

Abstract
Viral sequence classification has wide applications in clinical, epidemiological, structural and functional categorization studies. Most existing approaches rely on an initial alignment step followed by classification based on phylogenetic or statistical algorithms. Here we present an ultrafast alignment-free subtyping tool for human immunodeficiency virus type one (HIV-1) adapted from Prediction by Partial Matching compression. This tool, named COMET, was compared to the widely used phylogeny-based REGA and SCUEAL tools using synthetic and clinical HIV data sets (1 090 698 and 10 625 sequences, respectively). COMET's sensitivity and specificity were comparable to or higher than the two other subtyping tools on both data sets for known subtypes. COMET also excelled in detecting and identifying new recombinant forms, a frequent feature of the HIV epidemic. Runtime comparisons showed that COMET was almost as fast as USEARCH. This study demonstrates the advantages of alignment-free classification of viral sequences, which feature high rates of variation, recombination and insertions/deletions. COMET is free to use via an online interface.
INTRODUCTION
The human immunodeficiency virus type one (HIV-1) circulates as a number of distinct types, subtypes and recombinant forms. HIV-1 group M, the predominant type of the worldwide epidemic, is currently classified into nine pure subtypes (PURE) and 55 circulating recombinant forms (CRFs) recorded in the Los Alamos National Laboratory (LANL) database (1). Viral subtype impacts disease progression (2,3,4,5,6), treatment response (7,8,9,10) and vaccine development (11). Viral subtype identification is also an informative and reliable tool for epidemiological studies and surveillance, transmission follow-up and design of prevention strategies (12,13). Yet correct subtyping of clinical HIV-1 samples remains a challenging task, particularly in the case of CRFs and of unique recombinants forms (URFs) (14,15). This issue is of growing importance, as the prevalence of recombinant forms (15,16) has reached 20% of new infections (17) and continues to expand, fueling the complexity of the HIV pandemic.
Protocols for HIV-1 subtype determination share a common approach. The query sequence is compared to a set of reference sequences, generally using a sliding window, and the best match is returned as the putative subtype. The vast majority of currently used tools rely on a preliminary alignment step to measure similarity with the reference set. The REGA (18) and SCUEAL (19) tools extend the alignment with phylogenetic analyses, allowing the detection and identification of recombinant forms using either a sliding window with bootstrap support (REGA) or the phylogenetic likelihood of a mosaic (SCUEAL). Alternatives include position-specific scoring matrices (20) and profile Hidden Markov models (21,22).
The choice of one alignment as the correct alignment masks uncertainty as to how statistically distinct the chosen alignment is from other possible alignments. Determination of the best alignment also depends on the choice of algorithm and parameters (23). Alignment confidence can be measured (24). This, however, imposes additional computational overhead, particularly so for sequences comprising numerous mixtures and indels (25). While these factors may not have significant impact on classifications based on the highly conserved pol gene, the situation is less clear in more variable regions of the HIV genome, such as env, especially in the context of novel forms.
A small but growing literature demonstrates that sequence similarity can be quantified in terms of the amount of information shared between the two sequences. This can be measured by various data compression schemes, which do not require reference to alignment (26,27,28,29). For example, a Markov model expresses the probability of observing a given nucleotide given the previous k nucleotides in the sequence, i.e. its genomic context. The similarity between two sequences can be measured by how closely a Markov model built from the first sequence matches context-specific base frequencies in the second. Markov models are fast to build and to run, and have proved their worth in a variety of genomic sequence analysis contexts (30).
This work introduces COMET (COntext-based Modeling for Expeditious Typing), an alignment-free typing algorithm for HIV-1 and other viruses (HCV). The approach of COMET is inspired by the Prediction by Partial Matching compression algorithm (31), which has repeatedly and independently been shown to provide high classification accuracy for biological sequences (32,33). Benchmarking results for HIV on both synthetic and clinical data suggest significant improvements over existing tools in classification accuracy, ability to recognize known and novel recombinants, reproducibility and running time. COMET is freely available via an anonymous web-based interface at http://comet.retrovirology.lu, and for academic usage as a stand-alone Java jar file by request to the corresponding author.
The COMET algorithm
COMET begins by building variable-order Markov models over nucleotide frequencies for each of a set of reference sequences. Given a query, COMET uses these models to estimate, for each nucleotide in the query, the log likelihood of observing this base for each of the reference types. This procedure returns a matrix with rows corresponding to the reference types and columns representing the log likelihoods. A decision tree (see Figure 2) is applied to this matrix to determine the final subtype call. The final call is one of: a reference type (PURE or CRF); a PURE reference type that may come from a non-recombinant region of a CRF (both the PURE and potential CRF are indicated); or UNASSIGNED in the case of a novel recombinant form or a query sequence whose actual classification is ambiguous.
Figure 2.
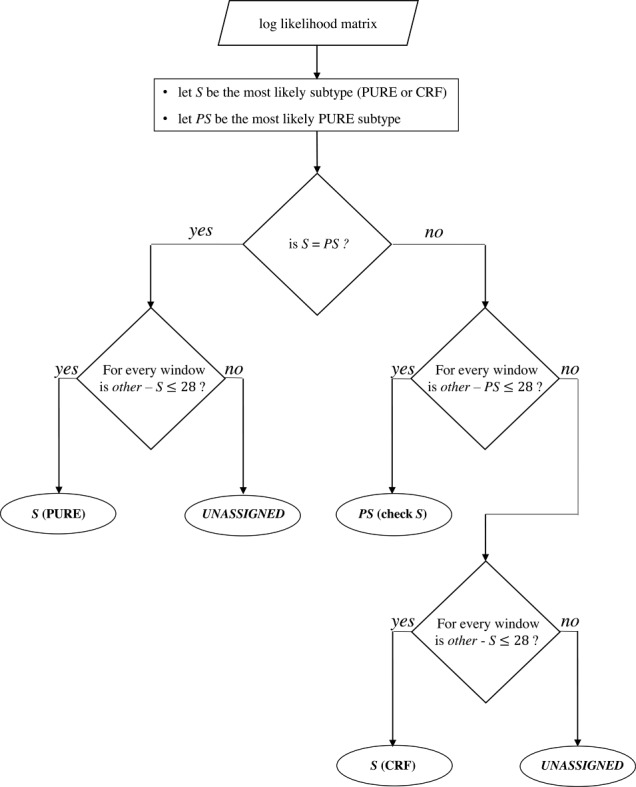
Subtype decision tree. The row sums of the log-likelihood matrix provide the overall likelihood of the query sequence to belong to each subtype. These sums are ordered to identify the most likely subtype (S) and the most likely pure subtype (PS). If the query sequence has the highest likelihood of belonging to a pure subtype (i.e. S = PS), this likelihood is challenged against the likelihoods of the sequence to be of any other subtype (other, PURE or CRF) by sliding over the matrix by 100-bp windows with a stepping size of 3 bp. If the difference between the row sums within the current window remains below the recombination threshold (i.e. 28) for each window, the pure subtype is assigned. Otherwise, COMET returns the result ‘UNASSIGNED’. If the query sequence has the highest likelihood of being a CRF, COMET performs a similar challenge, but only against the most likely pure subtype (PS) at first. If this difference remains below the recombination threshold (i.e. 28), COMET assigns the pure subtype (S) with an indication to check for the CRF, indicating a region where the CRF is pure. If the difference is higher than the recombination threshold, a second scan is performed as for the PURE situation, challenging each subtype against the initially assigned CRF.
