

Abstract
In the past decade, Clostridium difficile has emerged as an important gut pathogen. Symptoms of C. difficile infection range from mild diarrhea to pseudomembranous colitis. Besides the two main virulence factors toxin A and toxin B, other virulence factors are likely to play a role in the pathogenesis of the disease. In other Gram-positive and Gram-negative pathogenic bacteria, conserved high-temperature requirement A (HtrA)-like proteases have been shown to have a role in protein homeostasis and quality control. This affects the functionality of virulence factors and the resistance of bacteria to (host-induced) environmental stresses. We found that the C. difficile 630 genome encodes a single HtrA-like protease (CD3284; HtrA) and have analyzed its role in vivo and in vitro through the creation of an isogenic ClosTron-based htrA mutant of C. difficile strain 630Δerm (wild type). In contrast to the attenuated phenotype seen with htrA deletion in other pathogens, this mutant showed enhanced virulence in the Golden Syrian hamster model of acute C. difficile infection. Microarray data analysis showed a pleiotropic effect of htrA on the transcriptome of C. difficile, including upregulation of the toxin A gene. In addition, the htrA mutant showed reduced spore formation and adherence to colonic cells. Together, our data show that htrA can modulate virulence in C. difficile.
INTRODUCTION
The bacterial pathogen Clostridium difficile is the leading cause of infectious nosocomial diarrhea (1–3). Clostridium difficile infection (CDI) can cause disease with a wide variety of symptoms, ranging from mild diarrhea to severe pseudomembranous colitis (1, 4, 5). Since 2003, the global prevalence of reported CDI cases has escalated (5–8). In addition, the severity of the disease and the mortality have increased (1, 4, 9).
The main virulence factors of C. difficile are the two large clostridial toxins, toxin A (TcdA) and toxin B (TcdB) (10–12). These toxins are glycosyltransferases that inactivate Rho, Rac, and Cdc42, thereby disrupting the cytoskeleton and tight junctions of the colon epithelial cells, resulting in activation of the inflammasome and cellular apoptosis (10, 12). A third toxin, binary toxin, is produced by certain strains (e.g., PCR ribotypes 027 and 078) that have been associated with increased levels of mortality and morbidity (1, 9). It has been suggested that binary toxin may contribute to disease in hamsters (13). In vitro assays have demonstrated that the binary toxin affects adhesion of C. difficile to cells through protrusion formation by the target cells (14).
Besides the three toxins, little is known about other virulence factors and their role in colonization and establishment of an infection in the host. Presently, several surface proteins have been identified or hypothesized to play a role in colonic adhesion. These include the fibronectin-binding protein A (15), S-layer proteins (16), Cwp84 (17), flagellar proteins (18), and CD1581 (19).
Alongside colonization factors, adaptations to stresses in the host (including the antibacterial response, elevated temperatures, extreme pH, and osmotic stress) are likely to play a vital role in the establishment of an infection. These stresses can result in the accumulation of (partially) unfolded proteins that are nonfunctional or form poisonous aggregates (20). The well-conserved family of bacterial high-temperature requirement A (HtrA) proteases plays an important role in the protein quality control and homoeostasis by combining proteolytic and chaperone activities in a variety of microorganisms (21–23).
HtrA-like proteases are generally composed of three structurally distinct domains: a trypsin-like serine protease domain, one or two PDZ domains, and a transmembrane domain or a signal peptide (22–24). PDZ domains are highly flexible domains and are involved in oligomerization, substrate recognition, and/or the regulation of protease activity (21, 24). Membrane-anchored HtrA-like proteases are active as trimers, and soluble HtrA-like proteases form larger active oligomers (21, 22).
In this study, we describe the identification and characterization of a C. difficile HtrA-like protease (CD3284; here HtrA). We show that an htrA mutant has enhanced virulence in the Golden Syrian hamster model of acute C. difficile infection. These data contrast with those reported for other pathogens, in which mutation of htrA results in attenuation (25–27). Furthermore, the htrA mutation displayed a pleiotropic effect in a transcriptome analysis. Several differentially expressed genes were validated and are discussed in the context of C. difficile virulence.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The C. difficile and Escherichia coli strains and plasmids used in this study are listed in Table 1. E. coli strains were grown in Luria-Bertani medium (LB) (USB Corporation) supplemented with appropriate antibiotics when required. C. difficile strains were grown anaerobically in a microaerobic cabinet (Don Whitley DG 250) at 37°C in prereduced TTY medium (30 g/liter Bacto tryptone [BD], 20 g/liter yeast extract [Difco], and 0.1% thioglycolate, pH 7.4) or brain heart infusion broth (Oxoid) supplemented with 5 g/liter yeast extract and 0.01% l-cysteine (Sigma) (BHIS) (28). Logarithmic-growth-phase bacteria (optical density at 600 nm [OD600], 0.4 ± 0.08) from overnight precultures were used to inoculate prereduced TTY broth at a starting OD600 of 0.05 (±0.01). Optical density readings were taken hourly until the stationary growth phase (8 h postinoculation) and at 24 and 48 h postinoculation. Cultures of C. difficile strains harboring an expression plasmid were induced with anhydrotetracycline (100 ng/ml) at 1 h postinoculation. We routinely monitored the purity of cultures by performing control PCRs to confirm the identity of mutant strains and stability of conjugated expression plasmids.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Description | Reference or origin |
|---|---|---|
| Escherichia coli | ||
| DH5α | Laboratory stock | |
| CA434 | 34 | |
| C43 | 30 | |
| Clostridium difficile | ||
| WKS1241 | 630Δerm | 29 |
| DB0051 | 630Δerm, pRPF185 | This study |
| DB0002 | 630Δerm, cd3284::[ClosTron, ermB] | This study |
| DB0052 | 630Δerm, cd3284::[ClosTron, ermB] pRPF185 (catP) | This study |
| DB0047 | 630Δerm, cd3284::[ClosTron, ermB], pDB0031 (catP) | This study |
| WKS1237 | 630Δerm, spo0A::[ClosTron, ermB] | 32 |
| WKS1710 | 630Δerm, tcdA::[lsqb]ClosTron, ermB] tcdB::[ClosTron, catP] | 40 |
| Plasmids | ||
| pMTL007 | catP, ClosTron group II intron | 32 |
| pDB0001 | pMTL007::cd3284-162s, catP | This study |
| pRPF185 | tetR, Ptet-gusA, catP | 35 |
| pDB0031 | tetR, Ptet-cd3284, catP | This study |
| pVW001 | PT7-His10-Δ(1-30)-htrA, bla | This study |
| pJV001 | PT7-His10-Δ(1-30)-htrA-S217A, bla | This study |
Protein purification of His10-Δ(1-30)-HtrA and His10-Δ(1-30)-HtrA-S217A.
To facilitate purification from Escherichia coli, CD3284 was cloned without the N-terminal 30 amino acids (aa) into pET-16B, resulting in a fusion protein with an N-terminal 10×His tag but lacking the predicted transmembrane helix. To generate this clone, the CD3284 open reading frame was amplified using primers CD3284F2 and CD3284R2, Pfu polymerase (Fermentas), and chromosomal DNA from strain 630Δerm (29) as a template. The resulting product was digested with NdeI and XhoI and ligated into similarly digested pET-16B (Novagen), yielding pVW001. After sequence verification using conventional Sanger sequencing, the plasmid was transformed into E. coli C43(DE3), in which expression of toxic proteins/proteases is feasible (30). To induce expression of His10-Δ(1-30)-HtrA, an overnight culture grown in LB with 100 μg/ml ampicillin was diluted 1/100 in 500 ml fresh medium to an optical density at 600 nm of 0.6, after which 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) was added and left for 3 h. Thereafter, cells were harvested by centrifugation (10 min, 10,000 rpm, 4°C), and the protein was purified essentially as described for Spo0A (31), with the following modifications: an 0.5-ml Talon Superflow column was used, and the stepwise washing/elution was carried out with 8, 4, 4, 4, and 4 column volumes (20 mM, 50 mM, 100 mM, 250 mM, and 500 mM, respectively). After the addition of glycerol to a final concentration of 8%, protein was quantified by measuring the absorption at 280 nm on a NanoDrop ND-1000 machine (Thermo Scientific), using an extinction coefficient (ε) of 14,440 M−1 cm−1 and a molecular mass of 36.77 kDa, based on analysis of the protein sequence using the ProtParam tool (http://web.expasy.org/protparam/). Protein not used immediately was stored at −80°C.
To generate a catalytic mutant of HtrA, we mutated the conserved Ser217 to an alanine on the basis of published mutants of the E. coli DegS protease. To do so, oligonucleotide-directed mutagenesis was performed using primers CD3284S217AF and CD3284S217AR on plasmid pVW001, yielding plasmid pJV001. The mutant protein [His10-Δ(1-30)-HtrA-S217A] was purified and quantified as described for His10-Δ(1-30)-HtrA.
Protease assay.
Protease activity of the purified proteins was assayed as follows. In a 100-μl reaction mixture, 2.5 μM His10-Δ(1-30)-HtrA or His10-Δ(1-30)-HtrA-S217A was incubated with 20 μM casein or beta-casein (molecular mass, ∼24 kDa) in 1× HNE buffer (150 mM NaCl, 0.1 mM EDTA, 5 mM HEPES, pH 7.4), with the pH adjusted to pH 5.5 by the addition of 0.1× MES/A buffer (0.01 M MES [morpholineethanesulfonic acid], 0.01 M acetic acid, pH 5.0). Reaction mixtures were incubated on ice (0°C) or at 37°C for 18 h with a no-protein control included. After incubation, the reaction was terminated by the addition of 25 μl 5× SDS sample buffer (10% SDS, 10% beta-mercaptoethanol, 50% glycerol, 0.1% bromophenol blue, 250 mM Tris-HCl [pH 6.8], 50 mM EDTA) and heating to 96°C for 2 min. Twenty-microliter volumes of the samples were loaded on a 12% SDS-polyacrylamide gel and separated by electrophoresis at 80 mA. Gels were stained with Coomassie blue solution (0.1% Coomassie brilliant blue R-250, 20% [vol/vol] methanol, 10% [vol/vol] acetic acid) and destained with 50% methanol–10% (vol/vol) acetic acid.
Generation of an htrA (cd3284) mutant strain.
