

Abstract
The mitochondrial metabolism of β cells is thought to be highly specialized. Its direct comparison with other cells using isolated mitochondria is limited by the availability of islets/β cells in sufficient quantity. In this study, we have compared mitochondrial metabolism of INS1E/β cells with other cells in intact and permeabilized states. To selectively permeabilize the plasma membrane, we have evaluated the use of perfringolysin-O (PFO) in conjunction with microplate-based respirometry. PFO is a protein that binds membranes based on a threshold level of active cholesterol. Therefore, unless active cholesterol reaches a threshold level in mitochondria, they are expected to remain untouched by PFO. Cytochrome c sensitivity tests showed that in PFO-permeabilized cells, the mitochondrial integrity was completely preserved. Our data show that a time-dependent decline of the oligomycin-insensitive respiration observed in INS1E cells was due to a limitation in substrate supply to the respiratory chain. We predict that it is linked with the β cell-specific metabolism involving metabolites shuttling between the cytoplasm and mitochondria. In permeabilized β cells, the Complex l-dependent respiration was either transient or absent because of the inefficient TCA cycle. The TCA cycle insufficiency was confirmed by analysis of the CO2 evolution. This may be linked with lower levels of NAD+, which is required as a co-factor for CO2 producing reactions of the TCA cycle. β cells showed comparable OxPhos and respiratory capacities that were not affected by the inorganic phosphate (Pi) levels in the respiration medium. They showed lower ADP-stimulation of the respiration on different substrates. We believe that this study will significantly enhance our understanding of the β cell mitochondrial metabolism.
Keywords: Mitochondrial metabolism, Oxidative phosphorylation, OxPhos, Respiratory chain, Perfringolysin-O, Respirometry
Introduction
Impaired mitochondrial metabolism is implicated in numerous diseases including diabetes [1]. The failure of pancreatic β cells is considered to be the primary cause of Type 2 Diabetes [2]. One of the fundamental properties of β cells is their ability to appropriately release insulin in the blood stream in response to a wide range of glucose levels (3–20mM). The insulin secretion is linked with glucose metabolism via glycolysis, the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OxPhos) [3]. Fuel-stimulated insulin secretion is dependent on respiratory chain function [4]. Glucose-induced respiration appears to be a good predictor of the functional competence of pancreatic islets/β cells [5,6]. The absolute dependence of insulin secretion on redox shuttles indicates that β cell respiratory activity may be primarily supported by cytosolic NADH shuttling to mitochondria [7,8]. This may be essential for sustained glycolysis because of the very low level of lactate dehydrogenase activity in β cells [9]. In β cells, pyruvate cycles between the cytoplasm and mitochondria. This occurs via pyruvate/citrate, pyruvate/malate, pyruvate/iso-citrate/α-ketoglutarate and pyruvate/ phosphoenol pyruvate shuttles that are implicated in regulating insulin secretion [10–14]. Exits of citrate, iso-citrate and malate are expected to reduce NADH production within mitochondria. Therefore, β cell respiratory chain activity may depend on redox shuttles function. Alternatively the TCA cycle could be negatively regulated to facilitate metabolites export to cytosol.
The majority of cellular respiration supports OxPhos, which is carried out by multimenc enzyme Complexes (I to V) with the help of electron donors (NADH and FADH2) and electron carriers (ubiquinone and cytochrome c) [15,16]. Commonly, OxPhos is assessed using isolated mitochondria [17]. However, isolating mitochondria from a limited number of islets/β cells is not feasible. Further, isolation procedures may alter mitochondrial functions due to change in the microenvironment [18,19]. Therefore, respirometry methods using intact cells coupled with microplate-based respirometry offer excellent alternatives to isolated mitochondria particularly when the sample size is limited [5,6,20–22]. While assays with intact cells permit analyses of key bio-energetic features such as respiratory capacity, ATP-turnover rate, and proton (H+) leak, the permeability barrier of the plasma membrane to ADP and respiratory substrates limits the analyses of the spare respiratory and OxPhos capacities [16,23,24]. Analyses of direct vs. indirect effects of the compounds of interest on the respiratory chain are also not possible using intact cells. Selective permeabilization of the plasma membrane can overcome this limitation. Cholesterol-dependent detergents such as digitonin and sapponins have been commonly used to permeabilize the plasma membrane for mitochondrial function assays [21,25–28]. In our experience, the detergent-based assays are not applicable across different cell types under similar conditions.
In this study, we have evaluated the use of perfringolysin- O (PFO), a prototypical cholesterol-dependent cytolysin, to permeabilize β cells and enable study of their mitochondrial metabolism with minimal perturbations [29]. Cholesterol-dependent cytolysins are 50–70 kDa size proteins that form large ring- and arc shaped homo-oligomeric complexes that perforate plasma membranes of eukaryotic cells. Transmembrane pores formed by PFO are approximately 250Å in diameter and allow the passage of large molecules such as antibodies, β-amylase, and thyroglobulin [29]. Therefore, cells permeabilized with PFO will permit respiratory assays following supplementation with metabolic substrates and cytochrome c to test functional integrity of mitochondria while excluding the effects of glycolysis. We have compared the functional integrity of mitochondria in PFO- vs. digitonin-permeabilized cells. Our data show that cell permeabilization using PFO preserves mitochondrial integrity and that uniform conditions can be applied to compare mitochondrial metabolism of β cells with other cell types. In association with the intact cell respirometry, the PFO-based assays were employed to compare the mitochondrial metabolism of INS1E cell line, a commonly used β cell model, with others such as HEK293, a commonly used normal human cell line [30,31]. A few other cell types were also used for specific comparisons. The following parameters were investigated: (i) relative respiratory coupling on different substrates, (ii) stability of the oligomycin-insensitive respiration, (iii) differences in Complex I function, (iv) CO2 production, (v) relationship between the OxPhos and respiratory capacities, and (vi) the effects of inorganic phosphate (Pi) on respiration.
Materials and Methods
Reagents
Rotenone and digitonin were procured from Calbiochem. All other reagents were obtained from Sigma unless otherwise specified.
Preparation of functional PFO
The cDNAs encoding native PFO with wild type sequence (nPFO) and a cysteine-459 to alanine mutant (rPFO) with His6-tag at N-terminus were conditionally expressed in Escherichia coli strain BL21-DE3 and affinity purified [32]. The N-terminus of PFO does not affect protein structure or function [33]. After purification, PFOs were stored in a buffer containing 50 mM HEPES pH 7.5, 100 mM NaCl, and 10% (v/v) glycerol in the presence or absence of 5 mM DTT [(2S,3S)-l,4-bissulfanylbutane-2,3-diol]. The nPFO was stored with DTT to prevent oxidation of Cys459. Proteins were kept at −80°C until used. Their concentration was calculated using a molar absorptivity (ε280) of 74260 cm-1 M-1 [33]. No difference was noted in the respiratory response of cells permeabilized with nPFO vs. rPFO (data not shown). Unless otherwise specified the term PFO will be used for both nPFO and rPFO.
Cells and culture conditions
The rat insulinoma INS1E cell line was a generous gift from Dr. Martin Brand (Buck Institute for Research on Aging, Novato, CA) [20,30]. The cells were grown in RPMI1640 medium (Mediatech), which was supplemented with 11.1mM of glucose, 10% fetal bovine serum (FBS), 1 mM HEPES (Invitrogen), and 50 μM of β-mercapto ethanol (2-Sulfanylethan-1-ol). For starvation, cells were incubated for 24 hour in the above medium containing 4 mM (instead of 11.1 mM) glucose with 1 mM Na-pyruvate. The human embryonic kidney (HEK293) and hepatic (HepG2) cell lines were obtained from American Type Culture Collection (Manassas, VA). Cells were grown in DMEM medium (Invitrogen) supplemented with 10% FBS (Invitrogen), 1% nonessential amino acids (Mediatech), and 1% Pen Strep (Invitrogen). Primary astrocytes were prepared from embryonic day 18 (E18) cortical tissue obtained from Brain Bits LLC as per their protocol, grown and expanded in NbASTRO medium. All cells were grown at 37°C in a humidified atmosphere of 5% CO2/95% air unless otherwise indicated. Cells were harvested after washing once with Ca2+-and Mg2+-free phosphate buffered saline (PBS: pH 7.4) using 0.05% trypsin-EDTA (Invitrogen).
Respirometry
Respiration or oxygen consumption rates (OCR) were measured using Extracellular Flux (XF24-3) Analyzer (Seahorse Biosciences) as described [21,34]. Cells were grown to ~80% confluence in V7-PS culture plates unless otherwise stated. In some cases cell suspensions were spun down for assays on the same day as noted. Cells were seeded at the following densities: 25,000 (primary astrocytes)/well, 30,000 (HEK293); 50,000/well (INS1E); and 100,000 (primary β cells)/well. Following 24–72 hour growth (at ~80% confluence), the culture medium was replaced with the indicated respiration buffer and incubated in a non CO2 incubator at 37°C for ~30–60 min before the measurements. Respiration of intact cells was measured in regular low K+ buffer (LKB) containing 3.5 mM KCl, 120 mM NaCl, 0.4 mM KH2PO4, 1.2 mM Na2SO4, 2 mM MgCL2, 1.3 mM CaCL2, 20 mM Na-N-Tris-(hydroxymethyl)-methyl-2-amino-ethanesulfonic acid (TES, pH7.4) and 0.4% fat-free bovine serum albumin (BSA) unless otherwise noted. Respiration of permeabilized cells was measured in Ca2+-free LKB made by excluding CaCl2 and adding 1 mM ethylene-glycol-tetra-acetic acid (EGTA). Respiration media were supplemented with glucose and other substrates as indicated. XF24-3 cartridges were pre-hydrated for 24 hours and then injection ports (A–D) were loaded with indicated compounds. The oxygen and pH sensing probes were calibrated as per manufacturer’s instructions [21,34]. Following calibration, the V7 culture plates with cells were loaded into the XF24-3 analyzer to measure respiration rates. Cycles of 0.5–2 minute mixing, 1–2 minute waiting and 3–5 minute measurements were used. After 3–4 basal respiration rate measurements, cells were permeabilized with the indicated concentrations of digitonin or PFO. Respiratory substrates were added in the buffer either at the time of preparation with the permeabilizing agent or after the permeabilization as indicated. Unless otherwise indicated 1 mM ADP was adequate for all the cells used in this study. The protonophore carbonyl cyanide p-trifluoro methoxy phenyl hydrazone (FCCP) was used at 2 μM for INS1E and 3 μM for all other cells unless noted otherwise. FCCP concentrations were determined by titration using intact cells in the absence of oligomycin. The same FCCP concentrations were applied to permeabilized cells as well. Glutamate, pyruvate, isocitrate, a-ketoglutarate, malate, succinate, and glycerol-3-phosphate were used at 10 mM concentrations unless otherwise noted. 10 μM cytochrome c was used to test the outer mitochondrial membrane integrity. Respiratory activities are presented either as absolute values or as percent OCR by setting the basal respiration to 100% before any addition. All analyses are based on mid-point respiration rates calculated using the full correction algorithm (Akos) [21]. Data from at least 3 replicates for each group from the same plates are shown in each panel of figures. Different panels of figures may have data from single or different plates as specified. Each experiment was repeated at least two times. Student t-tests and one-way analysis of variance (1-ANOVA) were employed to determine the statistical significance (at p<0.05) for comparisons of two or more groups respectively using the Graph Pad Prism 5.
