

Abstract
Cancer is a disease regulated by the underlying gene networks. The emergence of normal and cancer states as well as the transformation between them can be thought of as a result of the gene network interactions and associated changes. We developed a global potential landscape and path framework to quantify cancer and associated processes. We constructed a cancer gene regulatory network based on the experimental evidences and uncovered the underlying landscape. The resulting tristable landscape characterizes important biological states: normal, cancer and apoptosis. The landscape topography in terms of barrier heights between stable state attractors quantifies the global stability of the cancer network system. We propose two mechanisms of cancerization: one is by the changes of landscape topography through the changes in regulation strengths of the gene networks. The other is by the fluctuations that help the system to go over the critical barrier at fixed landscape topography. The kinetic paths from least action principle quantify the transition processes among normal state, cancer state and apoptosis state. The kinetic rates provide the quantification of transition speeds among normal, cancer and apoptosis attractors. By the global sensitivity analysis of the gene network parameters on the landscape topography, we uncovered some key gene regulations determining the transitions between cancer and normal states. This can be used to guide the design of new anti-cancer tactics, through cocktail strategy of targeting multiple key regulation links simultaneously, for preventing cancer occurrence or transforming the early cancer state back to normal state.
Keywords: cancer, gene network, landscape, path
1. Introduction
Cancer is a disease involving unregulated cell growth. In 2007, cancer caused about 13% of all human deaths worldwide (7.9 million). Cancer rates are rising as more people live to older ages and as lifestyle changes occur in the developing world [1]. The chances of surviving cancer vary greatly by the type, the location and the extent of the disease when starting treatment. Taking breast cancer as an example, current therapies can delay tumour progression significantly, but recurrence is often inevitable, resulting in high mortality rates [2].
Cancer can be defined as a disease in which some abnormal cells ignore the normal rules of cell division and grow uncontrollably. Normal cells are frequently controlled by signals dictating whether the cell should divide, differentiate or die, while cancer cells develop a degree of autonomy from these control signals with uncontrolled growth and proliferation, which can be fatal. The foundation of modern cancer biology relies on a simple principle, which is that in fact all mammalian cells have similar molecular networks that control cell proliferation, differentiation and cell death. This suggests that the transformation from normal cells to cancer cells is caused by the changes in these networks at the molecular, biochemical and cellular levels [3–9].
Great efforts have been devoted to understand the mechanisms of cancerization. However, there are still challenges in many respects. For example, local probes of cancer by targeting individual genes are not all effective, so how do we combat cancer from the global perspectives of the network? How do we quantify the cancer and normal cell states? What is the quantitative measure for the capability of transforming from normal to cancer states and from cancer back to normal states? How do we understand the underlying mechanism and quantify the transformation process between normal and cancer states? How do we come up with the potential recipes for cancer prevention and treatment from the network perspective? How do we identify and quantify the key gene regulatory links for cancer?
Here, we develop a global yet physical landscape-path framework and try to address the above issues. The epigenetic landscape concept has been proposed to explain the development and differentiation of cells as a metaphor [10], and provided a quantitative way of understanding the dynamics of gene regulatory systems driving cell development. This picture has been quantitatively realized through the exploration of the global nature of the network in terms of probabilistic landscape framework [11–17]. The state space of gene regulatory networks contains states with different gene expression patterns (such as tumour repressor gene P53 and RB) in the cell, which further determine different cellular phenotypes. Employing landscape framework, cell types can be represented by the basins of attraction on the landscape, which reflect the probability of appearance of different cell types. States with lower potential or higher probability represent attractor or biological functional states, forming the basins of attraction. So, a biological process such as tumour genesis or apoptosis of cells can be understood as the transition from an attractor state to another one in the gene expression state space of the underlying gene regulatory network.
Conventional way for cancer studies has been focused on local properties of cancer networks (cancer as a disease of individual mutations). Many recent evidences show that cancer may be a disease at the network level [3–6], and can be understood as attractors in gene regulatory network state space [3,18–23]. However, global characterization and quantification for cancer attractors remains elusive. In this paper, from our physical and global landscape framework, we can quantify the underlying potential landscape of the cancer network and identify the attractor states in gene network state space as distinct biological functional states (normal, cancer and apoptosis states). Through the landscape topography in terms of barrier heights and kinetic transition rates between attractors, the global stability and the capability of the transitions between normal and cancer states can be quantified. By identifying the kinetic paths connecting the basins of normal, cancer and apoptosis attractors, we can uncover the underlying mechanism of the state transition and quantify the transition process. The kinetic paths from normal to cancer state and from cancer to normal state can be used to suggest potential recipes for cancer prevention and treatment. Furthermore, by global sensitivity analysis for parameters on the landscape topography, we will be able to identify and quantify the key regulations and genes for the function of the cancer network. This may provide multiple potential targets for curing or attenuating cancer.
Many approaches have been developed to construct cancer related protein or gene regulatory networks [24–26]. These network construction methods start from a variety of genomic or proteomic data. However, to model the dynamics of cancer regulatory systems, we believe that the networks constructed from mining genomic or proteomic data may not be able to provide enough accurate information in terms of regulation directions (activation or repression) and strengths due to the often insufficient statistics of samples. Some gene regulatory networks on cell fate decisions were constructed by directly searching regulation evidences from the literature [15,27–30]. We will start from some key gene markers characterizing the hallmarks of cancer [4,5], search for the connections between these genes and other cancer-associated genes from experimental evidences and construct a typical cancer network. In this way, we obtained a typical cancer gene regulatory network with 32 nodes (genes) and 111 edges (regulations).
Based on the cancer network built, we can explore the underlying landscape of the cancer network through the corresponding network dynamic equations to find out its global properties, uncover the functional mechanism of transitions among normal state, cancer state and apoptosis state. The barrier heights separating the basins of attraction and the transition rates can serve as the quantitative measures for global stability and kinetics of cell type transition process between normal attractor and cancer attractor. We explore two possible mechanisms of cancerization. One mechanism is by the change of the landscape topography through the changes in the genes and regulation strengths of the gene networks. The change of genes is often regarded as having genetic origin while the change of the regulations is often attributed to environmental changes. Upon gene or regulation changes, the cancer state attractor may become more and more stable and the normal state attractor may become less and less stable on the landscape, which characterizes the process of tumour genesis. When a dominant cancer state attractor forms on the landscape, it represents the formation of a tumour. A steep funnel-shaped cancer attractor landscape can guarantee the stability of the cancer state. It implies that at a certain stage the cancer could not be reversed. This may represent the formation of genome instability. We also explore another possible mechanism of cancerization: upon intrinsic or environmental fluctuations, the state transition from normal attractor to cancer attractor can occur by going over the barrier in between. In this scenario, landscape is fixed (for example, tristability, normal, cancer and apoptosis states coexist). A state in the normal attractor is stable against certain fluctuations. This indicates that for small fluctuations it is difficult for the system to escape from the normal state. However, when intrinsic or environmental fluctuations are large enough, the system can go over the barrier between normal and cancer basins and jump to the cancer state attractor. This characterizes the process of cancerization in another way.
We further quantify the kinetic paths for the transition process from normal to cancer state and cancer to normal state, as well as the apoptosis paths for both normal and cancer cells. The biological paths we acquired can be used to guide the design of new anti-cancer tactics. We show that both potential landscape and probabilistic flux are important to the dynamics of the cancer system. The force from the curl flux leads the kinetic paths of the system to deviate from the conventionally expected potential gradient paths. Consequently, the transition paths between normal and cancer states are irreversible. By the global sensitivity analysis of the genes or regulations among genes on the landscape topography, we will quantitatively predict which links (regulations) or nodes (genes) are critical to the transition between cancer and normal attractors, which can be directly tested from the experiments. This suggests a possible strategy for designing anti-cancer drugs from network perspectives by targeting multiple key genes and regulations, while traditional drug searches usually only target one molecule at a time and not for the regulation strength. Therefore, the results of our global sensitivity analysis may suggest a cocktail strategy of targeting multiple key genes of the key regulations, to prevent cancer occurrence or transform the early cancer state back to the normal state.