An htrA mutant was generated by insertional inactivation of the cd3284 gene in 630Δerm (here referred to as wild type) using ClosTron technology (32, 33). Briefly, the Perutka algorithm on the ClosTron website (http://www.clostron.com) was used to design primers (see Table S1 in the supplemental material) for retargeting the group II intron (Targetron; Sigma). The retargeted intron was cloned using the restriction enzymes BsrGI and HindIII into plasmid pMTL007, and the constructs were verified by sequencing (33). The sequenced verified plasmid (pDB001) was transformed into E. coli CA434 and transferred via conjugation to C. difficile (29, 34). The selection of C. difficile transconjugants was achieved by subculturing on prereduced BHIS agar supplemented with thiamphenicol (Sigma; 10 μg/ml) and C. difficile selective supplement (Oxoid). After this, several rounds of subculturing on prereduced BHIS agar supplemented with lincomycin (Sigma; 20 μg/ml) and C. difficile selective supplement were used to promote integration of the group II intron into the gene of interest. Chromosomal DNA isolated from the transconjugants (QIAamp blood kit; Qiagen) was analyzed by conventional PCR and sequencing of the PCR product to confirm the insertional inactivation of the cd3284 gene. Primers used for cloning and sequencing are listed in Table S1 in the supplemental material. In addition, Southern blot analysis with an ermB probe (see Fig. S1 in the supplemental material) was performed to verify a specific single integration into the genome as described previously (28).
Generation of strains carrying a plasmid-based inducible htrA gene.
Chromosomal DNA from the wild-type strain (WKS1241) was used to PCR amplify the open reading frame of cd3284 with primers oDB0067 and oDB0068. The resulting amplicon was digested with BamHI and SacI and cloned into pRPF185, thereby replacing the gusA gene (35). The sequence of the resulting inducible plasmid (pDB0031) was verified with primers NF_794, NF_1323 (35), oDB0067, and oDB0068. Plasmid pDB0031 was transferred into E. coli CA434 and conjugated into the htrA mutant, resulting in an htrA mutant that can be complemented by inducible expression of a plasmid-located copy of htrA. To address potential effects of the conjugated plasmid (pDB0031) in the phenotypic assays, we generated control strains by conjugating the pRPF185 plasmid into wild-type (DB0051) and htrA mutant (DB0052) strains (35). These strains served as controls in the in vitro assays. There was no significant difference between strains with and without pRPF185 (data not shown).
Animal experiments.
All procedures were conducted in strict accordance with the Animals (Scientific Procedures) Act of 1986 approved by the Home Office, United Kingdom (project license number PPL60/4218). Female Golden Syrian hamsters that weighed approximately 100 g (bred in-house) were housed individually and given water and food ad libitum. Telemetry chips (VitalView Emitter) were inserted by laparotomy into the body cavities of the animals at least 3 weeks before infection with C. difficile. Once the wounds healed, the animals were placed on receiver pads, and the body temperature and activity were monitored (Vital View software). Each animal received orogastrically 30 mg/kg of clindamycin phosphate in a single dose 5 days before infection. Six animals per group were inoculated by oral gavage with 104 spores of C. difficile. Animals were carefully monitored and culled when the core body temperature dropped below 35°C (28).
Statistical analyses were performed using GraphPad Prism 5.03 (GraphPad Prism Software). A Mantel-Cox log rank statistical test was used to determine significant differences in hamster survival times between C. difficile strains. P values of ≤0.05 were considered significant.
MIC determination.
The MICs of the C. difficile wild type and htrA mutant to erythromycin and clindamycin were determined by the doubling-dilution method described previously (85). Briefly, rows of preconditioned BHI broth (90 μl) were supplemented with a concentration range of 1,024 to 0.06 μg/ml of either antibiotic. Wells were inoculated with ∼5 × 103 spores/100 μl and incubated at 37°C for 48 h anaerobically. Wells to allow positive and negative evaluation of growth were included, in which either no antibiotic or no spores were added. After incubation, plates were visually inspected and compared to the controls; the MIC was determined as the lowest concentration of antibiotic at which growth was no longer visible. Results are given as the median MIC from three biological repeats.
RNA extraction.
Five milliliters of the C. difficile cultures was diluted with ice-cold methanol (1:1) and stored overnight at −80°C. Bacterial pellets, obtained by centrifugation (20 min, 3,000 × g, 4°C), were resuspended in 200 μl lysis buffer (100 mM EDTA, 200 mM Tris-HCl [pH 7.0], 50 mg/ml lysozyme) and incubated for 1 h at 37°C. RNA was isolated using Tri-pure reagent (Roche) as previously described (28). The RNA was treated twice with Turbo DNase (Ambion) according to the manufacturer's instructions, followed by another Tri-pure RNA isolation. The Agilent Bioanalyzer (Agilent Technologies Netherlands BV) and an ND-1000 spectrophotometer (NanoDrop Technologies) were used to analyze the quality and purity of the extracted RNA.
DNA microarray analysis.
Random nonamers were used to convert total RNA into amino allyl-modified cDNA using a Superscript III reverse transcriptase kit (Life Technologies), and purification was with the NucleoSpin Gel and PCR cleanup kit (Macherey-Nagel). The synthesized cDNA was labeled with DyLight 550 and DyLight 650 Amine/Reactive Dyes (Pierce Biotech USA) and purified again with the NucleoSpin Gel and PCR cleanup kit. The Agilent DNA microarrays (G2509F custom microarray GE 8x15K 60mer) were designed based on the available genome sequence of C. difficile 630 (36, 37). The custom-made microarray slides consist of 8 subarrays each with 15,208 60-mer probes (37).
The Agilent microarray slides were competitively hybridized with 300 ng labeled cDNA made from RNAs isolated from both the wild type and htrA mutant (DB0002) during logarithmic-phase (OD600, 0.5 ± 0.1) and stationary-phase (12 h postinoculation) growth. The microarray slides were scanned with a GenePix scanner, and the fluorescence intensities were quantified using GenePix 6.1. The raw data files were analyzed with a LimmaR package via the Genome2D pipeline (http://genome2d.molgenrug.nl) (38) using the following settings: (i) the microarray data were normalized using LOESS normalization, followed by quantile normalization between the slides, and (ii) a weight factor was determined based on the quality of each DNA microarray slide. For each time point (logarithmic or stationary phase), 3 biological replicates were used.
RT-qPCR.
For reverse transcriptase quantitative PCR (RT-qPCR), the RevertAid H Minus reverse transcriptase kit (Fermentas) was used to synthesize cDNA according to the manufacturer's instruction. Random hexamers were used to convert 750 ng RNA into cDNA. The synthesized cDNA was treated with RNase (Qiagen) for 1 h at 37°C and stored at −20°C. The software program Molecular Beacon (Premier Biosoft) was used to design primer pairs (see Table S1 in the supplemental material) for the quantitative PCRs (qPCRs), based on the available genome sequence of C. difficile strain 630 (36).
All primer pairs were first tested by conventional PCR to confirm specificity and amplicon sizes. The RT-qPCRs were then performed using a CFX96 real-time PCR detection system (Bio-Rad). The amplification efficiencies of the genes were determined using serially diluted genomic DNA. Expression levels of the tcdA and spo0A genes were normalized using the amplification efficiencies and the expression level of the reference gene rpsJ (39). The qPCR for tcdA and rpsJ was performed as previously described (28). The qPCR mixture for the spo0A gene contained 25 μl HotStar master mix (Qiagen), forward (oDB0117) and reverse primers (oDB0118) (80 nM each primer), 2.5 mM MgCl2, 0.06% SYBR green (Sigma), and 2 μl synthesized cDNA. The qPCR protocol included an enzyme activation step for 15 min at 95°C, followed by 50 cycles of amplification at 95°C for 30 s, 52°C for 30 s, and 72°C for 30 s.
To determine the specificity of the fluorescence, the fluorescence data were converted into melt curve peaks. For each biological replicate (n = 3), four technical replicates were tested. An independent Student t test was employed to compare the strains at different time points.
Western blot analysis of TcdA and Spo0A.
Quantitative Western blotting for TcdA and Spo0A were performed as follows. For TcdA, filtered (0.45-μm filter) bacterial supernatants from 48-h cultures were analyzed by SDS-PAGE (6% polyacrylamide) and transferred onto nitrocellulose membranes. For Spo0A protein, bacterial pellets from cultures in the logarithmic phase (OD600, 0.5 ± 0.1) and at 8, 12 and 24 h postinoculation were collected by centrifugation (10 min, 11,000 × g, 4°C). Pellets were resuspended in phosphate-buffered saline (PBS) containing protease inhibitor cocktail (Sigma) and lysed with a TissueLyser LT bead beater (50 Hz, 10 min) (Qiagen) using acid-wash glass beads (≤106 μm; Sigma). Equal amounts (OD600 corrected) of the lysed bacterial pellets were separated on 15% SDS-polyacrylamide gels and transferred onto nitrocellulose membrane.
For TcdA quantification, the membranes were probed with a mouse monoclonal anti-TcdA antibody (TCC8; tgcBiomics, Bingen, Germany), and for Spo0A quantification, the membranes were probed with a mouse polyclonal Spo0A serum (31). The probed membranes were analyzed by using a secondary anti-mouse biotin-labeled antibody (Dako) and a tertiary antibiotin antibody labeled with Cy3. A Typhoon 9410 scanner (GE Healthcare) was used to measure the fluorescence intensity. Clostridium difficile strains WKS1237 (spo0A mutant) and WKS1710 (tcdA tcdB mutant) were used as control strains for SpoA and TcdA production, respectively (32, 40). The software package ImageQuant TL (Amersham Biosciences) was used to quantify fluorescence intensity.
Relative quantification of toxin expression.
Quantification of the total toxin production was performed on filtered (0.45-μm filter) 24- and 48-h-postinoculation supernatants, using a Vero cell-based cytotoxicity assay (28). The endpoint titer was defined as the first dilution at which the Vero cell morphology was indistinguishable from that for the neutralized 200-fold diluted supernatant and untreated cells (28). Clostridium difficile strain WKS1710 (tcdA tcdB mutant) was used as a negative control (40). Each experiment was performed in duplicate on three separate occasions. An independent Student t test was employed to compare the strains at different time points.
Relative quantification of sporulation efficiency.