CO2 evolution assays
CO2 evolution rates (CDER) were measured using XF24-3 analyzer (Seahorse Biosciences) equipped with a sensor for CO2, O2, and pH. Although, the XF24-3 cartridges come in a pre-hydrated state, we followed calibration steps as described above in the section for respirometry. Cells grown in V7-PET plates up to ~80% confluence were used for measurements. Media were replaced with the modified Ca2+-free LKB (MLKB) buffer containing 3.5 mM KCl, 120 mM NaCl, 10 mM KH2PO4, 1.2 mM Na2SO4, 2 mM MgCl2, 1 mM EGTA, 20 mM 3-(N-morpholino)propane sulfonic acid (MOPS, pH7.0), 0.2% fat-free BSA and 15 mM glucose. Cells were incubated in a non CO2 incubator at 37°C for 45–60 minutes. Then after calibration of sensors was complete, the V7-PET plate with cells was loaded into the XF24-3 analyzer. Successive injections of different compounds (at downward arrows) were made using ports A to D. Repeated cycles of 0.5 minute mixing, 1.5 minute waiting and 3 minute measurements were used. Curve fit method of CO2 calculation was used for CDER analyses. Respiration and acidification rates were also monitored simultaneously as controls.
In-gel activity of Complex I
Mitochondria were isolated by differential centrifugation, solubilized with dodecyl-β-D-maltoside (Sigma) in 5 mM 6-aminohexanoic acid, 50 mM imidazole-HCl (pH 7.0), and 10% glycerol (2:1 detergent: protein ratio), and separated by blue native-polyacrylamide gel electrophoresis (BN-PAGE) as described earlier [35]. NADH dehydrogenase activity was assayed by incubating the gels with 0.1 mg/ml NADH and 2.5 mg/mi nitro blue tetrazolium, an artificial electron acceptor in 2 mM Tris-HCl (pH 7.4) until bands become clear (2–4 hour) [35].
Results
Respiratory responses of permeabilized β cells
To assess mitochondrial function in permeabilized β cells, our initial choice was digitonin to permeabilize the plasma membrane. We chose < 0.01% (w/v) digitonin concentration initiallybasedon ourprior studies using Clarke-electrode [21,26]. Experiments were performed with starved and non-starved cells because a recent study has suggested that stimulation of β cells with respiratory fuel can cause appearance of cytochrome c in the cytosol [36]. Because cytochrome c release into cytosol may limit its availability for respiratory chain function, we measured respiration in the presence of exogenous cytochrome c to test the functional integrity of mitochondria. First, respiration of starved INS1E cells was measured in Ca2+-free buffer (LKB) containing 2 mM glucose and 10 mM succinate. When 0.01% digitonin was added, a transient increase in respiration was observed (Figure S1A). Although addition of ADP after digitonin slowed respiratory decline, it did not increase respiration any further despite the presence of succinate in the medium (Figure S1A; Control vs. ADP). When 10 μM cytochrome c was added with ADP, a robust sustained respiratory response was observed (Figure S1A; ADP vs. ADP+CC). Using these conditions as a reference, we then determined whether lower digitonin concentrations could support stable respiration. (Figure S1B) shows a representative data set from non-starved INS1E cells. Addition of 0.0025–0.01% digitonin caused severe drop in respiration below the basal level (Figure S1B; DIG with arrow). When ADP and succinate were added together, the respiration could be rescued to different degrees depending upon the concentration of digitonin used (Figure S1B; Succ +ADP with arrow). However, the respiration was unstable even with the lowest digitonin concentration tested (Figure S1B; group 0.0025% DIG). Like starved cells, the non-starved permeabilized cells also required addition of exogenous cytochrome c for sustained respiration (Figure S1B; group 0.01% DIG+CC). A requirement for exogenous cytochrome c suggested that mitochondrial integrity of INS1E cells was compromised when attempts to permeate the plasma membrane were made, even with very low doses of digitonin.
As an alternative to digitonin, we tested whether the pore forming PFO could be used to permeabilize cells for respirometry. Like digitonin, PFO is also expected to be specific for the plasma membrane that is rich in cholesterol [29]. A pilot experiment with primary rat β cells showed better respiration rates in PFO-permeabilized cells compared to those permeabilized with digitonin under the same conditions (Figure S1C). In either conditions the succinate and ADP- supported respiration was found sensitive to oligomycin, a test of respiratory coupling with ATP synthesis. Oligomycin is an inhibitor of the ATP synthase (Complex V). To further evaluate the use of PFO, we performed PFO titration assays as shown in (Figure 1A). The ratio of maximal succinate and ADP-supported respiration over the basal rates were plotted against PFO concentration. As little as 1 nM PFO was found sufficient to obtain maximal respiratory response (Figure 1B). In permeabilized cells, addition of ADP alone was insufficient to support respiration even in the presence of 16.7 mM glucose in the medium (Figure 1C). This suggested that mitochondria were depleted of metabolites that could support respiratory chain function following plasma membrane permeabilization.
Figure 1.
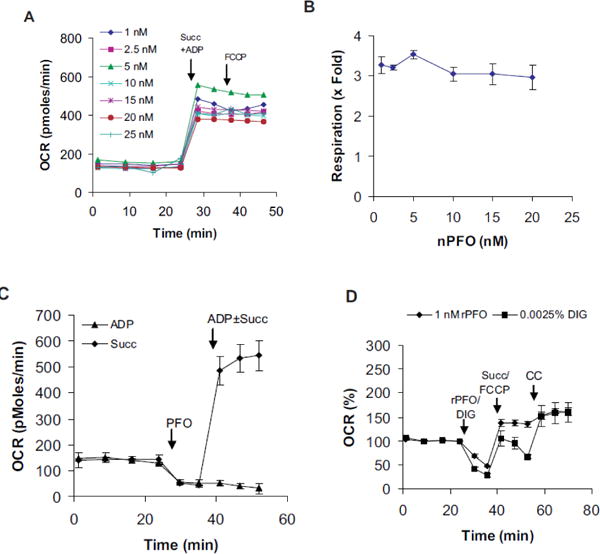
PFO titration with INS1E cells. Indicated concentrations of nPFO were used to permeabilize plasma membrane in Ca2+-free LKB containing 16.7 mM glucose. A) A typical respiration-based PFO titration experiment. Injections- Succ+ADP: 1–25 nM nPFO + 10 mM succinate + 1 mM ADP; FCCP: 2 μM FCCP. B) Plot of PFO concentration vs. respiratory stimulation over the basal rate. Ratio of data points 5/4 from panel A are plotted against PFO concentration. Data are mean ± SD (n=3). 1 nM PFO response was not significantly different from other concentrations (at p<0.05, 1-way ANOVA by Tukey- post hoc analysis). Response was similar for other cell lines also (not shown). C) Demonstration of respiratory substrate depletion following permeabilization. Injections- PFO: 1 nM nPFO; ADP±Succ: 1 m ADP with (Succ) or without 10 mM succinate (ADP). D) Cytochrome c test for functional integrity of mitochondria. Injections- rPFO/DIG: 1 nMr PFO or 0.0025% digitonin (DIG); Succ/FCCP: 10 mM Succinate with 2 μM FCCP; CC: 10 μM cytochrome c. Data mean ± SD (n= at least 3 wells/group) are shown. Graphs in panels A, C, and D were from data obtained with separate plates. CC: cytochrome c; DIG: digitonin, Succ: succinate.
Next we compared respiratory activities of INS1E cells permeabilized either with 1 nM PFO or the lowest digitonin concentration tested (0.0025%) side-by-side. In this assay, nPFO was replaced with the Cys-less rPFO to exclude any concerns with the use of DTT (See Materials and Methods). While 0.0025% digitonin-permeabilized cells showed respiratory decline, the PFO-permeabilized cells showed better and sustained respiratory activity (Figure 1D). When cytochrome c was added to digitonin-permeabilized cells, it brought respiration rates close to PFO-permeabilized cells (Figure 1D). Cytochrome c did not augment respiration of the cells permeabilized with PFO. These data suggest that mitochondrial integrity is preserved in PFO- permeabilized β cells and PFO can be safely used in a wide concentration range.
