

Abstract
The virulence of Candida albicans in a mouse model of invasive candidiasis is dependent on the phospholipids phosphatidylserine (PS) and phosphatidylethanolamine (PE). Disruption of the PS synthase gene CHO1 (i.e., cho1Δ/Δ) eliminates PS and blocks the de novo pathway for PE biosynthesis. In addition, the cho1Δ/Δ mutant's ability to cause invasive disease is severely compromised. The cho1Δ/Δ mutant also exhibits cell wall defects, and in this study, it was determined that loss of PS results in decreased masking of cell wall β(1-3)-glucan from the immune system. In wild-type C. albicans, the outer mannan layer of the wall masks the inner layer of β(1-3)-glucan from exposure and detection by innate immune effector molecules like the C-type signaling lectin Dectin-1, which is found on macrophages, neutrophils, and dendritic cells. The cho1Δ/Δ mutant exhibits increases in exposure of β(1-3)-glucan, which leads to greater binding by Dectin-1 in both yeast and hyphal forms. The unmasking of β(1-3)-glucan also results in increased elicitation of TNF-α from macrophages in a Dectin-1-dependent manner. The role of phospholipids in fungal pathogenesis is an emerging field, and this is the first study showing that loss of PS in C. albicans results in decreased masking of β(1-3)-glucan, which may contribute to our understanding of fungus-host interactions.
INTRODUCTION
The yeast Candida albicans is a commensal fungus that has the ability to cause systemic bloodstream infections in immunocompromised patients with neutropenia. C. albicans and other Candida species are, together, the fourth leading cause of nosocomial bloodstream infections, which have a mortality rate of approximately 30% (1). These fungi also have the ability to colonize prosthetic devices such as artificial heart valves, and complications arising from this colonization can have a mortality rate of greater than 60% (2). C. albicans also causes superficial mucosal infections such as thrush or oropharyngeal candidiasis, particularly in the immunocompromised, an affliction that can affect up to 50% of AIDS patients (3).
Currently, there are effective drugs available to treat fungal infections, but they are also limited, with some that have serious side effects and others that are less effective because of drug resistance (4–6). Therefore, it is crucial to discover new antifungal targets that are conserved in fungi but not in humans and characterize how inhibition or deletion of these targets affects the physiology and virulence of the pathogen.
The de novo pathway for synthesizing the phospholipids phosphatidylserine (PS) and phosphatidylethanolamine (PE) is crucial for virulence in a systemic mouse model of C. albicans infections (7). In general, PS makes up 6% and PE makes up 15% of the total cell membrane phospholipids (7). Occupying the first step in the de novo synthesis pathway, the Cho1p PS synthase, which is required for PS synthesis, is a good potential target for antifungal development, as it has no mammalian homolog (8), and its absence (cho1Δ/Δ mutant) leads to avirulence in a mouse model of candidemia (7). PE synthesis is also affected in this mutant because PS serves as a substrate for the de novo synthesis pathway, one of two pathways that synthesize PE. In this pathway, the partially redundant PS decarboxylases Psd1p and Psd2p convert PS to PE, and these enzymes are also required for virulence. When both enzymes are knocked out simultaneously in a psd1Δ/Δ psd2Δ/Δ double mutant, virulence is severely attenuated and PE levels drop by approximately half (7). The cho1Δ/Δ and psd1Δ/Δ psd2Δ/Δ mutants have in common a decrease in de novo PE synthesis, but only the cho1Δ/Δ mutant lacks PS (7).
Both the innate and adaptive immunological responses of the host play a role in controlling and eliminating fungal infections, but the innate response is the early responder and is crucial for controlling bloodstream infections (9, 10). The innate response is mediated largely by phagocytic cells such as macrophages and neutrophils, which recognize, phagocytose, and kill the pathogen (11). Recognition is dependent, in part, on pathogen-associated molecular patterns (PAMPs) that are present on the surface of the pathogen, which can then be recognized by phagocytic pattern recognition receptors (PRRs) (12–15).
Fungal cell walls contain the PAMP β(1-3)-glucan. The fungal wall is composed of two layers; mannosylated proteins (mannan) are enriched on the outer surface layer, and β(1-3)-glucan and chitin are enriched in the layer beneath (16, 17). β(1-3)-Glucan is a known PAMP that is recognized by Dectin-1, a C-type lectin PRR that is expressed by macrophages, neutrophils, and dendritic cells (12). Several cell wall mutants with increased exposure of β(1-3)-glucan also exhibit increased recognition by phagocytic cells, resulting in increased phagocytosis by macrophages and production of cytokines, inducing a strong response by the innate immune system that may play a role in the faster elimination of these mutants (18, 19).
In this communication, we report that the loss of PS in the C. albicans cho1Δ/Δ mutant results in changes in cell wall structure, notably, an increase in β(1-3)-glucan exposure on the cell surface. The full mechanism of how phospholipids affect the cell wall is not known, but it may relate to the manner in which phospholipids like PS modulate signaling pathways like calcineurin, protein kinase C, Hog1p, or Cek1p, which regulate the cell wall (20, 21). Increased β(1-3)-glucan exposure induces increased Dectin-1 binding, mediating macrophage release of the cytokine tumor necrosis factor alpha (TNF-α). The TNF-α response is important in protecting against systemic candidiasis, and when the TNF-α response is ablated by either knocking out the TNF-α response or neutralizing the response with an anti-TNF-α antibody, mice are more susceptible to systemic candidemia (22, 23). Interestingly, we find that the psd1Δ/Δ psd2Δ/Δ double mutant does not exhibit these phenotypes as strongly, suggesting that PS is specifically required for β(1-3)-glucan “masking” from immune recognition.
MATERIALS AND METHODS
Strains and growth media.
All of the strains used for these experiments are described in reference 7 or derived from these strains (Table 1). Strains were cultured in YPD liquid medium (1% yeast extract, 2% peptone, 2% glucose) or on YPD agar plates (24). The RAW264.7 mouse-derived macrophage cell line (25) and human neutrophils were both cultured in RPMI plus 10% fetal bovine serum and 1% penicillin-streptomycin (Gibco). The RAW-BLUE cells that expressed elevated levels of Dectin-1 were cultured in the same medium as RAW264.7 macrophages, only with the addition of 200 μg/ml Zeocin (Research Products International Corporation).
TABLE 1.
C. albicans strains used in this study
| Strain | Parent | Genotype | Source or reference |
|---|---|---|---|
| SC5314 | Clinical isolate | Prototrophic wild type | 43 |
| YLC337 | SC5314 | cho1Δ/cho1Δ | 7 |
| YLC344 | SC5314 | cho1Δ/cho1Δ::CHO1-SAT1 | 7 |
| YLC280 | SC5314 | psd1Δ/psd1Δ | 7 |
| YLC294 | SC5314 | psd1Δ/psd1Δ::PSD1-SAT1 | 7 |
| YLC375 | SC5314 | psd2Δ/psd2Δ psd1Δ/psd1Δ | 7 |
| SED022 | YLC337 | cho1Δ/Δ::CHO1 with SAT1 flipped out | This study |
| SED010 | SC5314 | Wild type with PENO1-GFP-SAT1 | This study |
| SED013 | YLC337 | cho1Δ/cho1Δ mutant with PENO1-GFP-SAT1 | This study |
| SED016 | YLC280 | psd1Δ/psd1Δ mutant with PENO1-GFP-SAT1 | This study |
| SED019 | YLC375 | psd2Δ/Δ psd1Δ/psd1Δ mutant with PENO1-GFP-SAT1 | This study |
| SED024 | SED022 | cho1Δ/cho1Δ::CHO1 mutant with PENO1-GFP-SAT1 | This study |
For the generation of strains that express cytoplasmic green fluorescent protein (GFP) constitutively under the control of the promoter of the gene for enolase (ENO1), we used the plasmid pENO1-yEGFP3-NAT (26) and transformation methods that have been previously described (27). To generate the cho1Δ/Δ::CHO1 reintegrant strain that expresses GFP, the SAT1 marker was flipped out of the CHO1 reintegration construct (pCHO1-SAT1-flipper) (strain SED022) and then transformed with the above-described GFP plasmid using nourseothricin selection at a concentration of 200 μg/ml (WERNER BioAgents).