Building and scoring the Markov models
The reference set for COMET version 1.0 is the full 2010 LANL subtype reference alignment, which includes 170 full-length sequences representing each pure subtype (A–J) and CRF (CRF 01_AG–49_cpx). This set was complemented by 45 full-length sequences from the LANL database (3 A1, 3 B, 2 C, 2 D, 2 F1, 1 G, 1 H and CRF 3 01_AE, 4 02_AG, 2 03_AB, 1 04_cpx, 2 06_cpx, 1 07_BC, 2 08_BC, 2 09_cpx, 1 11_cpx, 2 12_BF, 1 15_01B, 1 18_cpx, 2 20_BG, 1 25_cpx 1, 1 29_BF, 1 32_06A1, 2 34_01B, 1 42_BF and 1 44_BF) after manual verification that the sequence did indeed represent the stated type. Each reference type is represented by a variable-order Markov model that stores the probability of observing a given base conditional on the preceding k bases, with k ranging from 0 to 8. For k = 0, the model considers the frequency of each of the four bases in the reference sequences. The Markov models for each subtype are stored in an N-ary tree (Figure 1). When building and fitting the model, ambiguous codes are ignored.
Figure 1.
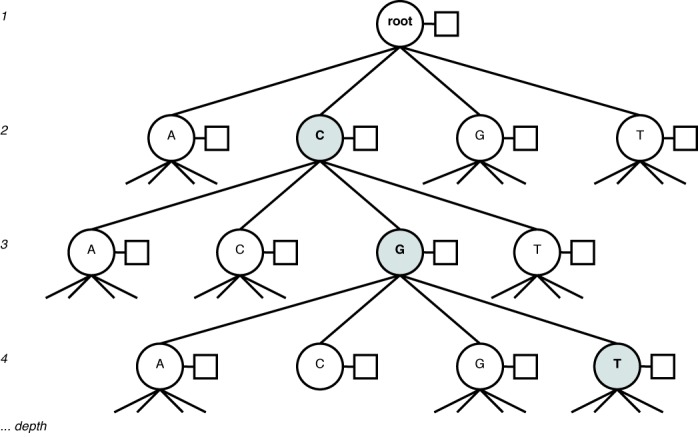
N-ary tree representation of a Markov model, with the context ‘CGT’ highlighted. Each node (circle) has an associated frequency table (box) over the next base in the sequence following the context.
The initial stage of COMET's algorithm uses these Markov models to compute a matrix giving the log likelihood of each reference subtype (row) at each position of the sequence (column). For a given model and query sequence, the model selects, for each base in the query, the k-mer, or context, which predicts the base with the best accuracy. This induces a bias toward the longest possible context, while allowing the prediction to jump to a shorter context if the latter provides higher accuracy.
The decision tree
The log-likelihood matrix is passed to COMET's decision tree. The decision tree incorporates a sliding window to perform fine-grained comparisons of reference subtype likelihoods. This allows more detailed investigation of potential recombination than simply outputting the most likely reference type. Finding that two different reference subtypes have similar likelihoods over one or more sections of the query provides evidence that the query represents a unique or novel recombinant form. Alternately, the query may come from an ambiguous genomic region. For example, the pol gene of CRF 14_BG originates entirely from subtype G. Here, the decision tree's sliding window allows COMET to report whether or not the query sequence has sufficient features to allow a clear distinction between the PURE and CRF types.
COMET begins by using the maximal row sum of the log-likelihood matrix to generate an initial indication of either a PURE or CRF subtype. An initial PURE indication is scanned for recombination by sliding over the matrix with a window of 100 base pairs (bp) and a stepping size of 3 bp. Log-likelihood differences are computed between all remaining reference subtypes (including CRFs) and the initial assignment S, considering only the 100 bp in the window, and using the difference in the log likelihoods to compute the ratio. As long as this difference remains less than a preset threshold (ℓℓ(other) − ℓℓ(S) ≤ 28, see below for determination of this level) for every window, COMET assigns the sequence to the initially determined PURE subtype. A potential recombination event is signaled if, in any window, the threshold is crossed. In this case COMET states UNASSIGNED.
An initial indication of CRF is given a more nuanced treatment. COMET begins by putatively assigning the most likely major PURE subtype (i.e. excluding CRFs) as determined by the row sums of the log-likelihood matrix. A scan for recombination is made as above. If the PURE type is confirmed, COMET returns the PURE type with an indication to check for the initially indicated CRF. If recombination is signaled in any window, a second scan is made. This second scan follows the same protocol, this time comparing each reference subtype to the initially indicated CRF. If the threshold is not surpassed in any window, COMET returns the initially indicated CRF. Otherwise COMET returns UNASSIGNED.
Determining the best model order, window size and threshold for recombination detection
The optimal context length, window size and selection thresholds were determined empirically. The sensitivities of COMET in assigning the correct subtype/CRF or identifying an URF were determined for kmax from 1 to 15, for different combinations of window sizes (50, 75, 100, 200, 300 and 400 bp), and for PURE and CRF likelihood ratio thresholds (difference in log likelihood varying from 18 to 38). The best tradeoff between sensitivities and Akaike information criterion (AIC) scores were observed for kmax = 8 with a window size of 100 bp and log-likelihood difference thresholded of 28 for both PURE and CRFs. These parameters were therefore chosen as COMET's default values. Sensitivity for detecting PURE and CRFs increases asymptotically toward 1.00 with the log-likelihood threshold, and is nearly level for the chosen thresholds for most subtypes. Specificity to PURE/CRFs varies only over the range 0.995–1.00 for all thresholds tested. Small variations to the thresholds should have minimal effect on results. The assays were conducted using synthetic variation and synthetic recombinant data sets (construction described below) containing 1–10% variation. Testing using only synthetic variation would have optimized the parameters solely for the detection of known categories (pure subtypes and common recombinant forms, CRF) and not for URFs. Parameter optimization was carried out on different data sets than those used for evaluation. Details of these investigations are reported in the Supplementary Notes, Supplementary Table S1 and Supplementary Figure S1.
Validation on synthetic data
COMET's performance was evaluated using a large panel of synthetically modified PURE and CRF sequences. COMET's performance was then compared with the REGAv2 (18) and SCUEAL (19) tools, commonly recognized as the current ‘best of breed’ of published subtyping tools (34). This comparison was based on additional synthetic data sets adopted to fit the limitations of these tools, constructed using parameters under which they are expected to perform best. For each evaluation, we report how the degree of variation and recombination affect sensitivity and specificity, per subtype. Running times are also given.
Synthetic variation
Synthetic sequences were created from the full length HIV-1 sequences in the LANL subtype reference alignment (1), including all recorded CRFs, after excluding non-M sequences, i.e. groups ‘O’, ‘N’, ‘P’ and ‘CPZ’, that is, 158 whole-genome sequences representing 58 subtypes and CRFs. Synthetic variation was introduced into each sample by randomly replacing an increasing proportion of nucleotides in the input reference sequence by a different nucleotide. The amount of variation increased from 0% to 20% with 1% increments. One full-genome synthetically varied sequence was generated from each input sequence at each noise level. Test sequences were extracted from the resulting 3318 full-genome sequences using sliding windows of increasing length (100, 200, 400, 600, 800, 1200 and 1600 bp) applied from sequence position HXB2 790 (gag start) to position 9417 (nef end), with stepping sizes half the size of the window length (e.g. for a window size of 400 bp, the stepping size was 200 bp). The resulting test set comprised 1 090 698 sequences of different lengths and noise levels covering the entire HIV genome.