We used the PFO-based assays to determine the differences in respiration induced by ADP on different substrates. We compared INS1E cells with HEK293 and HepG2 cells under similar conditions. Their respiratory activity in the permeabilized state without added substrate (State 1), following addition of substrate(s) (State 2 or S2), and then ADP (State 3) were measured. (Figure 2A) shows a representative respiratory trace for INS1E cells to assess State 1, 2 and 3 activities (S1, S2, & S3) serially. Because in permeabilized cells glycolysis is unable to support respiration, the residual respiration could be taken as State 1. In INS1E cells, the glutamate + malate addition did not increase respiration. Therefore, State 2/1 and 3/1 ratio are close to 1 (Figure 2B: GM; 3E: P_GM). Under the same conditions, the succinate and glycerol-3- phosphate increased respiration several folds (Figure 2A, B). Succinate and glycerol-3-phosphate modulated respiration by ~4 and 6 folds that approached to ~6 to 8 folds with ADP addition over State 1. INS1E State 3/2 (respiratory control) ratio were significantly lower on glutamate + malate (0.91 ± 0.18 vs. 2.01 ± 0.1*, HEK293; 1.68 ± 0.22*, HepG2), succinate (1.46 ± 0.03 vs. 2.58 ± 0.04*, HEK293; 2.45 ± .09*, HepG2), and glycerol-3-phospahte (1.36 ± 0.05 vs. 1.57 ± 0.05*, HEK293; 1.41 ± 0.10, HEpG2) at *p < 05 by student’s t-test. The lower respiratory control ratio in INS1E cells, particularly on succinate that gives higher proton motive force, suggested higher uncoupling in INS1E cells. To mimic State 4, oligomycin was added to cells treated with PFO, succinate and ADP together. The State 4o (in the presence of oligomycin) respiration on succinate was close to State 2 respiration (Figure 2E, F; PSA_0 vs. P_Succ). No significant differences in the State 2/4o ratio for INS1E, HEK293 and HepG2 cells were noted.
Figure 2.
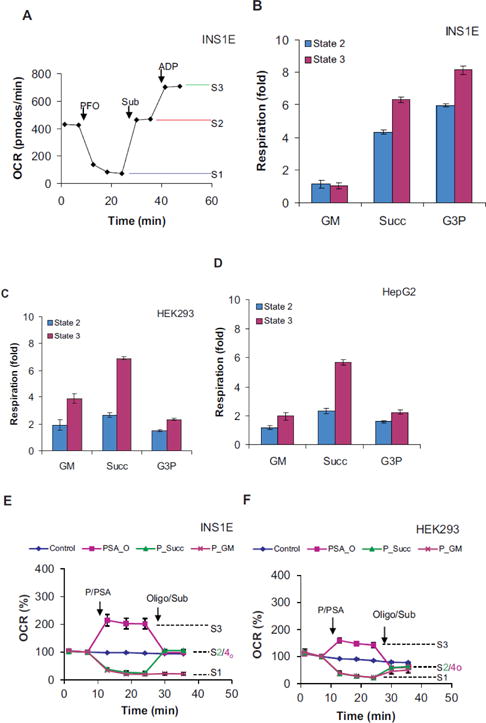
Respiratory coupling test on different substrates: ADP-stimulated respiration of INS1E (A, B, E), HEK293 (C, F) and HepG2 (D) cells were measured in Ca2+-free LKB containing 16.7 mM glucose. A) A typical trace showing State 1 (S1), 2 (S2) and 3 (S3) respiration in permeabilized cells. Injection- PFO: 1 nM nPFO; Sub: indicated substrates (GM: glutamate+malate, Succ: succinate; G3P: glycerol-3-phospahte); ADP: 1 mM ADP. B–D) State 2 vs. 3 respirations in INS1E (B), HEK293 (C) and HepG2 (D) cells. Fold stimulation over State 1 are shown. E, F) Respiratory trances showing State 2 vs. 4o. Oligomycin insensitive respiration in the presence of a substrate (e.g. succinate) and ADP in permeabilized cells was taken as State 4o. Injections- P/PSA: buffer (control) or 1 nM PFO (all groups but control) or 10 mM succinate with 1 mM ADP (PSA_O); Oligo/Sub: 10 mM succinate (P_Succ), glutamate with malate (10 mM, P_GM), 2μg/ml oligomycin (PSA_O). P, S, A and Sub denote PFO, succinate, ADP and substrate(s) respectively. Data mean ± SD (n= at least 3 wells/group) from one representative experiment for each cell type are shown. Average of averages from 3 independent replicates were used to determine statistical significance by student’s t tests for the differences in INS1E State 3/2 and State 2/4 ratio vs. HEK293 or HepG3 cells on each substrate as described in text.
Figure 3.
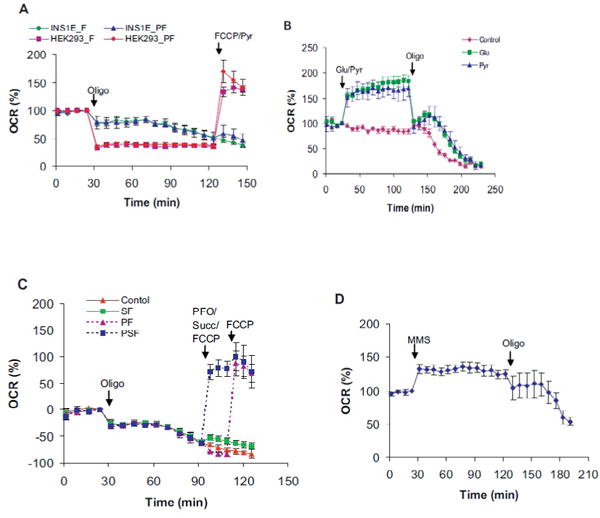
Gradual decline of the oligomycin-insensitive respiration in INS1E cells: A) Effect of oligomycin in INS1E vs. HEK293 cells in regular LKB containing 16.7 mM glucose. Injections- Oligo: 2μg/ml oligomycin; FCCP: 2 μM FCCP (INS1E) or 3 μM FCCP (HEK293) with (INS1E_PF, HEK293_PF) or without 10 mM pyruvate (INS1E_F, HEK293_F). B) Test of glucose-responsiveness and oligomycin-induced effects within the same assay. Regular LKB containing 2 mM glucose was used to measure respiration of 24 hr started cells. Injections- Glu/Pyr: buffer (control), 14.7 mM glucose (Glu), 10 mM Pyruvate (Pyr); Oligo: 2μg/ml oligomycin~90 min after the Glu/Pyr additions. C) Respiration rescue in permeabilized INS1E cells. Cells were exposed to oligomycin over 60 min before permeabilization in 16.7 mM glucose containing regular LKB without added CaCI2 Injections- Oligo: 2μg/ml oligomycin (all groups); PFO/Succ/FCCP: buffer (control), 10 mM succinate with 2 μM FCCP (SF), 1 nM rPFO with 2 μM FCCP (PF), 1 nM rPFO with 10 mM succinate and 2 μM FCCP (PSF); FCCP: 2 μM FCCP (PF). D) Oligomycin-induced respiratory decline in the presence of monomethyl sodium succinate (MMS). Injections- MMS: 10 mM MMS; Oligo: 2μg/ml oligomycin. Data mean ± SD of (4–5 wells/group) are shown. They are representative of 3 independent replicates.
Uncoupled respiration gradually declines in β cells
Using flow-through respirometry, we had noticed that it was difficult to get stable respiration rates in INS1E cells after exposure to oligomycin (data not shown; [16,37]). Therefore, we explored this phenomenon in detail by side-by-side comparison of INS1E with HEK293 using microplate-based respirometry. In comparison to HEK293 cells, the oligomycin addition caused a relatively smaller drop in INS1E cells under similar conditions (Figure 3A). The remaining oligomycin-insensitive respiration started to decline gradually in INS1E but not in the HEK293 cells. FCCP addition after ~90 min of oligomycin exposure did not increase INS1E cells respiration. In contrast, the HEK293 cells showed robust FCCP-induced respiration (Figure 3A; INS1E_F vs. HEK293_F). The addition of pyruvate with FCCP had a negligible effect (Figure 3A: INS1E_F vs. INS1E_PF). Like HEK293 cells, the oligomycin-insensitive respiration also did not decline in Chinese hamster lung fibroblasts CCL16 and V79 (data not shown) [38]. These data suggest that gradual decline of oligomycin-insensitive respiration could be a specific feature of β cells.
Glucose-induced respiration is one of the properties of β cells [5,6,20]. Therefore, we measured glucose-induced respiration and the gradual decline of oligomycin-insensitive respiration within the same assay. As expected, switching from 2 mM to 16.7 mM glucose increased INS1E cells respiration (Figure 3B) [39]. A comparable increase in respiration was also noted with pyruvate addition. Almost all glucose-or pyruvate-induced respiration was sensitive to oligomycin (Figure 3B; Control vs. Glu, Pyr). The oligomycin-insensitive respiration declined in all three conditions i.e. low glucose, high glucose and pyruvate media. Earlier start in the low glucose medium suggested a limitation in substrate supply to the respiratory chain (Figure 3B; Control vs. Glu, Pyr). Because similar response was observed in the pyruvate medium also, we concluded that a limitation in glycolysis alone was not responsible for the respiratory decline (Figure 3B; Glu vs. Pyr).
To distinguish between the limited substrate supplies vs. respiratory chain dysfunction, we performed respiration rescue experiments. INS1E cells treated with oligomycin for over 60 min in high glucose medium were permeabilized to assess respiratory chain function. As noted above, FCCP addition alone had minimal effect in intact cells (Figure 3C; Control vs. SF). Because succinate does not enter cells, its presence did not support respiration with FCCP in intact cells. Only when the cells were permeabilized with PFO, then it increased respiration with FCCP (Figure 3C; PF vs. PSF). Under similar settings glycerol-3-phosphate gave a better response than succinate (Figure S2B: PSF vs. PG3PSF). These data suggested that Complexes II-IV were functional. Complex I-dependent respiration under these conditions was not determined, because it was either minimal or lacking in permeabilized INS1E cells even when they were not treated with oligomycin (Figure 2B; 4A and B). In-gel activity assay of Complex I suggested that NADH oxidation was not impaired (Figure 4D). Together, these data suggest that substrate limitation rather than respiratory chain dysfunction is the underlying cause of the decline of oligomycin- insensitive respiration in INS1E cells.
Figure 4.