2. Results and discussion
2.1. Construction of the cancer network
Cancer is a complex and heterogeneous disease displaying a degree of complexity at physiological, tissue and cellular levels. Recent tumour genome sequencing efforts have demonstrated that there may be thousands of cancer driver-mutating genes. These genes are diverse and have little overlap between different tumours. Nevertheless, most cancer driver-mutated genes reflect cancer hallmarks or functional modules. Each hallmark or functional module is composed of a set of functionally linked pathways. Therefore, it is possible to map the functional modules and the mutated genes onto a cancer network. By doing so, we can use the network to represent complexity, compute biological relationships and seek to uncover the biological principles and insights of cancer [31].
Weinberg et al. put forward the 10 hallmarks of cancer [4–6]. These hallmarks are characterized by some key cancer marker genes, such as EGFR for proliferative signal, VEGF for angiogenesis, HGF for metastasis, hTERT for unlimited replication, HIF1 for glycolysis, CDK2 and CDk4 for evading growth suppressors. Starting from these cancer marker genes and some critical tumour suppressor genes such as P53, RB, P21 and PTEN, we made an extensive literature search [32] for the interactions among these key genes as well as the interactions among other cancer-associated genes, and constructed a cancer gene regulatory network with 32 gene nodes (figure 1) (see the electronic supplementary material for the key pathway description of the network). This cancer network includes 32 nodes (genes) and 111 edges (66 activation interactions and 45 repression interactions). In figure 1, arrows represent activation and short bars represent repression. The network mainly includes three kinds of marker genes: apoptosis marker genes (green nodes, including BAX, BAD, BCL2 and Caspase), cancer marker genes (magenta nodes, including AKT, MDM2, CDK2, CDK4, CDK1, NFKB, hTERT, VEGF, HIF1, HGF and EGFR), and tumour repressor genes (light blue nodes, including P53, RB, P21, PTEN, ARF and CDH1). The brown nodes represent other genes. We provide the order numbers and names of 32 genes, and also the related functions in the electronic supplementary material, table S1. We also show the evidences for the connections in constructing the network in the electronic supplementary material, table S2.
Figure 1.

The diagram for the cancer network including 32 nodes (genes) and 111 edges (66 activation interactions and 45 repression interactions). Red arrows represent activation and blue filled circles represent repression. The network includes mainly three kinds of marker genes: apoptosis (green nodes), cancer marker genes (magenta nodes) and tumour repressor genes (light blue nodes). The cancer marker genes include EGFR for proliferative signal, VEGF for angiogenesis, HGF for metastasis, hTERT for unlimited replication, HIF1 for glycolysis, CDK2 and CDk4 for evading growth suppressors. The solid black links represent the key links found by the global sensitivity analysis, and the octagon shape nodes represent key genes for the transition between normal and cancer states found by global sensitivity analysis. The brown nodes represent other genes.
For the 32-node gene regulatory network, we constructed the corresponding ordinary differential equations describing dynamics of the underlying system, in terms of Hill functions representing their activation or repression interaction strengths and cooperativity. The equations have the form
| 2.1 |
In the above equation, i = 1, 2, … , 32, so totally there are 32 equations. S represents the threshold (inflection point) of the explicitly sigmoidal functions, i.e. the strength of the regulatory interaction, and n is the Hill coefficient which determines the steepness of the sigmoidal function [33]. Here, parameters for Hill functions are specified as: S = 0.5, n = 4. In addition, k is self-degradation constant, b is repression constant and a is activation constant (see the electronic supplementary material for description of parameters).
Here, Xai and Xbi represent average interaction strength separately for activation and repression from other nodes to certain node i. For every node i, Xai is defined as 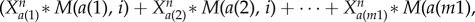
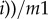 , and Xbi is defined as
, and Xbi is defined as 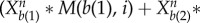
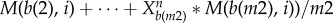 . Here a(1), a(2), … ,a(m1) is the number list of nodes which have activation interactions to node i, and b(1), b(2), … , b(m1) is the number list of nodes which have repression interactions to node i. M(j, i) (i, j = 1, 2, … , 32) is the element of interaction matrix M characterizing the interaction type and the interaction strength from node j to node i. M is acquired by multiplying interaction type matrix Mi (see table S6) with interaction strength matrix Ms (table S7): M(j, i) = Mi(j, i)
. Here a(1), a(2), … ,a(m1) is the number list of nodes which have activation interactions to node i, and b(1), b(2), … , b(m1) is the number list of nodes which have repression interactions to node i. M(j, i) (i, j = 1, 2, … , 32) is the element of interaction matrix M characterizing the interaction type and the interaction strength from node j to node i. M is acquired by multiplying interaction type matrix Mi (see table S6) with interaction strength matrix Ms (table S7): M(j, i) = Mi(j, i) Ms(j), i, j = 1, 2, … , 32. Here, we made an assumption that the regulation from one individual gene j to the other genes has the same interaction strength which is determined by Ms(j). Therefore, in equation (2.1), the first term represents self-degradation, the second term represents activation from other nodes (m1 nodes) to node i, and the last item denotes repression from other nodes (m2 nodes) to node i.
Ms(j), i, j = 1, 2, … , 32. Here, we made an assumption that the regulation from one individual gene j to the other genes has the same interaction strength which is determined by Ms(j). Therefore, in equation (2.1), the first term represents self-degradation, the second term represents activation from other nodes (m1 nodes) to node i, and the last item denotes repression from other nodes (m2 nodes) to node i.
2.2. Potential landscape of the cancer network
The above 32 ODEs (equation (2.1)) we constructed, as the driving force of the system, govern the network dynamics. We can further consider the corresponding stochastic dynamics [12]. By the self-consistent mean field approximation, we can obtain the steady-state probability distribution of 32 variables for the cancer regulatory system. According to U =−ln(Pss) [11–14], we can further acquire the potential landscape of the system. Here, Pss represents the probability distribution of the steady state, and U is the dimensionless potential energy.
For a 32-dimensional system, it is hard to visualize the landscape. So, we projected the landscape to a two-dimensional state space by integrating out the other 30 variables and leaving the two key variables AKT (an oncogene) and RB (a tumour repressor gene). Figure 2 shows three-dimensional and two-dimensional landscapes for the system in gene expression level state space in terms of AKT and RB. In figure 2a, we can see clearly that there are three stable states or basins of attraction on the landscape (tristability). Landscape reflects the steady-state probability distribution. Here, every basin of attraction (high probability states) represents a cell type in gene expression level state space, and they are separated by some barriers, which prevent easy transformation between different cell types. The bottom attractor represents apoptosis state, which has higher expression of tumour repressor gene RB, P21, PTEN, lower expression of oncogene AKT, EGFR, VEGF, HGF, HIF1, hTERT, MDM2, CDK2, CDK4 and higher expression level of apoptosis marker gene Caspase. The middle attractor represents the normal state, which has higher expression level of tumour repressor gene RB, P21, PTEN, higher expression level of oncogene AKT, EGFR, VEGF, HGF, HIF1, hTERT, MDM2, CDK2, CDK4, and lower expression level of apoptosis marker gene Caspase. The top attractor represents the cancer state, which has lower expression level of tumour repressor gene RB, P21, PTEN, much higher expression level of oncogene AKT, EGFR, VEGF, HGF, HIF1, hTERT, MDM2, CDK2, CDK4, and lower expression level of apoptosis marker gene Caspase (see the electronic supplementary material, table S3, for detailed relative gene expression levels of the three stable states). Biologically, this is consistent with our understanding for normal, cancer and apoptosis states [4–6], since for apoptosis state the apoptosis marker gene should be on and for cancer cells the oncogenes should be more highly expressed than for the normal cells. The three attractors or stable states are consistent with our biological understanding to cancer networks. In the following section, we will continue to investigate the transition paths between these three stable states. As far as we know, this is the first landscape for cancer gene regulatory network which can reflect the biological details for cancer regulations (such as cancerization and apoptosis process).
Figure 2.

The tristable landscape for the cancer network from self-consistent approximation method. The parameters are set as: degradation constant k = 1, activation constant a = 0.5 and repression constant b = 0.5 (see the electronic supplementary material for parameter depictions). Diffusion coefficient D = 0.01. (a) The three-dimensional landscape and dominant kinetic paths. The yellow path represents the path from normal state attractor to cancer state attractor, and the magenta path represents the path from cancer state attractor to normal state attractor. Black paths represent the apoptosis paths for normal and cancer states. (b) The corresponding two-dimensional landscape of cancer network. Red arrows represent the negative gradient of potential energy and white arrows represent the probabilistic flux.
