

Abstract
Purpose
Sunitinib is currently considered as the standard treatment for advanced renal cell carcinoma (RCC). We aimed to better understand the mechanisms of sunitinib action in kidney cancer treatment and in the development of acquired resistance.
Experimental Design
Gene expression profiles of RCC tumor endothelium in sunitinib-treated and -untreated patients were analyzed and verified by quantitative PCR and immunohistochemistry. The functional role of the target gene identified was investigated in RCC cell lines and primary cultures in vitro and in preclinical animal models in vivo.
Results
Altered expression of autotaxin (ATX), an extracellular lysophospholipase D, was detected in sunitinib-treated tumor vasculature of human RCC and in the tumor endothelial cells of RCC xenograft models when adapting to sunitinib. ATX and its catalytic product, lysophosphatidic acid (LPA), regulated the signaling pathways and cell motility of RCC in vitro. However, no marked in vitro effect of ATX-LPA signaling on endothelial cells was observed. Functional blockage of LPA receptor 1 (LPA1) using an LPA1 antagonist, Ki16425, or gene silencing of LPA1 in RCC cells attenuated LPA-mediated intracellular signaling and invasion responses in vitro. Ki16425 treatment also dampened RCC tumorigenesis in vivo. In addition, coadministration of Ki16425 with sunitinib prolonged the sensitivity of RCC to sunitinib in xenograft models, suggesting that ATX-LPA signaling in part mediates the acquired resistance against sunitinib in RCC.
Conclusions
Our results reveal that endothelial ATX acts through LPA signaling to promote renal tumorigenesis and is functionally involved in the acquired resistance of RCC to sunitinib.
Keywords: autotaxin, lysophosphatidic acid, sunitinib, angiogenesis
Introduction
The incidence of renal cell carcinoma (RCC) is arising throughout the world. It is estimated that 58,240 people in the US were diagnosed with kidney cancer in 2010, and 13,040 people died of the disease [1]. Although surgery may be curative for early-stage RCC, deaths from kidney cancer have not declined mainly because of recurrence and high mortality associated with metastatic disease [2]. Until recently, the only effective treatments for metastatic RCC have been high-dose interleukin-2 (IL-2) and interferon-α (IFN-α). However, the administration of these cytokines produced consistent responses only in a small percentage of patients with advanced RCC and had a limited effect on patient survival [3].
With advances in understanding RCC biology, the systemic management of RCC has improved over the past few years with FDA approval of seven agents targeted against the aberrant vascular endothelial growth factor (VEGF) or mammalian target of rapamycin (mTOR) pathways. Unfortunately, the vast majority of tumors eventually become refractory to these targeted therapies [4]. Sunitinib is a multitargeted tyrosine kinase inhibitor (TKI) that predominantly targets VEGF and is the first-line therapy for favorable- and intermediate-risk patients [5]. Resistance to sunitinib develops in most patients that is characterized by renewed angiogenesis after about one year of treatment, while a minority of patients have innate resistance and demonstrate no initial response [6]. Such drug resistance occurs through a variety of poorly understood mechanisms. Therefore, novel agents against distinct signaling pathways are needed to address the resistance mechanisms and to improve outcomes in RCC.
Autotaxin (ATX), encoded by the ecto-nucleotide pyrophosphatase/phosphodiesterase 2 (ENPP2) gene, was identified as a tumor motility-stimulating protein [7] and found to play a role in angiogenesis [8, 9]. ATX is an ecto-enzyme that hydrolyzes lysophosphatidylcholine (LPC), which is abundant in the circulation, to lysophosphatidic acid (LPA) [10]. ATX-deficient mice die at embryonic day 9.5 (E9.5) with profound vascular defects in the yolk sac and embryo [9]. Furthermore, ATX-deficient embryos exhibit allantois malformation, neural tube defects, and asymmetric headfolds. The bioactivities of ATX can be primarily explained by the production of LPA, a bioactive lipid mediator. LPA acts through specific G protein-coupled receptors (GPCRs) to promote cellular proliferation, migration, and survival [11]. Cumulative evidence points to a role of LPA in cancer progression [10, 12]. Aberrant expression of LPA receptors is detected in various human malignancies, including ovarian, colorectal, prostate, and gastric cancers. More importantly, a causal relationship between LPA signaling and tumor progression is emerging from studies using mouse models [13–17]. In addition to the catalytic activity of ATX that generates LPA, ATX was found to regulate cellular behaviors through non-enzymatic functions on platelets, lymphocytes, and oligodendrocytes [18–20]. The role, if any, of the ATX-LPA axis in RCC biology and drug resistance has not been elucidated. In this study, we aim to address the exact mechanism of sunitinib action against RCC through targeting the tumor endothelium. We show that ATX expression is upregulated in the tumor vasculature of human RCC and in the tumor endothelial cells of mouse models with the acquired resistance phenotype. Endothelial ATX acts through LPA signaling to promote renal tumorigenesis and is functionally involved in the acquired resistance of RCC to sunitinib.
Materials and Methods
Isolation of endothelial cells from fresh tissues
Isolation of endothelial cells from tissues was modified from a previously published protocol [21]. Briefly, the tissues were mechanically disrupted and digested using collagenase (Sigma-Aldrich), dispase (Sigma-Aldrich) and DNAse (Sigma-Aldrich), followed by filtration with 100-μm cell strainers (BD Biosciences) to create a single-cell suspension. Subsequently, human and mouse endothelial cells were selected from the single-cell suspension of human specimens and xenograft tumors, respectively, using anti-CD31-coated magnetic beads (Invitrogen).
