

Abstract
Background
Rhodococcus equi is an important pulmonary pathogen in foals and in immunocompromised individuals. Virulent R. equi strains carry an 80-90 kb virulence plasmid that expresses the virulence-associated protein A (VapA). VapA expression is regulated by temperature and pH. The LysR-type transcriptional regulator, VirR, is involved in the regulation of the vapA gene. To examine the mechanism underlying transcriptional regulation of vapA, we characterized an R. equi mutant in which another putative transcriptional regulator encoded on the virulence plasmid, VirS, was deleted.
Results
Deletion of virS reduced vapA promoter activity to non-inducible levels. Complementary expression of VirS in the virS deletion mutant restored transcription at the PvapA promoter, even under non-inducing conditions (30°C and pH 8.0). In addition, VirS expression increased PvapA promoter activity in the absence of functional VirR. Further, transcription of the icgA operon containing virS was regulated by pH and temperature in the same manner as vapA.
Conclusions
This study suggests that VirS is required for VapA expression and that regulation of PvapA-promoter activity may be achieved by controlling VirS expression levels.
Electronic supplementary material
The online version of this article (doi:10.1186/s12866-014-0243-1) contains supplementary material, which is available to authorized users.
Keywords: Opportunistic infections, Rhodococcus equi, Rhodococcus equi VapA protein virulence, VirS
Background
Rhodococcus equi is a Gram-positive bacterium and a facultative intracellular pathogen of alveolar macrophages. Rhodococcus equi can cause bronchopneumonia in foals up to five months of age [1,2]. This bacterium has further been identified as an opportunistic pathogen in individuals compromised by immunosuppressive drug therapy, lymphoma, or acquired immunodeficiency syndrome (AIDS) [3-6].
Isolates from pneumonic foals possess a large plasmid that varies in size from 80 to 90 kb [7-9]. This plasmid is present in most clinical R. equi isolates recovered from infected foals but it is absent from most environmental strains [10]. Importantly, plasmid-cured isogenic mutants of virulent strains lose their ability to survive in macrophages and are unable to cause pneumonia in foals [11-14]. A highly immunogenic 15–17 kDa protein of unknown function, designated as virulence-associated protein A (VapA), is encoded within a pathogenicity island of this virulence plasmid [15]. VapA is essential for intracellular growth in macrophages and for full virulence in an infected mouse model [16].
The expression of vapA is controlled by temperature and pH, where maximum expression occurs at 34–41°C with a pH of 5.0 [17,18]. These characteristics suggest that vapA expression is intracellularly upregulated in the mammalian host. Indeed, transcription of vapA is increased in ex vivo murine and equine macrophages [19]. Furthermore, expression of VapA can be detected in macrophages recovered from pulmonary lesions of infected foals [20].
The virR gene encodes a LysR-type transcriptional regulator that affects vapA gene expression [21]. DNA binding studies have shown that VirR binds to a DNA fragment that contains the vapA promoter (PvapA). VirR alone can induce vapA expression, but VapA expression is enhanced when four genes downstream of virR are also present. One of these genes is virS; it encodes a protein that shares homology with the OmpR/PhoB family of response regulators [22]. It has not yet been demonstrated whether VirS is involved in the regulation of the PvapA-promoter activity.
In the present study, we constructed a virS deletion mutant and analyzed PvapA promoter activity using a R. equi strain that harbored a PvapA-lacZ fusion virulence plasmid. Our results suggest that VirS contributes to the regulation of vapA transcription, and is thus a critical component of R. equi virulence.
Methods
Bacterial strains and culture conditions
The R. equi ATCC33701 strain, originally isolated from a pneumonic foal, was used as the genetic background for all experiments reported in this study. Rhodococcus equi was routinely grown on Luria–Bertani (LB) agar at 30°C. Apramycin (60 μg/mL) was added to LB agar to select for R. equi growth when necessary. All R. equi strains were stored at −80°C in 85% LB broth/15% glycerol (vol/vol). Escherichia coli DH5α was grown on LB agar or in LB broth. Antibiotics were used when necessary at the following concentrations: apramycin (60 μg/mL) or ampicillin (50 μg/mL). All E. coli strains were stored at −80°C in 85% LB broth/15% glycerol (vol/vol). Table 1 describes all strains and plasmids used in this study.
Table 1.
