

Abstract
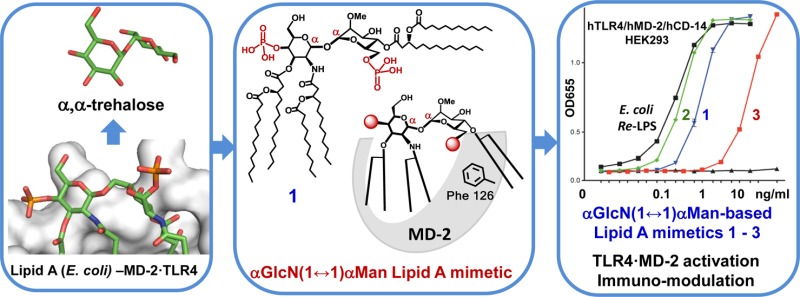
The endotoxic portion of lipopolysaccharide (LPS), a glycophospholipid Lipid A, initiates the activation of the Toll-like Receptor 4 (TLR4)–myeloid differentiation factor 2 (MD-2) complex, which results in pro-inflammatory immune signaling. To unveil the structural requirements for TLR4·MD-2-specific ligands, we have developed conformationally restricted Lipid A mimetics wherein the flexible βGlcN(1→6)GlcN backbone of Lipid A is exchanged for a rigid trehalose-like αGlcN(1↔1)αMan scaffold resembling the molecular shape of TLR4·MD-2-bound E. coli Lipid A disclosed in the X-ray structure. A convergent synthetic route toward orthogonally protected αGlcN(1↔1)αMan disaccharide has been elaborated. The α,α-(1↔1) linkage was attained by the glycosylation of 2-N-carbamate-protected α-GlcN-lactol with N-phenyl-trifluoroacetimidate of 2-O-methylated mannose. Regioselective acylation with (R)-3-acyloxyacyl fatty acids and successive phosphorylation followed by global deprotection afforded bis- and monophosphorylated hexaacylated Lipid A mimetics. αGlcN(1↔1)αMan-based Lipid A mimetics (α,α-GM-LAM) induced potent activation of NF-κB signaling in hTLR4/hMD-2/CD14-transfected HEK293 cells and robust LPS-like cytokines expression in macrophages and dendritic cells. Thus, restricting the conformational flexibility of Lipid A by fixing the molecular shape of its carbohydrate backbone in the “agonistic” conformation attained by a rigid αGlcN(1↔1)αMan scaffold represents an efficient approach toward powerful and adjustable TLR4 activation.
Introduction
Toll-like Receptor 4 (TLR4) is a mammalian transmembrane receptor protein which, in complex with a myeloid differentiation factor 2 (MD-2), detects picomolar concentrations of Gram-negative bacterial endotoxin (or lipopolysaccharide, LPS) (Figure 1A) and initiates an inflammatory signaling cascade aimed at the eradication of bacterial infection.1 Activation of the innate immune response through TLR4·MD-2-LPS complex was shown to contribute to the pathogenesis of numerous inflammatory, autoimmune, and chronic diseases, such as sepsis syndrome, asthma, arthritis, and cancer, which highlights the significance of TLR4-MD-2 complex as a therapeutic target.2−5 Therapeutic modulation of the innate immune response by intervention with TLR4·MD-2 signaling has grown to a “hot” topic in the past decade.4,6 Moreover, activation of TLR4 has been proposed to bridge the innate and adaptive immunity,7 emphasizing stimulation of the TLR4·MD-2 complex by nontoxic ligands as a straightforward way to efficient vaccine adjuvants.8−10
Figure 1.
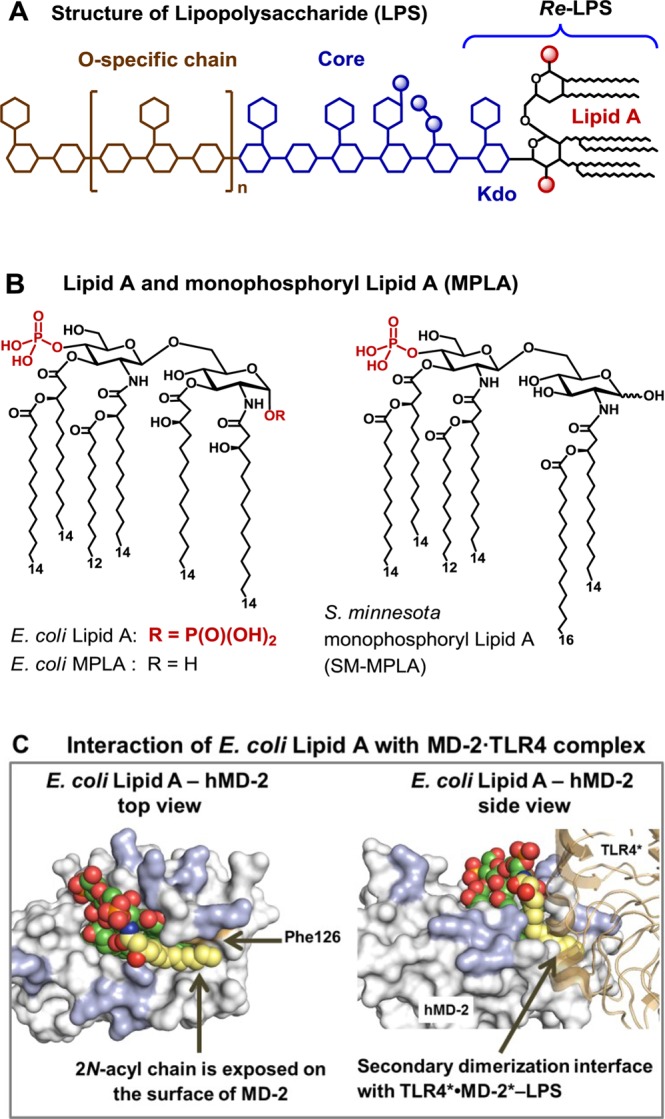
(A) Structure of LPS, with Re-LPS and Lipid A. (B) Structures of TLR4 agonist E. coli Lipid A and MPLA. (C) Co-crystal structure of E. coliRa-LPS-hMD-2·TLR4 (PDB code: 3FXI; only Lipid A portion of LPS is shown for clarity), top and side views. Phe126 (orange) together with 2-N-acyl chain (yellow) creates a hydrophobic patch at the dimerization interface with the second TLR4*·MD-2* complex (brown). Positively charged Arg and Lys (blue) at the rim of the binding pocket of MD-2 are involved in the ionic interactions with the Lipid A phosphates. Images were generated with PyMol.
Lipid A, an amphiphilic membrane-bound portion of LPS, represents the major pathogen-associated molecular pattern which drives the activation of TLR4 by binding to the co-receptor protein MD-2 and triggering the dimerization of two TLR4·MD-2·LPS complexes.11 Generally, the binding of hexaacylated bisphosphorylated Lipid A (such as from Escherichia coli, Figure 1B) by human TLR4·MD-2 complex results in the efficient activation of the innate immune response, whereas underacylated Lipid A variants are either inactive or antagonistic (such as tetraacylated lipid IVa, or the synthetic drug candidate Eritoran).12,13 The co-crystal structures of the E. coliRe- and Ra-LPS with mouse (m) or human (h) MD-2·TLR4 complex, respectively, unravel that only five long-chain acyl residues of the hexaacylated Lipid A are incorporated into the hydrophobic binding pocket of MD-2 whereas the sixth 2-N-acyl chain is exposed on the surface of MD-2 and is involved in the dimerization interface with the second TLR4*·MD-2*-LPS complex (Figure 1C).14,15 The Phe126 residue of MD-2 is proposed to stabilize the presentation of an acyl tail on the surface of the protein and to serve as hydrophobic switch allowing dimerization to occur.16,17 LPS-driven homodimerization of TLR4·MD-2-LPS complexes initiates recruitment of adaptor proteins to the intracellular TIR (Toll/interleukin-1 receptor) domains of TLR4 which ultimately results in the induction of the intracellular inflammatory signaling cascade.18 In contrast, submerging of all lipid chains of the ligand into the hydrophobic binding groove of MD-2 results in an efficient binding without initiation of signaling, which is a characteristic feature of TLR4·MD-2 antagonists.12,13
The presence of both 1- and 4′-phosphate groups of Lipid A was shown to be crucial for the efficiency of the dimerization and the potency of the initiated signaling.19 The absence of 1-phosphate leads to less efficient dimerization20,21 and dampened cytokine production while maintaining sufficient TLR4-mediated immune activation and full adjuvant activity, which guided the development of monophosphoryl Lipid A (MPLA), a licensed vaccine adjuvant (Figure 1B).8,10
Despite tremendous intensive research on the interaction of TLR4·MD-2 complex with isolated,22,23 genetically engineered,24 and synthetic Lipid A’s and analogues,25−29 the structure–activity relationships of the LPS-triggered TLR4 activation are not unambiguously established. Minor variations in the length and distribution pattern of fatty acyl chains in Lipid A typically result in dramatic amendment of TLR4-mediated immune signaling25−27 which cannot be rationally predicted.
We have addressed the challenges associated with the exploration of structural basis of LPS-induced TLR4 activation by development of a novel type of Lipid A mimetics wherein the flexible βGlcN(1→6)GlcN backbone of Lipid A is replaced by the conformationally restricted (1↔1)-connected disaccharide scaffolds. Notably, all Lipid A analogues synthesized so far were based either on the native β(1→6)-diglucosamine or on the more flexible backbones wherein one or both GlcN residues were replaced by a linear aglycon.9,10,27,30 Previously we reported on the synthesis and potent anti-endotoxic activity of tetraacylated Lipid A mimetics derived from the β,α(1↔1)-linked diglucosamine representing an “antagonistically” shaped scaffold.31
Taking advantage of a striking similarity between the conformation of the nonreducing sugar trehalose [αGlc(1↔1)αGlc] and the molecular shape of the β(1→6) diglucosamine backbone of TLR4·MD-2-bound agonist E. coli Lipid A disclosed in the X-ray structure, we have developed novel agonistic conformationally confined Lipid A mimetics based on the two-bond-linked rigid trehalose-type αGlcN(1↔1)αMan scaffold (Figure 2).
Figure 2.
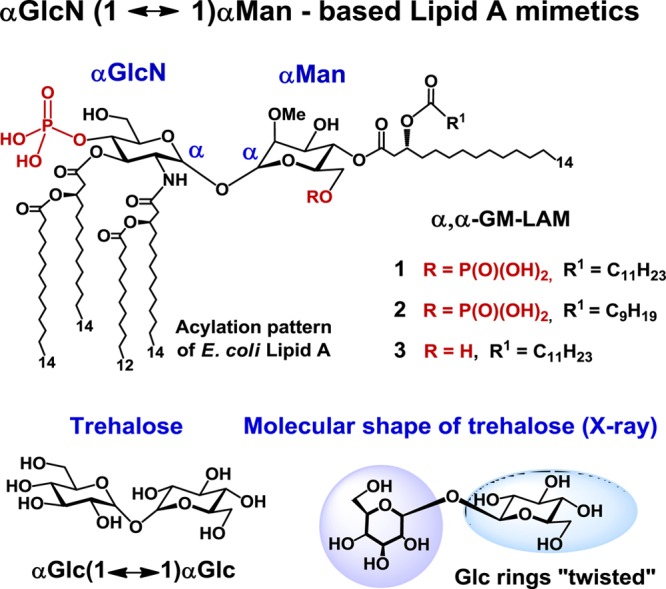
Structure of Lipid A mimetics (α,α-GM-LAMs) 1–3 based on the conformationally confined αGlcN(1↔1)αMan scaffold.
Results and Discussion
X-ray Structure-Guided Design of αGlcN(1↔1)αMan-Based Lipid A Mimetics
The β(1→6) diglucosamine backbone represents the most conserved part of Lipid A, whereas its acylation and phosphorylation pattern varies within bacterial species.22,32 The overall three-dimensional conformation of the intrinsically flexible three-bond-linked βGlcN(1→6)GlcN backbone of Lipid A is determined by the values of the dihedral angles ω, ϕ, and ψ about (1→6) glycosidic and oxymethyl linkages (Figure 3A). Thus, the relative orientation of GlcN rings can be easily adapted by rotation about glycosidic and oxymethyl linkages via altering the corresponding torsion angles. This permits spontaneous adjustment of the shape of Lipid A to the geometry of the binding pocket of MD-2, which complicates the estimation of the “active” conformation of the ligand in the [Lipid A-MD-2·TLR4]2 complex. As seen in the co-crystal structures, the proximal (reducing) GlcN ring of MD-2-bound hexaacylated Lipid A adopts an inclined (or “twisted”) orientation which, as we assume, is essential for the exposure of the long-chain 2-N-acyl residue on the surface of MD-2 followed by dimerization with the second MD-2·TLR4 complex (Figure 3A,B).31
Figure 3.
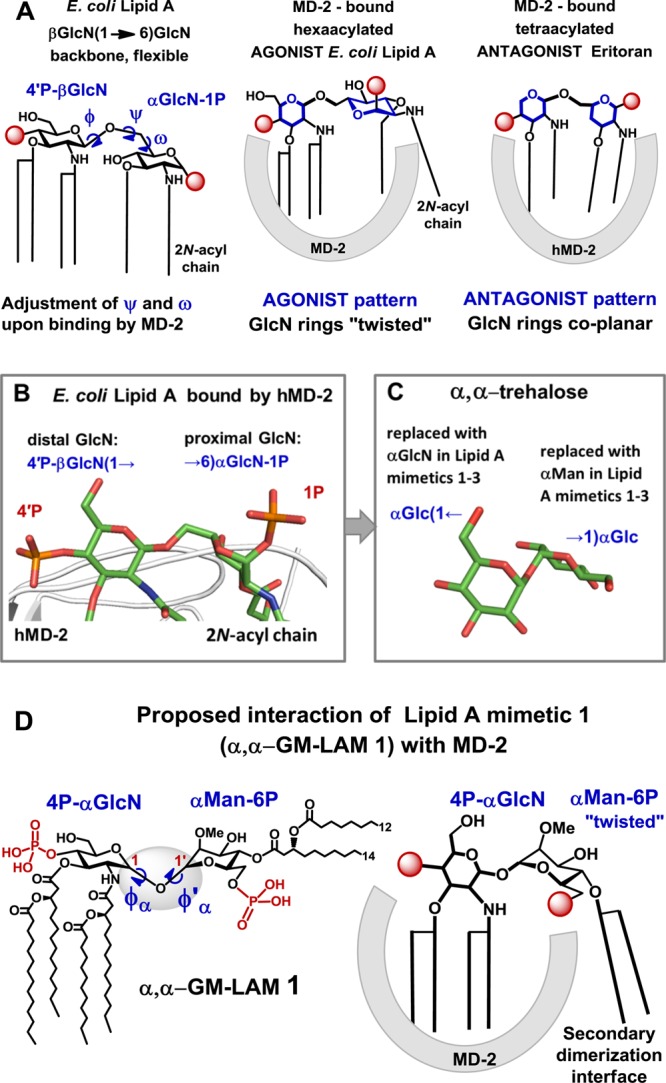
X-ray structure-based design of α,α-GM-LAMs. (A) Adjustment of the torsion angles about the (1→6) glycosidic linkage in the diglucosamine backbone of Lipid A upon binding by MD-2 results in a “twisted” orientation of the proximal GlcN ring for an agonist and in a coplanar orientation of the two GlcN rings for antagonist. (B) The proximal GlcN moiety of MD-2-bound E. coli Lipid A (PDB code 3FXI) adopts inclined orientation which allows the exposure of the 2-N-acyl chain.31 Image was generated with PyMol. (C) The molecular shape of α,α-trehalose (crystal structure)35,37 resembles the three-dimensional arrangement of βGlcN(1→6)GlcN backbone of the MD-2-bound E. coli Lipid A. (D) Structure of αGlcN(1↔1)αMan-based Lipid A mimetic (α,α-GM-LAM) 1 and proposed interaction of 1 with MD-2.
