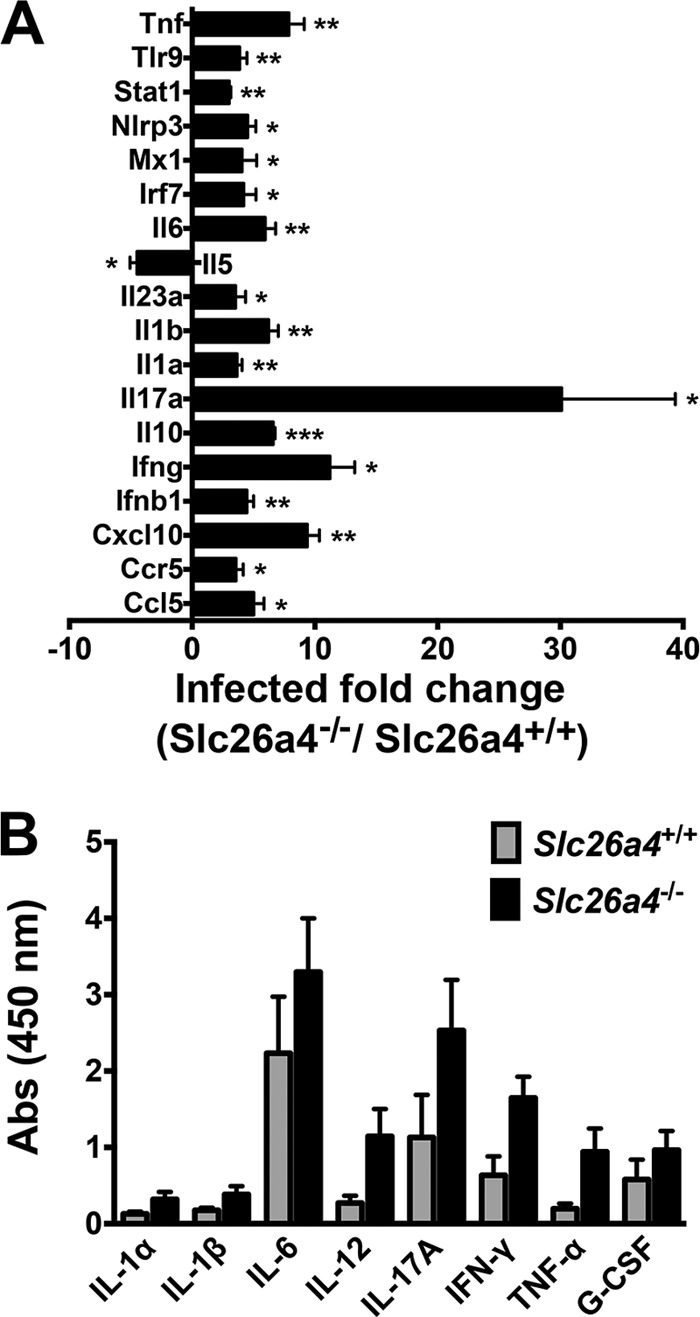
Cytokine and chemokine levels are higher in lungs of infected pendrin KO mice. (A) Pendrin-expressing and pendrin KO mice (n = 4) were infected with B. pertussis, and at 4 dpi lung cytokine gene mRNA levels were analyzed by RT Array (see Table S1 in the supplemental material for full data); those genes whose expression was significantly different in pendrin KO mice than in pendrin-expressing mice are displayed. (B) Protein levels for the indicated cytokines in BAL fluid were measured by ELISArray. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (relative to levels in infected pendrin-expressing mice).
Administration of a carbonic anhydrase inhibitor attenuates pertussis lung pathology.