Purification of sDectin-1–Fc.
HEK293T cells were transfected with the soluble Dectin-1–Fc (sDectin-1–Fc) construct previously described (28). After selection for stable transfection with Zeocin, the cells were seeded into a CELLine AD1000 bioreactor (Wheaton) and cell supernatants were harvested twice weekly. Supernatants were purified over HiTrap protein A columns (GE Healthcare) and dialyzed overnight with phosphate-buffered saline (PBS) in Slide-A-Lyzer Dialysis cassettes (Thermo Scientific). Dialyzed supernatants were concentrated with Amicon Ultra centrifugal filters (Millipore). Elution from the column directly into 1 M Tris (pH 9) neutralization buffer was performed under low-pH conditions with 100 mM glycine (pH 2.8). The concentration of sDectin-1–Fc was determined with the Bradford assay.
sDectin-1–Fc staining and flow cytometry.
C. albicans strains were streaked onto YPD agar plates and left at 37°C overnight. A single colony per strain was picked and transferred into 5 ml YPD liquid, which was put into a rotator wheel and left overnight at 37°C. A sample of culture was centrifuged and washed three times with PBS. Samples were blocked in PBS plus 2% bovine serum albumin for 1 h at room temperature. After blocking, samples were stained with sDectin-1–Fc at 16.5 μg/ml for 1.5 h on ice. Samples were washed with PBS five times and then stained with donkey anti-human IgG DyLight 488 antibody (Jackson ImmunoResearch) at 0.83 μg/ml for 20 min on ice. Samples were washed five times with PBS and then resuspended in 500 μl PBS for flow cytometry. Flow cytometry data were obtained for 10,000 gated events per strain, and statistics were calculated with the paired Student t test. For imaging, Candida cells were prepared as outlined above but were resuspended in 50 μl PBS and visualized with a Zeiss AxioVision Vivotome microscope (Carl Zeiss Microscopy, LLC). This experiment was repeated twice.
In other immunofluorescence experiments with sDectin-1–Fc staining, donkey anti-human IgG-Cy3 or rabbit-anti-human IgG-Cy2 (both from Jackson ImmunoResearch) was used as the secondary antibody.
Immunofluorescence of β(1-3)-glucan and complement.
Strains were grown overnight in YPD at 30°C. For yeast-form staining, 0.1 optical density unit of each strain was washed three times with PBS. Blocking and primary and secondary antibody incubations occurred on ice in PBS plus 3% bovine serum albumin. Blocking was for 1 h, primary-antibody incubation was for 1.5 h, and secondary-antibody incubation was for 20 min with five washes between antibodies. An anti-β(1-3)-glucan antibody (Biosupplies Australia) at a 1:800 dilution was used as the primary antibody, and a goat anti-mouse antibody conjugated to Cy3 (Jackson ImmunoResearch) at 1:600 was used as the secondary antibody. For hyphal-form staining (see Fig. 4), overnight cultures grown at 30°C were washed three times with PBS, added to 20% human serum in RPMI in Lab-Tek glass chamber slides (Thermo Scientific), and incubated at 37°C for 3 h to ensure adequate filamentation. Antibody staining was performed in the same way as yeast-form staining. A formaldehyde fixation step with 37% formaldehyde in PBS was used for yeast and hyphae in some cases before staining began, and this is noted in the figure legends if that was the case.
FIG 4.
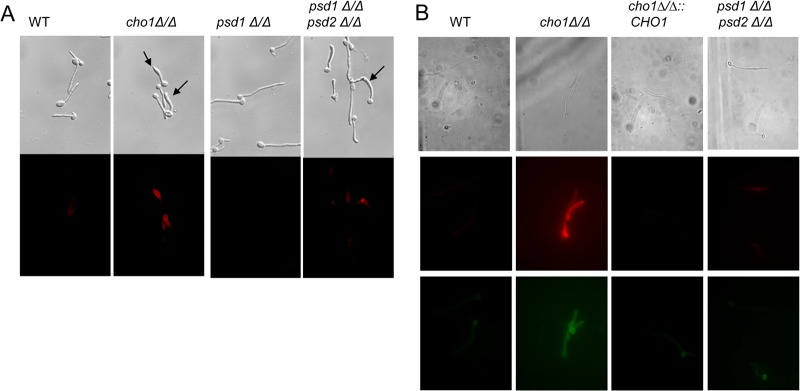
The hyphal form of the cho1Δ/Δ mutant has increased exposure of β(1-3)-glucan and binding by the Dectin-1 receptor. Cells were grown as hyphae in human serum. (A) Secondary immunofluorescence analysis was performed with an anti-β(1-3)-glucan antibody. Increased exposure of β(1-3)-glucan in the cho1Δ/Δ mutant and the psd1Δ/Δ psd2Δ/Δ double mutant appears to localize at malformed hyphal tips (indicated by arrows). Staining was performed after formaldehyde fixation. (B) In the absence of formaldehyde fixation, only the cho1Δ/Δ mutant revealed strong staining with either anti-β(1-3)-glucan antibody or sDectin-1–Fc. These probes exhibited overlapping staining patterns in the hyphal cell wall. Top row, bright-field optics; middle row, staining with anti-β(1-3)-glucan antibody and a Cy3 (red)-conjugated secondary antibody; bottom row, staining with sDectin-1–Fc and a Cy2 (green)-conjugated secondary antibody. WT, wild type.
Complement staining with the primary antibody against human C1q-c complement (Santa Cruz catalog no. sc-365301) (29) and a Cy3-conjugated anti-mouse secondary antibody (Jackson ImmunoResearch) was performed with overnight cultures grown at 30°C that were washed three times with PBS. The yeast cells were then incubated with 50% human serum for 30 min and then stained according to the yeast-form immunofluorescence protocol.
For all immunofluorescence studies, experiments were repeated in triplicate. In each experiment, n = ≥100 for yeast cells and ≥20 for hyphae.
Quantification of immunofluorescence signal of hyphae for β(1-3)-glucan and Dectin-1 immunofluorescence.
The fluorescence intensities of labeled hyphae were assessed with NIS-Elements AR 3.2 image analysis software (Nikon Instruments Inc., Melville, NY). Hyphae were viewed by using a rainbow display scale of pixel intensity and assigned a score based on fluorescence intensity (red = high [2], yellow = medium [1], green = low [0]). Averages were calculated for each strain (n = ≥20).
Killing assays.