To explore the structural prerequisites needed for an effective receptor complex homodimerization, we have manipulated the flexibility of the carbohydrate backbone of Lipid A by fixing its molecular shape in an “agonistic” conformation. Since the relative spatial arrangement of the two GlcN rings of MD-2-bound agonist E. coli Lipid A disclosed in the co-crystal structures14,15 resembles the arrangement of α,α-(1↔1)-connected glucoses in the nonreducing disaccharide trehalose (Figure 3B,C), we have developed predictably agonistic Lipid A mimetics based on the conformationally confined α,α-trehalose-like αGlcN(1↔1)αMan scaffold (Figure 3D).
The values for the torsion angles Φα and Φ′α, representing rings orientation about the α,α-(1↔1) glycosidic linkage (Figure 3D), are governed mostly by the anomeric and exo-anomeric effects and are only marginally dependent on the nature of functional groups in variably substituted α,α-trehaloses.33 The existence of a single conformational minimum with respect to the dihedrals about glycosidic linkage in α,α-trehaloses was confirmed by molecular dynamics simulations,33,34 whereas the preferred gauche–gauche conformation of the substituted α,α-trehalose35 and its αGlc(1↔1)αMan analogue36 was corroborated by X-ray and conformational analysis, respectively. Thus, conformationally restrained α,α-(1↔1) glycosidic linkage in αGlcN(1↔1)αMan-based Lipid A mimetics would impose a specific relative orientation of sugar rings resembling the molecular shape of the diglucosamine backbone of the agonistic MD-2-bound Lipid A.
αGlcN(1↔1)αMan-based Lipid A mimetics (α,α-GM-LAMs) 1–3 were designed such that the acylation and phosphorylation pattern of the nonreducing (distal) GlcN residue of E. coli Lipid A remains unaffected, whereas the β(1→6) glycosidic linkage is substituted by an α,α(1↔1) glycosidic bond and the reducing (proximal) GlcN moiety of natural Lipid A is exchanged for a nonreducing sugar (mannose) having a specific acylation and phosphorylation pattern (Figure 3D). The location of the phosphate functionality at C-6 of the Man residue was selected to closely resemble the positioning of a 1-phosphate group of E. coli lipid A at the secondary dimerization interface of the TLR4-MD-2-Lipid A complex (PDB code 3FXI). The site of acylation at the mannose moiety was chosen such that the attachment of the long-chain (R)-3-acyloxyacyl residue at Man C-4 would provide a sufficient hydrophobic patch to support the homodimerization and the interaction with the second TLR4*·MD-2*-ligand complex.
Upon interaction with the receptor complex, the tetraacylated GlcN unit of α,α-GM-LAMs 1–3 was supposed to be fully accommodated within the hydrophobic pocket of MD-2, whereas the “twisted” mannose ring should be excluded from the binding site on MD-2 such that the two lipid chains at Man C-4 are presented onto the surface of the protein and involved in the secondary dimerization interface (Figure 3D). The axial configuration at C-2 of mannose should provide, according to the crystal structures, a better fitting to the geometry of the binding pocket of MD-2 and, simultaneously, ensure easier stereocontrol in the 1,2-trans glycosylation step to α,α(1↔1)-linked disaccharide.
Synthetic Strategy
The assembly of αGlcN(1↔1)αMan, a 1,1-glycosidically connected (nonreducing) disaccharide, represents a formidable synthetic challenge with regard to simultaneous stereocontrol at two anomeric centers. Typically, approaches involving conventional glycosylation procedures for the synthesis of trehalose provide moderate stereoselectivity and low yields.38 Since we aimed to establish the α,α-(1↔1) glycosidic linkage between an amino sugar and a manno-configured monosaccharide, we could hardly rely on the intramolecular aglycon delivery approach39 or on the versatile synthetic desymmetrization of the natural trehalose.40
For the synthesis of bis- and monophosphorylated Lipid A mimetics 1–3 (Figure 2) having non-symmetrically distributed acyloxyacyl functional groups, a convergent approach involving first the preparation of the orthogonally protected αGlcN(1↔1)αMan disaccharide scaffold followed by regioselective phosphorylation and acylation with (R)-3-acyloxyacyl fatty acids of variable chain lengths was envisaged (Scheme 1). For the assembly of the αGlcN(1↔1)αMan backbone, a 2-N-carbamate-protected glucosamine-based lactol was chosen as acceptor, and a 2-O-levulinoyl (Lev, 4-oxopentanoyl)-protected mannose was selected to serve as glycosyl donor. The participating protecting group at C-2 (Lev) should allow for a preferable 1,2-trans mannosylation. Since the 2-O-Lev protection had to be exchanged for a 2-O-Me group later in the synthesis, an alternative glycosylation approach using non-participating methyl protection at C-2 of the mannose-based donor was planned to be explored as well.
Scheme 1. Synthetic Strategy toward Hexaacylated α,α-GM-LAMs 1–3.

The 2-N-carbamates of variably protected GlcN-based lactols revealed the highest α/β ratio (up to 9:1) of the anomeric 1-OH group, which highlighted these intermediates as the most “stereoselective” glycosyl acceptors. Apparently, the presence of a carbamate N–H capable of hydrogen bonding with the axial oxygen at C-1 is responsible for the substantial enrichment with the α-anomer.
Synthesis of αGlcN(1↔1)αMan Scaffold
To minimize the number of required orthogonal protecting groups, we intended the use of a GlcN acceptor with the pre-installed phosphate group at C-4, whereas C-6 was permanently and C-3 was temporarily protected. To this end, the 2-N-Troc-protected 4,6-di-O-benzylidene acetal 4(41) was first acetylated to provide 3-O-acetate 5, which was subjected to regioselective reductive opening of benzylidene acetal with Et3SiH/TfOH in CH2Cl2 to furnish 6-O-benzylated compound 6 (Scheme 2). Phosphitylation of 4-OH with bisbenzyl(diisopropylamino)phosphoramidite with 1H-tetrazole as catalyst followed by in situ oxidation with meta-chloroperbenzoic acid (mCPBA) afforded phosphotriester 7. Compoud 7 was either desilylated at C-1 by treatment with pyridinium hydrofluoride (HF·Py) in THF to provide a 2-N-Troc-protected axially configured lactol 8 (α/β = 9:1) or processed to the 2-N-(9-fluorenylmethyl)carbamate (Fmoc)-protected counterpart 9, which was similarly deprotected at C-1 to furnish anomeric lactol 10, again with a high preponderance of the α-anomer (α/β = 9:1).
Scheme 2. Synthesis of Glucosamine Lactol Acceptors 8 and 10.

Reagents and conditions: (a) Ac2O, DMAP, pyridine, 97%; (b) Et3SiH, TfOH, 4 Å MS, −78 °C, CH2Cl2, 68%; (c) 1. (BnO)2PN(iPr)2, 1H-tetrazole, CH2Cl2, 2. mCPBA, −78 °C, 90%; (d) HF·Py, THF, 91% for 8, 94% for 10; (e) 1. Zn, AcOH, CH2Cl2, 2. FmocCl, EtN(iPr)2, CH2Cl2, 89%.
The synthesis of the required mannose-based donors commenced with Zemplén deacetylation of the peracetylated thioethyl glycoside 11,42 followed by introduction of a 4,6-di-O-tert-butylsilylene (DTBS) group in 12 (Scheme 3). Regioselective benzylation at C-3 via stannylene acetal intermediate and the agency of benzyl bromide in the presence of (nBu)4NI furnished alcohol 13 in 93% yield. The 2-OH group was either levulinoyl-protected by reaction with 4-oxopentanoic acid, N,N′-diisopropylcarbodiimide (DIC), and a catalytic amount of 4-N,N-(dimethylamino)pyridine (DMAP) to give 2-O-levulinate ester 14 (90%) or methylated by reaction with MeI/NaH in DMF to provide 2-O-methyl ether 15 in 84% yield. Anomeric deprotection with N-bromosuccinimide (NBS) in aqueous acetone afforded lactols 16 and 17, respectively, which were converted to trichloroacetimidates (TCA) 18 and 20 or to N-phenyl-trifluoroacetimidate (NPTFA) donors4319 and 21.
Scheme 3. Synthesis of Mannose-Based Donors.

Reagents and conditions: (a) 1. NaOMe, MeOH, 2. (tBu)2Si(OTf)2, pyridine, DMF, −35 °C, 89%; (b) 1. (nBu)2SnO, toluene, 2. BnBr, (nBu)4NI, DMF, toluene, reflux, 93%; (c) →14: LevOH, DIC, DMAP, CH2Cl2, 90%; (d) →15: MeI, NaH, DMF, 84%; (e) NBS, acetone–H2O, 24:1, 0 °C, 77% for 16 and 89% for 17; (f) →18 and 20: CCl3CN, DBU, CH2Cl2, 0 °C, 90% for 18 and 94% for 20; (g) →19 and 21: CF3(NPh)CCl, K2CO3, acetone, 99% for 19 and 93% for 21.
The key point in our initial approach was to obtain a good double α-stereoselectivity in the glycosylation reaction between reducing acceptor 8 (α/β = 9:1) and the 2-O-levulinoyl-protected imidate donors 18 or 19. The participating levulinoyl group at C-2 of the manno-configured donors 18 and 19 should allow for a selective 1,2-trans glycosylation. A survey of the literature revealed that a complete α-manno selectivity upon application of 2-O-Lev-protected mannosyl donors could be obtained with a variety of acceptors.44,45
In an initial glycosylation attempt comprising coupling the TCA donor 18 and acceptor 8 using trimethylsilyl trifluoromethanesulfonate (TMSOTf) as promoter, an orthoester 23 was obtained as the major product (23%) along with a minor proportion of the target α,α-disaccharide 22 (8%) and a concurrently formed donor self-coupling product 24 (Scheme 4A, Supplementary SI-Table 1). With the less reactive NPTFA donor 19 or with the thioethyl donor 14 the formation of the orthoester was not observed; however, the desired product 22 was isolated only in trace amounts.
Scheme 4. Initial Attempts toward the Synthesis of αGlcN(1↔1)αMan Scaffold.

Reagents and conditions: (a) 1. HF·Py, THF, 2. Ac2O, DMAP, pyridine, 85%; (b) NBS, acetone–H2O, 24:1, 0 °C, 89%; (c) CCl3CN, DBU, CH2Cl2, 67%; (d) TMSOTf (0.05 equiv), 4 Å MS, CH2Cl2, 0 °C; (e) (CF3CO)2O, CF3COOH, Et3SiH, CH2Cl2, 0 °C, 91%; (f) 1. (BnO)2PN(iPr)2, 1H-tetrazole, CH2Cl2, 2. mCPBA, −78 °C, 91%; (g) 1. Zn, AcOH, CH2Cl2, 0 °C, 2. FmocCl, EtN(iPr)2, CH2Cl2, 88%; (h) 1. [Ir(COD)(Ph2MeP)2]PF6, H2, THF, 2. I2, aq. THF, 86%.
We have hypothesized that the diminished reactivity of the torsionally disarmed 4,6-di-O-cyclic-protected mannose donor was responsible for the glycosylation failure.46 Accordingly, the 4,6-di-O-DTBS group in 14 was cleaved and substituted for two acetates to provide 25, wherein the C5–C6 bond was unlocked from the disarming trans–gauche conformation.46,47 To provide consistency with the previously performed imidate-mediated glycosylations, the thioglycoside at C-1 was exchanged for a TCA group to furnish 27 (Scheme 4B). The coupling of 8 and the torsionally unlocked 4,6-di-O-acetyl donor 27 resulted in the isolation of the α,α-configured disaccharide 28, albeit in a similarly low yield (8%).
Thereafter, our attention was turned to the apparently low reactivity of the lactol acceptor 8 affected by the H-bonding between the α-1-OH and the carbamate NH groups. To increase the nucleophilicity of the lactol acceptor and to reduce steric constraints, the disarming acetate at C-3 was exchanged for a TBDMS group, and the sterically demanding 2-N-Troc group was replaced by Fmoc protection, which provided α-lactol acceptor 33. To this end, compound 30, made by a highly selective reductive opening of benzylidene acetal in the allyl glycoside 29,31 was phosphorylated at C-4 to give 31 (Scheme 4C). Reductive cleavage of 2-N-Troc protection with Zn in acetic acid followed by reaction with Fmoc chloride in the presence of a EtN(iPr)2 furnished 32, which was anomerically deprotected using [Ir(COD)(Ph2MeP)2]PF6-catalyzed isomerization of the allyl group followed by oxidative cleavage of the 1-propenyl group with I2 in THF–H2O to afford the “armed” acceptor 33. Reaction of the 2-O-levulinoyl TCA donor 18 with 33, however, reproducibly resulted in the formation of the orthoester 34 as the major product (Scheme 4C).
In retrospect, we assume that the failure of the 2-O-levulinoyl-protected mannose-based donors to provide the desired αGlcN(1↔1)αMan compound in a glycosylation reaction with GlcN-based lactol acceptors was rather related to a particular conformation of the arising trehalose-type α,α-disaccharide. A successful glycosylation would lead to a sterically hindered coupling product 22, having overlapping bulky 2-N-Troc/Fmoc groups at the GlcN moiety and a linear 4-oxopentanoate (levulinate) ester group at C-2 of mannose, while reciprocal repulsion of the N-carbamate and electron-rich levulinoyl groups could contribute as well.
Given the failure of 2-O-levulinoyl-protected mannosyl donors to provide the desired coupling products, the participating protecting group at C-2 of mannose was exchanged for the 2-O-Me group as in the donors 20 and 21 (Scheme 3). Successful application of non-participating groups in the α-selective mannosylation has been extensively reported.48,49 Gratifyingly, the coupling of the 2-O-Me-Man imidate donor 21 with the GlcN-lactol acceptor 8 allowed for a much higher isolated yield (51%) of the α,α-disaccharide 35 (Scheme 5, Table 1, entry 2). Owing to the low reactivity of acceptor 8, the NPTFA donor 21 was found to be superior to the TCA donor 20 due to the propensity of the latter to form substantial amounts of the rearranged glycosylamide 45 (52%), which is characteristic for glycosylations involving acceptors of diminished reactivity (Table 1, entries 1 and 2). The isolation of the α,α-disaccharide 35 was complicated by the concomitant formation of the co-migrating αGlcN−βMan 38 and βGlcN−αMan 41 byproducts. Glycosylation of the 2-N-Fmoc-protected acceptor 10 by the NPTFA donor 21 afforded a similar isolated yield (52%) of the target α,α-configured disaccharide 36 (Scheme 5, Table 1, entry 3).
Scheme 5. Synthesis of GlcN(1↔1)Man Disaccharides.
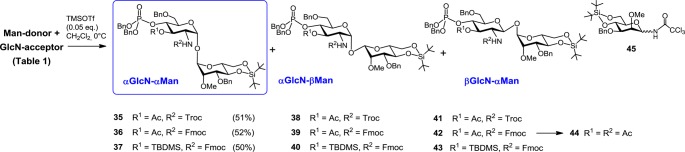
Table 1. Outcome of (1↔1) Glycosylation Utilizing 2-O-Me-Protected Imidate Donors 20 and 21 (Scheme 5).