The synthetic variation data set used for comparison to REGAv2 and SCUEAL was limited as follows. Since SCUEAL only analyzes the pol gene (19), sequences were taken from this region, including the RNaseH and integrase (start position: HXB2 2085; end position: 5096). Comparisons were limited to subtypes recognized by REGAv2: the 10 PURE subtypes and the first 14 listed CRFs (CRF 01_AE - 14_BG). To contain the size of the final data set within the bounds imposed by the running times of the other algorithms, only one recorded example sequence of each type was used, and five levels of synthetic variation: 0%, 2.5%, 5%, 7.5% and 10%. Samples were taken from each input reference sequence by sliding over the sequences with window sizes of 200, 400, 800, 1200 and 1600, and with stepping sizes half the window size, as above, except for the smallest window. The resulting test set comprised 5125 sequences.
Synthetic recombination
Synthetic recombinant sequences were generated from the 39 sequences that represent pure subtypes in the LANL reference set. One sequence was taken as background, the second as insertion. A sliding window was applied to the sequence pair starting at position 790 (gag start) and ending at position 9417 (nef end), and the portion of the background sequence covered by the window was replaced by the corresponding insert sequence. The following window lengths and step sizes were used: 100:100; 200:200; 300:200; 400:200; 600:200; 800:200. Each sequence was used once as a background for inserts from all remaining sequences; the 1482 possible permutations yielded 405 132 full-genome synthetic recombinants.
Synthetic recombination data for comparison to REGAv2 and SCUEAL were limited by choosing only one representative sequence for each subtype. In addition, only the pol gene was included. Insert window lengths and steps were as above. The resulting data set contained 10 780 synthetically recombined pol gene sequences representing the 110 possible permutations of the source genotypes.
Validation on clinical data
COMET's reliability and practical utility was assessed by testing its ability to identify clinical sequences downloaded from the LANL database, alone and in comparison with REGAv2 and SCUEAL. Sequence selection was based on the following inclusion/exclusion criteria applied to the 174 894 pol sequences retrieved from the LANL database: (i) select all sequences with a minimum fragment length of 800 bp and a maximum of 8000 bp. (ii) Exclude sequences used to train COMET and all duplicate sequences (identical and subsets of longer sequences) (remaining n = 112 997). (iii) Exclude subtypes represented by less than 50 samples. For subtypes with more than 1000 sequences, select 1000 at random (remaining n = 11 341). Clinical samples represented by less than 50 pol sequences were excluded because comparisons based on such small sample sizes are not representative. For example, referenced subtypes other than A1, B, C, D, F, G, 01_AE, 02_AG represent only 0.59% of new infections in Europe (13), and only 3.31% of the sequences in the pol LANL data set. (iv) Retain only PURE subtypes and the first 14 listed CRFs (CRF01_AE - 14_BG) (remaining n = 10 625). The subtype/CRF assigned by the submitting author and stored in the LANL HIV database was considered to be correct. Comparison was limited to the first 14 listed CRFs as REGAv2 does not recognize other subtypes.
Comparison to USEARCH
To demonstrate the claim of ultrafast subtyping, we compared the performance and running times of COMET and USEARCH (35), one of the fastest sequence search tools. Given a query sequence, USEARCH can quickly find the closest matching subtype reference sequence in its internal reference set. Sequence search is generally not recommended for subtyping (it cannot identify novel forms, among other limitations), but its use to provide preliminary subtype indications has been endorsed (36). To ensure equitable comparisons, USEARCH's reference set was here created using the same training data used for COMET, and COMET's recombination detection module was de-activated. The sensitivity, specificity and running times of COMET and USEARCH were compared using both the full-length synthetic variation (1 090 698 sequences) data set and the pol clinical data set (105 752 sequences). The clinical data set was as above, additionally excluding URFs as these cannot be classified by the USEARCH methodology. Sensitivity and specificity on the clinical data set were calculated by considering the subtype stored in the LANL database as correct. Reported running times are a mean of 10 independent runs. Tests were made using USEARCH version 7.0.1090 with the following parameters: usearch_global, id=0.8, strand=plus. Preliminary investigations showed that these settings gave USEARCH the highest sensitivities.
RESULTS
Synthetic variation
COMET showed high specificity (>93% for PURE subtypes and >99% for CRFs) for all sequence lengths and even for high levels (20%) of noise (Table 1). The sensitivity of COMET was very high (>99% for PURE subtypes and >87% for CRFs) for sequences longer than 800 bp and containing up to 10% noise (Table 1). For shorter sequences, the sensitivity of COMET remained >97% and >78% for PURE and CRFs, respectively, for 400-bp-long sequences (Table 1). When more noise was introduced (up to 14%), sensitivity was >97% for PURE and >69% for CRFs for 1200-bp-long sequences (Table 1). For PURE sequences, sensitivity remained >83% for 1200-bp-long sequences even when sequences contained 18% noise. These figures indicate that COMET features very high sensitivity and is able to correctly classify sequences with the lengths and variation typically encountered in clinical practice (circa 800–1200 bp).
Table 1. Sensitivity and specificity of COMET to type known sequences with varying levels of noise.
| (a1) PURE sensitivities | Noise | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fragment size | n | 0.0% | 2.0% | 4.0% | 6.0% | 8.0% | 10.0% | 12.0% | 14.0% | 16.0% | 18.0% | 20.0% |
| 100 | 494 256 | 98.4% | 96.5% | 92.6% | 87.2% | 80.8% | 72.9% | 63.1% | 53.6% | 44.2% | 38.3% | 32.3% |
| 200 | 242 235 | 99.9% | 99.8% | 99.0% | 97.5% | 94.9% | 89.5% | 82.1% | 71.0% | 61.3% | 52.9% | 44.1% |
| 400 | 128 604 | 100.0% | 100.0% | 100.0% | 99.9% | 98.9% | 97.6% | 93.1% | 87.0% | 78.1% | 68.8% | 57.2% |
| 600 | 87 402 | 100.0% | 100.0% | 100.0% | 100.0% | 99.9% | 98.9% | 96.3% | 91.9% | 86.2% | 75.2% | 63.5% |
| 800 | 65 352 | 100.0% | 100.0% | 100.0% | 100.0% | 99.8% | 99.5% | 97.1% | 95.4% | 89.5% | 78.4% | 66.6% |
| 1200 | 43 008 | 100.0% | 100.0% | 100.0% | 100.0% | 99.5% | 100.0% | 99.1% | 97.4% | 92.9% | 83.4% | 70.3% |
