

Abstract
The effector protein Yersinia outer protein M (YopM) of Yersinia enterocolitica has previously been identified and characterized as the first bacterial cell-penetrating protein (CPP). We found that recombinant YopM (rYopM) enters different eukaryotic cell types and downregulates the expression of several pro-inflammatory cytokines (e.g., tumor necrosis factor-α [TNF-α]) after autonomous translocation. After infection with Y. enterocolitica or transfection of host cells, YopM interacts with isoforms of the two kinases ribosomal S6 protein kinase (RSK) and protein kinase C-related kinase (PRK). This interaction caused sustained RSK activation due to interference with dephosphorylation.
Here we demonstrate by co-immunoprecipitation that rYopM interacts with RSK and PRK following cell-penetration. We show that autonomously translocated rYopM forms a trimeric complex with different RSK and PRK isoforms. Furthermore, we constructed a series of truncated versions of rYopM to map the domain required for the formation of the complex. The C-terminus of rYopM was identified to be essential for the interaction with RSK1, whereas any deletion in rYopM’s leucin-rich repeat domains abrogated PRK2 binding. Moreover, we found that the interaction of cell-penetrating rYopM with RSK led to enhanced autophosphorylation of this kinase at serine 380. Finally, we investigated whether downstream signaling of the trimeric rYopM-RSK/PRK complex modulates the expression of pro-inflammatory TNF-α. Here, we could exclude that interaction with RSK1 and PRK2 is essential for the anti-inflammatory effects of rYopM.
Keywords: anti-inflammatory, bacterial cell-penetrating proteins, pro-inflammatory cytokines, protein kinases, Yersinia outer protein, YopM
Introduction
The genus Yersinia of gram-negative bacteria comprises 11 different species. In addition to Y. pestis, which is infamous for being the causative agent of plague, both Y. pseudotuberculosis and Y. enterocolitica are human pathogens. Infections with the two species usually occur through ingestion of contaminated food or water that typically results in inflammation of glands and lymph nodes partially associated with inflammation of the terminal ileum. Furthermore, infections with Y. enterocolitica are often accompanied with acute enteritis or enterocolitis.1 The pathogenicity of all human pathogenic Yersinia species is mediated by a virulence plasmid, which encodes for both, the type III secretion system (T3SS) and several secreted effector proteins. Among these, a set of so-called Yersinia outer proteins (Yops), namely YopO, YopH, YopM, YopJ, and YopE, was identified. These effector proteins are directly inserted by the Yersinia T3SS during infection into the host cell cytoplasm where they modulate multiple signaling responses. Thereby numerous key immune defensive mechanisms are subverted. For instance, several Yops antagonize phagocytic uptake of Yersinia or the production of pro-inflammatory chemokines and cytokines during infection.2
According to the current model of Yersinia infection, YopM is translocated via the T3SS into the host cell cytoplasm.2 It was shown that YopM is essential for full virulence as ΔyopM mutant strains of Y. enterocolitica revealed a reduced ability to replicate within the infected host.3 Moreover, this mutant was unable to establish a systemic infection in mice.4 Interestingly, a YopM-dependent depletion of NK cells and a significant reduction of pro-inflammatory cytokines in isolated macrophages have been observed in murine models of Y. pestis infection.5 However, thus far it is not known by which mechanism a locally translocated effector protein might be responsible for systemic effects on innate immunity. Interestingly, interactions between YopM and the abundant serum proteins α-thrombin and α1-anti-trypsin have been described suggesting an additional extracellular role of the effector protein (Fig. 1).6,7
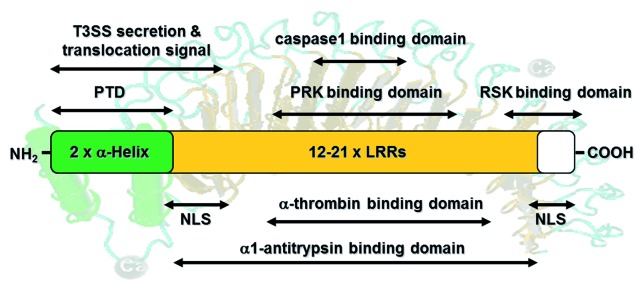
Figure 1. Schematic overview of YopM’s functional domains. YopM consists of two N-terminal α-helices (indicated in green) and 12–20 leucine rich repeats (LRRs; orange). The N-terminal amino acids (aa) encode a secretion signal (aa 1–40) and a translocation signal (aa 41–100). Furthermore, each α-helix harbors a protein transduction domain (PTD) for autonomous translocation of YopM into target cells. Two nuclear localization signals (NLSs) are encoded by LRRs 1–3 and the 32 C-terminal amino acid residues. Interactions with the serum proteins α1-thrombin and α1-antitrypsin depend on conserved regions within the LRRs. YopM proteins from Y. pestis and the Y. pseudotuberculosis YPIII can bind to caspase-1 via a 4 aa loop within its LRR domain. Additionally, it was shown that YopM from Y. pseudotuberculosis interacts with RSK1 via a domain comprising LRRs 12-C and PRK2 via LRRs 6–15 to form a novel trimeric complex. This figure summarizes the literature described in the introduction.
YopM is composed of two N-terminal helices followed by variable numbers of an approximately 20 amino acids-containing leucine-rich repeat (LRR) motif (12–21 LRRs in YopM of different Yersinia strains), thus forming horseshoe-shaped proteins of 42 kDa to 57 kDa (Fig. 1).8-10 A putative significance of these variations for pathogenicity has not been addressed. Furthermore, a short C-terminal tail with unknown conformation is highly conserved among all YopM isoforms.11 In contrast to other Yops, YopM is the only effector protein of Yersinia that apparently does not harbor any known enzymatic activity and whose mode of molecular action is still unknown.1 It has been shown that after translocation into the host cell cytoplasm by the T3SS, YopM traffics to the nucleus via a vesicle-associated pathway.12 Accordingly, two putative nuclear localization signals (NLSs) have been identified within the YopM sequence comprising LRR 1–3 and the 32 C-terminal amino acid (aa) residues. However, they do not resemble any known NLSs (Fig. 1).12 The role of the nuclear localization of this effector protein is still unclear.
Since the discovery of YopM the molecular mechanisms of the effector protein have been studied extensively, and recent studies indicate that YopM proteins of Y. pestis and Y. pseudotuberculosis (YPIII strain) inhibit caspase-1 to promote Yersinia survival.13 The YopM protein of these strains binds to caspase-1 via a 4 aa loop within its LRR domain (Fig. 1) thereby avoiding the activation of the enzyme.13 Moreover, an interaction of YopM with the ribosomal S6 protein kinase 1 (RSK1) and the protein kinase C-related kinase 2 (PRK2) could be identified in several studies.11,14-16 Additionally, it was shown that YopM can also interact with the isoforms RSK2–4 and PRK1/3.15 YopM serves as a scaffolding protein which promotes the autophosphorylation of RSK by blocking the dephosphorylation of its activatory phosphorylation site.15 The sustained activation of RSK subsequently leads to the phosphorylation of PRK.14 Interestingly, an interaction of the two kinases under normal cellular conditions has not been described. Nearly ten years after the discovery of YopM-RSK/PRK-trimers, the substrates of the newly formed kinase complex still remain enigmatic. Moreover, several endogenous substrates of the kinases such as protein kinase B (PKB, also known as Akt) of PRK or BAD, Jun, and CREB of RSK could be excluded.14,15 Thus, it can be assumed that YopM causes a change in the substrate specificity of the kinases. Nevertheless, the interaction of YopM with RSK and PRK appears to be important for Yersinia to promote pathogenesis, tissue colonization, and full virulence.16 Further evidence is provided by the finding that Y. pseudotuberculosis strains expressing YopM mutants, which are unable to interact with either RSK1 (YopM Δ12-C) or PRK2 (YopM Δ6–15), were defective in virulence after intravenous infection of mice.11 The virulence attenuation of the YopM mutants was linked to reduced levels of interleukin-18 (IL-18) and IL-10 in the serum of infected mice compared with those that had been infected with the wild-type strain, suggesting an alteration of the balance of these key cytokines by YopM.11 Although these two kinases are involved in many cellular processes, a link to the observed downregulation of pro-inflammatory cytokines by YopM has not yet been established.