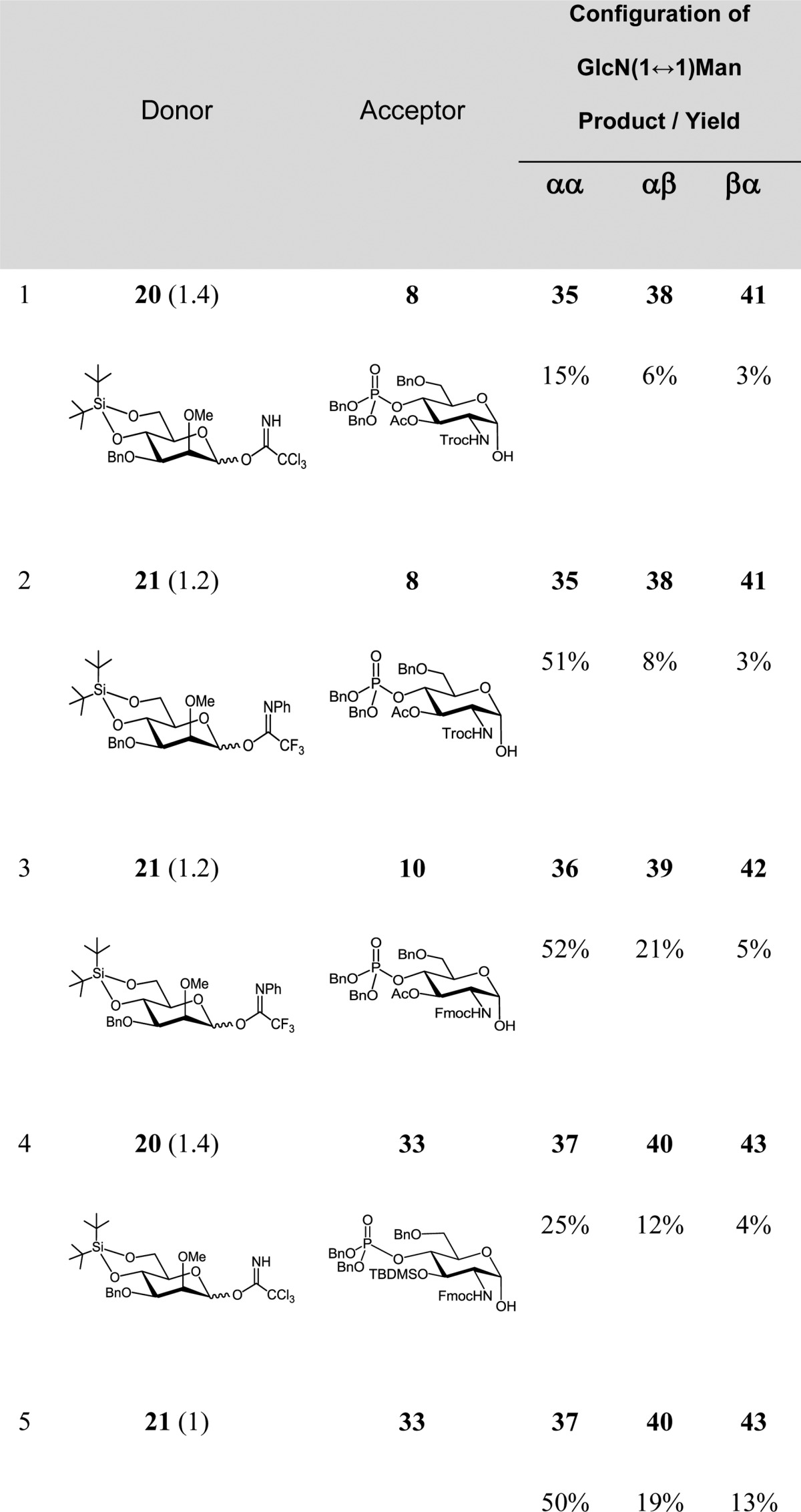
Enhancement of the acceptor reactivity by the use of 3-O-TBDMS-protected “armed” lactol 33 did not improve the yields in the coupling reactions with either donor 20 or 21 (Table 1, entries 4 and 5), furnishing αGlcN(1↔1)αMan disaccharide 37 in 25% and 50% yield, respectively. The yield of the αGlcN(1↔1)αMan disaccharide was related to the ease of its chromatographic purification, which, in turn, was strongly dependent on the protection group pattern. Since isolation of the 2-N-Fmoc-3-O-Ac-protected 36 from the mixture of anomeric products was the most straightforward, this disaccharide was chosen for further transformation to the target α,α-GM-LAMs 1–3.
The configurations at the anomeric centers of the (1↔1)-linked disaccharides were assigned on the basis of 1H and 13C NMR shifts at the anomeric positions and the 1JC1,H1 coupling constants50 (SI-Table 2). To circumvent the severe peak broadening in the 1H NMR spectrum, the Fmoc group in 42 was replaced by a 2-N-acetate to give acetamide 44 appropriate for the unambiguous signal assignment. The α,α-linkage in 35–37 was confirmed by the large 1JC1,H1 coupling constant values of the anomeric carbons (1JC1,H1 = 170–174 Hz for α-manno- and 1JC1,H1 = 173–178 Hz for α-gluco-anomers) and by the downfield shifts and the corresponding vicinal proton coupling constants 3JH1,H2 of the anomeric H-1 signals (δ 5.02–5.12, 3J1,2 = 1.5 Hz for α-manno- and 5.08–5.23, 3J1,2 ≈ 3.7 Hz for α-gluco-anomers).51 The β-manno linkage in 38–40 was corroborated by the typically smaller 1JC,H coupling constants for the anomeric carbons (1JC1,H1 = 155–157 Hz) and by the upfield shifts of the anomeric H-1 signals (4.6 ppm) and H-5 signals (3.28 ppm), characteristic for β-mannosides.52
Synthesis of Lipid A Mimetics 1–3 Based on αGlcN(1↔1)αMan Scaffold
Having orthogonally protected αGlcN(1↔1)αMan disaccharide scaffold 36 in hand, we next approached the stepwise deprotection and acylation at C-2 and C-3 of the GlcN fragment. The feasibility of regioselective deacetylation at C-3 in the presence of the adjacent 2-N-Fmoc carbamate and the base-labile phosphotriester group at C-4 in 36 was first examined (SI-Scheme 1). Among a variety of tested conditions, application of aqueous hydroxylamine provided the best reproducible results for the exclusive removal of 3-O-acetate (SI-Table 3). Cleavage of the 3-O-Ac group to furnish 46 was accompanied by the migration of the phosphate from C-4 to the liberated hydroxyl group at C-3 to give 47 (Scheme 6). Besides, a partial hydrolytic loss of one benzyl protecting group in the phosphotriester 46 leading to formation of the phosphodiester 48 was also observed. Further improvements could be achieved through optimization of reaction conditions. Indeed, when the reaction was terminated prior to completion (48 h), the formation of the undesired byproducts 47 and 48 could be largely avoided (2% and 9%, respectively), providing 3-O-deacylated compound 46 in 53% yield (SI-Table 3). Repeated chromatographic purifications of the disaccharide 46 partly account for the relatively low yield.
Scheme 6. Synthesis of αGlcN(1↔1)αMan-Based Lipid A Mimetics (α,α-GM-LAMs) 1–3.

Reagents and conditions: (a) aq. 50% NH2OH, THF, 0 °C: 46 (53%), 47 (2%), 48 (9%), recovered 36 (15%); (b) 49, DIC, DMAP, CH2Cl2, 0 °C, 83%; (c) DBU, CH2Cl2; (d) 50, EDC, CHCl3; (e) HF·Py, THF; (f) →57 + 59, DIC, DMAP, 50, CH2Cl2, 0 °C; (g) →58 + 60, DIC, DMAP, 51, CH2Cl2, 0°C; (h) 1. (BnO)2PN(iPr)2, 1H-tetrazole, CH2Cl2, 2) mCPBA, −78 °C, 91% for 61, 89% for 62; (i) Pd black, toluene–MeOH, 1:1, 72% for 1, 87% for 2, 81% for 3.
The first (R)-3-(tetradecanoyloxy)tetradecanoyl residue at C-3 of the GlcN moiety was introduced by reaction of 46 with β-acyloxyacyl acid 49 under the agency of DIC and a catalytic amount of DMAP to provide 52 in 83% yield (Scheme 6). Notably, strictly equimolar amounts of DIC and fatty acid 49, and a catalytic quantity of DMAP at 0 °C, had to be applied to suppress the concomitant formation of the co-migrating 3-O-tetradecanoyl (53) and 3-O-alkenoyl (54) byproducts (11% and 3%, respectively). Application of higher amounts of DIC and/or fatty acid aimed to accelerate the transformation resulted in augmented formation of 53 and 54, which could be rationalized by a probable β-elimination or rearrangement53 of the in situ-formed O-acyl-oxyacylisourea intermediate. Formation of a 3-O-alkanoyl side product similar to 53 in the DIC/DMAP-mediated condensation had been previously reported, though no elimination byproduct such as 54 was detected.30 Since esterification with β-acyloxyacyl fatty acids under Steglich conditions is routinely applied in the synthesis of Lipid A’s and analogues which often display high bioactivity at picomolar doses, this finding is of importance for the preparation of Lipid A-based compounds of the uppermost purity.
Subsequent Fmoc cleavage in 52 with DBU followed by EDC-mediated N-acylation with (R)-3-(dodecanoyloxy)tetradecanoic acid 50 provided tetraacylated disaccharide 55. Conversion of 4,6-di-O-DTBS derivative 55 into diol 56 was performed under standard conditions with HF·Py in THF. To introduce acyl and phosphate functional groups at mannose C-4 and C-6, respectively, without additional protecting group manipulation, the regioselectivity of the acylation of the diol 56 with the acyloxyacyl acids 50 or 51 was first examined. Since hydroxyl groups in the substituted trehaloses are known to differ in reactivity due to both steric and electronic effects,38,40 we expected that, in a heavily substituted αGlcN(1↔1)αMan disaccharide 56, positions C-4 and C-6 at the Man moiety could be discriminated in a subsequent acylation procedure.
Indeed, the major outcome of DIC/DMAP-mediated acylation of 56 with the acids 50 or 51 was not the intrinsically expected primary 6-OH-derived acylation products, but the 4-O-acyloxyacyl derivatives 57 and 58 in 51% and 65% yield, respectively, having a 6-OH group at the mannose unit accessible for the ensuing phosphorylation. Minor amounts of the 4,6-bis-O-acylated derivatives 59 and 60 (24% and 13%, respectively) were isolated as well. Although the 6-OH group of the mannose residue in αGlcN(1↔1)αMan disaccharide 56 is somewhat remote from the 2-NH of the GlcN moiety, the crystal structures of α,α-trehalose-based compounds indicate spatial proximity of the two groups. Thus, it could be assumed that the intramolecular hydrogen bonding (2-NHGlcN–6-OHMan) exerts an adverse effect on the reactivity of the primary hydroxyl group at Man C-6. Furthermore, the bulkiness of the in situ-formed acyloxyacyl-activated ester resulting from the reaction of the fatty acids 50 or 51 with DIC/DMAP could also explain the limited access to the sterically hindered 6-OH group in the α,α-trehalose-like disaccharide 56.
Next, the free 6-OH group in the hexaacylated disaccharides 57 and 58 was phosphorylated by reaction with dibenzyl-(N,N′-diisopropylamino)phosphoramidite in the presence of a mild acid catalyst, 1H-tetrazole, and subsequent oxidation with mCPBA at −78 °C to furnish the bisphosphorylated hexaacylated products 61 and 62 in 91% and 89% yield, respectively. Final debenzylation by hydrogenation of 57, 61, and 62 on Pd-black followed by purification with gel permeation chromatography on Sephadex SX1 in toluene–methanol (2:1) afforded target bisphosphorylated Lipid A mimetics 1 and 2 and a monophosphorylated counterpart 3. In contrast to native Lipid A, compounds 1 and 2 do not possess a labile anomeric phosphate functionality; consequently, they were isolated and biologically assessed as free acids at the phosphates and could be stored in aqueous solution at 4 °C for several months without any noticeable sign of degradation, which was confirmed by MALDI-TOF analysis.
Activation of TLR4·MD-2 Complex by α,α-GM-LAMs 1–3
The propensity of the α,α-GM-LAMs 1–3 to stimulate TLR4-mediated immune signaling was first assessed in the hTLR4/hMD-2/CD14 transfected human embryonic kidney (HEK) 293 cells (HEK-Blue). Since we were particularly interested in the molecular recognition mechanisms implicated in the binding of LPS by the MD-2·TLR4 complex wherein the Lipid A/Re-LPS portion of LPS is exclusively involved, we evaluated the activities of α,α-GM-LAMs 1–3 compared to E. coliRe-LPS (Figure 1A). It has been previously shown that the minimum structural requirement for the expression of the highest cytokine-inducing potency resides in Re-LPS, which entails two 3-deoxy-d-manno-oct-2-ulosonic acid (Kdo) residues in addition to Lipid A (Supporting Information, SI-Figure 1A).54,55 Moreover, the Re-LPS has a defined molecular weight (MW) similar to the MW range for Lipid A mimetics (1.8–2.2 kDa), in contrast to wild-type LPS having variable MW (10–15 kDa), so that the direct comparison of a dose-dependent response between compounds 1–3 and Re-LPS is more appropriate. The TLR4-stimulating activity of Lipid A mimetics 1–3 was examined over a wide concentration range by monitoring of the activation of the NF-κB regulated signal transduction pathway via measuring the induction of secreted embryonic alkaline phosphatase (SEAP) and compared to the responses elicited by E. coliRe-LPS and SM-MPLA.
Remarkably, the glycosylation at C-6′ of Lipid A with Kdo residues was reported to be responsible for the 10–20-fold enhancement of the activity of the Kdo/Kdo2-Lipid A (Re-LPS) compared to Lipid A alone in the nanomolar concentration range.28,54 Notably, conformationally confined α,α-GM-LAM 2 based on just a disaccharide scaffold displayed NF-κB activation (EC50 = 0.08 nM) similar to those of Re-LPS (EC50 = 0.04 nM) and E. coli LPS (EC50 = 0.08 nM, SI-Figure 1A), and the TLR4 saturation plateau was reached at a concentration of 1 ng/mL for both ligands (Figure 4). Compound 1, having a 2×CH2-longer acyl side chain at Man C-4 compared with α,α-GM-LAM 2, was a less efficient activator of NF-κB (EC50 = 0.4 nM), so that its TLR4 saturation plateau was reached at a concentration of 5 ng/mL. Thus, shortening of a lipid side chain at C-4 of the mannose moiety by 2×CH2 resulted in a 5-fold increase of TLR-4 stimulating activity, which underlines the significance of hydrophobic interactions at the dimerization interface. Along this line, synthetic manipulation of the length of the acyl side chain at Man C-4 could be used for fine-tuning of the hTLR4-mediated activity in α,α-GM-LAMs. The monophosphate 3 was, as expected, significantly less active (EC50 = 31 nM) than its bisphosphorylated counterpart 1, but it showed a more potent activation profile than SM-MPLA at concentrations above 10 ng/mL.
Figure 4.
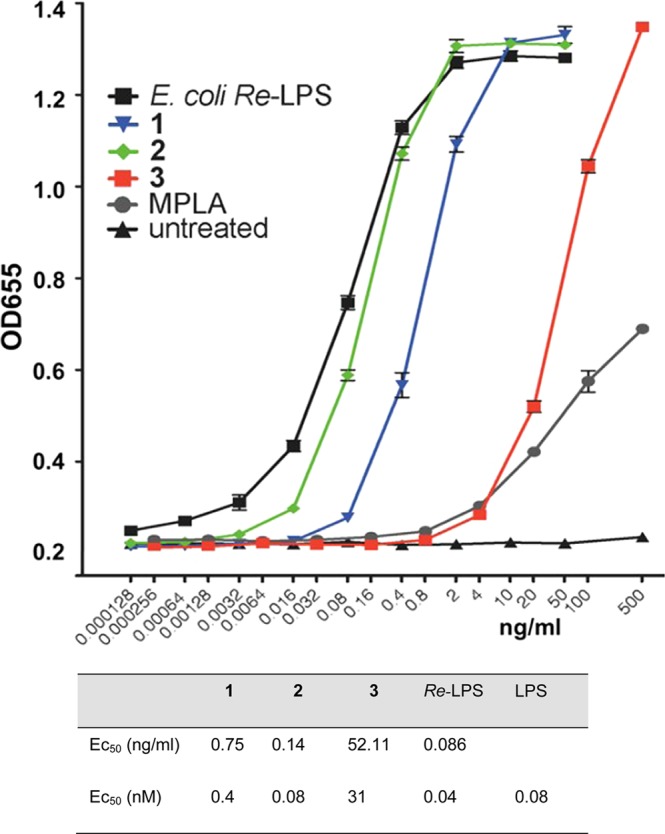
Dose-dependent activation of TLR4 signaling in hTLR4/hMD-2/hCD14-transfected HEK293 cells (HEK-Blue) by Lipid A mimetics 1–3 compared to E. coliRe-LPS and S. minnesota MPLA.
