

Abstract
Antifungal therapy failure can be associated with increased resistance to the employed antifungal agents. Candida glabrata, the second most common cause of invasive candidiasis, is intrinsically less susceptible to the azole class of antifungals and accounts for 15% of all Candida bloodstream infections. Here, we show that C. glabrata MED2 (CgMED2), which codes for a tail subunit of the RNA polymerase II Mediator complex, is required for resistance to azole antifungal drugs in C. glabrata. An inability to transcriptionally activate genes encoding a zinc finger transcriptional factor, CgPdr1, and multidrug efflux pump, CgCdr1, primarily contributes to the elevated susceptibility of the Cgmed2Δ mutant toward azole antifungals. We also report for the first time that the Cgmed2Δ mutant exhibits sensitivity to caspofungin, a constitutively activated protein kinase C-mediated cell wall integrity pathway, and elevated adherence to epithelial cells. The increased adherence of the Cgmed2Δ mutant was attributed to the elevated expression of the EPA1 and EPA7 genes. Further, our data demonstrate that CgMED2 is required for intracellular proliferation in human macrophages and modulates survival in a murine model of disseminated candidiasis. Lastly, we show an essential requirement for CgMed2, along with the Mediator middle subunit CgNut1 and the Mediator cyclin-dependent kinase/cyclin subunit CgSrb8, for the high-level fluconazole resistance conferred by the hyperactive allele of CgPdr1. Together, our findings underscore a pivotal role for CgMed2 in basal tolerance and acquired resistance to azole antifungals.
INTRODUCTION
Candida species are the most common cause of opportunistic fungal infections worldwide (1) and, although predominantly associated with intensive care unit (ICU) patients, have been found in non-ICU patients as well (1, 2). Candidiasis accounts for about 15% of nosocomial infections, which are associated with high mortality rates of 25 to 60% (3). The frequencies of isolation of Candida species causing candidemia vary among care centers, with Candida albicans being the leading causative agent in most of the centers (2, 4). C. glabrata is an emerging fungal pathogen which shows a varied prevalence on the basis of the geographical region and ranges from the second to the fourth most commonly isolated Candida species among common Candida isolates in bloodstream infections (BSIs) (2, 4–6).
Fluconazole, a triazole compound, is the most widely used drug for the treatment of candidiasis. Fluconazole inhibits ergosterol biosynthesis in yeast cells by targeting the cytochrome P450-dependent C14-lanosterol demethylase enzyme, which is encoded by the ERG11 gene (7). The antifungal activity of fluconazole is due to depletion of ergosterol in the cell membrane and its replacement by 14α-methyl-3,6-diol, resulting in altered membrane structure and function (7). The effect of fluconazole is mainly fungistatic and not fungicidal for Candida spp. (7).
The occurrence of C. glabrata in BSIs has increased over the last 2 decades, and this increase has partly been attributed to the increased use of fluconazole and the high intrinsic and acquired azole resistance shown by C. glabrata isolates (3, 8). Common azole resistance mechanisms associated with C. glabrata are amino acid substitutions in the lanosterol demethylase enzyme leading to a reduced affinity to azoles, increased expression of the C. glabrata ERG11 (CgERG11) gene, dysfunctional mitochondria, increased drug efflux, and mutations in genes encoding sterol biosynthetic enzymes (9–11). However, the most prevalent drug resistance mechanism is the increased efflux of azoles by the membrane-located multidrug efflux (MDR) pumps CgCdr1 and CgCdr2 (9–11). These pumps belong to the ATP-binding cassette (ABC) class of transporters and are under the transcriptional control of CgPdr1, a Zn2-Cys6 zinc cluster-containing transcription factor (9–15). CgPdr1 is a single-copy ortholog of Saccharomyces cerevisiae Pdr1 and Pdr3 transcription factors, and the CgPDR1 gene is known to be transcriptionally autoregulated via binding to two pleiotropic drug resistance elements (PDREs; TCCACGGA) in its promoter (15). Pdr1 has been shown to bind directly to xenobiotics and activate drug efflux pump genes in a manner similar to that for ligand-activated nuclear hormone receptor-governed gene expression (16). Gain-of-function (GOF)/activating mutations in the CgPDR1 gene result in high levels of expression of ABC transporters, leading to elevated drug resistance and enhanced virulence in murine models (17). So far, studies have identified CgPdr1 to be the sole regulatory determinant of multidrug resistance gene expression in C. glabrata (12–15).
The Mediator complex is an important and highly conserved part of the transcription machinery. It interacts with the carboxy-terminal domain of the largest subunit of RNA polymerase II and acts as a bridge between upstream gene-specific regulatory proteins and core RNA polymerase II complex to activate target gene transcription (18). The Mediator complex in S. cerevisiae has 25 subunits in three different modules, the head, middle, and tail (19). A kinase module composed of Cdk8, cyclin C, Med12, and Med13 can associate transiently and reversibly with the Mediator complex to regulate transcription initiation (18, 20, 21). Functions of the individual Mediator complex subunits are variable; while some components contribute to regulated expression of all genes, others are essential only for a certain subset of genes (20, 21). Consistent with this, on the basis of the mutated subunit, mutations in individual subunits of the Mediator complex result in enhanced or diminished transcription of a certain subset of genes (20, 21). The head and middle modules of the Mediator complex primarily interact with universal transcription factors and RNA polymerase II subunits (18, 20, 21).
The tail domain is the largest, structurally least conserved module and serves as a target for gene-specific activators (18). Compared to the head and middle modules, the tail module of the S. cerevisiae Mediator complex consists of nonessential subunits Med2, Med3, Med14, Med15 (Gal11), and Med16; preferentially regulates SAGA complex-dependent genes; and has been postulated to relay information from gene-specific regulatory proteins, through the head and middle modules, to the RNA polymerase II transcription machinery (20, 21). Recruitment of the tail subunit CgMed15 to the promoters of the CDR genes via KIX domain-mediated binding of the CgPdr1-fluconazole complex to activate CDR gene transcription in an azole-dependent manner epitomizes the gene-specific role of CgMed15 (16). Further, Med2 in S. cerevisiae is required for utilization of galactose as the sole carbon source (22) and known to be relocalized to the cytosol under hypoxic conditions (23). Additionally, MED2 deletion has recently been reported to lead to short telomeres, elevated acetylation of histone H4 at lysine 16, and reduced Sir2 binding at the telomeres in S. cerevisiae (24).
We have recently reported two putative C. glabrata tail module subunits, CgMed2 and CgPgd1, to be essential for retaining viability during azole stress (25). Here, using a combined approach of deletion, molecular, and biochemical analyses, we show that CgMED2 is required for both basal and acquired resistance to azole antifungals in C. glabrata. Further, we implicate for the first time a tail subunit of the Mediator complex in adherence to epithelial cells and survival and/or replication in human macrophages and a murine model of disseminated candidiasis.
MATERIALS AND METHODS
Strains and culture conditions.
Bacterial and yeast strains were routinely maintained in LB medium at 37°C and yeast extract-peptone-dextrose (YPD) medium at 30°C, respectively, unless otherwise stated. C. glabrata wild-type (wt) and mutant strains were grown either in rich medium (YPD; 1% yeast extract, 2% peptone, 2% dextrose) or in CAA medium (0.67% yeast nitrogen base [YNB] without amino acids, 0.6% Casamino Acids, 2% dextrose) at 30°C with shaking at 200 rpm for different experiments. Logarithmic-phase cells were collected by growing overnight cultures in fresh medium for 4 h at 30°C with shaking at 200 rpm. The wt strain and the Cgmed2Δ mutant transformed with vector alone are referred to as wt/V and Cgmed2Δ/V, respectively. The strains used in this study are listed in Table S1 in the supplemental material.
Generation of Cgmed2Δ deletion strain.
The C. glabrata Cgmed2Δ deletion strain was generated using the homologous recombination-based strategy, as described previously (25), by replacing the CgMED2 open reading frame (ORF) (CAGL0C04477g, 1.1 kb) with a cassette-containing the nat1 gene, which codes for nourseothricin acetyltransferase and confers resistance to nourseothricin. Nourseothricin-resistant yeast transformants were checked for gene replacement by PCR after colony purification. Primers for creation of the deletion strain were designed using Primer3 Plus software (http://bioinfo.ut.ee/primer3-0.4.0/primer3/) and are listed in Table S2 in the supplemental material.