We stress that the tristability appears in some parameter range for regulation strength (table S7). The tristable landscape provides a relatively balanced case for the three (normal, cancer and apoptosis) state coexistence so that we can explore the transition among these three attractors. Changing the regulation strengths mimicking the non-genetic environmental changes leads to the change of landscape topography, for example, from single dominant basin to bistable basin and to tristable basin or vice versa. This helps to provide a hint and a quantitative basis of how environmental changes may lead to or prevent the cancer state formation.
In order to show the landscape of the complete 32-dimensional system, we applied stochastic Langevin dynamics method to obtain the quantitative information on the landscape (see the electronic supplementary material for detailed methods). We can uncover the landscapes using RMSD coordinates based on Langevin dynamics (see the electronic supplementary material, figure S1). It gives similar dynamics to the one using AKT and RB as the coordinates (figure 2) based on the self-consistent approximation. This shows that the two-dimensional projection of landscape in AKT and RB state space can reflect the main dynamics of the full 32-dimensional gene network, and the three attractor landscape is not affected by choosing which gene pairs to display the results.
2.3. Kinetic paths between normal, cancer and apoptosis states
Based on our path integral method [13,14,34], we also acquired the quantitative kinetic paths between the normal cell state and the cancer cell state as well as the apoptosis paths for both normal and cancer states. In figure 2, the yellow path represents the kinetic path from normal to cancer attractor and the magenta path represents the kinetic path from cancer to normal attractor. We can see that the kinetic paths between normal and cancer states are irreversible. In addition, we also show, respectively, the apoptosis paths for normal and cancer states (black paths), which separately characterize the path for the death of normal cells and cancer cells. We also obtained the probabilistic flux of the cancer system, which is shown on the landscape (figure 2b). The white and red arrows, respectively, represent the direction of probabilistic flux and the negative gradient of the potential energy. We found that the dynamics of the cancer system is determined by both the force from the gradient of potential and the force from the curl flux [11,14]. The force from the curl flux leads the paths of the system to deviate from the steepest descent path calculated from the gradient of potential; thus, as we can see the two kinetic paths of normal to cancer state and cancer to normal state are irreversible (yellow line and magenta line are not identical).
The landscape in figure 2 is only a two-dimensional projection of the whole 32-dimensional state space. In order to demonstrate the cell states and the transitions between different cell types in the complete state space, we projected the expression level of the 21 major marker genes to binary states of each gene with high and low expressions (221 cell states totally). Here, to analyse the dynamics of the system, we chose the key 21 marker genes (table S3) to explore the underlying landscape and transition jumps between two nodes (cell types) based on Langevin dynamics. The reason for choosing the 21 marker genes is that the 32-dimensional state space is huge, and it will have 232 states even in the discrete form, which cannot be easily handled computationally. We believe employing key 21 maker genes can capture major regulatory dynamics or paths without losing the essential information, since our purpose is to explore the dynamical mechanism of the cancer system. In this way, the normal state is represented by the binary number 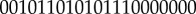 (representing expression level from gene 1 to gene 21, 1 for high expression, 0 for low expression), and for the cancer state, it is represented by 100010000101110111100. For the apoptosis state, it is represented by 001101011010000000001. Figure 3 (see Methods section for detailed methods) shows the discrete cancer landscape represented by 247 cell states (nodes, characterized by expression patterns of the 21 marker genes) and 334 transition jumps (edges) between the different cell states (produced by Cytoscape [35]). The sizes of nodes and edges are, respectively, proportional to the occurrence probability of the corresponding states and paths. Blue nodes represent cell states closer to normal cell states, red nodes represent cell states closer to cancer states and green nodes represent cell states closer to apoptosis states. The largest blue node (high RB/high AKT/low Caspase) represents the most significant normal state, the largest red node (low RB/high AKT/low Caspase) represents the most significant cancer state and the largest green node (high RB/low AKT/high Caspase) represents the most significant apoptosis state.
(representing expression level from gene 1 to gene 21, 1 for high expression, 0 for low expression), and for the cancer state, it is represented by 100010000101110111100. For the apoptosis state, it is represented by 001101011010000000001. Figure 3 (see Methods section for detailed methods) shows the discrete cancer landscape represented by 247 cell states (nodes, characterized by expression patterns of the 21 marker genes) and 334 transition jumps (edges) between the different cell states (produced by Cytoscape [35]). The sizes of nodes and edges are, respectively, proportional to the occurrence probability of the corresponding states and paths. Blue nodes represent cell states closer to normal cell states, red nodes represent cell states closer to cancer states and green nodes represent cell states closer to apoptosis states. The largest blue node (high RB/high AKT/low Caspase) represents the most significant normal state, the largest red node (low RB/high AKT/low Caspase) represents the most significant cancer state and the largest green node (high RB/low AKT/high Caspase) represents the most significant apoptosis state.
Figure 3.

Discrete landscape for cancer network with 247 nodes (every node denotes a cell state, characterized by expression patterns of the 21 marker genes) and 334 edges (paths) at default parameter values (a = 0.5, b = 0.5, k = 1). The sizes of nodes and edges are proportional to the occurrence probability of the corresponding states and paths, respectively. Blue nodes represent states closer to normal cell states, red nodes represent states closer to cancer cell states and green nodes represent states closer to apoptosis cell states. The green and magenta paths denote dominant kinetic paths from path integrals separately for normal to cancer attractor and cancer to normal attractor, and black paths represent the apoptosis paths for normal and cancer states. Here, we demonstrate only the states and paths with higher probability by setting a probability cutoff. The largest blue node (high RB/high AKT/low Caspase) represents the major normal state, the largest red node (low RB/high AKT/low Caspase) represents the major cancer state, and the largest green node (high RB/low AKT/high Caspase) represents the major apoptosis state. Some key intermediate states along the kinetic paths also have been labelled with high or low expression level.
We also calculated kinetic paths from path integral methods (see Methods for the details of path integral) in terms of 21 key marker genes (not only two-dimensional projection). Figure 2 shows the two-dimensional projection of the kinetic paths. In particular, we displayed the 21-dimensional kinetic paths (biological paths), which are shown as green and magenta paths separately for normal to cancer process and cancer to normal process, and black paths for apoptosis paths of normal and cancer states. In tables S8, S9, S10 and S11, we also provide the detailed 21-dimensional discrete kinetic paths. Again, we can see that the path from normal to cancer attractor and the path from cancer to normal attractor are irreversible.
From table S8, monitoring the transition process from normal to cancer state according to certain vital marker genes RB (column 6), MDM2 (column 16) and CDK2 (column 17), we can see that the differentiation process experiences a successive process (also reflected in the green path from normal to cancer attractor in figure 3) from MDM2 on (from 0 to 1), CDK2 on (from 0 to 1), RB off (from 1 to 0) and finally to cancer state. This provides a possible mechanism for the cancerization process as follows. First, the on state of MDM2 represses the tumour repressor gene P53 and P21, which releases the gene CDK2 and CDK4 (CDK2 and CDK4 on) in charge of cell growth due to the inhibition of P21 to CDK. Then RB is off because of suppression of activated CDK2 and CDK4 to it, and system gets into cancer state. This indicates the importance of oncogene MDM2 to induce tumour genesis.
For the reverse transition path of cancerization (from cancer to normal state) in table S9, we can see that the cell experiences a process (also reflected in the magenta path from cancer to normal attractor in figure 3) of RB on, off of CDK2 and CDK4, and off of MDM2. This indicates that in the transition process from cancer state to normal state the cell may first switch on the key tumour repressor genes RB, then growth genes CDK2 and CDK4 are gradually inactivated due to the repression of RB to them. Finally, oncogene MDM2 gene is switched off, and the cell goes back to the normal state. Experimentally, inhibiting expression of TCTP (encoding translationally controlled tumour protein) has been suggested as an important mechanism for tumour reversion, which activates the expression of P53, because TCTP promotes MDM2-mediated degradation of P53 [36–38]. This confirms the role of oncogene MDM2 as an important drug target for tumour reversion, and provides some verification for our predictions. Meanwhile, this also shows the importance of restoring the function of tumour repressor gene RB as an anti-cancer tactic. Making comparisons between tables S8 and S9, we can also confirm the irreversibility of paths from normal to cancer state and from cancer to normal state.