Sporulation efficiency was determined by analyzing culture samples at 24 and 48 h postinoculation. Serially diluted non-heat-shocked and heat-shocked (20 min, 65°C) culture samples were plated onto BHI plates supplemented with 0.1% taurocholate to stimulate germination and enhance spore recovery (41). Plates were incubated for 48 h before CFU were enumerated. The C. difficile strain WKS1237 was included as a negative control for sporulation (32). Sporulation efficiency was normalized using the CFU count of the nontreated sample. Three biological replicates were analyzed in duplicate. An independent Student t test was employed to compare the strains at different time points.
Relative quantification of adhesion to Caco-2 cells.
Adhesion of C. difficile to Caco-2 cells was performed essentially as described previously with minor modifications (42). Briefly, Caco-2 cells were grown in RPMI (GE Healthcare), 10% fetal calf serum, penicillin (100 μg/ml), and streptomycin (100 U/ml) in a humidified 5% CO2 atmosphere at 37°C. Caco-2 cells were seeded into a 24-well plate at a density of 1 × 105 cells per well and incubated overnight at 37°C with 5% CO2. Confluent Caco-2 monolayers were washed once with PBS before transfer into the anaerobic cabinet.
Mid-logarithmic-phase bacterial cultures (OD600, 0.5 ± 0.08) were collected by centrifugation (1 min, 4,000 × g, 4°C) and washed once with prereduced PBS. To each well of Caco-2 cells, approximately 1 × 108 bacteria were added. Exact numbers of viable bacteria within the inoculums for each well were calculated by serial dilution and plating of cultures on BHIS plates. After 3 h of incubation under anaerobic conditions, the Caco-2 cell monolayers with the adhered bacteria were washed 5 times with prereduced PBS. Adhered bacteria were released using100 μl 1× trypsin solution (PAA Laboratories, Coelbe, Germany), serially diluted and plated on prereduced BHIS plates. After 48 h, CFU were enumerated to determine the percentage of adherent C. difficile relative to the initial inoculum. Each experiment was carried out in duplicate and repeated three times. An independent sample t test was employed to compare the strains at different time points.
Microarray data accession number.
The complete microarray data set, including experimental raw data and analyzed data, has been submitted to the GEO database (accession number GSE55926).
RESULTS
Bioinformatics analysis of CD3284.
In this study, we identified a single HtrA-like protease in C. difficile 630Δerm. CD3284 was identified using the BLASTP (http://blast.ncbi.nlm.nih.gov/Blast.cgi), Smart (http://smart.embl-heidelberg.de/), and Merops (http://merops.sanger.ac.uk/) databases (36, 43–45). Further bioinformatics analyses were performed with a conserved-domain prediction web server (http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml), the transmembrane prediction webserver TMHMM2.0 (http://www.cbs.dtu.dk/services/TMHMM/) and the signal peptide cleavage site web server SignalP4.1 (http://www.cbs.dtu.dk/services/SignalP/). These analyses revealed the presence of a transmembrane domain (amino acids [aa] 7 to 29), a trypsin-like serine protease domain (aa 92 to 231; E value, 3.96 × 10−29), and a PDZ domain (aa 269 to 357; E value, 2.17 × 10−19) (Fig. 1A). The N terminus of CD3284 was predicted by TMHMM to be highly hydrophobic, most likely transmembrane, whereas SignalP analysis revealed no signal sequence. This suggests that the protease and PDZ domains are located extracellularly but membrane associated (46). Comparison of the CD3284 protein sequence of our wild-type strain with the NCBI nonredundant protein sequence database using BLASTP resulted in identical protein sequence hits (query coverage, 100%; E value, 0.0) in other epidemic (e.g., PCR ribotype 027) and nonepidemic (e.g., PCR ribotype 001) C. difficile strains.
FIG 1.

HtrA is a protease. (A) Schematic representation of the predicted structural domains of HtrA. The box on the top line indicates the position (nucleotide 163) at which the group II intron has inserted to create the htrA mutant (not to scale). The predicted N-terminal transmembrane helix is shown in blue, the serine protease domain in red, and the PDZ domain in yellow. The inset shows the conserved catalytic triad (in green) of the serine protease domain. The predicted structure of C. difficile HtrA is overlaid with the structure of E. coli DegS (1TE0A) in gray. (B) HtrA shows protease activity toward β-casein. A 2.5 μM concentration of the indicated protein was incubated for 18 h with 20 μM beta-casein at 0°C or 37°C. wt, His10-Δ(1–30)-HtrA; S217A, catalytic mutant His10-Δ(1–30)-HtrA-S217A. The asterisk indicates a proteolytic product of β-casein.
As protein three-dimensional (3D) structure is more conserved in evolution than its equivalent amino acid sequence, we validated the homology of CD3284 using the I-TASSER (http://zhanglab.ccmb.med.umich.edu/I-TASSER/) and Phyre2 three-dimensional structure prediction server (http://www.sbg.bio.ic.ac.uk/phyre2/) (47, 48). These analyses revealed with 100% confidence, over 81% alignment coverage and 40% amino acid identity, the structural homology of this protein to the HtrA-like protease DegQ (49, 50), and the predicted structure of C. difficile HtrA overlaid with the structure of E. coli DegS demonstrated a high level of structure homology (Fig. 1A). Homologs of DegQ and DegS are generally referred to as HtrA-like proteases in Gram-positive prokaryotes. Considering the above, we will refer here to the CD3284-encoding gene as htrA and to the CD3284 protein as HtrA.
C. difficile HtrA shows protease activity toward β-casein.
To investigate if C. difficile HtrA possesses proteolytic activity similar to that of other HtrA-like proteases (51) we performed a protease assay. Wild-type HtrA [His10-Δ(1-30)-HtrA] and a catalytic HtrA mutant [His10-Δ(1-30)-HtrA-S217A] protein were incubated with β-casein, a highly unfolded protein substrate (Fig. 1B). After 18 h of incubation at 37°C, casein was degraded only by wild-type HtrA and not by the catalytic HtrA mutant. This effect was temperature dependent, as no degradation was seen when the proteins were incubated at 0°C (Fig. 1B). These results demonstrate the proteolytic activity of C. difficile HtrA.
Construction of an htrA mutant.
To determine the contribution of HtrA to C. difficile virulence, we constructed an htrA mutant of the 630Δerm (wild-type) (29) strain by insertion of a group II intron upstream of the sequence encoding the predicted trypsin-like serine protease domain using ClosTron technology (Fig. 1A). Insertional inactivation of genes using the ClosTron technology results in stable mutants and nonfunctional proteins (32, 40). The genotype of the insertional inactivation was confirmed by PCR (Fig. 2), sequence analysis, and Southern blotting (see Fig. S1 in the supplemental material), which verified that the ClosTron group II intron was inserted at a single site.
FIG 2.
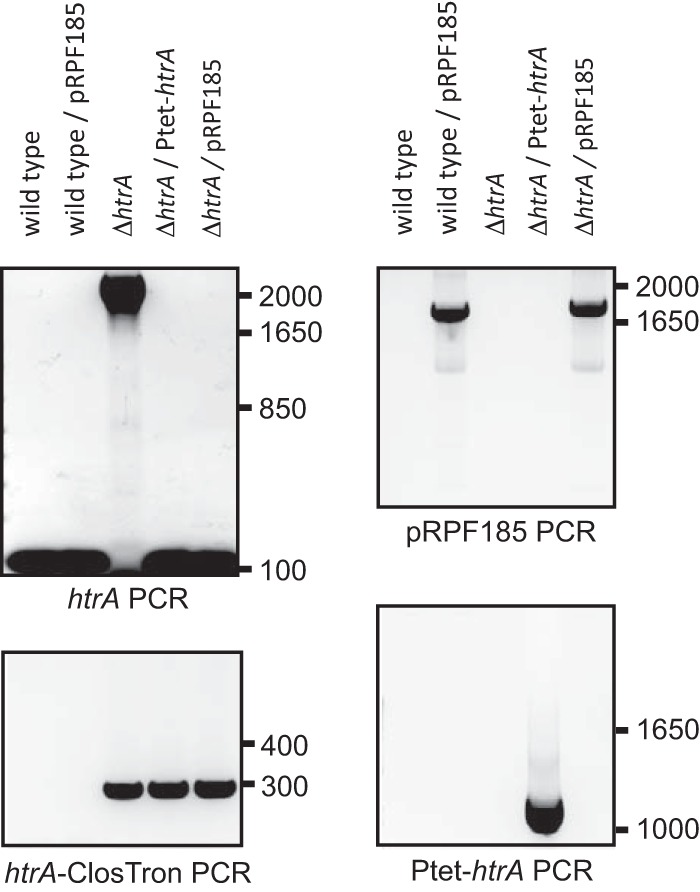
PCR screening of the strains used in this study. For htrA PCR, the primers oDB0123 and oDB0124 generated a PCR product of approximately 2,200 bp in the htrA mutant (ΔhtrA, DB0002) and a PCR product of 150 bp in the wild type (WKS1241), wild type/pRPF185 (DB0051), complemented htrA mutant (ΔhtrA/htrA+, DB0047), and ΔhtrA/pRPF185 (DB0052). For pRPF185 PCR, the primers oWKS-1177 and NF794 generated a PCR product of approximately 1,800 bp in the DB0051 and DB0052 strains. For htrA-ClosTron PCR, the primers oDB0123 and EBSuni generated a PCR product of approximately 280 bp in the DB0002, DB0047, and DB0052 strains. For Ptet-htrA PCR, the primers oDB0123 and NF794 generated a PCR product of approximately 1,200 bp in DB0047.
Initially, the impact of the mutation on growth in vitro was considered. This revealed that postinoculation both the mutant and wild-type strains showed logarithmic growth for the first 8 h, and by 12 h postinoculation both strains had entered the stationary growth phase (see Fig. S2 in the supplemental material). Though growth kinetics were not significantly different from those of wild-type cells, we noted that the htrA mutant strain generally showed a slight growth delay in logarithmic phase and reached lower optical density in stationary phase (see Fig. S2 in the supplemental material).
The htrA mutant is more virulent in hamsters.