Immunohistochemistry
Immunohistochemical analyses were performed on formalin-fixed, paraffin-embedded tissue sections using VECTASTAIN Universal Elite ABC Kit (Vector Labs). High pH Antigen Unmasking Solution (Vector Labs) was used to heat tissue sections for antigen retrieval and the Avidin/Biotin Blocking Kit (Vector Labs) was utilized to avoid non-specific background before the 1st antibody hybridization. Primary antibodies against von Willebrand Factor (MAB3442, Millipore) and ATX (ab77104, Abcam) were used to assess protein expression on human specimens. The endothelial ATX in normal kidney vasculature and RCC tumor vessels was evaluated semiquantitatively by measuring the mean digital density of immunohistochemically labeled ATX with the Pro Plus 4.5.1 image analysis program (Media Cybernetic, MD). Data from at least three vWF-positive, lumen-containing vessels of RCC tumors and their corresponding normal counterparts from four patients were assessed and averaged to give mean ATX expression. Cancer proliferation of xenograft tumors in vivo was calculated as the ratio of the number of nuclei immunostained for Ki67 using a mouse monoclonal antibody (M7240, Dako) to the total number of nuclei per field and expressed as the percentage of Ki67-positive nuclei. For determining the microvascular density, a rat anti-mouse CD34 antibody (ab8158, Abcam) was used to stain the vascular structures of xenograft tumors. Three areas with the greatest amount of neovascularization from each tumor section were selected and the microvessels in these areas were counted under 10× objective lens. Any brown-stained endothelial cell or endothelial cell cluster that could be clearly separated from the adjacent structures, with or without a vessel lumen, was considered as a single, countable microvessel. The numbers of microvessels in the three areas were averaged to give mean microvascular density.
Impedance-based cellular assay
Real-time impedance assays were performed with the xCELLigence system (Roche). Briefly, 50 μl of selective medium (RPMI-1640 for RCC; M199 for HUVEC) was added to wells of E-Plates (Roche) to obtain background readings, and then 150 μl of cell suspension with 10,000 cells was added. The E-Plate containing the cells was placed on the device station (in the 37°C CO2 incubator). In each tested well, viable adherent cells attached to the surface of the sensor electrodes will lead to an increase in impedance that is read by the instrument. Impedance was recorded every 5 minutes, and the relative change in impedance is given in a unitless parameter termed cell index [CI = (Zi − Z0)/15 Ω; Zi – impedance at a given time point of the experiment; Z0 – impedance at the start of the experiment (background value)]. The value of CI is directly proportional to the number and size (morphology) of attached cells. The cells were allowed to attach and spread to reach a stable baseline before treatments were applied.
Migration assay
Migration assays were performed in a modified Boyden chamber using 24-well polycarbonate filters (Corning Inc., NY) with 8 μm pores. The filters were coated with 50 μg/ml of collagen type 1 (BD Biosciences, CA) in 0.2 N acetic acid overnight and air dried. The bottom wells were filled with conditioned media containing approximately 1 ug/ml ATX, T210A, or ATX plus 200 ng/ml of pertussis toxin (PTX) (Sigma-Aldrich). RCC primary culture cells and HUVECs were suspended in RPMI1640 and M199, respectively. 20,000 cells in 100 μl of medium were added to each well in the upper chamber. To test the inhibitory activity of PTX on cell migration, cells were pre-incubated with 200 ng/ml of PTX for 10 minutes before adding to the upper chambers. The assembled chemotaxis chambers were incubated for 4 hours at 37°C in a 5% CO2 humidified incubator and then non-migrated cells on the upper surface of the filter were mechanically removed. The cells that migrate to the lower surface of the filter were fixed and stained with 0.1% toluidine blue (Sigma-Aldrich) in 30% methanol. Cell migration was performed in quadruplicate and quantified under light microscopy by counting cells in three randomly chosen high-power fields.
Invasion assay
Type 1 collagen matrices (BD Biosciences, CA) were prepared at 2.5 mg/ml with 20 μM LPC, 1 μM LPA, or 50 μM S1P (Avanti Polar Lipids, AL) as described (Bayless 2003 BBRC). Gels (25 μl) were added to half-area (A/2) 96-well plates (Costar) and allowed to equilibrate for 45 minutes at 37 °C with 5% CO2. Cells were suspended at a density of 30,000 cells per 50 μl in media (MEM for RCC; M199 for HUVEC) and allowed to attach for 30 minutes before adding growth media (50 μl/well) containing a 1:250 dilution of RSII (Transferrin, oleic acid, BSA, and insulin) (Sigma-Aldrich), 50 μg/ml ascorbic acid (Sigma-Aldrich), and 40 ng/ml VEGF and bFGF (R&D Systems). RCC and HUVEC were allowed to invade for 48 and 24 hours, respectively. For imaging and quantifying the average numbers of invading cells, conditioned media were removed, and invasion cultures were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) in PBS for 10 minutes and stained with 0.1% toluidine blue. For visualization of invasion responses, photographs of invading cells were taken from the side view using an Olympus CKX41 microscope equipped with a Q color 3 Olympus camera. For quantification of invading cells, eyepieces mounted with a vertical reticle displaying a 10×10 grid, covering an area of 6.25, 1, and 0.25 mm2 at 4×, 10×, and 20×, respectively, were used for quantifying average numbers of invading cells per standardized field. For each data set, four separate fields from each treatment were recorded and averaged.