Similarly, observing the apoptosis paths for both normal cells and cancer cells (table S10 and S11), we can find that they both experience a process of AKT first being turned off and then Caspase being switched on, which demonstrates the vital role of AKT in inducing cell apoptosis. This provides another possible anti-cancer strategy by repressing AKT to induce cell apoptosis.
The biological paths for cancerization and reverse process as well as for the apoptosis of normal and cancer cells acquired here can be validated by related experiments, and we expect that they can be used to guide the design of new anti-cancer strategies.
2.4. Landscape topography quantifies the global stability of cancer network
Our simulation results showed that the landscape is critically influenced by the activation regulation strength a or repression regulation strength b. Figure 4 shows the landscape of the cancer network in terms of AKT and RB when activation constant a and repression constant b are changed, separately corresponding to a = 0.4, 0.46, 0.5, 0.52, 0.6 (from left column to right column), and b = 0.4, 0.5, 0.7, 0.8, 1 (from top row to bottom row). Landscape comparisons (second row at b = 0.5) illustrate that with activation a gradually increasing from 0.4 to 0.6, the landscape of cancer network experiences a change in topography from monostable state (apoptosis), to bistable state (apoptosis and normal coexist), to tristable state (normal, cancer and apoptosis coexist) and finally to another monostable cancer state (see the electronic supplementary material, figure S2, for landscape of more different a). So, by the change of landscape topography, we found that the process of a increasing characterizes the transition process from dominant normal state to dominant cancer state, or cancerization process. We do not have direct evidences so far that in tumour-genesis process the activation regulation strength between genes is enhanced. Nevertheless, we suggest here (the first mechanism we suggested) that cancerization process can be understood as the change of cancer network landscape topography caused by regulation strength changes among different genes (here represented by the increase in a). As we can see from above landscape comparisons (here we focus on the second row at b = 0.5, other rows give similar trends), in the tristable landscape the normal state has the deepest potential well (most stable state among three states) and it is hard for the system to make a transition from the normal attractor to the other two attractors (cancer and apoptosis). This represents normal cells performing normal cell functions, which are stable against fluctuations. With a increasing, the normal attractor gradually disappears, and cancer attractor becomes more and more stable. This represents the process of cancerization for a normal cell caused by different kinds of mutations in genes and regulation strengths. When activation strength a is large enough, the system shows a landscape with only one dominant cancer attractor, which represents the formation of stable cancer cells. At this time, a funnel-shaped landscape guarantees the stability of cancer state, which means that it is hard for the system to escape from cancer attractor. This reflects a fact that at a certain stage, it is very difficult for cancer cells to be reversed to normal cells. This stage may correspond to the formation of genome instability. Genome instability refers to a high frequency of mutations within the genome. In our landscape view, genome instability could mean that the change of network structure (network wiring) caused by mutations leads to the change of topography of landscape, e.g. the barrier from normal state to cancer state decreases. This makes the transition easier from the normal state attractor to the cancer state attractor, and cancerization is more likely to occur. We expect our suggestion be tested by further experiments.
Figure 4.

The change of landscape when activation a and repression strength b are changed. From left to right is the change corresponding to: a = 0.4, 0.46, 0.5, 0.52, 0.6 and from top to bottom is the change corresponding to: b = 0.4, 0.5, 0.7, 0.8, 1. The labels C, N and A, respectively, represent cancer attractor, normal attractor and apoptosis attractor.
We also propose another possible mechanism of cancerization: upon fluctuations, the state transition from normal attractor to cancer attractor happens by going over the barrier in between. In this scenario, the underlying landscape is fixed (tristability, normal, cancer and apoptosis states coexist). A state in the normal attractor is stable against certain fluctuations. At small fluctuations it is difficult for the system to escape from normal state attractor. However, when fluctuation is large enough, the system will be able to go over the barrier between normal and cancer basins and reach the cancer attractor. This gives another possibility of realizing cancerization.
Similarly, observing the landscape change for b increasing (top down direction), we can see that with b increasing the landscape topography changes from a monostable apoptosis state (central column at a = 0.5), to a tristable state, and finally into a monostable normal state (see the electronic supplementary material, figure S3, for three-dimensional landscape of different b) when b is big enough (b = 1).
To quantify the global stability of the cancer network in terms of landscape topography, we define global barrier height, representing potential difference between each attractor minimum and the corresponding saddle point on the landscape. We define USN as the potential energy difference between the normal state and the saddle point, Usaddle − UN, and USC as the potential energy difference between cancer state and the saddle point, Usaddle − UC. Here, UN and UC denote, respectively, the potential at the minimum for the normal state attractor and the cancer state attractor and Usaddle denotes the potential at the saddle point between these two basins of attraction. The results of barrier heights are from Langevin dynamics in terms of RMSD coordinates (see the electronic supplementary material, figure S1). We projected the whole network landscape to two dimensions (RMSD1 and RMSD2). For this two-variable landscape, we can acquire saddle points, local minimums and barrier heights.
In this way, USN and USC measure the relative global stability of the normal state and the cancer state, respectively. When the system has larger USN and smaller USC, the normal cell state is more stable and the system is inclined to stay in the normal state. The transition from normal to cancer state (cancerization process) is hard to realize, because the system must go across a large barrier in order to escape from the normal attractor to cancer attractor. The reverse process, the transition from cancer state to normal state is relatively easy to realize in this case (small USC). In contrast, if USN is small and USC is larger for the cancer system, it will be advantageous for the transition process of normal attractor to cancer attractor and difficult for the process of cancer attractor to normal attractor, because the system only needs to overcome a small barrier to go from the normal state to the cancer state (small USN), but a large barrier from cancer state to normal state (large USC).
Figure 5a shows the barrier heights for normal attractor (USN) and cancer attractor (USC) when activation strength a changes. We can see that with the activation strength a increasing, relatively USC becomes larger and USN declines (figure 5b gives the relative change of USN versus USC). This indicates that the enhancement of activation regulation in the network leads to a more stable cancer state, making it easier for the transition from the normal state to the cancer state. Meanwhile, when activation links are strengthened, the normal state becomes less stable relatively, and the system is not inclined to stay at normal state with a smaller barrier height USN. This implies that changing the strength of the activation links in the cancer gene regulatory network provides a way to modulate the relative stability of normal and cancer attractors and make the system inclined to stay at the normal state or inclined to stay at the cancer state. The stability of the two attractors can be quantified by the landscape topography (the barrier height). Figure 5c,d shows the barrier heights for normal attractor (USN) and cancer attractor (USC) when repression strength b changes. It can be seen that with b going up relatively USN declines and USC increases, which means that increasing repression strength makes cancer attractor more stable and normal attractor less stable. However, when b is very large (b = 1), the normal attractor becomes stable again. This shows that repression interaction could have a complicated influence on the landscape and the dynamics of the system.
Figure 5.

The change of barrier height when activation a, repression strength b and noise level D are changed. Here, USN represents the potential difference between the saddle point (between normal attractor and cancer attractor) and the local minimum in normal attractor. USC represents the potential difference between the saddle point (between normal attractor and cancer attractor) and the local minimum in cancer attractor.
We also show how the fluctuations measured by the diffusion coefficient D influence the barriers and mean first passage time (MFPT; figure 5e,f and see the electronic supplementary material, figure S6e,f, for the detailed method of calculating MFPT). It shows that at a small noise level D, barrier is large and MFPT (escape time) is slower, which demonstrates that the landscape of the cancer system is stable against certain fluctuations. We can also see that when D increases, the barriers decline and the MFPT become faster. This indicates that larger noise destroys the stability of the system (landscape becomes flat) and transitions between different attractors become easy, so barriers and escape time between normal and cancer attractors both decline. Additionally, we also found that with noise D changing, the barriers and MFPT do not change monotonically, which means that the noise has a complicated influence on the relative stability of normal and cancer attractors, i.e. we cannot say that the increase in noise is advantageous to normal or cancer state.