| 1600 | 29 841 | 100.0% | 100.0% | 100.0% | 100.0% | 99.5% | 100.0% | 99.2% | 97.8% | 93.7% | 84.8% | 71.0% |
| (a2) PURE specificities | Noise | |||||||||||
| 100 | 494 256 | 93.8% | 93.8% | 93.7% | 93.6% | 93.5% | 93.5% | 93.5% | 93.5% | 93.5% | 93.7% | 93.8% |
| 200 | 242 235 | 96.9% | 96.1% | 95.2% | 94.6% | 94.2% | 93.9% | 93.7% | 93.6% | 93.6% | 93.7% | 93.9% |
| 400 | 128 604 | 98.6% | 97.9% | 97.0% | 96.1% | 95.4% | 94.7% | 94.3% | 94.0% | 93.8% | 93.8% | 94.0% |
| 600 | 87 402 | 99.3% | 98.8% | 98.1% | 97.2% | 96.3% | 95.5% | 94.8% | 94.3% | 94.1% | 94.0% | 94.2% |
| 800 | 65 352 | 99.5% | 99.2% | 98.6% | 97.9% | 97.0% | 96.0% | 95.3% | 94.7% | 94.3% | 94.1% | 94.3% |
| 1200 | 43 008 | 99.8% | 99.6% | 99.3% | 98.8% | 98.1% | 97.0% | 96.2% | 95.4% | 94.8% | 94.4% | 94.5% |
| 1600 | 29 841 | 99.9% | 99.7% | 99.5% | 99.2% | 98.7% | 97.8% | 96.8% | 96.0% | 95.2% | 94.6% | 94.7% |
| (b1) CRF sensitivities | Noise | |||||||||||
| 100 | 494 256 | 91.0% | 85.4% | 77.2% | 66.7% | 56.1% | 46.1% | 37.0% | 28.7% | 22.1% | 16.6% | 12.6% |
| 200 | 242 235 | 97.6% | 95.6% | 90.7% | 83.7% | 75.3% | 64.7% | 52.2% | 41.6% | 31.2% | 21.9% | 15.8% |
| 400 | 128 604 | 99.5% | 99.1% | 97.3% | 93.4% | 88.2% | 78.3% | 66.6% | 54.4% | 41.3% | 28.9% | 19.7% |
| 600 | 87 402 | 99.7% | 99.7% | 98.9% | 96.4% | 92.6% | 84.3% | 74.4% | 61.4% | 46.9% | 32.6% | 21.3% |
| 800 | 65 352 | 99.9% | 99.8% | 99.5% | 97.0% | 94.5% | 87.6% | 78.3% | 64.8% | 48.7% | 34.2% | 22.8% |
| 1200 | 43 008 | 99.8% | 99.9% | 100.0% | 97.3% | 96.1% | 91.2% | 82.9% | 69.0% | 52.8% | 36.9% | 23.0% |
| 1600 | 29 841 | 100.0% | 100.0% | 100.0% | 97.7% | 97.8% | 91.9% | 84.7% | 71.7% | 53.8% | 38.1% | 25.4% |
| (b2) CRF specificities | Noise | |||||||||||
| 100 | 494 256 | 99.7% | 99.7% | 99.6% | 99.6% | 99.5% | 99.4% | 99.4% | 99.4% | 99.3% | 99.3% | 99.3% |
| 200 | 242 235 | 99.9% | 99.9% | 99.8% | 99.8% | 99.7% | 99.7% | 99.7% | 99.6% | 99.6% | 99.6% | 99.6% |
| 400 | 128 604 | 99.9% | 99.9% | 99.9% | 99.9% | 99.9% | 99.8% | 99.8% | 99.8% | 99.7% | 99.7% | 99.7% |
| 600 | 87 402 | 100.0% | 100.0% | 99.9% | 99.9% | 99.9% | 99.9% | 99.8% | 99.8% | 99.8% | 99.8% | 99.8% |
| 800 | 65 352 | 100.0% | 100.0% | 99.9% | 99.9% | 99.9% | 99.9% | 99.9% | 99.9% | 99.8% | 99.8% | 99.8% |
| 1200 | 43 008 | 100.0% | 100.0% | 100.0% | 99.9% | 99.9% | 99.9% | 99.9% | 99.9% | 99.8% | 99.8% | 99.8% |
| 1600 | 29 841 | 100.0% | 100.0% | 100.0% | 100.0% | 99.9% | 99.9% | 99.9% | 99.9% | 99.9% | 99.8% | 99.8% |
A synthetic data set was generated from reference sequences from the LANL HIV database, by randomly introducing mutations throughout the genome (‘noise’). The sensitivity and specificity of COMET was calculated for varying degrees of noise (0–20%) introduced into PURE subtypes (A–J), (Tables a1 and a2) or CRFs (CRF01_AE-CRF49_cpx) (Tables b1 and b2). Sequences of different lengths were submitted to COMET.
The performance of COMET was also compared to that of REGAv2 and of SCUEAL using synthetic sequences ranging from 200 to 1600 bp in length and comprising 0–10% noise. COMET outperformed both REGAv2 and SCUEAL on PURE and CRF sequences in terms of sensitivity. Specificity was high and comparable for all three tools for both PURE and CRFs, regardless of sequence length and noise level (Figure 3).
Figure 3.
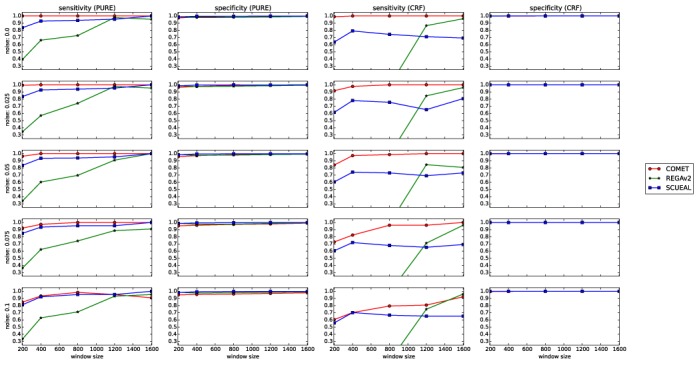
Sensitivities and specificities of COMET, REGAv2 and SCUEAL assessed using the synthetic variation data set spanning the pol region.
Synthetic recombination
The synthetic recombination data set was generated to assess the capacity of COMET to classify URFs. On a data set of 405 132 synthetic recombinant sequences, COMET correctly identified >99% of synthetic recombinant forms for inserts longer than 400 bp, identifying the correct mixture in 96.5% of the cases (Table 2). For shorter inserts (200 bp), COMET detected and correctly identified 97% (Table 2).
Table 2. Sensitivity and specificity of COMET to detect and identify synthetic recombinants.
| Insert size | n | URF found | Composition found |
|---|---|---|---|
| 100 | 118 508 | 84.5% | 84.1% |
| 200 | 59 254 | 96.9% | 96.8% |
| 300 | 57 876 | 98.7% | 98.7% |
| 400 | 57 876 | 99.6% | 99.6% |
| 600 | 56 498 | 99.9% | 99.9% |
| 800 | 55 120 | 100.0% | 100.0% |
| Mean | 96.6% | 96.5% |
A synthetic recombination data set was generated by replacing DNA sequences by the same sequence from another subtype. Different sizes of insert (from 100 bp to 800 bp) were introduced throughout the genome, generating 405 132 different recombinants. In the table, ‘URF found’ means that COMET recognized the sequence as ‘UNASSIGNED’. ‘Composition found’ means that COMET correctly identified the subtypes composing the background and the insert.
When compared to the two other subtyping tools using a similar synthetic recombinant data set restricted to the pol gene, COMET clearly outperformed both REGAv2 and SCUEAL, detecting 92% of the recombinant forms where SCUEAL detected 84% and REGAv2 only 58% (Table 3). COMET and SCUEAL reliably detected recombinants composed of 200-bp-long inserts (90% and 75%, respectively), whereas REGAv2 required inserts of 400 bp to reach comparable performance (78.1% detection rate). Over 99% of the synthetic recombinants were correctly identified by COMET for 800-bp- and 600-bp-long fragments against 92% and 91% for REGAv2 and SCUEAL, respectively. COMET correctly identified 90% of short inserts (200 bp), compared to 59% for SCUEAL and 18% for REGAv2 (Table 3).
Table 3. Detection and identification of unknown recombinants by COMET, REGAv2 and SCUEAL.
| COMET | REGAv2 | SCUEAL | |||||
|---|---|---|---|---|---|---|---|
| Insert size | n | URF found | Composition found | URF found | Composition found | URF found | Composition found |
| 100 | 3300 | 68.6% | 67.9% | 1.0% | 0.9% | 49.5% | 31.5% |
| 200 | 1650 | 90.4% | 90.1% | 18.5% | 17.5% | 75.3% | 58.7% |
| 300 | 1540 | 96.2% | 96.1% | 56.5% | 54.2% | 88.3% | 74.8% |
| 400 | 1540 | 97.9% | 97.9% | 78.1% | 74.2% | 94.6% | 84.4% |
| 600 | 1430 | 99.7% | 99.7% | 96.5% | 92.0% | 97.5% | 91.5% |
| 800 | 1320 | 99.9% | 99.9% | 96.9% | 94.9% | 99.1% | 95.5% |
| Mean | 92.1% | 91.9% | 57.9% | 55.6% | 84.1% | 72.7% | |
The synthetic recombination data set was restricted to the pol region, and one recombinant was selected for each pattern, leading to 10 780 sequences for this analysis. In the table, ‘URF found’ means that COMET assigned the sequence as ‘unassigned’, REGAv2 assigned the sequence as ‘check the bootscan’ or ‘check the report’ and SCUEAL assigned the sequence as ‘complex’ or ‘recombinant’. ‘Composition found’ means that the tool correctly identified the subtype of the background and of the insert composing the synthetic recombinant.