Epitope tagging of proteins.
Proteins were tagged with an epitope by amplifying the full-length gene sequence from the genomic DNA of the C. glabrata Bg2 strain utilizing primers with epitope-coding sequences. To add the c-myc epitope (EQKLISEEDL) at the N terminus or the C terminus, the sequences ATGGAACAAAAACTTATTTCTGAAGAAGATCTG and CAGATCTTCTTCAGAAATAAGTTTTTGTTC were added to the forward and reverse primers for CgMED2 amplification, respectively. To tag the CgPdr1 protein with hemagglutinin (HA) protein epitope YPYDVPDYA at the C terminus, the sequence AGCGTAGTCTGGGACGTCGTATGGGTA was added to the reverse primer. The primer sequences are listed in Table S2 in the supplemental material.
Replacement of CgPDR1 with the CgPDR1L280F GOF allele.
The fragment containing a GOF allele consisting of CgPDR1 with an L-to-F change at position 280 (CgPDR1L280F) flanked by the SAT1 gene (which confers resistance to nourseothricin) was excised by double digesting the pBRK949 plasmid with KpnI and SacI, run on an agarose gel, and purified. Five hundred nanograms of the excised DNA fragment was transformed into yeast strains following standard procedures, and the transformed yeast cells were grown in YPD medium for 4 h at 30°C with constant agitation. Cells were spun down, resuspended in 200 μl YPD medium, and spread plated on YPD agar medium supplemented with 200 μg/ml nourseothricin. Plates were incubated at 30°C for 48 h, and transformants were colony purified. The replacement of CgPDR1 with the CgPDR1L280F-SAT1 allele was confirmed by PCR using CgPDR1 untranslated region (UTR)-specific primers (see Table S2 in the supplemental material).
Growth and phenotypic characterization.
The C. glabrata wild type and mutants were grown overnight in YPD medium at 30°C. For time course analyses, yeast cultures were inoculated in CAA medium containing 16 μg/ml fluconazole at an initial optical density at 600 nm (OD600) of 0.1 in a 96-well plate. At regular time intervals, the culture absorbance at 600 nm was documented and OD600 values were plotted with respect to time. For CFU-based cell viability analysis, appropriate culture dilutions were plated on YPD medium, and the number of viable colonies that appeared on the YPD plates after 1 to 2 days of incubation at 30°C was counted. Phenotypic characterization of the C. glabrata mutants was conducted via serial dilution plate spotting analysis.
qPCR.
Primers for quantitative real-time PCR (qPCR) analysis were designed using Primer3 Plus software and are listed in Table S2 in the supplemental material. Total RNA was isolated using the acid phenol extraction method, and quantitative reverse transcription-PCR was performed as described previously (25). The C. glabrata glyceraldehyde-3-phosphate dehydrogenase (CgGAPDH) gene transcript level was used as a normalization control, and the threshold cycle (CT) values either between the wild-type and mutant cultures or between cultures grown in the presence and absence of fluconazole were compared.
Human macrophage infection assay.
Human THP-1 macrophages were infected with C. glabrata cells at a multiplicity of infection (MOI) of 0.1 as described previously (26). Briefly, 50 μl of suspensions of C. glabrata cells that had been grown overnight, normalized to an OD600 of 0.1, and washed in phosphate-buffered saline (PBS), were added to phorbol 12-myristate 13-acetate (PMA)-activated THP-1 cells in a 24-well plate to attain an MOI of 0.1. At 2 h postinfection, the THP-1 cells were washed thrice with PBS to remove the nonphagocytosed yeast cells and lysed in water to calculate the phagocytosis rate. Appropriate dilutions of lysates were plated on YPD medium, and yeast colonies appearing after 1 to 2 days of incubation at 30°C were counted. The phagocytosis rate was determined by dividing the number of C. glabrata cells harvested from THP-1 macrophages at 2 h postinfection by the number of C. glabrata cells added to THP-1 cells. Fold replication was calculated by dividing the number of C. glabrata cells harvested from THP-1 macrophages at 24 h postinfection by the number of C. glabrata cells internalized by THP-1 cells at 2 h postinfection. As a control, C. glabrata cells were grown in RPMI complete medium, and the CFUs were enumerated by plating appropriate culture dilutions during a 24-h time course experiment.
Adherence assay.
The adherence of C. glabrata cells to Lec2 Chinese hamster ovary (CHO) epithelial cells was measured as described previously (26). Lec2 CHO epithelial cells were seeded in a 24-well tissue culture plate at a seeding density of 5 × 105 cells per well. After 12 h of incubation, Lec2 cells were washed thrice with PBS and fixed in 3.7% paraformaldehyde for 15 min. Fixed Lec2 epithelial cells were washed twice with PBS and incubated with 2 × 106 C. glabrata cells grown in CAA medium containing 200 μCi of 35S (Met/Cys, 65:25) In Vivo Protwin label mix (Jonaki, India) for 16 to 20 h at 30°C. After 30 min, the nonadherent C. glabrata cells were removed with three PBS washes, and adherent yeast cells were collected by lysing Lec2 cells in 5% SDS. The radioactive counts measured in 2 × 106 labeled C. glabrata cells and Lec2 cell lysates were considered the input and output values, respectively. Percent adherence was calculated by dividing the output numbers by the input numbers and multiplying that value by a factor of 100.
Mouse infection assay.
Experiments involving mice were conducted at the CDFD animal facility, Vimta Labs Ltd., Hyderabad, India, in strict accordance with the guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Government of India. The protocol was approved by the Institutional Animal Ethics Committee (IAEC) of Vimta Labs Ltd. (IAEC protocol approval number PCD/CDFD/05). YPD-grown C. glabrata cells (4 × 107; 100-μl volume) were injected into 6- to 8-week-old female BALB/c mice through the tail vein. At 7 days postinfection, mice were sacrificed; the brain, kidneys, liver, and spleen were harvested; and suitable dilutions of organ homogenates were plated on YPD medium containing penicillin and streptomycin. The fungal load per organ was enumerated by counting the number of colonies that appeared on YPD medium after 1 to 2 days of incubation at 30°C.
Zymolyase digestion assay.
The susceptibility of C. glabrata cells to Zymolyase digestion was assessed as described previously (27). Log-phase cells were collected, washed with PBS, and suspended in Tris-HCl (10 mM, pH 7.5). The OD600 of the cultures was adjusted to 1.0, and Zymolyase-20T was added to a final concentration of 50 μg/ml. Every 10 min, the culture OD600 was measured in a multimode plate reader (Synergy H1 hybrid; Biotech). The initial (0-min) absorbance of the cultures (1.0) was considered to be 100%, and results are presented as the percent decrease in the absorbance compared to the initial absorbance.
Immunoblotting.
Western analysis was performed with the log-phase cells as described previously (25). Anti-phospho-p44/42 mitogen-activated protein kinase (MAPK; Thr202/Tyr204) antibody (1:5,000 dilution; catalog number 4370; Cell Signaling Technology, Inc.), anti-P44/42 MAPK (Erk1/2) antibody (1:3,000 dilution; catalog number 9102; Cell Signaling Technology, Inc.), and anti-GAPDH antibody (1:10,000 dilution; catalog number ab22555; Abcam) were used to detect CgSlt2 phosphorylation, CgSlt2 protein levels, and CgGAPDH levels, respectively. Quantification of protein band intensity was performed using ImageJ software.
Other procedures.
Trypan blue exclusion, drug susceptibility testing by Clinical and Laboratory Standards Institute (CLSI)/National Committee for Clinical Laboratory Standards (NCCLS) broth microdilution assay (M27-A), C. glabrata transformation, and rhodamine 6G (R6G) efflux analyses were performed as described previously (25, 28).
RESULTS
Disruption of the CgMED2 ORF impairs the transcriptional activation of the CgPDR1 and CgCDR1 genes.