Animal studies
All animal procedures were approved by the MDACC Institutional Animal Care and Use Committee. Sunitinib-resistant xenograft models were established as previously described [22]. Briefly, 6-week-old female BALB/c nude mice were given s.c. injections of 5 ×106 786-O or UMRC3 in the right flank. Tumor size was measured twice per week using a digital caliper. Treatment began when the average tumor volume reached 100 to 200 mm3. For Ki16425 (Selleckchem) treatment, Ki16425 was first prepared in DMSO at a concentration of 100 mg/ml and then diluted in PBS at final concentration of 5 mg/ml. Mice were treated with the vehicle control (PBS) and Ki16425 (20 mg/kg in PBS) daily by s.c. injections. Sunitinib (40 mg/kg in water) was administered once daily by oral gavage as previously described [22]. For combination group, xenografts were treated with reduced dosages of Ki16425 (10 mg/kg in PBS) daily by s.c. injections while reduced dosages of sunitinib (20 mg/kg in water) were delivered everyday by oral gavage. Serum samples were collected after the course of each treatment groups. Tumors were removed, cleaned from adjacent tissues, fixed in 4% paraformaldehyde, and embedded in paraffin for sections.
Cytokine analysis
Concentrations of bFGF, IL-8, GROα, and MCP-1 in sera and conditioned media were determined by using the Bio-Plex Pro assays (Bio-Rad Laboratories) according to the manufacturer’s instructions.
Statistical Analysis
Student’s t-test analyses were carried out on the raw or normalized data by using the SPSS software (Version 10·0). Data of multiple observations were presented as the mean ± S.D. or as representative results, unless otherwise stated. p < 0.05 was considered statistically significant.
Other materials and methods are in supplementary information for this article at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/)
Results
Altered ATX expression in sunitinib-treated endothelial cells of RCC tumor vessels
In order to search endothelial markers that potentially regulate the angiogenesis and progression of RCC, we undertook a microarray screen, in which the gene expression profiles of endothelial cells isolated from RCC tumors in sunitinib-treated and -untreated patients were analyzed. The expression levels of a panel of known endothelial markers were examined to verify the endothelial isolation (Supplemental Figure 1A). A cohort of endothelial genes was differentially expressed between sunitinib-treated and -untreated RCC endothelium, one of which is autotaxin (ATX/ENPP2), an ecto-enzyme with lysophospholipase D activity (Supplemental Figure 1B). While sunitinib acts on RCC tumor progression and angiogenesis, endothelial ATX expression is downregulated, suggesting a potential role for ATX in RCC tumorigenesis and response to anti-angiogenic treatment. To verify the finding from microarray screening, we isolated endothelial cells from an additional set of 13 human RCC specimens and their corresponding normal kidney tissues using anti-CD31-coated magnetic beads, and utilized RT-PCR and quantitative PCR to assess the expression of ENPP2 mRNA (Figure 1A & Supplemental Table 1). Using quantitative PCR, all the samples examined showed higher levels of ENPP2 mRNA in endothelial cells from RCC than from the corresponding normal kidneys except those treated with sunitinib (Supplemental Table 1). However, ENPP2 expression was very low or undetectable in bead-unbound cells from either tumor or normal tissues (data not shown). ATX is known as a secreted protein and the level of serum ATX has been reported to be elevated in many pathological conditions [23, 24]. We observed that the levels of serum ATX in RCC patients were higher than those in healthy subjects (Figure 1B). In addition, immunohistochemical analysis revealed that ATX protein was highly expressed in tumor vessels but not in normal kidney vasculature (Figure 1C and D). These results indicate that ATX is expressed in endothelial cells of tumor vessels but not in tumor cells or in endothelial cells of normal renal capillaries. This endothelial ATX expression in RCC is downregulated with sunitinib treatment.
Figure 1.
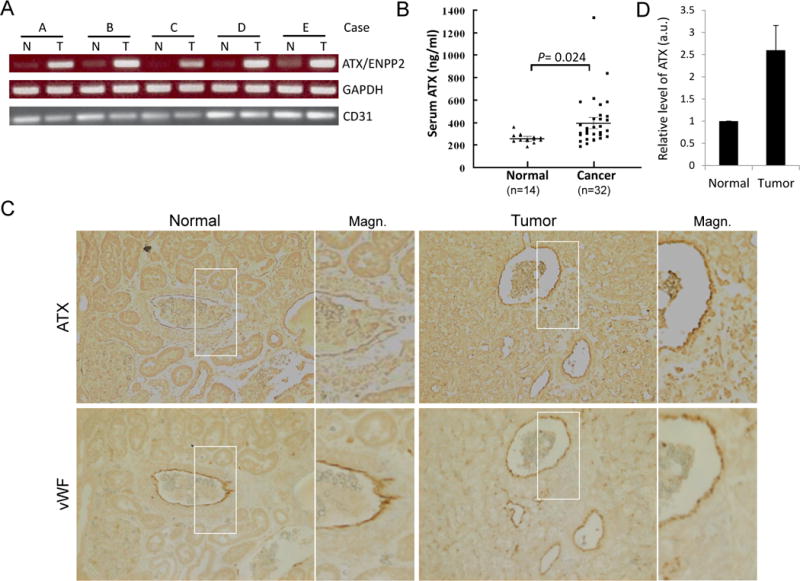
Elevated ATX expression in endothelial cells (ECs) of tumor vessels, but not in tumor cells or in ECs of the normal kidney vasculature. A, expression of ATX (ENPP2) mRNA in endothelial cells of RCC and corresponding normal kidneys. ECs from human RCC specimens (T) and their normal counterparts (N) were isolated from a single-cell suspension using anti-CD31-coated magnetic beads. Total RNA was prepared and RT-PCR was performed for ENPP2, GAPDH, and CD31. B, serum ATX levels of RCC patients (Cancer) and healthy subjects (Normal) were measured by the enzyme-linked immunosorbent assay (ELISA). The difference in serum ATX levels between two groups was statistically significant (by Student’s t-test). C, immunohistochemical staining of RCC and normal kidney tissues. Human RCC specimens and their normal counterparts were stained using specific antibodies against autotaxin (ATX) and von Willebrand factor (vWF). A total of six samples were examined and a photograph of one representative case is shown. The right panels are magnified images of the areas boxed in the left panels (Magn.). D, densitometric analyses of endothelial ATX expression in normal and tumor kidney vasculature. The levels of immunohistochemically labeled ATX in vWF-positive vessels of RCC (Tumor) and their corresponding normal counterparts (Normal) from each of four patients were measured and expressed as means ± S.D. in arbitrary units (a.u.).