Interestingly, it was shown that recombinant YopM (rYopM) has the capacity to translocate into host cells independently of the T3SS. However, during bacterial infection the major translocation route of YopM is the injection via the T3SS system.17,18 Using truncated versions of YopM, the protein transduction domain (PTD) was identified within the α-helices of the protein (Fig. 1). Although one α-helix is sufficient for membrane transduction, both α-helices together promote more efficient uptake. In addition, this domain was shown to deliver heterologous cargo into host cells as well.17 Recent analyses suggest that the cell-penetrating ability of rYopM is predominantly attributed to escape of the protein from endosomes after initial induction of endocytosis, whereas direct membrane penetration appears to play a minor role.17,18 Once inside the host cell, rYopM is functional and efficiently downregulates the expression of pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukins 12, 15, and 18.19 This feature indicates that autonomously translocated rYopM retains its effect of repressing pro-inflammatory cytokines as it was also observed during Y. pestis infection.5 Since the PTD of rYopM alone was not able to exert such effects, the LRR domains appear to be crucial for the immunomodulatory properties of rYopM.17
In order to explain the controversial effects of YopM on the host cell kinases RSK1 and PRK2 as well as on regulating the expression levels of pro-inflammatory cytokines, we investigated the correlation of the different findings and in particular sought to unravel the involvement of YopM’s LRR domains. In this regard, we were interested to assess whether the interaction of rYopM with RSK1 and PRK2 directly affects the YopM-mediated downregulation of pro-inflammatory cytokines.5,11,17 For this purpose, we initially showed that rYopM derived from Y. enterocolitica (pYV8081) also forms a complex with RSK1 and PRK2 after penetration of target cells. It was necessary to provide this evidence, because the intracellular route of the isolated recombinant effector protein after cell-penetration might differ from YopM injected via the T3SS. After confirmation of the interaction with the two kinases, we constructed a series of LRR-truncated versions of rYopM and analyzed their ability to interact and activate RSK1 and PRK2. Furthermore, the impact of these constructs on expression of pro-inflammatory TNF-α was evaluated.
Results
Cell-penetrating rYopM interacts with RSK and PRK host cell kinases
Previously it was reported that YopM derived from different pathogenic Yersinia spp. interacts with the host kinase families RSK and PRK after transfection of a plasmid carrying the yopM gene or following infection with the pathogen.11,14,15 Recently, we described the ability of the effector protein to enter eukaryotic cells independently of Yersinia’s T3SS.17,18 Thus, the central question of this study was whether recombinant YopM (rYopM) interacts with RSK and PRK following autonomous penetration into the cytosol. To verify this property, HeLa cells were incubated with rYopM (Y. enterocolitica [pYV8081]), and the protein was precipitated using a YopM-specific polyclonal antibody. Western blot analysis showed that the full-length rYopM co-precipitated with RSK1 as well as with PRK2 (Fig. 2A). This finding confirms the formation of the trimeric YopM-RSK1/PRK2 complex after autonomous translocation of rYopM, whereas no association of RSK1 and PRK2 was found in the absence of rYopM (Fig. 2A). In addition, autonomously translocated rYopM interacts with the isoforms RSK2 and PRK1 (Fig. 2C). Using the fusion protein r2αH-GFP for co-immunoprecipitation demonstrated that the N-terminal α-helices of rYopM, which mediate the translocation,17 are not contributing to the complex formation with RSK1 and PRK2 since both kinases were not precipitated together with the fusion construct (Fig. 2B).

Figure 2. After cell-penetration rYopM interacts with RSK1 and PRK2. (A) Immunoprecipitation (IP) of RSK1, PRK2, and rYopM. HeLa cells were incubated with 25 µg/mL rYopM for 1 h at 37 °C. rYopM, RSK1 and PRK2 were precipitated using specific antibodies. Specificity of the IP was monitored using isotype controls (rabbit IgG and goat IgG) as well as protein A/G agarose without IgG. rYopM, RSK1 and PRK2 were co-precipitated in a complex. In absence of rYopM PRK2 was not co-precipitated with RSK1. Different western blots are separated by dashed lines. *heavy chain rabbit IgG. (B) RSK1 and PRK2 are not co-precipitated with r2αH-GFP. HeLa cells were incubated with 25 µg/mL r2αH-GFP for 1 h at 37 °C. GFP was precipitated using a specific antibody. Different western blots are separated by dashed lines. IP, immunoprecipitation; SN, supernatant. (C) rYopM interacts with RSK and PRK isoforms. HeLa and A549 cells were incubated with 25 µg/mL rYopM for 1 h at 37 °C. rYopM was precipitated using a specific antibody. PRK1 and RSK2 are co-precipitated with rYopM. Different western blots are separated by dashed lines. *heavy chain rabbit IgG. IP, immunoprecipitation; SN, supernatant.
The C-terminal tail of rYopM is mediating the interaction with RSK1
The individual LRR motifs of YopM from Y. enterocolitica, Y. pseudotuberculosis, and Y. pestis show a high sequence identity, particularly in the N- and C-terminal region (Fig. 3A; Fig. S1; 95% and 90% identity).20 The size of YopM differs between different strains and serotypes due to a variable number and composition of the LRRs. The central domain of the 20–22-residue long LRR motif (yellow boxes) exhibits additional LRRs in Y. pestis (409 aa) and Y. pseudotuberculosis (529 aa; orange and blue boxes) and deletions in Y. enterocolitica (367 aa; Fig. 3A).

Figure 3. Analysis of domains involved in the interaction of YopM with RSK1 and PRK2. (A) Schematic comparison of YopM from Y. pseudotuberculosis PB1/+, Y. pestis KIM 10+, and Y. enterocolitica 8081. The yellow boxes represent the LRR domains. The central LRR domain has additional LRRs in Y. pestis and Y. pseudotuberculosis. The N-terminal α-helical region (green boxes) and the C-terminal region (red box) flanking the LRRs are highly conserved (95% and 90% identity, respectively) between the different species. The N-terminal domain is necessary for secretion by the T3SS and autonomous translocation across eukaryotic cell membranes. (B) Schematic overview of different truncated YopM versions used in this study. Dark green, α-helices; yellow, LRRs; red, C-terminus; light green, GFP. (C) Cell fractionation and detection of truncated YopMΔLRR1–3, YopMΔLRR4–6, YopMΔLRR7–9, YopMΔLRR10–13, YopMΔC, and rYopMΔNLS1+2 by cellular fractionation of HeLa cells after 30 min incubation with the respective recombinant protein. Note that cell fractionations of YopM, YopMN-239, YopM87-C, and 2αH-GFP have been performed in previous studies.17 CF, cytosolic fraction; MF, membrane fraction. (D) rYopM interacts with RSK1 and PRK2. HeLa cells were incubated with 25 µg/mL rYopM and rYopM deletion proteins for 1 h at 37 °C. Recombinant proteins were precipitated using specific antibodies. rYopM deletion proteins missing the C-terminus do not interact with RSK1. Any deletion in rYopM abrogates interaction with PRK2. Different western blots are separated by dashed lines.