HtrA-like proteases have been shown to be important for the virulence of pathogenic bacteria (25, 52, 53). To determine the contribution of HtrA in C. difficile virulence we performed in vivo experiments with the htrA mutant and wild type in the commonly used Golden Syrian hamster model (54, 55). Six clindamycin-pretreated hamsters were orally infected with either the wild type or the htrA mutant. The mean time to death for hamsters challenged with the wild type was 53 h 39 min (±5 h 3 min; n = 6). Hamsters infected with the htrA mutant revealed a significantly decreased survival time (31 h 20 min ± 44 min; n = 6; P = 0.0005) compared to that with the wild type (Fig. 3), suggesting that disruption of htrA causes enhanced virulence in this strain. Our experience evaluating a number of mutants in the hamster model suggests that animals challenged with other mutants created using ClosTron technology have a shorter survival time than the wild type (unpublished data). We hypothesize that this is due to the greater resistance to clindamycin (as a consequence of ClosTron insertion) and hence greater survival rates in vivo following pretreatment of animals with clindamycin to induce disease. Indeed, we find that the htrA mutant (and other ClosTron mutants) in vitro is more resistant to clindamycin (MIC, >256 μg/ml) than the wild type (630Δerm; MIC, 16 μg/ml). Nevertheless, if we compare the htrA mutant to the original parental 630 isolate (56) (43 h 43 min [n = 11; P < 0.0001]) or to other mutants (ClosTron 1, 36 h 41 min [n = 7, P = 0.0049]; ClosTron 2, 36 h 47 min [n = 9, P < 0.0001]) infected in the same way, the enhanced mortality rate remains significant. We therefore conclude that in the Golden Syrian hamster model, the htrA mutant of Clostridium difficile demonstrates an enhanced virulence phenotype.
FIG 3.
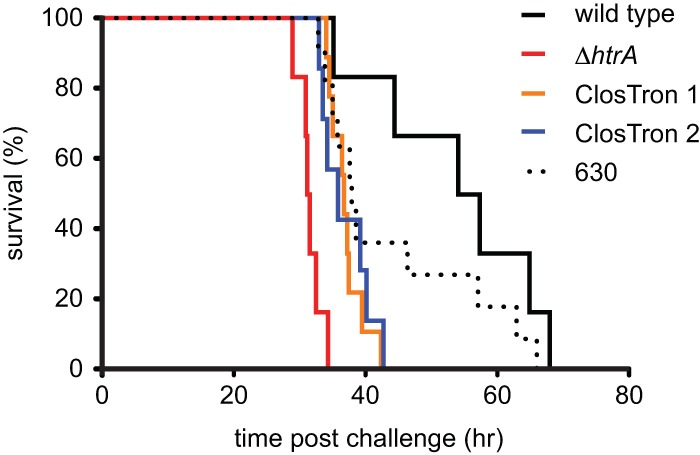
The htrA mutant has increased virulence in a hamster model of infection. Survival curves of hamsters challenged with either 104 spores of C. difficile 630Δerm (solid black line), the htrA mutant (red line), ClosTron mutants 1 and 2 (blue and green lines, respectively), or the erythromycin-resistant 630 strain (dashed black line) are shown. Hamsters challenged with the htrA mutant (31 h 20 min; n = 6) showed a significantly decreased survival time compared to those challenged with 630Δerm (53 h 39 min; n = 6; P = 0.0005), ClosTron mutant 1 (36 h 41 min; n = 9; P = 0.049), ClosTron mutant 2 (36 h 47 min; n = 7; P < 0.0001), or 630 (43 h 43 min; n = 11; P < 0.0001). Hamsters were culled when the core body temperature declined below 35°C (clinical endpoint), and data were analyzed with Mantel-Cox log rank statistical tests.
Comparative transcriptional microarray data analysis of htrA mutant and wild-type strains.
To understand the enhanced virulence of the htrA mutant in the Golden Syrian hamster model, we performed comparative transcriptome analyses of the wild type and the htrA mutant. The htrA mutant displayed a pleiotropic effect in the transcriptome analyses (see Table S2 in the supplemental material). The most interesting genes that were differentially regulated in the htrA mutant were tcdA, spo0A, and several genes encoding surface-associated proteins.
The transcriptional profiles of the htrA mutant strain and the wild-type strain were studied at two different time points, the logarithmic and stationary growth phases. The DNA microarray raw data sets and the normalized ratio data are available from the NCBI GEO database (http://www.ncbi.nlm.nih.gov/geo/; accession number GSE55926) (57). In the logarithmic growth phase and in the stationary growth phase, 551 genes and 567 genes were significantly differentially expressed, respectively (fold change of ≥1.5; P ≤ 0.05) (see Table S2 in the supplemental material). In total, 248 genes in the logarithmic growth phase and 263 genes in the stationary growth phase were upregulated. Two hundred eight genes were upregulated in both logarithmic and stationary growth phases. Among these, several genes were involved in the stress adaptation response, such as the ctsR and hrcA genes (58). The tcdA gene, encoding the well-characterized virulence factor toxin A, was 3.3-fold and 3.0-fold upregulated in the logarithmic and stationary growth phases, respectively.
A total of 303 genes in the logarithmic growth phase and 304 genes in the stationary growth phase were downregulated in the htrA mutant. Two hundred sixty-five genes were downregulated in both logarithmic and stationary growth phases. Among these are genes involved in cell metabolism, such as ribosomal proteins, ABC transporters, and phosphotransferase system (PTS) transporters. The spo0A gene, encoding the master regulator of sporulation (59, 60), was 2.4-fold and 2.3-fold downregulated in the logarithmic and stationary growth phases, respectively, and many other sporulation genes showed a similar effect (see Table S2 in the supplemental material). Furthermore, several genes encoding surface-associated proteins (such as slpA and 5 other paralogs of slpA) (16), flagellum-associated genes (18, 61), and cd1581 (a gene with a potential effect on adhesion and intestinal colonization) (19) were significantly downregulated.
Overall, transcriptional profiling of the htrA mutant showed a plethora of differences compared to the wild type. The most prominent differentially regulated genes were tcdA, spo0A, and several genes encoding surface-associated proteins that were further investigated phenotypically.
Increased TcdA production of the htrA mutant compared to the wild type.
Golden Syrian hamsters are exquisitely sensitive to C. difficile toxins, and increased levels of tcdA transcription might explain the observed enhanced virulence. To validate the increased level of tcdA transcription in the comparative DNA microarray analysis, we determined the tcdA transcription by RT-qPCR, quantitative Western blot analysis of TcdA, and a cytotoxicity assay.
First, we confirmed the effect on transcription of the tcdA gene in the htrA mutant. We also complemented the htrA mutant (DB0047) to confirm that the observed effect on tcdA transcription in the htrA mutant was specific. RT-qPCR was used to compare the normalized tcdA transcription profiles in the wild-type, htrA mutant, and complemented htrA mutant strains in the logarithmic and stationary growth phases.
In the logarithmic phase and the stationary phase, the normalized tcdA transcription of the htrA mutant was significantly increased, 2.1-fold (P < 0.000001) and 8.1-fold (P < 0.000001), respectively (Fig. 4A). Expressing HtrA from a plasmid in the mutant strain (DB0047) partially restored tcdA transcript levels in logarithmic phase (1.4-fold; P = 0.0022) toward the wild-type level, while in stationary phase the tcdA transcript level was indistinguishable from wild-type levels (Fig. 4A). These data suggest that the reduction in transcription of tcdA in the htrA mutant is a consequence of the htrA mutation.
FIG 4.
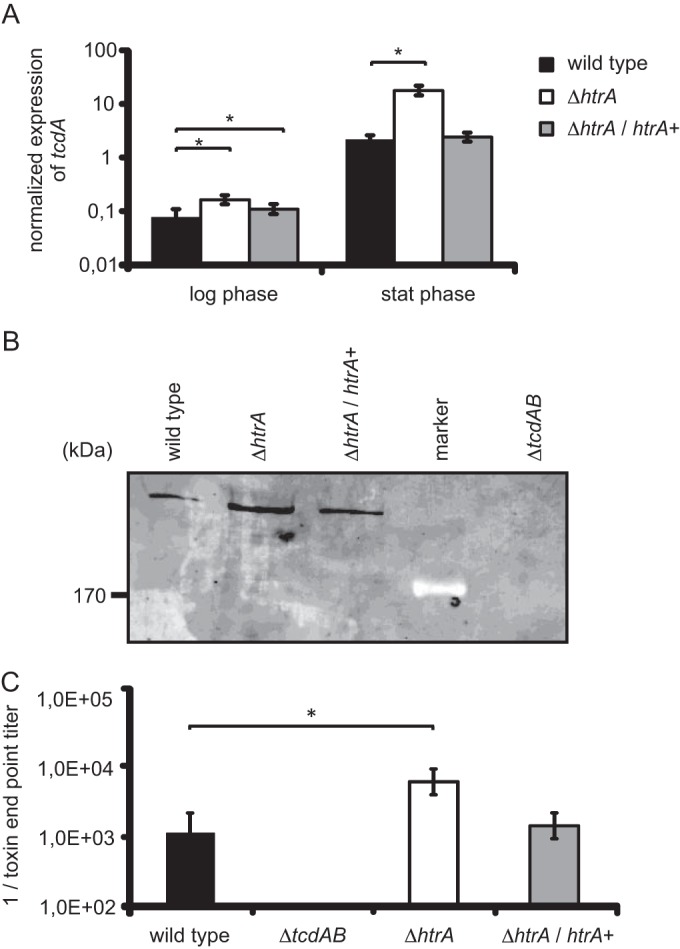
Increased TcdA production by the htrA mutant compared to the wild type. (A) Relative tcdA gene expression profiles of C. difficile strains in logarithmic and stationary phases. The asterisks indicate a significant difference between the wild type and the htrA mutant (P < 0.000001) and the complemented htrA mutant (P < 0.000001). (B) Quantitative Western blot analysis of TcdA in culture supernatants of the wild-type, htrA mutant, and complemented htrA mutant strains at 48 h postinoculation. (C) Toxin endpoint titers of the various C. difficile strains. The asterisk indicates a significant difference between the wild-type and htrA mutant strains (P = 0.001) at the same time point. The error bars indicate the standard errors of the mean from three experiments. The wild type is WKS1241, the htrA mutant is DB0002, the complemented htrA mutant is DB0047, and the tcdAB mutant is WKS1710.