In a previous screen for mutants that lose viability upon fluconazole exposure, we identified a mutant carrying a Tn7 insertion in the 5′ region of the CgMED2 gene (25). This mutant displayed elevated susceptibility to the azole antifungals fluconazole, clotrimazole, and ketoconazole (25). In the current study, we have attempted to delineate the molecular basis underlying the increased sensitivity of the Cgmed2::Tn7 mutant toward azole antifungal drugs. For this, we first generated the Cgmed2Δ deletion strain, wherein the whole CgMED2 ORF was replaced with the nat1 gene. Consistent with the Tn7 insertion mutant phenotypes, serial dilution spotting analysis revealed attenuated growth of the Cgmed2Δ mutant in the presence of fluconazole, ketoconazole, and clotrimazole (Fig. 1A). Further, the Cgmed2Δ mutant failed to grow in medium supplemented with the protein synthesis inhibitor drug cycloheximide, a known substrate of the fungal drug transporters (Fig. 1A). The elevated sensitivity of the Cgmed2Δ mutant to azoles and cycloheximide compared to that in drug-free medium was verified by determination of the MIC80 (Table 1). Importantly, ectopic expression of CgMED2 from a plasmid restored the growth defects of the Cgmed2Δ mutant in drug-containing medium (Fig. 1A and Table 1). Further, in liquid culture growth analyses, during the exponential phase, the Cgmed2Δ mutant was observed to have a cell density approximately 2-fold less in the fluconazole-supplemented YPD medium than in YPD medium (see Fig. S1 in the supplemental material). Trypan blue (TPB) exclusion analysis to assess the viability of the Cgmed2Δ mutant in medium containing fluconazole revealed that only 30% of the cells were viable (Fig. 1B). In contrast, the survival rate of wild-type (wt) cells was found to be 70% under similar conditions (Fig. 1B). As expected, CgMED2 expression complemented the loss of viability of the Cgmed2Δ mutant upon fluconazole treatment (Fig. 1B).
FIG 1.

The Cgmed2Δ mutant displays impaired activation of the CgCDR1 and CgPDR1 genes upon fluconazole exposure. (A) Spotting assays of the indicated C. glabrata strains. Cells were grown in YPD medium for 16 h, the OD600 was normalized to 1.0, and the cells were spotted in 10-fold serial dilutions on YPD agar plates supplemented or not with 8 μg/ml fluconazole (FLC), 10 μg/ml ketoconazole (KTZ), 15 μg/ml clotrimazole (CTZ), and 1.5 μg/ml cycloheximide (CHX). Plates were imaged after 1 to 2 days of incubation at 30°C. (B) Trypan blue assay-based cell viability enumeration. The indicated C. glabrata strains were grown in either CAA medium (CAA) or CAA medium containing 128 μg/ml fluconazole (FLC) for 24 h at 30°C. Cells were collected, washed with PBS, and stained with 0.4% trypan blue for 10 min. For each strain, a minimum of 300 cells (stained [dead] and unstained [viable]) were counted microscopically, and cell viability data (means of three to five independent analyses ± SEMs) were plotted as the percentage of trypan blue exclusion. Statistical significance was determined using Student's unpaired t test. The significance of the difference between untreated and fluconazole-treated cells is indicated. **, P ≤ 0.01; ***, P ≤ 0.001. (C) qPCR-based quantification of the indicated mRNA levels in wild-type and Cgmed2Δ cells after 4 h treatment with 16 μg/ml fluconazole. Data were normalized to the levels of CgGAPDH mRNA and represent the means ± SEMs of three independent experiments. Statistical significance was determined using Student's unpaired t test. The significance of the difference between fluconazole-treated wild-type and Cgmed2Δ cells is indicated. *, P ≤ 0.05; **, P ≤ 0.01. (D) Measurement of rhodamine 6G efflux in the indicated C. glabrata strains. Cells were grown to log phase in the absence or presence of 16 μg/ml fluconazole and incubated with rhodamine 6G (10 μM) and 2′-deoxyglucose (5 mM) for 2 h at 30°C, followed by a pulse with 2 mM glucose. After 20 min, rhodamine 6G fluorescence was measured at excitation and emission wavelengths of 529 and 553 nm, respectively. Each value represents the mean and standard deviation from at least three independent experiments. The significance of the difference between fluconazole-treated wild-type and Cgmed2Δ cells is indicated. *, P ≤ 0.05; ***, P ≤ 0.001. AU, arbitrary units.
TABLE 1.
MIC80 values of fluconazole, ketoconazole, clotrimazole, and cycloheximide for individual C. glabrata strains
| Strain | MIC80a (μg/ml) |
|||
|---|---|---|---|---|
| FLC | KTZ | CTZ | CHX | |
| wt/V | 16 | 2 | 2 | 1.5 |
| Cgmed2Δ/V | 4 | 1 | 0.25 | 0.19 |
| Cgmed2Δ/CgMED2 | 16 | 2 | 2 | 1.5 |
FLC, fluconazole; KTZ, ketoconazole; CTZ, clotrimazole; CHX, cycloheximide.
C. glabrata cells are known to respond to fluconazole exposure via transcriptional upregulation of the CgPDR1 and CgCDR1 genes (9–15). To examine whether the elevated fluconazole susceptibility of the Cgmed2Δ mutant is due to a defective transcriptional response, we checked the levels of the CgCDR1 and CgPDR1 transcripts in the Cgmed2Δ mutant after 4 h treatment with fluconazole. As shown in Fig. 1C, fluconazole induced a 5- and 2-fold induction of CgCDR1 and CgPDR1 gene expression, respectively, in wt cells. Contrary to this, Cgmed2Δ cells exhibited only a 2-fold increase in CgCDR1 transcript levels (Fig. 1C). Further, no appreciable change in CgPDR1 gene expression in the Cgmed2Δ mutant from that in wt cells was observed upon fluconazole exposure (Fig. 1C). These results are consistent with the lack of fluconazole-mediated induction of CgCDR1 and CgPDR1 in a Cggal11aΔ mutant disrupted for the CgMed15 tail subunit of the Mediator complex (16). Importantly, both the wt and the Cgmed2Δ mutant displayed 3- to 4-fold increases in CgERG11 transcript levels upon fluconazole exposure (Fig. 1C), thereby indicating that transcription of other genes may not be affected upon CgMED2 deletion. Lower CgCDR1 transcript levels correlated well with no detectable efflux of rhodamine 6G (R6G), a CgCdr1 substrate, in the Cgmed2Δ mutant (Fig. 1D). Notably, both wt and reconstituted Cgmed2Δ cells displayed significant extrusion of R6G upon fluconazole exposure (Fig. 1D). Altogether, these data indicate that the inability to properly activate the genes implicated in drug efflux may largely account for the enhanced susceptibility of the Cgmed2Δ mutant to fluconazole.
The PKC-mediated CWI pathway is constitutively active in the Cgmed2Δ mutant.
We have previously shown that the protein kinase C (PKC)-mediated cell wall integrity (CWI) signaling pathway is required for the transcriptional activation of multidrug efflux pumps in response to fluconazole exposure (25). To investigate if a dysfunctional PKC signaling cascade is a cause for the diminished activation of the CgCDR1 and CgPDR1 genes in the Cgmed2Δ mutant, we examined the activation status of CgSlt2, the terminal mitogen-activated protein kinase (MAPK) of the PKC signaling pathway. Immunoblotting using an antibody specific for the phosphorylated form of CgSlt2 (CgSlt2-P) revealed 2-fold higher basal levels of CgSlt2-P in the Cgmed2Δ mutant (Fig. 2A). As expected, a 4- to 6-fold increase in the phosphorylation of CgSlt2 was observed in wt cells after fluconazole treatment (Fig. 2A). Surprisingly, Cgmed2Δ cells were also able to respond to the fluconazole signal via further activation of CgSlt2 (Fig. 2A). Hence, constitutively active CgSlt2 in the Cgmed2Δ mutant may be indicative of cell wall-related defects and/or impaired azole tolerance.
FIG 2.
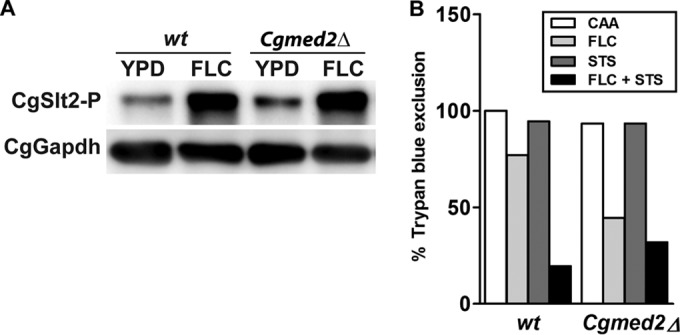
CgSlt2 kinase is constitutively active in the Cgmed2Δ mutant. (A) Representative Western blot analysis of CgSlt2 phosphorylation in the indicated C. glabrata strains. Log-phase cells were harvested after either 4 h growth in YPD medium or treatment with fluconazole (FLC; 16 μg/ml). Protein extracts of each strain were separated by SDS-12% PAGE and immunoblotted with antibodies directed against phosphorylated CgSlt2 and against glyceraldehyde-3-phosphate dehydrogenase (CgGapdh) as the loading control. (B) wt and Cgmed2Δ cells were grown in the indicated medium for 24 h, and cell viability was determined by the trypan blue exclusion assay. Fluconazole (FLC) and staurosporine (STS) were used at concentrations of 128 μg/ml and 2 μg/ml, respectively. Data represent the means of two independent experiments.