Although the sequences of the various YopM isoforms of human pathogenic Yersinia strains are quite homologous (Fig. S1), contradictory data were published concerning the interactions of YopM from different species with the eukaryotic kinases RSK and PRK. McPhee et al. showed that YopM from Y. pseudotuberculosis interacts with RSK1 via a domain from LRR 12 to the C-terminus and, moreover, that the interaction with PRK2 is mediated by LRRs 6–15.11 In contrast, McCoy et al. showed that YopM from Y. pseudotuberculosis (YPIIIpIB1) did not interact with PRK2 following infection of RAW 264.7 cells and that only the last 6 aa of the C-terminus of YopM from Y. pseudotuberculosis were essential for the interaction with RSK1.16
In order to identify and analyze the domains of rYopM from Y. enterocolitica (pYV8081) that are essential for the interactions with RSK and PRK, we constructed a series of truncated proteins (rYopMΔLRR1–3, rYopMΔLRR4–6, rYopMΔLRR7–9, rYopMΔLRR10–13, rYopMΔC, and rYopMΔNLS1+2 [Fig. 3B]). Along with already described constructs (rYopM, rYopMN-239, rYopM87-C, and r2αH-GFP)17 the new truncated proteins were tested for their interaction with RSK and PRK after autonomous cell-penetration. As all new constructs harbor the N-terminal PTDs of YopM, they are expected to enter target cells independently of the T3SS. To confirm their translocation into the cytosol, HeLa cells were incubated with the recombinant proteins and subsequently fractionated. As shown in Figure 3C, all new truncated rYopM proteins lacking LRRs 1–3, LRRs 4–6, LRRs 7–9, or LRRs 10–13, respectively, were detected in the cytosolic fraction (CF) but not in the membrane fraction (MF) following incubation of HeLa cells for 30 min. Interestingly, rYopM variants with a deletion of either one of the two NLSs (Fig. 3B; rYopMΔLRR1–3 and rYopMΔC) penetrate the host cell membrane (Fig. 3C) and are still able to translocate to the nucleus after 3 h incubation (Fig. S2). This indicates that both NLSs function independently of each other in enabling rYopM to enter the nucleus, whereas the deletion of both NLSs in combination (rYopMΔNLS1+2) abolishes the nuclear localization of the effector protein (Fig. S2).
After confirming the cell-penetrating abilities, the different truncated versions of rYopM were analyzed for co-immunoprecipitation with RSK and PRK. In this regard, RSK1 was found to be co-precipitated with rYopM∆LRR1–3, rYopM∆LRR4–6, rYopM∆LRR7–9, and rYopM∆LRR10–13, indicating that the LRR domain is not required for interaction with RSK1 (Fig. 3D). Interestingly, rYopM∆LRR10–13, which lacks four LRRs proximal to the C-terminus, exhibits a reduced binding to RSK1 suggesting that these LRRs contribute to some extent to RSK1-interaction. However, deletion of the 32 C-terminal amino acids of rYopM (rYopM∆C, rYopM∆NLS1+2, and rYopMN-239) prevents binding to RSK1 entirely (Fig. 3D). This indicates that the C-terminal tail is essential for the interaction of rYopM with RSK1. Surprisingly, all truncated versions of rYopM appeared to lose their ability for interaction with PRK2 (Fig. 3D), possibly due to a (slightly) altered conformation that apparently influences PRK2- but not RSK1-binding.
rYopM enhances autophosphorylation of RSK after autonomous translocation
The family of RSKs is involved in various cellular processes such as translation, apoptosis, and phagocytosis, and regulates cell proliferation, growth, and motility.21,22 Following activation by the extracellular signal regulated kinase 1/2 (ERK1/2) signaling cascade, they, among others, phosphorylate transcription factors such as cAMP response element binding protein (CREB) and serum response factor (SRF).23 YopM has been described to induce hyperactivation of RSK by inhibiting the dephosphorylation of serine 380 (S380).15 However, since in this particular study a YopM isoform with 20 LRRs was introduced into the cells via transfection, the effect on RSK-phosphorylation of a shorter YopM isoform with only 13 LRRs after autonomous translocation remains unclear. In order to confirm the enhanced phosphorylation, we determined the phosphorylation status of RSK at S380 which correlates with kinase activity in the presence or absence of rYopM.24 To this end, HeLa cells were incubated with rYopM under serum starvation conditions to minimize basal ERK1/2 activation, and RSK was precipitated from cell lysates using a specific antibody. For inhibition of the ERK1/2 signaling cascade, the MAPK inhibitor U0126 was added to the cells. In cells that were stimulated with fetal bovine serum (FBS), less phosphorylation of S380 of RSK was detected when U0126 was present, indicating that the inhibitor efficiently reduced basal ERK1/2 activation (Fig. 4A). Strikingly, we found that rYopM enhances phosphorylation of RSK at S380 independently of a Yersinia infection (*2 in Fig. 4A). Furthermore, we conclude that rYopM’s impact on RSK is independent of the upstream kinases MEK and ERK since RSK-phosphorylation was enhanced by rYopM even in the presence of the MAPK inhibitor (*1 in Fig. 4A). So far, altered activation of downstream targets of RSK in response to YopM binding has not been reported. To investigate whether downstream signaling of RSK is influenced by the interaction with rYopM we determined in a first step the amount of phosphoinositide-dependent kinase 1 (PDK1), bound to phosphorylated S380 of RSK, in the presence or absence of rYopM. However, the amount of PDK1 was not influenced by rYopM (Fig. S3A). This suggests that the physiological downstream signaling of RSK is probably not altered. However, other non-physiological targets might be affected by the YopM-RSK/PRK-trimer.