Bacteria and plasmids used in this study
| Bacterial species and plasmids | Bacterial strains and plasmid names | Relevant characteristics | Source or reference |
|---|---|---|---|
| R. equi | ATCC33701 | virulent strain | |
| TKR255 | PvapA-lacZ fusion strain of ATCC33701 | This study | |
| TKR303 | ΔvirS of TKR255 | This study | |
| TKR474 | virRΔHTH of TKR255 | This study | |
| E. coli | DH5α | F-, Φ80dlacZΔM15, Δ(lacZYA-rgF)U169, deoR, recA1, endA1, hsdR17(rK-, mK+), phoA, supE44, λ-, thi-1, gyrA96, relA1 | |
| plasmid | pBluescript | Ampr | |
| pTKR131 | pBluescript::aac(3)IV | This study | |
| pECO101 | E. coli-C. jejuni shuttle vector | [23] | |
| pTKR144 | pTKR131::oriT | This study | |
| pTKR159 | pTKR144::PaphII | This study | |
| pDelta | pTKR159::codA-upp | This study | |
| pINT | pUC57::aac(3)IV-integrase | This study | |
| pGEM-T Easy | Ampr | Promega | |
| pTKR130 | pGEM, 3.5 kb fragment containg vapA | This study | |
| pTKR139 | ΔvapA (codon4-189) of pTKR130 | This study | |
| pTKR148 | pTKR139::lacZ | This study | |
| pTKR169 | pDela::PvapA-lacZ | This study | |
| pTKR333 | pGEM, 3.9 kb fragment containg virR | This study | |
| pTKR223 | pGEM, 3.9 kb fragment containg virS | This study | |
| pTKR361 | vapRΔHTH(codon2-50) of pTKR333 | This study | |
| pTKR226 | ΔvirS (codon2-252) of pTKR223 | This study | |
| pTKR265 | pDelta::vapRΔHTH | This study | |
| pTKR391 | pDelta::ΔvirS | This study | |
| pTKR174 | pGEM::PaphII | This study | |
| pTKR340 | pGEM::PaphII-virS | This study | |
| pTKR344 | pINT::PaphII-virS | This study | |
| . | pTKR445 | pINT::PaphII-virSD57A | This study |
| pTKR509 | pGEM::PvirR-VirR | This study | |
| pTKR528 | pINT::PvirR-VirR | This study | |
Western blot analysis
Cell extracts were boiled for 5 min in sodium dodecyl sulfate (SDS) solution (62.5 mM Tris–HCl [pH 6.8], 10% [vol/vol] glycerol, 2% [wt/vol] SDS, 5% [vol/vol] 2-mercaptoethanol, and 0.02% [wt/vol] bromophenol blue). SDS-polyacrylamide gel electrophoresis was performed using a 15% polyacrylamide gel according to the method described previously by Laemmli [24]. After electrophoresis, proteins were transferred to a nitrocellulose membrane (Protoran; GE Healthcare, Piscataway, NJ, USA), according to the manufacturer’s instructions. A monoclonal antibody against VapA (Mab10G5) was used for immunoblotting procedures [25].
Plasmid construction
To construct mutants with unmarked in-frame gene deletions within R. equi, a plasmid containing the codA-upp cassette was constructed to facilitate positive selection of targeted gene deletion mutants. Briefly, an apramycin resistance gene [aac(3)IV] was synthesized and cloned into pUC57at the EcoRI and HindIII sites. Next, the apramycin resistance gene cassette was excised by digestion with EcoRI and HindIII, then cloned into pBluescript II SK(+) digested with EcoRI and HindIII to create pTKR131. oriT was amplified from pEco101 by polymerase chain reaction (PCR) using primers oriT-F and oriT-R. The PCR product was digested with SpeI and EcoRI, and cloned into pTKR131 digested with SpeI and EcoRI to create pTKR144. The aphII promoter (PaphII) region was amplified using primers aph2-F and aph2-R. The amplified DNA fragment was digested with HindIII and ClaI, and cloned into pTKR144 digested with HindIII and ClaI to create pTKR159. Finally, the codA-upp cassette was excised from pORF-codA-upp (InvivoGen, San Diego, CA, USA) by digesting with NcoI and HindIII, and then cloned into pTKR159 digested with NcoI and HindIII to create pDelta. Primers used in this study are listed in Additional file 1: Table S1.
A plasmid containing the Streptomyces ϕC31 integrase gene was constructed to generate the integration vector for the complementation experiments [26]. The ϕC31 integrase gene flanked by ApaI sites was synthesized and cloned into pUC57 digested with EcoRV. The ϕC31 integrase gene was excised with ApaI and cloned into ApaI-digested pTKR131 to create pINT.
Construction of a vapA::lacZ fusion R. equi ATCC33701 strain
To construct the transcriptional fusion product containing the vapA promoter and the lacZ open reading frame (ORF), the primer pair vapA-LF and vapA-LR was designed according to the published sequence of pRE701 [22] and used for PCR amplification of a 3.5 kb fragment that included approximately 1,500 nucleotides upstream and downstream of vapA. This fragment was cloned into the pGEM-T Easy vector (Promega, Tokyo, Japan) to create pTKR130. PCR-mediated mutagenesis was used to delete the vapA gene and to create BglII and MfeI sites within the coding sequence, and to produce pTKR139 using pTKR130 as the template with the primer pair ΔvapA-1 and ΔvapA-2. The deleted region in the vapA gene comprised codons 4–189. The promoterless lacZ gene was excised from pORF-lacZ (InvivoGen) by digesting with BamHI and EcoRI, and then ligated to pTKR139 digested with BclI and MfeI to create pTKR148. The DNA fragment that contained the PvapA-lacZ fusion was excised from pTKR148 by digesting with EcoRI, and then ligated to EcoRI-digested pDelta to create pTKR169. pTKR169 was electroporated into R. equi ATCC33701 as described previously [27]. Transformants (single crossovers) were selected on LB agar containing apramycin (60 μg/mL). 5-Fluorocytosine (5-FC) positive selection was performed as described previously [28]. Briefly, R. equi transformants were inoculated into LB liquid medium and grown overnight at 30°C. 5-FC selection of double crossovers was performed by plating 100-μL aliquots of a dilution series [10−1 to 10−3 in mineral acetate (MM-Ac) medium] of the culture onto MM-Ac agar plates supplemented with 5-FC (100 μg/mL). Plates were incubated at 30°C for 2–3 days. Virulence plasmids were isolated from 5-FC-resistant and apramycin-sensitive mutants, and analyzed by digestion with EcoRI. Mutants that produced the expected digestion pattern were selected (Additional file 2: Figure S1). The mutated locus was further analyzed by PCR and sequencing. One mutant strain was selected, designated TKR255, and used for further characterization.