Modulation of the Expression of Cytokines by α,α-GM-LAMs 1–3 in Human and Mouse Macrophages
Lipid A mimetics 1–3 were examined for the ability to initiate the expression of tumor necrosis factor-α (TNF-α), interleukin-8 (IL-8), and monocyte chemotactic protein-1 (MCP-1) in the human monocytic macrophage-like cell line THP-1, which expresses MD-2, CD14, and a variety of cell surface receptors, including TLR4.
The dose-dependent stimulating activity of synthetic Lipid A mimetics was cytokine-specific, revealing higher potency in the induction of the expression of TNF-α and IL-8 by α,α-GM-LAMs 1 and 2 than by Re-LPS/LPS (Figure 5A,B, SI-Figures 2 and 3). The release of MCP-1 induced by α,α-GM-LAM 2 was clearly more effective than the production of this chemokine by Re-LPS and compound 1 (Figure 5C). Expression of MyD88-dependent chemokine MCP-1 is associated with the activation of the intracellular TLR4·MD-2 complex.56 The dampened induction of the expression of cytokines by α,α-GM-LAM 3 correlates to its chemical structure missing a phosphate group at Man C-6. Our results indicate that both α,α-GM-LAMs 1 and 2 are more potent activators of the MyD88 signaling pathway than Re-LPS/LPS (Figure 5, SI-Figure 4).
Figure 5.
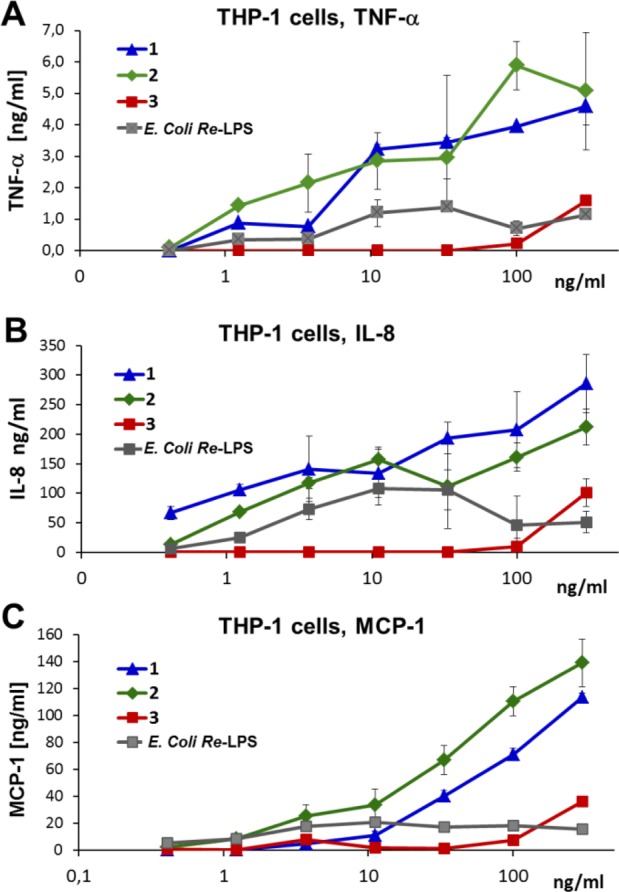
Dose-dependent expression of cytokines induced by α,α-GM-LAMs 1–3 in human macrophage cell line THP-1 compared to E. coliRe-LPS. (A) Production of TNF-α. (B) Induction of the expression of IL-8. (C) Induction of release of MCP-1.
The ability of Lipid A mimetics 1–3 to induce the production of TNF-α and IL-6 from bone marrow-derived macrophages (BMDM) in mice was subsequently examined and compared to that of synthetic E. coli Lipid A and E. coli MPLA, which are reliable positive controls due to their chemical purity and homogeneity (Figure 6). The maximum level of TNF-α (4000 pg/mL) was detected in BMDM cultivated in the presence of 1 nM α,α-GM-LAM 1 or 2, whereas the same quantity of the “parent” Lipid A resulted in release of half the amount of TNF-α (2000 pg/mL). On the other hand, monophosphoryl α,α-GM-LAM 3 exhibited a dampened ability to express TNF-α (500 pg/mL) at a concentration of 1 nM. Likewise, 1 nM (1.8 ng/mL) of 1 or 2 triggered the release of a 3-fold higher amount of IL-6 (1500 pg/mL) compared to Lipid A (500 pg/mL). Monophosphate 3 revealed a dose-dependent cytokine induction profile showing only marginal expression level of IL-6 at a concentration of 1 nM but higher IL-6 release than MPLA at concentrations above 1 nM.
Figure 6.

Induction of expression of TNF-α and IL-6 by Lipid A mimetics 1–3 in mBMDM compared to synthetic E. coli Lipid A and E. coli MPLA. E. coli O111:B4 LPS was used as positive control (1 nM).
TLR4 Stimulating Activities of α,α-GM-LAMs 1 and 3 in Human Dendritic Cells
To test the impact of selected α,α-GM-LAMs 1 and 3 on maturation of human dendritic cells (hDCs), immature monocyte-derived hDCs were stimulated with 1 and 3 in a wide concentration range or with E. coli LPS as positive control. DCs are able to persistently sense pathogen-associated molecular patterns and present antigens to T lymphocytes, thereby initiating an adaptive immune response.7 DCs treated with LPS acquired a distinctive morphologic phenotype and, when analyzed by flow cytometry, displayed characteristic markers of mature DCs.
Stimulation of DCs with 1 (1 mg/mL) was as potent as that with LPS in inducing DCs maturation and up-regulation of the co-stimulatory molecules CD86, as well as the antigen-presenting structures MHC class I and MHC class II, which are necessary for the induction of an adaptive immune response (SI-Figure 5). None of the α,α-GM-LAMs exerted cytotoxic effects on DCs, as determined by propidium iodide staining (data not shown).
Activated DCs were examined for the production of pro-inflammatory cytokines TNF-α, IL-6, and IL-12, which contribute to the modulation of the T cell response and innate effector functions.57 The release of TNF-α and IL-6 reached nearly maximum levels (attained with 10 ng/mL LPS) at the α,α-GM-LAM 1 concentration of 1 ng/mL (Figure 7A). Also the expression of IL-12, which promotes the development of adaptive immune cells and is involved in coordinating innate and adaptive immunity,58 was efficiently induced by α,α-GM-LAM 1, indicating its potential adjuvant capacity. The release of TNF-α, IL-6, and IL-10, a unique cytokine with a wide spectrum of anti-inflammatory effects, in DCs induced by monophosphate 3 was, in agreement with the experiments in the recombinant hTLR4/hMD-2 signaling system, less efficient compared to that induced by 1 (Figure 7B).
Figure 7.

Expression of cytokines induced by α,α-GM-LAMs 1 and 3 in human dendritic cells. (A) Induction of cytokine production by 1 in DCs. (B) Expression of TNF-α, IL-6, and IL-10 induced by monophoshorylated α,α-GM-LAM 3 compared to its bisphosphorylated counterpart 1. E. coli LPS (10 ng/mL) was used as a positive control.
Conclusions and Perspectives
We have rationally designed, synthesized, and biologically evaluated an entirely novel type of TLR4 agonists based on the conformationally restrained disaccharide scaffold which mimics the spatial arrangement of (1→6) diglucosamine backbone of MD-2-bound E. coli Lipid A, revealed in the co-crystal structures of the TLR4·MD-2-LPS complexes. A convergent synthetic approach toward the αGlcN(1↔1)αMan scaffold and hexaacylated Lipid A mimetics based thereon has been developed. Orthogonally protected nonreducing αGlcN(1↔1)αMan disaccharide was assembled via imidate-mediated glycosylation by taking advantage of the axial orientation of the anomeric OH group in the 2-N-carbamate-protected GlcN-lactol acceptors. The protecting group pattern was fine-tuned to ensure stereospecific α,α-(1↔1) glycosylation and to afford efficient isolation of the α,α-disaccharide from the anomeric mixtures. Replacement of the labile anomeric 1-phosphate functionality (as in natural Lipid A) for a primary phosphate group in α,α-GM-LAMs 1 and 2 provides unambiguous advantages with respect to hydrolytic stability.
In spite of the lack of Kdo moieties which were demonstrated to be responsible for a 10–20-fold enhancement of the activity of Re-LPS (Kdo-Kdo-Lipid A) compared to Lipid A alone, the NF-κB activation efficacy and cytokine inducing capacity of bisphosphorylated Lipid A mimetics 1 and 2 in DCs and human macrophages were comparable to or higher than those of Re-LPS/LPS. The inherent rigidity of the α,α-(1↔1) glycosidic linkage in 1–3 would not allow for extensive conformational adjustment of the αGlcN(1↔1)αMan backbone to the shape of the binding pocket of MD-2, such that the three-dimensional arrangement of an α,α-GM-LAM molecule should remain preserved also in the protein-bound state. Accordingly, our results indicate that restricting the flexibility of the carbohydrate backbone of Lipid A in an “agonistic” conformation, as in α,α-GM-LAMs, allows the prearrangement of the phosphate and acyl groups in the “flipped” Man moiety in a defined conformation, which results in a very potent TLR4 activation (SI-Figure 6). The shortening of a secondary acyl chain at Man C-4 and the presence of a phosphate group at Man C-6 significantly enhanced the TLR-4 stimulating activity, which indicates the involvement of these functionalities at the dimerization interface with the second TLR4*·MD-2*-ligand complex and opens opportunities for fine-tuning the activities of α,α-GM-LAMs by chemical modifications.
Since the molecular shape of αGlcN(1↔1)αMan-based Lipid A mimetics is believed to resemble the conformation of the MD-2-bound E. coli Lipid A in the active [TLR4·MD-2-LPS]2 complex, further immuno-biological studies of the interaction of α,α-GM-LAMs with TLR4·MD-2 would provide deeper insight into the molecular basis of TLR4 activation by LPS.
Along these lines, application of a conformationally restricted “agonistically” shaped disaccharide scaffold in place of the native βGlcN(1→6)GlcN Lipid A backbone appears to provide a useful tool for modulation of TLR4·MD-2-mediated immune signaling. Thus, synthetic αGlcN(1↔1)αMan-based Lipid A mimetics represent the key structures for the advanced development of pharmaceutically applicable immuno-therapeutics or vaccine adjuvant candidates.
Experimental Section
General Synthetic Methods
Reagents and solvents were purchased from commercial suppliers and used without further purification unless otherwise stated. Dichloromethane was distilled from CaH2 and stored over activated 4 Å molecular sieves (MS). THF was distilled over Na/benzophenone directly prior to use. Other solvents were dried by storage over activated MS for at least 48 h prior to use [toluene (4 Å), acetonitrile (3 Å), and DMF (3 Å)]. Residual moisture was determined by colorimetric titration on a Mitsubishi CA-21 Karl Fischer apparatus and did not exceed 20 ppm for dry solvents. Reactions were monitored by TLC performed on silica gel 60 F254 HPTLC precoated glass plates with a 25 mm concentration zone (Merck). Spots were visualized by UV light followed by dipping into a H2SO4–p-anisaldehyde solution or a ninhydrin–EtOH solution and subsequent charring at 250 °C. Solvents were removed under reduced pressure at ≤30 °C. Preparative HPLC was performed with linear solvent gradients on a YMC Pack SIL-06 250 × 20 mm, S-5 μm, 6 nm column (Column A, loadings 50–150 mg), or on a YMC Pack SIL-06 250 × 10 mm, S-5 μm, 6 nm column (Column B, loadings 5–50 mg). Preparative MPLC was performed on silica gel 60 (230–400 mesh, Merck). Size exclusion chromatography was performed on Sephadex LH20 or Bio-Beads SX1 (BioRad) supports. NMR spectra were recorded at 298 K on a Bruker Avance III 600 spectrometer (1H at 600.22 MHz; 13C at 150.92 MHz; 31P at 242.97 MHz) or on a Bruker DPX 400 spectrometer (1H at 400.13 MHz; 13C at 100.61 MHz; 31P at 161.68 MHz) using standard Bruker NMR software. Chemical shifts are reported in ppm, where 1H NMR spectra in CDCl3 are referenced to internal TMS and 13C-spectra are referenced to the corresponding solvent signal (77.0 ppm for CDCl3). NMR spectra in other solvents are referenced to residual solvent signals (for acetone-d6, 2.05 and 29.84 ppm; for MeOD, 3.31 and 49.00 ppm, 1H and 13C NMR, respectively). 31P NMR spectra in CDCl3 are referenced according to IUPAC recommendations from 2001 from a referenced 1H NMR spectrum. In the disaccharides, the mannose NMR signals are indicated by primes. The purity (>95%) was determined by LC-MS and HRMS. HPLC-MS was performed by injections of 0.01–0.1% CH3CN solutions into a Shimadzu LC-10AD VP system equipped with two gradient pumps, a degasser, a Shimadzu LCMS 2020 detector, and an AllTech 3300 ELSD detector. Analytes were eluted over a Phenomenex Jupiter 5μ C4 300A column using mobile phase A = H2O (0.1% HCOOH) and mobile phase B = CH3CN (0.1% HCOOH) in linear gradients from 5% B to 100% B and a flow rate of 0.5 mL/min. High-resolution mass spectrometry (HRMS) was carried out on 1–10 mg/L acetonitrile solutions via LC-TOF MS (Agilent 1200SL HPLC and Agilent 6210 ESI-TOF, Agilent Technologies). The mass spectrometer was tuned with Agilent tune mix to provide a mass accuracy below 2 ppm. The data were analyzed using Agilent Mass Hunter Software. MALDI-TOF MS was performed in the negative-ion mode using a Bruker Autoflex Speed instrument with 6-aza-2-thiothymine (ATT) as matrix. Optical rotation was measured on a PerkinElmer 243B polarimeter equipped with a Haake water circulation bath and a Haake D1 immersion circulator for temperature control. All [α]D20 values are reported in units of deg·dm–1·cm3·g–1.