Next, to investigate whether abrogation of the PKC signaling pathway counteracts the increased azole susceptibility of the Cgmed2Δ mutant, we assessed the viability of wt and Cgmed2Δ cells in the presence of fluconazole and the PKC inhibitor staurosporine (STS). As shown in Fig. 2B, ∼20% and 75% of wt cells could efflux TPB when grown in medium containing fluconazole plus STS and containing fluconazole, respectively. Conversely, the survival of Cgmed2Δ cells was only slightly lower upon growth in fluconazole- and STS-supplemented medium than upon growth in the fluconazole-containing medium (Fig. 2B). Together, these results suggest that the diminished tolerance of Cgmed2Δ cells to fluconazole could partly be attributed to the constitutively activated PKC-mediated CWI pathway.
Azole resistance conferred by a GOF allele of CgPDR1 is dependent on CgMed2.
The Zn2-Cys6 transcription factor CgPdr1 is a master regulator of CgCDR gene expression in C. glabrata (12). Many gain-of-function (GOF) point mutations which result in high levels of basal multidrug transporter-encoding gene expression and confer drug resistance have been identified in the CgPDR1 gene (17, 29). An apparent requirement for CgMed2 in the transcriptional activation of multidrug efflux pumps upon fluconazole exposure prompted us to investigate whether the constitutive high level of expression of CgCDR1, CgCDR2, and CgPDR1 often observed in GOF CgPDR1 alleles would be abolished in the Cgmed2Δ mutant. To this end, we expressed the hyperactive allele of CgPDR1, which carries the substitution of a cytosine for a guanine at nucleotide 840 in both the wt and the Cgmed2Δ mutant. This base substitution results in the replacement of leucine with phenylalanine (L280F; CgPdr1 of the azole-resistant C. glabrata isolate DSY565) in the putative inhibitory domain of CgPdr1 (17). Expression of this CgPDR1 allele has previously been associated with high levels of azole resistance due to 2- to 60-fold increased levels of transcription of the CgPDR1, CgCDR1, CgCDR2, and CgSNQ2 genes (17). Consistent with earlier reports, wt cells expressing the CgPDR1 allele containing the L280F substitution exhibited robust growth even in the presence of 64 μg/ml fluconazole, while the growth of wt cells carrying vector alone was attenuated in medium containing 16 μg/ml fluconazole (Fig. 3A). This GOF CgPDR1 allele was also able to confer high levels of resistance to ketoconazole, clotrimazole, and cycloheximide (data not shown). Intriguingly, no discernible differences in the growth profiles between Cgmed2Δ cells carrying the vector and those carrying the hyperactive CgPDR1 allele on medium containing different azole compounds and cycloheximide were recorded (Fig. 3A and data not shown). Further, although 2- and 4-fold upregulation of CgPDR1 and CgCDR1 gene expression, respectively, was observed in wt cells expressing the GOF CgPDR1 allele, no such induction was seen in Cgmed2Δ cells carrying the CgPDR1 allele containing the L280F substitution (Fig. 3B). It is noteworthy here that fluconazole exposure led to no further activation of CgCDR1 and CgPDR1 gene expression in wt and Cgmed2Δ cells transformed with the GOF CgPDR1 allele (data not shown). Together, these data accentuate an essential role for CgMed2 in the CgPDR1 GOF allele-mediated transcriptional activation of the CgCDR1 and CgPDR1 genes.
FIG 3.
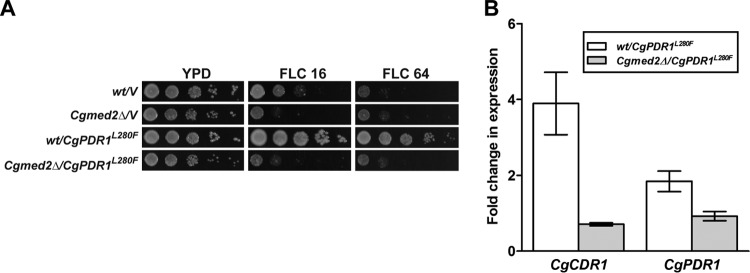
Expression of the GOF CgPDR1 allele did not confer fluconazole resistance in Cgmed2Δ cells. (A) Spotting assays of the indicated C. glabrata strains. The OD600 of overnight cultures grown in YPD medium was normalized to 1.0, and 10-fold serial culture dilutions were spotted onto YPD plates lacking or containing 16 μg/ml fluconazole (FLC 16) and 64 μg/ml fluconazole (FLC 64). Plates were imaged after 1 day of incubation at 30°C. (B) qPCR-based quantification of CgCDR1 and CgPDR1 mRNA levels in the indicated log-phase C. glabrata strains grown in YPD medium. Data (means ± SEMs of 3 to 4 independent experiments) represent the fold change in expression in wt and Cgmed2Δ cells expressing the GOF CgPDR1 allele compared to the level of expression by the respective strains carrying the vector alone.
Two other subunits of the tail module of the RNA polymerase II Mediator complex, CgPgd1 and CgRgr1, are not required for azole resistance conferred by the GOF CgPDR1 allele.
In our previous screens for mutants with altered fluconazole susceptibility profiles, we identified Tn7 insertions in genes coding for RNA polymerase II coactivators CgSrb8, CgRgr1, CgNut1, and CgPgd1, which rendered C. glabrata cells sensitive to fluconazole (25, 28). Of these, CgPGD1 (CgMED3) and CgRGR1 (CgMED14) code for putative components of the tail module of the RNA polymerase II Mediator complex, while CgSRB8 (CgMED12) and CgNUT1 (CgMED5) encode putative components of cyclin-dependent kinase (CDK)/cyclin and the middle module of the RNA polymerase II Mediator complex, respectively. To examine if these RNA polymerase II coactivators are also required for the high levels of azole resistance conferred by the CgPDR1 allele carrying the L280F substitution, we replaced the endogenous CgPDR1 locus with the GOF CgPDR1 allele in the genomes of Cgsrb8::Tn7, Cgrgr1::Tn7, Cgnut1::Tn7 and Cgpgd1::Tn7 mutants. As a control, this exchange of the CgPDR1 allele was also performed in the wt and the Cgmed2::Tn7 mutant. The mutants exhibited a modest increase in susceptibility to fluconazole relative to that of wt cells (Fig. 4). Intriguingly, expression of the hyperactive allele of CgPDR1 led to elevated resistance to azole antifungals in the Cgrgr1::Tn7 and Cgpgd1::Tn7 mutants (Fig. 4). Further, no growth advantage was conferred by the hyperactive allele of CgPDR1 to the Cgsrb8::Tn7, Cgnut1::Tn7, and Cgmed2::Tn7 mutants on fluconazole-supplemented medium (Fig. 4). These results were quite unexpected and indicate differential roles for the tail subunits of the RNA polymerase II Mediator complex in interaction with the CgPdr1 zinc finger transcription factor. Furthermore, these data may imply a prerequisite for functional CgMed2, CgNut1, and CgSrb8 proteins for azole resistance acquired via mutations in the CgPDR1 gene during azole antifungal therapy.
FIG 4.