Construction of R. equi ΔvirS and virRΔHTH strains harboring the PvapA-lacZ fusion
To construct in-frame virR and virS deletion mutants, 3.9 kb and 3.8 kb fragments including approximately 1,500 nucleotides upstream and downstream of virR and virS, respectively, were amplified by PCR using the primer pairs virR-LF and virR-LR, and virS-LF and virS-LR. These fragments were cloned into the pGEM-T Easy vector to create pTKR333 and pTKR223. PCR-mediated mutagenesis was employed to delete the virR and virS genes using primer pairs ΔvirR-1 and ΔvirR-2, and ΔvirS-1 and ΔvirS-2, respectively. pTKR333 and pTKR223 were used as templates to create pTKR361 and pTKR226, respectively. The deleted region in the virR gene comprised codons 2–50 (virRΔHTH). The deleted region in the virS gene comprised codons 2–252. Fragments that contained ΔvirS and virRΔHTH were excised from pTKR361 and pTKR226 by EcoRI digestion and ligated to EcoRI-digested pDelta to create pTKR265 and pTKR391, respectively. pTKR265 and pTKR391 were separately electroporated into the PvapA-lacZ strain (TKR255), and the ΔvirS and virRΔHTH mutants (TKR303 and TKR474, respectively) were selected and confirmed as described above (Additional file 3: Figure S2 and Additional file 4: Figure S3).
Complementation of R. equi mutants
The virS ORF was amplified using the primer pair virS-NcoF and virS-HindR. The PCR product was digested with NcoI and HindIII, and cloned into pTKR174 digested with NcoI and HindIII to create pTKR340. pTKR340 was digested with NotI and ligated to NotI-digested pINT to create pTKR344. pTKR344 was electroporated into TRK303. The transformants were recovered on LB agar containing 60 μg/mL apramycin. PCR-mediated mutagenesis was used to introduce point mutations into the coding sequence of virS in pTKR344, and pTKR445 (pINT::virSD57A) was produced using the primer pair virS D57A-1 and virS D57A-2. This plasmid was electroporated into TKR303. Transformants were recovered on LB agar containing 60 μg/mL apramycin.
The fragment that contained the virR ORF and promoter region was amplified using the primer pair PvirR-F and virR-R. The DNA fragment was cloned into the pGEM-T vector to create pTKR509. pTKR509 was digested with NotI and ligated to NotI-digested pINT to create pTKR528. pTKR528 was electroporated into TKR474. Transformants were recovered on LB agar containing 60 μg/mL apramycin.
β-Galactosidase assays
Cells were grown overnight at 30°C in brain-heart infusion (BHI) broth with shaking. Cultures were diluted to 1: 10 with 60 mM Tris-buffered BHI, and the pH was adjusted to pH 6.5 or pH 8.0. Cultures were grown until they reached an optical density at 600 nm (OD600) of 0.5–0.7. Cells were washed twice with 0.9% NaCl and resuspended in 500 μL Z buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol, pH 7.6) [29]. Next, cells were permeabilized by adding 20 μL chloroform and 35 μL 0.1% SDS. One hundred microliters of 13 mM 2-nitrophenyl beta-D-galactopyranoside (Sigma-Aldrich, St Louis, MO, USA) was added to each sample, followed by incubation at 28°C for 5 min. The reaction was stopped by adding 250 μL 1 M Na2CO3, and absorbance was read at 420 nm using a spectrophotometer (GENESYS 20; Thermo Fisher Scientific, Waltham, MA, USA). The activity of each sample was calculated in Miller units as follows: 1,000 × OD420/OD600 × reaction time × volume. Assays were performed in triplicate at least three times. Graphs were created using GraphPad PRISM software.
Transcriptional analysis of the operon containing virS
Total bacterial RNA was isolated from 5 mL cultures grown to the mid-logarithmic phase (OD600 = 0.25). Next, 10 mL of RNAprotect Bacteria Reagent (Qiagen, Hilden, Germany) was added to the bacterial cultures, immediately mixed, and incubated for 5 min at room temperature. Cells were harvested by centrifugation for 10 min at 5,000 × g at 4°C. Following this, cells were resuspended in 1 mL of RLT buffer (RNeasy Mini Kit; Qiagen) and added to 0.5 mL of 0.1 mm diameter zirconia-silica beads (μT-01; TAITEC, Saitama, Japan). Samples were lysed three times for 1 min with a bead beater (TAITEC) at 4,600 rpm. Total RNA was isolated using an RNeasy RNA mini kit (Qiagen), according to the manufacturer’s instructions. To eliminate DNA contamination, RNA was treated with 10 U of RNase-free DNase for 30 min at 37°C. DNase was inactivated by incubating the mixture for 5 min at 75°C. Next, 200 ng RNA was mixed with random 6-mers and cDNA was synthesized using a PrimeScript RT–PCR kit (Takara, Tokyo, Japan), according to the manufacturer’s instructions. Real-time RT-PCR analysis was performed in a 20-μL volume that contained 1× PowerSYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA), 200 nM forward and reverse primers, and the sample cDNA. The primer pairs used to amplify vapH, orf7, and virS were vapH-RTF and vapH-RTR, orf7-RTF, and orf7-RTR, and virS-RTF and virS-RTR, respectively. Reactions were performed with StepOne Real-Time PCR System (Applied Biosystems) using the following conditions: 95°C for 10 min, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. Results were normalized using 16S rRNA as a control and analyzed with the ΔΔCT method. Graphs were created using GraphPad PRISM software.