3-O-Benzyl-4,6-O-di-tert-butylsilylene-2-O-methyl-α-d-mannopyranosyl-(1↔1)-6-O-benzyl-4-O-[bis(benzyloxy)phosphoryl]-2-deoxy-2-(9-fluorenylmethoxycarbonylamino)-α-d-glucopyranoside (36)
A solution of glycosyl donor 21 (613 mg, 1.03 mmol) and acceptor 10 (596 mg, 0.751 mmol) in dry CH2Cl2 (22 mL) was stirred with powdered activated 4 Å molecular sieves at room temperature (r.t.) for 1 h under an atmosphere of Ar. The mixture was cooled to 0 °C, and a solution of TMSOTf (9 μL, 52 μmol) in dry CH2Cl2 (450 μL of a stock solution prepared from 20 μL of TMSOTf in 1 mL of CH2Cl2) was added. The mixture was stirred at 0 °C for 30 min, and the reaction was quenched by addition of saturated aqueous (sat. aq.) NaHCO3 (2 mL). The mixture was warmed to r.t. and diluted with EtOAc (100 mL), the solids were removed by filtration over a pad of Celite, and the filtrate was concentrated. The residue was purified by MPLC (two successive columns: toluene–EtOAc, 4:1→1:1→0:1 followed by hexane–EtOAc, 3:1→2:1) to afford 36 (469 mg, 391 μmol, 52%), 39 (186 mg, 155 μmol, 20%), and 42 (44 mg, 37 μmol, 5%). Disaccharide 36: Rf = 0.32 (hexane–EtOAc, 1:1); [α]D20 = +61 (c 0.9, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.76–7.75 (m, 2H, arom), 7.57–7.56 (m, 1H, arom.), 7.52–7.51 (m, 1H, arom), 7.45–7.23 (m, 24H, arom), 5.23 (t, 1H, 3J3,2 = 3J3,4 = 10.0 Hz, H-3), 5.20 (d, 1H, 3J1,2 = 3.5 Hz, H-1), 5.12 (s, 1H, H-1′), 5.03 (AB, 1H, 2J = 11.9 Hz, CH2Ph), 4.99–4.93 (m, 4H, OP(O)(OCH2Ph)2), 4.90 (d, 1H, 3JNH,2 = 9.6 Hz, NH), 4.82 (AB, 1H, 2J = 11.9 Hz, CH2Ph), 4.53–4.47 (m, 2H, H-4, CH2, Fmoc), 4.52 (AB, 1H, J = 11.7 Hz, CH2Ph), 4.44 (AB, 1H, J = 11.8 Hz, CH2Ph), 4.32 (t, 1H, 3J4′,3′ = 3J4′,5′ = 9.5 Hz, H-4′), 4.20 (t, 1H, 3J = 7.4 Hz, CH, Fmoc), 4.10–4.07 (m, 2H, H-2, CH2, Fmoc), 4.05 (dd, 1H, 3J6a′,5′ = 4.8 Hz, 2J6a′,6b′ = 10.3 Hz, H-6a′), 3.96 (t, 1H, 3J6b′,5′ = 2J6b′,6a′= 10.3 Hz, H-6b′), 3.79–3.76 (m, 2H, H-5, H-6a), 3.72–3.68 (m, 2H, H-3′, H-5′), 3.63 (dd, 1H, J = 5.9 Hz, 11.0 Hz, H-6b), 3.41 (s, 1H, H-2′), 3.40 (s, 3H, CH3, Me), 1.87 (s, 3H, CH3, Ac), 1.06 (s, 9H, 3×CH3, DTBS), 0.96 (s, 9H, 3×CH3, DTBS); 13C NMR (151 MHz, CDCl3) δ 171.58 (CO, Ac), 155.69 (CO, Fmoc), 143.79, 143.47, 141.23, 141.20 (4×Cq, Fmoc), 138.73, 137.78 (2×Cq, CH2Ph), 135.46 (Cq, 3JC,P = 6.8 Hz, OP(O)(OCH2Ph)2), 135.39 (Cq, 3JC,P = 7.3 Hz, OP(O)(OCH2Ph)2), 128.72, 128.63, 128.36, 128.32, 128.01, 127.94, 127.79, 127.77, 127.70, 127.67, 127.61, 127.14, 125.12, 125.02, 120.00 (28×CH, arom), 93.70 (C-1′, 1JC,H = 173 Hz), 93.07 (C-1, 1JC,H = 172 Hz), 79.59 (C-2′), 77.84 (C-3′), 74.78 (C-4′), 74.03 (CH2Ph), 73.55 (CH2Ph), 73.49 (C-4, 2JC4,P = 5.9 Hz), 71.35 (C-3), 70.63 (C-5, 3JC5,P = 5.9 Hz), 69.70 (OP(O)(OCH2Ph)2), 69.66 (OP(O)(OCH2Ph)2), 69.19 (C-5′), 68.29 (C-6), 67.70 (CH2, Fmoc), 66.36 (C-6′), 59.68 (CH3, Me), 53.65 (C-2), 46.91 (CH, Fmoc), 27.41, 27.04 (6×CH3, DTBS), 22.56 (Cq, DTBS), 20.70 (CH3, Ac), 19.79 (Cq, DTBS); 31P NMR (243 MHz, CDCl3) δ −2.02; HRMS (ESI) m/z calcd for C66H78NO16PSi+H+ 1200.4900 [M+H+], found 1200.4900.
3-O-Benzyl-4,6-O-di-tert-butylsilylene-2-O-methyl-α-d-mannopyranosyl-(1↔1)-6-O-benzyl-4-O-[bis(benzyloxy)phosphoryl]-2-deoxy-2-(9-fluorenylmethoxycarbonylamino)-α-d-glucopyranoside (46)
A biphasic mixture of 36 (192 mg, 160 μmol) in THF (4 mL) and aq. hydroxylamine (50%, 4 mL) was vigorously stirred at 0 °C for 48 h. The mixture was diluted with EtOAc (50 mL) and washed with aq. citric acid (0.25 M, 70 mL), sat. aq. NaHCO3 (50 mL), and brine (50 mL). The organic layer was dried over MgSO4, filtered, and concentrated. The residue was purified by HPLC (two successive columns: toluene–EtOAc, 1:1, and hexane–EtOAc, 3:1→1:1, Column A) to afford 46, unreacted 36 (56%), 47, and 48. The recovered 36 (108 mg, 90 μmol) was subjected to two additional reaction cycles to afford, after isolation by HPLC, compounds 46 (overall yield: 99 mg, 85 μmol, 53%), 47 (4 mg, 3.5 μmol, 2%), and 48 (16 mg, 15 μmol, 9%). Disaccharide 46: Rf = 0.60 (toluene–EtOAc, 1:1); [α]D20 = +72 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.76–7.75 (m, 2H, arom), 7.61–7.58 (m, 2H, arom), 7.41–7.23 (m, 24H, arom), 5.25 (s, 1H, H-1), 5.09–4.99 (m, 6H, H-1′, CH2Ph, OP(O)(OCH2Ph)2), 4.79–4.74 (m, 2H, CH2Ph, NH), 4.63–4.56 (m, 1H, CH2, Fmoc), 4.47 (s, 2H, CH2Ph), 4.32 (t, 1H, J = 9.4 Hz, H-4′), 4.28–4.22 (m, 2H, H-4, CH, Fmoc), 4.19–4.13 (m, 1H, CH2, Fmoc), 4.07–4.05 (m, 1H, H-6a′), 3.98–3.88 (m, 1H, H-2), 3.95 (t, 1H, J = 10.4 Hz, H-6b′), 3.81–3.75 (m, 1H, H-3), 3.71 (td, J = 4.8 Hz, J = 10.1 Hz, H-5′), 3.69–3.60 (m, 3H, H-3′, H-5, H-6a), 3.55 (dd, J = 5.2 Hz, J = 10.9 Hz, H-6b), 3.40 (s, 3H, CH3, Me), 3.36 (s, 1H, H-2′), 1.06 (s, 9H, 3×CH3, DTBS), 0.95 (s, 9H, 3×CH3, DTBS); 13C NMR (151 MHz, CDCl3) δ 143.87, 143.78, 141.27, 141.25 (4×Cq, Fmoc), 138.84, 137.77 (2×Cq, CH2Ph), 135.28, 135.24 (2×Cq, OP(O)(OCH2Ph)2), 128.91, 128.78, 128.73, 128.65, 128.36, 128.24, 127.93, 127.84, 127.73, 127.71, 127.67, 127.58, 127.17, 127.08, 125.32, 125.19, 119.96, 119.95 (28×CH, arom), 93.81 (C-1′), 93.48 (C-1), 79.81 (C-2′), 77.69 (C-3′), 74.83 (C-4′), 73.84, 73.63 (2×CH2Ph), 70.98 (C-3), 70.49, 70.32 (C-5, JC,P =8.9 Hz, OP(O)(OCH2Ph)2, JC,P = 5.7 Hz,), 70.04 (OP(O)(OCH2Ph)2, 2JC,P = 5.6 Hz), 69.20 (C-5′), 68.36 (C-6), 67.59 (CH2, Fmoc), 66.40 (C-6′), 59.61 (CH3, Me), 54.79 (C-2), 47.14 (CH2, Fmoc), 27.46, 27.06 (6×CH3, DTBS), 22.59, 19.81 (2×Cq, DTBS); 31P NMR (243 MHz, CDCl3) δ 0.43; HRMS (ESI) m/z calcd for C64H767NO15PSi+H+ 1158.4795 [M+H+], found 1158.4790.
3-O-Benzyl-4,6-O-di-tert-butylsilylene-2-O-methyl-α-d-mannopyranosyl-(1↔1)-6-O-benzyl-4-O-[bis(benzyloxy)phosphoryl]-2-deoxy-2-(9-fluorenylmethoxycarbonylamino)-3-O-[(R)-3-(tetradecanoyloxy)tetradecanoyl]-α-d-glucopyranoside (52)
To a stirred solution of 46 (95 mg, 82 μmol) in dry CH2Cl2 (500 μL) were added solutions of 49 (37 mg, 82 μmol) in dry CH2Cl2 (200 μL), DMAP (1 mg, 8 μmol) in dry CH2Cl2 (90 μL of a stock solution 11 mg of DMAP in 1 mL of CH2Cl2), and DIC (10 mg, 82 μmol) in dry CH2Cl2 (150 μL) at 0 °C, and the mixture was stirred for 2.5 h. Then additional portions of 49 (37 mg, 82 μmol) and DIC (10 mg, 82 μmol) were added, and the mixture was stirred for 4.5 h at 0 °C, diluted with EtOAc (50 mL), and washed with aq. citric acid (0.25 M, 50 mL), sat. aq. NaHCO3 (50 mL), and brine (50 mL). The organic layer was dried over MgSO4, filtered, and concentrated. The residue was purified by HPLC (hexane–EtOAc, 5:1→3:1) to afford 52 (109 mg, 68 μmol, 83%) as a colorless syrup. Fractions containing byproducts were purified by HPLC (hexane–EtOAc, 4:1→3:1, Column B) to afford 53 (12 mg, 9 μmol, 11%) and 54 (3 mg, 2.2 μmol, 3%). Compound 52: Rf = 0.66 (toluene–EtOAc, 1:1); [α]D20 = +44 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.75–7.74 (m, 2H, arom), 7.59–7.58 (m, 1H, arom), 7.54–7.53 (m, 1H, arom), 7.45–7.43 (m, 2H, arom), 7.40–7.23 (m, 22H, arom), 5.32 (d, 1H, 3JNH,2 = 8.5 Hz, NH), 5.27 (d, 1H, 3J1,2 = 3.0 Hz, H-1), 5.24 (t, 1H, 3J3,4 = 3J3,2 = 10.0 Hz, H-3), 5.20–5.16 (m, 1H, βMyr-CH), 5.11 (s, 1H, H-1′), 5.04 (AB, 1H, 2J = 11.9 Hz, CH2Ph), 4.98–4.94 (m, 4H, OP(O)(OCH2Ph)2), 4.81 (AB, 1H, 2J = 11.8 Hz, CH2Ph), 4.50 (AB, 1H, 2J = 11.9 Hz, CH2Ph), 4.47–4.41 (m, 2H, H-4, CH2, Fmoc), 4.41 (AB, 1H, 2J = 11.9 Hz, CH2Ph), 4.32 (t, 1H, 3J4′,5′ = 3J4′,3′ = 9.5 Hz, H-4′), 4.18 (t, 1H, J = 7.4 Hz, CH, Fmoc), 4.15–4.12 (m, 1H, CH2, Fmoc), 4.08–4.04 (m, 2H, H-2, H-6a′), 3.96 (t, 1H, 3J6b′,5′ = 2J6b′,6a′ =10.2 Hz, H-6b′), 3.80–3.73 (m, 4H, H-3′, H-5′, H-5, H-6a), 3.61 (dd, 1H, J = 6.0 Hz, J = 11.1 Hz, H-6b), 3.39 (s, 1H, H-2′), 3.38 (s, 3H, CH3, Me), 2.49 (dd, 1H, 3J = 8.3 Hz, 2J = 16.1 Hz, αMyr-CH2), 2.40 (dd, 1H, 3J = 4.4 Hz, 2J = 16.1 Hz, αMyr-CH2), 2.19–2.10 (m, 2H, αMyr-CH2), 1.54–1.50 (m, 2H, βMyr-CH2), 1.42–1.35 (m, 2H, γMyr-CH2), 1.30–1.06 (m, 38H, 19×CH2), 1.05 (s, 9H, 3×CH3, DTBS), 0.94 (s, 9H, 3×CH3, DTBS), 0.88 (t, 6H, J = 7.1 Hz, 2×ωMyr-CH3); 13C NMR (151 MHz, CDCl3) δ 173.23, 171.16 (2×CO), 155.91 (CO, Fmoc), 143.74, 143.66, 141.22, 141.19 (4×Cq, Fmoc), 138.83, 137.87 (2×Cq, CH2Ph), 135.55, 135.51 (2×Cq, OP(O)(OCH2Ph)2), 128.71, 128.64, 128.38, 128.33, 128.16, 128.04, 127.94, 127.80, 127.68, 127.66, 127.62, 127.21, 127.18, 125.29, 125.18, 119.95 (28×CH, arom), 94.02 (C-1′), 93.25 (C-1), 79.76 (C-2′), 78.15 (C-3′), 74.84 (C-4′), 74.23 (CH2Ph), 73.65 (C-4, 2JC4,P = 5.7 Hz), 73.51 (CH2Ph), 71.21 (C-3), 70.69 (C-5, 3JC5,P = 5.3 Hz), 69.79 (βMyr-CH or C-5′), 69.78 (OP(O)(OCH2Ph)2, 2JC,P = 6.5 Hz), 69.74 (OP(O)(OCH2Ph)2, 2JC,P = 6.1 Hz), 69.21 (βMyr-CH or C-5′), 68.36 (C-6), 67.92 (CH2, NHFmoc), 66.40 (C-6′), 59.70 (CH3, Me), 53.80 (C-2), 47.01 (CH, Fmoc), 38.96 (αMyr-CH2), 34.37 (αMyr-CH2), 34.09 (γMyr-CH2), 31.91, 29.68, 29.65, 29.62, 29.52, 29.49, 29.38, 29.35, 29.30, 29.17, 27.46, 27.07, 24.98, 22.68 (20×CH2, 6×CH3, DTBS), 22.59, 19.79 (2×Cq, DTBS), 14.10 (2×ωMyr-CH3); 31P NMR (243 MHz, CDCl3) δ −1.71; HRMS (ESI) m/z calcd for C92H128NO18PSi+Na+ 1616.8513 [M+Na+], found 1616.8526.