We can find that the global barrier heights and the MFPT have a similar trend for quantifying the transition between normal and cancer states, and both of them can serve as a quantitative measure of the global stability of the two attractors and kinetic speeds.
2.5. Global sensitivity analysis finding key regulations for normal and cancer state transitions
We explored cancer as a network disease. Changes in both the nodes (the individual genes, local probes) and links (the connections between genes) can be important to cancer dynamics. We did a global sensitivity analysis of the parameters for the cancer network in order to uncover the key parameters or connections in the network affecting the stability and kinetic transitions of both the normal state and the cancer state. Giving parameters (the strength of 111 links in the cancer network) a perturbation level pl, we can explore the influence of these parameters on the stability of the system by comparing the change of landscape topography quantified by the barrier heights.
We first exploited the self-consistent approximation method [12,14] to obtain those most important parameters—that is, by finding those parameters affecting barrier heights of the system significantly. Specifically, we changed the value of each of the activation and repression strengths Mji, by giving a percentage change Δs/s (here, s represents parameter Mji, Δs represents the change of parameter s, the value of Δs/s is controlled as between −1 and 1) as the degree of change. Then for perturbation of every parameter we compared the change of the landscape topography in terms of calculating the change of barrier heights for both normal state (ΔUSN) and cancer state (ΔUSC). In this way, we acquired 25 most important parameters or connections (15 of them are activation links and 10 of the others are repression links; see the electronic supplementary material, figure S4, for details).
In the following, we employed the stochastic Langevin dynamics to further obtain the change of barrier heights when these 25 parameters are changed, because by the Langevin dynamics the landscape of the system can be acquired directly by the statistics of the trajectories of the system—not through approximation. Figure 6 shows the results of the global sensitivity analysis for the 25 parameters or connections (see the electronic supplementary material for details). Figure 6a shows the results for 10 repression links, and figure 6b shows the results for 15 activation links. Blue bars represent the change of the barrier for normal state (USN) and the magenta bars represent the change of the barrier for cancer state (USC).
Figure 6.

The effects of key parameters (10 repression parameters and 15 activation parameters) on the barrier (USN and USC) and MFPT (mean first passage time, τNC and τCN) from Langevin dynamics. Y-axis represents the percentage of barrier or MFPT changed. Here, USN represents the potential difference between the saddle point (between normal attractor and cancer attractor) and the local minimum in normal attractor. USC represents the potential difference between the saddle point (between normal attractor and cancer attractor) and the local minimum in cancer attractor. τNC represents the MFPT from normal attractor to cancer attractor. τCN represents the MFPT from the cancer attractor to the normal attractor.
In figure 6a, x-axis represents 10 repression parameters or connections. The 10 links, respectively, correspond to: (link R1, 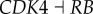 ), (link R2,
), (link R2, 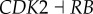 ), (link R3,
), (link R3, 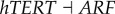 ), (link R4,
), (link R4, 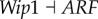 ), (link R5,
), (link R5, 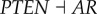 ), (link R6,
), (link R6, 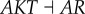 ), (link R7,
), (link R7, 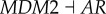 ), (link R8,
), (link R8, 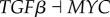 ), (link R9,
), (link R9,  ) and (link R10,
) and (link R10, 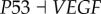 ). Here,
). Here, 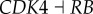 represents the repression regulation from gene CDK4 to gene RB (table S4). We can see that when the repression of CDK4 to RB increases, USN (normal state barrier) decreases and USC (cancer state barrier) increases significantly, making it easier to jump from normal state attractor to cancer state attractor, or easier for tumour genesis. In the same way, we can find quantitatively from sensitivity analysis that the regulations promoting cancer state significantly include (link R1,
represents the repression regulation from gene CDK4 to gene RB (table S4). We can see that when the repression of CDK4 to RB increases, USN (normal state barrier) decreases and USC (cancer state barrier) increases significantly, making it easier to jump from normal state attractor to cancer state attractor, or easier for tumour genesis. In the same way, we can find quantitatively from sensitivity analysis that the regulations promoting cancer state significantly include (link R1, 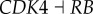 ), (link R2,
), (link R2, 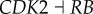 ), (link R3,
), (link R3, 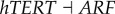 ), (link R4,
), (link R4, 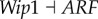 ), which means that the strengthening of these links promotes the formation of cancer, and the regulations attenuating cancer state significantly include (link R6,
), which means that the strengthening of these links promotes the formation of cancer, and the regulations attenuating cancer state significantly include (link R6, 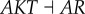 ), (link R9,
), (link R9,  ), (link R10,
), (link R10, 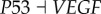 ), which means that the strengthening of these links inhibits the formation of cancer. By analysing these repression links, we can find that the results from global sensitivity analysis are reasonable. RB is a famous tumour repressor gene [4,5], so the repression of RB will promote the formation of cancer (link R1 and R2). ARF serves as a tumour repressor gene [39] by inhibiting oncogene MDM2, so the repression of ARF will promote the formation of cancer (link R3 and R4). In addition, AKT and VEGF are both key oncogenes, so the repression of AKT and VEGF will inhibit the formation of cancer state (link R9 and R10). Our results also predict that AKT and VEGF are valid anti-cancer target genes [40–42].
), which means that the strengthening of these links inhibits the formation of cancer. By analysing these repression links, we can find that the results from global sensitivity analysis are reasonable. RB is a famous tumour repressor gene [4,5], so the repression of RB will promote the formation of cancer (link R1 and R2). ARF serves as a tumour repressor gene [39] by inhibiting oncogene MDM2, so the repression of ARF will promote the formation of cancer (link R3 and R4). In addition, AKT and VEGF are both key oncogenes, so the repression of AKT and VEGF will inhibit the formation of cancer state (link R9 and R10). Our results also predict that AKT and VEGF are valid anti-cancer target genes [40–42].
Moreover, link R6 shows that the increase in the repression of AKT to AR (androgen receptor) will weaken the cancer state, which is consistent with some experiments showing that the inhibition of AR activity may delay prostate cancer progression [43]. We need to stress that our predictions for parameter changes are thoroughly from the network topology (connections). However, by looking at the network links we found that the network topology does not necessarily reflect clearly the role of AR as oncogene (due to the limited experimental evidences for regulations between genes, we cannot guarantee our network contains all regulations). In this case, our simulations can still obtain consistent results for gene AR with experiments, which shows the power of our theoretical framework and the associated global approach (probabilistic landscape and global sensitivity analysis).
Figure 6b shows the global sensitivity analysis results for 15 activation regulations, in which the x-axis represents separately: A1(E2F1 → ARF), A2(RB → AR), A3(EGFR → MYC), A4(HGF → MYC), A5(NFKB → MYC), A6(RAS → MYC), A7(EGFR → AKT), A8(VEGF → AKT), A9(HGF → AKT), A10(MYC → VEGF), A11(AKT → VEGF), A12(EGFR → VEGF), A13(HGF → VEGF), A14(HIF1 → VEGF), A15(hTERT → VEGF) (arrows represent activation regulation, A1, … , A15 represents the name of the activation links; see electronic supplementary material, table S5 for details).
Analysing the relative change of normal state barrier USN and cancer state barrier USC, we can uncover the regulations critically affecting the relative stability of the two attractors. The key regulations promoting cancer state significantly (cancer barrier increases and normal barrier decreases, the transition from normal to cancer attractor becomes easier) include A7(EGFR → AKT), A8(VEGF → AKT), A9(HGF → AKT), A11(AKT → VEGF) and A13(HGF → VEGF), which means that the strengthening of these links will promote the transition from normal state to cancer state. Among these five activation links, we can find that the target genes are AKT and VEGF, which is consistent with our above analysis for key repression links, demonstrating again that AKT and VEGF could be valid anti-cancer target genes [40–42]. In the meantime, we can see that these key regulations are all involved in the mutual activation process between oncogenes. This indicates that mutual activations between oncogenes or self-activations of cancer marker genes play an important role in the process of cancerization.