Distinct effects of secreted ATX on RCC primary cultures and endothelial cells
ATX is known as a potent stimulator of cell invasion and tumor progression. In addition to its lysophospholipase D activity that generates LPA, ATX was found to interact with specific cell-surface receptors to regulate cell responses in platelets and lymphocytes as well as to facilitate the adhesive state and cellular remodeling in oligodendrocytes [18–20]. To examine the effects of ATX on RCC cells and endothelial cells, conditioned media of HEK293 cells overexpressing ATX, T210A (a catalytically inactive mutant of ATX), and GFP (as a control) were prepared for various assays. We found that partially-purified conditioned media containing ATX activated Akt and ERK in RCC primary cells, but not in HUVECs (Figure 2A). Moreover, conditioned media with ATX mutant, T210A, failed to induce Akt and ERK activation in RCC cells or HUVECs. We further conducted an impedance-based assay that covers a wide range of applications in live cell experiments, including monitoring the activation of endogenous receptors, morphological change, cell adhesion, and cell proliferation [25, 26]. We observed a significant change in electrical impedance following exposure of RCC primary cells to conditioned media containing ATX (Figure 2B) as an indication of alternations in cell adhesion or morphology likely due to regulated reorganization of the cytoskeleton [25]. This alternation was not seen in HUVECs. In addition, RCC cells exhibited an enhancement of migratory response to conditioned media with ATX, whereas HUVECs did not (Figure 2C). This increase in RCC mobility was abolished in the presence of pertussis toxin (PTX), suggesting that the ATX-induced RCC motility is mediated by GPCRs. Furthermore, neither RCC nor HUVECs migrated to conditioned media containing T210A when compared with the control media (GFP). Similar results were obtained by using additional lines of RCC primary cultures and human dermal microvascular endothelial cells (HDMECs) (data not shown). These results indicate that ATX regulates the intracellular signaling and cell motility of RCC through its enzymatic activity. Surprisingly, no marked in vitro effect of ATX on endothelial cells was observed.
Figure 2.
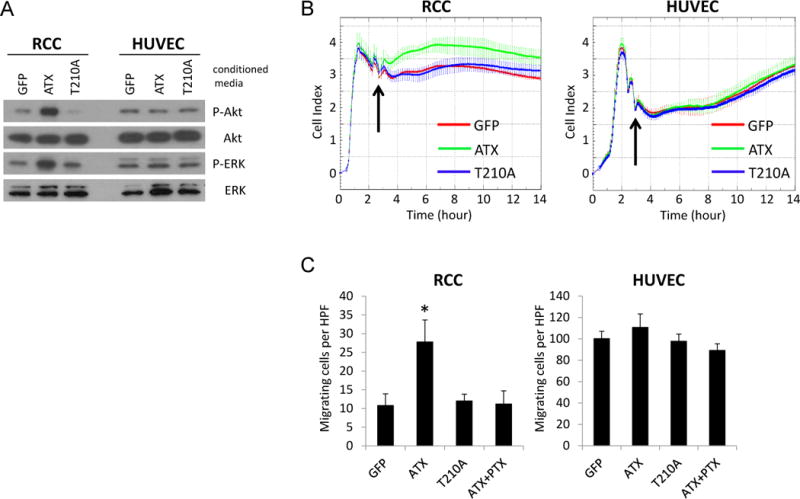
Effects of ATX and its catalytically inactive mutant (T210A) on RCC and endothelial cells. A, HRC-223 (RCC) and HUVECs were serum-starved for 4 hours and treated with conditioned media containing ATX or its mutant for 30 minutes. Cell lysates were collected and analyzed by immunoblotting using the indicated antibodies. B, HRC-223 and HUVECs were seeded on E-Plates at 10,000 cells per well and continuously monitored for impedance using The xCELLigence System. Arrowhead indicates the time point at which conditioned media from HEK293 cells transfected with vectors encoding the indicated proteins were added. C, HRC-223 and HUVECs were assessed for their chemotactic response to conditioned media containing ATX, T210A, or ATX plus pertussis toxin (PTX) using a modified Boyden chamber assay. The assays were performed in triplicate and quantified under light microscopy by counting three randomly chosen HPFs for each replicate (Student’s t-test; *, P<0.01).
RCC, but not endothelial cells, responds to LPA
We next examined the responses of RCC and HUVECS to the substrate and product of ATX. LPC is abundantly present in plasma and serum (at >100 μM), yet LPA levels in plasma or freshly-isolated blood are very low [27]. The physiological/pathological concentrations of LPA will largely depend on the local availability of LPC and the levels of ATX expressed within nearby tissues. Similar with the effects of ATX on RCC and HUVECs, LPA significantly activated Akt and ERK and augmented cell proliferation in RCC, but not in HUVECs (Figure 3A, B and Supplemental Figure 2). ATX substrate, LPC, had no or slight effect on the activation of Akt and ERK or on cell proliferation in RCC. Unexpectedly, LPC dramatically triggered Akt and ERK activation but not cell proliferation in HUVECs, while VEGF served as a positive activator of endothelial proliferation. In addition, we utilized a three-dimensional culture system to study the effects of LPA on RCC cell invasion [28]. Various RCC cell lines and primary cultures were placed on the surface of collagen matrices and allowed to invade in response to LPA. We found that most RCC lines tested were stimulated by LPA to invade, as few lines (Caki-1, ACHN, and MDA-RCC-M62) were naturally unable to penetrate into three-dimensional collagen matrices (Figure 3C). We did not observe robust endothelial invasion induced by either LPA or LPC. However, another bioactive phospholipid, sphingosine-1-phosphate, efficiently elicited the invasion response of endothelial cells. These data indicate that LPA is a modulator of processes that contribute to RCC progression, such as cell proliferation and invasion, but argue for a direct role for LPA in tumor angiogenesis.