Since a dramatic reduction in B. pertussis-induced pathology was observed in Slc26a4−/− mice, we next asked whether ablating bicarbonate secretion by pendrin could also attenuate B. pertussis-driven lung inflammatory pathology. The carbonic anhydrase inhibitor acetazolamide (ACTZ) blocks IL-17A-induced HCO3−-dependent short-circuit current in human bronchial epithelial cells (47), inhibits apical Cl−/HCO3− exchange in type B intercalated kidney cells (50), and reduces pendrin expression in murine kidneys (31, 51) and rat kidneys (52). Therefore, we examined lung pathology in B. pertussis-infected mice treated daily with ACTZ. The lung bacterial burden in ACTZ-treated mice was higher than that in vehicle-treated mice, though this difference was not quite statistically significant (Fig. 8A). While ACTZ treatment did not affect airway levels of pendrin in infected mice (Fig. 8B), assessment of lung pathology revealed a significant decrease in airway inflammation in the ACTZ-treated group compared with vehicle-treated mice (Fig. 8C, F, and G). Alveolar spaces in ACTZ-treated mice had fewer inflammatory cell infiltrates, reduced inflammatory exudate, and a reduction in BALT hyperplasia. ACTZ treatment did not significantly affect cytokine or chemokine levels in infected mice (data not shown). In addition, ACTZ treatment had no significant effect on the low level of lung inflammatory pathology in pendrin KO mice (data not shown), indicating the specificity of this inhibitor for a function of pendrin. These findings with ACTZ treatment indicate that inhibition of pendrin activity results in ameliorated lung pathology associated with B. pertussis infection without significantly increasing bacterial loads, suggesting a potential novel therapeutic target for pertussis.
FIG 8.
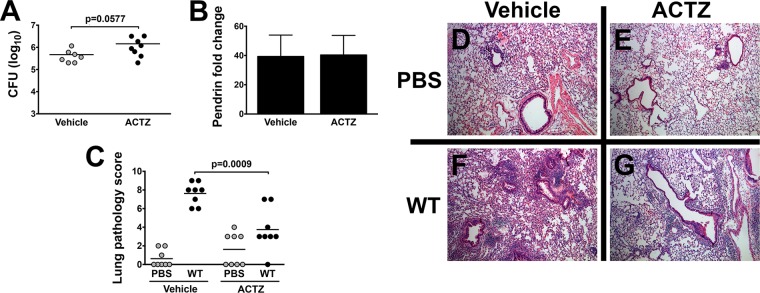
B. pertussis-induced pathology is reduced by acetazolamide (ACTZ) treatment. C57BL/6 mice (n = 8) were inoculated with B. pertussis and treated daily with vehicle or ACTZ via subcutaneous injection. (A to C) ACTZ treatment caused a slight increase in bacterial lung burden (A) and no change in pendrin protein production (B). However, lung pathology was significantly lower in ACTZ-treated mice as determined by pathology score (C). Bars represent mean ± SEM. (D to G) Representative H&E-stained histology images.
DISCUSSION
In this study, we have shown that B. pertussis infection in mouse models upregulates lung expression of the epithelial anion exchanger pendrin. Slc26a4/pendrin was enhanced at the level of both mRNA and protein, was dependent upon PT production by the bacteria, and was independent of mouse genetic background. In addition, pendrin upregulation was not dependent on the transcription factor STAT6 but was dependent upon the cytokine IL-17A. Importantly, pendrin appears to make a significant contribution to lung pathology during B. pertussis infection, since pendrin KO mice suffered significantly lower levels of lung inflammatory pathology and treatment of B. pertussis-infected mice with the carbonic anhydrase inhibitor ACTZ also significantly reduced lung inflammatory pathology. However, inflammatory cytokine and chemokine levels were higher in pendrin KO B. pertussis-infected mice than in pendrin-expressing infected mice, but the mechanism of pendrin-dependent enhancement of inflammatory pathology remains to be determined.
We identified the pendrin gene as a PT-dependent upregulated gene in B. pertussis-infected BALB/c mouse lungs in our recently reported microarray analysis (27). In a previous microarray study of upregulated genes in B. pertussis-infected C3H mice, Banus et al. also found upregulation of lung Slc26a4 expression (3.3-fold at 5 dpi versus uninfected mice) (53), and since we have observed pendrin upregulation in both BALB/c and C57BL/6 mice, this appears to be common to different genetic backgrounds. Pendrin production was apparently upregulated on the bronchial but not the tracheal epithelium in infected mice. However, preliminary immunohistochemistry analysis indicated that pendrin may also be produced by other cells in the lung tissue during B. pertussis infection (data not shown), although the identity of these cells and the role that their pendrin expression plays in inflammatory pathology remain to be determined.