Results
Development of a reporter system to analyze vapA gene expression
Previous studies have reported that vapA gene expression is regulated by temperature and pH [17,18]. To confirm these regulatory effects, we performed western blot analysis on cellular extracts of the R. equi ATCC33701 strain grown under four different conditions: 30°C at pH 6.5, 30°C at pH 8.0, 37°C at pH 6.5, and 37°C at pH 8.0 (Figure 1A). Maximal expression of VapA occurred at 37°C and pH 6.5. At 37°C and pH 8.0, VapA expression was lower but still detectable. When bacteria were grown at 30°C; however, VapA expression was undetectable. To measure the promoter activity of the vapA gene using a β-galactosidase assay, we constructed a mutant strain wherein the virulence plasmid contained a PvapA-lacZ fusion. The highest β-galactosidase activity was detected when this strain was grown at 37°C and pH 6.5 (Figure 1B). At 37°C and pH 8.0, β-galactosidase activity was lower. Thus, these results agreed with the results of the western blot analysis. At 30°C, β-galactosidase activity was approximately 12-fold lower than that at 37°C and pH 6.5. Importantly, these results indicated that this reporter strain could be used to analyze vapA gene expression.
Figure 1.
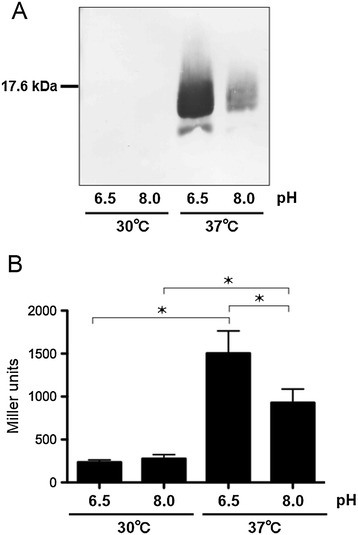
Regulation of vapA transcription and VapA expression in R. equi . (A) Western blot analysis performed with an anti-VapA antibody on cell extracts prepared from R. equi grown under conditions indicated in the Figure (B) β-galactosidase activity assays of PvapA –lacZ fusions in the R. equi TKR255 strain grown at different temperatures and pH indicated in the Figure TKR255 was grown overnight at 30°C, diluted to an OD60th0 of 0.05 in fresh medium at the pH indicated, and incubated at the temperature indicated. β-galactosidase activity was measured in Miller units and error bars represent standard deviations for each data set (n = 3). Data were evaluated for statistical significance using one-way ANOVA followed by the Dunnett’s multiple comparison test, *p < 0.001.
VirS is required for vapA expression
A previous study reported that VapA expression was higher when four genes including virS were present in addition to virR, when compared with virR alone (Figure 2) [21]. To examine whether this increase could be attributed to VirS, the virS gene was deleted from the virulence plasmid in the PvapA-lacZ fusion strain. In the ΔvirS mutant, vapA promoter activity was reduced to a non-detectable level (Figure 3). Complementation of the ΔvirS mutant with virS expressed via from the PaphII promoter on the bacterial chromosome increased PvapA-lacZ expression by more than two-fold when this strain was grown under inducing conditions. Moreover, when the complemented mutant was grown at 30°C (under non-inducing conditions), the transcription level of PvapA-lacZ was the same as that of the strain grown under inducing conditions. These results suggest that VirS is required for vapA expression and that vapA transcription can be induced if VirS is expressed, even when grown under non-inducing conditions.
Figure 2.
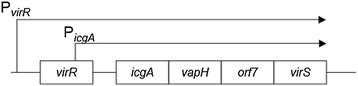
Schematic of the virR and icgA operons. Open boxes represent ORFs. Arrows indicate the transcript and its orientation.
Figure 3.
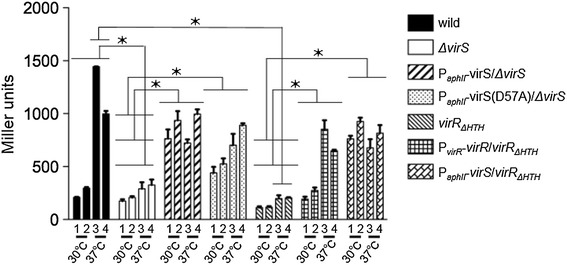
β-Galactosidase activity assays of P vapA -acZ virS and virR deletion mutant strains. β-Galactosidase activity (measured in Miller units) of the wild-type strain (TKR255), ΔvirS, pINT::PaphII-virS-integrated ΔvirS (PaphII-virS/ΔvirS), pINT::PaphII-virS (D57A)-integrated ΔvirS [PaphII-virS (D57A)/ΔvirS], virR ΔHTH, pINT::PvirR-virR-integrated virR ΔHTH (PvirR-virR/virR ΔHTH), and pINT::PaphII-virS-integrated virR ΔHTH (PaphII-virS/virR ΔHTH) strains grown at pH 6.5 (lanes 1 and 3) or pH 8.0 (lanes 2 and 4). Error bars represent standard deviations for each data set (n = 3). Data were evaluated for statistical significance using one-way ANOVA followed by the Dunnett’s multiple comparison test, *p < 0.001.