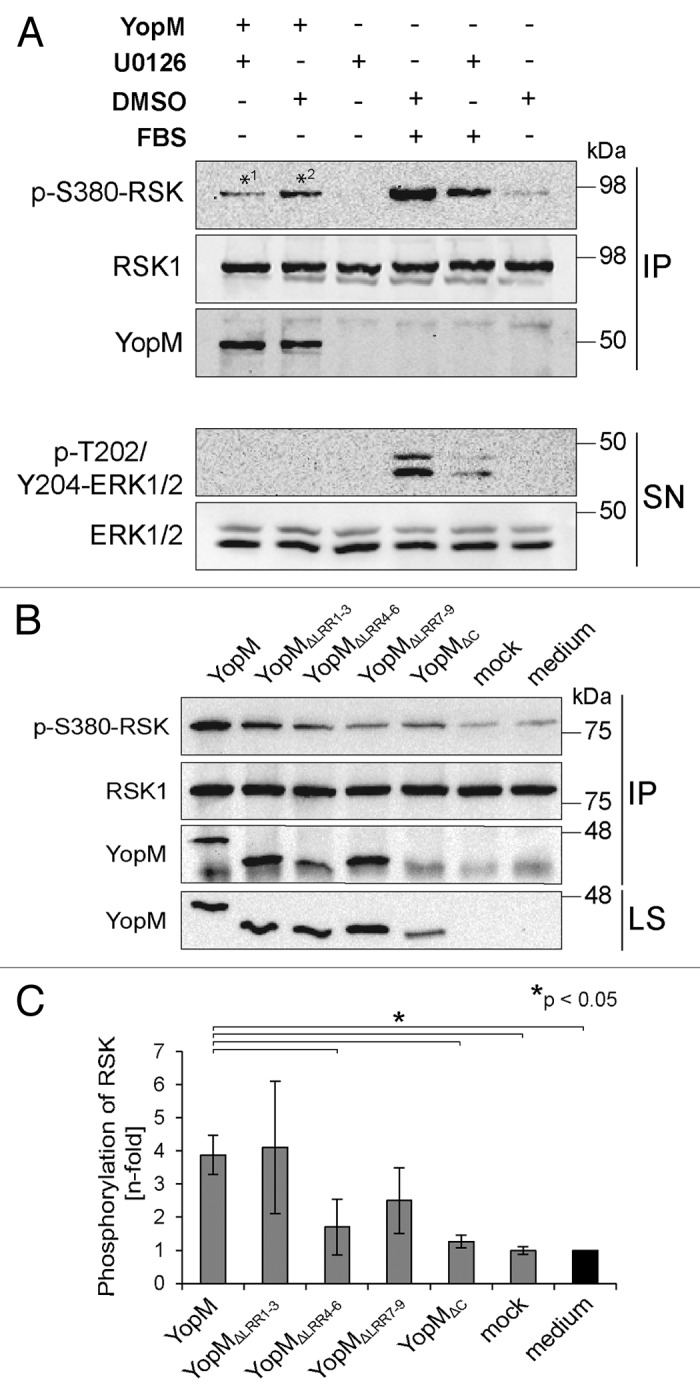
Figure 4. rYopM induces autophosphorylation of RSK. (A) HeLa cells were starved and incubated with 25 µg/mL rYopM for 16 h at 37 °C. For inhibition of ERK1/2 signaling, cells were pre-incubated for 1 h with U0126 or DMSO as control. For stimulation of ERK1/2 signaling, cells were incubated with 20% FBS for 10 min. RSK1 was immunoprecipitated by a specific antibody. Phosphorylation of RSK and ERK1/2 was analyzed by western blotting. rYopM induces phosphorylation of RSK1 at S380 (*2), independent of ERK1/2 (*1). IP, immunoprecipitation; SN, supernatant. (B) LRRs 1–3 of rYopM are not essential for induction of RSK autophosphorylation. HeLa cells were starved and incubated with 25 µg/mL of the indicated recombinant proteins for 16 h at 37 °C. RSK1 was immunoprecipitated by a specific antibody. Phosphorylation of RSK was analyzed by western blotting. rYopM and rYopM∆LRR1–3 but not rYopM∆C induce autophosphorylation of RSK1 at S380. IP, immunoprecipitation; LS, lysate. (C) Densitometric quantification of RSK phosphorylation. rYopM and rYopM∆LRR1–3 induce a 4-fold increase in phosphorylation of RSK. n = 3 independent experiments. The asterisk (*) indicates significant differences (P < 0.05) in the phosphorylation status of RSK normalized to untreated cells.
In addition, we analyzed the impact of rYopM on cell migration and proliferation, two processes that are mediated by RSK and PRK.21,22,25 For this purpose, HeLa cells were incubated in the presence or absence of rYopM and the RSK-interaction-deficient construct rYopM∆C, respectively. The cells did not show a significantly altered proliferation, when incubated either with rYopM or with rYopM∆C (Fig. S3B). In addition, cell migration within the cell-free “window” was not influenced by the presence of rYopM inside the cells (Fig. S3C and D). Taken together, these results suggest that autonomously translocated rYopM from Y. enterocolitica (pYV8081) enhances autophosphorylation of RSK independently of the ERK1/2 signaling cascade but does not affect cell proliferation and motility mediated by RSK.
To further analyze domains of rYopM required for the enhancing effect on RSK-phosphorylation, we investigated the phosphorylation status of RSK at S380 after incubation of HeLa cells with truncated versions of rYopM. Incubation with the full-length rYopM led to significantly enhanced phosphorylation of RSK at S380 compared with the medium and mock controls as well as to the incubation with the interaction-deficient truncated rYopM∆C protein (Fig. 4B and C). Furthermore, interaction of rYopM∆LRR1–3 with RSK induced its phosphorylation to a similar extent (Fig. 4B and C), indicating that the LRRs 1–3 are not crucial for RSK-interaction (Fig. 3D) and for enhanced RSK-phosphorylation. In contrast, incubation of cells with truncated versions lacking either LRRs 4–6 or LRRs 7–9 reduced the enhancing effect on the phosphorylation of RSK (Fig. 4B and C). These results suggest that LRRs 4–9 are essential for rYopM’s effect on sustained RSK-activation. In summary, we could show that LRRs 4–9, but not LRRs 1–3, contribute to the effect of rYopM on enhanced RSK-phosphorylation.
The anti-inflammatory effect of rYopM on expression of TNF-α is independent of its interaction with host cell kinases RSK1 and PRK2
To investigate whether downstream signaling of the trimeric YopM-RSK/PRK complex modulates the expression of pro-inflammatory TNF-α, the truncated versions of rYopM were applied on HL-60-derived macrophages for 3 h. The expression of the cytokine was measured by qRT-PCR. Except for the non-cell-penetrating control protein rYopM87-C, only those versions of rYopM were used, which are still able to penetrate host cell membranes (Fig. 3B and C), as the cell-penetration ability is required for immunomodulation.17 While the control protein rYopM87-C, which does not penetrate cells, did not reduce transcription of TNF-α, the cell-penetrating versions rYopMN-239, rYopMΔLRR4–6, rYopMΔLRR7–9, rYopMΔLRR10–13, and rYopMΔC (Fig. 3B and C) were still able to reduce TNF-α transcription (Fig. 5A). The fact that only the protein rYopMΔLRR1–3 lacking the first NLS was no longer able to reduce transcription of TNF-α (Fig. 5A) indicates that this domain of rYopM is involved in the regulation of TNF-α mRNA transcription. Interestingly, rYopMΔLRR1–3 is the only construct that is cell-penetrating but not immunomodulatory. However, it was excluded in our previous work that the penetration event itself mediates the downregulation of pro-inflammatory cytokines. Hence, the cell-penetrating r2αH-GFP fusion construct, harboring the PTD of YopM, did not influence TNF-α transcription.17

Figure 5. Characterization of domains involved in the immunomodulatory activity of rYopM. (A) HL-60 cells were differentiated into macrophages and incubated with rYopM and its truncated versions for 3 h. After total RNA extraction and cDNA synthesis, qRT-PCR analysis of TNF-α mRNA was performed. The asterisk (*) indicates significant differences (P < 0.05). (B) Knockdown of RSK1 and PRK2 by siRNA. HeLa cells were transfected with siRNA against RSK1 and PRK2. Control cells were treated with Lipofectamine 2000 without addition of siRNA. qRT-PCR analysis of the relative mRNA expression of RSK1 and PRK2 72 h after siRNA transfection revealed that the expression of both genes was reduced by the siRNA by approximately ten times. (C) Western blot analysis of the protein expression of RSK1 and PRK2 after 72 h siRNA transfection. α-tubulin was used as loading control. The knockdown resulted in a greatly reduced protein expression of RSK1 and PRK2. (D) Anti-inflammatory effects of rYopM are independent of its interaction with the two kinases. After knock-down of RSK1 and PRK2 by siRNA, cells were pre-incubated with rYopM for 3 h before stimulation with 10 ng/mL TNF-α for 16 h. Representative results are shown. The asterisk (*) indicates that rYopM reduces TNF-α expression significantly after TNF-α stimulation in comparison to untreated cells (P < 0.05).