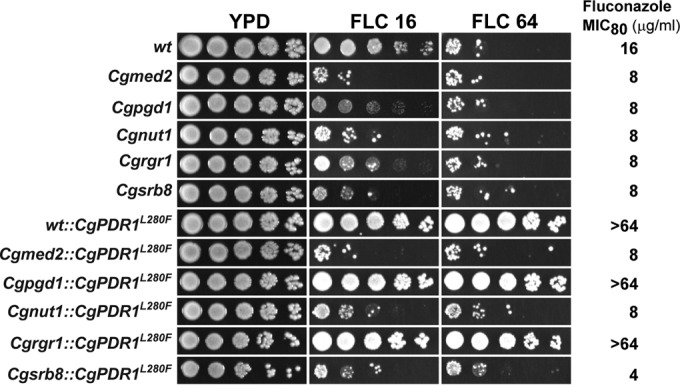
Expression of the GOF CgPDR1 allele conferred fluconazole resistance in Cgpgd1::Tn7 and Cgrgr1::Tn7 mutants disrupted for tail subunits of the RNA polymerase II Mediator complex. The serial dilution spot assay of the indicated C. glabrata strains was performed as described in the legend to Fig. 3A. Plates were imaged after 1 to 2 days of incubation at 30°C. The susceptibility of each strain toward fluconazole was also tested by determination of the fluconazole MIC80s, which are indicated on the right.
Next, to check whether the GOF CgPDR1 allele confers resistance to caspofungin, which inhibits β-glucan synthesis, we examined the viability of wt and fluconazole-sensitive Tn7 insertion mutants expressing either the normal or the hyperactive allele of CgPDR1 after 24 h treatment with caspofungin. Caspofungin exposure led to no significant growth inhibition for the Cgpgd1::Tn7, Cgnut1::Tn7, Cgrgr1::Tn7, and Cgsrb8::Tn7 mutants compared to the growth of wt cells (see Fig. S3 in the supplemental material). However, the Cgmed2::Tn7 and the Cgmed2Δ mutants exhibited exquisite sensitivity to caspofungin and rapidly lost viability (data not shown; see Fig. S3 in the supplemental material). Importantly, expression of the GOF CgPDR1 allele rendered neither the wt nor RNA polymerase II coactivator-defective mutants resistant to caspofungin (see Fig. S3 in the supplemental material). Since caspofungin is not a substrate for multidrug transporters, these data are in accord with the increased efflux of azole antifungals being the basis for the hyperactive CgPDR1 allele-mediated azole resistance.
The Cgmed2Δ mutant exhibits increased adherence to Lec2 epithelial cells.
To more closely investigate the cell wall-related phenotypes of the Cgmed2Δ mutant, we checked its sensitivity to digestion with Zymolyase, which hydrolyzes β-glucan in the cell wall. Altered susceptibility to Zymolyase treatment is indicative of a different cell wall architecture. As shown in Fig. 5A, in contrast to wt cells, the Cgmed2Δ mutant displayed resistance to Zymolyase digestion, which was reversed in Cgmed2Δ cells expressing CgMED2 from a plasmid. During our phenotypic analyses, we noticed that Cgmed2Δ cells always formed a loose pellet after centrifugation, which could be reflective of altered cell-cell interactions. As adherence to host tissues is an important virulence attribute of fungal pathogens (30), we sought to examine the potential of the Cgmed2Δ mutant to adhere to epithelial cells. Interestingly, CAA medium-grown Cgmed2Δ cells exhibited ∼2.5-fold higher levels of adherence to Lec2 ovary epithelial cells than wt cells (Fig. 5B). Importantly, ectopic expression of CgMED2 in the Cgmed2Δ mutant restored the adherence capability to wt levels, thus attributing the increased adherence of Cgmed2Δ cells to Lec2 cells to the lack of the CgMED2 gene (Fig. 5B). Of note, expression of the hyperactive CgPDR1L280F allele has recently been associated with the increased adherence of C. glabrata cells to Lec2 CHO, HeLa, and Caco-2 epithelial cells (31).
FIG 5.
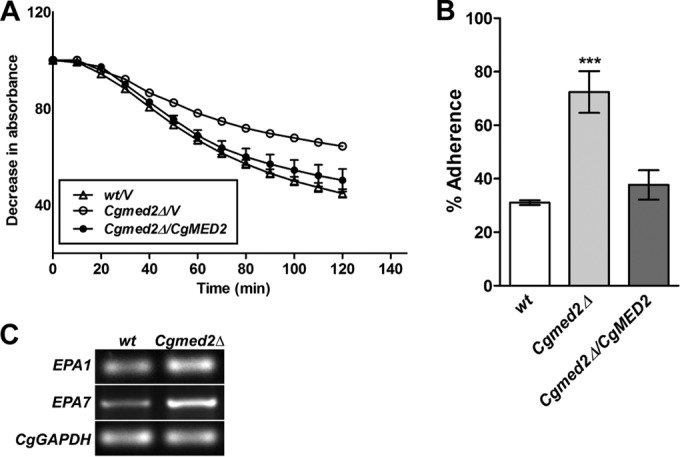
The Cgmed2Δ mutant exhibits increased adherence to Lec2 epithelial cells. (A) The indicated C. glabrata strains were grown to log phase in YPD medium, and cells corresponding to an OD600 of 1.0 were collected. After washing with PBS, cells were suspended in Tris-HCl (10 mM; pH 7.5) and treated with 50 μg/ml Zymolyase. The absorbance of the culture was recorded at 600 nm at the indicated regular time intervals. The initial OD600 of the cultures was considered 100%, and the decrease in the OD600, which represents cell lysis, was expressed as a percentage of the starting OD600. Data are means ± SEMs of three independent experiments. (B) Enumeration of adherence of 35S-labeled yeast cells to Lec2 ovary epithelial cells. Data were analyzed using an unpaired Student's t test (***, P ≤ 0.001) and represent the means ± SEMs of three to four independent experiments. (C) Reverse transcription-semiquantitative PCR analysis of EPA1, EPA7, and CgGAPDH gene expression in the indicated C. glabrata strains. wt and Cgmed2Δ cells were grown for 24 h in YPD medium, followed by inoculation into fresh YPD medium. After 30 min incubation, cells were collected, RNA was isolated, and transcript levels were examined. A representative gel image from three independent experiments with similar results is shown. CgGAPDH gene expression was used to normalize all data.
In vitro epithelial cell adherence in C. glabrata is primarily mediated by the glycosylphosphatidylinositol (GPI)-anchored cell wall adhesin Epa1 (32). Epa1 is a member of a family of about 23 adhesins (30). Of these, many adhesins are encoded by genes located at subtelomeric regions and not expressed under in vitro conditions owing to subtelomeric silencing (30). EPA7 is one such gene whose expression is known to be regulated by the telomere position effect (33). To investigate whether increased adherence of the Cgmed2Δ mutant is due to enhanced transcription of the EPA1 and EPA7 genes, we monitored the expression of the EPA genes via reverse transcription-semiquantitative PCR (RT-PCR) analysis. We observed ∼1.5-fold higher basal levels of EPA1 and EPA7 transcripts in Cgmed2Δ mutant cells than wt cells (Fig. 5C), indicating the possible derepression of EPA1 and EPA7 transcription upon disruption of the CgMED2 gene. These data are in accord with reports from C. albicans and S. cerevisiae wherein Med13 and Med31 Mediator subunits have been shown to act as activators of the adhesin-encoding ALS gene family in C. albicans, while their S. cerevisiae counterparts repress transcription of the FLO genes, which code for cell wall flocculins/adhesins (34).
CgMED2 is required for intracellular proliferation in THP-1 macrophages.
Elevated adherence of the Cgmed2Δ mutant incited us to examine the role of CgMED2 in other virulence strategies of C. glabrata. C. glabrata cells are known to proliferate in mammalian macrophages (26), which represent the first line of host defense against fungal pathogens. To investigate the intracellular behavior of the Cgmed2Δ mutant, we infected macrophages derived from the human monocytic cell line THP-1 with wt and Cgmed2Δ cells and monitored their survival. Both wt and Cgmed2Δ cells were ingested by THP-1 macrophages at a similar rate of 65 to 70% (see Fig. S2A in the supplemental material). Further, consistent with earlier findings (26), wt cells exhibited 6-fold replication in macrophages over a period of 24 h (Fig. 6A). In contrast, no appreciable increase in the number of CFUs of the Cgmed2Δ mutant was recorded over a similar duration of time (Fig. 6A). Notably, ectopic expression of CgMED2 in Cgmed2Δ cells led to wild-type-like 6-fold proliferation in THP-1 cells (Fig. 6A). Further, an inability of the Cgmed2Δ mutant to replicate in THP-1 macrophages was not due to diminished growth under tissue culture conditions, as similar numbers of CFU were obtained when wt and Cgmed2Δ cells were grown in RPMI medium at 37°C in 5% CO2 (see Fig. S2B in the supplemental material). It is noteworthy here that C. glabrata cells expressing the GOF CgPDR1 allele are known to survive and proliferate similarly to wild-type CgPDR1 allele-expressing cells (31).