Second, we determined if the increased level of tcdA transcription was reflected in elevated levels of TcdA protein. Quantitative Western blot analysis with a TcdA-specific monoclonal antibody on filtered supernatants of 48-h-postinoculation cultures showed a 7-fold increase of TcdA in the htrA mutant compared to the wild type (Fig. 4B). Similar to tcdA transcription, complementation of the htrA mutant resulted in TcdA levels that were partially restored toward the wild-type level (2-fold) (Fig. 4B). The signals detected were specific for TcdA, as they were absent in the supernatant of the control strain WKS1710 (40).
We also determined the effect of the htrA mutant in a cytotoxicity assay on Vero cells, which detects the cumulative effects of both toxins A and B (28, 40) (Fig. 4C). We observed a 5-fold increase in toxin endpoint titer for the htrA mutant (P = 0.001) compared to the wild type (Fig. 4C). Complementing the htrA mutant with an inducible copy of htrA on a plasmid (DB0047) restored the toxin endpoint titer to near-wild-type levels. To determine these effects as toxin specific, Vero cells were exposed to filter-sterilized supernatant of strain WKS1710, which produces no toxin A and B (40), which as expected did not result in a cytotoxic effect. Moreover, preincubation of the filter-sterilized bacterial supernatants with antitoxin (Techlab, Blacksburg, VA, USA) resulted in complete neutralization of the observed cytotoxic effects on the Vero cell monolayers (data not shown). Thus, the effects of the supernatants on Vero cells were toxin A and/or toxin B specific.
In summary, we found increased tcdA transcription levels, increased TcdA expression levels, and elevated toxin endpoint titers of supernatants derived from htrA mutant cells compared to the wild type. Taken together, these data contribute to an explanation for the enhanced virulence of the htrA mutant strain in vivo.
The htrA mutant displays decreased sporulation compared to that of the wild type.
Our transcriptome analysis revealed pleiotropic effects that included a decreased transcription of spo0A, the master regulator of sporulation, in the htrA mutant compared to the wild type (59, 60). Sporulation is highly relevant for transmission, persistence, and biofilm formation of C. difficile (62, 63). To validate the microarray results, we performed RT-qPCR, quantitative Western blot analysis of Spo0A, and a sporulation assay.
We used RT-qPCR to determine the normalized spo0A transcript levels in the wild-type, htrA mutant, and complemented htrA mutant strains in the logarithmic and stationary growth phases. The spo0A transcript level in the htrA mutant was significantly decreased in the logarithmic phase (4.3-fold; P < 0.000001) and stationary phase (4.1-fold; P < 0.000001) compared to that of the wild type (Fig. 5A). The complemented htrA mutant failed to reach wild-type levels of spo0A transcript in the log phase (5.2-fold difference; P < 0.000001), but in the stationary phase the complemented strain reached wild-type levels of spo0A transcript. (Fig. 5A). Therefore, we believe that the reduced spo0A transcript level in the htrA mutant is partially, if not completely, due to the htrA mutation.
FIG 5.
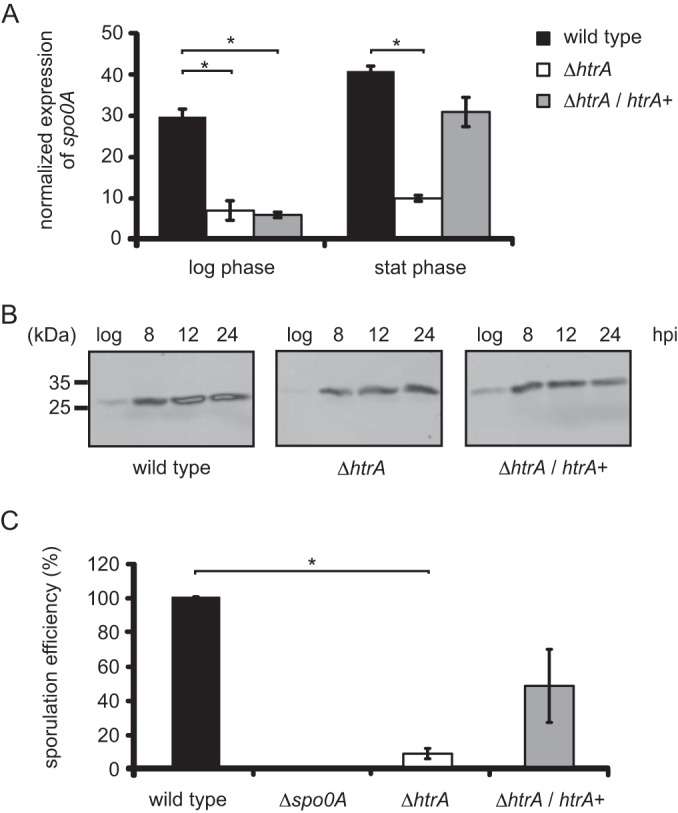
Decreased sporulation of the htrA mutant compared to the wild type. (A) Relative spo0A gene expression profiles of C. difficile strains in logarithmic and stationary phases. The asterisks indicate a significant difference between the wild type and the htrA mutant (P < 0.000001) and the complemented htrA mutant (P < 0.000001) at the same time point. (B) Quantitative Western blot analysis of Spo0A in the wild-type, htrA mutant, and complemented htrA mutant strains over time. (C) Normalized sporulation efficiencies (heat-resistant CFU compared to wild type) of the different strains at 24 h postinoculation. The asterisk indicates a significant difference between the wild type and the htrA mutant (P = 0.0006). The error bars indicate the standard errors of the mean from three experiments. The wild type is WKS1241, the htrA mutant is DB0002, the complemented htrA mutant is DB0047, and the spo0A mutant is WKS1237.
Next, we quantified the decreased Spo0A levels by quantitative Western blot analysis (31). In wild-type cells, the Spo0A levels increased during growth and signals were absent from the spo0A mutant strain, as previously reported (Fig. 5B) (31). In the htrA mutant, Spo0A levels were on average 3-fold lower than those in the wild type (Fig. 5B). Complementation of the htrA mutant restored Spo0A protein levels throughout the growth to near-wild-type levels (Fig. 5B).
We also investigated the sporulation efficiencies of the various strains by determining heat-resistant CFU. The efficiency of heat treatment on spore recovery was initially tested using cultures of the WKS1237 mutant (32). No colonies could be recovered using this strain, indicating that this is a good way to measure actual spore formation. At 24 h postinoculation, we observed (12-fold) decreased sporulation efficiency for the htrA mutant compared to the wild type (significant, P = 0.0006) (Fig. 5C). Expressing htrA from a plasmid in the htrA mutant strain (DB0047) partly restored sporulation efficiency toward wild-type levels (Fig. 5C). In contrast, sporulation efficiency at 48 h postinoculation appeared to be comparable in all strains tested, including the htrA mutant (data not shown).
In summary, we showed that the htrA mutant has a decreased spo0A transcript level, a delayed expression of Spo0A protein, and a significant reduction in the number of spores formed at 24 h postinoculation.
Decreased adherence of the htrA mutant to a Caco-2 cell monolayer compared to the wild type.
In addition to spores, it has been suggested that surface proteins on vegetative cells are important factors for adherence of C. difficile to the colonic epithelium and colonization of the intestine (16, 19, 64). Prompted by the downregulation of several important cell surface proteins in our transcriptome analyses, we determined the capability of the htrA mutant strains to adhere to colon epithelial cells.
We incubated the various strains on a monolayer of Caco-2 cells and determined the fraction of bacterial cells that remained attached after rigorous washing. We observed only 10% adherence of the htrA mutant to the Caco-2 cell monolayer (P = 0.000001) compared to the wild type (Fig. 6). Inducing DB0047 resulted in a slightly, but not significantly (P = 0.17), increased capability to adhere to a monolayer of Caco-2 cells compared to wild-type levels, indicating that the adherence defect of the htrA mutant is specific.
FIG 6.
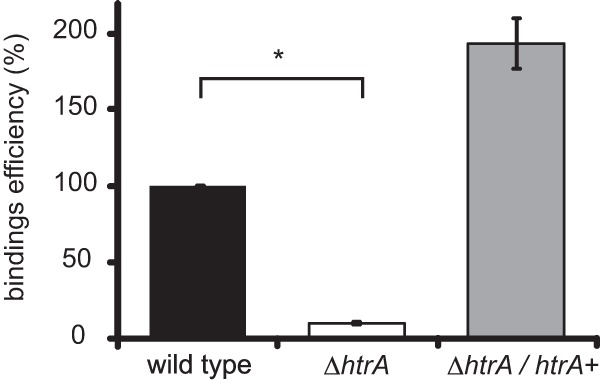
Decreased adhesion of the htrA mutant to Caco2 cells. The asterisk indicates a significant difference between the wild type and the htrA mutant (P = 0.000001). The error bars indicate the standard errors of the mean from three experiments. The wild type is WKS1241, the htrA mutant is DB0002, and the complemented htrA mutant is DB0047.
DISCUSSION
Role for HtrA in virulence of C. difficile.
In addition to the prevalence, the severity and mortality of disease caused by C. difficile infections have increased significantly in the last decade (1, 6, 9). It is unclear whether this is a consequence of more diligent screening or a consequence of acquisition of additional virulence traits by the bacteria. The refractory nature of the organism to genetic manipulation has limited opportunities to investigate the roles of many genes in the control of virulence traits, including the two large clostridial toxins, the binary toxin, and other factors that contribute to the virulence of C. difficile, such as those affecting colonization and survival of the bacteria in the host (15, 16, 18, 19). In multiple organisms, HtrA-like foldase/proteases play a considerable role in virulence by controlling protein homeostasis (21, 22, 26, 52, 65). Here, we found that C. difficile encodes a single HtrA-like protease. In contrast to observations in other organisms, we have found that a C. difficile htrA mutant shows enhanced virulence in the Golden Syrian hamster model of acute C. difficile infection (Fig. 3).
The observed increase in virulence is likely to be a consequence of elevated toxin levels. Toxin measurements from the hamsters showed no differences in measurement of toxin production in filtered gut samples; however, these samples were taken at the endpoint of infection, when toxin levels reflect significant symptoms. In vitro, we find increased transcription of tcdA, increased levels of TcdA protein in the htrA mutant, and increased cytotoxicity of the filtered supernatant derived from htrA mutant cells, all of which could be substantially reversed by prolonged induction of a plasmid-located copy of htrA (Fig. 4). Shorter induction did not result in full complementation, most likely because expression levels or the temporal pattern of expression does not match that of the wild type.