Killing assays were performed with macrophages and neutrophils. Neutrophil isolation was done by a method adapted from a previously published protocol (30). Neutrophils were obtained from whole blood from a human donor. The blood was diluted with an equal volume of PBS, Lymphocyte Separation medium (BioWhittaker) was underlaid, and the cells were centrifuged to pellet neutrophils and erythrocytes. The pellet was subjected to 3% dextran sedimentation to isolate neutrophils and further treated with 0.2% sodium chloride to lyse any residual erythrocytes. Yeast-form cells (from an overnight culture grown in YPD) and phagocytes were coincubated at a 1:40 ratio for 2 h at 37°C and 5% CO2, and afterward, yeast cells were plated on YPD plates for total CFU counting. The experiment was performed three separate times, and for each strain in an individual experiment, n = 5, for a total of n = 15 for each strain.
ELISA of TNF-α.
RAW264.7 macrophages were plated the day prior at 5 × 105/well in a 24-well plate. Macrophages and UV-killed yeast-form Candida cells were coincubated at a 1:10 ratio for 8 h at 37°C and 5% CO2. The enzyme-linked immunosorbent assay (ELISA) kit instructions of the manufacturer (R&D Systems) were followed. To determine if the release of TNF-α was due to Dectin-1 binding, a commercial anti-Dectin-1 neutralizing antibody (InvivoGen MAb-mDect) was administered to the macrophages for 1 h prior to coincubation at a concentration of 100 ng/ml. Three individual experiments were performed. For each of these experiments, three individual wells were stimulated for 8 h or for 4 h when RAW-BLUE cells were used. From each well, the sample was taken and measured in triplicate, for a total of nine data points per group. Statistical analysis of the data was performed by the unpaired-sample t test with two-tailed P values.
β-Defensin.
By using a protocol adapted from reference 31, strains were grown overnight in YPD at 30°C, washed with 10 mM sodium phosphate buffer (NaPB), and counted with a hemacytometer and 105 cells were incubated with various concentrations of human β-defensin 3 (AnaSpec catalog no. 60741) for 1 h in 10 mM NaPB in a shaking incubator at 37°C. After incubation, cells were washed with 10 mM NaPB, diluted 1:100, and plated for CFU counting. The assay was performed three separate times. For each strain in each assay, n = 4. Statistical analysis of the data was performed by the unpaired-sample t test with two-tailed P values.
Time-lapse microscopy of hyphal formation.
A 2% agarose gel was overlaid on the surface of a Delta T dish (Bioptechs, Inc.) with wells formed in the gel to contain medium and yeast. Strains were grown overnight in YPD at 30°C and washed with PBS, and cells were counted with a hemacytometer. RPMI plus 25% human serum was added to the wells, and 5 × 103 yeast cells were added to each well. The Delta T dish was placed on a heated stage set to 37°C and imaged automatically to determine hyphal length over a 3-h period. Hyphal length was determined by software analysis (NIS-Elements version 3.1; Nikon Instruments). Quantification was performed by monitoring 20 cells of each strain.
In vivo hyphal formation.
GFP-expressing strains were washed with PBS and stained with Alexa Fluor 555 (Invitrogen) in 100 mM NaCO3 buffer (pH 10) for 30 min at room temperature. After cells were washed three times in PBS, 107 cells were injected per mouse via the tail vein. After 6 h, the mice were sacrificed and their kidneys were removed and immediately homogenized with a Dounce tissue homogenizer and visualized by microscopy. This experiment was performed in triplicate with two or three mice per genotype, so there were eight or nine mice per strain. For each experiment, both kidneys of each mouse were homogenized together and visualized to look for yeast cells. A minimum of 3 cells from each pair of kidneys were imaged, for a minimum of 24 to 27 cells of each strain.
RESULTS
Loss of PS results in increased exposure of β(1-3)-glucan on the C. albicans yeast-form cell surface.
In a previous report, the cho1Δ/Δ and psd1Δ/Δ psd2Δ/Δ mutants' cell wall defects were studied in the context of yeast-form cells grown at 30°C in rich medium (YPD). In these studies, the cho1Δ/Δ mutant exhibited stronger cell wall defects than the psd1Δ/Δ psd2Δ/Δ mutant, including a much greater increase in chitin levels than in wild-type cells and a thicker, more irregular cell wall, as shown by transmission electron microscopy (7). It was hypothesized that these cell wall defects would correlate with phenotypes that impact virulence, such as increased exposure of β(1-3)-glucan, which affects host-pathogen interactions. We examined the effects of these mutations on β(1-3)-glucan exposure by immunofluorescence with an anti-β(1-3)-glucan antibody (18). β(1-3)-Glucan is noticeably more exposed in the log phase of cho1Δ/Δ mutant cells than in wild-type, cho1Δ/Δ::CHO1, psd1Δ/Δ, or even psd1Δ/Δ psd2Δ/Δ mutant cells (Fig. 1). This phenomenon appears to be independent of the growth phase, as the results were also observed in stationary phase after 3 h of growth in YPD (see Fig. S1 in the supplemental material). We also found that these staining patterns held true for the yeast-form cells in both growth phases regardless of whether formaldehyde fixation was used prior to staining with antibodies (data not shown).
FIG 1.
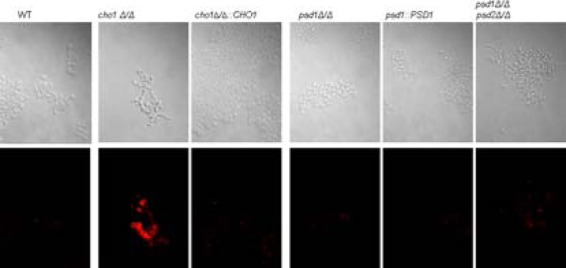
Yeast-form cells of the cho1Δ/Δ mutant exhibit increased exposure of β(1-3)-glucan. Secondary immunofluorescence with an anti-β(1-3)-glucan primary antibody and a Cy3-conjugated secondary antibody reveals that the cho1Δ/Δ mutant has greater β(1-3)-glucan exposure than the other strains. Strains were grown to log phase in YPD at 30°C. WT, wild type.
β(1-3)-Glucan on the surface of C. albicans is bound and detected by Dectin-1, a C-type lectin that is expressed on innate immune effector cells such as macrophages, neutrophils, and dendritic cells (18, 32). This binding and recognition aids macrophages and neutrophils in the phagocytosis of yeast and results in the secretion of cytokines like TNF-α (18, 33). Fluorescent staining with sDectin-1–Fc reveals that cho1Δ/Δ mutant yeast-form cells exhibit greater binding by Dectin-1 than the other strains (Fig. 2A). Flow cytometry to quantify the binding of sDectin-1–Fc to the surface of yeast-form C. albicans supports the microscopy results and indicates that the cho1Δ/Δ mutant has significantly greater binding by Dectin-1 than the other strains and that the psd1Δ/Δpsd2Δ/Δ double mutant (solid pink line) does not differ from the wild type (black) (Fig. 2B). The cho1Δ/Δ cells exhibit an average increase in binding by Dectin-1 over wild type of approximately 58% (Fig. 2C). These results support those showing increased anti-β(1-3)-glucan staining exposure on the cho1Δ/Δ mutant cell surface (Fig. 1).
FIG 2.