Phosphorylation of Asp57 is not required for the function of VirS
Activation of OmpR/PhoB family response regulators requires phosphorylation of a conserved aspartate residue [30]. Interestingly, VirS contains an aspartate residue (Asp57) that represents a putative phosphorylation site (Figure 4). To determine whether Asp57 in VirS is necessary for VirS function, Asp57 was replaced with alanine via site-directed mutagenesis. Transcription of PvapA-lacZ in the virS (Asp57Ala) mutant was comparable to that of the strain expressing wild-type VirS when they were both grown under inducing conditions (Figure 3). These results suggest that the putative phosphorylation site Asp57 is not necessary for VirS function.
Figure 4.

Primary sequence alignment of VirS and other OmpR/PhoB subfamily members ( E. coli CheY, PhoB, and OmpR). Similarity between homologous proteins is highlighted by differences in shading: black, all amino acids in a column are identical; light gray, over half of the amino acids in a column are identical. The putative phosphorylation site (Asp57), conserved catalytic residues (Asp12, Asp13, and Lys109), and conserved conformational switch residues (Thr87 and Tyr106) are indicated by an asterisk, closed stars, and open stars, respectively. Proteins were aligned according to GENETYX-MAC software.
VirS can increase PvapA promoter activity in the absence of a functional VirR
Previous reports suggest that VirR is required for the expression of both vapA and virS [21,31], and our results support the hypothesis that VirS can increase vapA expression in the presence of virR. To examine whether VirS function is VirR dependent, we constructed a virR deletion mutant. As the promoter of the icgA operon containing virS is located within the virR ORF (Figure 2), we did not delete the entire virR gene. Instead, only the locus that corresponded to the helix-turn-helix region (codons 2–50) was deleted and the promoter of the icgA operon was kept intact. The virRΔHTH mutation reduced the transcription level of PvapA-lacZ to undetectable levels under non-inducing conditions (Figure 3). When virS was expressed from the chromosomal PaphII promoter in the virRΔHTH mutant, the PvapA promoter was activated to comparable levels detected in the presence of virR. These results suggest that VirS can activate transcription of the PvapA promoter in the absence of VirR.
Transcription of the icgA operon is regulated by temperature and pH
A previous study reported that transcription of the icgA operon (Figure 2) was VirR dependent and was induced at 37°C and pH 6.5 [31]. Although inducing (37°C at pH 8.0) and non-inducing (30°C at pH 6.5) conditions were compared, it was still not clear whether the transcription of the icgA operon was regulated by temperature, pH, or both. Thus, we semi-quantitatively determined the transcriptional level of the icgA operon by real-time RT-PCR. As shown previously, transcription of all the genes in this operon was induced when the wild-type strain was grown at 37°C and pH 6.5 (Figure 5A). However, their transcriptional levels were lower when cells were grown at 37°C and pH 8.0. Transcription was even lower when cells were grown at 30°C. In agreement with the previous study, we did not observe an increase in the transcription of the icgA operon in the virRΔHTH mutant (Figure 5B). These results demonstrate that transcription of the icgA operon is VirR dependent and that it is regulated by both temperature and pH.
Figure 5.
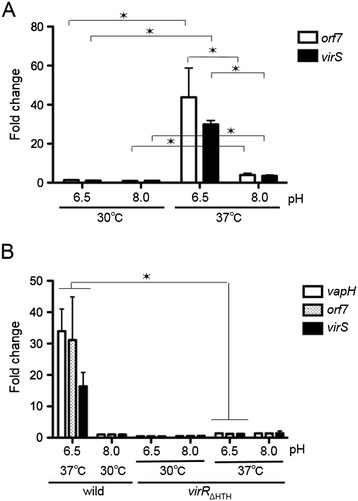
Effects of temperature and pH on P icgA promoter activity. mRNA isolated from R. equi wild-type (A) and virR ΔHTH (B) strains were grown under the conditions indicated, followed by semi-quantitative analysis of transcription using real-time RT-PCR. Values represent the fold increase in expression relative to expression in the wild-type grown at 30°C and pH 8.0. Data were evaluated for statistical significance using one-way ANOVA followed by the Dunnett’s multiple comparison test, *p < 0.001.
Discussion
Pneumonia-causing virulence by the bacterial pulmonary pathogen R. equi has not been fully elucidated. However, a previous study has demonstrated that VirR is involved in the regulation of the vapA gene located on the R. equi virulence plasmid [21]. To examine the contribution of virS (located downstream of virR) to R. equi virulence and the expression of VapA, we constructed an R. equi virS deletion strain paired with a PvapA-lacZ reporter virulence plasmid. In the current study, we demonstrate that VirS is another transcriptional regulator encoded on the virulence plasmid that is required for vapA transcription.
With this work, we show that deletion of virS reduced the transcriptional activation of the vapA promoter to non-inducible levels. Further, previous studies have demonstrated that virR is constitutively expressed and that VirS has no effect on the transcription of virR [31]. Together, these data suggested that VirR expression would be unaltered in a ΔvirS mutant, and that VirR alone is not sufficient to activate the transcription of vapA in the absence of VirS. In addition, deletion of virR completely abolished vapA promoter activity. However, it has previously been demonstrated that VirR is required for VirS expression [31]. Thus, we hypothesized that VirS has an indirect effect on vapA gene transcription. Indeed, VirS expression via the PaphII promoter restored expression of the PvapA-lacZ fusion plasmid in the virRΔHTH mutant, thereby supporting this hypothesis. Furthermore, when VirS was expressed via the PaphII promoter, there was no difference in transcription from the vapA promoter in the presence or absence of VirR. These results indicate that VirS can activate vapA transcription via the vapA promoter in the absence of functional VirR.