As shown in this study, the interaction of rYopM with RSK is mediated by its C-terminus, while the interaction with PRK is dependent on LRR domains (Fig. 3D). Any LRR deletion construct of rYopM unable to bind PRK, except rYopMΔLRR1–3, was still able to downregulate TNF-α expression. Moreover, the C-terminal deletion construct rYopMΔC, which failed to bind both kinases, inhibited the expression of the pro-inflammatory cytokine to the same extent as full-length rYopM. Thus, it is evident that there is no direct correlation between the interaction of rYopM with the kinases RSK and PRK and the inhibitory effects of the recombinant effector protein on cytokine expression (Fig. 5A). This conclusion was confirmed by silencing of RSK and PRK (Fig. 5B–D). Employing siRNA-mediated knock-down of RSK1 and PRK2, we investigated whether the interaction of YopM with the two kinases is essential for the inhibition of TNF-α transcription. For this purpose, the silencing efficiency of specific siRNAs on RSK1 and PRK2 was examined. HeLa cells were cultured in 6-well dishes and transfected with 25 pmol of RSK1-siRNA and 12 pmol PRK2-siRNA per well. The transfection efficiency was analyzed using qRT-PCR and western blotting. Relative mRNA expression of RSK1 and PRK2 upon transfection with the specific siRNA was reduced by about 90% in each case (Fig. 5B). Furthermore, protein expression levels of RSK1 and PRK2 in total cell lysates confirmed the efficient knockdown of the kinases upon transfection with the specific siRNAs (Fig. 5C). Finally, we investigated whether rYopM is still able to inhibit pro-inflammatory cytokine expression after silencing of RSK1 and PRK2. Therefore, the expression of pro-inflammatory TNF-α was induced in siRNA-transfected HeLa cells by incubation with 10 ng/mL TNF-α. Subsequently, the cells were incubated with 25 µg/mL rYopM. To evaluate the effect of rYopM on TNF-α mRNA transcription, qRT-PCR was performed. While TNF-α stimulation showed an induction of TNF-α compared with unstimulated cells, the transcription of the pro-inflammatory cytokine was reduced to its basal level in presence of rYopM (Fig. 5D). Taken together, these results clearly show that interactions with RSK1 and PRK2 are not essential for the anti-inflammatory effects of rYopM.
Discussion
Beside the well-known translocation of YopM into host cells by the Yersinia T3SS, the isolated recombinant YopM effector protein has the ability to enter eukaryotic cells autonomously, without the need of the injection apparatus or any other bacterial factors. Hence, YopM is a cell-penetrating peptide (protein) or CPP. Within the target cell, the cell-penetrating rYopM of Y. enterocolitica (pYV8081) is functional and interacts with various RSK and PRK isoforms. It could be shown that rYopM is essential for the formation of the RSK-PRK kinase complex, since the two kinases do not interact in the absence of the effector protein. The screening of domains which mediate the interaction of the effector with the two kinases revealed that any of the rYopM variants truncated in distinct LRRs used in this study lost the ability to interact with PRK2. This observation corresponds with the data of McPhee et al., showing that a YopM LRR 6–15 deletion variant of Y. pseudotuberculosis was unable to interact with PRK2.11 Presumably, the tertiary structure of the effector protein appears to be of decisive importance for its interaction with this kinase. In contrast, only the short 32 aa long C-terminus of rYopM is essential for the interaction with RSK1. This is in line with previously published data on YopM from Y. pseudotuberculosis.11,16
So far, it is only known that the formation of the YopM/kinase complex during infection with Yersinia is crucial for the virulence of these human pathogenic bacteria.5,16 The mechanism by which the interaction of YopM with the two kinases contributes to virulence is still largely unclear. One possibility to explain this phenomenon could be that the interaction with the kinases influences the subcellular localization of YopM. For example, the kinase PRK1 is recruited to endosomal membranes after activation by the GTPase RhoB.26 Since a “piggy back” transport for YopM has been suggested by Skryzpek et al.,12 activated PRK isoforms might shuttle YopM from the cell periphery toward the nucleus. However, variants of rYopM that did not interact with RSK1 or PRK2 were still able to translocate into the nucleus, indicating a kinase-independent shuttling mechanism. This observation is in accordance with the nuclear localization of mutant YopM constructs from Y. pseudotuberculosis that are unable to bind RSK1, indicating that YopM is not shuttled by RSK1 to the nucleus.16 Moreover, it was shown that the translocation of RSK1 and PRK2 into the nucleus is not influenced by the presence of rYopM (Fig. S4). Thus, translocation of the effector into the nucleus is probably not affected by the two kinases.
Furthermore, the formation of the rYopM/kinase complex does not seem to be directly linked to the suppression of cytokine expression by the effector protein. The non-scaffolding variant rYopMΔC was still able to inhibit the expression of the pro-inflammatory cytokine TNF-α to the same extent as full-length rYopM. Obviously, the first three LRRs of rYopM are involved in this process. The analysis of the effects of different LRR deletion proteins on the transcription of TNF-α in macrophages identified an essential function of the first three LRRs in regulating the transcription of the cytokine. Whether the first three LRRs have a direct effect on the transcription of the cytokine is still unknown and has to be addressed in future investigations.
Finally, knock down of RSK1 and PRK2 by siRNA did not influence the immunomodulatory effect of rYopM on TNF-α transcription, further suggesting that there is no relevance of the rYopM/kinase complex for the modulation of pro-inflammatory cytokines by the effector protein. In this context, McCoy et al. did not publish results regarding a correlation between complex formation and cytokine expression.16 McPhee et al. showed that the interaction of YopM from Y. pseudotuberculosis with RSK and PRK led to an increased expression of the anti-inflammatory cytokine IL-10 in vivo.11 However, these are controversial data, as in several independent studies no evidence was found for increased IL-10 expression in vitro and in vivo.5,17,27,28
Taken together, our data strongly suggest that YopM may have multiple functions on host cell signaling. It modulates innate immune responses regardless of its ability to interact with RSK1 and PRK2. YopM might interact with other so far unknown cellular components during its trafficking resulting in potential pathogenic consequences.
Materials and Methods
Cell culture
HeLa cells were grown in DMEM, low glucose (1 g/L) with l-glutamine (Sigma-Aldrich, D6046) supplemented with 10% fetal bovine serum (FBS), 5% non-essential amino acids, and penicillin/streptomycin. Lung epithelial A549 cells were grown in DMEM/Ham’s F12 with l-glutamine (PAA, E15-813) supplemented with 10% FBS, and penicillin/streptomycin. HL-60 cells were grown in RPMI 1640 with l-glutamine (PAA, E15–840) supplemented with 10% FBS, 5% non-essential amino acids, and penicillin/streptomycin. All cell lines were maintained at 37 °C in a 5% CO2 atmosphere. The growth medium was replaced every 2–3 d.