The cho1Δ/Δ mutant has increased binding to the Dectin1 receptor. (A) C. albicans strains grown overnight in YPD were stained with sDectin-1–Fc and a fluorescently labeled secondary antibody, showing that the cho1Δ/Δ mutant exhibits greater staining than all of the other strains. (B) Flow cytometry reveals that the cho1Δ/Δ mutant (solid red line) has greater binding to the Dectin-1 receptor than the other strains. (C) A graph of the relative mean staining intensity of each sample reveals that the cho1Δ/Δ mutant exhibits significantly greater staining with sDectin-1–Fc than the other strains. *, P < 0.05.
β(1-3)-Glucan is bound by Dectin-1, but there are data that suggest that anti-β(1-3)-glucan antibody and sDectin-1–Fc may bind to somewhat distinct epitopes (18). If so, then it should be possible to see colocalization of staining with both probes. To assess this, yeast-form cells were double stained with an anti-β(1-3)-glucan antibody and sDectin-1–Fc and secondary antibodies with differential fluorophores. It is clear that the signals colocalized and are very prominent for the cho1Δ/Δ mutant (see Fig. S2 in the supplemental material).
Phospholipid mutants generate hyphae when growing in human serum.
Since cho1Δ/Δ, but not psd1Δ/Δ psd2Δ/Δ, mutant cells exhibit increased exposure of β(1-3)-glucan in the yeast form, the effects of these mutations on the exposure of β(1-3)-glucan in hyphae needed to be addressed to determine if there were any cell wall differences that are dependent on morphology. Previous work has shown that the cho1Δ/Δ and psd1Δ/Δ psd2Δ/Δ mutants show poor hyphal formation on plates in response to a neutral pH and Spider medium. However, the cho1Δ/Δ and psd1Δ/Δ psd2Δ/Δ mutants could both form hyphae in response to mouse serum (7). These previous data were only qualitative, and it was not clear whether the mutants grew as hyphae at a rate similar to that of the wild type in response to serum. Consequently, the hyphal extension rates of cho1Δ/Δ and psd1Δ/Δ psd2Δ/Δ mutants were compared to those of the wild type and psd1Δ/Δ mutants in human serum by time-lapse microscopy. Through this assay, it was found that all of the strains except the cho1Δ/Δ mutant exhibited similar hyphal extension rates, but the cho1Δ/Δ mutant was slightly slower, although the difference was not statistically significant (Fig. 3A).
FIG 3.

Hyphal formation by the cho1Δ/Δ mutant is not significantly impaired in serum or host tissue. (A) C. albicans strains were incubated in 100% human serum and observed by time-lapse microscopy on thermally regulated glass slides. After 3 h, hyphal length was measured as an indicator of growth over time. (B) C. albicans strains expressing GFP were stained with Alexa Fluor dye during yeast-form growth. Stained yeast cells were then injected into mice via the tail vein, and after 6 h, the mice were sacrificed and their kidneys were removed and then homogenized and viewed by microscopy. Hyphae expressing GFP indicate new growth that occurred within mouse tissue. (C) Percentages of yeast cells inoculated in vivo that generated hyphae in mouse kidneys. A minimum of 24 cells were analyzed per strain. WT, wild type.
In order to determine if these mutants could also form hyphae in response to more in vivo stimuli, such as host tissue, the strains were compared for growth after they infected mice and were localized to the kidneys. Each strain was transformed with a construct that would express cytoplasmic GFP from the constitutive ENO1 promoter (26). Then yeast-form cells of each strain were stained on the exterior with Alexa Fluor 555 dye and administered to mice via tail vein injection. Kidney homogenates were analyzed by microscopy 6 h after infection to determine if hyphal growth occurred in the kidney. The Alexa Fluor stain allows the identification of injected yeast-form cells, and GFP-expressing hyphae that lack Alexa Fluor staining indicate new growth after infection of the kidney. Analysis of kidney homogenates revealed that all four strains were able to survive and grow similarly within the mouse (Fig. 3B and C). Thus, when grown under host-simulating conditions, the mutants appear to form hyphae at a rate similar to that of the wild type, at least for several hours.
Loss of PS causes increased exposure of β(1-3)-glucan in hyphae.
The cho1Δ/Δ mutant exhibits a greater exposure of β(1-3)-glucan than the wild-type, psd1Δ/Δ psd2Δ/Δ double mutant, and other strains (Fig. 1 and 2; see Fig. S1 and S2 in the supplemental material). In order to determine if this held true for hyphae as well, cells were grown in human serum at 37°C to induce hyphal growth and then stained for β(1-3)-glucan exposure with an anti-β(1-3)-glucan antibody.
When grown as hyphae in human serum and fixed with formaldehyde during the process of immunofluorescence staining, the cho1Δ/Δ and psd1Δ/Δ psd2Δ/Δ mutants both exhibited greater exposure of β(1-3)-glucan than the wild-type, psd1Δ/Δ mutant, and reintegrant strains (Fig. 4A). In addition, a number of the hyphae of the cho1Δ/Δ and psd1Δ/Δ psd2Δ/Δ mutants were also malformed compared to those of the wild-type and psd1Δ/Δ strains; they were enlarged at the tips in comparison and in many cases were somewhat curved near the tip (Fig. 4A). Approximately 48% of the cho1Δ/Δ mutant hyphae and 52% of the psd1Δ/Δ psd2Δ/Δ mutant hyphae showed this phenotype, whereas none of the other strains exhibited this phenotype (Table 2).
TABLE 2.
Hyphae of the cho1Δ/Δ and psd1Δ/Δ psd2Δ/Δ mutants are malformed
| Strain | No. of cells with deformity | Total no. of cells | % with deformity |
|---|---|---|---|
| SC5314 | 0 | 37 | 0.00 |
| cho1Δ/Δ mutant | 11 | 23 | 47.83 |
| cho1::CHO1 mutant | 0 | 13 | 0.00 |
| psd1Δ/Δ mutant | 0 | 29 | 0.00 |
| psd1::PSD1 mutant | 0 | 19 | 0.00 |
| psd1Δ/Δ psd2Δ/Δ double mutant | 11 | 21 | 52.38 |
However, when costaining of hyphae with sDectin-1–Fc and an anti-β(1-3)-glucan antibody was performed without fixation with formaldehyde, only the cho1Δ/Δ mutant exhibited strong staining with either probe, and the staining, like that in yeast-form cho1Δ/Δ mutant cells, was found in overlapping patterns (Fig. 4B). The frequency of colocalization of anti-β(1-3)-glucan antibody and Dectin-1 staining was quantified for all of the strains, revealing that they bind to unfixed hyphal cells similarly for the most part (Fig. 4B; see Fig. S3 in the supplemental material). The psd1Δ/Δ and psd1Δ/Δ psd2Δ/Δ mutants did have stronger staining than the wild type, but it was generally much weaker than that of the cho1Δ/Δ mutant strain. Interestingly, for many of the mutants, staining with sDectin-1–Fc tended to be stronger for the mother yeast cells when they were grown in serum (Fig. 4B).
Surprisingly, when sDectin-1–Fc staining was performed with hyphal-form cells from these strains that had been fixed with formaldehyde, the psd1Δ/Δ psd2Δ/Δ mutant did not exhibit noticeable staining compared to the wild type, although the cho1Δ/Δ mutant continued to stain much more brightly than the wild type (data not shown). Thus, formaldehyde fixation may reveal anti-β(1-3)-glucan-specific epitopes under some conditions with specific probes.