Figure 3.
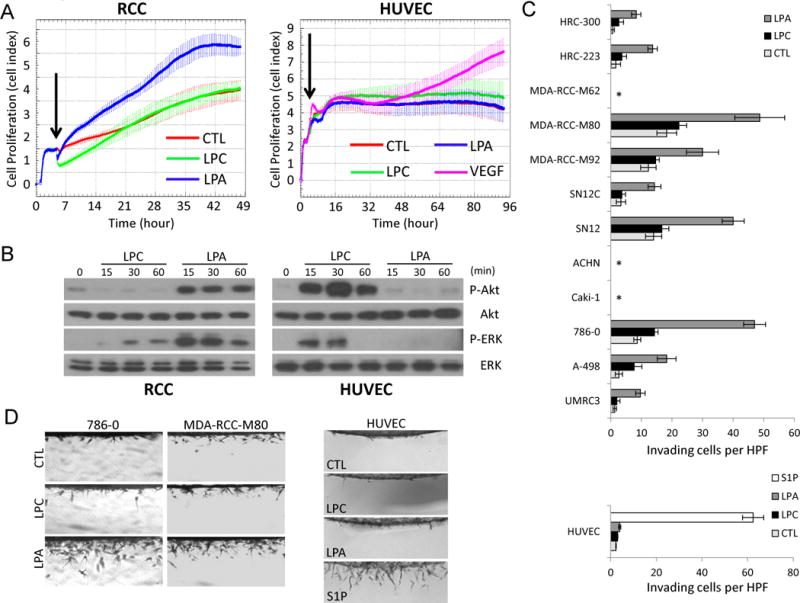
Effects of LPA on cell proliferation, signaling and invasion of RCC and endothelial cells. A, HRC-223 (RCC) and HUVECs were seeded on E-Plates at 10,000 cells per well and continuously monitored for cell proliferation using The xCELLigence System. Arrowhead indicates the time point at which medium only (CTL), 20 μM LPC, 1 μM LPA, or 40 ng/ml VEGF was added. B, HRC-223 and HUVECs were serum-starved for 4 hours and treated with 20 μM LPC or 1 μM LPA for the indicated time points. Cell lysates were collected and analyzed by immunoblotting using the indicated antibodies. C-D, cell lines and primary cultures of RCC and HUVECs were induced to invade into 3D collagen matrices (2.5 mg/ml) under serum-free conditions in the absence (CTL) or the presence of 20 μM LPC, 1 μM LPA, or 1 μM S1P. After 48 hours, cultures were fixed, stained, quantitated for cell invasion (C), and photographed (D). Data are presented as mean numbers of invading cells per standardized field ± S.D. (n=4 fields) (*, no invasion). HUVECs were fixed and analyzed 24 hours after seeding.
LPA1 mediates LPA-induced cell signaling and invasion in RCC
LPA has been shown to bind and signal through a group of GPCRs [11]. Therefore, we next characterized which receptors were involved in LPA-induced responses in RCC. To address this, we have examined the spectrum of LPA receptors (LPARs) expressed on RCC and determined that RCC cell lines and primary cultures preferentially express LPA1 and LPA2 (Supplemental Table 2). We further tested various LPA receptor antagonists, such as Ki16425, TDPA and BrP-LPA [29–32], and found that only Ki16425, a selective LPA1 and LPA3 antagonist, effectively attenuated LPA-induced cell signaling and invasion in 786-O cells (Figure 4A–C). Similar results were observed in UMRC3 cells (data not shown). Together, the data from the expression profile of LPARs and the use of LPAR inhibitors indicate that LPA1 mediates LPA-stimulated responses in RCC. Moreover, we specifically knocked down the expression of LPA1 in 786-O cells by using recombinant lentiviruses that express shRNAs against LPA1 and non-target shRNA control to verify the result from the use of Ki16425. Silencing of LPA1 blocked LPA-induced cell invasion and activation of Akt and ERK in 786-O cells (Figure 4D–F), supporting the finding that LPA-induced cell responses in RCC are mediated by LPA1.
Figure 4.