Our transcriptome analysis did not reveal a significant change in tcdB levels in the htrA mutant. This has been observed before and is probably due to lower transcription levels of tcdB than of tcdA in general, though our data cannot exclude differential regulation of the two toxin genes (28, 66, 67).
We did notice changes in transcription of several other virulence-associated genes. We validated a number of phenotypes that could be related to these genes experimentally, including a reduction in sporulation frequency and a decrease in colon epithelial cell adherence. In agreement with the observation for TcdA, these effects could be reversed by complementation (Fig. 5 and 6). While the most significant impact of this mutation in the hamster model appears to be the change of toxin expression, this does not reflect the potential role of HtrA more globally in gene expression.
The delay in sporulation, as measured by enumeration of colonies on taurocholate-containing plates after heat shock, does not necessarily reflect a block in sporulation but could also be the result of a defect in germination. However, the complementary observation of a decrease in the spo0A transcript level and a delay in the Spo0A protein level in the mutant strain would suggest that sporulation is most likely affected. Interestingly, the delay in sporulation results in a prolonged vegetative state during which the bacteria can produce more toxin than wild-type cells.
Structure of C. difficile HtrA.
The HtrA family of proteases can be distinguished from other serine proteases by the presence of at least one PDZ domain (21, 23). PDZ domains are involved in the regulation of the activity of the protein through protein-protein interactions or binding of specific substrates (22, 50). HtrA proteins in other organisms can be either membrane associated or secreted (49, 50). A detailed bioinformatics analysis of C. difficile HtrA suggests that the protein is a membrane-associated protein with a single PDZ domain (Fig. 1A). Consistent with this, we did not identify HtrA in the exoproteome of strain 630Δerm (68).
HtrA-like proteases of E. coli are among the best-characterized members of the family. DegP and DegQ consist of single protease domains with two associated PDZ domains (22, 49). Both proteins are foldase/proteases that have relatively broad substrate specificity. DegS is a protease with a single PDZ domain that acts specifically as a site 1 protease in a regulatory pathway for the extracytoplasmic stress response (49). Considering the domain structure, one might expect HtrA of C. difficile to be a site 1 protease. However, we would argue against this based on four findings. First, our protease assay with a non-C. difficile unfolded protein suggests a nonspecific proteolytic activity. Second, our transcriptome analyses (see Table S2 in the supplemental material) suggest a pleiotropic effect, rather than a specific pathway to be affected. Third, homology modeling of the predicted structure of C. difficile HtrA using I-TASSER and Phyre2 suggests that the overall fold of the catalytic domain and PDZ domain is more similar to that of the DegQ/P subfamily of HtrA-like proteases (22, 24, 49, 50). Finally, site 1 proteases in Gram-positive bacteria generally belong to the PrsW family (69–71). Indeed, in C. difficile, PrsW (CD0552) is involved in the activation of the extracytoplasmic function sigma factor CsfT (72).
HtrA and virulence.
In the majority of pathogenic bacteria, HtrA is directly or indirectly implicated in virulence. In many bacteria, htrA mutants show a complete loss of virulence or at least a significant level of attenuation (21, 22, 25, 26, 52). To our knowledge, our study is the first example of increased virulence for an htrA mutant.
Only a few studies have found little or no effect of htrA on virulence in animal models. In Staphylococcus aureus COL, the deletion of htrA led to minimal changes in surface protein expression and virulence (73). In Brucella abortus, the htrA mutant was not found to be attenuated in a BALB/c mouse model (74). For Porphyromonas gingivalis, only late effects were observed in a competition assay in mice (75). For Streptococcus pyogenes, no effects were seen in a murine model of subcutaneous infection (76). In these cases, htrA mutant strains did display an increased sensitivity to stresses or an altered expression of virulence factors. However, this may reflect the choice of model and the environmental niche tested.
It should be noted that the effects of HtrA can be strain dependent (26, 73) and may be masked by additional htrA-like genes that can compensate in site-specific deletion mutants (77). In the case of C. difficile, we have not identified any additional HtrA-like protease genes (data not shown), but we cannot exclude the possibility that another (serine) protease(s) might be able to substitute for certain functions of HtrA. The contribution of the sporulation and adhesion phenotypes of the htrA mutant of C. difficile to virulence therefore awaits validation in a model other than the hamster, in which toxin production is so dominant that observation of changes to other traits can be limited. For example, analysis of this mutant in mice, which can be colonized but are less susceptible to toxin production, may be a good alternative to study the effects of htrA on colonization, transmission, and persistence (15, 35).
Mode of action of HtrA.
In most pathogens, attenuation as a consequence of mutation in htrA appears to be the result of a reduced capacity of the cells to deal with environmental or host-dependent stresses (e.g., heat or oxidative stress), a reduced level of virulence factors that depend on HtrA for processing or activation, and/or a reduced invasiveness of the bacterium (21, 24, 50). In some cases, the effects of HtrA have not been traced to a specific protein or pathway (78, 79).
Our study revealed broad effects of the mutation of htrA on multiple pathways. Interestingly, several of these pathways have been linked to each other in previous work. For instance, it was noted that the asporogenic spo0A mutant showed elevated levels of toxin production in PCR ribotype 027 but not in 630Δerm (62, 80). Similarly, there is a clear link between motility and toxin production, as the alternative sigma factor SigD is required for both (81). Interestingly, flagellar operon mutants showed changes in both toxin levels (82) and their capacity to adhere to Caco-2 cells (61). Finally, more general effects of stress (nutrient depletion or temperature) on toxin levels were noted (83, 84).
Our study shows that htrA of C. difficile can modulate toxin levels, the formation of spores, and the adhesion to colonic cells, all of which are highly relevant for virulence. However, the high connectivity of the networks regulating these processes does not allow us to draw any conclusions as to the direct targets of HtrA in C. difficile. Future research will be aimed at identifying which proteins are direct targets of the foldase/protease activity of C. difficile HtrA.
Supplementary Material
ACKNOWLEDGMENTS
Wiep Klaas Smits was supported by Veni grant 016.116.043 from NWO-ZonMW and an LUMC Gisela Thier fellowship. Anthony M. Buckley is funded by Wellcome Trust grant 086418.
We thank N. Minton and S. Kuehne for providing the ClosTron system and the toxin A/B mutant. We thank R. Fagan and N. Fairweather for providing the pRF185 plasmid.
Footnotes
Published ahead of print 21 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02336-14.
REFERENCES
- 1. Loo VG, Poirier L, Miller MA, Oughton M, Libman MD, Michaud S, Bourgault AM, Nguyen T, Frenette C, Kelly M, Vibien A, Brassard P, Fenn S, Dewar K, Hudson TJ, Horn R, Rene P, Monczak Y, Dascal A. 2005. A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N. Engl. J. Med. 353:2442–2449. 10.1056/NEJMoa051639 [DOI] [PubMed] [Google Scholar]
- 2. Kyne L, Hamel MB, Polavaram R, Kelly CP. 2002. Health care costs and mortality associated with nosocomial diarrhea due to Clostridium difficile. Clin. Infect. Dis. 34:346–353. 10.1086/338260 [DOI] [PubMed] [Google Scholar]
- 3. Polage CR, Solnick JV, Cohen SH. 2012. Nosocomial diarrhea: evaluation and treatment of causes other than Clostridium difficile. Clin. Infect. Dis. 55:982–989. 10.1093/cid/cis551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goorhuis A, van der KT, Vaessen N, Dekker FW, van den BR, Harmanus C, van den HS, Notermans DW, Kuijper EJ. 2007. Spread and epidemiology of Clostridium difficile polymerase chain reaction ribotype 027/toxinotype III in The Netherlands. Clin. Infect. Dis. 45:695–703. 10.1086/520984 [DOI] [PubMed] [Google Scholar]
- 5. Kuijper EJ, Coignard B, Tull P. 2006. Emergence of Clostridium difficile-associated disease in North America and Europe. Clin. Microbiol. Infect. 12(Suppl 6):S2–S18. 10.1111/j.1469-0691.2006.01580.x [DOI] [PubMed] [Google Scholar]
- 6. Kuijper EJ, Barbut F, Brazier JS, Kleinkauf N, Eckmanns T, Lambert ML, Drudy D, Fitzpatrick F, Wiuff C, Brown DJ, Coia JE, Pituch H, Reichert P, Even J, Mossong J, Widmer AF, Olsen KE, Allerberger F, Notermans DW, Delmee M, Coignard B, Wilcox M, Patel B, Frei R, Nagy E, Bouza E, Marin M, Akerlund T, Virolainen-Julkunen A, Lyytikainen O, Kotila S, Ingebretsen A, Smyth B, Rooney P, Poxton IR, Monnet DL. 2008. Update of Clostridium difficile infection due to PCR ribotype 027 in Europe, 2008. Euro Surveill. 13(31):pii=18942 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=18942 [PubMed] [Google Scholar]
- 7. Paltansing S, van den Berg RJ, Guseinova RA, Visser CE, van der Vorm ER, Kuijper EJ. 2007. Characteristics and incidence of Clostridium difficile-associated disease in The Netherlands, 2005. Clin. Microbiol. Infect. 13:1058–1064. 10.1111/j.1469-0691.2007.01793.x [DOI] [PubMed] [Google Scholar]