Interestingly, chromosomal integration of the PaphII-virS fusion did not restore PvapA-lacZ expression to wild-type levels in the ΔvirS and virRΔHTH deletion mutants. Furthermore, activation of the PvapA promoter was observed under non-inducing conditions. It is possible that expression of the PaphII-virS fusion, which was present as a monocopy on the chromosome, may have resulted in lower VirS expression levels when compared with VirS expression from the virulence plasmid, as each cell harbors two or more plasmid copies [32]. In addition, this may have caused disordered regulation when VirS was expressed from the PaphII promoter but not from the original promoter found on the virulence plasmid.
VirS is an orphan response regulator, and its cognate sensor is not found on the virulence plasmid [22]. VirS can activate the PvapA promoter under non-inducing conditions; thus, pH and temperature are unlikely to be the stimuli that are responsible for VirS activation. We further observed that the Asp57Ala mutation did not affect activity of the PvapA promoter when compared with that of wild-type VirS under inducing conditions. These data suggest that this putative phosphorylation site is not necessary for function of this domain. Although most residues that are critical for canonical functions in response regulators are well conserved in VirS, Asp13, which chelates the Mg2+ necessary for aspartic acid phosphorylation, is substituted with arginine [33-35]. Substitution of this conserved residue in the E. coli CheY protein means that it cannot chelate Mg2+ effectively and phosphorylation of Asp57 is blocked [30]. However, other OmpR/PhoB subfamily members such as Myxococcus xanthus FrzS and Helicobacter pylori HP1043 retain their functional activity in the absence of phosphorylation [36-38]. Therefore, VirS may be another atypical response regulator that does not require a sensor protein for activation.
In the present study, we demonstrated that the PicgA promoter is regulated by both temperature and pH, and this corresponds to regulation of the PvapA promoter. Further, expression of VirS from the PaphII promoter could induce the vapA transcription under non-inducing conditions. These results suggest that regulation of vapA by temperature and pH may be achieved by controlling VirS expression levels, and a proposed model for this regulation is presented in Figure 6. As such, mechanisms that regulate expression of the icgA operon are likely crucial in controlling vapA gene expression via environmental stimuli in the bacterial pathogen R. equi.
Figure 6.
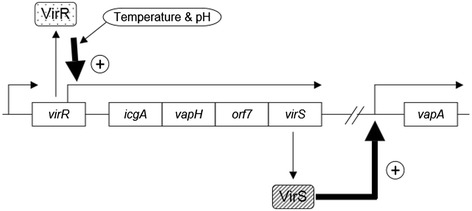
Proposed model of vapA transcriptional regulation in R. equi . Transcription of the icgA operon is VirR dependent and is regulated by temperature and pH via unknown mechanisms. VirS activates transcription of the vapA promoter.
Conclusions
VirS is required for the expression of vapA, and VirS expression is regulated by both temperature and pH. We hypothesize that regulation of the PvapA promoter is influenced by VirS expression levels. Future studies are required to examine the mechanisms that regulate transcription of the virS-containing icgA operon, and how this operon contributes to R. equi virulence.
Acknowledgments
This research was partly supported by Grants-in-Aid for Scientific Research (No. 24580452 to TK) from the Japan Society for the Promotion of Science.
Additional files
Primer used in this study.
Construction of the R. equi vapA::lacZ fusion strain by homologous recombination.
Targeted mutagenesis of virS by homologous recombination.
Targeted mutagenesis of virR by homologous recombination.
Footnotes
Competing interest
The authors declare that they have no competing financial interests.
Authors’ contributions
Conceived and designed the experiments: TK, ST. Performed the experiments: TH, TT, HH, SM, and TK. Data analysis: TK and ST. Contributed reagents/materials/analysis tools: TK. Manuscript preparation: TK and ST. All authors read and approved the final manuscript.
Contributor Information
Tsutomu Kakuda, Email: kakuda@vmas.kitasato-u.ac.jp.
Takuya Hirota, Email: 12345roncha-wa-good-taste@ezweb.ne.jp.
Tatsuya Takeuchi, Email: taketatu-tatuya-0915@ezweb.ne.jp.
Hirofumi Hagiuda, Email: hahaha3500@gmail.com.
Shiko Miyazaki, Email: wwe-stone.cold316_slipknot_mth@ezweb.ne.jp.
Shinji Takai, Email: takai@vmas.kitasato-u.ac.jp.