Antibodies
The following antibodies were used in the experiments described: β-actin (Sigma-Aldrich, A2228), p-S380-RSK (Cell Signaling Technology, 9335), RSK1 (Santa Cruz Biotechnology, sc-231-G), RSK2 (Cell Signaling Technology, 9340), PRK1 (BD Transduction Laboratories, 610687), PRK2 (Cell Signaling Technology, 2612), LSD1 (Cell Signaling Technology, 2139), α-tubulin (Sigma-Aldrich, T8203), PDK1 (Cell Signaling Technology, 3062), ERK1/2 (Cell Signaling Technology, 4696), p-T202/Y204-ERK1/2 (Cell Signaling Technology, 9101), YopM-specific polyclonal rabbit antibody, YopM-specific monoclonal rabbit antibody,29 goat IgG, rabbit IgG, Cy2-labeled goat anti-rabbit (Dianova), HRP-labeled goat anti-rabbit (Dianova, 111-035-003), HRP-labeled goat anti-mouse (Dianova, 115-035-003), HRP-labeled donkey anti-goat (Santa Cruz Biotechnology, sc-2020).
Plasmid construction and protein purification
The generation of YopMN-367 from Y. enterocolitica 8081 strain (serotype O:8, biotype 1B, virulence plasmid pYVe8081), its truncated versions YopMN-239, YopM87-C, YopM∆LRR1–3, YopM∆C, and the 2αH-GFP fusion protein has been described previously.6,17,30 The deletion constructs YopM∆LRR4–6, YopM∆LRR7–9, and YopM∆LRR10–13 with a C-terminal 6xHis tag were constructed by deleting the DNA sequence coding for indicated LRRs by inverse PCR of the plasmid pET-yopM using primer pairs F-yopM∆LRR4–6 and R-yopM∆LRR4–6, F-yopM∆LRR7–9 and R-yopM∆LRR7–9, and F-yopM∆LRR10–13 and R-yopM∆LRR10–13 (Table 1), respectively.31 For the expression and purification of YopM∆NLS1+2 with a C-terminal 6xHis tag primers F-yopM∆NLS1+2 and R-yopM∆NLS1+2 were used to delete the DNA sequence coding for the LRR domain 1–3 (NLS1) amino acids by inverse PCR of the plasmid pET-yopM∆C (NLS2). Expression and purification of the recombinant proteins were performed as described.6 Affinity-purified proteins were dialyzed against PBS and concentrated using Centricon centrifugal filters (Merck Millipore).
Table 1. Primer pairs used in this study.
| Primer | Sequence (5′→ 3′) | Description |
|---|---|---|
| F-yopMΔLRR4–6 | CGGGGTACCC AGTGAAGGAG GTAAATCGGA | “Forward primer” for yopMΔLRR4-6 from Y. enterocolitica pYV8081 with KpnI-linker |
| R-yopMΔLRR4–6 | CGGGGTACCA CTGAGATTCA TGCTGATAAC | “Reverse primer” for yopMΔLRR4-6 from Y. enterocolitica pYV8081 with KpnI-linker |
| F-yopMΔLRR7–9 | CGGGGTACCT ATTATCTCGAT GCATCCAGC | “Forward primer” for yopMΔLRR7-9 aus Y. enterocolitica pYV8081 mit KpnI-linker |
| R-yopMΔLRR7–9 | CGGGGTACCC AAGAATGGCA AGTTTTGTAA | “Reverse primer” for yopMΔLRR7-9 from Y. enterocolitica pYV8081 with KpnI-linker |
| F-yopMΔLRR10–13 | CGGGGTACCG AAGATCTTCG GATGGACTCT | “Forward primer” for yopMΔLRR10-13 from Y. enterocolitica pYV8081 with KpnI-linker |
| R-yopMΔLRR10–13 | CGGGGTACCC AAGTTTGGTG GCAATTCCGA | “Reverse primer” for yopMΔLRR10-13 from Y. enterocolitica pYV8081 with KpnI-linker |
| F-yopMΔNLS1+2 | CGGGGTACCG AATTTCTTGC TGCTGGTAAT | “Forward primer” of yopMΔLRR10-13 from pET: yopMΔC with KpnI-linker |
| R-yopMΔNLS1+2 | GGGGTACCGGC TTGTCGCTCC AGGCAATG | “Reverse primer” of yopMΔLRR10-13 from pET: yopMΔC with KpnI-linker |
Immunoprecipitation
HeLa or A549 cells were plated in 145-mm cell culture dishes and grown for 2–3 d to reach confluence. For co-immunoprecipitation studies, cells were incubated with the different proteins (25 µg/mL) for 1 h at 37 °C. For analysis of RSK auto-phosphorylation, cells were serum starved, and incubated with different recombinant proteins (25 µg/mL) for 16 h at 37 °C. Prior to harvest, cells were pre-incubated for 1 h with U0126 (10 µM, Cell Signaling Technology) for inhibition of ERK1/2 signaling or for 10 min with 20% FBS for stimulation of ERK1/2 signaling. Cells were washed 3 times in ice-cold PBS and lysed in lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 2 mM EDTA, 1% [v/v] Triton X-100, HALT protease and phosphatase inhibitor cocktail [Thermo Scientific, 78441]) followed by sonication (3 × 20 s) and incubation for 30 min at 15 rpm and 4 °C. After centrifugation at 20 000 × g for 30 min at 4 °C, lysates were incubated with 5 µg of antibody and 30 µL of Protein A/G PLUS-Agarose (Santa Cruz, sc-2003) for 16–18 h at 15 rpm and 4 °C. Beads were washed three times with lysis buffer, and precipitated proteins were analyzed by immunoblot analysis using specific antibodies. Experiments were repeated at least three times. Data are expressed as means ± s.d. For significance analysis, a Student t test was performed applying the unpaired two-sided test using Excel (Microsoft) to calculate P values.
Fractionation of eukaryotic cells
HeLa cells were plated in 100-mm cell culture dishes and grown until confluence was reached. For cytosolic and membrane protein extraction, cells were washed with PBS and incubated with recombinant proteins (25 µg/mL) in fresh medium for 30 min at 37 °C. After incubation, cells were washed two times in PBS, followed by an acid buffer (0.2 M glycine, pH 2) wash and PBS wash. Fractionation of the cells was performed as described,19 and protein extracts were analyzed by immunoblot analysis. In order to assess the purity of the fractions, both were analyzed using a specific antibody against cytosolic β-actin. The presence of rYopM in the cytosolic and membrane fractions was determined using a YopM-specific polyclonal rabbit antibody. Immunoblots were analyzed using Lumi-Imager T1 and Lumi Analyst software (Roche Diagnostics).
For cytoplasmic and nuclear protein extraction, cells were washed with PBS and incubated with recombinant protein (25 µg/mL) for 1 h, 3 h, or overnight at 37 °C in fresh medium. Prior to fractionation, cells were washed twice with PBS and protein extracts were prepared using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific) and analyzed by immunoblot analysis. As internal controls for the purity of the fractions, all extracts were analyzed using antibodies for cytosolic α-tubulin and nuclear lysine-specific demethylase 1 (LSD-1). Analysis of the presence of rYopM in the fractions and immunoblot analysis was performed in the same manner as described for cytosolic and membrane fractionation.