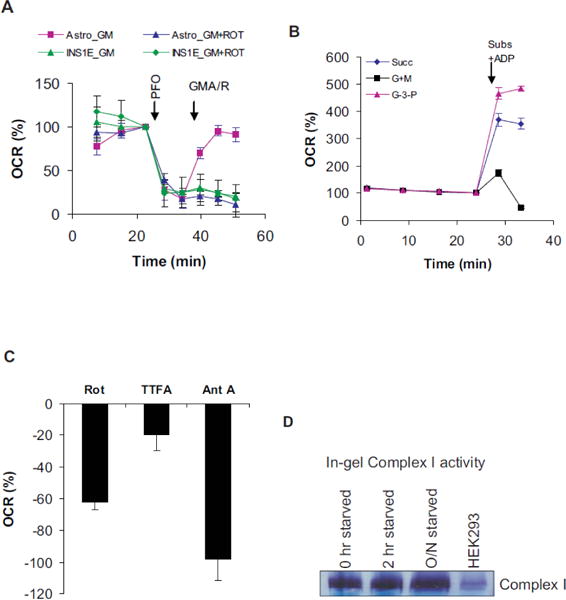
Permeabilized INS1E cells lack or show minimal Complex l-dependent respiration: A) Rotenone-sensitive respiration in INS1E v/s. astrocytes. Primary rat astrocytes were used as positive homologous controls. Respiration was assessed in Ca2+-free LKB containing 16.7 mM glucose. Injections- PFO: 1 nM rPFO; GMA/R: 10 mM gutamate and 10 mM malate (GM) with 1 mM ADP in the presence (INS1 E_GM+Rot, HEK293_GM+Rot) or absence of 1 μM rotenone (INS1E_GM, HEK293_GM). B) Relative Complex I to Ill-dependent respirations in INS1E cells. Respiration medium was the same as in panel A. Injections- Subs+ADP: 1 nM rPFO and 1 mM ADP were added with substrates supporting Complex I- (10 mM glutamate and 10 mM malate, G+M), Complex II- (10 mM succinate, Succ), Complex III- (10 mM glycerol-3-phosphate, G-3-P)-dependent respiration. C) Complex I, II and Ill-dependent respirations in intact INS1E cells measured using specific inhibitors, the rotenone (1μ M), TTFA (400μM) and antimycin-A (4μg/ml) respectively. D) ln-gel activity of Complex I. NADH- dehydrogenase activity was assayed following BN-PAGE. Mitochondria isolated from the INS1E cells starved for indicated time periods were used for BN-PAGE analyses. HEK293 mitochondria served as positive controls. Data mean ± SD (n= at least 3 wells/group) are shown in panels A & B. They are representative of >3 independent replicates.
To determine if a prolonged inhibition of OxPhos with oligomycin could slow down overall cellular metabolism in INS1E cells, we tested the effects of substrates that were capable of inducing respiration in starved INS1E cells. These were the pyruvate, glutamine and monomethyl sodium succinate (MMS) that increased respiration by 61 ± 8%, 22 ± 8 and 31 ± 7%, respectively over the basal levels (Figure 3B: Pyr; 3D; S2A: Gln). Despite their ability to increase respiration in starved cells, they could not rescue respiration in the cells treated with oligomycin for over 60 min (Figure S2B: Pyr, Gln, MMS). Pre-incubation with these substrates also did not prevent the decline of oligomycin-insensitive respiration (Figure 3B, D). Even monomethyl sodium succinate, which is expected to support Complex II-dependent respiration by raising intracellular succinate level, was unable to rescue respiration (Figure 3D). Therefore, we conclude that long- term inhibition of OxPhos can slowdown the overall metabolism of INS1E cells, which in turn can limit substrates supply to the respiratory chain.
Complex I physiology in permeabilized and intact β cells
When Complex I substrates, the glutamate and malate were added to PFO-permeabilized INS1E cells, no increase in respiration was observed (Figure 2B: GM; 2E: P_GM). The ADP presence did not make any difference. Under the same conditions HEK293 and HepG2 cells showed variable levels of respiration (Figure 2C, D and F: GM). In side-by-side comparison while the homologous primary rat astrocytes showed Complex I-dependent respiration, the INS1E cells did not (Figure 4A). Pyruvate, isocitrate, and α-ketoglutarate with malate did not support respiration in permeabilized INS1E cells (Figure 5E; and data not shown). Complex I-dependent respiration was detected only transiently when the substrates and ADP were added along with PFO (Figure 4B). Even under this setting, it was very low compared to Complex II and III supported respirations (Figure 4B). Like INS1E cells, primary β cells also showed either no or transient Complex I dependent respiration depending on whether substrates were added after or with PFO (data not shown). Therefore, we conclude that Complex I physiology in permeabilized β cells is different from other cells.
Figure 5.
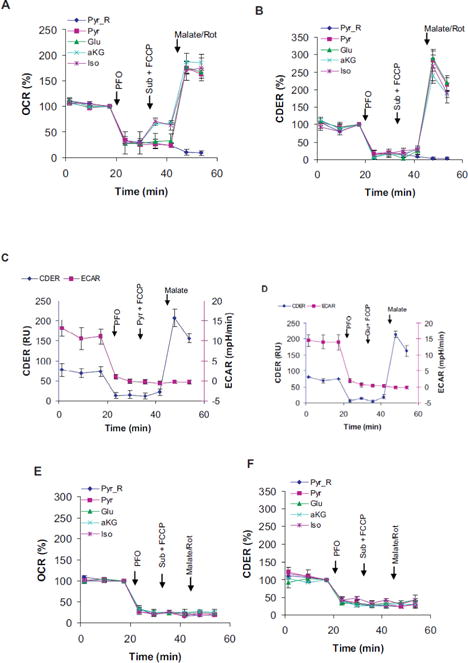
Measurements of CO2 evolution rates (CDER) in permeabilized cells. Assays were performed in Ca2+-free MLKB containing 15 mM glucose using XF24-3 analyzer. Respiration (OCR) and acidification (ECAR) were also simultaneously monitored as controls. A, E), Respiration on different substrates. B, F) CO2 evolution on different substrates. C, D) Relationship between the acidification and CO2 evolution. HEK293 cells served as positive controls (A–D) for the negative responses of INS1E cells (E, F). Injections- PFO: 1 nMrPFO; Sub+FCCP: 10 mM of indicated substrate with 2–3 μM FCCP; Malate/Rot: 10 mM malate alone (all groups but Pyr_R) or with 2 μM rotenone (Pyr_R). Pyruvate (Pyr), glutamate (Glu), αketoglutarate (aKG), and isocitrate (Iso) were used with malate (added afterward) as substrates. Data mean ± SD of 3–4 wells/group are shown. They are representative of 3 independent replicates.
Because Complex I function is known to be crucial for β cell function, it is unlikely that Complex I is not functioning in β cells [4]. Therefore, we measured the portion of respiration sensitive to rotenone, a specific inhibitor of Complex I [26]. Complexes II and III-dependent respirations were also determined using the inhibitors TTFA and antimycin A, respectively [34]. In high glucose medium, the rotenone, TTFA and antimycin-sensitive respirations were found to be ~62 ± 5%, 20 ± 10% and 98 ± 13% respectively (Figure 4C). Relatively higher rotenone sensitivity (~70%) was noted in low glucose medium in intact cells. Measurements of Complex I activity by in-gel assay after BN-PAGE separation was performed as described elsewhere [35]. The BN- PAGE is a native gel electrophoretic technique commonly used to separate OxPhos complexes. Activity staining showed that Complex I from INS1E mitochondria efficiently oxidized NADH (Figure 4D). Therefore, the possibility that Complex I becomes inactive following permeabilization can be excluded because the conditions are milder than the mitochondrial isolation procedure.
Because Complex I substrates are expected to support both respiration and CO2 production; we measured CO2 evolution rates (CDER) on them in permeabilized cells. Simultaneous measurements of respiration and CO2 production rates were made possible by the XF24 Analyzer equipped with O2, CO2 and pH sensors. Since HEK293 cells showed Complex I- dependent respiration, they served as positive controls. To permeabilized cells, pyruvate, isocitrate, a-ketoglutarate or glutamate was added along with FCCP and then followed by malate addition later. The maximal respiration was detected only after malate addition in HEK293 cells (Figure 5A). The respiratory increase following malate addition corresponded with an increase in CO2 production (Figure 5A vs. B). Because no change in acidification of the medium was observed in the same assays, the possibility of changes in CO2 solubility was excluded (Figure 5C,D). The CO2 production was sensitive to Complex I inhibition (Figure 5B: Pyr_Rot). Since Complex I function is thermodynamically regulated by mitochondrial membrane potential, we tested how oligomycin pre-treatment affected respiration and CO2 production. In oligomycin pre-treated cells, pyruvate and malate did not support respiration or CO2 production without FCCP (Figure S3A, B: Pyr+Mal). These data suggest that respiratory chain activity modulates the functions of Complex I and the TCA cycle in real-time. Under the conditions that showed robust response in HEK293 cells, no increase in respiration and CO2 production were observed in INS1E cells (Figure 5A vs. E; 5B vs. F). This suggested that in β cells the TCA cycle was inefficient in producing adequate NADH to support Complex I-dependent respiration.
To determine if NADH production was indeed lower in INS1E cells, we measured NAD(P)H fluorescence. INS1E mitochondria incubated with glutamate and malate in the presence of rotenone showed about 40.7 ± 9.51% lower NAD(P)H levels compared to HEK293 mitochondria. Analysis using the Fluoro NAD™ kit also gave similar results in PFO- permeabilized cells. The NADH was lower by 36–43% in INS1E cells compared to HEK293 cells (Figure S4A). However, the total nucleotide (NADH+NAD+) pool was also 55.54 ± 1.55% lower in INS1E (Figure S4B). No significant difference in the NADH/NAD+ ratio was noted (not shown). The lower levels of NAD+ and NADH can limit activities of both the TCA cycle and Complex I, respectively.