The mechanism of pendrin gene upregulation during B. pertussis infection is unclear. In mouse models of asthma, pendrin upregulation is dependent on the TH2 cytokines IL-4 and IL-13 acting through the transcription factor STAT6 (34, 40). However, these cytokines are not produced at significant levels during B. pertussis infection, and our data rule out STAT6 involvement. PT is necessary for full pendrin upregulation, but it is not sufficient, indicating that other host factors associated with PT production during B. pertussis infection are involved. Our finding that pendrin upregulation is significantly lower in Il-17a−/− mice suggests that IL-17A is one such host factor. These results are consistent with our previous finding that IL-17A upregulation is associated with PT production during B. pertussis infection (22). IL-17A was also found to induce bicarbonate secretion (a pendrin activity) in human bronchial epithelial cells (47), suggesting a role for this cytokine in pendrin upregulation in human airways. However, other host factors may also be involved, such as IL-1β and IFN-γ, since both are upregulated during B. pertussis infection, are associated with PT production (27), and have been linked with pendrin upregulation in other studies (40, 54).
As well as exchanging chloride for bicarbonate ions at the lung epithelium, pendrin exports thiocyanate (SCN−) ions, which can be converted to microbicidal hypothiocyanite (OSCN−) ions (39). Therefore, pendrin upregulation may be an antimicrobial host defense mechanism at epithelial surfaces. Our observation that bacterial loads were higher in pendrin KO mice than in pendrin-expressing mice is consistent with this idea. However, our data demonstrating reduced inflammatory pathology in pendrin KO mice (despite the higher bacterial loads) and in ACTZ-treated mice strongly implicate pendrin as a contributor to lung inflammatory pathology in B. pertussis infection. The mechanism of pendrin-enhanced inflammatory pathology is unclear. Surprisingly, levels of inflammatory cytokines and chemokines were higher in the lungs of infected pendrin KO mice than in those of infected pendrin-expressing mice. One possibility is that pendrin increases the pH and decreases the salinity of the airway surface liquid (ASL) through its export of bicarbonate ions and import of chloride ions, respectively. These conditions may favor increased activity of antimicrobial factors, such as defensins, lysozyme, or lactoferrin, as suggested in recent study that employed a porcine model (55). Such an environment may also favor increased activity of cytokines, chemokines, and their receptors, leading to the increased inflammatory pathology in B. pertussis infection. Consistent with this, Dairaghi et al. found that the activity of chemokine receptor CCR3 in its interaction with CCL11 was greatly increased by a modest increase in pH and decrease in salinity (56). In pendrin KO mice, the higher levels of cytokines and chemokines may therefore not result in inflammatory pathology because the ASL pH is correspondingly lower and the activity of these molecules is reduced. Interestingly, this situation may be similar in cystic fibrosis, where reduced function of the chloride channel CFTR may lower ASL pH and increase susceptibility to lung bacterial infection (55). Evidence exists that CFTR and SLC transporters may colocalize and interact (57) and that CFTR and pendrin may act coordinately to control ASL conditions in the lung (37, 58).
Our finding that treatment of B. pertussis-infected mice with ACTZ significantly reduced lung inflammatory pathology associated with the infection highlights pendrin as a possible therapeutic target for treatment of pertussis. ACTZ is a carbonic anhydrase inhibitor that is in clinical use for treatment of a variety of medical conditions, including as a respiratory stimulant in patients with COPD and metabolic alkalosis (59). Treatment from the outset of bacterial infection, as in our study, is somewhat artificial, but in future work we will investigate the therapeutic potential of this drug by treatment of infected mice at later time points of infection and by different routes of administration, since systemic inhibition of pendrin activity may lead to unwanted deleterious effects on other organs. Whether pendrin is upregulated in pertussis infection in humans remains to be determined, but it is upregulated in human asthma (35) as well as in mouse asthma models (33, 40), suggesting that the mouse model may be representative. Whether the inflammatory lung pathology seen in mouse models of pertussis is associated with the severe cough in human pertussis is unknown, but severe inflammatory pathology is a hallmark of fatal pertussis infections in young infants (9, 60). Therefore, therapeutic targeting of pendrin may be beneficial in treatment of infants with critical pertussis, for whom there are currently few, if any, effective treatments (1).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Public Health Service grant AI-101055 from the National Institute of Allergy and Infectious Diseases.