Additionally, we also quantified the global sensitivity of parameters through MFPT, since MFPT reflects the average transition time from one basin of attraction to another, and therefore provides another quantitative measure for the stability of the system. Figure 6c,d shows the influence of parameter change on the MFPT, respectively, for 10 repression links and 15 activation links. Comparing figure 6a with 6c, and figure 6b with 6d, we can find that MFPT and barrier height give consistent results on the global sensitivity analysis. Larger USN makes the transition from normal state to cancer state harder, and thus means larger MFPT for normal to cancer transition. In contrast, larger USC makes the transition from cancer state to normal state harder, and thus larger MFPT for cancer to normal transition. Therefore, USN corresponds to MFPT for normal to cancer transition, and USC corresponds to MFPT for cancer to normal transition.
In order to uncover the key factors determining the transition from cancer attractor to apoptosis attractor (the death of cancer cells), we also quantified the effects of key regulations (15 activation strength parameters and 10 repression strength parameters) on the MFPT from cancer state to apoptosis state (τCA, characterizing the kinetics of the apoptosis process of cancer cells) and from normal to apoptosis state (τNA, characterizing the kinetics of the apoptosis process of normal cells) from stochastic dynamics approach (see the electronic supplementary material, figure S5). The results can provide some insights for suppressing cancer cells. In the electronic supplementary material, figure S5, most of the regulations lead to longer τCA. This means that the cancer attractor becomes more stable and more time is needed for the system to escape from cancer attractor to apoptosis attractor. In the meantime, we find that link R9 ( ) and R10 (
) and R10 (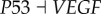 ) can lead to significantly shorter τCA. This shows that the cancer attractor loses its stability and leads to cell death. This provides another mechanism that AKT and VEGF can serve as potential anti-cancer target genes by inducing cancer cells to death (through suppressing AKT or VEGF). We also find that link R9 (
) can lead to significantly shorter τCA. This shows that the cancer attractor loses its stability and leads to cell death. This provides another mechanism that AKT and VEGF can serve as potential anti-cancer target genes by inducing cancer cells to death (through suppressing AKT or VEGF). We also find that link R9 ( ) and R10 (
) and R10 (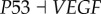 ) can lead to significantly shorter τNA. This indicates that repressing AKT and VEGF can also lead the normal cells to death.
) can lead to significantly shorter τNA. This indicates that repressing AKT and VEGF can also lead the normal cells to death.
In summary, the global sensitivity analysis in terms of the barriers and MFPT provides a way to identify the key factors determining the process of transition between normal and cancer state (key regulation connections are highlighted in black solid links in figure 1). Some of our predictions are consistent with the experimental evidences. More importantly, we provided certain predictions about which regulation links in the cancer network are critical to the relative stability of normal and cancer states (figure 6), which can be directly validated from relevant experiments in terms of MFPT. We need to emphasize that compared with the conventional sensitivity analysis which is usually local, our sensitivity analysis is global since it is based on the global landscape topography quantified by the barrier height or MFPT. Our results of global sensitivity analysis on the landscape topography may provide a cocktail strategy of targeting multiple key regulation links, to prevent cancer occurrence or transform the early cancer state back to normal state.
Based on the results of global sensitivity analysis, we picked out some key regulation parameters (activation and repression parameters, Mji), and visualized the change of landscape when these regulation strengths are changed (figure 7). In figure 7, the vertical axis of every sub-figure represents negative probability (−P corresponding to potential energy U according to U =−ln(P)). The four rows separately correspond to four specific parameters (here we picked out two key activation parameters and two key repression parameters for illustration). Observing the change of landscape, we can see that with the increase of activations on AKT and VEGF (first two rows), the landscape changes from tristability with dominant normal state gradually to bistability (cancer and apoptosis coexist), and finally to a dominant cancer state. This again demonstrates the role of AKT and VEGF to induce cancer, which is consistent with the sensitivity analysis. When the repression on RB is strengthened (the third column), we can see that the landscape changes from bistability (dominant normal state) gradually to tristability and finally to a dominant cancer state. This indicates the role of suppressing RB in inducing cancer. From the fourth row, with the repression on AKT enhanced, we can see that the landscape changes from a cancer dominant bistability, to tristability with dominant normal state and finally to a dominant apoptosis state. This implies that repressing AKT can attenuate cancer by inducing cancer to normal transition or inducing cell apoptosis.
Figure 7.

The change of landscape when some key regulation strengths (activation and repression parameters, Mji) are changed. The vertical axis of every sub-figure represents –P ×1000 (P is probability and −P corresponds to potential energy U). From left column to right column separately corresponds to: s =−50%, 0%, 50% and 100% (here s represents the percentage of parameters changed). The four rows separately correspond to the change of four parameters. As labelled, the first row represents activation strength from VEGF to AKT, the second row represents the activation strength from AKT to VEGF, the third row represents the repression strength from CDK2 to RB and the fourth row represents the repression strength from PTEN to AKT. The labels C, N and A represent cancer attractor, normal attractor and apoptosis attractor, respectively.
In the meantime, the changes of landscape with parameters provide a possible explanation for the mechanisms of cancerization, which is reflected by the change of landscape topography caused by changing regulation strength among different genes (such as the increase in activation on AKT in the first row). We can see that at small AKT activation, the tristable landscape has a normal state with the deepest potential basin and it is difficult for the system to make a transition from the normal attractor to the other two attractors (cancer and apoptosis). This represents normal cells performing normal cell functions, which are stable against fluctuations. With the activation on AKT increasing, the normal attractor gradually becomes less stable, and the cancer attractor becomes more and more stable. This represents the process of cancerization for normal cells caused by the change of AKT activation strength. Finally, the system displays a landscape with only one dominant cancer attractor, which represents the finish of transformation from normal cells to cancer cells. At this time, a funnel-shaped landscape guarantees the stability of cancer state, and it is difficult for the system to escape from the cancer attractor. In the same way, the landscape topography changes for changing repression on AKT (the fourth row) providing a strategy for inducing the death of cancer cells which is reflected by the landscape topography changes from a dominant cancer state gradually to a dominant apoptosis state. We need to stress that here we only show some examples for changing regulation strength which can induce the landscape topography change of cancer network. As a matter of fact, there are all kinds of combinations for changing different regulation strengths in the network which can lead to the change of landscape topography and further the change of network function. Owing to the limitation of computational cost, we only did single-factor sensitivity analysis. Ideally, a multi-factor sensitivity analysis is expected to find more realistic and interesting anti-cancer recipes by inducing the transition from cancer state to normal state or the apoptosis of cancer cells.
3. Conclusion
We uncovered the landscape of a cancer gene regulatory network reconstructed from a literature search. Landscape shows that the cancer gene regulatory network has three stable basins of attraction at specific parameter regions, which represent the normal cell state, cancer cell state and apoptosis cell state, respectively. In terms of the path integral approach, we acquired the kinetic paths for the transformation among normal, cancer and apoptosis states. Both landscape and curl flux determine the dynamics of the cancer network. Flux leads the kinetic paths of the system deviating from the steepest descent path from gradient of potential, and the transition paths between normal state to cancer state are irreversible. Barrier heights based on landscape topography provide quantitative measures for the global stability and kinetic transition of the attractors. MFPT provides an avenue to acquire the information of transition rates or kinetic speeds for the system to jump from one attractor to another one. By the global sensitivity analysis in terms of barrier heights and MFPT, we provided some predictions about the key genes and connections affecting the transition between normal and cancer states significantly, which can be tested by experiments. Importantly, the key links and genes from global sensitivity analysis and biological paths we acquired can be used to guide designing anti-cancer tactics by targeting multiple key nodes or regulations.
Throughout the paper, we have used different ways to vary regulation strengths to explore broader parameter space (activation constant a, repression constant b, as well as specific regulation strengths in sensitivity section). As the regulation strengths vary, the shape of the underlying landscape also varies as found in the sensitivity analysis section. The changes in regulation strengths reflect the changes from the environments. This leads to the changes of the network structure. In this respect, we are able to quantify how the environmental changes or network changes lead to the changes of the landscape topography, and therefore the appearance or disappearance of the cancer state in our approach. In other words, by changing the regulation strengths, we are effectively exploring different network structures and how those influence the functions.
We need to stress that our current cancer network is merely a typical one, in which we include certain biological markers for cancer and their interactions. With more biological details added into the network, we expect to obtain more accurate network structures/topology guided by the relevant experiments and construct a more realistic cancer regulatory network. On the other hand, in consideration of some specific cancer marker genes, some specific cancer networks can also be constructed, such as a breast cancer network including key marker genes BRCA1 and BRCA2 [26]. It can be anticipated that by exploring the landscape and paths of some more accurate and specific cancer networks, we can obtain more intricate mechanisms and predictions about cancer regulatory networks.