Relative OxPhos and respiratory capacities
The OxPhos and respiratory capacities provide estimates of the ATP synthesis and respiration. To determine whether OxPhos capacity matched respiratory capacity, we measured ADP- vs. FCCP-stimulated respirations serially. No further increase in INS1E cells respiration by FCCP over the ADP-stimulated rate was observed (Figure 6A). Under similar conditions in HEK293 cells, the OxPhos capacity was lower than the respiratory capacity (Figure 6A). By manipulating the composition of the respiration buffer, we found that Pi concentration affected both the OxPhos and respiratory capacities in HEK293 cells. Higher Pi levels increased both (Figure 6B; KH2PO4 group). Irrespective of the Pi concentration, the difference between OxPhos and respiratory capacities persisted in HEK293 cells. The Pi level did not affect INS1E cells respiration (Figure 6C; INS1E_LKB vs. KH2PO4). These results indicate cell type differences in the relationship between OxPhos and respiratory capacities and the effect of Pi concentration on mitochondrial bioenergetics.
Figure 6.
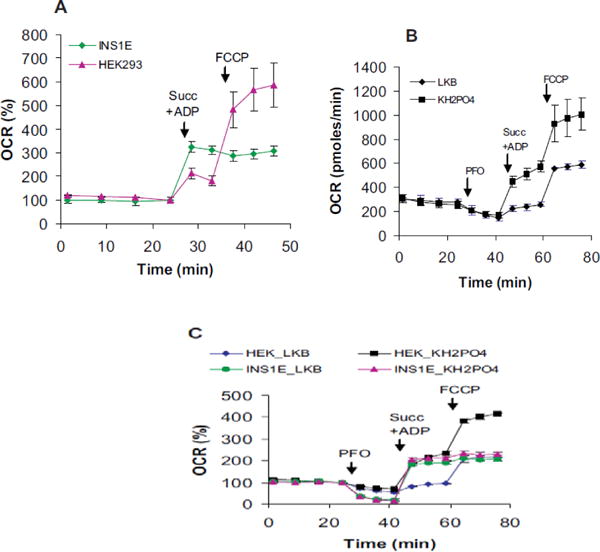
Relationship between the OxPhos and respiratory capacities. ADP-and FCCP-induced respirations were successively assessed in Ca2+-free LKB buffer containing 15 mM glucose. Maximal respiration in the presence of ADP alone and ADP+FCCP were taken as OxPhos and respiratory capacities, respectively. A) OxPhos vs. respiration capacity in INS1E and HEK293 cells. Injections-Succ+ADP: 1nM rPFO with 10 mM succinate and 1 mM ADP; FCCP: 2–3 μM FCCP. B) Effects of Pi concentration on the OxPhos and respiratory capacities of HEK293 cells. Injections- PFO: 1 nM rPFO; Succ+ADP: 10 mM succinate and 1 mM ADP; FCCP: 3 μM FCCP. LKB and KH2PO4 differ only in their KH2PO4 content, which were have 0.4 and 10 mM respectively. C) Relative effect of Pi in HEK293 v/slNS1E cells. Mean ± SD from at least 3 well/group are shown. They are representative of 3 independent replicates.
Discussion
In this study, we have performed comparative analyses of the mitochondrial metabolism of pancreatic β cells with others. A novel method to permeabilize the plasma membrane in concert with microplate-based respirometry for side-by-side comparisons with other cells is also described. Measurements involving intact and permeabilized cells have enabled us to compare the mitochondrial physiology of β cells with other cells. Comparisons are made under experimental conditions that give robust glucose-stimulated respiration, a key feature of β cells.
Use of PFO to permeabilize β cells for assessing mitochondrial metabolism
The selective permeabilization of the plasma membrane with PFO is based on its requirement for high cholesterol content [29,33,40]. Binding of PFO to membranes is triggered when cholesterol is accessible to the membrane surface i.e. in the presence of active cholesterol, [41]. Considering ~90% of cellular cholesterol is concentrated in the plasma membrane, PFO is less likely to bind mitochondria under normal conditions because active cholesterol will be minimal [41–43]. Compared to digitonin, the cholesterol-dependent cytolysins are expected to require more cholesterol for pore formation in membranes [44]. PFO is not expected to bind membranes indiscriminately. Detergents can bind membranes even at very low levels of cholesterol and their binding depends on cholesterol content rather than cholesterol activity [40,45]. Therefore, it may become difficult to find a safe working concentration range with detergents. This could be the reason for our difficulty in getting stable respiration in digitonin permeabilized INS1E cells without exogenous cytochrome c addition (Figures 1D, S1). Because PFO is a protein and its activity depends on active cholesterol content, the pore formation is more controlled. In our experience, it can be safely used in a wide concentration range (1–20 nM) without any detrimental effects (Figure 1B). The mitochondrial integrity is completely preserved in PFO-permeabilized cells (Figure 1D). Direct exposure of isolated mouse liver mitochondria with ~10 nM PFO did not show any evidence of damage, while digitonin exposure dose-dependently reduced respiration up to 46.92 ± 11.92% at ≤0.01% concentration (data not shown) [46,47].
In the permeabilized cells, the mitochondrial function was solely dependent on direct delivery of metabolites to mitochondria. The presence of glucose in the respiration medium did not support respiration possibly because dilution of cytosolic enzymes supporting glycolysis. Therefore, respiratory responses could be determined in relation to basal respiration following permeabilization in the presence of exogenous substrates. The use of PFO allowed respirometry under similar conditions with different cells, which was very important for our study because we wanted to carry out in a low K+ buffer that supported robust glucose-stimulated respiration in INS1E (Figure 3B) and dispersed islet cells (data not shown). Even though the PFO-based assays were equally applicable to high K+ and/or mannitol and sucrose containing buffers, they were not suitable for assays of glucose-stimulated respiration, a key feature of β cells [5,6]. Data in Figure 2 clearly show that PFO-based assays are applicable to different cells for assessing various bioenergetic parameters. While in β cells the glycerol-3-phosphate-dependent respiration was maximal, in HEK293 and HepG2 cells, it was the succinate-dependent respiration.
Gradual decline of the oligomycin-insensitive respiration in β cells
Pancreatic β cells have high degree of uncoupled respiration [5,20,48,49], which is also supported by this study (Figures 2,3). However, the uncoupled respiration gradually declines after 30–45 min of oligomycin exposure. FCCP fails to increase respiration in INS1E cells treated with oligomycin for a longer time (60–90 min). This decline is linked with limitations in substrate supply to respiratory chain rather than its dysfunction (Figures 3B, C; 4D). We predict this is because of the unique mitochondrial metabolism in β cells. Very low lactate dehydrogenase activity, high redox shuttling, and the operation of different metabolites cycles may contribute to this phenomenon. In the presence of low lactate dehydrogenase activity, β cells will mostly depend on OxPhos for their bioenergetic needs [9]. Thus, when OxPhos is blocked for a prolonged period and glycolysis can not meet cellular ATP demand, the overall β cell metabolism will be halted. Therefore, a gradual decline of uncoupled respiration will occur because of the limitation in substrates. Faster kinetics of respiratory decline in low glucose supports this (Figure 3B). The observations that pyruvate, glutamine and monomethyl sodium succinate were unable to rescue respiration in oligomycin-treated cells or prevent respiratory decline further provide real-time functional data to support our conclusion (Figures 3,S2). A metabolic shutdown is indicated by a preliminary metabolomics analysis of oligomycin-treated INS1E cells (R. Kibbey and N. Yadava, unpublished results). Impairments in glycolysis, pentose phosphate pathway, the TCA cycle and amino acids metabolism were noted. Decrease in dihydroxy acetone phosphate, glycerol-3-phospahte, phosphoenol pyruvate, malate and succinate levels, and increase in glutamate and aspartate levels are interesting with respect to respiratory chain function and metabolites shuttling in β cells. Dihydroxy acetone phosphate, glycerol-3-phospahte, malate, aspartate, and glutamate are components of the NADH redox shuttles.
Many aspects of β cell metabolism may be responsible for this phenomenon. A prolonged inhibition of OxPhos may negatively inhibit cytosolic NADH shuttling to mitochondria, which occurs via malate-aspartate and glycerol phosphate shuttles. If the β cell metabolic design is such that its respiratory activity is primarily dependent on redox shuttles (Figure 7A), then prolonged oligomycin treatment that reduces dihydroxy acetone phosphate, glycerol-3-phospahte, malate can impair the function of redox shuttles. Malate-aspartate shuttle positively modulates pyruvate oxidation ratherthan the rate of glycolysis [8,50,51]. In β cells, pyruvate cycles between the cytoplasm and mitochondria via the pyruvate/citrate, pyruvate/isocitrate/α-ketoglutarate, pyruvate/malate, and pyruvate/phosphoenol pyruvate shuttles [10,14,52]. Malate plays key roles in pyruvate/malate and pyruvate/citrate cycles, and equilibrations of Pi and metabolites that support respiration (Figure 5A). In β cells, a major fraction (up to 41%) of phosphoenol pyruvate is generated from mitochondrial oxaloacetate [14]. Pyruvate cycling will prevent its complete oxidation inside mitochondria because the TCA cycle intermediates will exit the cycle. Thus, a lower yield of NADH/FADH2 per pyruvate can be expected in β cells. In cells such as HEK293 with minimal pyruvate cycling a prolonged blockage of is not expected to severely limit respiratory activity because pyruvate will be oxidized completely, albeit at a slower rate. The operations of metabolites shuttles consuming ATP will be eventually halted if ATP becomes limiting. For example pyruvate carboxylase and ATP-citrate lyase reactions are ATP-dependent. Oxaloacetate export as phosphoenol pyruvate and citrate, into cytoplasm also supports redox shuttles. The phosphoenol pyruvate can generate glycerol-3-phosphate generation via reverse glycolysis. NADH availability is not expected to restrict phosphoenol pyruvate conversion to glycerol-3-phosphate because cytosolic glycerol-3-phosphate dehydrogenase can also use NADPH [53]. The glycerol-3-phosphate level increases in fuel-responsive INS1 cells following stimulation with glucose [54]. However, it must be noted that only ~15.5% of INS1E cells respiration is supported by FADH2 oxidation at the level of Complex III (Figure 4C) and the glycerol-3-phosphate shuttle is not essential for β cell function [8,55]. The role of malate-aspartate shuttle is more critical and it may depend upon efficient clearance of the mitochondrial oxaloacetate. If pyruvate/phosphoenol pyruvate shuttle function is critical for the operation of malate-aspartate shuttle, then GTP supply should not become limiting. Because the kinetics of respiratory decline in cells with >50% lower pyruvate carboxylase was similar to control cells, we predict that the observed phenomenon is linked with metabolites’ exit rather than the anaplerosis at the level of oxaloacetate (Figure S5).