Clinical sequences
The performance of COMET, REGAv2 and SCUEAL was further compared using 10 625 clinical patient-derived pol sequences belonging to PURE subtypes A–J or CRF01_AE to CRF14_BG. Specificity was comparably high for all three tools (mean >=99.7%). Sensitivities were also comparable for PURE subtypes (COMET: 87.8%; REGAv2: 86.6%; SCUEAL: 84.8%), whereas for CRFs the sensitivity of COMET was remarkably higher (COMET: 92.7%; REGAv2: 72.3%; SCUEAL: 44.1%) [Table 4(a) and (b)].
Table 4. Sensitivity and specificity of COMET, REGAv2 and SCUEAL to type clinical patient-derived sequences retrieved from the LANL database.
| COMET | REGAv2 | SCUEAL | |||||
|---|---|---|---|---|---|---|---|
| (a) PURE | n | Sensitivity | Specificity | Sensitivity | Specificity | Sensitivity | Specificity |
| A1 | 1000 | 97.0% | 99.9% | 95.7% | 99.4% | 80.2% | 100.0% |
| A2 | 142 | 71.1% | 100.0% | 72.5% | 100.0% | 76.1% | 100.0% |
| B | 1000 | 99.6% | 99.8% | 96.2% | 99.9% | 97.6% | 99.9% |
| C | 1000 | 98.9% | 100.0% | 99.5% | 99.6% | 91.8% | 99.9% |
| D | 1000 | 92.4% | 100.0% | 87.2% | 100.0% | 86.8% | 100.0% |
| F1 | 1000 | 93.1% | 99.8% | 96.8% | 99.4% | 87.8% | 99.8% |
| F2 | 184 | 75.0% | 100.0% | 53.8% | 100.0% | 77.7% | 100.0% |
| G | 1000 | 89.0% | 99.9% | 89.9% | 98.6% | 79.5% | 98.9% |
| H | 97 | 74.2% | 100.0% | 87.6% | 100.0% | 85.6% | 100.0% |
| Mean | 87.8% | 99.9% | 86.6% | 99.7% | 84.8% | 99.8% | |
| (b) CRF | n | Sensitivity | Specificity | Sensitivity | Specificity | Sensitivity | Specificity |
| 01_AE | 1000 | 97.5% | 100.0% | 93.7% | 100.0% | 68.3% | 100.0% |
| 02_AG | 1000 | 95.6% | 99.5% | 14.8% | 100.0% | 26.6% | 99.9% |
| 06_cpx | 823 | 92.5% | 100.0% | 84.2% | 100.0% | 40.0% | 100.0% |
| 07_BC | 581 | 97.4% | 100.0% | 97.2% | 100.0% | 14.8% | 100.0% |
| 08_BC | 365 | 95.9% | 100.0% | 91.0% | 100.0% | 77.5% | 100.0% |
| 11_cpx | 116 | 82.8% | 100.0% | 75.0% | 100.0% | 72.4% | 100.0% |
| 12_BF | 317 | 87.1% | 100.0% | 50.2% | 100.0% | 9.1% | 100.0% |
| Mean | 92.7% | 99.9% | 72.3% | 100.0% | 44.1% | 100.0% | |
This data set includes 10 625 sequences spanning pol.
All three tools had the lowest sensitivities in detecting subtypes labeled as A2 and F2 in the LANL HIV database (see Table 4). In 92.7% of cases where COMET and the LANL label disagreed over an A2 assignment, COMET indicated that the sequences were recombinants (CRFs or URFs) containing A2 sequences. Both REGAv2 and SCUEAL agreed with COMET in 73% of the cases.
Likewise, for subtype F2, 39.1% of the sequences not identified as F2 by COMET were recombinants also identified as such by both other tools, and 43.5% corresponded to F1 sequences according to all three tools. In five cases (10.8%), COMET identified a recombinant where SCUEAL assigned F2 and REGA either F1 or a recombinant as well, and in two cases COMET did not identify an F2 sequence identified as such by REGAv2 and SCUEAL. These results suggest that many of the disagreements between the subtype assigned by COMET and the subtype stored in LANL are due to an incorrect labeling by the submitting author.
For CRFs, COMETs sensitivity was >87% for all tested samples except CRF11_cpx (82.8%) [Table 4(b)]. For this CRF, COMET agreed with both other tools in identifying a URF, whereas in 30.4% of the cases, REGAv2 and SCUEAL agreed with the subtype stored in the LANL HIV database and in one case, all three tools disagreed with the LANL label. REGAv2 had difficulty identifying CRF02_AG and CRF12_BF (Table 4b), perhaps partially reflecting the fact that these CRFs do not feature recombination in the pol region.
The low sensitivity of SCUEAL in typing CRFs is mainly due to the fact that the algorithm often designated sequences as ‘recombinant’ without providing further specificity. This was mostly observed for CRF02_AG, CRF07_BC and CRF12_BF and to some extent for CRF06_cpx. In a three-way comparison, the three tools agreed for 7130 (67.1%) pol sequences. The three tools disagreed in 233 cases (2.2%). In all other cases, two tools agreed and disagreed with the third (Figure 4).
Figure 4.
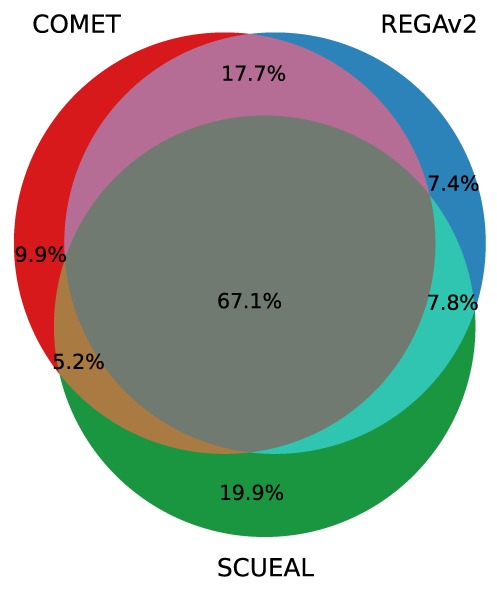
Agreement between the three subtyping tools on the subtype assigned to clinical patient-derived sequences retrieved from the LANL database. This data set includes 10 625 sequences spanning pol.
Figure 5.