We thank Achsah Keegan for STAT6 knockout mice, Mark Shirtliff for IL-17A knockout mice, and Lindsay Goicochea for help with assessing pathology scores.
Footnotes
Published ahead of print 28 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02222-14.
REFERENCES
- 1. Berger JT, Carcillo JA, Shanley TP, Wessel DL, Clark A, Holubkov R, Meert KL, Newth CJ, Berg RA, Heidemann S, Harrison R, Pollack M, Dalton H, Harvill E, Karanikas A, Liu T, Burr JS, Doctor A, Dean JM, Jenkins TL, Nicholson CE, Eunice Kennedy Shriver National Institute of Child Health and Human Development Collaborative Pediatric Critical Care Research Network 2013. Critical pertussis illness in children: a multicenter prospective cohort study. Pediatr. Crit. Care Med. 14:356–365. 10.1097/PCC.0b013e31828a70fe [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mooi FR, Van Der Maas NA, De Melker HE. 2014. Pertussis resurgence: waning immunity and pathogen adaptation—two sides of the same coin. Epidemiol. Infect. 142:685–694. 10.1017/S0950268813000071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ausiello CM, Urbani F, la Sala A, Lande R, Cassone A. 1997. Vaccine- and antigen-dependent type 1 and type 2 cytokine induction after primary vaccination of infants with whole-cell or acellular pertussis vaccines. Infect. Immun. 65:2168–2174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Klein NP, Bartlett J, Rowhani-Rahbar A, Fireman B, Baxter R. 2012. Waning protection after fifth dose of acellular pertussis vaccine in children. N. Engl. J. Med. 367:1012–1019. 10.1056/NEJMoa1200850 [DOI] [PubMed] [Google Scholar]
- 5. Ross PJ, Sutton CE, Higgins S, Allen AC, Walsh K, Misiak A, Lavelle EC, McLoughlin RM, Mills KH. 2013. Relative contribution of Th1 and Th17 cells in adaptive immunity to Bordetella pertussis: towards the rational design of an improved acellular pertussis vaccine. PLoS Pathog. 9:e1003264. 10.1371/journal.ppat.1003264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Smits K, Pottier G, Smet J, Dirix V, Vermeulen F, De Schutter I, Carollo M, Locht C, Ausiello CM, Mascart F. 2013. Different T cell memory in preadolescents after whole-cell or acellular pertussis vaccination. Vaccine 32:111–118. 10.1016/j.vaccine.2013.10.056 [DOI] [PubMed] [Google Scholar]
- 7. Warfel JM, Zimmerman LI, Merkel TJ. 2014. Acellular pertussis vaccines protect against disease but fail to prevent infection and transmission in a nonhuman primate model. Proc. Natl. Acad. Sci. U. S. A. 111:787–792. 10.1073/pnas.1314688110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cherry JD. 2012. Epidemic pertussis in 2012—the resurgence of a vaccine-preventable disease. N. Engl. J. Med. 367:785–787. 10.1056/NEJMp1209051 [DOI] [PubMed] [Google Scholar]
- 9. Paddock CD, Sanden GN, Cherry JD, Gal AA, Langston C, Tatti KM, Wu KH, Goldsmith CS, Greer PW, Montague JL, Eliason MT, Holman RC, Guarner J, Shieh WJ, Zaki SR. 2008. Pathology and pathogenesis of fatal Bordetella pertussis infection in infants. Clin. Infect. Dis. 47:328–338. 10.1086/589753 [DOI] [PubMed] [Google Scholar]
- 10. Carbonetti NH. 2010. Pertussis toxin and adenylate cyclase toxin: key virulence factors of Bordetella pertussis and cell biology tools. Future Microbiol. 5:455–469. 10.2217/fmb.09.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Witvliet MH, Burns DL, Brennan MJ, Poolman JT, Manclark CR. 1989. Binding of pertussis toxin to eucaryotic cells and glycoproteins. Infect. Immun. 57:3324–3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wong WS, Rosoff PM. 1996. Pharmacology of pertussis toxin B-oligomer. Can. J. Physiol. Pharmacol. 74:559–564. 10.1139/y96-045 [DOI] [PubMed] [Google Scholar]