Figure 7.
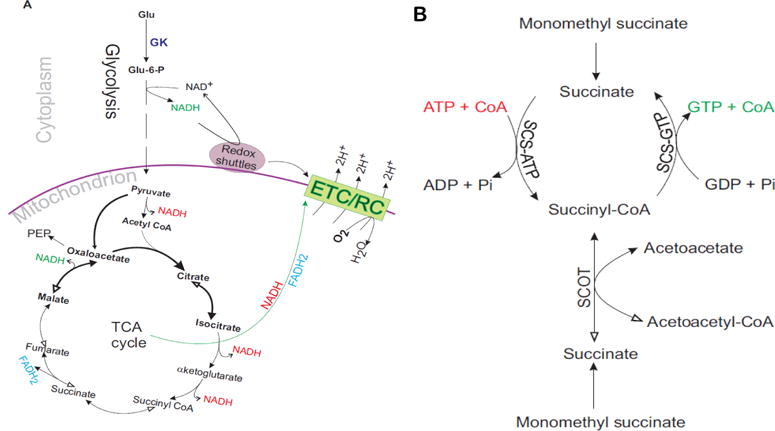
Models for βcell metabolism. A) Dependence of the respiratory chain activity on cytosolic NADH shuttling to mitochondria. It requires limited NADH production inside mitochondria. NADH in red font indicates limitation in production. Malate-aspartate and glycerol phosphate redox shuttles support respiration at the levels of Complex I and III respectively. Malate-aspartate shuttle consumes 1 NADH in converting oxaloacetate to malate in the cytosol and it generates 1 NADH inside mitochondria by importing and converting malate to oxaloacetate. Glycerol phosphate shuttle consumes 1 NADH in conversion of dihydroxyacetone phosphate to glycerol-3-phosphate in the cytosol and it produces 1 FADH2 in converting glycerol-3-phosphate into dihydroxyacetone phosphate in mitochondrial inter membranes space. GK: glucokinase; PEP: phosphoenol pyruvate; ETC/RC: electron transport chain/respiratory chain. B) Fates of monomethyl succinate (MMS)-derived succinate in βcells. It can be converted into fumarate (see Panel A) and succinyl-CoA. SCS-ATP: ATP-dependent succinyl-CoA synthase, SCS-GTP: GTP-dependent succinyl-CoA synthase, and SCOT: succinyl-CoA:3-ketoacid- CoA transferase.
Further evidence that metabolites cycling controls β cell respiration comes from the observations made with monomethyl sodium succinate that is expected to support respiration at the level of Complex II by providing succinate (Figure 3D). A respiratory decline on monomethyl sodium succinate is not possible, if it supports respiration at the level of Complex II by raising succinate level. However, if it supports respiration indirectly by facilitating metabolites export to the cytoplasm and consuming ATP, then it is possibly. Succinate may be converted into succinyl-CoA at the expense of ATP and then back to succinate as shown in Figure 7B. This will generate more GTP to support conversion of oxaloacetate to phosphoenol pyruvate via the mitochondrial phosphoenol pyruvate carboxy kinase reaction [10,14]. An ATP limitation may halt this succinate/succinyl-CoA cycling (Figure 7B). Knockdowns of the ATP- vs. GTP-dependent isoforms of succinyl-CoA synthase were found to increase and decrease insulin secretion in INS1 cells, respectively [10]. The increase in insulin secretion following the knockdown of ATP-dependent isoform may have occurred by favoring an alternative route of succinate utilization to generate succinyl-CoA involving succinyl-CoA:3-ketoacid-CoA transferase (Figure 7B) [56]. Higher levels of succinyl-CoA may inhibit α-ketoglutarate dehydrogenase, which will favor truncated TCA cycle via facilitating exits of citrate/isocitrate. Although, our study does not identify a specific pathway responsible for metabolic slow down causing respiratory decline when OxPhos is blocked, it points toward a critical relationship among NADH shuttles, pyruvate/phosphoenol pyruvate cycle, and succinate/succinyl-CoA cycle. It would be interesting to determine whether the lack of a gradual decline of oligomycin-insensitive respiration could predict β cell failure. In association with glucose-stimulated respiration, it may enable better prediction of β cell function for transplantation studies.
Complex I-physiology of β cells is different
Complex I-dependent respiration is dependent on the NADH availability inside mitochondria. Rotenone sensitivity of respiration in intact cells, and the transient or minimal Complex I- dependent respiration in permeabilized β cells suggest that Complex I function is mainly supported by cytosolic NADH shuttling to mitochondria. This may be the reason why insulin secretion is critically dependent on cytosolic NADH shuttling to mitochondria [7,8]. In the absence of glycerol-3-phosphate shuttle function, the insulin secretion is completely blocked with an aspartate amino-transferase inhibitor [8]. The aspartate amino-transferase is a component of the malate-aspartate shuttle. The fuel-stimulated insulin secretion is also blocked by rotenone [4]. Slow dilution of the soluble cytosolic components of the malate-aspartate NADH shuttle (e.g. the aspartate amino-transferase, malate dehydrogenase and NADH) following permeabilization could be responsible for transient Complex I-dependent respiration. Although our data do not completely rule out the inactivation of Complex I at other steps of electron/proton transfer, it is very unlikely that it happens specifically in β cells following permeabilization. The response is same whether cells are permeabilized with digitonin or if PFO is removed from the medium before Complex I assays. The presence of free Ca2+ up to 300 nM does not make any difference (not shown). Over expression of the aspartate/glutamate carrier ACC1, a component of the malate-aspartate shuttle was found to increase insulin secretion via elevating NAD(P)H and ATP levels and mitochondrial membrane potential in β cells [50]. While the ACC1 knockdown did not affect mitochondrial membrane potential, it did decrease insulin secretion that correlated with reduced NAD(P)H and ATP levels [51]. Reconstitution of the malate-aspartate shuttle in permeabilized cells would be required to clearly address its role in Complex I physiology of β cells (a subject of separate study).
The TCA cycle and OxPhos activities are interdependent due to regulatory loops involving NADH metabolism (Figure 7A) [57,58]. While a deficit in NADH supply can limit OxPhos, a severe respiratory chain defect can block the TCA cycle via NADH build-up [57–60]. Therefore, a delicate balance is maintained between these processes to meet the metabolic demands of cells. It is thought that in most cells respiration is primarily controlled by ATP demand rather than substrate supply [15,24]. In β-cells it is regulated by the substrate supply. This "feed-forward" regulation favors a model of limited production of reducing equivalents (NADH, FADH2) inside mitochondria (Figure 7A). The significantly less NADH production on Complex I substrates in INS1E mitochondria/cells supports this model (Figure S4A). This would ensure that cytosolic NADH shuttling to mitochondria plays a major role in supporting β cell respiration.
The measurements of CO2 evolution suggest that the TCA cycle is inefficient in β cell mitochondria (Figure 5). It cannot supply sufficient NADH to support Complex I-dependent respiration. However, other studies have reported glucose-stimulated CO2 production in intact β cells [50]. There is no information available regarding relative level of CO2 production from β cells compared to other cells. It is possibly that part of the TCA cycle may be running in the cytoplasm following citrate/isocitrate exit via cytosolic isocitrate dehydrogenase. Our studies report only mitochondrial CO2 along with the changes in respiratory activity at the Complex I level. In most cells, the major portion of CO2 produced comes from the de-carboxylation of three metabolites: pyruvate, isocitrate, a-ketoglutarate by their respective dehydrogenases that are also allosterically regulated by NADH. Only a minor amount of CO2 is derived from other processes including 6-phosphogluconate conversion to ribulose-5-phosphate in the pentose phosphate pathway and decarboxylations of amino acids, malate and oxaloacetate [61]. In β cell mitochondria, malate and oxaloacetate can produce CO2 via the malic enzymes and phosphoenol pyruvate carboxy kinase, respectively. Pyruvate conversion into oxaloacetate can consume CO2 via pyruvate carboxylase. Because none of the substrates (pyruvate, isocitrate, a-ketoglutarate and glutamate) produced CO2 in INS1E cells, the possibility of CO2 consumption by pyruvate carboxylase can be excluded.
Lower NAD+- pool is expected to compromise the TCA cycle efficiency of β cells by affecting NAD+-dependent dehydrogenases (Figure S4). Further, succinyl-CoA and acetyl-CoA build-ups may also negatively regulate a-ketoglutarate and pyruvate dehydrogenases, respectively. Oxaloacetate accumulation can cause reversal of the malate dehydrogenase and possibly inhibition of the succinate dehydrogenase [62]. Therefore, clearance of the oxaloacetate would be essential for the operation of malate-aspartate supported NADH shuttling. This underscores the importance of the pyruvate/phosphoenol pyruvate cycling in insulin secretion [10,14]. Since the ATP yield would be limiting due to less NADH/FADH2 produced per glucose molecule, a fraction of the phosphoenol pyruvate may be also involved in ATP generation in the cytosol. Our studies have identified a clear difference in the physiology of Complex I between β cells and others. We predict that lower NAD+ levels could be responsible for inefficient TCA cycle. However, additional factors such as inefficient transport of Complex I substrates into mitochondria can not be excluded [47].