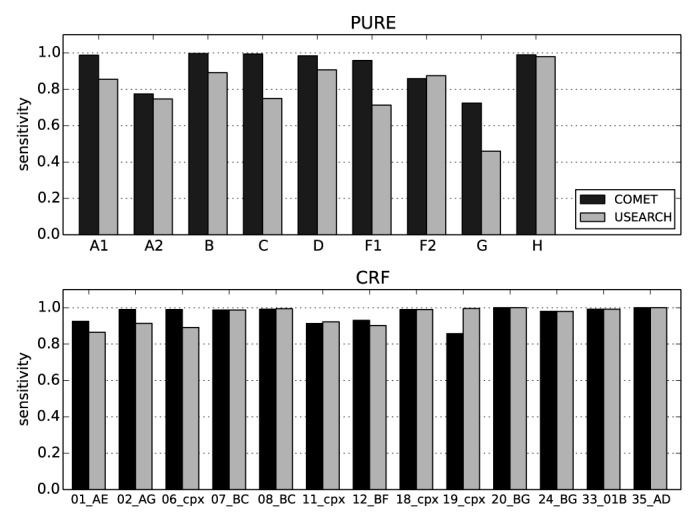
Sensitivity and specificity of COMET and USEARCH on clinical pol sequences. A data set of 105 752 clinical pol sequences from the LANL HIV database was used for this analysis. The data set includes all PURE and CRF sequences longer than 800 bp, represented by at least 50 sequences, but excludes URFs, which are not classifiable by USEARCH. The USEARCH database was built using the COMET training set. The recombination module of COMET was disabled for this analysis.
Taken together, these results highlight the inherent difficulties of typing viral sequences in the clinical setting, particularly regarding the confidence of the result issued by each tool. Different approaches should be used in parallel, calling for manual inspection when different tools disagree, as no guidelines exist to date.
Running times
Running times of COMET on the different data sets were as follows. The 1 090 698 synthetic variation sequences were processed in 320 s, the 405 132 synthetic recombination sequences in 3404 s and the 161 901 sequences from LANL in 119 s. All measured times are the mean time over 10 runs of the algorithm. These timing results cannot be neatly summarized into a mean throughput rate since COMET's run time is linear in sequence length while the sequences in these data sets ranged from 100 bp to 8769 bp in length. Mean throughput is better measured by observing that COMET took 0.1 s to process the 100 random sequences from the synthetic variation pol gene sequences (each 1200 bp), indicating a throughput of 0.001 seconds/sequence. SCUEAL required 2811 s to analyze the same 100 sequences (28.1 s/sequence, for details see Supplementary Notes). In comparison, REGAv2 claims to take 28.8 s to analyze one pol sequence (34). It is not specified, however, if these reported timings are based on the entire pol gene or only the 1300-bp protease-reverse transcriptase region.
Comparison to USEARCH
For the synthetic data set, mean sensitivities over all noise levels and window sizes of COMET and USEARCH were 76.5% and 82.2%, respectively. The sensitivity of USEARCH was slightly higher than that of COMET (up to 0.9%) for sequences containing 0–8% noise; for higher noise levels, USEARCH had higher sensitivities than COMET regardless of window size (Supplementary Figure S3). For synthetic CRFs, COMETs sensitivity was lower than that of USEARCH (Supplementary Figure S3). Manual inspection showed that for 39% of the misassignments, COMET assigned a component of the CRF rather than the CRF itself. This behavior was particularly prominent in regions where the CRFs comprise no breakpoints. This observation highlights the importance of the recombination module in discriminating between PURE subtypes and CRFs.
In contrast, when challenged with the clinical pol data set, the sensitivity of COMET was higher than that of USEARCH for PURE subtypes and CRFs (Supplementary Figure S3). For both tools, the lowest sensitivities were recorded for subtypes A2 and G, as well as for subtype F1 for USEARCH. Inspection of the most common disagreements between the subtype assigned by COMET or USEARCH and the subtype stored in the LANL HIV database again showed that the subtyping tools assigned a CRF comprising the subtype stored in the LANL database.
COMET analyzed the synthetic variation data set in 284 s and USEARCH in 179 s. For the clinical data set, COMET required 78.7 s and USEARCH 33.6 s. Notably, further testing on synthetic data suggests that COMET's time requirements increased only slightly and in a linear fashion in response to noise, while USEARCH's grew quasi-exponentially to nearly double COMET's time at a noise of 20% (Supplementary Figure S4). Taken together, these results suggest that in this setup, running times for COMET and USEARCH are on the same order of magnitude.
COMET online tool
COMET can be accessed via an on-line interface hosted at http://comet.retrovirology.lu. The interface allows direct uploading of sequences in fasta format, or for sequences to be pasted into a web form. COMET returns the subtype for each sample. The results can be copy-pasted or downloaded in comma-separated value format.
DISCUSSION
The benchmarking tests show that COMET was able to accurately and reliably subtype more sequences and more types of sequences orders of magnitude faster than current ‘best of breed’ tools. COMET was robust in the face of noise and well able to identify both rare and novel recombinant forms. Its results are consistent both on short sequences of a few hundred base pairs and on whole genome sequences. These results agree with a recent comparison of eight subtyping algorithms conducted by an unrelated research group that concluded that COMET is one of the best performing subtyping tools (34).
COMET's speed results from dispensing with both full alignment and with tree-building. This makes it blind to evolutionary effects such as variable substitution rates, which tend to be well captured by alignment-based methods. Yet it also implies that COMET will not suffer from artifacts induced during an alignment step. COMET inherently performs a more detailed recombination analysis than allowed for by tree-based methods, regardless of how the tree is built. Accurate tree reconstruction requires a minimum sequence length, estimated at ∼800 base-pairs for HIV (18). The approach taken by COMET, in contrast, accurately identifies the closest matching reference type within a sliding window of 100 base-pairs. A caveat, however, is in order. COMET bases its classification of query sequences solely on the closest matches stored in COMET's internal model. Generalizing the approach to classify other viral families or applications is dependent on the availability of sufficient training sequences for those applications. Additionally, the parameters used to determine the closest match will need to be empirically tuned to the new species. This was the case for Hepatitis C, and the version of the COMET algorithm prepared for this virus has been referenced as a valid subtyping tool in the latest update to the consensus HCV classification (37). Further extension to other virus families or other microbial typing, however, will have to contend with limits imposed by reference sequence availability. Phylogenetic methods may better handle limited availability of training sequences by leveraging information in the tree structure.
USEARCH's speed comes from dispensing with a full alignment. It only attempts alignment with the best matching sequences in its database and terminates alignments that are going poorly (35). This helps explain the variation in its sensitivity on simulated versus clinical data. Simulated query sequences were noisy versions of sequences in USEARCH's internal database. The author of USEARCH reports that its speed is strongly dependent on the data analyzed and does not report asymptotic behavior (35). COMET's worst runtime requirement, however, can be quantified as O(nc) where n is the length of the input sequence and c is the number of categories trained. This allows analytical comparison to tools that require phylogenetic analysis. Creating a single tree even via neighbor-joining takes O(n2c3) operations, in addition to the cost of measuring the required sequence similarities. COMET's speed advantage comes at a cost in memory, with the strongest memory limit imposed by the maximal context length. Within this limit, the memory requirement increases linearly with the number of reference categories trained (see Supplement for details). The current implementation of COMET had a memory requirement of 487 MB for the N-ary tree. Thus the time-memory tradeoff of COMET should constitute a fading challenge for modern workstations.
COMET's speed allows its use in large-scale studies. Several large publically available data sets do not have complete subtype information or have not rigorously quality-controlled the recorded subtypes. Subtype assignments in the LANL HIV database (1) represent only the best estimate of the submitter at the time of submissions. Given the inherent difficulties in subtyping and that not every submitting author has accurate subtype determination as their primary research interest, it is likely that URFs and CRFs are under-reported. We observed that COMET, REGAv2 and SCUEAL all indicated that many of the sequences labeled as subtype A2 or H were more likely to be recombinant forms. Likewise, other large clinical cohorts that may not be publically accessible due to privacy concerns still need to be subtyped for epidemiological studies. It has been observed that up to 12% of UK sequences cannot be confidently assigned to any previously defined subtype (15). Successfully subtyping large data sets requires a tool that is both fast and sensitive to novel recombinant forms.