- 13. Hazes B, Read RJ. 1997. Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry 36:11051–11054. 10.1021/bi971383p [DOI] [PubMed] [Google Scholar]
- 14. Plaut RD, Carbonetti NH. 2008. Retrograde transport of pertussis toxin in the mammalian cell. Cell. Microbiol. 10:1130–1139. 10.1111/j.1462-5822.2007.01115.x [DOI] [PubMed] [Google Scholar]
- 15. Worthington ZE, Carbonetti NH. 2007. Evading the proteasome: absence of lysine residues contributes to pertussis toxin activity by evasion of proteasome degradation. Infect. Immun. 75:2946–2953. 10.1128/IAI.02011-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Burns DL. 1988. Subunit structure and enzymic activity of pertussis toxin. Microbiol. Sci. 5:285–287 [PubMed] [Google Scholar]
- 17. Katada T, Ui M. 1982. ADP ribosylation of the specific membrane protein of C6 cells by islet-activating protein associated with modification of adenylate cyclase activity. J. Biol. Chem. 257:7210–7216 [PubMed] [Google Scholar]
- 18. Carbonetti NH, Artamonova GV, Mays RM, Worthington ZE. 2003. Pertussis toxin plays an early role in respiratory tract colonization by Bordetella pertussis. Infect. Immun. 71:6358–6366. 10.1128/IAI.71.11.6358-6366.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carbonetti NH, Artamonova GV, Van Rooijen N, Ayala VI. 2007. Pertussis toxin targets airway macrophages to promote Bordetella pertussis infection of the respiratory tract. Infect. Immun. 75:1713–1720. 10.1128/IAI.01578-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Andreasen C, Carbonetti NH. 2008. Pertussis toxin inhibits early chemokine production to delay neutrophil recruitment in response to Bordetella pertussis respiratory tract infection in mice. Infect. Immun. 76:5139–5148. 10.1128/IAI.00895-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kirimanjeswara GS, Agosto LM, Kennett MJ, Bjornstad ON, Harvill ET. 2005. Pertussis toxin inhibits neutrophil recruitment to delay antibody-mediated clearance of Bordetella pertussis. J. Clin. Invest. 115:3594–3601. 10.1172/JCI24609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Andreasen C, Powell DA, Carbonetti NH. 2009. Pertussis toxin stimulates IL-17 production in response to Bordetella pertussis infection in mice. PLoS One 4:e7079. 10.1371/journal.pone.0007079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carbonetti NH, Artamonova GV, Andreasen C, Dudley E, Mays RM, Worthington ZE. 2004. Suppression of serum antibody responses by pertussis toxin after respiratory tract colonization by Bordetella pertussis and identification of an immunodominant lipoprotein. Infect. Immun. 72:3350–3358. 10.1128/IAI.72.6.3350-3358.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wolfe DN, Mann PB, Buboltz AM, Harvill ET. 2007. Delayed role of tumor necrosis factor-alpha in overcoming the effects of pertussis toxin. J. Infect. Dis. 196:1228–1236. 10.1086/521303 [DOI] [PubMed] [Google Scholar]