FIG 6.

Measurement of Cgmed2Δ mutant survival in THP-1 macrophages and murine model of systemic candidiasis. (A) CFU assay-based measurement of replication of the indicated C. glabrata strains in THP-1 macrophages. (B) Fungal burden analysis. Groups of mice (n = 12 to 20) were infected by tail vein injection of 4 × 107 C. glabrata cells, and the indicated organs were harvested at 7 days postinfection. Triangles, number of CFUs recovered from the kidneys, liver, spleen, and brain of individual mice; bars, geometric mean number of CFUs per organ. Statistically significant differences in the numbers of CFU between the wt and the Cgmed2Δ mutant are indicated (**, P ≤ 0.01; ***, P ≤ 0.001; two-tailed Student's unpaired t test).
Next, to examine if CgMed2 is required for survival in vivo, we assessed the fungal burden in the kidneys, livers, spleens, and brains of BALB/c mice infected intravenously with Cgmed2Δ cells. As shown in Fig. 6B, we recovered 5- and 15-fold lower numbers of yeast CFU from the kidneys and brain, respectively, of Cgmed2Δ-infected mice than the wt-infected mice. No statistically significant differences in the numbers of CFU were observed in spleens harvested from mice infected with the wt and the Cgmed2 mutant (Fig. 6B). Quite surprisingly, the hepatic fungal load of Cgmed2Δ-infected mice was 4-fold higher than that of the wt-infected mice (Fig. 6B). The molecular basis underlying this effect is not understood and warrants further closer inspection. Importantly, mice infected with the Cgmed2Δ-complemented strain displayed an organ fungal burden similar to that of the wt-infected mice (Fig. 6B). Taken together, these data implicate CgMed2 in the replication of C. glabrata in human macrophages and point toward an organ-specific role for CgMed2 in survival in a murine model of systemic candidiasis.
DISCUSSION
C. glabrata is emerging to be an important pathogen in clinical settings and is associated with a mortality rate of up to 40% (2). The azole class of drugs is still the mainstay of antifungal therapy in developing countries, primarily owing to cost-effectiveness. Besides high levels of innate and acquired resistance toward azole antifungals, C. glabrata isolates have recently been reported to exhibit cross-resistance to echinocandins which inhibit the synthesis of β-(1,3)-glucan by noncompetitive inhibition of β-(1,3)-glucan synthase (35). Hence, identifying new genes and pathways that are pivotal to antifungal drug tolerance/resistance may aid in the discovery of novel therapeutic strategies. In the current study, we have elucidated the molecular basis of the elevated fluconazole susceptibility of the Cgmed2Δ mutant which is disrupted for a tail subunit of the RNA polymerase II Mediator complex and implicate CgMed2 in antifungal tolerance, adherence, and intracellular replication. We also report for the first time differential roles for the tail subunits of the C. glabrata Mediator complex in the high level of azole resistance conferred by the hyperactive allele of CgPDR1.
The protein kinase C-mediated cell wall integrity pathway, a linear MAPK cascade of a MAPK kinase kinase (Bck1), a pair of redundant MAPK kinases (Mkk1 and Mkk2), and a MAP kinase (Slt2), is activated through phosphorylation of specific serine and threonine residues in S. cerevisiae (36). Fluconazole exposure is known to result in the activation of CgSlt2, which is pivotal to the cellular drug response exemplified by the upregulated multidrug transporters (25). High basal levels of phosphorylated CgSlt2 in the Cgmed2Δ mutant could be reflective of both an altered cell wall structure and a malfunctional CgPdr1 regulon. Intriguingly, combined treatment with STS and fluconazole did not significantly reduce the survival of Cgmed2Δ cells (Fig. 2B). Since STS is an ATP-competitive kinase inhibitor and not selective for PKC, this observed effect could also be attributed to the inhibition of kinases outside the CWI pathway. Of note, disruption of the RhoGAP domain-containing protein CgBem2, a negative regulator of CgRho1, leads to elevated fluconazole sensitivity, constitutively active CgSlt2, and impaired activation of multidrug efflux pumps (25). However, unlike the Cgmed2Δ mutant, expression of the GOF CgPDR1 allele conferred high levels of fluconazole resistance in the Cgbem2Δ mutant (see Fig. S4 in the supplemental material), suggesting that despite the phenotypic similarity, a CgPdr1-regulatory mechanism(s) is probably disparate in these mutants.
Further, in our attempts to examine whether the CgMed2-mediated transcriptional activation of CgCDR1 is via its interaction with CgPdr1 and/or CgSlt2, we tagged CgMed2 with the myc epitope at both the N and C termini and CgPdr1 with the human influenza virus hemagglutinin (HA) epitope at the C terminus (data not shown; see Fig. S5A in the supplemental material). The functionality of N- and C-terminally myc-tagged CgMed2 proteins was verified by their ability to complement the fluconazole sensitivity of the Cgmed2Δ mutant (see Fig. S5B in the supplemental material). In contrast, CgPdr1 tagged with HA at the C terminus was found to be hyperactive and conferred high levels of fluconazole resistance in both the wt and the Cgpdr1Δ mutant (see Fig. S5C in the supplemental material). These data are in accord with the finding of the epitope fusion at the N and C termini leading to the hyperactive CgPdr1 (S. Ferrari and D. Sanglard, unpublished data). Importantly, no conclusive evidence of an interaction between CgPdr1 and CgMed2, CgMed2 and CgSlt2, and CgPdr1 and CgSlt2 was observed in pairwise coimmunoprecipitation experiments (data not shown). Despite this lack of experimental evidence, direct physical interaction between the aforementioned proteins could not be precluded due to the high likelihood of transient CgSlt2 kinase-substrate interaction and impeded CgPdr1 transcription factor-partner interactions owing to the C-terminal HA tag.
Our data suggest that CgMed2 is required for the GOF CgPDR1 allele-mediated transcriptional activation of the multidrug efflux pumps (Fig. 3B and 7). Since another tail subunit of the RNA polymerase II Mediator complex, CgMed15A, is known to bind to CgPdr1 and enhance the transcription of CgCDR genes (16), a simplistic explanation would be an essential requirement for a structurally and stoichiometrically intact tail module of the RNA polymerase II Mediator complex. However, three lines of evidence disfavor this notion. First, disruption of two other tail subunits of the Mediator complex, CgPgd1 and CgRgr1, did not render the hyperactive CgPDR1L280F allele quiescent. Second, azole resistance-conferring CgPDR1 GOF mutations have varied consequences on the expression of major drug efflux pumps (17). For example, contrary to the CgPDR1L280F-mediated upregulation of all three primary drug transporters, CgCdr1, CgCdr2, and CgSnq2, CgPDR1S316I allele expression resulted in the activation of only CgCDR1 (17), alluding to the differential binding of CgPdr1 to PDREs present in the promoter regions of its target genes. Third, CgPdr1-mediated transcription of CgCDR1 has been found to be dependent upon CgGal11A (CgMed15A) in response to fluconazole but not to loss of the mitochondrial genome (15), raising a possibility of CgPdr1-CgCdr1 activation through multiple regulatory circuits. Together, these observations suggest a complex interplay between regulatory components of the drug response network in C. glabrata. Consistent with this notion, the Pdr1 and Pdr3 transcription factors in S. cerevisiae are known to bind to different subunits of the Mediator complex (37). Furthermore, of five tail subunits of the S. cerevisiae Mediator complex, Med2, Med3, and Med15 (Gal11) are known to form a discrete complex which can activate RNA polymerase II transcription, under certain conditions, independently of other Mediator complex components (38). Hence, it will be intriguing to investigate whether CgMed2 constitutes a distinct subcomplex in conjunction with CgPdr1, CgSrb8, and CgNut1, required specifically for azole tolerance/resistance via upregulation of multidrug efflux pumps. It is worth noting that despite several attempts, we could not generate a strain with the CgMED2 and CgPDR1 double deletion, which may be reflective of an essential regulatory role for the CgMed2-CgPdr1 complex in the expression of the genes implicated in cellular growth. Future studies will be directed to address this and examine if drug-dependent and drug-independent functions of CgMed2 are conserved across fungal species.