COMET's ability to handle short read lengths makes it highly applicable for analysis of next-generation sequencing outputs. This could indicate, for example, if a patient has a single or multiple infections, or allow better resolution of quasispecies diversity circulating in the patient, including detection of minority variants. Similar alignment-free methods have been successfully used for metagenomic assembly from next-generation sequencing (30). Interpolated Markov models with a variable-sized context-based approach have previously been applied for microbial genome annotation (38).
A close examination of the results from the synthetic variation data shows that the level of variation between the different referenced subtypes is not constant across the genome. COMET's sensitivity tends to be higher for the second half compared to the first half of the genome (see Supplementary Figure S2). This suggests that COMET could be further improved by using a variable-sized window when determining the log likelihood of the query sequence. A variable window might also be important for recombination detection, as the probability of recombination appears to be in part a function of genetic location, be it constant portions of the genome (39) or locations where recombination may give a selective advantage (40). Additional improvements may be possible from a more sophisticated approach to model discrimination than the simple likelihood ratio, for example Vuong's suggestion of first normalizing or otherwise adjusting the likelihoods before computing the ratio (41).
A number of the benchmarking results are based on synthetically generated variation or recombination. These synthetically altered sequences were derived assuming uniform random noise. Biological change, however, is not uniform, such as the just noted hot and cold regions for both variation and recombination (39,40). The use of uniform random noise was a deliberate choice designed to mask as much biological structure as possible, increasing the difficulty of correct classification. The additional testing on clinical sequences downloaded from LANL demonstrates that the method is equally applicable in clinical settings.
CONCLUSION
A context-based, rather than alignment-based, approach to HIV-1 viral subtype determination is substantially faster and more robust than previous methods. The COMET tool can be applied in both clinical and epidemiological studies.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The initial spark for COMET came from discussions with Francisco Azuaje from CRP-Santé Luxembourg on using compression for clustering.
FUNDING
EU Project CHAIN [HEALTH-F7-2009-223131 to G.L.]. Funding for open access charge: Luxembourg Ministry of Health [REC-LRTV-20081203].
Conflict of interest statement. CRP-Santé has entered into a licensing agreement with Advanced Biological Laboratories (ABL) S.A. for the COMET software. The software remains however freely available for non-commercial research use through a dedicated website: http://comet.retrovirology.lu.
REFERENCES
- 1.Robertson D.L., Anderson J.P., Bradac J.A., Carr J.K., Foley B., Funkhouser R.K., Gao F., Hahn B.H., Kalish M.L., Kuiken C., et al. HIV-1 nomenclature proposal. Science. 2000;288:55–56. doi: 10.1126/science.288.5463.55d. [DOI] [PubMed] [Google Scholar]
- 2.Kanki P.J., Hamel D.J., Sankalé J.L., Hsieh C., Thior I., Barin F., Woodcock S.A., Guèye-Ndiaye A., Zhang E., Montano M., et al. Human immunodeficiency virus type 1 subtypes differ in disease progression. J. Infect. Dis. 1999;179:68–73. doi: 10.1086/314557. [DOI] [PubMed] [Google Scholar]
- 3.Kaleebu P., French N., Mahe C., Yirrell D., Watera C., Lyagoba F., Nakiyingi J., Rutebemberwa A., Morgan D., Weber J., et al. Effect of human immunodeficiency virus (HIV) type 1 envelope subtypes A and D on disease progression in a large cohort of HIV-1-positive persons in Uganda. J. Infect. Dis. 2002;185:1244–1250. doi: 10.1086/340130. [DOI] [PubMed] [Google Scholar]
- 4.Kiwanuka N., Laeyendecker O., Robb M., Kigozi G., Arroyo M., McCutchan F., Eller L.A., Eller M., Makumbi F., Birx D., et al. Effect of human immunodeficiency virus Type 1 (HIV-1) subtype on disease progression in persons from Rakai, Uganda, with incident HIV-1 infection. J. Infect. Dis. 2008;197:707–713. doi: 10.1086/527416. [DOI] [PubMed] [Google Scholar]
- 5.Vasan A., Renjifo B., Hertzmark E., Chaplin B., Msamanga G., Essex M., Fawzi W., Hunter D. Different rates of disease progression of HIV type 1 infection in Tanzania based on infecting subtype. Clin. Infect. Dis. 2006;42:843–852. doi: 10.1086/499952. [DOI] [PubMed] [Google Scholar]
- 6.Laurent C., Bourgeois A., Faye M.A., Mougnutou R., Seydi M., Gueye M., Liégeois F., Kane C.T., Butel C., Mbuagbaw J., et al. No difference in clinical progression between patients infected with the predominant human immunodeficiency virus type 1 circulating recombinant form (CRF) 02_AG strain and patients not infected with CRF02_AG, in Western and West-Central Africa: a four-year prospective multicenter study. J. Infect. Dis. 2002;186:486–492. doi: 10.1086/341833. [DOI] [PubMed] [Google Scholar]
- 7.Atlas A., Granath F., Lindström A., Lidman K., Lindbäck S., Alaeus A. Impact of HIV type 1 genetic subtype on the outcome of antiretroviral therapy. AIDS Res. Hum. Retroviruses. 2005;21:221–227. doi: 10.1089/aid.2005.21.221. [DOI] [PubMed] [Google Scholar]
- 8.Geretti A.M., Harrison L., Green H., Sabin C., Hill T., Fearnhill E., Pillay D., Dunn D., UK Collaborative Group on HIV Drug Resistance Effect of HIV-1 subtype on virologic and immunologic response to starting highly active antiretroviral therapy. Clin. Infect. Dis. 2009;48:1296–1305. doi: 10.1086/598502. [DOI] [PubMed] [Google Scholar]
- 9.Pillay D., Walker A.S., Gibb D.M., de Rossi A., Kaye S., Ait-Khaled M., Muñoz-Fernandez M., Babiker A. Impact of human immunodeficiency virus type 1 subtypes on virologic response and emergence of drug resistance among children in the Paediatric European Network for Treatment of AIDS (PENTA) 5 trial. J. Infect. Dis. 2002;186:617–625. doi: 10.1086/342680. [DOI] [PubMed] [Google Scholar]
- 10.Theys K., Vercauteren J., Snoeck J., Zazzi M., Camacho R.J., Torti C., Schülter E., Clotet B., Sönnerborg A., De Luca A., et al. HIV-1 subtype is an independent predictor of reverse transcriptase mutation K65R in HIV-1 patients treated with combination antiretroviral therapy including tenofovir. Antimicrob. Agents Chemother. 2013;57:1053–1056. doi: 10.1128/AAC.01668-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taylor B.S., Sobieszczyk M.E., McCutchan F.E., Hammer S.M. The challenge of HIV-1 subtype diversity. N. Engl. J. Med. 2008;358:1590–1602. doi: 10.1056/NEJMra0706737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paraskevis D., Magiorkinis E., Magiorkinis G., Sypsa V., Paparizos V., Lazanas M., Gargalianos P., Antoniadou A., Panos G., Chrysos G., et al. Increasing prevalence of HIV-1 subtype A in Greece: estimating epidemic history and origin. J. Infect. Dis. 2007;196:1167–1176. doi: 10.1086/521677. [DOI] [PubMed] [Google Scholar]