Transfection of HeLa cells with siRNA
Transfection with siRNA was performed using Lipofectamine 2000 according to manufacturer’s instructions (Invitrogen, 11668027). Briefly, HeLa cells were cultured in 6-well plates to a confluence of about 50%. The evening before transfection, the culture medium was removed and cells were incubated overnight with the transfection medium Opti-MEM I. For transfection, 25 pmol of RSK1 siRNA or 12 pmol of PRK2 siRNA (RSK1-siRNA ACGGCUACGU GGUAAAGGA; PRK2 GGAUCUUCAA AGGAUCGGA) per batch were separately incubated with 5 µL Lipofectamine 2000 transfection reagent and 250 µL Opti-MEM I for 5 min at RT. Subsequently, the two solutions were combined and incubated for another 20 min at RT. The cells were incubated with 2 mL Opti-MEM I mixed with 500 µL of transfection solution. After 6 h at 37 °C and 5% CO2, the transfection medium was replaced with culture medium and cells were incubated for 72 h before additional experiments were performed.
Cytokine expression analysis by qRT-PCR
For identification of the immune modulatory domain of rYopM, 5 × 106 HL-60 cells were plated into 100-mm cell culture dishes and differentiated into macrophages by incubation with 1 µM TPA (Sigma-Aldrich, P1585) for 24 h. Following a PBS wash, cells were incubated with the different recombinant proteins (25 µg/mL) in fresh medium for 6 h at 37 °C. For induction of TNF-α expression, HL-60 derived macrophages were stimulated with 1 µg/mL LPS from E. coli O111:B4 (Sigma-Aldrich, L3012) for 16 h at 37 °C. Subsequently, cells were washed twice with PBS and total RNA was extracted using the High Pure RNA Isolation Kit (Roche Applied Science, 11828665001) following the manufacturer’s instructions and transcribed into cDNA. Changes in TNF-α expression levels were determined by qRT-PCR with specific primers for human TNF-α using the Universal ProbeLibrary (UPL, Roche Applied Science). As reference the HPRT1 (hypoxanthine-guanine phosphoribosyltransferase 1) housekeeping gene was used. PCR was performed using the LightCycler amplification and detection system (Roche Diagnostics) in a Lightcycler 1.5 (Roche Applied Science). Data were analyzed with the Roche quantification program software (version 3.5, Roche Applied Science) and expressed as means ± s.d. For significance analysis, a Student t test was performed applying the unpaired two-sided test using Excel (Microsoft) to calculate P values.
Cell migration assay
For studying cell migration the cell-free “window” assay was performed as described.32 Briefly, 1 × 105 HeLa cells were seeded in each well of a 48-well plate coated with extracellular matrix (ECM) (Tebu-bio) and containing a 1-mm steel plate to produce the cell-free “window”. Following 3 h incubation at 37 °C, the steel plate was removed and cells were washed once with PBS and incubated for 24 or 48 h at 37 °C in presence or absence of rYopM (25 µg/mL). Prior to analysis, cells were washed three times with PBS and stained with Wright staining solution (0.3% Wright stain in methanol) for 3 min. Subsequently, the staining solution was diluted 1:3 by adding water followed by additional incubation for 7 min. Cells were washed three times with water and analyzed using an inverse microscope (Axiovert 100, Carl Zeiss).
Cell proliferation assay
Cell proliferation was analyzed using the Cell Proliferation ELISA, BrdU (colorimetric) (Roche Applied Science, version 16, 11647229001), following the manufacturer’s instructions. Briefly, 4 × 103 HeLa cells were seeded in each well of a 96-well plate and incubated for 48 h with the indicated recombinant proteins (25 µg/mL and 50 µg/mL, respectively), performing each condition in triplicate. Cells were incubated with BrdU, anti-BrdU-POD antibody and substrate solution and the reaction product was quantified by measuring the absorbance at 370 nm using a scanning multiwell spectrophotometer (SpectraMax M2, Molecular Devices). Values were correlated with cells grown in absence of recombinant protein and expressed as mean percentage of proliferating cells ± s.d. For significance analysis, a Student t test was performed applying the unpaired two-sided test using Excel (Microsoft) to calculate P values.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This study was supported in part by a grant of the Deutsche Forschungsgemeinschaft (DFG, PA689/13-1), the SFB1009 (TP B03), by the DFG Graduiertenkolleg GRK 1409, and by a grant of the Innovative Medizinische Forschung (IMF) (I-RÜ11106). This study is part of the PhD thesis of S.H. We would like to thank the group of Infectiology for critical reading and useful comments on the manuscript.
Glossary
Abbreviations:
- aa
amino acids
- BAD
Bcl-2-associated death promoter protein
- CF
cytosolic fraction
- CREB
cAMP response element binding protein
- CPP
cell-penetrating peptide
- ERK1/2
extracellular signal regulated kinase 1/2
- FBS
fetal bovine serum
- GFP
green fluorescent protein
- IL
interleukin
- IP
immunoprecipitation
- LRR
leucine-rich repeat
- LS
lysate
- MAPK
mitogen-activated protein kinase
- MF
membrane fraction
- NLS
nuclear localization signal
- PDK1
phosphoinositide-dependent kinase 1
- PKB(Akt)
protein kinase B
- PRK
protein kinase C-related kinase
- PTD
protein transduction domain
- qRT-PCR
quantitative real-time polymerase chain reaction
- RSK
ribosomal S6 protein kinase
- rYopM
recombinant YopM
- SD
standard deviation
- siRNA
small interfering ribonucleic acid
- SN
supernatant
- SRF
serum response factor
- T3SS
type III secretion system
- TNF-α
tumor necrosis factor-α
- YopM
Yersinia outer protein M
- Yops
Yersinia outer proteins
References
- 1.Viboud GI, Bliska JB. Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu Rev Microbiol. 2005;59:69–89. doi: 10.1146/annurev.micro.59.030804.121320. [DOI] [PubMed] [Google Scholar]
- 2.Cornelis GR. The Yersinia Ysc-Yop ‘type III’ weaponry. Nat Rev Mol Cell Biol. 2002;3:742–52. doi: 10.1038/nrm932. [DOI] [PubMed] [Google Scholar]
- 3.Mulder B, Michiels T, Simonet M, Sory MP, Cornelis G. Identification of additional virulence determinants on the pYV plasmid of Yersinia enterocolitica W227. Infect Immun. 1989;57:2534–41. doi: 10.1128/iai.57.8.2534-2541.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trülzsch K, Sporleder T, Igwe EI, Rüssmann H, Heesemann J. Contribution of the major secreted yops of Yersinia enterocolitica O:8 to pathogenicity in the mouse infection model. Infect Immun. 2004;72:5227–34. doi: 10.1128/IAI.72.9.5227-5234.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kerschen EJ, Cohen DA, Kaplan AM, Straley SC. The plague virulence protein YopM targets the innate immune response by causing a global depletion of NK cells. Infect Immun. 2004;72:4589–602. doi: 10.1128/IAI.72.8.4589-4602.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heusipp G, Spekker K, Brast S, Fälker S, Schmidt MA. YopM of Yersinia enterocolitica specifically interacts with alpha1-antitrypsin without affecting the anti-protease activity. Microbiology. 2006;152:1327–35. doi: 10.1099/mic.0.28697-0. [DOI] [PubMed] [Google Scholar]