3-O-Benzyl-4,6-O-di-tert-butylsilylene-2-O-methyl-α-d-mannopyranosyl-(1↔1)-6-O-benzyl-4-O-[bis(benzyloxy)phosphoryl]-2-deoxy-2-[(R)-3-(dodecanoyloxy)tetradecanonylamino]-3-O-[(R)-3-(tetradecanoyloxy)tetradecanoyl]-α-d-glucopyranoside (55)
To a stirred solution of 52 (104 mg, 65 μmol) in dry CH2Cl2 (1 mL) was added DBU (20 μL, 134 μmol) at 0 °C, and the stirring was continued for 1.5 h at r.t. The mixture was diluted with EtOAc (25 mL) and washed with aq. citric acid (0.25 M, 25 mL), sat. aq. NaHCO3 (25 mL), and brine (25 mL). The organic phase was dried over MgSO4, filtered, and concentrated. The mixture was purified by silica gel chromatography (hexane–EtOAc, 3:1 (0.1% Et3N)→1:1 (0.1% Et3N) to afford the crude amine (78 mg), Rf = 0.28 (hexane–EtOAc, 1:1). To a stirred solution of the intermediate amine (78 mg) in dry CHCl3 (400 μL) were added a solution of 50 (29 mg, 68 μmol) in dry CHCl3 (150 μL) and a solution of EDC·HCl (13 mg, 68 μmol) in dry CHCl3 (200 μL) at r.t. The mixture was stirred for 1.5 h, and then additional amounts of 50 (29 mg, 68 μmol) and EDC·HCl (13 mg, 68 μmol) were added. The mixture was stirred for 3 h, diluted with CHCl3 (30 mL), and washed with water–brine (1:1, 50 mL). The organic phase was dried over MgSO4, filtered, and concentrated. The residue was purified by MPLC (hexane–EtOAc, 4:1→3:1) to afford 55 (91 mg, 51 μmol, 78% over two steps) as a colorless syrup: Rf = 0.44 (hexane–EtOAc, 2:1); [α]D20 = +35 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.43–7.42 (m, 2H, CH2Ph), 7.35–7.22 (m, 18H, CH2Ph), 6.10 (d, 1H, 3JNH,2 = 7.9 Hz, NH), 5.23–5.12 (m, 2H, 2×βMyr-CH), 5.20 (d, 1H, 3J1,2 = 3.6 Hz, H-1), 5.17 (dd, 3J3,4 = 9.0 Hz, 3J3,2 = 11.1 Hz, H-3), 5.08 (d, 1H, 3J1′,2′ = 1.5 Hz, H-1′), 5.01 (AB, 1H, 2J = 11.9 Hz, CH2Ph), 4.96–4.92 (m, 4H, 2×CH2, OP(O)(OCH2Ph)2), 4.77 (AB, 1H, 2J = 11.9 Hz, CH2Ph), 4.48 (AB, 1H, 2J = 11.8 Hz, CH2Ph), 4.39 (AB, 1H, 2J = 11.8 Hz, CH2Ph), 4.38–4.35 (m, 1H, H-4), 4.30 (t, 3J4′,3′ = 3J4′,5′ = 9.5 Hz, H-4′), 4.25 (ddd, 1H, 3J2,1 = 3.6 Hz, 3J2,NH = 7.8 Hz, 3J2,3 = 11.2 Hz, H-2), 4.03 (dd, 1H, 3J6a′,5′ = 5.0 Hz, 2J6a′,6b′ = 10.3 Hz, H-6a′), 3.93 (t, 1H, 3J6b′,5′ = 2J6b′,6a′ = 10.4 Hz, H-6b′), 3.76–3.74 (m, 2H, H-5, H-6a), 3.71 (dd, 1H, 3J3′,2′ = 3.3 Hz, 3J3′,4′ = 9.6 Hz, H-3′), 3.63 (td, 1H, 3J5′,6a′ = 4.9 Hz, 3J5′,4′ = 3J5′,6b′ = 9.9 Hz, H-5′), 3.56 (dd, 1H, J = 6.1 Hz, J = 10.8 Hz, H-6b), 3.39 (dd, 1H, 3J2′,1′ = 1.6 Hz, 3J2′,3′ = 3.2 Hz, H-2′), 3.34 (s, 3H, CH3, Me), 2.48–2.32 (m, 4H, 2×αMyr-CH2), 2.28–2.19 (m, 4H, αMyr-CH2, αLau-CH2), 1.60–1.46 (m, 8H, 2×γMyr-CH2, βMyr-CH2, βLau-CH2), 1.28–1.23 (m, 72H, 36×CH2, fatty acids), 1.07 (s, 9H, 3×CH3, DTBS), 1.01 (s, 9H, 3×CH3, DTBS), 0.89–0.86 (m, 12H, 3×ωMyr-CH3, ωLau-CH3); 13C NMR (151 MHz, CDCl3) δ 173.53, 173.26, 171.26, 169.79 (4×CO), 138.90, 137.89 (2×Cq, CH2Ph) 135.56, 135.51 (2×Cq, OP(O)(OCH2Ph)2) 128.71, 128.63, 128.36, 128.31, 128.18, 128.05, 127.85, 127.63, 127.62, 127.59 (20×CH, CH2Ph), 93.80 (C-1′), 93.11 (C-1), 79.74 (C-2′), 78.25 (C-3′), 74.65 (C-4′), 74.11 (CH2Ph), 73.86 (C-4), 73.47 (CH2Ph), 70.90, 70.76 (C-3, C-5), 70.09, 70.01 (2×βMyr-CH), 69.77, 69.75, 69.74, 69.72 (2×CH2, OP(O)(OCH2Ph)2), 69.33 (C-5′), 68.44 (C-6), 66.36 (C-6′), 59.57 (CH3, Me), 51.66 (C-2), 40.95, 39.29 (2×αMyr-CH2), 34.58, 34.46 (αMyrCH2, αLauCH2, 2×γMyrCH2), 31.93, 31.92, 29.72, 29.69, 29.66, 29.62, 29.57, 29.52, 29.39, 29.36, 29.25, 29.22, 25.26, 25.19, 25.05, 25.04, 22.68 (38×CH2) 27.53, 27.29 (6×CH3, DTBS), 22.64, 19.87 (2×Cq, DTBS), 14.09 (3×ωMyr-CH3, ωLau-CH3); 31P NMR (242.97 MHz, CDCl3) δ −1.67; HRMS (ESI) m/z calcd for C103H166NO19PSi+Na+ 1780.1453 [M+Na+], found 1780.1430.
3-O-Benzyl-2-O-methyl-α-d-mannopyranosyl-(1↔1)-6-O-benzyl-4-O-[bis(benzyloxy)phosphoryl]-2-deoxy-2-[(R)-3-(dodecanoyloxy)tetradecanoylamino]-3-O-[(R)-3-(tetradecanoyloxy)tetradecanoyl]-α-d-glucopyranoside (56)
To a stirred solution of 55 (91 mg, 51 μmol) in dry THF (3 mL) in a PTFE vial was added a solution of HF·Py (70%, 50 μL) at 0 °C. The mixture was stirred for 30 min at r.t., diluted with EtOAc (20 mL), and washed with sat. aq. NaHCO3 (30 mL) and brine (30 mL). The organic phase was dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel chromatography (toluene–EtOAc, 3:1) to afford 56 (78 mg, 47 μmol, 92%): Rf = 0.22 (toluene–EtOAc, 1:1); [α]D20 = +41 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.43–7.22 (m, 20H, CH2Ph), 6.34 (d, 1H, 3JNH,2 = 8.2 Hz, NH), 5.26 (d, 1H, 3J1,2 = 3.7 Hz, H-1), 5.22 (dd, 1H, 3J3,4 = 9.1 Hz, 3J3,2 = 11.0 Hz, H-3), 5.14–5.10 (m, 1H, βMyr-CH), 5.13 (d, 1H, 3J1′,2′ = 1.6 Hz, H-1′), 5.05–5.01 (m, 1H, βMyr-CH), 4.97–4.93 (m, 4H, 2×CH2, OP(O)(OCH2Ph)2), 4.79 (AB, 1H, 2J = 11.6 Hz, CH2Ph), 4.73 (AB, 1H, 2J = 11.7 Hz, CH2Ph), 4.49 (AB, 1H, 2J = 11.8 Hz, CH2Ph), 4.42 (q, 1H, 3J4,3 = 3J4,5 = 3JH,P = 9.2 Hz, H-4), 4.40 (AB, 1H, 2J = 11.9 Hz, CH2Ph), 4.26 (ddd, 1H, 3J1,2 = 3.5 Hz, 3J2,NH = 8.0 Hz, 3J2,3 = 11.2 Hz, H-2), 3.92–3.88 (m, 1H, H-4′), 3.84–3.79 (m, 2H, H-3′, H-6a′), 3.76–3.74 (m, 3H, H-5, H-6a, H-6b′), 3.60–3.56 (m, 2H, H-5′, H-6b), 3.30 (s, 3H, CH3, Me), 3.30–3.28 (m, 1H, H-2′), 2.70 (s, 1H, OH), 2.48–2.42 (m, 3H, αMyr-CH2), 2.39 (dd, 1H, J = 6.2 Hz, J = 15.3 Hz, αMyr-CH2), 2.34–2.26 (m, 2H, αLau-CH2), 2.23–2.20 (m, 2H, αMyr-CH2), 1.65–1.46 (m, 8H, 2×γMyrCH2, βMyrCH2, βLauCH2), 1.31–1.21 (m, 72H, 36×CH2), 0.89–0.86 (m, 12H, 3×ωMyr-CH3, ωLau-CH3); 13C NMR (151 MHz, CDCl3) δ 174.65, 173.48, 171.15, 170.08 (4×CO), 138.10, 137.87 (2×Cq, CH2Ph), 135.47 (2×Cq, OP(O)(OCH2Ph)2), 128.72, 128.63, 128.60, 128.31, 128.17, 128.04, 128.01, 127.63, 127.59 (20×CH, CH2Ph), 93.14 (C-1′), 92.56 (C-1), 78.78 (C-3′), 77.38 (C-2′), 73.76 (C-4, C-5′), 73.48, 72.76 (2×CH2Ph), 71.33 (βMyr-CH), 71.02 (C-3), 70.62 (C-5, 3JC5,P = 5.0 Hz), 70.01 (βMyr-CH), 69.82 (2×CH2, OP(O)(OCH2Ph)2), 68.42 (C-6), 67.84 (C-4′), 63.04 (C-6′), 59.03 (CH3, Me), 51.70 (C-2), 41.50, 39.09 (2×αMyr-CH2), 34.74, 34.60, 34.41, 34.23 (αMyrCH2, αLauCH2, 2×γMyrCH2), 31.92, 31.91, 29.70, 29.68, 29.65, 29.61, 29.58, 29.54, 29.47, 29.43, 29.37, 29.34, 29.20, 29.18, 25.28, 25.17, 25.01, 24.99, 22.67 (38×CH2), 14.08 (3×ωMyr-CH3, ωLau-CH3); 31P NMR (243 MHz, CDCl3) δ −1.80; HRMS (ESI) m/z calcd for C95H150NO19P+Na+ 1663.0432 [M+Na+], found 1663.0424.
3-O-Benzyl-4-O-[(R)-3-(dodecanoyloxy)tetradecanoyl]-2-O-methyl-α-d-mannopyranosyl-(1↔1)-6-O-benzyl-4-O-[bis(benzyloxy)phosphoryl]-2-deoxy-2-[(R)-3-(dodecanoyloxy)tetradecanoylamino]-3-O-[(R)-3-(tetradecanoyloxy)tetradecanoyl]-α-d-glucopyranoside (57)
To a stirred solution of 56 (21 mg, 13 μmol) and DMAP (0.15 mg, 1.3 μmol) in dry CH2Cl2 (500 μL) was added a solution of fatty acid 50 (6.5 mg, 15 μmol) in CH2Cl2 (50 μL of a stock solution prepared from 26 mg of 50 in 200 μL of CH2Cl2). The mixture was cooled to 0 °C, and DIC (1.9 mg, 15.4 μmol) from a 50 mg/mL stock solution in toluene was added dropwise over a period of 1 h. Then additional amounts of 50 (1.3 mg, 3.1 μmol) and DIC (0.4 mg, 0.3 μmol) from the indicated stock solutions were successively added (DIC was added dropwise over 30 min) at 0 °C. The mixture was diluted with EtOAc (20 mL) and washed with aq. 2 M HCl (20 mL), aq. sat. NaHCO3 (20 mL), and brine (20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by HPLC (two consecutive columns: toluene–EtOAc, 3:1→2:1, and hexane–EtOAc, 3:1→1:1, column B) to afford 57 (13.5 mg, 6.6 μmol, 51%) as a syrup. Fractions containing byproduct 59 were purified by HPLC (hexane–EtOAc, 5:1→3:1) to afford 59 (7.6 mg, 3.1 μmol, 24%) as a syrup. Compound 57: Rf = 0.27 (hexane–EtOAc, 1:1); [α]D20 = +68 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.38–7.21 (m, 20H, CH2Ph), 6.37 (d, 1H, 3JNH,2 = 8.2 Hz, NH), 5.24–5.15 (m, 5H, H-1, H-3, H-4′, 2×βMyr-CH), 5.12 (d, 1H, 3J1′,2′ = 2.0 Hz, H-1′), 5.07–5.03 (m, 1H, βMyr-CH), 4.97–4.94 (m, 4H, OP(O)(OCH2Ph)2), 4.69 (AB, 1H, 2J = 11.9 Hz, CH2Ph), 4.64 (AB, 1H, 2J = 12.1 Hz, CH2Ph), 4.48 (AB, 1H, 2J = 11.8 Hz, CH2Ph), 4.42 (ddd, 1H, 3J4,3 = 3J4,5 = 3JH,P = 9.3 Hz, H-4), 4.40 (AB, 1H, 2J = 11.9 Hz, CH2Ph), 4.24 (ddd, 1H, 3J2,1 = 3.6 Hz, 3J2,NH = 8.1 Hz, 3J2,3 = 11.1 Hz, H-2), 3.94 (dd, 1H, 3J3′,2′ = 3.1 Hz, 3J3′,4′ = 9.4 Hz, H-3′), 3.75–3.72 (m, 2H, H-5, H-6a), 3.62–3.55 (m, 4H, H-5′, H-6a′, H-6b′, H-6b), 3.30–3.29 (m, 1H, H-2′), 3.29 (s, 3H, CH3, Me), 2.60 (dd, 1H, J = 7.6 Hz, 15.5 Hz, αMyr-CH2), 2.54–2.47 (m, 3H, αMyr-CH2), 2.44 (dd, 1H, J = 7.2 Hz, 15.0 Hz, αMyr-CH2), 2.37 (dd, 1H, J = 5.6 Hz, 15.1 Hz, αMyr-CH2), 2.30–2.27 (m, 2H, α-CH2), 2.24–2.19 (m, 4H, 2×α-CH2), 1.62–1.48 (m, 12H, βMyr-CH2, 2×βLau-CH2, 3×ωMyr-CH2), 1.30–1.22 (m, 106H, 53×CH2), 0.89–0.86 (m, 18H, 4×ωMyr-CH3, 2×ωLau-CH3); 13C NMR (151 MHz, CDCl3) δ 173.64, 173.40, 173.10, 171.43, 169.93, 169.88 (6×CO), 138.01, 137.88 (2×Cq, CH2Ph), 135.55, 135.51 (2×Cq, OP(O)(OCH2Ph)2), 128.71, 128.64, 128.53, 128.32, 128.12, 128.11, 128.04, 127.93, 127.65, 127.59 (20×CH, CH2Ph), 94.03 (C-1′), 93.51 (C-1), 77.81 (C-2′), 75.89 (C-3′), 73.67 (C-4, 2JC4,P = 5.8 Hz), 73.49 (CH2Ph), 72.74 (C-5′), 72.52 (CH2Ph), 71.09 (C-3), 70.84 (βMyr-CH), 70.69 (C-5, 3JC5,P = 5.3 Hz), 69.95, 69.88 (2×βMyr-CH), 69.77, 69.73 (2×CH2, OP(O)(OCH2Ph)2), 68.36 (C-6), 68.35 (C-4′), 61.71 (C-6′), 59.10 (CH3, Me), 51.81 (C-2), 41.38, 39.16, 39.02 (3×αMyr-CH2), 34.60, 34.53, 34.44, 34.28, 34.05 (αMyr-CH2, 2×αLau-CH2, 3×γMyr-CH2), 31.93, 29.73, 29.70, 29.67, 29.62, 29.60, 29.57, 29.55, 29.54, 29.52, 29.48, 29.42, 29.39, 29.37, 29.36, 29.33, 29.28, 29.20, 25.36, 25.17, 25.10, 25.01, 22.69 (56×CH2), 14.10 (4×ωMyr-CH3, 2×ωLau-CH3); 31P NMR (243 MHz, CDCl3) δ −1.71; HRMS (ESI) m/z calcd for C121H198NO22P+Na+ 2071.4035 [M+Na+], found 2071.4036.