Phospholipid mutants are not more susceptible to killing by innate immune responses.
As our cho1Δ/Δ mutant has increased β(1-3)-glucan exposure and increased binding by sDectin-1–Fc, it was hypothesized that increased binding of Dectin-1 would result in increased susceptibility to killing by macrophages. However, when in vitro killing assays were performed with macrophages (RAW264.7 or RAW-BLUE), it was found that there was no difference in killing for any of the strains (Fig. 5A, data not shown for RAW-BLUE). Neutrophils are typically more efficient at killing C. albicans than macrophages, so the susceptibility of the cho1Δ/Δ and psd1Δ/Δ psd2Δ/Δ mutants to neutrophils was also tested. However, the mutants were not more readily killed by neutrophils either (Fig. 5B).
FIG 5.
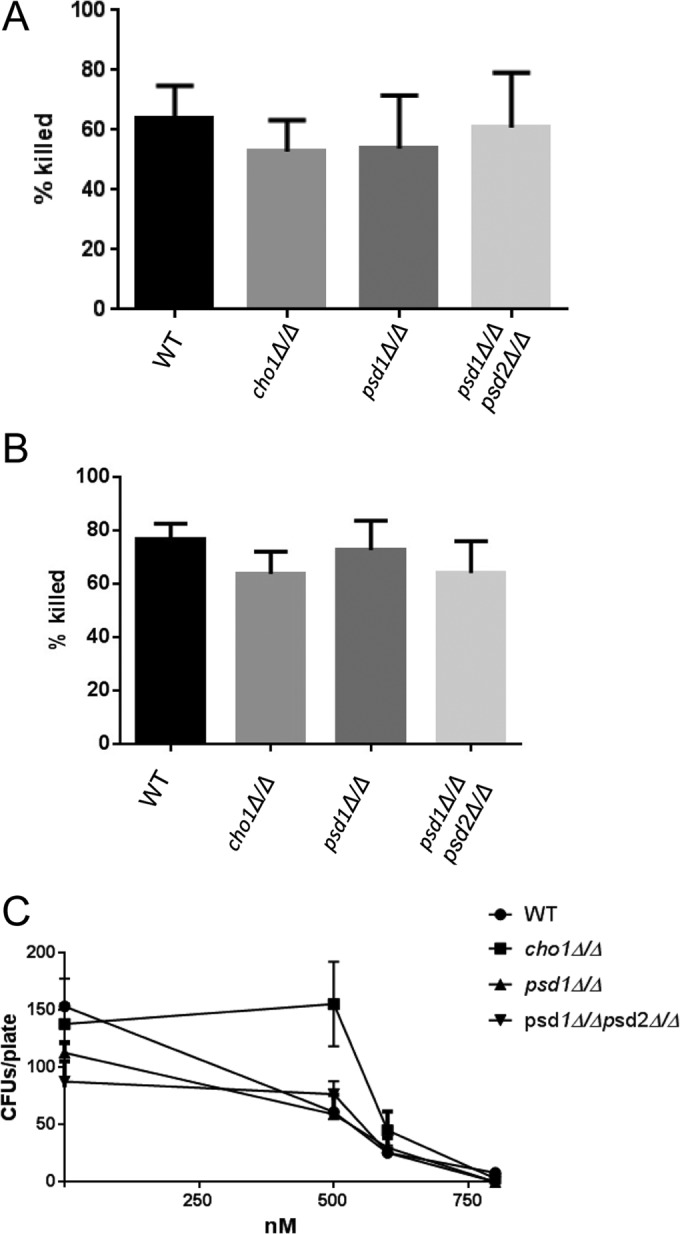
There are no differences in killing by phagocytes or defensins. (A) RAW264.7 macrophages and C. albicans cells were coincubated for 6 h, and C. albicans was plated to measure the number of viable CFU. C. albicans strains mock challenged with macrophages were used to calculate the percentages of C. albicans cells killed. (B) Human neutrophils were tested for the ability to kill the different strains by an approach similar to that used for panel A, except that they were coincubated for only 2 h. (C) The cho1Δ/Δ mutant was incubated with β-defensin or mock challenged, and then the viable CFU were counted. WT, wild type.
In addition to cellular innate immune responses, it is also possible that the mutants might be more susceptible to noncellular innate immune response elements like cationic peptides, as their membranes are defective. We tested the susceptibility of our mutants to the cationic peptide β-defensin, which is known to act against the negatively charged membranes of invading pathogens (34). Surprisingly, the cho1Δ/Δ mutant was more resistant than the wild type to β-defensin at a concentration of 500 nM. The psd1Δ/Δ and psd1Δ/Δ psd2Δ/Δ mutants showed no differences (Fig. 5C) from the wild type. In addition, the mutants showed no difference in binding by complement C (data not shown).
Loss of PS in C. albicans causes increased elicitation of cytokines from macrophages.
Increased exposure of β(1-3)-glucan in C. albicans cell wall mutants has been shown to cause an increase in TNF-α release from macrophages (18). The differences in β(1-3)-glucan exposure reported in the cho1Δ/Δ mutant was hypothesized to cause an increase in TNF-α production. A TNF-α-specific ELISA revealed that the cho1Δ/Δ mutant elicits greater TNF-α production from RAW-BLUE macrophages than the wild-type strain (Fig. 6). This is not the case for the reintegrant, psd1Δ/Δ, or psd1Δ/Δ psd2Δ/Δ mutant strain.
FIG 6.
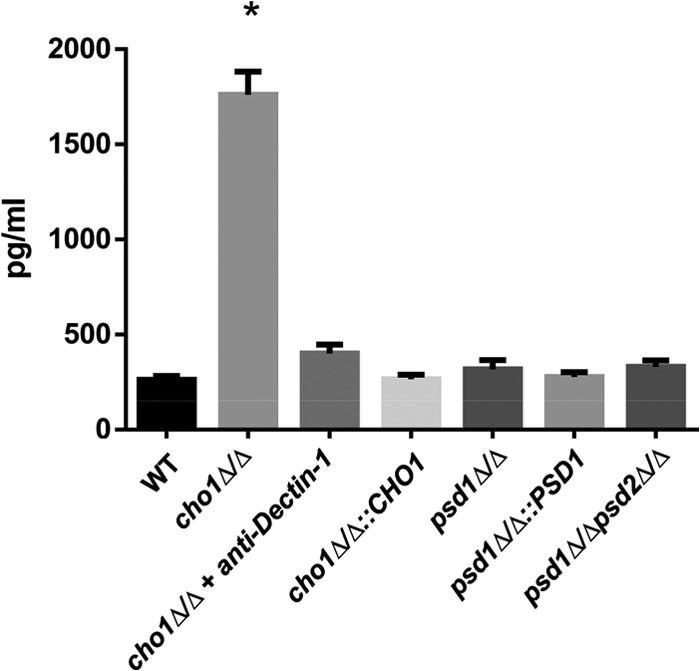
The cho1Δ/Δ mutant elicits TNF-α from macrophages in a Dectin-1-dependent manner. A TNF-α-specific ELISA was used to measure TNF-α elicited from RAW-BLUE macrophages in response to a C. albicans challenge after 4 h. Pretreatment of RAW-BLUE macrophages with an anti-Dectin-1 neutralizing antibody reveals that the TNF-α response is Dectin-1 mediated. *, P < 0.0001. WT, wild type.