Relationship between the spare OxPhos and respiratory capacities
Spare respiratory capacity is a critical parameter that determines capability of cells to meet ATP demand under acute conditions [16,37,63]. Whether spare respiratory capacity is indeed a surrogate for OxPhos capacity in all cells is unclear. In this study, we show that while OxPhos capacity can match respiratory capacity in β cells, it is lower in other cells under the tested conditions. Our data suggest that the relationship between spare OxPhos capacity and respiratory capacity varies with cell type and experimental conditions [7]. When respiratory substrate and ADP are not limiting, a limitation in Pi concentration drastically affects both OxPhos and respiratory capacity depending upon the cell type. This may be a physiologically relevant finding as impaired Pi homeostasis is implicated in premature aging in mice [64]. Multiple posS1Bilities exist by which Pi can affect mitochondrial bioenergetics including its transport, action on Complex V, and altered equilibration with substrates (e.g. succinate) and ions (Ca2+, H+). A difference in the expression of Pi carrier isoform PiC-A may also be responsible [65]. Like HEK293 cells, the Chinese hamster lung fibroblasts (V79 and CCL16) also showed difference in their OxPhos and respiratory capacities. Cells such as C2C12 (myoblasts/myotubes) and SHSY-5Y (neuorblasts) showed comparable OxPhos and respiratory capacities (not shown).
In summary, using in situ respirometry with intact and permeabilized cells, we have studied the comparative mitochondrial physiology of β cells. In this process, we have applied a novel PFO-based assay for analysis of the mitochondrial metabolism. PFO is a cholesterol-dependent cytolysin that can permeabilize plasma membrane selectively without damaging mitochondria. In permeabilized β cells, the maximal respiration was obtained on glycerol-3- phosphate while in others it was on succinate. Permeabilized β cells showed either transient or no Complex I-dependent respiration, which we predict is due to dependence of Complex I function on cytosolic NADH shuttling to mitochondria and an inefficient TCA cycle. In β cells, the oligomycin-insensitive respiration gradually declines over time due to limitation in substrate supply. Interactions among the NADH shuttles, pyruvate/phosphoenol pyruvate and succinate/succinyl-CoA cycles may play important role in this phenomenon. Gradual decline of the uncoupled respiration, and a transient or no Complex I-dependent respiration in permeabilized β cell cells appear to be specific features. The relationship between the OxPhos and respiratory capacities are cell type dependent, which is close to 1 in β cells. We believe that this study will lead to a better understanding β cell mitochondrial metabolism by permitting further comparative analyses with other cells close to physiological conditions.
Supplementary Material
Acknowledgments
We thank David Nicholls, Martin Brand, David Ferrick, George Rogers and Richard Kibbey for their insightful feedbacks and helps at different stages of this study, and Maureen Lahti and Marcia D. Woods for critical reading of the manuscript.
Funding
This work was supported by grants from National Institutes of Health (R21NS057224), Rays of Hope from the Baystate Health Foundation, and start-up and translational funds from CEAR at the PVLSI supported by an award (A00000000004448) from Massachusetts Technology Collaborative as administrator of the John Adams Innovation Institute to N. Y. Seahorse Bioscience Inc. provided in-kind support to the Translational grant. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prentki M, Nolan CJ. lslet β cell failure in type 2 diabetes. J Clin Invest. 2006;116:1802–1812. doi: 10.1172/JCI29103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nolan CJ, Prentki M. The islet β-cell: fuel responsive and vulnerable. Trends Endocrinol Metab. 2008;19:285–291. doi: 10.1016/j.tem.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 4.Antinozzi PA, Ishihara H, Newgard CB, Wollheim CB. Mitochondrial metabolism sets the maximal limit of fuel-stimulated insulin secretion in a model pancreatic β cell: a survey of four fuel secretagogues. J Biol Chem. 2002;277:11746–11755. doi: 10.1074/jbc.M108462200. [DOI] [PubMed] [Google Scholar]
- 5.Wikstrom JD, Sereda SB, Stiles L, Elorza A, Allister EM, et al. A novel high-throughput assay for islet respiration reveals uncoupling of rodent and human islets. PLoSOne. 2012;7:e33023. doi: 10.1371/journal.pone.0033023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malmgren S, Nicholls DG, Taneera J, Bacos K, Koeck T, et al. Tight coupling between glucose and mitochondrial metabolism in clonal β-cells is required for robust insulin secretion. J Biol Chem. 2009;284:32395–32404. doi: 10.1074/jbc.M109.026708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dukes ID, Mclntyre MS, Mertz RJ, Philipson LH, Roe MW, et al. Dependence on NADH produced during glycolysis for β-cell glucose signaling. J BiolChem. 1994;269:10979–10982. [PubMed] [Google Scholar]
- 8.Eto K, Tsubamoto Y, Terauchi Y, Sugiyama T, Kishimoto T, et al. Role of NADH shuttle system in glucose-induced activation of mitochondrial metabolism and insulin secretion. Science. 1999;283:981–985. doi: 10.1126/science.283.5404.981. [DOI] [PubMed] [Google Scholar]
- 9.Sekine N, Cimili V, Regazzi R, Brown LJ, Gine E, et al. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic β-cells. Potential role in nutrient sensing. J BiolChem. 1994;269:4895–4902. [PubMed] [Google Scholar]
- 10.Kibbey RG, Pongratz RL, Romanelli AJ, Wollheim CB, Cline GW, et al. Mitochondrial GTP regulates glucose-stimulated insulin secretion. Cell Metab. 2007;5:253–264. doi: 10.1016/j.cmet.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cline GW, Lepine RL, Papas KK, Kibbey RG, Shulman GI. 13C NMR isotopomer analysis of anaplerotic pathways in INS-1 cells. J BiolChem. 2004;279:44370–44375. doi: 10.1074/jbc.M311842200. [DOI] [PubMed] [Google Scholar]
- 12.Ronnebaum SM, Ilkayeva O, Burgess SC, Joseph JW, Lu D, et al. A pyruvate cycling pathway involving cytosolic NADP-dependent isocitrate dehydrogenase regulates glucose-stimulated insulin secretion. J Biol Chem. 2006;281:30593–30602. doi: 10.1074/jbc.M511908200. [DOI] [PubMed] [Google Scholar]
- 13.Jensen MV, Joseph JW, RonnebaumSM, Burgess SC, Sherry AD, et al. Metabolic cycling in control of glucose-stimulated insulin secretion. AmJ Physiol Endocrinol Metab. 2008;295:1287–1297. doi: 10.1152/ajpendo.90604.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stark R, Pasquel F, Turcu A, Pongratz RL, Roden M, et al. Phosphoenolpyruvate cycling via mitochondrial phosphoenolpyruvatecarboxykinase links anaplerosis and mitochondrial GTP with insulin secretion. J Biol Chem. 2009;284:26578–26590. doi: 10.1074/jbc.M109.011775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nicholls DG, Ferguson SJ. Bioenergetics. Academic Press; London: 2002. [Google Scholar]
- 16.Jekabsons MB, Nicholls DG. In situ respiration and bioenergetic status of mitochondria in primary cerebellar granule neuronal cultures exposed continuously to glutamate. J Biol Chem. 2004;279:32989–33000. doi: 10.1074/jbc.M401540200. [DOI] [PubMed] [Google Scholar]
- 17.Chance B, Williams GR. A simple and rapid assay of oxidative phosphorylation. Nature. 1955;175:1120–1121. doi: 10.1038/1751120a0. [DOI] [PubMed] [Google Scholar]
- 18.Picard M, Taivassalo T, Ritchie D, Wright KJ, Thomas MM, et al. Mitochondrial structure and function are disrupted by standard isolation methods. PLoS ONE. 2011;6:e18317. doi: 10.1371/journal.pone.0018317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Picard M, Ritchie D, Wright KJ, Romestaing C, et al. Mitochondrial functional impairment with aging is exaggerated in isolated mitochondria compared to permeabilized myofibers. Aging Cell. 2010;9:1032–1046. doi: 10.1111/j.1474-9726.2010.00628.x. [DOI] [PubMed] [Google Scholar]
- 20.Affourtit C, Brand MD. Measuring mitochondrial bioenergetics in INS-1 E insulinoma cells. Methods Enzymol. 2009;457:405–424. doi: 10.1016/S0076-6879(09)05023-X. [DOI] [PubMed] [Google Scholar]
- 21.Gerencser AA, Neilson A, Choi SW, Edman U, Yadava N, et al. Quantitative microplate-based respirometry with correction for oxygen diffusion. Anal Chem. 2009;81:6868–6878. doi: 10.1021/ac900881z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Riordan TC, Zhdanov AV, Ponomarev GV, Papkovsky DB. Analysis of intracellular oxygez and metabolic responses of mammalian cells by time-resolved fluorometry. Anal Chem. 2007;79:9414–9419. doi: 10.1021/ac701770b. [DOI] [PubMed] [Google Scholar]
- 23.Dranka BP, Benavides GA, Diers AR, Giordano S, Zelickson BR. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic Biol Med. 2011;51:1621–1635. doi: 10.1016/j.freeradbiomed.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gnaiger E, Kuznetsov AV, Lassnig B, Fuchs A, Reck M, et al. High-resoultion respirometry-optimum permeabilization of the cell memebrane by digitonin. Bio Thermo Kinetics in the Post Genomic Era. 1998:89–95. [Google Scholar]
- 26.Yadava N, Potluri P, Smith EN, Bisevac A, Scheffler IE. Species-specific and mutant MWFE proteins. Their effect on the assembly of a functional mammalian mitochondrial complex I. J Biol Chem. 2002;277:21221–21230. doi: 10.1074/jbc.M202016200. [DOI] [PubMed] [Google Scholar]