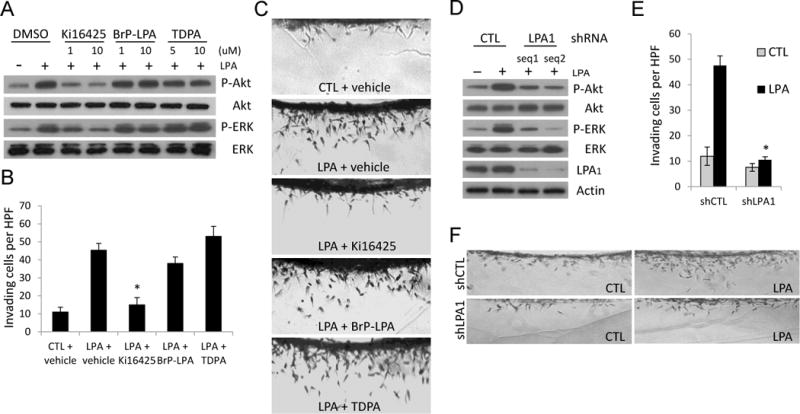
LPA1 regulates LPA-induced cell signaling and invasion in RCC. A, 786-O (RCC) cells were serum-starved overnight and pre-treated with the indicated concentrations of LPA antagonists for 10 minutes before the incubation with or without 1 μM LPA for 30 minutes. Cell lysates were collected and analyzed by immunoblotting using the indicated antibodies. B–C, 786-O cells were induced to invade into 3D collagen matrices (2.5 mg/ml) in the presence or absence of 1 μM LPA combined with the indicated LPA antagonist. After 48 hours, cultures were fixed, stained, quantitated for cell invasion (B), and photographed (C). Data are presented as mean numbers of invading cells per standardized field ± S.D. (n=4 fields; Student’s t-test; *, P<0.001 compared with LPA + vehicle). D, 786-O cells were transduced with lentiviruses expressing the indicated shRNAs for 3 days and treated with or without 1 μM LPA for 30 minutes. Cell lysates were collected and analyzed by immunoblotting using the indicated antibodies. E–F, 786-O cells were transduced with lentiviruses expressing the indicated shRNAs and selected with puromycin prior to seeding on the surface of collagen matrices. Cultures with or without 1 μM LPA were fixed at 48 hours, stained, quantitated for cell invasion (E), and photographed (F). (Student’s t-test; *, P<0.001 compared with shCTL + LPA).
LPA1 regulates RCC tumorigenesis through the production of specific cytokines
LPA signaling regulates RCC proliferation and invasion in vitro (Figure 3). We next investigated whether the ATX-LPA axis plays a role in RCC tumorigenesis through LPA1. To assess the role of LPA1 in RCC tumorigenesis, subcutaneous xenograft models using RCC cell lines (786-O and UMRC3) were generated and treated with Ki16425. Previously, the potential off-target effect of Ki16425 has been addressed, and the in vivo efficacy of Ki16425 in blocking the action of the LPA1 receptor subtype has been demonstrated [17]. Pharmacological blockade of LPA1 markedly inhibited RCC tumor growth for both lines (Figure 5A). In addition, the suppression of tumor growth by Ki16425 was associated with reduced tumor proliferation and angiogenesis as judged by Ki67 nuclear antigen and CD34 staining, respectively (Figure 5B–D). From our in vitro studies, ATX-LPA signaling failed to elicit substantial effects on endothelial cells directly. We hypothesized that the decrease in vascularization of Ki16425-treated tumors may be mediated by tumor-derived angiogenic factors. To test this hypothesis, we searched for LPA-upregulated angiogenic factors in RCC cells using quantitative PCR and further detected for the changes in secreted angiogenic factors between the serum from Ki16425-treated and untreated xenograft models. Among the angiogenic factors examined, LPA upregulated the mRNA expression of interleukin-8 (IL-8), growth-related oncogene α (GROα), macrophage chemoattactant protein-1 (MCP-1), and colony stimulating factor 2 (CSF2) in RCC (Supplemental Figure 3).
Figure 5.
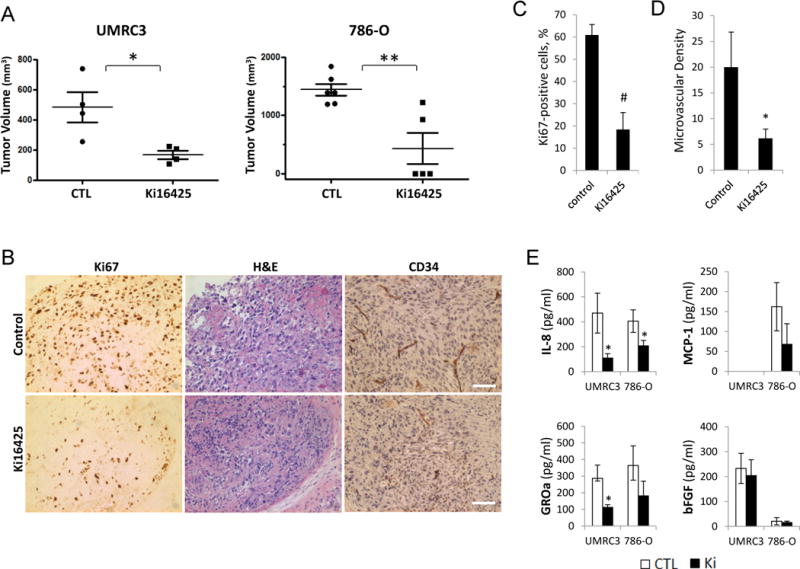
Effects of Ki16425 on tumor growth and angiogenesis in mouse models of RCC. A, BALB/c nude mice were given s.c. injections of RCC cells (UMRC3 and 786-O) in the right flank, randomized into two groups, and administered either PBS (control, CTL) or 20 mg/kg Ki16425 given subcutaneously once daily. Treatment began when the average tumor volume reached approximately 100 mm3. Mice were killed on day 48 and day 37 after treatment for UMRC3 and 786-O, respectively. Tumor volume was assessed and expressed as the mean ± S.D. (Student’s t-test; *, P<0.05; **, P<0.01). B–D, UMRC3 tumors were collected on the final day of study, fixed, and embedded in paraffin. Tissue sections were subjected to immunohistochemical analysis by using monoclonal antibodies against the nuclear Ki67 antigen and CD34 (B). The Ki67 positivity (C) and microvascular density (D) were calculated from four independent tumor sections per group and expressed as the mean ± S.D. (Student’s t-test; *, P<0.05; #, P<0.001); scale bars, 50 μm. E, production of specific cytokines secreted by xenograft tumors in animals treated with and without Ki16425. The levels of cytokine release in serum of untreated (CTL) and Ki16425-treated (Ki) mice were measured. Data are presented as mean ± S.D. (Student’s t-test; *, P<0.05). Results are representative of two independent experiments.