- 13.Abecasis A.B., Wensing A. M.J., Paraskevis D., Vercauteren J., Theys K., Van de Vijver D. A. M.C., Albert J., Asjö B., Balotta C., Beshkov D., et al. HIV-1 subtype distribution and its demographic determinants in newly diagnosed patients in Europe suggest highly compartmentalized epidemics. Retrovirology. 2013;10:7. doi: 10.1186/1742-4690-10-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holguín A., López M., Soriano V. Reliability of rapid subtyping tools compared to that of phylogenetic analysis for characterization of human immunodeficiency virus type 1 non-B subtypes and recombinant forms. J. Clin. Microbiol. 2008;46:3896–3899. doi: 10.1128/JCM.00515-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gifford R., de Oliveira T., Rambaut A., Myers R.E., Gale C.V., Dunn D., Shafer R., Vandamme A.-M., Kellam P., Pillay D., et al. Assessment of automated genotyping protocols as tools for surveillance of HIV-1 genetic diversity. AIDS. 2006;20:1521–1529. doi: 10.1097/01.aids.0000237368.64488.ae. [DOI] [PubMed] [Google Scholar]
- 16.Zhang M., Foley B., Schultz A.-K., Macke J.P., Bulla I., Stanke M., Morgenstern B., Korber B., Leitner T. The role of recombination in the emergence of a complex and dynamic HIV epidemic. Retrovirology. 2010;7:25. doi: 10.1186/1742-4690-7-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hemelaar J., Gouws E., Ghys P.D., Osmanov S., WHO-UNAIDS Network for HIV Isolation and Characterisation Global trends in molecular epidemiology of HIV-1 during 2000-2007. AIDS. 2011;25:679–689. doi: 10.1097/QAD.0b013e328342ff93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Oliveira T., Deforche K., Cassol S., Salminen M., Paraskevis D., Seebregts C., Snoeck J., van Rensburg E.J., Wensing A. M.J., van de Vijver D.A., et al. An automated genotyping system for analysis of HIV-1 and other microbial sequences. Bioinformatics. 2005;21:3797–3800. doi: 10.1093/bioinformatics/bti607. [DOI] [PubMed] [Google Scholar]
- 19.Kosakovsky Pond S.L., Posada D., Stawiski E., Chappey C., Poon A. F.Y., Hughes G., Fearnhill E., Gravenor M.B., Leigh Brown A.J., Frost S.D.W. An evolutionary model-based algorithm for accurate phylogenetic breakpoint mapping and subtype prediction in HIV-1. PLoS Comput. Biol. 2009;5:e1000581. doi: 10.1371/journal.pcbi.1000581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Myers R.E., Gale C.V., Harrison A., Takeuchi Y., Kellam P. A statistical model for HIV-1 sequence classification using the subtype analyser (STAR) Bioinformatics. 2005;21:3535–3540. doi: 10.1093/bioinformatics/bti569. [DOI] [PubMed] [Google Scholar]
- 21.Schultz A.-K., Zhang M., Leitner T., Kuiken C., Korber B., Morgenstern B., Stanke M. A jumping profile Hidden Markov Model and applications to recombination sites in HIV and HCV genomes. BMC Bioinformatics. 2006;7:265. doi: 10.1186/1471-2105-7-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dwivedi S.K., Sengupta S. Classification of HIV-1 sequences using profile Hidden Markov Models. PLoS ONE. 2012;7:e36566. doi: 10.1371/journal.pone.0036566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Löytynoja A. Alignment methods: strategies, challenges, benchmarking, and comparative overview. Methods Mol. Biol. 2012;855:203–235. doi: 10.1007/978-1-61779-582-4_7. [DOI] [PubMed] [Google Scholar]
- 24.Lassmann T., Sonnhammer E.L.L. Automatic assessment of alignment quality. Nucleic Acids Res. 2005;33:7120–7128. doi: 10.1093/nar/gki1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abecasis A., Vandamme A.M., Lemey P. Sequence alignment in HIV computational analysis. In: Thomas Leitner T., Foley B., Hahn B., Marx P., McCutchan F., Mellors J., Wolinsky S., Korber B., editors. HIV Sequence Compendium 2006/2007. Theoretical Biology and Biophysics Group. Los Alamos, NM: Los Alamos National Laboratory; 2007. p. 216. LA-UR 07-4826. [Google Scholar]
- 26.Vinga S., Almeida J. Alignment-free sequence comparison-a review. Bioinformatics. 2003;19:513–523. doi: 10.1093/bioinformatics/btg005. [DOI] [PubMed] [Google Scholar]
- 27.Cilibrasi R., Vitanyi P. Clustering by compression. IEEE Trans. Inf. Theory. 2005;51:1523–1545. [Google Scholar]
- 28.Didier G., Debomy L., Pupin M., Zhang M., Grossmann A., Devauchelle C., Laprevotte I. Comparing sequences without using alignments: application to HIV/SIV subtyping. BMC Bioinformatics. 2007;8:1. doi: 10.1186/1471-2105-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corel E., Pitschi F., Laprevotte I., Grasseau G., Didier G., Devauchelle C. MS4–Multi-Scale Selector of Sequence Signatures: an alignment-free method for classification of biological sequences. BMC Bioinformatics. 2010;11:406. doi: 10.1186/1471-2105-11-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Narlikar L., Mehta N., Galande S., Arjunwadkar M. One size does not fit all: on how Markov model order dictates performance of genomic sequence analyses. Nucleic Acids Res. 2013;41:1416–1424. doi: 10.1093/nar/gks1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cleary J.G., Witten I. Data compression using adaptive coding and partial string matching. IEEE Trans. Commun. 1984;32:396–402. [Google Scholar]
- 32.Begleiter R., El-Yaniv R., Yona G. On prediction using variable order Markov models. J. Artif. Intell. Res. 2004;22:385–421. [Google Scholar]
- 33.Ferragina P., Giancarlo R., Greco V., Manzini G., Valiente G. Compression-based classification of biological sequences and structures via the Universal Similarity Metric: experimental assessment. BMC Bioinformatics. 2007;8:252. doi: 10.1186/1471-2105-8-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pineda-Peña A.-C., Faria N.R., Imbrechts S., Libin P., Abecasis A.B., Deforche K., Gómez-López A., Camacho R.J., de Oliveira T., Vandamme A.-M. Automated subtyping of HIV-1 genetic sequences for clinical and surveillance purposes: performance evaluation of the new REGA version 3 and seven other tools. Infect. Genet. Evol. 2013;19:337–348. doi: 10.1016/j.meegid.2013.04.032. [DOI] [PubMed] [Google Scholar]
- 35.Edgar R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 36.Rozanov M., Plikat U., Chappey C., Kochergin A., Tatusova T. A web-based genotyping resource for viral sequences. Nucleic Acids Res. 2004;32:W654–W659. doi: 10.1093/nar/gkh419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith D.B., Bukh J., Kuiken C., Muerhoff A.S., Rice C.M., Stapleton J.T., Simmonds P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment Web resource. Hepatology. 2014;59:318–327. doi: 10.1002/hep.26744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salzberg S.L., Delcher A.L., Kasif S., White O. Microbial gene identification using interpolated Markov models. Nucleic Acids Res. 1998;26:544–548. doi: 10.1093/nar/26.2.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baird H.A., Galetto R., Gao Y., Simon-Loriere E., Abreha M., Archer J., Fan J., Robertson D.L., Arts E.J., Negroni M. Sequence determinants of breakpoint location during HIV-1 intersubtype recombination. Nucleic Acids Res. 2006;34:5203–5216. doi: 10.1093/nar/gkl669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Archer J., Pinney J.W., Fan J., Simon-Loriere E., Arts E.J., Negroni M., Robertson D.L. Identifying the important HIV-1 recombination breakpoints. PLoS Comput. Biol. 2008;4:e1000178. doi: 10.1371/journal.pcbi.1000178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vuong Q.H. Likelihood ratio tests for model selection and non-nested hypotheses. Econometrica. 1989;57:307–333. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.