- 8. Pepin J, Valiquette L, Alary ME, Villemure P, Pelletier A, Forget K, Pepin K, Chouinard D. 2004. Clostridium difficile-associated diarrhea in a region of Quebec from 1991 to 2003: a changing pattern of disease severity. CMAJ 171:466–472. 10.1503/cmaj.1041104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goorhuis A, Bakker D, Corver J, Debast SB, Harmanus C, Notermans DW, Bergwerff AA, Dekker FW, Kuijper EJ. 2008. Emergence of Clostridium difficile infection due to a new hypervirulent strain, polymerase chain reaction ribotype 078. Clin. Infect. Dis. 47:1162–1170. 10.1086/592257 [DOI] [PubMed] [Google Scholar]
- 10. Just I, Gerhard R. 2004. Large clostridial cytotoxins. Rev. Physiol. Biochem. Pharmacol. 152:23–47. 10.1007/s10254-004-0033-5 [DOI] [PubMed] [Google Scholar]
- 11. Voth DE, Ballard JD. 2005. Clostridium difficile toxins: mechanism of action and role in disease. Clin. Microbiol. Rev. 18:247–263. 10.1128/CMR.18.2.247-263.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jank T., Giesemann T., Aktories K. 2007. Rho-glucosylating Clostridium difficile toxins A and B: new insights into structure and function. Glycobiology 17:15R–22R. 10.1093/glycob/cwm004 [DOI] [PubMed] [Google Scholar]
- 13. Kuehne SA, Collery MM, Kelly ML, Cartman ST, Cockayne A, Minton NP. 2014. Importance of toxin A, toxin B, and CDT in virulence of an epidemic Clostridium difficile strain. J. Infect. Dis. 209:83–86. 10.1093/infdis/jit426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schwan C, Stecher B, Tzivelekidis T, HMvan Rohde M, Hardt WD, Wehland J, Aktories K. 2009. Clostridium difficile toxin CDT induces formation of microtubule-based protrusions and increases adherence of bacteria. PLoS Pathog. 5:e1000626. 10.1371/journal.ppat.1000626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barketi-Klai A, Hoys S, Lambert-Bordes S, Collignon A, Kansau I. 2011. Role of fibronectin-binding protein A in Clostridium difficile intestinal colonization. J. Med. Microbiol. 60:1155–1161. 10.1099/jmm.0.029553-0 [DOI] [PubMed] [Google Scholar]
- 16. Calabi E, Calabi F, Phillips AD, Fairweather NF. 2002. Binding of Clostridium difficile surface layer proteins to gastrointestinal tissues. Infect. Immun. 70:5770–5778. 10.1128/IAI.70.10.5770-5778.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Janoir C, Pechine S, Grosdidier C, Collignon A. 2007. Cwp84, a surface-associated protein of Clostridium difficile, is a cysteine protease with degrading activity on extracellular matrix proteins. J. Bacteriol. 189:7174–7180. 10.1128/JB.00578-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tasteyre A, Barc MC, Collignon A, Boureau H, Karjalainen T. 2001. Role of FliC and FliD flagellar proteins of Clostridium difficile in adherence and gut colonization. Infect. Immun. 69:7937–7940. 10.1128/IAI.69.12.7937-7940.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Janoir C, Deneve C, Bouttier S, Barbut F, Hoys S, Caleechum L, Chapeton-Montes D, Pereira FC, Henriques AO, Collignon A, Monot M, Dupuy B. 2013. Adaptive strategies and pathogenesis of Clostridium difficile from in vivo transcriptomics. Infect. Immun. 81:3757–3769. 10.1128/IAI.00515-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fang FC. 2004. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat. Rev. Microbiol. 2:820–832. 10.1038/nrmicro1004 [DOI] [PubMed] [Google Scholar]
- 21. Clausen T, Kaiser M, Huber R, Ehrmann M. 2011. HTRA proteases: regulated proteolysis in protein quality control. Nat. Rev. Mol. Cell. Biol. 12:152–162. 10.1038/nrm3065 [DOI] [PubMed] [Google Scholar]
- 22. Malet H, Canellas F, Sawa J, Yan J, Thalassinos K, Ehrmann M, Clausen T, Saibil HR. 2012. Newly folded substrates inside the molecular cage of the HtrA chaperone DegQ. Nat. Struct. Mol. Biol. 19:152–157. 10.1038/nsmb.2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hansen G, Hilgenfeld R. 2013. Architecture and regulation of HtrA-family proteins involved in protein quality control and stress response. Cell. Mol. Life Sci. 70:761–775. 10.1007/s00018-012-1076-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dalbey RE, Wang P, van Dijl JM. 2012. Membrane proteases in the bacterial protein secretion and quality control pathway. Microbiol. Mol. Biol. Rev. 76:311–330. 10.1128/MMBR.05019-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chitlaru T, Zaide G, Ehrlich S, Inbar I, Cohen O, Shafferman A. 2011. HtrA is a major virulence determinant of Bacillus anthracis. Mol. Microbiol. 81:1542–1559. 10.1111/j.1365-2958.2011.07790.x [DOI] [PubMed] [Google Scholar]
- 26. Ibrahim YM, Kerr AR, McCluskey J, Mitchell TJ. 2004. Role of HtrA in the virulence and competence of Streptococcus pneumoniae. Infect. Immun. 72:3584–3591. 10.1128/IAI.72.6.3584-3591.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ingmer H, Brondsted L. 2009. Proteases in bacterial pathogenesis. Res. Microbiol. 160:704–710. 10.1016/j.resmic.2009.08.017 [DOI] [PubMed] [Google Scholar]
- 28. Bakker D, Smits WK, Kuijper EJ, Corver J. 2012. TcdC does not significantly repress toxin expression in Clostridium difficile 630ΔErm. PLoS One 7:e43247. 10.1371/journal.pone.0043247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hussain HA, Roberts AP, Mullany P. 2005. Generation of an erythromycin-sensitive derivative of Clostridium difficile strain 630 (630Deltaerm) and demonstration that the conjugative transposon Tn916DeltaE enters the genome of this strain at multiple sites. J. Med. Microbiol. 54:137–141. 10.1099/jmm.0.45790-0 [DOI] [PubMed] [Google Scholar]
- 30. Miroux B, Walker JE. 1996. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260:289–298. 10.1006/jmbi.1996.0399 [DOI] [PubMed] [Google Scholar]
- 31. Rosenbusch KE, Bakker D, Kuijper EJ, Smits WK. 2012. C. difficile 630Δerm Spo0A regulates sporulation, but does not contribute to toxin production, by direct high-affinity binding to target DNA. PLoS One 7:e48608. 10.1371/journal.pone.0048608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Heap JT, Pennington OJ, Cartman ST, Carter GP, Minton NP. 2007. The ClosTron: a universal gene knock-out system for the genus Clostridium. J. Microbiol. Methods 70:452–464. 10.1016/j.mimet.2007.05.021 [DOI] [PubMed] [Google Scholar]
- 33. Heap JT, Kuehne SA, Ehsaan M, Cartman ST, Cooksley CM, Scott JC, Minton NP. 2010. The ClosTron: mutagenesis in Clostridium refined and streamlined. J. Microbiol. Methods 80:49–55. 10.1016/j.mimet.2009.10.018 [DOI] [PubMed] [Google Scholar]
- 34. Purdy D, O'Keeffe TA, Elmore M, Herbert M, McLeod A, Bokori-Brown M, Ostrowski A, Minton NP. 2002. Conjugative transfer of clostridial shuttle vectors from Escherichia coli to Clostridium difficile through circumvention of the restriction barrier. Mol. Microbiol. 46:439–452. 10.1046/j.1365-2958.2002.03134.x [DOI] [PubMed] [Google Scholar]
- 35. Fagan RP, Fairweather NF. 2011. Clostridium difficile has two parallel and essential Sec secretion systems. J. Biol. Chem. 286:27483–27493. 10.1074/jbc.M111.263889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sebaihia M, Wren BW, Mullany P, Fairweather NF, Minton N, Stabler R, Thomson NR, Roberts AP, Cerdeno-Tarraga AM, Wang H, Holden MT, Wright A, Churcher C, Quail MA, Baker S, Bason N, Brooks K, Chillingworth T, Cronin A, Davis P, Dowd L, Fraser A, Feltwell T, Hance Z, Holroyd S, Jagels K, Moule S, Mungall K, Price C, Rabbinowitsch E, Sharp S, Simmonds M, Stevens K, Unwin L, Whithead S, Dupuy B, Dougan G, Barrell B, Parkhill J. 2006. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat. Genet. 38:779–786. 10.1038/ng1830 [DOI] [PubMed] [Google Scholar]
- 37. Wolber PK, Collins PJ, Lucas AB, De WA, Shannon KW. 2006. The Agilent in situ-synthesized microarray platform. Methods Enzymol. 410:28–57.:28–57. 10.1016/S0076-6879(06)10002-6 [DOI] [PubMed] [Google Scholar]
- 38. Smyth GK. 2004. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3:Article3. [DOI] [PubMed] [Google Scholar]
- 39. Metcalf D, Sharif S, Weese JS. 2010. Evaluation of candidate reference genes in Clostridium difficile for gene expression normalization. Anaerobe 16:439–443. 10.1016/j.anaerobe.2010.06.007 [DOI] [PubMed] [Google Scholar]
- 40. Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. 2010. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467:711–713. 10.1038/nature09397 [DOI] [PubMed] [Google Scholar]
- 41. Burns DA, Heap JT, Minton NP. 2010. SleC is essential for germination of Clostridium difficile spores in nutrient-rich medium supplemented with the bile salt taurocholate. J. Bacteriol. 192:657–664. 10.1128/JB.01209-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cerquetti M, Serafino A, Sebastianelli A, Mastrantonio P. 2002. Binding of Clostridium difficile to Caco-2 epithelial cell line and to extracellular matrix proteins. FEMS Immunol. Med. Microbiol. 32:211–218. 10.1111/j.1574-695X.2002.tb00556.x [DOI] [PubMed] [Google Scholar]
- 43. Ponting CP, Schultz J, Milpetz F, Bork P. 1999. SMART: identification and annotation of domains from signalling and extracellular protein sequences. Nucleic Acids Res. 27:229–232. 10.1093/nar/27.1.229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schultz J, Milpetz F, Bork P, Ponting CP. 1998. SMART, a simple modular architecture research tool: identification of signaling domains. Proc. Natl. Acad. Sci. U. S. A. 95:5857–5864. 10.1073/pnas.95.11.5857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rawlings ND, Barrett AJ, Bateman A. 2012. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 40:D343–D350. 10.1093/nar/gkr987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Petersen TN, Brunak S, HGvon Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8:785–786. 10.1038/nmeth.1701 [DOI] [PubMed] [Google Scholar]
- 47. Roy A, Kucukural A, Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5:725–738. 10.1038/nprot.2010.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4:363–371. 10.1038/nprot.2009.2 [DOI] [PubMed] [Google Scholar]