- 25. Zhang X, Hester SE, Kennett MJ, Karanikas AT, Bendor L, Place DE, Harvill ET. 2011. Interleukin-1 receptor signaling is required to overcome the effects of pertussis toxin and for efficient infection- or vaccination-induced immunity against Bordetella pertussis. Infect. Immun. 79:527–541. 10.1128/IAI.00590-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weiss AA, Goodwin MS. 1989. Lethal infection by Bordetella pertussis mutants in the infant mouse model. Infect. Immun. 57:3757–3764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Connelly CE, Sun Y, Carbonetti NH. 2012. Pertussis toxin exacerbates and prolongs airway inflammatory responses during Bordetella pertussis infection. Infect. Immun. 80:4317–4332. 10.1128/IAI.00808-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Royaux IE, Suzuki K, Mori A, Katoh R, Everett LA, Kohn LD, Green ED. 2000. Pendrin, the protein encoded by the Pendred syndrome gene (PDS), is an apical porter of iodide in the thyroid and is regulated by thyroglobulin in FRTL-5 cells. Endocrinology 141:839–845. 10.1210/endo.141.2.7303 [DOI] [PubMed] [Google Scholar]
- 29. Royaux IE, Wall SM, Karniski LP, Everett LA, Suzuki K, Knepper MA, Green ED. 2001. Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc. Natl. Acad. Sci. U. S. A. 98:4221–4226. 10.1073/pnas.071516798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Scott DA, Wang R, Kreman TM, Sheffield VC, Karniski LP. 1999. The Pendred syndrome gene encodes a chloride-iodide transport protein. Nat. Genet. 21:440–443. 10.1038/7783 [DOI] [PubMed] [Google Scholar]
- 31. Alesutan I, Daryadel A, Mohebbi N, Pelzl L, Leibrock C, Voelkl J, Bourgeois S, Dossena S, Nofziger C, Paulmichl M, Wagner CA, Lang F. 2011. Impact of bicarbonate, ammonium chloride, and acetazolamide on hepatic and renal SLC26A4 expression. Cell. Physiol. Biochem. 28:553–558. 10.1159/000335114 [DOI] [PubMed] [Google Scholar]
- 32. Kuperman DA, Lewis CC, Woodruff PG, Rodriguez MW, Yang YH, Dolganov GM, Fahy JV, Erle DJ. 2005. Dissecting asthma using focused transgenic modeling and functional genomics. J. Allergy Clin. Immunol. 116:305–311. 10.1016/j.jaci.2005.03.024 [DOI] [PubMed] [Google Scholar]
- 33. Nakao I, Kanaji S, Ohta S, Matsushita H, Arima K, Yuyama N, Yamaya M, Nakayama K, Kubo H, Watanabe M, Sagara H, Sugiyama K, Tanaka H, Toda S, Hayashi H, Inoue H, Hoshino T, Shiraki A, Inoue M, Suzuki K, Aizawa H, Okinami S, Nagai H, Hasegawa M, Fukuda T, Green ED, Izuhara K. 2008. Identification of pendrin as a common mediator for mucus production in bronchial asthma and chronic obstructive pulmonary disease. J. Immunol. 180:6262–6269. 10.4049/jimmunol.180.9.6262 [DOI] [PubMed] [Google Scholar]
- 34. Nofziger C, Vezzoli V, Dossena S, Schonherr T, Studnicka J, Nofziger J, Vanoni S, Stephan S, Silva ME, Meyer G, Paulmichl M. 2011. STAT6 links IL-4/IL-13 stimulation with pendrin expression in asthma and chronic obstructive pulmonary disease. Clin. Pharmacol. Ther. 90:399–405. 10.1038/clpt.2011.128 [DOI] [PubMed] [Google Scholar]
- 35. Yick CY, Zwinderman AH, Kunst PW, Grunberg K, Mauad T, Dijkhuis A, Bel EH, Baas F, Lutter R, Sterk PJ. 2013. Transcriptome sequencing (RNA-Seq) of human endobronchial biopsies: asthma versus controls. Eur. Respir. J. 42:662–670. 10.1183/09031936.00115412 [DOI] [PubMed] [Google Scholar]
- 36. Ishida A, Ohta N, Suzuki Y, Kakehata S, Okubo K, Ikeda H, Shiraishi H, Izuhara K. 2012. Expression of pendrin and periostin in allergic rhinitis and chronic rhinosinusitis. Allergol. Int. 61:589–595. 10.2332/allergolint.11-OA-0370 [DOI] [PubMed] [Google Scholar]