3-O-Benzyl-6-O-[bis(benzyloxy)phosphoryl]-4-O-[(R)-3-(dodecanoyloxy)tetradecanoyl]-2-O-methyl-α-mannopyranosyl-(1↔1)-6-O-benzyl-4-O-[bis(benzyloxy)phophoryl]-2-deoxy-2[(R)-3-(dodecanoyloxy)tetradecanoylamino]-3-O-[(R)-3-(tetradecanoyloxy)tetradecanoyl]-α-d-glucopyranoside (61)
To a stirred solution of 57 (11 mg, 6 μmol) and dibenzyl N,N-diisopropylphosphoramidite (10 μL, 25 μmol, 90%) in dry CH2Cl2 (400 μL) was added a solution of 1H-tetrazole (54 μL, 24 μmol, 0.45 M in CH3CN) under an atmosphere of Ar. The mixture was stirred for 1 h and then cooled to −78 °C, and a solution of mCPBA (7 mg, 70%) in CH2Cl2 (54 μL) was added. After being stirred for 45 min, the reaction was quenched by addition of Et3N (5 μL), and then the mixture was warmed to r.t., diluted with CH2Cl2 (10 mL), and washed with sat. aq. NaHCO3 (5 mL) and brine (5 mL). The organic phase was dried over Na2SO4 and concentrated. The residue was purified by HPLC (two consecutive columns: toluene–EtOAc, 10:1→5:1→1:1 followed by hexane–EtOAc, 2:1→1:1, column B) to afford 61 (11 mg, 5 μmol, 89%) as a syrup: Rf = 0.53 (hexane–EtOAc, 1:1); [α]D20 = +31 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.38–7.21 (m, 30H, CH2Ph), 6.46 (d, 1H, 3JNH,2 = 8.3 Hz, NH), 5.30 (t, 1H, 3J4′,5′ = 3J4′,3′ = 9.8 Hz, H-4′), 5.23–5.16 (m, 3H, H-3, 2×βMyr-CH), 5.15 (d, 1H, 3J1,2 = 3.7 Hz, H-1), 5.10 (d, 1H, 3J1′,2′ = 2.0 Hz, H-1′), 5.10–5.06 (m, 1H, βMyr-CH), 5.03–4.93 (m, 8H, 2×OP(O)(OCH2Ph)2), 4.67 (AB, 1H, 2J = 12.1 Hz, CH2Ph), 4.63 (AB, 1H, 2J = 12.1 Hz, CH2Ph), 4.45 (AB, 1H, 2J = 11.8 Hz, CH2Ph), 4.43–4.39 (m, 1H, H-4), 4.38 (AB, 1H, 2J = 11.9 Hz, CH2Ph), 4.30–4.26 (m, 1H, H-2), 4.07–4.04 (m, 2H, H-6a′, H-6b′), 4.01 (dd, 1H, 3J3′,2′ = 3.0 Hz, 3J3′,4′ = 9.6 Hz, H-4′), 3.89–3.86 (m, 1H, H-5′), 3.74 (dd, 1H, 3J6a,5 = 1.7 Hz, 2J6a,6b = 10.8 Hz, H-6a), 3.72–3.69 (m, 1H, H-5), 3.56 (dd, 1H, 3J6b,5 = 5.9 Hz, 2J6b,6a = 10.8 Hz, H-6b), 3.28 (t, 1H, J = 2.6 Hz, H-2′), 3.23 (s, 3H, CH3, Me), 2.58–2.52 (m, 3H, αMyr-CH2), 2.48–2.44 (m, 2H, αMyr-CH2), 2.41–2.38 (m, 1H, αMyr-CH2), 2.34–2.24 (m, 2H, αLau-CH2), 2.20–2.16 (m, 4H, αLau-CH2, αMyr-CH2), 1.61–1.49 (m, 12H, βMyr-CH2, 2×βLau-CH2, 3×γMyr-CH2), 1.29–1.22 (m, 106H, 53×CH2), 0.89–0.86 (m, 18H, 4×ωMyr-CH3, 2×ωLau-CH3); 13C NMR (151 MHz, CDCl3) δ 173.89, 173.25, 172.94, 171.20, 169.77, 169.07 (6×CO), 137.99, 137.85 (2×Cq, CH2Ph), 135.98, 135.94, 135.56, 135.51 (4×Cq, OP(O)(OCH2Ph)2), 128.69, 128.63, 128.54, 128.46, 128.44, 128.30, 128.28, 128.14, 128.02, 127.96, 127.92, 127.88, 127.64, 127.60 (30×CH, CH2Ph), 93.36 (C-1′), 93.18 (C-1), 77.55 (C-2′), 75.96 (C-3′), 73.69 (C-4, 2JC4,P = 6.1 Hz), 73.49, 72.47 (2×CH2, Ph), 71.24, 70.96, 70.66, 70.60, 69.82, 69.76 (C-3, C-5, C-5′, βMyr-CH, 2×βLau-CH), 69.78, 69.74, 69.70, 69.32 (4×CH2, OP(O)(OCH2Ph)2), 68.48 (C-6), 67.50 (C-4′), 66.15 (C-6′), 59.02 (CH3, Me), 51.44 (C-2), 41.56, 38.96, 38.77 (3×αMyr-CH2), 34.77, 34.55, 34.41, 34.20, 34.00 (αMyr-CH2, 2×αLau-CH2, 3×γMyr-CH2), 31.94, 31.92, 29.75, 29.73, 29.69, 29.67, 29.65, 29.63, 29.56, 29.54, 29.51, 29.47, 29.39, 29.36, 29.31, 29.19, 25.37, 25.17, 25.12, 25.01, 22.68 (56×CH2), 14.09 (4×ωMyr-CH3, 2×ωLau-CH3); 31P NMR (243 MHz, CDCl3) δ −1.61, −1.69; HRMS (ESI) m/z calcd for C135H211NO25P2+Na+ 2331.4638 [M+Na+], found 2331.4618.
4-O-[(R)-3-(Dodecanoyloxy)tetradecanoyl]-2-O-methyl-6-O-phosphoryl-α-d-mannopyranosyl-(1↔1)-2-deoxy-2-[(R)-3-(dodecanoyloxy)tetradecanoylamino]-4-O-phosphoryl-3-O-[(R)-3-(tetradecanoyloxy)tetradecanoyl]-α-d-glucopyranoside (1)
A solution of 61 (10 mg, 4.3 μmol) in toluene–MeOH (1 mL, 1:1) was hydrogenated over Pd-black (5 mg, 47 μmol) for 20 h. The mixture was diluted with CH2Cl2–EtOH (1:1, 10 mL), and the solids were removed by filtration (syringe filter, regenerated cellulose, 45 μm). The filtrate was concentrated, and the residue was purified by size exclusion chromatography on Bio-Beads SX1 (200–400 mesh, 2 × 60 cm, toluene–CH2Cl2, 3:1). Appropriate fractions were concentrated, and the residue was dissolved in DMSO (2 mL) and freeze-dried to afford 1 (6.0 mg, 3.4 μmol, 79%) as a white solid: Rf = 0.29 (CHCl3–MeOH–H2O, 50:28:6); 1H NMR (600 MHz, CDCl3–MeOD, 3:1) δ 5.29 (d, 1H, 3J1,2 = 3.8 Hz, H-1), 5.27–5.22 (m, 4H, H-1′, H-3, 2×βMyr-CH), 5.14–5.09 (m, 1H, βMyr-CH), 5.00 (t, 1H, 3J4′,5′ = 3J4′,3′ = 9.7 Hz, H-4′), 4.31 (dt, 1H, 3J = 9.7 Hz, 3J = 9.9 Hz, H-4), 4.17 (dd, 1H, 3J2,1 = 3.7 Hz, 3J2,3 = 10.9 Hz, H-2), 4.03 (dd, 1H, 3J3′,2′ = 3.6 Hz, 3J3′,4′ = 9.5 Hz, H-3′), 4.02–3.98 (m, 1H, H-6a′), 3.94–3.87 (m, 2H, H-6a, H-6b′), 3.79–3.75 (m, 2H, H-5′, H-6b), 3.73–3.69 (m, 1H, H-5), 3.49 (dd, 1H, 3J2′,1′ = 1.8 Hz, 3J2′,3′ = 3.5 Hz, H-2′), 3.48 (s, 3H, CH3, Me), 2.68–2.60 (m, 4H, 2×αMyr-CH2), 2.52–2.45 (m, 2H, αMyr-CH2), 2.33–2.29 (m, 6H, αMyr-CH2, 2×αLau-CH2), 1.67–1.54 (m, 12H, βMyr-CH2, 2×βLau-CH2, 3×γMyr-CH2), 1.35–1.23 (m, 106H, 53×CH2), 0.89 (t, 18H, J = 7.0 Hz, 4×ωMyr-CH3, 2×ωLau-CH3); 31P NMR (243 MHz, CDCl3–MeOD, 3:1) δ 0.38, 0.01; MS (MALDI) m/z calcd for C93H175NO25P–H 1767.186 [M-H]−, found 1767.058. For MALDI-TOF MS, 1 μL of a solution of 1 (0.5 mg/mL in CH2Cl2–EtOH, 1:1) was mixed with 6-aza-2-thiothymine matrix (3 mg/mL EtOH–20 mM aq. ammonium citrate buffer, 1:1).
4-O-[(R)-3-(Decanoyloxy)tetradecanoyl]-2-O-methyl-6-O-phosphoryl-α-mannopyranosyl-(1↔1)-2-deoxy-2-[(R)-3-(dodecanoyloxy)tetradecanoylamino]-4-O-phosphoryl-3-O-[(R)-3-(tetradecanoyloxy)tetradecanoyl]-α-d-glucopyranoside (2)
Compound 2 (12 mg, 5.3 μmol) was prepared from 62 in a manner similar to the synthesis of 1, which afforded 2 (8.0 mg, 4.6 μmol, 87%) as a white fluffy solid: Rf = 0.38 (CHCl3–MeOH–H2O, 50:28:6); 1H NMR (600 MHz, CDCl3–MeOD, 3:1) δ 5.29 (d, 1H, 3J1,2 = 3.6 Hz, H-1), 5.27–5.21 (m, 4H, H-1′, H-3, 2×βMyr-CH), 5.14–5.09 (m, 1H, βMyr-CH), 5.00 (t, 1H, 3J4′,5′ = 3J3′,4′ = 9.7 Hz, H-4′), 4.31 (dt, 1H, 3J4,5 = 3J4,3 = 3J4,P = 9.7 Hz, H-4), 4.19–4.13 (m, 1H, H-2), 4.03 (dd, 1H, 3J3′,2′ = 3.5 Hz, 3J3′,4′ = 9.5 Hz, H-3′), 4.01–3.98 (m, 1H, H-6a′), 3.94–3.88 (m, 2H, H-6a, H-6b′), 3.79–3.74 (m, 2H, H-5′, H-6b), 3.73–3.69 (m, 1H, H-5), 3.49 (dd, 1H, 3J2′,1′ = 1.8 Hz, 3J2′,3′ = 3.4 Hz, H-2′), 3.48 (s, 3H, CH3, Me), 2.70–2.60 (m, 4H, 2×α Myr-CH2), 2.52–2.45 (m, 2H, αMyr-CH2), 2.33–2.29 (m, 6H, αMyr-CH2, αLau-CH2, αCap-CH2), 1.65–1.55 (m, 12H, 3×γ Myr-CH2, βMyr-CH2, βLau-CH2, βCap-CH2), 1.37–1.22 (m, 102H, 51×CH2), 0.89 (t, 18H, J = 7.0 Hz, 4×ω Myr-CH3, ωLau-CH3, ωCap-CH3); 31P NMR (243 MHz, CDCl3–MeOD, 3:1) δ 0.42, 0.01; MS (MALDI) m/z calcd for C91H171NO25P 2 −H 1739.154 [M-H]−, found 1739.160.
4-O-[(R)-3-(Dodecanoyloxy)tetradecanoyl]-2-O-methyl-α-d-mannopyranosyl-(1↔1)-2-deoxy-2-[(R)-3-(dodecanoyloxy)tetradecanoylamino]-4-O-phosphoryl-3-O-[(R)-3-(tetradecanoyloxy)tetradecanoyl]-α-d-glucopyranoside (3)
Compound 3 (5.6 mg, 2.7 μmol) was prepared from 57 in a manner similar to the synthesis of 1, which afforded 3 (3.7 mg, 2.2 μmol, 81%) as a white fluffy solid: Rf = 0.47 (CHCl3–MeOH–H2O, 50:28:6); 1H NMR (600 MHz, CDCl3–MeOD, 3:1) δ 7.16 (d, 1H, 3JNH,2 = 8.3 Hz, NH), 5.26–5.17 (m, 5H, H-1, H-1′, H-3, 2×βMyr-CH), 5.12–5.08 (m, 1H, βMyr-CH), 5.04 (t, 1H, 3J4′,3′ = 3J4′,5′ = 9.8 Hz, H-4′), 4.30 (dt, 1H, 3J4,5 = 3J4,3 = 3J4,P = 9.9 Hz, H-4), 4.24–4.19 (m, 1H, H-2), 4.04 (dd, 1H, 3J3′,2′ = 3.5 Hz, 3J3′,4′ = 9.6 Hz, H-3′), 3.94 (dd, 1H, J = 2.9 Hz, J = 13.0 Hz, H-6a), 3.75–3.71 (m, 1H, H-6b), 3.71–3.67 (m, 1H, H-5), 3.66–3.62 (m, 1H, H-5′), 3.57–3.54 (m, 2H, H-6a′, H-6b′), 3.51–3.48 (m, 1H, H-2′), 3.49 (s, 3H, CH3, Me), 2.69–2.58 (m, 4H, 2×αMyr-CH2), 2.49 (dd, 1H, J = 6.9 Hz, J = 14.9 Hz, αMyr-CH2), 2.43 (dd, 1H, J = 5.9 Hz, J = 14.9 Hz, αMyr-CH2), 2.32–2.28 (m, 6H, αMyr-CH2, 2×αLau-CH2), 1.67–1.54 (m, 12H, βMyr-CH2, 2×βLau-CH2, 3×γMyr-CH2), 1.38–1.22 (m, 106H, 53×CH2), 0.89 (t, 18, J = 7.0 Hz, 4×ωMyr-CH3, 2×ωLau-CH3); 31P NMR (243 MHz, CDCl3–MeOD, 3:1) δ 0.68; MS (ESI) m/z calcd for C93H174NO22P–H 1687.219 [M-H]−, found 1687.205.
Biological Assays
Reagents
HEK293 stably expressing human TLR4, MD-2, CD14, and a secreted NF-κB-dependent reporter (HEKBlue hTLR4), E. coli O111:B4 LPS, E. coli serotype R515 Re-LPS, S. minnesota R595 MPLA (SM-MPLA), and synthetic E. coli MPLA were purchased from InvivoGen. Synthetic E. coli lipid A was purchased from Peptide Institute. The THP-1 human monocyte-like cell line was obtained from Dr. Rene Devos (Roche Research Ghent) and originally purchased from ATCC. The phorbol ester 12-O-tetradecanoylphorbol-13-acetate (TPA) was purchased from Sigma. Lipid A mimetics 1–3 were reconstituted in DMSO to provide 1 mg/mL stock solutions. Further dilutions were made with cell medium (RPMI or DMEM) supplemented with 10% FCS so that the final amount of DMSO in the cell culture did not exceed 0.01%.
hTLR4/MD-2/CD14-Transfected HEK293 Cells (Hek-Blue) Activation Assay
Growth conditions and activation assay were set as recommended by InvivoGen. The cells were stimulated with the solutions of compounds 1–3 or LPS/Re-LPS in DMEM supplemented by 10% FCS at the indicated concentrations. The final amount of DMSO in the cell culture did not exceed 0.005% for 1 and 2 or 0.01% for 3. The compounds were added in a total volume of 20 μL to 25000 HEK-Blue hTLR4 cells in 180 μL plates and were incubated for 20–24 h at 37 °C and 5% CO2. SEAP levels were determined by incubation of 20 μL of challenged cell supernatants with 180 μL of detection reagent (QUANTI-Blue), and the color development was measured at 650 nm using a spectrophotometer (SpectraMAX 190). Data were combined from n = 3 independent experiments; error bars indicate standard error of the mean.
Differentiation and Stimulation of THP-1 Cells
THP-1 cells were grown in RPMI-1640 cell-culture medium (Life Technologies) that was supplemented with 2 mM l-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% FCS. Cells were seeded in a 96-well plate at 105 cells/well in 150 μL of complete medium and stimulated by 200 nM TPA for 24 h to induce the differentiation into macrophage-like cells.59 On the next day the cells were washed twice with complete culture medium to discard the cells that did not adhere, refreshed with 200 μL of complete medium, and left for 1 h to recover. Cells were stimulated with α,α-GM-LAMs 1–3 at the indicated concentration and with E. coliRe-LPS (or E. coli O111:B4 LPS), which were added as solutions in 10 μL of complete medium. The total volume of the well after stimulation reached 220 μL. The cells were incubated for 18 h, and the supernatants were analyzed for TNF-α, IL-8, and MCP-1 by ELISA (BD Biosciences).
Activation of Mouse Bone Marrow-Derived Macrophages by α,α-GM-LAMs 1–3
Bone-marrow-derived microphages (BMDMs) were isolated and differentiated from the bone marrow of C57BL/6J mice. The bone marrow was flushed from femur and fibia with RPMI media. The erythrocytes were lysed with 0.88% ammonium chloride, 15 min at 37 °C. A single-cell suspension of the bone marrow cells was then seeded in cell culture flasks at a concentration of 1 × 106 cells/mL in RPMI supplemented with 20% FBS and 40 ng/mL recombinant M-CSF. The cell culture medium was changed on day 3. On day 6, the differentiated cells were trypsinized, counted, and seeded in 24-well plates at a concentration of 1 × 106 cells/mL in RPMI supplemented with 10% FBS. After 24 h, the cells were stimulated with Lipid A mimetics 1–3 and with synthetic E. coli Lipid A/MPLA (E. coli O111:B4 LPS was used as positive control) for 16 h. The nanomolar concentrations of 1–3 were calculated according to the MW of the synthetic compounds. The supernatants were then tested for cytokines using Ready-Set-Go ELISA kits (eBioscience).