The Dectin-1 receptor expressed on immune effector cells binds to β(1-3)-glucan on the C. albicans cell surface, and this has been reported to trigger TNF-α release (32). Thus, Dectin-1 was presumed to be responsible for the increase in TNF-α release. In order to rule out other non-Dectin-1 receptors as mediators of increased TNF-α production by macrophages in response to the cho1Δ/Δ mutant, an anti-Dectin-1 antibody was administered to the macrophages prior to their exposure to cho1Δ/Δ mutant cells in order to block Dectin-1 activation during coincubation with yeast cells. The results in Fig. 6 show that blocking of Dectin-1 reduces TNF-α production to nearly wild-type levels, revealing that Dectin-1 is responsible for cytokine release. When the RAW 264.7 cell line was used, a similar elicitation of TNF-α in a Dectin-1-dependent manner was observed as well (see Fig. S4 in the supplemental material).
DISCUSSION
Loss of the PS synthase gene CHO1 results in changes in immune recognition, most notably, increased exposure of β(1-3)-glucan in the cell wall. The increased exposure of β(1-3)-glucan can be seen from the increased binding of anti-β(1-3)-glucan antibodies (Fig. 1, 2, and 4; see Fig. S1 to S3 in the supplemental material). On the basis of current models, this may be due to glycosylated cell surface proteins on the cho1Δ/Δ mutant cell wall not adequately concealing the layer of β(1-3)-glucan underneath, causing “chinks” in the masking of this important yeast cell wall PAMP. These differences in β(1-3)-glucan exposure in the cho1Δ/Δ mutant are also supported by Dectin-1 receptor binding to both yeast-form (Fig. 2; see Fig. S2 in the supplemental material) and hyphal cells (Fig. 4; see Fig. S3 in the supplemental material) and elicitation of TNF-α (Fig. 6; see Fig. S4 in the supplemental material). These data reveal that the immune response to the cho1Δ/Δ mutant is stronger than that to all of the other strains tested. The more severe defects in immune evasion in the cho1Δ/Δ mutant (lacks PS and has deficient PE) than the psd1Δ/Δ psd2Δ/Δ double mutant, which has deficient PE but abundant PS, reflect the stronger cell wall deficiencies of the cho1Δ/Δ mutant (7).
The correlation between increased β(1-3)-glucan exposure and increased TNF-α production in the cho1Δ/Δ mutant is notable, as Wheeler and Fink (18) found some mutants of Saccharomyces cerevisiae that exhibit increased binding by anti-β(1-3)-glucan antibody but did not necessarily bind better to Dectin-1 or hyperelicit TNF-α. Thus, while increased staining by anti-β(1-3)-glucan antibody does not always correlate with increased immune recognition, the increase that occurs in the cho1Δ/Δ mutant does yield a more productive immune response (Fig. 6; see Fig. S4 in the supplemental material).
These data suggest that the loss of PS alone is responsible for unmasking in C. albicans, and this conclusion is drawn from the following observations. (i) Loss of CHO1 results in a complete loss of PS and a 40% drop in PE levels, since this population of PE molecules is derived from PS by decarboxylation via Psd1p or Psd2p. (ii) Loss of PSD1 and PSD2 simultaneously (psd1Δ/Δ psd2Δ/Δ) causes a 40% drop in PE similar to that observed in the cho1Δ/Δ mutant, but there is an increase in PS, since it is not converted to PE by decarboxylation. (iii) Although the cho1Δ/Δ and psd1Δ/Δ psd2Δ/Δ mutants have similar drops in PE, only the cho1Δ/Δ mutant, with its lack of PS, shows a strong increase in β(1-3)-glucan exposure by staining with anti-β(1-3)-glucan antibody or sDectin-1–Fc (Fig. 1, 2, and 4; see Fig. S1 to S3 in the supplemental material) and increased elicitation of TNF-α from macrophages (Fig. 6; see Fig. S4 in the supplemental material). Thus, it appears that the loss of PS is responsible for the majority of the increased exposure of β(1-3)-glucan.
There is a subtle contrast between the hyphal form and the yeast form in regard to β(1-3)-glucan exposure. In particular, although cho1Δ/Δ mutant hyphae have the strongest exposure of β(1-3)-glucan, the psd1Δ/Δ and psd1Δ/Δ psd2Δ/Δ mutants also exhibit greater exposure of β(1-3)-glucan and binding of sDectin-1–Fc than the wild type, but exposure is lower than that of the cho1Δ/Δ mutant (Fig. 4B; see Fig. S3 in the supplemental material). Thus, for hyphae, the defect in masking is affected primarily by PS but is impacted by PE as well. We did not test to see if this difference in the hyphal form leads to greater TNF-α release in macrophages because of technical difficulties when working with hyphae in this assay.
In addition, when hyphae are fixed with formaldehyde, the psd1Δ/Δ psd2Δ/Δ double mutant, but not the psd1Δ/Δ mutant, exhibits increased exposure of β(1-3)-glucan (Fig. 4A). This does not hold true for yeast-form cells (see Fig. S1 in the supplemental material). Thus, fixation may cause some increases in exposure that do not necessarily reflect other conditions. Alternatively, these data may indicate that β(1-3)-glucan is more readily unmasked in the psd1Δ/Δ psd2Δ/Δ mutant along with the cho1Δ/Δ mutant, than in the psd1Δ/Δ mutant. This could be of interest, as the unmasking that occurs under these conditions correlates with these strains' defects in virulence.
The precise defect that leads to increased exposure of β(1-3)-glucan in yeast-form cells of the PS synthase mutant alone is unclear. This increase does not appear to resemble that of the kre5Δ/Δ mutant, which has increased exposure all over the cell surface (18). Instead, the increased exposure we observe is localized to foci at the periphery of yeast-form cells (see Fig. S2 in the supplemental material). These increases also do not correlate with bud scars or septa (data not shown). Transmission electron microscopy images of cho1Δ/Δ mutant cells reveal irregularities in the membrane and cell wall that look like invaginations (7). The increased exposure of β(1-3)-glucan in the cell wall of the cho1Δ/Δ mutant may correspond to these irregularities, although that remains to be determined.
The mechanism by which PS controls β(1-3)-glucan masking could be related to effects of PS on cell wall signal transduction pathways, general endocytic trafficking, or alterations of the plasma membrane (35, 36). A number of cell wall signaling pathways are directly affected by PS, including the protein kinase C (Pkc1p) and calcineurin pathways (37–41). In S. cerevisiae, PS is a cofactor for ScPkc1p activity in vitro. In addition, calcineurin is recruited to membranes by PS in vitro. Therefore, these pathways could potentially affect the cell wall because of PS loss.
The role that β(1-3)-glucan exposure in the cho1Δ/Δ mutant plays in compromising virulence in the host is not clear. The yeast form of the cho1Δ/Δ mutant elicits a greater release of cytokines from macrophages than the psd1Δ/Δ psd2Δ/Δ mutant and has more exposure of β(1-3)-glucan as yeast cells or hyphae, suggesting a stronger immune response. However, these mutants are pleiotropic, so there are likely other mechanisms at work in addition to unmasking to affect virulence. For example, both mutants are ethanolamine auxotrophs (7). Even in YPD, which contains ethanolamine, the cho1Δ/Δ mutant has a 2.5-fold lower growth rate than the wild type, which suggests that the growth rate could play a role in the cho1Δ/Δ mutant's ability to be pathogenic; however, it can clearly grow for several hours in serum or in the host (Fig. 3), but this may not translate into growth over several days, which is required to overwhelm the host during sepsis. Thus, the mutants may not be getting enough ethanolamine to make sufficient PE, which is essential (7), as it is in other yeasts like S. cerevisiae and Schizosaccharomyces pombe (42). Alternatively, auxotrophy may play a role, but the increased exposure of β(1-3)-glucan may result in more rapid clearance of the mutant.