- 37. Garnett JP, Hickman E, Burrows R, Hegyi P, Tiszlavicz L, Cuthbert AW, Fong P, Gray MA. 2011. Novel role for pendrin in orchestrating bicarbonate secretion in cystic fibrosis transmembrane conductance regulator (CFTR)-expressing airway serous cells. J. Biol. Chem. 286:41069–41082. 10.1074/jbc.M111.266734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pedemonte N, Caci E, Sondo E, Caputo A, Rhoden K, Pfeffer U, Di Candia M, Bandettini R, Ravazzolo R, Zegarra-Moran O, Galietta LJ. 2007. Thiocyanate transport in resting and IL-4-stimulated human bronchial epithelial cells: role of pendrin and anion channels. J. Immunol. 178:5144–5153. 10.4049/jimmunol.178.8.5144 [DOI] [PubMed] [Google Scholar]
- 39. Ratner AJ, Prince A. 2000. Lactoperoxidase. New recognition of an “old” enzyme in airway defenses. Am. J. Respir. Cell Mol. Biol. 22:642–644. 10.1165/ajrcmb.22.6.f186 [DOI] [PubMed] [Google Scholar]
- 40. Nakagami Y, Favoreto S, Jr, Zhen G, Park SW, Nguyenvu LT, Kuperman DA, Dolganov GM, Huang X, Boushey HA, Avila PC, Erle DJ. 2008. The epithelial anion transporter pendrin is induced by allergy and rhinovirus infection, regulates airway surface liquid, and increases airway reactivity and inflammation in an asthma model. J. Immunol. 181:2203–2210. 10.4049/jimmunol.181.3.2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bart MJ, van Gent M, van der Heide HG, Boekhorst J, Hermans P, Parkhill J, Mooi FR. 2010. Comparative genomics of prevaccination and modern Bordetella pertussis strains. BMC Genomics 11:627. 10.1186/1471-2164-11-627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kaplan MH, Schindler U, Smiley ST, Grusby MJ. 1996. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity 4:313–319. 10.1016/S1074-7613(00)80439-2 [DOI] [PubMed] [Google Scholar]
- 43. Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y. 2002. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity 17:375–387. 10.1016/S1074-7613(02)00391-6 [DOI] [PubMed] [Google Scholar]
- 44. Amlal H, Petrovic S, Xu J, Wang Z, Sun X, Barone S, Soleimani M. 2010. Deletion of the anion exchanger Slc26a4 (pendrin) decreases apical Cl(−)/HCO3(−) exchanger activity and impairs bicarbonate secretion in kidney collecting duct. Am. J. Physiol. Cell Physiol. 299:C33–C41. 10.1152/ajpcell.00033.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Knauf F, Yang CL, Thomson RB, Mentone SA, Giebisch G, Aronson PS. 2001. Identification of a chloride-formate exchanger expressed on the brush border membrane of renal proximal tubule cells. Proc. Natl. Acad. Sci. U. S. A. 98:9425–9430. 10.1073/pnas.141241098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kasuga T, Nakase Y, Ukishima K, Takatsu K. 1954. Studies on Haemophilus pertussis. V. Relation between the phase of bacilli and the progress of the whooping-cough. Kitasato Arch. Exp. Med. 27:57–62 [PubMed] [Google Scholar]
- 47. Kreindler JL, Bertrand CA, Lee RJ, Karasic T, Aujla S, Pilewski JM, Frizzell RA, Kolls JK. 2009. Interleukin-17A induces bicarbonate secretion in normal human bronchial epithelial cells. Am. J. Physiol. 296:L257–L266. 10.1152/ajplung.00344.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Higgins SC, Jarnicki AG, Lavelle EC, Mills KH. 2006. TLR4 mediates vaccine-induced protective cellular immunity to Bordetella pertussis: role of IL-17-producing T cells. J. Immunol. 177:7980–7989. 10.4049/jimmunol.177.11.7980 [DOI] [PubMed] [Google Scholar]