References
- 1.Takai S. Epidemiology of Rhodococcus equi infections: a review. Vet Microbiol. 1997;56:167–176. doi: 10.1016/S0378-1135(97)00085-0. [DOI] [PubMed] [Google Scholar]
- 2.Prescott JF. Rhodococcus equi: an animal and human pathogen. Clin Microbiol Rev. 1991;4:20–34. doi: 10.1128/cmr.4.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takai S, Sasaki Y, Ikeda T, Uchida Y, Tsubaki S, Sekizaki T. Virulence of Rhodococcus equi isolates from patients with and without AIDS. J Clin Microbiol. 1994;32:457–460. doi: 10.1128/jcm.32.2.457-460.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takai S, Imai Y, Fukunaga N, Uchida Y, Kamisawa K, Sasaki Y, Tsubaki S, Sekizaki T. Identification of virulence-associated antigens and plasmids in Rhodococcus equi from patients with AIDS. J Infect Dis. 1995;172:1306–1311. doi: 10.1093/infdis/172.5.1306. [DOI] [PubMed] [Google Scholar]
- 5.Takai S, Tharavichitkul P, Takarn P, Khantawa B, Tamura M, Tsukamoto A, Takayama S, Yamatoda N, Kimura A, Sasaki Y, Kakuda T, Tsubaki S, Maneekarn N, Sirisanthana T, Kirikae T. Molecular epidemiology of Rhodococcus equi of intermediate virulence isolated from patients with and without acquired immune deficiency syndrome in Chiang Mai, Thailand. J Infect Dis. 2003;188:1717–1723. doi: 10.1086/379739. [DOI] [PubMed] [Google Scholar]
- 6.Takai S, Tharavichitkul P, Sasaki C, Onishi Y, Yamano S, Kakuda T, Tsubaki S, Trinarong C, Rojanasthien S, Sirimalaisuwan A, Tesaprateep T, Maneekarn N, Sirisanthana T, Kirikae T. Identification of virulence-associated antigens and plasmids in Rhodococcus equi from patients with acquired immune deficiency syndrome and prevalence of virulent R. equi in soil collected from domestic animal farms in Chiang Mai, Thailand. Am J Trop Med Hyg. 2002;66:52–55. doi: 10.4269/ajtmh.2002.66.52. [DOI] [PubMed] [Google Scholar]
- 7.Takai S, Sekizaki T, Ozawa T, Sugawara T, Watanabe Y, Tsubaki S. Association between a large plasmid and 15- to 17-kilodalton antigens in virulent Rhodococcus equi. Infect Immun. 1991;59:4056–4060. doi: 10.1128/iai.59.11.4056-4060.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takai S, Shoda M, Sasaki Y, Tsubaki S, Fortier G, Pronost S, Rahal K, Becu T, Begg A, Browning G, Nicholson VM, Prescott JF. Restriction fragment length polymorphisms of virulence plasmids in Rhodococcus equi. J Clin Microbiol. 1999;37:3417–3420. doi: 10.1128/jcm.37.10.3417-3420.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takai S, Murata N, Kudo R, Narematsu N, Kakuda T, Sasaki Y, Tsubaki S. Two new variants of the Rhodococcus equi virulence plasmid, 90 kb type III and type IV, recovered from a foal in Japan. Vet Microbiol. 2001;82:373–381. doi: 10.1016/S0378-1135(01)00398-4. [DOI] [PubMed] [Google Scholar]
- 10.Takai S, Watanabe Y, Ikeda T, Ozawa T, Matsukura S, Tamada Y, Tsubaki S, Sekizaki T. Virulence-associated plasmids in Rhodococcus equi. J Clin Microbiol. 1993;31:1726–1729. doi: 10.1128/jcm.31.7.1726-1729.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hondalus MK, Mosser DM. Survival and replication of Rhodococcus equi in macrophages. Infect Immun. 1994;62:4167–4175. doi: 10.1128/iai.62.10.4167-4175.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wada R, Kamada M, Anzai T, Nakanishi A, Kanemaru T, Takai S, Tsubaki S. Pathogenicity and virulence of Rhodococcus equi in foals following intratracheal challenge. Vet Microbiol. 1997;56:301–312. doi: 10.1016/S0378-1135(97)00098-9. [DOI] [PubMed] [Google Scholar]
- 13.Giguere S, Hondalus MK, Yager JA, Darrah P, Mosser DM, Prescott JF. Role of the 85-kilobase plasmid and plasmid-encoded virulence-associated protein A in intracellular survival and virulence of Rhodococcus equi. Infect Immun. 1999;67:3548–3557. doi: 10.1128/iai.67.7.3548-3557.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toyooka K, Takai S, Kirikae T. Rhodococcus equi can survive a phagolysosomal environment in macrophages by suppressing acidification of the phagolysosome. J Med Microbiol. 2005;54:1007–1015. doi: 10.1099/jmm.0.46086-0. [DOI] [PubMed] [Google Scholar]
- 15.Sekizaki T, Takai S, Egawa Y, Ikeda T, Ito H, Tsubaki S. Sequence of the Rhodococcus equi gene encoding the virulence-associated 15–17-kDa antigens. Gene. 1995;155:135–136. doi: 10.1016/0378-1119(95)00009-U. [DOI] [PubMed] [Google Scholar]
- 16.Jain S, Bloom BR, Hondalus MK. Deletion of vapA encoding Virulence Associated Protein A attenuates the intracellular actinomycete Rhodococcus equi. Mol Microbiol. 2003;50:115–128. doi: 10.1046/j.1365-2958.2003.03689.x. [DOI] [PubMed] [Google Scholar]
- 17.Takai S, Iie M, Watanabe Y, Tsubaki S, Sekizaki T. Virulence-associated 15- to 17-kilodalton antigens in Rhodococcus equi: temperature-dependent expression and location of the antigens. Infect Immun. 1992;60:2995–2997. doi: 10.1128/iai.60.7.2995-2997.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takai S, Fukunaga N, Kamisawa K, Imai Y, Sasaki Y, Tsubaki S. Expression of virulence-associated antigens of Rhodococcus equi is regulated by temperature and pH. Microbiol Immunol. 1996;40:591–594. doi: 10.1111/j.1348-0421.1996.tb01113.x. [DOI] [PubMed] [Google Scholar]