FIG 7.
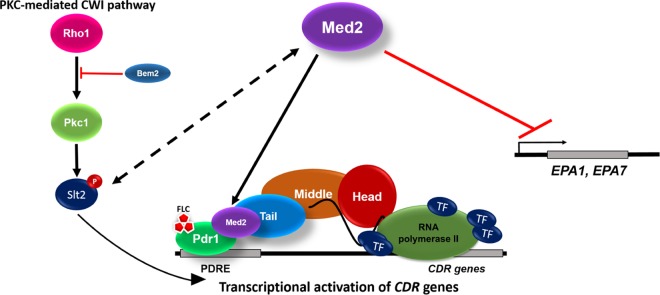
A model for fluconazole-dependent CgCDR gene activation by CgMed2. Upon fluconazole exposure, the activated PKC-mediated cell wall integrity pathway results in phosphorylation of the terminal MAPK, CgSlt2, and CgMed2 upregulates the expression of CDR genes probably through an association of the tail module of the Mediator complex with CgPdr1. In addition, CgMed2 is involved in the transcriptional silencing of the adhesin-encoding genes at subtelomeric regions, thereby regulating adherence to epithelial cells and survival in the mammalian host. TF, transcription factor; EPA1 and EPA7, two adhesin-encoding genes.
An important finding of our study is an essential role for CgMed2 in the intracellular replication of C. glabrata. C. glabrata strains expressing hyperactive CgPDR1 alleles have recently been shown to exhibit a higher virulence potential than wild-type CgPDR1 allele-expressing strains in immunocompetent and immune-suppressed mice (17). Further, expression of the hyperactive CgPDR1L280F allele is known to lead to diminished uptake by bone marrow-derived macrophages (31). Additionally, expression of FLO10 (EPA1) and other flocculin-encoding FLO genes was found to be dependent on CgPdr1 (14, 39). Although the molecular mechanism underlying the hyperadherent and altered survival phenotype of the Cgmed2Δ mutant remains to be deciphered, the inability to maintain subtelomeric silencing (for adhesin expression regulation) and the heterochromatin architecture (for survival in macrophages), two critical determinants of C. glabrata pathogenesis, may largely contribute to the effects observed. Notably, the tail domain of the Mediator complex in S. cerevisiae has recently been found to be essential for the sustenance of telomere silencing and the closed chromatin structure (40). Specifically, deletion of Med16, a tail subunit of the Mediator complex, resulted in increased transcription of the subtelomeric region-resident genes (40). Furthermore, while the Mediator complex has conventionally been considered a transcriptional coactivator complex, acting as a dynamic interface between eukaryotic transcription factors and RNA polymerase II, recent evidence suggests a direct interaction between the Med5 and Med17 constituents of the Mediator complex with the histone H4 tail (41). Consistently, Mediator is required for the stability of the epigenetically repressed (white) and epigenetically active (opaque) morphological states of C. albicans (42). Hence, it is plausible that desilencing of several genes coding for members of the 23 EPA gene family, including EPA1 and EPA7, may account for the hyperadherence to the Lec2 cells observed upon deletion of the CgMED2 gene in C. glabrata (Fig. 5 and 7).
Lastly, although our data are indicative of a role for the hyperactivated PKC MAPK cascade in the regulation of multidrug transporter-encoding genes, it remains to be investigated whether the cellular drug response is effectuated by CgSlt2-mediated phosphorylation of CgPdr1 or tail subunits of the Mediator complex.
In conclusion, we report an essential role for CgMed2, a tail subunit of the RNA polymerase II Mediator complex, in resistance to azole antifungal drugs in C. glabrata.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the Department of Biotechnology, Government of India (BT/PR13289/BRB/10/745/2009, BT/PR5145/MED/29/470/2012, and BT/PR7388/MED/29/650/2012), and by core funds of the Centre for DNA Fingerprinting and Diagnostics, Hyderabad, India (www.cdfd.org.in). S.B. and V.K.S. are the recipients of junior and senior research fellowships of the Department of Biotechnology and Council of Scientific and Industrial Research, respectively, toward the pursuit of a Ph.D. degree of Manipal University.
We are indebted to Suman Komjeti and Jayant Pundalikrao Hole for their help in the mouse experiments. We thank R. Raj Kumar for his help in epitope tagging of the CgMed2 protein.
Footnotes
Published ahead of print 28 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02786-14.
REFERENCES
- 1.Pfaller MA, Diekema DJ. 2007. Epidemiology of invasive candidiasis: a persistent public health problem. Clin. Microbiol. Rev. 20:133–163. 10.1128/CMR.00029-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pfaller MA, Moet GJ, Messer SA, Jones RN, Castanheira M. 2011. Candida bloodstream infections: comparison of species distributions and antifungal resistance patterns in community-onset and nosocomial isolates in the SENTRY Antimicrobial Surveillance Program, 2008-2009. Antimicrob. Agents Chemother. 55:561–566. 10.1128/AAC.01079-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bassetti M, Mikulska M, Viscoli C. 2010. Bench-to-bedside review: therapeutic management of invasive candidiasis in the intensive care unit. Crit. Care 14:244. 10.1186/cc9239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wisplinghoff H, Ebbers J, Geurtz L, Stefanik D, Major Y, Edmond MB, Wenzel RP, Seifert H. 2014. Nosocomial bloodstream infections due to Candida spp. in the USA: species distribution, clinical features and antifungal susceptibilities. Int. J. Antimicrob. Agents 43:78–81. 10.1016/j.ijantimicag.2013.09.005 [DOI] [PubMed] [Google Scholar]
- 5.Montagna MT, Lovero G, Borghi E, Amato G, Andreoni S, Campion L, Lo Cascio G, Lombardi G, Luzzaro F, Manso E, Mussap M, Pecile P, Perin S, Tangorra E, Tronci M, Iatta R, Morace G. 2014. Candidemia in intensive care unit: a nationwide prospective observational survey (GISIA-3 study) and review of the European literature from 2000 through 2013. Eur. Rev. Med. Pharmacol. Sci. 18:661–674 [PubMed] [Google Scholar]
- 6.Yang ZT, Wu L, Liu XY, Zhou M, Li J, Wu JY, Cai Y, Mao EQ, Chen EZ, Lortholary O. 2014. Epidemiology, species distribution and outcome of nosocomial Candida spp. bloodstream infection in Shanghai. BMC Infect. Dis. 14:241. 10.1186/1471-2334-14-241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marie C, White T. 2009. Genetic basis of antifungal drug resistance. Curr. Fungal Infect. Rep. 3:163–169. 10.1007/s12281-009-0021-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pfaller MA, Diekema DJ, Jones RN, Messer SA, Hollis RJ, SENTRY Participants Group 2002. Trends in antifungal susceptibility of Candida spp. isolated from pediatric and adult patients with bloodstream infections: SENTRY Antimicrobial Surveillance Program, 1997 to 2000. J. Clin. Microbiol. 40:852–856. 10.1128/JCM.40.3.852-856.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanglard D, Ischer F, Calabrese D, Majcherczyk PA, Bille J. 1999. The ATP binding cassette transporter gene CgCDR1 from Candida glabrata is involved in the resistance of clinical isolates to azole antifungal agents. Antimicrob. Agents Chemother. 43:2753–2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Izumikawa K, Kakeya H, Tsai HF, Grimberg B, Bennett JE. 2003. Function of Candida glabrata ABC transporter gene, PDH1. Yeast 20:249–261. 10.1002/yea.962 [DOI] [PubMed] [Google Scholar]
- 11.Tscherner M, Schwarzmüller T, Kuchler K. 2011. Pathogenesis and antifungal drug resistance of the human fungal pathogen Candida glabrata. Pharmaceuticals 4:169–186. 10.3390/ph4010169 [DOI] [Google Scholar]
- 12.Tsai HF, Krol AA, Sarti KE, Bennett JE. 2006. Candida glabrata PDR1, a transcriptional regulator of a pleiotropic drug resistance network, mediates azole resistance in clinical isolates and petite mutants. Antimicrob. Agents Chemother. 50:1384–1392. 10.1128/AAC.50.4.1384-1392.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vermitsky JP, Earhart KD, Smith WL, Homayouni R, Edlind TD, Rogers PD. 2006. Pdr1 regulates multidrug resistance in Candida glabrata: gene disruption and genome-wide expression studies. Mol. Microbiol. 61:704–722. 10.1111/j.1365-2958.2006.05235.x [DOI] [PubMed] [Google Scholar]