PS clearly affects the cell wall and masking independently of PE, but PE appears to at least subtly affect the cell wall in hyphae. These defects compromise an important defensive virulence factor of fungi, “masking” of the cell wall PAMP β(1-3)-glucan. This report sets the stage for a better understanding of how PS and PE regulate pathways that ultimately affect virulence traits and cell wall physiology in this pathogen.
Supplementary Material
ACKNOWLEDGMENTS
We thank Gordon Brown for providing the sDectin-1–Fc expression construct. We also thank Greg Christianson in the Roopenian laboratory for technical advice on the purification of sDectin-1–Fc.
This work was funded by NIH grants 5P20RR016463, 8P20GM103423, R15AI094406 (R.T.W.), NSF MBC-0919787, and NIH-1R01AL105690 (T. B. R.). Research reported in this publication was supported by NIDCR award F31DE022512 (S.E.D.).
The content of this report is solely our responsibility and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Published ahead of print 11 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01612-14.
REFERENCES
- 1. Morrell M, Fraser VJ, Kollef MH. 2005. Delaying the empiric treatment of Candida bloodstream infection until positive blood culture results are obtained: a potential risk factor for hospital mortality. Antimicrob. Agents Chemother. 49:3640–3645. 10.1128/AAC.49.9.3640-3645.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ellis ME, Al-Abdely H, Sandridge A, Greer W, Ventura W. 2001. Fungal endocarditis: evidence in the world literature, 1965-1995. Clin. Infect. Dis. 32:50–62. 10.1086/317550 [DOI] [PubMed] [Google Scholar]
- 3. Cassone A, Cauda R. 2012. Candida and candidiasis in HIV-infected patients: where commensalism, opportunistic behavior and frank pathogenicity lose their borders. AIDS 26:1457–1472 [DOI] [PubMed] [Google Scholar]
- 4. Bates DW, Su L, Yu DT, Chertow GM, Seger DL, Gomes DR, Dasbach EJ, Platt R. 2001. Mortality and costs of acute renal failure associated with amphotericin B therapy. Clin. Infect. Dis. 32:686–693. 10.1086/319211 [DOI] [PubMed] [Google Scholar]
- 5. Kontoyiannis DP, Lewis RE. 2002. Antifungal drug resistance of pathogenic fungi. Lancet 359:1135–1144. 10.1016/S0140-6736(02)08162-X [DOI] [PubMed] [Google Scholar]
- 6. Sanglard D. 2002. Clinical relevance of mechanisms of antifungal drug resistance in yeasts. Enferm. Infecc. Microbiol. Clin. 20:462–469. 10.1016/S0213-005X(02)72842-5 [DOI] [PubMed] [Google Scholar]
- 7. Chen YL, Montedonico AE, Kauffman S, Dunlap JR, Menn FM, Reynolds TB. 2010. Phosphatidylserine synthase and phosphatidylserine decarboxylase are essential for cell wall integrity and virulence in Candida albicans. Mol. Microbiol. 75:1112–1132. 10.1111/j.1365-2958.2009.07018.x [DOI] [PubMed] [Google Scholar]
- 8. Braun BR, van Het Hoog M, d'Enfert C, Martchenko M, Dungan J, Kuo A, Inglis DO, Uhl MA, Hogues H, Berriman M, Lorenz M, Levitin A, Oberholzer U, Bachewich C, Harcus D, Marcil A, Dignard D, Iouk T, Zito R, Frangeul L, Tekaia F, Rutherford K, Wang E, Munro CA, Bates S, Gow NA, Hoyer LL, Kohler G, Morschhauser J, Newport G, Znaidi S, Raymond M, Turcotte B, Sherlock G, Costanzo M, Ihmels J, Berman J, Sanglard D, Agabian N, Mitchell AP, Johnson AD, Whiteway M, Nantel A. 2005. A human-curated annotation of the Candida albicans genome. PLoS Genet. 1(1):36–57. 10.1371/journal.pgen.0010001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Romani L. 2004. Immunity to fungal infections. Nat. Rev. Immunol. 4:1–23. 10.1038/nri1290 [DOI] [PubMed] [Google Scholar]
- 10. Ashman RB. 2008. Protective and pathologic immune responses against Candida albicans infection. Front. Biosci. 13:3334–3351. 10.2741/2929 [DOI] [PubMed] [Google Scholar]
- 11. Vonk AG, Wieland CW, Netea MG, Kullberg BJ. 2002. Phagocytosis and intracellular killing of Candida albicans blastoconidia by neutrophils and macrophages: a comparison of different microbiological test systems. J. Microbiol. Methods 49:55–62. 10.1016/S0167-7012(01)00348-7 [DOI] [PubMed] [Google Scholar]
- 12. Herre J, Marshall AS, Caron E, Edwards AD, Williams DL, Schweighoffer E, Tybulewicz V, Reis e Sousa C, Gordon S, Brown GD. 2004. Dectin-1 uses novel mechanisms for yeast phagocytosis in macrophages. Blood 104:4038–4045. 10.1182/blood-2004-03-1140 [DOI] [PubMed] [Google Scholar]
- 13. Doyle SE, O'Connell RM, Miranda GA, Vaidya SA, Chow EK, Liu PT, Suzuki S, Suzuki N, Modlin RL, Yeh WC, Lane TF, Cheng G. 2004. Toll-like receptors induce a phagocytic gene program through p38. J. Exp. Med. 199:81–90. 10.1084/jem.20031237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Blander JM, Medzhitov R. 2004. Regulation of phagosome maturation by signals from Toll-like receptors. Science 304:1014–1018. 10.1126/science.1096158 [DOI] [PubMed] [Google Scholar]
- 15. Jouault T, Sarazin A, Martinez-Esparza M, Fradin C, Sendid B, Poulain D. 2009. Host responses to a versatile commensal: PAMPs and PRRs interplay leading to tolerance or infection by Candida albicans. Cell. Microbiol. 11:1007–1015. 10.1111/j.1462-5822.2009.01318.x [DOI] [PubMed] [Google Scholar]
- 16. Gow NA, van de Veerdonk FL, Brown AJ, Netea MG. 2012. Candida albicans morphogenesis and host defence: discriminating invasion from colonization. Nat. Rev. Microbiol. 10:112–122. 10.1038/nrmicro2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ruiz-Herrera J, Elorza MV, Valentin E, Sentandreu R. 2006. Molecular organization of the cell wall of Candida albicans and its relation to pathogenicity. FEMS Yeast Res. 6:14–29. 10.1111/j.1567-1364.2005.00017.x [DOI] [PubMed] [Google Scholar]
- 18. Wheeler RT, Fink GR. 2006. A drug-sensitive genetic network masks fungi from the immune system. PLoS Pathog. 2(4):e35. 10.1371/journal.ppat.0020035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Klippel N, Cui S, Groebe L, Bilitewski U. 2010. Deletion of the Candida albicans histidine kinase gene CHK1 improves recognition by phagocytes through an increased exposure of cell wall beta-1,3-glucans. Microbiology 156:3432–3444. 10.1099/mic.0.040006-0 [DOI] [PubMed] [Google Scholar]