We further examined the serum level of tumor-secreted IL-8, GROα, MCP-1, and basic fibroblast growth factor (bFGF) in xenograft models treated with and without Ki16425. Despite the fact that the secretion of each angiogenic factor from RCC tumor cells can be regulated by a variety of mechanisms in vivo, a significant decrease in secreted human IL-8 and GROα was observed in both Ki16425-treated RCC models as compared with untreated controls (Figure 5E). Tumor-derived MCP-1 was slightly lower in 786-O xenografts with LPA1 blockade by Ki16425 but was undetectable in UMRC3 xenografts. In contrast, bFGF was barely detectable in 786-O xenograft-bearing mice and its level remained unchanged in the UMRC3 xenograft model. Altogether, these results suggest that LPA1 expressed by tumor cells mediates LPA action that leads to RCC tumorigenesis.
ATX-LPA axis is functionally associated with the development of acquired resistance to targeted therapies
Our results showed that LPA signaling promotes RCC tumor growth, invasion, and angiogenesis, all of which are phenomena that commonly accompany acquired tumor resistance to anti-angiogenic therapies [33]. We next examined whether the ATX-LPA axis is involved in acquired resistance of RCC to sunitinib. To explore the potential association of ATX-LPA signaling with drug resistance in RCC, we have established mouse xenograft models of RCC with acquired resistance to sunitinib, which mimics the clinical resistance phenotype (Figure 6A and Supplemental Figure 4) [22]. For our purposes, the treatment of xenograft tumors can be split into two sequential phases, sensitive and resistant phase. After treatment, the tumor stopped growing and then began to actively shrink, and the angiogenesis declined. This period of active shrinkage is the sensitive phase. With time, the tumor began to regrow, and the neovascularization is reactivated. During this regrowth, further treatment with sunitinib is ineffective. This period of active regrowth is referred to as the resistant phase.
Figure 6.
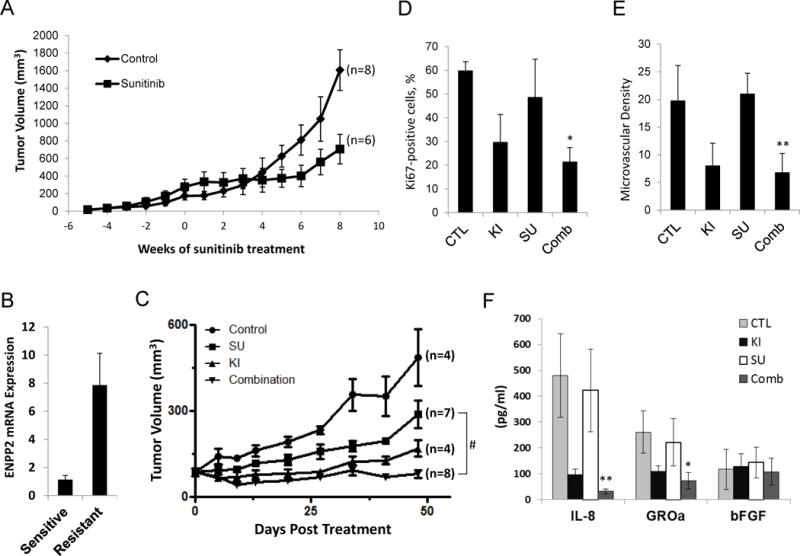
Effects of Ki16425 treatment on RCC xenograft models with acquired resistance to sunitinib. A, establishment of RCC xenograft models with the acquired resistance to sunitinib. BALB/c nude mice were given s.c. injections of UMRC3 cells in both flanks. Mice were treated daily with sunitinib (40 mg/kg per day) by oral gavage, and tumor size was measured twice per week. Tumor growth curve was shown as averaged tumor size. The treatment began at time point 0. The individual tumor growth was shown in Supplemental Figure 4. B, endothelial cells from xenograft tumors were isolated using anti-CD31-coated magnetic beads when tumors reached the sensitive or resistant phase. Total RNA was prepared and real-time RT-PCR was performed to assess the expression level of ENPP2. Data are expressed as mean ± S.D. of three replicates and are representative of two separate experiments. C, UMRC3 xenograft models were generated, randomized into four groups and given water (Control), sunitinib (SU) (40 mg/kg per day) by oral gavage, Ki16425 (KI) (20 mg/kg per day) subcutaneously, and a combination of sunitinib (20 mg/kg) plus Ki16425 (10 mg/kg). Tumor volume was calculated at the indicated time point and expressed as the mean ± S.D. (Student’s t-test; #, P<0.001 on day 48 after treatment). D–E, tumors were fixed and embedded in paraffin after 48 days of treatment. Tissue sections were subjected to immunohistochemical analysis by using monoclonal antibodies against the nuclear Ki67 antigen and CD34. The Ki67 positivity (D) and microvascular density (E) were calculated from four independent tumor sections per group and expressed as the mean ± S.D. (Student’s t-test; *, P<0.05; **, P<0.01 compared with sunitinib alone). F, secretion of tumor-derived cytokines in serum from mice of each treatment arm was measured. Data are presented as mean ± S.D. (Student’s t-test; *, P<0.05; **, P<0.01 compared with sunitinib alone). Results are representative of two independent experiments.