Induction of Cytokine Production by α,α-GM-LAMs 1 and 3 in hDCs
Human peripheral blood monocytes were cultured for 6 days in GM-CSF and IL-4 to receive immature monocyte-derived DCs and were then stimulated with the indicated concentrations of 1 and 3 using 10 ng/mL LPS as positive control (solutions in PRMI containing 10% FCS and 0.005–0.01% DMSO). The amounts of IL-6, IL-12, IL-10, and TNF-α in the supernatants of the cells were analyzed after 24 h by Luminex. Results are representative of three independent experiments for 1 and of two experiments for 3. Mean values of duplicate examinations ± SD are presented.
Acknowledgments
Financial support from Austrian Science Foundation (grant FWF P-22116) is gratefully acknowledged. We thank Dr. Andreas Hofinger for support with NMR measurements.
Glossary
Abbreviations Used
- TLR4
Toll-like Receptor 4
- MD-2
myeloid differentiation factor 2
- α,α-GM-LAM
αGlcN(1↔1)αMan-based Lipid A mimetic
- MPLA
synthetic E. coli monophosphoryl Lipid A
- SM-MPLA
S. minnesota monophosphoryl Lipid A
- MyD88
myeloid differentiation primary response gene 88
Supporting Information Available
Supplementary tables and figures, experimental procedures, and 1H NMR, 13C NMR, and MALDI-TOF spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Kawai T.; Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 2010, 5, 373–384. [DOI] [PubMed] [Google Scholar]
- Opal S. M. The host response to endotoxin, antilipopolysaccharide strategies, and the management of severe sepsis. Int. J. Med. Microbiol. 2007, 5, 365–377. [DOI] [PubMed] [Google Scholar]
- Hammad H.; Chieppa M.; Perros F.; Willart M. A.; Germain R. N.; Lambrecht B. N. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat. Med. 2009, 4, 410–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisser D.; Jeannin J.-F.. Lipid A in cancer therapies: preclinical studies. In Lipid A in cancer therapy; Jeannin J.-F., Ed.; Springer, Landes Bioscience: New York, 2009; pp 101–110. [Google Scholar]
- Gangloff M.; Gay N. J. MD-2: the Toll ‘gatekeeper’ in endotoxin signalling. Trends Biochem. Sci. 2004, 6, 294–300. [DOI] [PubMed] [Google Scholar]
- Hotchkiss R. S.; Monneret G.; Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat. Rev. Immunol. 2013, 12, 862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A.; Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 10, 987–995. [DOI] [PubMed] [Google Scholar]
- Casella C.; Mitchell T. Putting endotoxin to work for us: Monophosphoryl lipid A as a safe and effective vaccine adjuvant. Cell. Mol. Life Sci. 2008, 20, 3231–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizaka S. T.; Hawkins L. D. E6020: a synthetic Toll-like receptor 4 agonist as a vaccine adjuvant. Exp. Rev. Vaccines 2007, 5, 773–784. [DOI] [PubMed] [Google Scholar]
- Johnson D. A. Synthetic TLR4-active glycolipids as vaccine adjuvants and stand-alone immunotherapeutics. Curr. Top. Med. Chem. 2008, 2, 64–79. [DOI] [PubMed] [Google Scholar]
- Bryant C. E.; Spring D. R.; Gangloff M.; Gay N. J. The molecular basis of the host response to lipopolysaccharide. Nat. Rev. Microbiol. 2010, 1, 8–14. [DOI] [PubMed] [Google Scholar]
- Ohto U.; Fukase K.; Miyake K.; Satow Y. Crystal structures of human MD-2 and its complex with antiendotoxic lipid IVa. Science 2007, 5831, 1632–1634. [DOI] [PubMed] [Google Scholar]
- Kim H. M.; Park B. S.; Kim J.-I.; Kim S. E.; Lee J.; Oh S. C.; Enkhbayar P.; Matsushima N.; Lee h.; Yoo O. J.; Lee J.-O. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 2007, 906–917. [DOI] [PubMed] [Google Scholar]
- Park B. S.; Song D. H.; Kim H. M.; Choi B. S.; Lee H.; Lee J. O. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 2009, 1191–1195. [DOI] [PubMed] [Google Scholar]
- Ohto U.; Fukase K.; Miyake K.; Shimizu T. Structural basis of species-specific endotoxin sensing by innate immune receptor TLR4/MD-2. Proc. Natl. Acad. Sci. U.S.A. 2012, 19, 7421–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L.; Phillips R. L.; Zhang D.; Teghanemt A.; Weiss J. P.; Gioannini T. L. NMR studies of hexaacylated endotoxin bound to wild-type and F126A mutant MD-2 and MD-2-TLR4 ectodomain complexes. J. Biol. Chem. 2012, 20, 16346–16355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teghanemt A.; Re F.; Prohinar P.; Widstrom R.; Gioannini T. L.; Weiss J. P. Novel roles in human MD-2 of Phenylalanines 121 and 126 and Tyrosine 131 in activation of Toll-like receptor 4 by endotoxin. J. Biol. Chem. 2008, 3, 1257–1266. [DOI] [PubMed] [Google Scholar]
- Kumar H.; Kawai T.; Akira S. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 2009, 4, 621–625. [DOI] [PubMed] [Google Scholar]
- Meng J.; Lien E.; Golenbock D. T. MD-2-mediated ionic interactions between Lipid A and TLR4 are essential for receptor activation. J. Biol. Chem. 2010, 12, 8695–8702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casella C. R.; Mitchell T. C. Inefficient TLR4/MD-2 heterotetramerization by monophosphoryl lipid A. PLoS One 2013, 4, e62622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimura N.; Saitoh S. i.; Ohto U.; Akashi-Takamura S.; Fujimoto Y.; Fukase K.; Shimizu T.; Miyake K. The attenuated inflammation of MPL is due to the lack of CD14-dependent tight dimerization of the TLR4/MD2 complex at the plasma membrane. Int. Immunol. 2014, 6, 307–314. [DOI] [PubMed] [Google Scholar]
- Trent M. S.; Stead C. M.; Tran A. X.; Hankins J. V. Diversity of endotoxin and its impact on pathogenesis. J. Endotoxin Res. 2006, 4, 205–223. [DOI] [PubMed] [Google Scholar]
- Zughaier S. M.; Zimmer S. M.; Datta A.; Carlson R. W.; Stephens D. S. Differential induction of the Toll-Like Receptor 4-MyD88-dependent and -independent signaling pathways by endotoxins. Infect. Immun. 2005, 5, 2940–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needham B. D.; Carroll S. M.; Giles D. K.; Georgiou G.; Whiteley M.; Trent M. S. Modulating the innate immune response by combinatorial engineering of endotoxin. Proc. Natl. Acad. Sci. U.S.A. 2013, 4, 1464–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto Y.; Adachi Y.; Akamatsu M.; Fukase Y.; Kataoka M.; Suda Y.; Fukase K.; Kusumoto S. Synthesis of lipid A and its analogues for investigation of the structural basis for their bioactivity. J. Endotoxin Res. 2005, 6, 341–347. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Gaekwad J.; Wolfert M. A.; Boons G. J. Modulation of innate immune responses with synthetic Lipid A derivatives. J. Am. Chem. Soc. 2007, 16, 5200–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akamatsu M.; Fujimoto Y.; Kataoka M.; Suda Y.; Kusumoto S.; Fukase K. Synthesis of lipid A monosaccharide analogues containing acidic amino acid: exploring the structural basis for the endotoxic and antagonistic activities. Bioorg. Med. Chem. 2006, 19, 6759–6777. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Gaekwad J.; Wolfert M. A.; Boons G. J. Innate immune responses of synthetic lipid A derivatives of Neisseria meningitidis. Chem.—Eur. J. 2007, 2, 558–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peri F.; Calabrese V. Toll-like Receptor 4 (TLR4) modulation by synthetic and natural compounds: an update. J. Med. Chem. 2013, 9, 3612–3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazin H. G.; Bess L. S.; Livesay M. T.; Ryter K. T.; Johnson C. L.; Arnold J. S.; Johnson D. A. New synthesis of glycolipid immunostimulants RC-529 and CRX-524. Tetrahedron Lett. 2006, 13, 2087–2092. [Google Scholar]
- Artner D.; Oblak A.; Ittig S.; Garate J. A.; Horvat S.; Arrieumerlou C.; Hofinger A.; Oostenbrink C.; Jerala R.; Kosma P.; Zamyatina A. Conformationally constrained Lipid A mimetics for exploration of structural basis of TLR4/MD-2 activation by lipopolysaccharide. ACS Chem. Biol. 2013, 11, 2423–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needham B. D.; Trent M. S. Fortifying the barrier: the impact of lipid A remodelling on bacterial pathogenesis. Nat. Rev. Microbiol. 2013, 7, 467–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French A. D.; Johnson G. P.; Kelterer A. M.; Dowd M. K.; Cramer C. J. Quantum mechanics studies of the intrinsic conformation of trehalose. J. Phys. Chem. A 2002, 106, 4988–4997. [Google Scholar]
- Nunes S. C. C.; Jesus A. J. L.; Moreno M. J.; Eusebio M. E. Conformational preferences of α,α-trehalose in gas phase and aqueous solution. Carbohydr. Res. 2010, 14, 2048–2059. [DOI] [PubMed] [Google Scholar]
- Färnbäck M.; Eriksson L.; Widmalm G. Octa-O-acetyl-α,α-trehalose ethanol disolvate. Acta Crystallogr. Sect. E 2004, 1483–1485. [Google Scholar]
- Bock K.; Defaye J.; Driguez H.; Bar-Guilloux E. Conformations in solution of α,α-trehalose, α-D-glucopyranosyl α-D-mannopyranoside, and their 1-thioglycosyl analogs, and a tentative correlation of their behaviour with respect to the enzyme trehalase. Eur. J. Biochem. 1983, 595–600. [DOI] [PubMed] [Google Scholar]
- Brown G. M.; Rohrer D. C.; Berking B.; Beevers C. A.; Gould R. O.; Simpson R. The crystal structure of α,α-trehalose dihydrate from three independent X-ray determinations. Acta Crystallogr. Sect. B 1972, 3145–3158. [Google Scholar]
- Chaube M. A.; Kulkarni S. S. Stereoselective construction of 1,1-α,α-glycosidic bond. Trends Carbohydr. Res. 2012, 2, 1–19. [Google Scholar]
- Leigh C. D.; Bertozzi C. R. Synthetic studies toward Mycobacterium tuberculosis Sulfolipid-1. J. Org. Chem. 2008, 3, 1008–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul N. K.; Twibanire J. d.; Grindley T. B. Direct synthesis of maradolipids and other trehalose 6-monoesters and 6,6′-diesters. J. Org. Chem. 2012, 2, 363–369. [DOI] [PubMed] [Google Scholar]
- Kunz H.; Zimmer J. Glycoside synthesis via electrophile-induced activation of N-allyl carbamates. Tetrahedron Lett. 1993, 18, 2907–2910. [Google Scholar]
- Saibal K. D.; Roy N. An improved method for the preparation of some ethyl 1-thioglycosides. Carbohydr. Res. 1996, 1–4, 275–277. [Google Scholar]
- Yu B.; Sun J. Glycosylation with glycosyl N-phenyltrifluoroacetimidates (PTFAI) and a perspective of the future development of new glycosylation methods. Chem. Commun. 2010, 26, 4668–4679. [DOI] [PubMed] [Google Scholar]
- Hölemann A.; Stocker B. L.; Seeberger P. H. Synthesis of a core arabinomannan oligosaccharide of Mycobacterium tuberculosis. J. Org. Chem. 2006, 21, 8071–8088. [DOI] [PubMed] [Google Scholar]
- Boltje T. J.; Zhong W.; Park J.; Wolfert M. A.; Chen W.; Boons G. J. Chemical synthesis and immunological evaluation of the inner core oligosaccharide of Francisella tularensis. J. Am. Chem. Soc. 2012, 134, 14255–14262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen H. H.; Nordstrom L. U.; Bols M. The disarming effect of the 4,6-acetal group on glycoside reactivity: torsional or electronic?. J. Am. Chem. Soc. 2004, 126, 9205–9213. [DOI] [PubMed] [Google Scholar]
- Walvoort M. T. C.; de Witte W.; van Dijk J.; Dinkelaar J.; Lodder G.; Overkleeft H. S.; Codée J. D. C.; van der Marel G. A. Mannopyranosyl uronic acid donor reactivity. Org. Lett. 2011, 16, 4360–4363. [DOI] [PubMed] [Google Scholar]
- Tanifum C. T.; Chang C. W. Sonication-assisted oligomannoside synthesis. J. Org. Chem. 2008, 2, 634–644. [DOI] [PubMed] [Google Scholar]
- Geng X.; Dudkin V. Y.; Mandal M.; Danishefsky S. J. In pursuit of carbohydrate-based HIV vaccines. Part 2: the total synthesis of high-mannose-type gp120 fragments: evaluation of strategies directed to maximal convergence. Angew. Chem., Int. Ed. 2004, 19, 2562–2565. [DOI] [PubMed] [Google Scholar]
- Bock K.; Lundt I.; Pedersen C. Assignment of anomeric structure to carbohydrates through geminal 13C-H coupling constants. Tetrahedron Lett. 1973, 13, 1037–1040. [Google Scholar]
- Bubb W. A. NMR spectroscopy in the study of carbohydrates: Characterizing the structural complexity. Concepts Magn. Reson., Part A 2003, 1, 1–19. [Google Scholar]
- Coxon B.Developments in the Karplus equation as they relate to the NMR coupling constants of carbohydrates. In Advances in Carbohydrate Chemistry and Biochemistry, Vol. 62; Derek H., Ed.; Academic Press: New York, 2009; pp 17–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duynstee H. I.; van Vliet M. J.; van der Marel G. A.; van Boom J. H. An efficient synthesis of (R)-3-{(R)-3-[2-O-(α-L-rhamnopyranosyl)-α-L-rhamnopyranosyl] oxydecanoyl}oxydecanoic acid, a rhamnolipid from Pseudomonas Aeruginosa. Eur. J. Org. Chem. 1998, 2, 303–307. [Google Scholar]
- Kusumoto S.; Fukase K. Synthesis of endotoxic principle of bacterial lipopolysaccharide and its recognition by the innate immune systems of hosts. Chem. Rec. 2006, 6, 333–343. [DOI] [PubMed] [Google Scholar]
- Raetz C. R. H.; Garrett T. A.; Reynolds C. M.; Shaw W. A.; Moore J. D.; Smith D. C. Jr.; Ribeiro A. A.; Murphy R. C.; Ulevitch R. J.; Fearns C.; Reichart D.; Glass C. K.; Benner C.; Subramaniam S.; Harkewicz R.; Bowers-Gentry R. C.; Buczynski M. W.; Cooper J. A.; Deems R. A.; Dennis E. A. Kdo2-Lipid A of E. coli, a defined endotoxin that activates macrophages via TLR-4. J. Lipid Res. 2006, 5, 1097–1111. [DOI] [PubMed] [Google Scholar]
- Shibata T.; Motoi Y.; Tanimura N.; Yamakawa N.; Akashi-Takamura S.; Miyake K. Intracellular TLR4/MD-2 in macrophages senses Gram-negative bacteria and induces a unique set of LPS-dependent genes. Int. Immunol. 2011, 8, 503–510. [DOI] [PubMed] [Google Scholar]
- Re F.; Strominger J. L. Heterogeneity of TLR-induced responses in dendritic cells: from innate to adaptive immunity. Immunobiology 2004, 1–2, 191–198. [DOI] [PubMed] [Google Scholar]
- Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003, 2, 133–146. [DOI] [PubMed] [Google Scholar]
- Tsuchiya S.; Kobayashi Y.; Goto Y.; Okumura H.; Nakae S.; Konno T.; Tada K. Induction of maturation in cultured human monocytic leukemia cells by a phorbol diester. Cancer Res. 1982, 4, 1530–1536. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.