- 49. Meltzer M, Hasenbein S, Mamant N, Merdanovic M, Poepsel S, Hauske P, Kaiser M, Huber R, Krojer T, Clausen T, Ehrmann M. 2009. Structure, function and regulation of the conserved serine proteases DegP and DegS of Escherichia coli. Res. Microbiol. 160:660–666. 10.1016/j.resmic.2009.07.012 [DOI] [PubMed] [Google Scholar]
- 50. Kim DY, Kim KK. 2005. Structure and function of HtrA family proteins, the key players in protein quality control. J. Biochem. Mol. Biol. 38:266–274. 10.5483/BMBRep.2005.38.3.266 [DOI] [PubMed] [Google Scholar]
- 51. Sawa J, Malet H, Krojer T, Canellas F, Ehrmann M, Clausen T. 2011. Molecular adaptation of the DegQ protease to exert protein quality control in the bacterial cell envelope. J. Biol. Chem. 286:30680–30690. 10.1074/jbc.M111.243832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wilson RL, Brown LL, Kirkwood-Watts D, Warren TK, Lund SA, King DS, Jones KF, Hruby DE. 2006. Listeria monocytogenes 10403S HtrA is necessary for resistance to cellular stress and virulence. Infect. Immun. 74:765–768. 10.1128/IAI.74.1.765-768.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chatfield SN, Strahan K, Pickard D, Charles IG, Hormaeche CE, Dougan G. 1992. Evaluation of Salmonella typhimurium strains harbouring defined mutations in htrA and aroA in the murine salmonellosis model. Microb. Pathog. 12:145–151. 10.1016/0882-4010(92)90117-7 [DOI] [PubMed] [Google Scholar]
- 54. Buckley AM, Spencer J, Candlish D, Irvine JJ, Douce GR. 2011. Infection of hamsters with the UK Clostridium difficile ribotype 027 outbreak strain R20291. J. Med. Microbiol. 60:1174–1180. 10.1099/jmm.0.028514-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Goulding D, Thompson H, Emerson J, Fairweather NF, Dougan G, Douce GR. 2009. Distinctive profiles of infection and pathology in hamsters infected with Clostridium difficile strains 630 and B1. Infect. Immun. 77:5478–5485. 10.1128/IAI.00551-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wust J, Sullivan NM, Hardegger U, Wilkins TD. 1982. Investigation of an outbreak of antibiotic-associated colitis by various typing methods. J. Clin. Microbiol. 16:1096–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Muertter RN, Edgar R. 2009. NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res. 37:D885–D890. 10.1093/nar/gkn764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Alsaker KV, Papoutsakis ET. 2005. Transcriptional program of early sporulation and stationary-phase events in Clostridium acetobutylicum. J. Bacteriol. 187:7103–7118. 10.1128/JB.187.20.7103-7118.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Errington J. 2003. Regulation of endospore formation in Bacillus subtilis. Nat. Rev. Microbiol. 1:117–126. 10.1038/nrmicro750 [DOI] [PubMed] [Google Scholar]
- 60. Paredes CJ, Alsaker KV, Papoutsakis ET. 2005. A comparative genomic view of clostridial sporulation and physiology. Nat. Rev. Microbiol. 3:969–978. 10.1038/nrmicro1288 [DOI] [PubMed] [Google Scholar]
- 61. Baban ST, Kuehne SA, Barketi-Klai A, Cartman ST, Kelly ML, Hardie KR, Kansau I, Collignon A, Minton NP. 2013. The role of flagella in Clostridium difficile pathogenesis: comparison between a non-epidemic and an epidemic strain. PLoS One 8:e73026. 10.1371/journal.pone.0073026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Deakin LJ, Clare S, Fagan RP, Dawson LF, Pickard DJ, West MR, Wren BW, Fairweather NF, Dougan G, Lawley TD. 2012. The Clostridium difficile spo0A gene is a persistence and transmission factor. Infect. Immun. 80:2704–2711. 10.1128/IAI.00147-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ethapa T, Leuzzi R, Ng YK, Baban ST, Adamo R, Kuehne SA, Scarselli M, Minton NP, Serruto D, Unnikrishnan M. 2013. Multiple factors modulate biofilm formation by the anaerobic pathogen Clostridium difficile. J. Bacteriol. 195:545–555. 10.1128/JB.01980-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Deneve C, Janoir C, Poilane I, Fantinato C, Collignon A. 2009. New trends in Clostridium difficile virulence and pathogenesis. Int. J. Antimicrob. Agents 33(Suppl 1):S24–S28. 10.1016/S0924-8579(09)70012-3 [DOI] [PubMed] [Google Scholar]
- 65. Biswas S, Biswas I. 2005. Role of HtrA in surface protein expression and biofilm formation by Streptococcus mutans. Infect. Immun. 73:6923–6934. 10.1128/IAI.73.10.6923-6934.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mani N, Lyras D, Barroso L, Howarth P, Wilkins T, Rood JI, Sonenshein AL, Dupuy B. 2002. Environmental response and autoregulation of Clostridium difficile TxeR, a sigma factor for toxin gene expression. J. Bacteriol. 184:5971–5978. 10.1128/JB.184.21.5971-5978.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mani N, Dupuy B. 2001. Regulation of toxin synthesis in Clostridium difficile by an alternative RNA polymerase sigma factor. Proc. Natl. Acad. Sci. U. S. A. 98:5844–5849. 10.1073/pnas.101126598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hensbergen PJ, Klychnikov OI, Bakker D, van Winden VJ, Ras N, Kemp AC, Cordfunke RA, Dragan I, Deelder AM, Kuijper EJ, Corver J, Drijfhout JW, van Leeuwen HC. 2014. A novel secreted metalloprotease (CD2830) from Clostridium difficile cleaves specific proline sequences in LPXTG cell surface proteins. Mol. Cell. Proteomics 13:1231–1244. 10.1074/mcp.M113.034728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Schobel S, Zellmeier S, Schumann W, Wiegert T. 2004. The Bacillus subtilis sigmaW anti-sigma factor RsiW is degraded by intramembrane proteolysis through YluC. Mol. Microbiol. 52:1091–1105. 10.1111/j.1365-2958.2004.04031.x [DOI] [PubMed] [Google Scholar]
- 70. Chen G, Zhang X. 2010. New insights into S2P signaling cascades: regulation, variation, and conservation. Protein Sci. 19:2015–2030. 10.1002/pro.496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ellermeier CD, Losick R. 2006. Evidence for a novel protease governing regulated intramembrane proteolysis and resistance to antimicrobial peptides in Bacillus subtilis. Genes Dev. 20:1911–1922. 10.1101/gad.1440606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ho TD, Ellermeier CD. 2011. PrsW is required for colonization, resistance to antimicrobial peptides, and expression of extracytoplasmic function sigma factors in Clostridium difficile. Infect. Immun. 79:3229–3238. 10.1128/IAI.00019-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rigoulay C, Entenza JM, Halpern D, Widmer E, Moreillon P, Poquet I, Gruss A. 2005. Comparative analysis of the roles of HtrA-like surface proteases in two virulent Staphylococcus aureus strains. Infect. Immun. 73:563–572. 10.1128/IAI.73.1.563-572.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Phillips RW, Roop RM. 2001. Brucella abortus HtrA functions as an authentic stress response protease but is not required for wild-type virulence in BALB/c mice. Infect. Immun. 69:5911–5913. 10.1128/IAI.69.9.5911-5913.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yuan L, Rodrigues PH, Belanger M, Dunn WA, Jr, Progulske-Fox A. 2008. Porphyromonas gingivalis htrA is involved in cellular invasion and in vivo survival. Microbiology 154:1161–1169. 10.1099/mic.0.2007/015131-0 [DOI] [PubMed] [Google Scholar]
- 76. Lyon WR, Caparon MG. 2004. Role for serine protease HtrA (DegP) of Streptococcus pyogenes in the biogenesis of virulence factors SpeB and the hemolysin streptolysin S. Infect. Immun. 72:1618–1625. 10.1128/IAI.72.3.1618-1625.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Farn J, Roberts M. 2004. Effect of inactivation of the HtrA-like serine protease DegQ on the virulence of Salmonella enterica serovar Typhimurium in mice. Infect. Immun. 72:7357–7359. 10.1128/IAI.72.12.7357-7359.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sebert ME, Patel KP, Plotnick M, Weiser JN. 2005. Pneumococcal HtrA protease mediates inhibition of competence by the CiaRH two-component signaling system. J. Bacteriol. 187:3969–3979. 10.1128/JB.187.12.3969-3979.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tsui HC, Keen SK, Sham LT, Wayne KJ, Winkler ME. 2011. Dynamic distribution of the SecA and SecY translocase subunits and septal localization of the HtrA surface chaperone/protease during Streptococcus pneumoniae D39 cell division. mBio 2(5):e00202-211. 10.1128/mBio.00202-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mackin KE, Carter GP, Howarth P, Rood JI, Lyras D. 2013. Spo0A differentially regulates toxin production in evolutionarily diverse strains of Clostridium difficile. PLoS One 8:e79666. 10.1371/journal.pone.0079666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. McKee RW, Mangalea MR, Purcell EB, Borchardt EK, Tamayo R. 2013. The second messenger c-di-GMP regulates Clostridium difficile toxin production by controlling expression of sigD. J. Bacteriol. 195:5174–5185. 10.1128/JB.00501-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Aubry A, Hussack G, Chen W, KuoLee R, Twine SM, Fulton KM, Foote S, Carrillo CD, Tanha J, Logan SM. 2012. Modulation of toxin production by the flagellar regulon in Clostridium difficile. Infect. Immun. 80:3521–3532. 10.1128/IAI.00224-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Dupuy B, Sonenshein AL. 1998. Regulated transcription of Clostridium difficile toxin genes. Mol. Microbiol. 27:107–120. 10.1046/j.1365-2958.1998.00663.x [DOI] [PubMed] [Google Scholar]
- 84. Karlsson S, Lindberg A, Norin E, Burman LG, Akerlund T. 2000. Toxins, butyric acid, and other short-chain fatty acids are coordinately expressed and down-regulated by cysteine in Clostridium difficile. Infect. Immun. 68:5881–5888. 10.1128/IAI.68.10.5881-5888.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Andrews JM. 2001. Determination of minimal inhibitory concentrations. J. Antimicrob. Chemother. 48(Suppl 1):5–16. 10.1093/jac/48.suppl_1.5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.