Our approach provides a general way to investigate the global properties (landscape topography, transition rate, and kinetic path) of large gene regulatory networks which have information only on interaction directions (activation or repression) without interaction strengths, and can be applied to other disease related gene regulatory networks or protein networks.
4. Methods
4.1. Self-consistent mean field approximation
The time evolution of dynamical systems is governed by the diffusion equations. Given the system state P(X1, X2, … ,Xn,t), where X1, X2, … ,Xn represents the concentrations or populations of molecules or species, we expected to have N-coupled differential equations, which are difficult to solve. Following a self-consistent mean field approach [12,14,44,45], we split the probability into the products of individual ones: 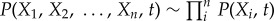 and solve the probability self-consistently. This can effectively reduce the dimensionality from MN to M × N, and thus make the computation of the problem tractable.
and solve the probability self-consistently. This can effectively reduce the dimensionality from MN to M × N, and thus make the computation of the problem tractable.
However, for the multi-dimensional system, it is still hard to solve diffusion equations directly. We start from moment equations and simply assume specific probability distribution based on physical argument, i.e. we give some specific connections between moments. In principle, once we know all moments, we can acquire the probability distribution. In this work, we use Gaussian distribution as an approximation, which means we need two moments, mean and variance.
When the diffusion coefficient D is small, the moment equations can be approximated to [46,47]
| 4.1 |
and
| 4.2 |
Here, x, σ(t) and A(t) are vectors and tensors, and AT(t) is the transpose of A(t). The matrix elements of A are Aij = ∂Fi[X(t)]/∂xj(t). According to this equation, we can solve x(t) and σ(t). Here, we consider only diagonal elements of σ(t) from mean field splitting approximation. Therefore, the evolution of probabilistic distribution for each variable could be acquired using the mean and variance based on Gaussian approximation:
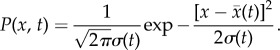 |
4.3 |
The probability obtained above corresponds to one fixed point or basin of attraction. If the system has multistability, then there are several probabilistic distributions localized at every basin of attraction, with different variations. Therefore, the total probability is the weighted sum of all these probability distributions. Finally, once we have the total probability, we can construct the potential landscape by the relationship with the steady-state probability: U(x) =−lnPss(x).
For non-equilibrium dynamical systems, the driving force F cannot be written as the gradient of potential U as in the equilibrium case. We have shown previously that, in general, F can be decomposed into a gradient of the potential and a curl flux force linking the steady-state flux Jss and the steady-state probability Pss [11,12] (F = +D/Pss · (∂/∂x)Pss + Jss(x)/Pss = −D(∂/∂x)U + Jss(x)/Pss). The probability flux vector J of the system in concentration or gene expression level space x is defined as [46]: J(x, t) = FP − D · (∂/∂x)P. From our theory, both the barriers (from potential landscape) and curl flux determine the total force. Therefore, both barrier and curl flux are important for the dynamics of the system.
In the 32-dimensional protein concentration space, it is hard to visualize 32-dimensional probabilistic flux. Approximately, we explored the associated two-dimensional projection of flux vector: J1(x1, x2, t) = F1(x1, x2)P − D(∂/∂x1)P and J2(x1, x2, t) = F2(x1, x2)P − D(∂/∂x2)P.
4.2. Kinetic path from path integral
Within the cell, there exists intrinsic noise from statistical fluctuations of the finite number of molecules and external noise from highly dynamical and inhomogeneous environments, which can be significant to the dynamics of the system [48–50]. Therefore, a network of chemical reactions in noisy fluctuating environments can be addressed by: 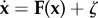 . Here, x = (x1(t), x2(t), … , x32(t)) represents the vector of protein concentration or gene expression level. F(x) is the vector for the driving force of chemical reaction. ζ is the Gaussian noise term whose autocorrelation function is
. Here, x = (x1(t), x2(t), … , x32(t)) represents the vector of protein concentration or gene expression level. F(x) is the vector for the driving force of chemical reaction. ζ is the Gaussian noise term whose autocorrelation function is 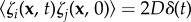 and D is the diffusion coefficient matrix.
and D is the diffusion coefficient matrix.
The dynamics for the probability of starting from initial configuration xinitial at t = 0 and ending at the final configuration xfinal at time t, in terms of the Onsager–Machlup functional, can be formulated [13,51] as
D(x) is the diffusion coefficient matrix. The integral over Dx denotes the sum over all possible paths from the state xinital at time t = 0 to xfinal at time t. The exponent factor gives the weight of each path. Therefore, the probability of network dynamics from initial state xinitial to the final state xfinal is equal to the sum of all possible paths with different weights. S(x) is the action and L(x(t)) is the Lagrangian or the weight for each path.
The path integrals can be approximated with a set of dominant paths, since each path is exponentially weighted, and the other subleading path contributions are often small and can be neglected. So, the dominant path with the optimal weights can be acquired through minimization of the action or Lagrangian. In our case, we identify the optimal paths as the biological paths (normal to cancer and cancer to normal).
Supplementary Material
Funding statement
This work was supported by the National Science Foundation.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. 2011. Global cancer statistics. CA Cancer J. Clin. 61, 69–90. ( 10.3322/caac.20107) [DOI] [PubMed] [Google Scholar]
- 2.Cowin P, Rowlands TM, Hatsell SJ. 2005. Cadherins and catenins in breast cancer. Curr. Opin. Cell Biol. 17, 499–508. ( 10.1016/j.ceb.2005.08.014) [DOI] [PubMed] [Google Scholar]
- 3.Kauffman S. 1971. Differentiation of malignant to benign cells. J. Theor. Biol. 31, 429–451. ( 10.1016/0022-5193(71)90020-8) [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. 2000. The hallmarks of cancer. Cell 100, 57–70. ( 10.1016/S0092-8674(00)81683-9) [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144, 646–674. ( 10.1016/j.cell.2011.02.013) [DOI] [PubMed] [Google Scholar]
- 6.Weinberg RA. 2007. The biology of cancer. New York, NY: Garland Science. [Google Scholar]
- 7.Hejmadi M. 2010. Introduction to cancer biology. Holstebro, Denmark: Ventus Publishing.