- 14.Caudle KE, Barker KS, Wiederhold NP, Xu L, Homayouni R, Rogers PD. 2011. Genome-wide expression profile analysis of the Candida glabrata Pdr1 regulon. Eukaryot. Cell 10:373–383. 10.1128/EC.00073-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paul S, Schmidt JA, Moye-Rowley WS. 2011. Regulation of the CgPdr1 transcription factor from the pathogen Candida glabrata. Eukaryot. Cell 10:187–197. 10.1128/EC.00277-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thakur JK, Arthanari H, Yang F, Pan SJ, Fan X, Breger J, Frueh DP, Gulshan K, Li DK, Mylonakis E, Struhl K, Moye-Rowley WS, Cormack BP, Wagner G, Näär AM. 2008. A nuclear receptor-like pathway regulating multidrug resistance in fungi. Nature 452:604–609. 10.1038/nature06836 [DOI] [PubMed] [Google Scholar]
- 17.Ferrari S, Ischer F, Calabrese D, Posteraro B, Sanguinetti M, Fadda G, Rohde B, Bauser C, Bader O, Sanglard D. 2009. Gain of function mutations in CgPDR1 of Candida glabrata not only mediate antifungal resistance but also enhance virulence. PLoS Pathog. 5:e1000268. 10.1371/journal.ppat.1000268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Casamassimi A, Napoli C. 2007. Mediator complexes and eukaryotic transcription regulation: an overview. Biochimie 89:1439–1446. 10.1016/j.biochi.2007.08.002 [DOI] [PubMed] [Google Scholar]
- 19.Guglielmi B, van Berkum NL, Klapholz B, Bijma T, Boube M, Boschiero C, Bourbon HM, Holstege FC, Werner M. 2004. A high resolution protein interaction map of the yeast Mediator complex. Nucleic Acids Res. 11:5379–5391. 10.1093/nar/gkh878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gustafsson CM, Samuelsson T. 2001. Mediator—a universal complex in transcriptional regulation. Mol. Microbiol. 41:1–8. 10.1046/j.1365-2958.2001.02481.x [DOI] [PubMed] [Google Scholar]
- 21.Biddick R, Young ET. 2005. Yeast mediator and its role in transcriptional regulation. C. R. Biol. 328:773–782. 10.1016/j.crvi.2005.03.004 [DOI] [PubMed] [Google Scholar]
- 22.Myers LC, Gustafsson CM, Bushnell DA, Lui M, Erdjument-Bromage H, Tempst P, Kornberg RD. 1998. The Med proteins of yeast and their function through the RNA polymerase II carboxy-terminal domain. Genes Dev. 12:45–54. 10.1101/gad.12.1.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dastidar RG, Hooda J, Shah A, Cao TM, Henke RM, Zhang L. 2012. The nuclear localization of SWI/SNF proteins is subjected to oxygen regulation. Cell Biosci. 2:30. 10.1186/2045-3701-2-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peng J, Zhou JQ. 2012. The tail-module of yeast Mediator complex is required for telomere heterochromatin maintenance. Nucleic Acids Res. 40:581–593. 10.1093/nar/gkr757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Borah S, Shivarathri R, Kaur R. 2011. The Rho1 GTPase-activating protein CgBem2 is required for survival of azole stress in Candida glabrata. J. Biol. Chem. 286:34311–34324. 10.1074/jbc.M111.264671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaur R, Ma B, Cormack BP. 2007. A family of glycosylphosphatidylinositol-linked aspartyl proteases is required for virulence of Candida glabrata. Proc. Natl. Acad. Sci. U. S. A. 104:7628–7633. 10.1073/pnas.0611195104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bairwa G, Kaur R. 2011. A novel role for a glycosylphosphatidylinositol-anchored aspartyl protease, CgYps1, in the regulation of pH homeostasis in Candida glabrata. Mol. Microbiol. 79:900–913. 10.1111/j.1365-2958.2010.07496.x [DOI] [PubMed] [Google Scholar]
- 28.Kaur R, Castaño I, Cormack BP. 2004. Functional genomic analysis of fluconazole susceptibility in the pathogenic yeast Candida glabrata: roles of calcium signaling and mitochondria. Antimicrob. Agents Chemother. 48:1600–1613. 10.1128/AAC.48.5.1600-1613.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsai HF, Sammons LR, Zhang X, Suffis SD, Su Q, Myers TG, Marr KA, Bennett JE. 2010. Microarray and molecular analyses of the azole resistance mechanism in Candida glabrata oropharyngeal isolates. Antimicrob. Agents Chemother. 54:3308–3317. 10.1128/AAC.00535-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaur R, Domergue R, Zupancic ML, Cormack BP. 2005. A yeast by any other name: Candida glabrata and its interaction with the host. Curr. Opin. Microbiol. 8:378–384. 10.1016/j.mib.2005.06.012 [DOI] [PubMed] [Google Scholar]
- 31.Vale-Silva L, Ischer F, Leibundgut-Landmann S, Sanglard D. 2013. Gain-of-function mutations in PDR1, a regulator of antifungal drug resistance in Candida glabrata, control adherence to host cells. Infect. Immun. 81:1709–1720. 10.1128/IAI.00074-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cormack BP, Ghori N, Falkow S. 1999. An adhesin of the yeast pathogen Candida glabrata mediating adherence to human epithelial cells. Science 285:578–582. 10.1126/science.285.5427.578 [DOI] [PubMed] [Google Scholar]
- 33.Castaño I, Pan S-J, Zupancic M, Hennequin C, Dujon B, Cormack BP. 2005. Telomere length control and transcriptional regulation of subtelomeric adhesins in Candida glabrata. Mol. Microbiol. 55:1246–1258. 10.1111/j.1365-2958.2004.04465.x [DOI] [PubMed] [Google Scholar]
- 34.Uwamahoro N, Qu Y, Jelicic B, Lo TL, Beaurepaire C, Bantun F, Quenault T, Boag PR, Ramm G, Callaghan J, Beilharz TH, Nantel A, Peleg AY, Traven A. 2012. The functions of Mediator in Candida albicans support a role in shaping species-specific gene expression. PLoS Genet. 8:e1002613. 10.1371/journal.pgen.1002613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alexander BD, Johnson MD, Pfeiffer CD, Jimenez-Ortigosa C, Catania J, Booker R, Castanheira M, Messer SA, Perlin DS, Pfaller MA. 2013. Increasing echinocandin resistance in Candida glabrata: clinical failure correlates with presence of FKS mutations and elevated minimum inhibitory concentrations. Clin. Infect. Dis. 56:1724–1732. 10.1093/cid/cit136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levin DE. 2005. Cell wall integrity signaling in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 69:262–291. 10.1128/MMBR.69.2.262-291.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shahi P, Gulshan K, Näär AM, Moye-Rowley WS. 2010. Differential roles of transcriptional mediator subunits in regulation of multidrug resistance gene expression in Saccharomyces cerevisiae. Mol. Biol. Cell 21:2469–2482. 10.1091/mbc.E09-10-0899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang F, Sumibcay L, Hinnebusch AG, Swanson MJ. 2004. A triad of subunits from the Gal11/tail domain of Srb mediator is an in vivo target of transcriptional activator Gcn4p. Mol. Cell. Biol. 24:6871–6886. 10.1128/MCB.24.15.6871-6886.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferrari S, Sanguinetti M, Torelli R, Posteraro B, Sanglard D. 2011. Contribution of CgPDR1-regulated genes in enhanced virulence of azole-resistant Candida glabrata. PLoS One 6:e17589. 10.1371/journal.pone.0017589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu X, Liu B, Carlsten JO, Beve J, Nyström T, Myers LC, Gustafsson CM. 2011. Mediator influences telomeric silencing and cellular life span. Mol. Cell. Biol. 31:2413–2421. 10.1128/MCB.05242-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Z, Myers LC. 2012. Med5 (Nut1) and Med17 (Srb4) are direct targets of mediator histone H4 tail interactions. PLoS One 7:e38416. 10.1371/journal.pone.0038416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang A, Liu Z, Myers LC. 2013. Differential regulation of white-opaque switching by individual subunits of Candida albicans mediator. Eukaryot. Cell 12:1293–1304. 10.1128/EC.00137-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.