- 49. Khelef N, Bachelet CM, Vargaftig BB, Guiso N. 1994. Characterization of murine lung inflammation after infection with parental Bordetella pertussis and mutants deficient in adhesins or toxins. Infect. Immun. 62:2893–2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Milton AE, Weiner ID. 1998. Regulation of B-type intercalated cell apical anion exchange activity by CO2/HCO3. Am. J. Physiol. 274:F1086–F1094 [DOI] [PubMed] [Google Scholar]
- 51. Pech V, Pham TD, Hong S, Weinstein AM, Spencer KB, Duke BJ, Walp E, Kim YH, Sutliff RL, Bao HF, Eaton DC, Wall SM. 2010. Pendrin modulates ENaC function by changing luminal HCO3. J. Am. Soc. Nephrol. 21:1928–1941. 10.1681/ASN.2009121257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zahedi K, Barone S, Xu J, Soleimani M. 2013. Potentiation of the effect of thiazide derivatives by carbonic anhydrase inhibitors: molecular mechanisms and potential clinical implications. PLoS One 8:e79327. 10.1371/journal.pone.0079327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Banus S, Pennings J, Vandebriel R, Wester P, Breit T, Mooi F, Hoebee B, Kimman T. 2007. Lung response to Bordetella pertussis infection in mice identified by gene-expression profiling. Immunogenetics 59:555–564. 10.1007/s00251-007-0227-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hogmalm A, Bry M, Strandvik B, Bry K. 2014. IL-1beta expression in the distal lung epithelium disrupts lung morphogenesis and epithelial cell differentiation in fetal mice. Am. J. Physiol. 306:L23–L34. 10.1152/ajplung.00154.2013 [DOI] [PubMed] [Google Scholar]
- 55. Pezzulo AA, Tang XX, Hoegger MJ, Alaiwa MH, Ramachandran S, Moninger TO, Karp PH, Wohlford-Lenane CL, Haagsman HP, van Eijk M, Banfi B, Horswill AR, Stoltz DA, McCray PB, Jr, Welsh MJ, Zabner J. 2012. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 487:109–113. 10.1038/nature11130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dairaghi DJ, Oldham ER, Bacon KB, Schall TJ. 1997. Chemokine receptor CCR3 function is highly dependent on local pH and ionic strength. J. Biol. Chem. 272:28206–28209. 10.1074/jbc.272.45.28206 [DOI] [PubMed] [Google Scholar]
- 57. Ko SB, Zeng W, Dorwart MR, Luo X, Kim KH, Millen L, Goto H, Naruse S, Soyombo A, Thomas PJ, Muallem S. 2004. Gating of CFTR by the STAS domain of SLC26 transporters. Nat. Cell Biol. 6:343–350. 10.1038/ncb1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Garnett JP, Hickman E, Tunkamnerdthai O, Cuthbert AW, Gray MA. 2013. Protein phosphatase 1 coordinates CFTR-dependent airway epithelial HCO3− secretion by reciprocal regulation of apical and basolateral membrane Cl(−)-HCO3− exchangers. Br. J. Pharmacol. 168:1946–1960. 10.1111/bph.12085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Heming N, Urien S, Faisy C. 2012. Acetazolamide: a second wind for a respiratory stimulant in the intensive care unit? Crit. Care 16:318. 10.1186/cc11323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sawal M, Cohen M, Irazuzta JE, Kumar R, Kirton C, Brundler MA, Evans CA, Wilson JA, Raffeeq P, Azaz A, Rotta AT, Vora A, Vohra A, Abboud P, Mirkin LD, Cooper M, Dishop MK, Graf JM, Petros A, Klonin H. 2009. Fulminant pertussis: a multi-center study with new insights into the clinico-pathological mechanisms. Pediatr. Pulmonol. 44:970–980. 10.1002/ppul.21082 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.