- 8.Vogelstein B, Kinzler KW. 2004. Cancer genes and the pathways they control. Nat. Med. 10, 789–799. ( 10.1038/nm1087) [DOI] [PubMed] [Google Scholar]
- 9.Ao P, Galas D, Hood L, Zhu X. 2008. Cancer as robust intrinsic state of endogenous molecular–cellular network shaped by evolution. Med. Hypotheses 70, 678–684. ( 10.1016/j.mehy.2007.03.043) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waddington CH. 1957. The strategy of the genes: a discussion of some aspects of theoretical biology. London, UK: Allen and Unwin. [Google Scholar]
- 11.Wang J, Xu L, Wang EK. 2008. Potential landscape and flux framework of non-equilibrium networks: robustness, dissipation and coherence of biochemical oscillations. Proc. Natl Acad. Sci. USA 105, 12 271–12 276. ( 10.1073/pnas.0800579105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang J, Li CH, Wang EK. 2010. Potential and flux landscapes quantify the stability and robustness of budding yeast cell cycle network. Proc. Natl Acad. Sci. USA 107, 8195–8200. ( 10.1073/pnas.0910331107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang J, Zhang K, Xu L, Wang EK. 2011. Quantifying the Waddington landscape and biological paths for development and differentiation. Proc. Natl Acad. Sci. USA 108, 8257–8262. ( 10.1073/pnas.1017017108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li CH, Wang J. 2013. Quantifying cell fate decisions for differentiation and reprogramming of a human stem cell network: landscape and biological paths. PLoS Comput. Biol. 9, e1003165 ( 10.1371/journal.pcbi.1003165) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li CH, Wang J. 2013. Quantifying Waddington landscapes and paths of non-adiabatic cell fate decisions for differentiation, reprogramming and transdifferentiation. J. R. Soc. Interface 10, 20130787 ( 10.1098/rsif.2013.0787) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liao C, Lu T. 2013. A minimal transcriptional controlling network of regulatory T cell development. J. Phys. Chem. B 117, 12 995–13 004. ( 10.1021/jp402306g) [DOI] [PubMed] [Google Scholar]
- 17.Qi H, Blanchard A, Lu T. 2013. Engineered genetic information processing circuits. WIREs Syst. Biol. Med. 5, 273–287. ( 10.1002/wsbm.1216) [DOI] [PubMed] [Google Scholar]
- 18.Huang S, Ernberg I, Kauffman S. 2009. Cancer attractors: a systems view of tumors from a gene network dynamics and developmental perspective. Semin. Cell Dev. Biol. 20, 869–876. ( 10.1016/j.semcdb.2009.07.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ao P. 2009. Global view of bionetwork dynamics: adaptive landscape. J. Genet. Genom. 36, 63–73. ( 10.1016/S1673-8527(08)60093-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi M, Shi J, Jung SH, Chen X, Cho KH. 2012. Attractor landscape analysis reveals feedback loops in the p53 network that control the cellular response to DNA damage. Sci. Signal. 5, ra83 ( 10.1126/scisignal.2003363) [DOI] [PubMed] [Google Scholar]
- 21.Creixell P, Schoof EM, Erler JT, Linding R. 2012. Navigating cancer network attractors for tumorspecific therapy. Nat. Biotechnol. 30, 842–848. ( 10.1038/nbt.2345) [DOI] [PubMed] [Google Scholar]
- 22.Davies P, Lineweaver C. 2011. Cancer tumors as metazoa 1.0: tapping genes of ancient ancestors. Phys. Biol. 8, 015001 ( 10.1088/1478-3975/8/1/015001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bar-Yam Y, Harmon D, de Bivort B. 2009. Attractors and democratic dynamics. Science 323, 1016C1017 ( 10.1126/science.1163225) [DOI] [PubMed] [Google Scholar]
- 24.Chu LH, Chen BS. 2008. Construction of a cancer-perturbed protein–protein interaction network for discovery of apoptosis drug targets. BMC Syst. Biol. 2, 56 ( 10.1186/1752-0509-2-56) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X, Gotoh O. 2010. Inference of cancer-specific gene regulatory networks using soft computing rules. Gene Regul. Syst. Biol. 4, 19–34. ( 10.4137/GRSB.S4509) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pujana MA, et al. 2007. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat. Genet. 39, 1338–1349. ( 10.1038/ng.2007.2) [DOI] [PubMed] [Google Scholar]
- 27.Wang P, Song C, Zhang H, Wu Z, Tian X-J, Xing J. 2014. Epigenetic state network approach for describing cell phenotypic transitions. Interface Focus 4, 20130068 ( 10.1098/rsfs.2013.0068) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sasai M, Kawabata Y, Makishi K, Itoh K, Terada T. 2013. Time scales in epigenetic dynamics and phenotypic heterogeneity of embryonic stem cells. PLoS Comput. Biol. 9, e1003380 ( 10.1371/journal.pcbi.1003380) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang B, Wolynes P. 2014. Stem cell differentiation as a many-body problem. Proc. Natl Acad. Sci. USA 111, 10 185–10 190. ( 10.1073/pnas.1408561111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang R, Shoemaker R, Wang W. 2011. Systematic search for recipes to generate induced pluripotent stem cells. PLoS Comput. Biol. 7, e1002300 ( 10.1371/journal.pcbi.1002300) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang E, Lenferink A, O'Connor-McCourt M. 2007. Cancer systems biology: exploring cancer-associated genes on cellular networks. Cell Mol. Life Sci. 64, 1752–1762. ( 10.1007/s00018-007-7054-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Landeghem S, et al. 2013. Large-scale event extraction from literature with multi-level gene normalization. PLoS ONE 8, e55814 ( 10.1371/journal.pone.0055814) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang S, Guo Y, May G, Enver T. 2007. Bifurcation dynamics of cell fate decision lineage-commitment in bipotent progenitor cells. Dev. Biol. 305, 695–713. ( 10.1016/j.ydbio.2007.02.036) [DOI] [PubMed] [Google Scholar]
- 34.Wang J, Zhang K, Wang EK. 2010. Kinetic paths, time scale, and underlying landscapes: a path integral framework to study global natures of nonequilibrium systems and networks. J. Chem. Phys. 133, 125103 ( 10.1063/1.3478547) [DOI] [PubMed] [Google Scholar]
- 35.Cline M, Smoot M, Cerami E, Kuchinsky A, Landys N. 2007. Integration of biological networks and gene expression data using Cytoscape. Nat. Prot. 2, 2366–2382. ( 10.1038/nprot.2007.324) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amson R, Karp J, Telerman A. 2013. Lessons from tumor reversion for cancer treatment. Curr. Opin. Oncol. 25, 59–65. ( 10.1097/CCO.0b013e32835b7d21) [DOI] [PubMed] [Google Scholar]
- 37.Amson R, et al. 2011. Reciprocal repression between p53 and TCTP. Nat. Med. 18, 91–99. ( 10.1038/nm.2546) [DOI] [PubMed] [Google Scholar]
- 38.Tuynder M, Susini L, Prieur S, Besse S, Fiucci G, Amson R, Telerman A. 2002. Biological models and genes of tumor reversion: cellular reprogramming through tpt1/TCTP and SIAH-1. Proc. Natl Acad. Sci. USA 99, 14 976–14 981. ( 10.1073/pnas.222470799) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lowe SW, Sherr CJ. 2003. Tumor suppression by Ink4a–Arf: progress and puzzles. Curr. Opin. Genet. Dev. 13, 77–83. ( 10.1016/S0959-437X(02)00013-8) [DOI] [PubMed] [Google Scholar]
- 40.Altomare D, Testa J. 2005. Perturbations of the AKT signaling pathway in human cancer. Oncogene 24, 7455–7464. ( 10.1038/sj.onc.1209085) [DOI] [PubMed] [Google Scholar]
- 41.Cardones A, Banez L. 2006. VEGF inhibitors in cancer therapy. Curr. Pharm. Des. 12, 387–394. ( 10.2174/138161206775201910) [DOI] [PubMed] [Google Scholar]
- 42.Ellis L, Hicklin D. 2008. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat. Rev. Cancer 8, 579–591. ( 10.1038/nrc2403) [DOI] [PubMed] [Google Scholar]
- 43.Heinlein CA, Chang C. 2004. Androgen receptor in prostate cancer. Endocr. Rev. 25, 276–308. ( 10.1210/er.2002-0032) [DOI] [PubMed] [Google Scholar]
- 44.Sasai M, Wolynes P. 2003. Stochastic gene expression as a many-body problem. Proc. Natl Acad. Sci. USA 100, 2374–2379. ( 10.1073/pnas.2627987100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li CH, Wang J, Wang EK. 2011. Potential landscape and probabilistic flux of a predator prey network. PLoS ONE 6, e17888 ( 10.1371/journal.pone.0017888) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van Kampen NG. 1992. Stochastic processes in chemistry and physics, 1st edn Amsterdam, The Netherlands: North Holland. [Google Scholar]
- 47.Hu G. 1994. Stochastic forces and nonlinear systems. Shanghai, China: Shanghai Scientific and Technological Education Press. [Google Scholar]
- 48.Swain PS, Elowitz MB, Siggia ED. 2002. Intrinsic and extrinsic contributions to stochasticity in gene expression. Proc. Natl Acad. Sci. USA 99, 12 795–12 800. ( 10.1073/pnas.162041399) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaern M, Elston TC, Blake WJ, Collins JJ. 2005. Stochasticity in gene expression: from theories to phenotypes. Nat. Rev. Genet. 6, 451–464. ( 10.1038/nrg1615) [DOI] [PubMed] [Google Scholar]
- 50.Thattai M, Van OA. 2001. Intrinsic noise in gene regulatory networks. Proc. Natl Acad. Sci. USA 98, 8614–8619. ( 10.1073/pnas.151588598) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hunt K, Ross J. 1981. Path integral solutions of stochastic equations for nonlinear irreversible processes: the uniqueness of the thermodynamic Lagrangian. J. Chem. Phys. 75, 976–984. ( 10.1063/1.442098) [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.