- 7.Hines J, Skrzypek E, Kajava AV, Straley SC. Structure-function analysis of Yersinia pestis YopM’s interaction with alpha-thrombin to rule on its significance in systemic plague and to model YopM’s mechanism of binding host proteins. Microb Pathog. 2001;30:193–209. doi: 10.1006/mpat.2000.0424. [DOI] [PubMed] [Google Scholar]
- 8.Evdokimov AG, Anderson DE, Routzahn KM, Waugh DS. Overproduction, purification, crystallization and preliminary X-ray diffraction analysis of YopM, an essential virulence factor extruded by the plague bacterium Yersinia pestis. Acta Crystallogr D Biol Crystallogr. 2000;56:1676–9. doi: 10.1107/S0907444900013378. [DOI] [PubMed] [Google Scholar]
- 9.Kobe B, Kajava AV. The leucine-rich repeat as a protein recognition motif. Curr Opin Struct Biol. 2001;11:725–32. doi: 10.1016/S0959-440X(01)00266-4. [DOI] [PubMed] [Google Scholar]
- 10.Boland A, Havaux S, Cornelis GR. Heterogeneity of the Yersinia YopM protein. Microb Pathog. 1998;25:343–8. doi: 10.1006/mpat.1998.0247. [DOI] [PubMed] [Google Scholar]
- 11.McPhee JB, Mena P, Bliska JB. Delineation of regions of the Yersinia YopM protein required for interaction with the RSK1 and PRK2 host kinases and their requirement for interleukin-10 production and virulence. Infect Immun. 2010;78:3529–39. doi: 10.1128/IAI.00269-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skrzypek E, Cowan C, Straley SC. Targeting of the Yersinia pestis YopM protein into HeLa cells and intracellular trafficking to the nucleus. Mol Microbiol. 1998;30:1051–65. doi: 10.1046/j.1365-2958.1998.01135.x. [DOI] [PubMed] [Google Scholar]
- 13.LaRock CN, Cookson BT. The Yersinia virulence effector YopM binds caspase-1 to arrest inflammasome assembly and processing. Cell Host Microbe. 2012;12:799–805. doi: 10.1016/j.chom.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McDonald C, Vacratsis PO, Bliska JB, Dixon JE. The yersinia virulence factor YopM forms a novel protein complex with two cellular kinases. J Biol Chem. 2003;278:18514–23. doi: 10.1074/jbc.M301226200. [DOI] [PubMed] [Google Scholar]
- 15.Hentschke M, Berneking L, Belmar Campos C, Buck F, Ruckdeschel K, Aepfelbacher M. Yersinia virulence factor YopM induces sustained RSK activation by interfering with dephosphorylation. PLoS One. 2010;5:5. doi: 10.1371/journal.pone.0013165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCoy MW, Marré ML, Lesser CF, Mecsas J. The C-terminal tail of Yersinia pseudotuberculosis YopM is critical for interacting with RSK1 and for virulence. Infect Immun. 2010;78:2584–98. doi: 10.1128/IAI.00141-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rüter C, Buss C, Scharnert J, Heusipp G, Schmidt MA. A newly identified bacterial cell-penetrating peptide that reduces the transcription of pro-inflammatory cytokines. J Cell Sci. 2010;123:2190–8. doi: 10.1242/jcs.063016. [DOI] [PubMed] [Google Scholar]
- 18.Scharnert J, Greune L, Zeuschner D, Lubos ML, Alexander Schmidt M, Rüter C. Autonomous translocation and intracellular trafficking of the cell-penetrating and immune-suppressive effector protein YopM. Cell Mol Life Sci. 2013;70:4809–23. doi: 10.1007/s00018-013-1413-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Michgehl S, Heusipp G, Greune L, Rüter C, Schmidt MA. Esp-independent functional integration of the translocated intimin receptor (Tir) of enteropathogenic Escherichia coli (EPEC) into host cell membranes. Cell Microbiol. 2006;8:625–33. doi: 10.1111/j.1462-5822.2005.00655.x. [DOI] [PubMed] [Google Scholar]
- 20.Benabdillah R, Mota LJ, Lützelschwab S, Demoinet E, Cornelis GR. Identification of a nuclear targeting signal in YopM from Yersinia spp. Microb Pathog. 2004;36:247–61. doi: 10.1016/j.micpath.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 21.Woo MS, Ohta Y, Rabinovitz I, Stossel TP, Blenis J. Ribosomal S6 kinase (RSK) regulates phosphorylation of filamin A on an important regulatory site. Mol Cell Biol. 2004;24:3025–35. doi: 10.1128/MCB.24.7.3025-3035.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanimura S, Hashizume J, Kurosaki Y, Sei K, Gotoh A, Ohtake R, Kawano M, Watanabe K, Kohno M. SH3P2 is a negative regulator of cell motility whose function is inhibited by ribosomal S6 kinase-mediated phosphorylation. Genes Cells. 2011;16:514–26. doi: 10.1111/j.1365-2443.2011.01503.x. [DOI] [PubMed] [Google Scholar]
- 23.Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–62. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- 24.Frödin M, Antal TL, Dümmler BA, Jensen CJ, Deak M, Gammeltoft S, Biondi RM. A phosphoserine/threonine-binding pocket in AGC kinases and PDK1 mediates activation by hydrophobic motif phosphorylation. EMBO J. 2002;21:5396–407. doi: 10.1093/emboj/cdf551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lachmann S, Jevons A, De Rycker M, Casamassima A, Radtke S, Collazos A, Parker PJ. Regulatory domain selectivity in the cell-type specific PKN-dependence of cell migration. PLoS One. 2011;6:e21732. doi: 10.1371/journal.pone.0021732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Symons M, Rusk N. Control of vesicular trafficking by Rho GTPases. Curr Biol. 2003;13:R409–18. doi: 10.1016/S0960-9822(03)00324-5. [DOI] [PubMed] [Google Scholar]
- 27.Auerbuch V, Isberg RR. Growth of Yersinia pseudotuberculosis in mice occurs independently of Toll-like receptor 2 expression and induction of interleukin-10. Infect Immun. 2007;75:3561–70. doi: 10.1128/IAI.01497-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bubeck SS, Cantwell AM, Dube PH. Delayed inflammatory response to primary pneumonic plague occurs in both outbred and inbred mice. Infect Immun. 2007;75:697–705. doi: 10.1128/IAI.00403-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rüter C, Silva MR, Grabowski B, Lubos ML, Scharnert J, Poceva M, von Tils D, Flieger A, Heesemann J, Bliska JB, et al. Rabbit monoclonal antibodies directed at the T3SS effector protein YopM identify human pathogenic Yersinia isolates. Int J Med Microbiol. 2014;304:444–51. doi: 10.1016/j.ijmm.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 30.Rüter C, Hardwidge PR. ‘Drugs from bugs’: bacterial effector proteins as promising biological (immune-) therapeutics. FEMS Microbiol Lett. 2014;351:126–32. doi: 10.1111/1574-6968.12333. [DOI] [PubMed] [Google Scholar]
- 31.Jones DH, Howard BH. A rapid method for recombination and site-specific mutagenesis by placing homologous ends on DNA using polymerase chain reaction. Biotechniques. 1991;10:62–6. [PubMed] [Google Scholar]
- 32.Samson T, Smyth N, Janetzky S, Wendler O, Müller JM, Schüle R, von der Mark H, von der Mark K, Wixler V. The LIM-only proteins FHL2 and FHL3 interact with alpha- and beta-subunits of the muscle alpha7beta1 integrin receptor. J Biol Chem. 2004;279:28641–52. doi: 10.1074/jbc.M312894200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.