- 20. Sanglard D, Ischer F, Marchetti O, Entenza J, Bille J. 2003. Calcineurin A of Candida albicans: involvement in antifungal tolerance, cell morphogenesis and virulence. Mol. Microbiol. 48:959–976. 10.1046/j.1365-2958.2003.03495.x [DOI] [PubMed] [Google Scholar]
- 21. Monge RA, Roman E, Nombela C, Pla J. 2006. The MAP kinase signal transduction network in Candida albicans. Microbiology 152:905–912. 10.1099/mic.0.28616-0 [DOI] [PubMed] [Google Scholar]
- 22. Marino MW, Dunn A, Grail D, Inglese M, Noguchi Y, Richards E, Jungbluth A, Wada H, Moore M, Williamson B, Basu S, Old LJ. 1997. Characterization of tumor necrosis factor-deficient mice. Proc. Natl. Acad. Sci. U. S. A. 94:8093–8098. 10.1073/pnas.94.15.8093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Louie A, Baltch AL, Smith RP, Franke MA, Ritz WJ, Singh JK, Gordon MA. 1994. Tumor necrosis factor alpha has a protective role in a murine model of systemic candidiasis. Infect. Immun. 62:2761–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Styles C. 2002. How to set up a yeast laboratory. Methods Enzymol. 350:42–71. 10.1016/S0076-6879(02)50955-1 [DOI] [PubMed] [Google Scholar]
- 25. Raschke WC, Baird S, Ralph P, Nakoinz I. 1978. Functional macrophage cell lines transformed by Abelson leukemia virus. Cell 15:261–267. 10.1016/0092-8674(78)90101-0 [DOI] [PubMed] [Google Scholar]
- 26. Wheeler RT, Kombe D, Agarwala SD, Fink GR. 2008. Dynamic, morphotype-specific Candida albicans beta-glucan exposure during infection and drug treatment. PLoS Pathog. 4(12):e1000227. 10.1371/journal.ppat.1000227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. De Backer MD, Maes D, Vandoninck S, Logghe M, Contreras R, Luyten WH. 1999. Transformation of Candida albicans by electroporation. Yeast 15:1609–1618. [DOI] [PubMed] [Google Scholar]
- 28. Graham LM, Tsoni SV, Willment JA, Williams DL, Taylor PR, Gordon S, Dennehy K, Brown GD. 2006. Soluble dectin-1 as a tool to detect beta-glucans. J. Immunol. Methods 314:164–169. 10.1016/j.jim.2006.05.013 [DOI] [PubMed] [Google Scholar]
- 29. Peltz G, Zaas AK, Zheng M, Solis NV, Zhang MX, Liu HH, Hu Y, Boxx GM, Phan QT, Dill D, Filler SG. 2011. Next-generation computational genetic analysis: multiple complement alleles control survival after Candida albicans infection. Infect. Immun. 79:4472–4479. 10.1128/IAI.05666-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oh H, Siano B, Diamond S. 2008. Neutrophil isolation protocol. J. Vis Exp. 17:745. 10.3791/745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Edgerton M, Koshlukova SE, Araujo MW, Patel RC, Dong J, Bruenn JA. 2000. Salivary histatin 5 and human neutrophil defensin 1 kill Candida albicans via shared pathways. Antimicrob. Agents Chemother. 44:3310–3316. 10.1128/AAC.44.12.3310-3316.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Taylor PR, Tsoni SV, Willment JA, Dennehy KM, Rosas M, Findon H, Haynes K, Steele C, Botto M, Gordon S, Brown GD. 2007. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat. Immunol. 8:31–38. 10.1038/ni1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Drummond RA, Brown GD. 2011. The role of dectin-1 in the host defence against fungal infections. Curr. Opin. Microbiol. 14:392–399. 10.1016/j.mib.2011.07.001 [DOI] [PubMed] [Google Scholar]
- 34. White SH, Wimley WC, Selsted ME. 1995. Structure, function, and membrane integration of defensins. Curr. Opin. Struct. Biol. 5:521–527. 10.1016/0959-440X(95)80038-7 [DOI] [PubMed] [Google Scholar]
- 35. Dagley MJ, Gentle IE, Beilharz TH, Pettolino FA, Djordjevic JT, Lo TL, Uwamahoro N, Rupasinghe T, Tull DL, McConville M, Beaurepaire C, Nantel A, Lithgow T, Mitchell AP, Traven A. 2011. Cell wall integrity is linked to mitochondria and phospholipid homeostasis in Candida albicans through the activity of the post-transcriptional regulator Ccr4-Pop2. Mol. Microbiol. 79:968–989. 10.1111/j.1365-2958.2010.07503.x [DOI] [PubMed] [Google Scholar]
- 36. Qu Y, Jelicic B, Pettolino F, Perry A, Lo TL, Hewitt VL, Bantun F, Beilharz TH, Peleg AY, Lithgow T, Djordjevic JT, Traven A. 2012. Mitochondrial sorting and assembly machinery subunit Sam37 in Candida albicans: insight into the roles of mitochondria in fitness, cell wall integrity, and virulence. Eukaryot. Cell 11:532–544. 10.1128/EC.05292-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Politino M, King MM. 1987. Calcium- and calmodulin-sensitive interactions of calcineurin with phospholipids. J. Biol. Chem. 262:10109–10113 [PubMed] [Google Scholar]
- 38. Orr JW, Newton AC. 1992. Interaction of protein kinase C with phosphatidylserine. 2. Specificity and regulation. Biochemistry 31:4667–4673 [DOI] [PubMed] [Google Scholar]
- 39. Martin BA, Oxhorn BC, Rossow CR, Perrino BA. 2001. A cluster of basic amino acid residues in calcineurin b participates in the binding of calcineurin to phosphatidylserine vesicles. J. Biochem. 129:843–849. 10.1093/oxfordjournals.jbchem.a002928 [DOI] [PubMed] [Google Scholar]
- 40. Kennedy MT, Brockman H, Rusnak F. 1997. Determinants of calcineurin binding to model membranes. Biochemistry 36:13579–13585. 10.1021/bi9716027 [DOI] [PubMed] [Google Scholar]
- 41. Kamada Y, Qadota H, Python CP, Anraku Y, Ohya Y, Levin DE. 1996. Activation of yeast protein kinase C by Rho1 GTPase. J. Biol. Chem. 271:9193–9196. 10.1074/jbc.271.16.9193 [DOI] [PubMed] [Google Scholar]
- 42. Matsuo Y, Fisher E, Patton-Vogt J, Marcus S. 2007. Functional characterization of the fission yeast phosphatidylserine synthase gene, pps1, reveals novel cellular functions for phosphatidylserine. Eukaryot. Cell 6:2092–2101. 10.1128/EC.00300-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gillum AM, Tsay EY, Kirsch DR. 1984. Isolation of the Candida albicans gene for orotidine-5′-phosphate decarboxylase by complementation of S. cerevisiae ura3 and E. coli pyrF mutations. Mol. Gen. Genet. 198:179–182. 10.1007/BF00328721 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.