- 19.Ren J, Prescott JF. Analysis of virulence plasmid gene expression of intra-macrophage and in vitro grown Rhodococcus equi ATCC 33701. Vet Microbiol. 2003;94:167–182. doi: 10.1016/S0378-1135(03)00099-3. [DOI] [PubMed] [Google Scholar]
- 20.Madarame H, Takai S, Morisawa N, Fujii M, Hidaka D, Tsubaki S, Hasegawa Y. Immunohistochemical detection of virulence-associated antigens of Rhodococcus equi in pulmonary lesions of foals. Vet Pathol. 1996;33:341–343. doi: 10.1177/030098589603300312. [DOI] [PubMed] [Google Scholar]
- 21.Russell DA, Byrne GA, O’Connell EP, Boland CA, Meijer WG. The LysR-type transcriptional regulator VirR is required for expression of the virulence gene vapA of Rhodococcus equi ATCC 33701. J Bacteriol. 2004;186:5576–5584. doi: 10.1128/JB.186.17.5576-5584.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takai S, Hines SA, Sekizaki T, Nicholson VM, Alperin DA, Osaki M, Takamatsu D, Nakamura M, Suzuki K, Ogino N, Kakuda T, Dan H, Prescott JF. DNA sequence and comparison of virulence plasmids from Rhodococcus equi ATCC 33701 and 103. Infect Immun. 2000;68:6840–6847. doi: 10.1128/IAI.68.12.6840-6847.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wiesner RS, Hendrixson DR, DiRita VJ. Natural transformation of Campylobacter jejuni requires components of a type II secretion system. J Bacteriol. 2003;185:5408–5418. doi: 10.1128/JB.185.18.5408-5418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 25.Takai S, Iie M, Kobayashi C, Morishita T, Nishio T, Ishida T, Fujimura T, Sasaki Y, Tsubaki S. Monoclonal antibody specific to virulence-associated 15- to 17-kilodalton antigens of Rhodococcus equi. J Clin Microbiol. 1993;31:2780–2782. doi: 10.1128/jcm.31.10.2780-2782.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hong Y, Hondalus MK. Site-specific integration of Streptomyces PhiC31 integrase-based vectors in the chromosome of Rhodococcus equi. FEMS Microbiol Lett. 2008;287:63–68. doi: 10.1111/j.1574-6968.2008.01298.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sekizaki T, Tanoue T, Osaki M, Shimoji Y, Tsubaki S, Takai S. Improved electroporation of Rhodococcus equi. J Vet Med Sci. 1998;60:277–279. doi: 10.1292/jvms.60.277. [DOI] [PubMed] [Google Scholar]
- 28.van der Geize R, de Jong W, Hessels GI, Grommen AW, Jacobs AA, Dijkhuizen L. A novel method to generate unmarked gene deletions in the intracellular pathogen Rhodococcus equi using 5-fluorocytosine conditional lethality. Nucleic Acids Res. 2008;36:e151. doi: 10.1093/nar/gkn811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1972. [Google Scholar]
- 30.Bourret RB, Hess JF, Simon MI. Conserved aspartate residues and phosphorylation in signal transduction by the chemotaxis protein CheY. Proc Natl Acad Sci U S A. 1990;87:41–45. doi: 10.1073/pnas.87.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Byrne GA, Russell DA, Chen X, Meijer WG. Transcriptional regulation of the virR operon of the intracellular pathogen Rhodococcus equi. J Bacteriol. 2007;189:5082–5089. doi: 10.1128/JB.00431-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodríguez-Lázaro D, Lewis DA, Ocampo-Sosa AA, Fogarty U, Makrai L, Navas J, Scortti M, Hernández M, Vázquez-Boland JA. Internally controlled real-time PCR method for quantitative species-specific detection and vapA genotyping of Rhodococcus equi. Appl Environ Microbiol. 2006;72:4256–4263. doi: 10.1128/AEM.02706-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Djordjevic S, Stock AM. Structural analysis of bacterial chemotaxis proteins: components of a dynamic signaling system. J Struct Biol. 1998;124:189–200. doi: 10.1006/jsbi.1998.4034. [DOI] [PubMed] [Google Scholar]
- 34.Zundel CJ, Capener DC, McCleary WR. Analysis of the conserved acidic residues in the regulatory domain of PhoB. FEBS Lett. 1998;441:242–246. doi: 10.1016/S0014-5793(98)01556-7. [DOI] [PubMed] [Google Scholar]
- 35.Lewis RJ, Brannigan JA, Muchová K, Barák I, Wilkinson AJ. Phosphorylated aspartate in the structure of a response regulator protein. J Mol Biol. 1999;294:9–15. doi: 10.1006/jmbi.1999.3261. [DOI] [PubMed] [Google Scholar]
- 36.Fraser JS, Merlie JP, Jr, Echols N, Weisfield SR, Mignot T, Wemmer DE, Zusman DR, Alber T. An atypical receiver domain controls the dynamic polar localization of the Myxococcus xanthus social motility protein FrzS. Mol Microbiol. 2007;65:319–332. doi: 10.1111/j.1365-2958.2007.05785.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hong E, Lee HM, Ko H, Kim DU, Jeon BY, Jung J, Shin J, Lee SA, Kim Y, Jeon YH, Cheong C, Cho HS, Lee W. Structure of an atypical orphan response regulator protein supports a new phosphorylation-independent regulatory mechanism. J Biol Chem. 2007;282:20667–20675. doi: 10.1074/jbc.M609104200. [DOI] [PubMed] [Google Scholar]
- 38.Schär J, Sickmann A, Beier D. Phosphorylation-independent activity of atypical response regulators of Helicobacter pylori. J Bacteriol. 2005;187:3100–3109. doi: 10.1128/JB.187.9.3100-3109.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]