- 27.Jonckheere AI, Huigsloot M, Janssen AJ, Kappen AJ, Smeitink JA, et al. High-throughput assay to measure oxygen consumption in digitonin-permeabilized cells of patients with mitochondrial disorders. Clin Chem. 2010;56:424–431. doi: 10.1373/clinchem.2009.131441. [DOI] [PubMed] [Google Scholar]
- 28.Clerc P, Polster BM. Investigation of mitochondrial dysfunction by sequential microplate-based respiration measurements from intact and permeabilized neurons. PLoS One. 2012;7:e34465. doi: 10.1371/journal.pone.0034465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heuck AP, Moe PC, Johnson BB. The cholesterol-dependent cytolysin family of gram-positive bacterial toxins. Subcell Biochem. 2010;51:551–577. doi: 10.1007/978-90-481-8622-8_20. [DOI] [PubMed] [Google Scholar]
- 30.Merglen A, Theander S, Rubi B, Chaffard G, Wollheim CB, et al. Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology. 2004;145:667–678. doi: 10.1210/en.2003-1099. [DOI] [PubMed] [Google Scholar]
- 31.Graham FL, Smiley J, Russell WC, Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol. 1977;36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 32.Flanagan JJ, Tweten RK, Johnson AE, Heuck AP. Cholesterol exposure at the membrane surface is necessary and sufficient to trigger perfringolysin O binding. Biochemistry. 2009;48:3977–3987. doi: 10.1021/bi9002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moe PC, Heuck AP. Phospholipid Hydrolysis Caused by Clostridium perfringens alpha-Toxin Facilitates the Targeting of Perfringolysin O to Membrane Bilayers. Biochemistry. 2010;49:9498–9507. doi: 10.1021/bi1013886. [DOI] [PubMed] [Google Scholar]
- 34.Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol. 2007;292:125–136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
- 35.Yadava N, Houchens T, Potluri P, Scheffler IE. Development and characterization of a conditional mitochondrial complex I assembly system. J Biol Chem. 2004;279:12406–12413. doi: 10.1074/jbc.M313588200. [DOI] [PubMed] [Google Scholar]
- 36.Jung SR, Kuok IT, Couron D, Rizzo N, Margineantu DH, et al. Reduced cytochrome C is an essential regulator of sustained insulin secretion by pancreatic islets. J Biol Chem. 2011;286:17422–17434. doi: 10.1074/jbc.M110.202820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yadava N, Nicholls DG. Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. J Neuro sci. 2007;27:7310–7317. doi: 10.1523/JNEUROSCI.0212-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scheffler IE, Yadava N, Potluri P. Molecular genetics of complex l-deficient Chinese hamster cell lines. Biochim Biophys Acta. 2004;1659:160–171. doi: 10.1016/j.bbabio.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 39.Affourtit C, Jastroch M, Brand MD. Uncoupling protein-2 attenuates glucose- stimulated insulin secretion in INS-1E insulinoma cells by lowering mitochondrial reactive oxygen species. Free Radic Biol Med. 2010;50:609–619. doi: 10.1016/j.freeradbiomed.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson BB, Moe PC, Wang D, Rossi K, Trigatti BL, et al. Modifications in perfringolysin O domain 4 alter the cholesterol concentration threshold required for binding. Biochemistry. 2012;51:3373–3382. doi: 10.1021/bi3003132. [DOI] [PubMed] [Google Scholar]
- 41.Steck TL, Lange Y. Cell cholesterol homeostasis: mediation by active cholesterol. Trends Cell Biol. 2010;20:680–687. doi: 10.1016/j.tcb.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lange Y, Swaisgood MH, Ramos BV, Steck TL. Plasma membranes contain half the phospholipid and 90% of the cholesterol and sphingomyelin in cultured human fibroblasts. J Biol Chem. 1989;264:3786–3793. [PubMed] [Google Scholar]
- 43.Yu W, Gong JS, Ko M, Garver WS, Yanagisawa K, et al. Altered cholesterol metabolism in Niemann-Pick type C1 mouse brains affects mitochondrial function. J Biol Chem. 2005;280:11731–11739. doi: 10.1074/jbc.M412898200. [DOI] [PubMed] [Google Scholar]
- 44.Rosenqvist E, Michaelsen TE, Vistnes AI. Effect of streptolysin O and digitonin on egg lecithin/cholesterol vesicles. Biochim Biophys Acta. 1980;600:91–102. doi: 10.1016/0005-2736(80)90414-9. [DOI] [PubMed] [Google Scholar]
- 45.Nishikawa M, Nojima S, Akiyama T, Sankawa U, Inoue K. Interaction of digitonin and its analogs with membrane cholesterol. J Biochem. 1984;96:1231–1239. doi: 10.1093/oxfordjournals.jbchem.a134941. [DOI] [PubMed] [Google Scholar]
- 46.Rogers GW, Brand MD, Petrosyan S, Ashok D, Elorza AA, et al. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS ONE. 2011;6:e21746. doi: 10.1371/journal.pone.0021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Divakaruni AS, Wiley SE, Rogers GW, Andreyev AY, Petrosyan S, et al. Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proc Natl Acad Sci USA. 2013;110:5422–5427. doi: 10.1073/pnas.1303360110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Affourtit C, Brand MD. Uncoupling protein-2 contributes significantly to high mitochondrial proton leak in INS-1E insulinoma cells and attenuates glucose-stimulated insulin secretion. Biochem J. 2008;409:199–204. doi: 10.1042/BJ20070954. [DOI] [PubMed] [Google Scholar]
- 49.Affourtit C, Brand MD. Stronger control of ATP/ADP by proton leak in pancreatic β-cells than skeletal muscle mitochondria. Biochem J. 2006;393:151–159. doi: 10.1042/BJ20051280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rubi B, del AA, Bartley C, Satrustegui J, Maechler P. The malate-aspartate NADH shuttle member Aralar1 determines glucose metabolic fate, mitochondrial activity, and insulin secretion in β cells. J BiolChem. 2004;279:55659–55666. doi: 10.1074/jbc.M409303200. [DOI] [PubMed] [Google Scholar]
- 51.Casimir M, Rubi B, Frigerio F, Chaffard G, Maechler P. Silencing of the mitochondrial NADH shuttle component aspartate-glutamate carrier AGC1/Aralar1 in INS-1E cells and rat islets. Biochem J. 2009;424:459–466. doi: 10.1042/BJ20090729. [DOI] [PubMed] [Google Scholar]
- 52.Jitrapakdee S, Wutthisathapornchai A, Wallace JC, MacDonald MJ. Regulation of insulin secretion: role of mitochondrial signalling. Diabetologia. 2010;53:1019–1032. doi: 10.1007/s00125-010-1685-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fahien LA, Laboy JI, Din ZZ, Prabhakar P, Budker T, et al. Ability of cytosolic malate dehydrogenase and lactate dehydrogenase to increase the ratio of NADPH to NADH oxidation by cytosolic glycerol-3-phosphate dehydrogenase. Arch Biochem Biophys. 1999;364:185–194. doi: 10.1006/abbi.1999.1117. [DOI] [PubMed] [Google Scholar]
- 54.Spegel P, Malmgren S, Sharoyko VV, Newsholme P, Koeck T, et al. Metabolomic analyses reveal profound differences in glycolytic and tricarboxylic acidcycle metabolism in glucose-responsive and -unresponsive clonal β-cell lines. Biochem J. 2011;435:277–284. doi: 10.1042/BJ20100655. [DOI] [PubMed] [Google Scholar]
- 55.MacDonald MJ, Marshall LK. Mouse lacking NAD+-linked glycerol phosphate dehydrogenase has normal pancreatic β cell function but abnormal metabolite pattern in skeletal muscle. Arch Biochem Biophys. 2000;384:143–153. doi: 10.1006/abbi.2000.2107. [DOI] [PubMed] [Google Scholar]
- 56.Hasan NM, Longacre MJ, Seed AM, Kendrick MA, Gu H, et al. Lower succinyl-CoA:3-ketoacid-CoA transferase (SCOT) and ATP citrate lyase in pancreatic islets of a rat model of type 2 diabetes: knockdown of SCOT inhibits insulin release in rat insulinoma cells. Arch Biochem Biophys. 2010;499:62–68. doi: 10.1016/j.abb.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nicholls DG, Shepherd D, Garland PB. A continuous recording technique for the measurement of carbon dioxide, and its application to mitochondrial oxidation and decarboxylation reactions. Biochem J. 1967;103:677–691. doi: 10.1042/bj1030677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Breen GA, Scheffler IE. Respiration-deficient Chinese hamster cell mutants: biochemical characterization. Somatic Cell Genet. 1979;5:441–451. doi: 10.1007/BF01538879. [DOI] [PubMed] [Google Scholar]
- 59.Au HC, Seo BB, Matsuno-Yagi A, Yagi T, Scheffler IE. The NDUFA1 gene product (MWFE protein) is essential for activity of complex I in mammalian mitochondria. Proc Natl Acad Sci USA. 1999;96:4354–4359. doi: 10.1073/pnas.96.8.4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Soderberg KL, Ditta GS, Scheffler IE. Mammalian cells with defective mitochondrial functions: a Chinese hamster mutant cell line lacking succinate dehydrogenase activity. Cell. 1977;10:697–702. doi: 10.1016/0092-8674(77)90103-9. [DOI] [PubMed] [Google Scholar]
- 61.Chang TW, Goldberg AL. A relationship between the rates of acetyl group oxidation and the oxygen consumption of cellls. J Biol Chem. 1978;253:3693–3695. [PubMed] [Google Scholar]
- 62.Moser MD, Matsuzaki S, Humphries KM. Inhibition of succinate-linked respiration and complex II activity by hydrogen peroxide. Arch Biochem Biophys. 2009;488:69–75. doi: 10.1016/j.abb.2009.06.009. [DOI] [PubMed] [Google Scholar]
- 63.Nicholls DG. Spare respiratory capacity, oxidative stress and excitotoxicity. Biochem Soc Trans. 2009;37:1385–1388. doi: 10.1042/BST0371385. [DOI] [PubMed] [Google Scholar]
- 64.Kuro-o M. A potential link between phosphate and aging–lessons from Klotho- deficient mice. Mech Ageing Dev. 2010;131:270–275. doi: 10.1016/j.mad.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nishi Y, Fujimoto S, Sasaki M, Mukai E, Sato H, et al. Role of mitochondrial phosphate carrier in metabolism-secretion coupling in rat insulinoma cell line INS-1. Biochem J. 2011;435:421–430. doi: 10.1042/BJ20101708. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.