We have isolated endothelial cells from implanted tumors at sensitive and resistant stages and observed elevated expression of ENPP2 in endothelial cells of resistant tumors compared with sensitive tumors (Figure 6B). To investigate whether replenished ATX-LPA signaling contributes to tumor regrowth while the tumors adapt to sunitinib, we cotreated UMRC3 xenografts with sunitinib and Ki16425. To reduce the potential toxicity, a combination treatment of half doses of sunitinib and Ki16425 was used to compare with sunitinib treatment alone. We found that the combination treatment showed more effective and persistent effects on the inhibition of tumor growth than treatment with individual agent alone (Figure 6C). More importantly, the addition of Ki16415 to sunitinib treatment, compared with sunitinib treatment alone, resulted in a trend of decreased tumor cell proliferation and microvascular density (Figure 6D and E), indicating that functional blockage of LPA1 prevented the restoration of tumor growth and angiogenesis in RCC xenografts adapting to sunitinib treatment. We also noted that the serum levels of tumor-derived IL-8 and GROα were lower in UMRC3 xenografts treated with both sunitinib and Ki16425 than in animals bearing sunitinib-resistant tumors, while secretion of tumor-derived bFGF appeared unrelated to tumor growth in vivo (Figure 6F). Intriguingly, IL-8 has been shown to mediate sunitinib resistance in mouse models of RCC using various cell lines [34]. We found that IL-8 is capable of bypassing sunitinib to induce endothelial sprouting in an in vitro angiogenesis model (Supplemental Figure 5). Our data suggest that coadministration of Ki16425 with sunitinib may prevent or delay the development of acquired resistance against sunitinib in RCC.
Discussion
RCC, a hypervascular neoplasm thought to be highly dependent on aberrant angiogenesis, is one of the most common non-cutaneous adult malignancies in the United States. Interactions between RCC and the tumor-associated endothelium may play an important role both in tumorigenesis and treatment. Sunitinib recently became the standard care for treatment of advanced renal cancer and is reported to act primarily on the tumor endothelium rather than on tumor cells [35]. To better understand the mechanisms of sunitinib action against RCC, we focused on the change in gene expression profile of tumor endothelium instead of the whole RCC tumor in patients administered with or without sunitinib. A set of endothelial genes was identified between sunitinib-treated and -untreated RCC endothelium, among which elevated expression of RGS5 in tumor vasculature of RCC has been previously reported [36]. In this study, we demonstrated that ATX is differentially expressed in sunitinib-treated tumor vasculature of human RCC and in the tumor endothelial cells of mouse models with the acquired resistance to Sunitinib. We further showed that endothelial ATX acts through LPA signaling to promote renal tumorigenesis and is functionally involved in the acquired resistance of RCC to sunitinib. Our results implicate the ATX-LPA signaling axis as an important element not only for RCC pathogenesis, but also for response to targeted therapy.
Regulation of ATX expression is controlled by various growth factors, cytokines, and oncogenes, which appears to differ among cell types [37, 38]. VEGF and bFGF have been shown to stimulate ATX expression in endothelial cells [8, 39], but the duration of treatment with two growth factors varies, suggesting that a secondary effect may be involved. VEGF signaling pathway has long been implicated in RCC angiogenesis and is the primary target of sunitinib. Moreover, bFGF has been shown to be upregulated in a murine pancreatic neuroendocrine tumor model with phenotypic resistance to VEGF blockade [40] and can stimulate angiogenic responses in endothelial cells despite the presence of sunitinib [41]. In this study, we found that ATX expression is elevated in the tumor vasculature of human RCC but declined with the treatment of sunitinib. Furthermore, ATX is highly expressed in the tumor endothelial cells of RCC mouse models while adapting to sunitinib. Determining the mechanism by which ATX expression is regulated in RCC-associated endothelium will require further investigation.
Originally proposed to act as a cell motility-stimulating protein [7], most ATX functions are linked to its ability to produce LPA and consequent activation of LPA receptors [42]. However, it has been intriguingly reported that ATX facilitates morphological change and adhesive state in oligodendrocytes in a catalytic activity-independent manner [20]. Using a cell impedance technology that was designed for biophysically monitoring the cell number, cell morphology, and degree of cell adhesion, we detected an ATX-mediated augmentation of cell impedance in RCC but did not observe a non-catalytic effect of ATX on either RCC or endothelial cells. In addition, ATX-induced migration in RCC was suppressed by pertussis toxin while the catalytically inactive form of ATX failed to stimulate RCC motility, suggesting that the effect of ATX on RCC is mainly dependent on LPA signaling.
Increasing evidence has pointed out a vital role for ATX and LPA signaling in the modulation of processes that contribute to cancer progression, such as cell proliferation, invasion, and angiogenesis [10, 43]. LPA levels are markedly elevated in malignant effusions, and its receptors are aberrantly expressed in several human cancers. In addition, overexpression of ATX or individual LPA receptors promotes tumorigenesis and metastasis in mouse models, whereas silencing of these genes produces the opposite effects. ATX overexpression has previously been seen in human RCC tissues but not in any of RCC cell lines [44]. In the present study, we showed that ATX is exclusively expressed in RCC tumor vessels but neither in tumor cells nor in normal renal capillaries. It has been reported that ATX expression is elevated in high endothelial venules of lymphoid organs, which potentiates actin rearrangement, chemotaxis, and entry of lymphocytes into lymph nodes, spleen, and Peyer’s patches through the production of LPA [19, 45]. Similarly, we found that ATX, produced by RCC tumor endothelium, acts through LPA signaling to promote renal tumorigenesis. In addition, pharmacological inhibition of LPA1 with Ki16425 reduced tumor growth in RCC xenograft models. Inhibition of LPA1 by Ki16425 has been shown to suppress tumor growth and bone metastasis in breast and ovarian cancer, whereas another antagonist targeting LPA1 inhibited only metastasis but showed no effect on primary tumor growth [17, 46]. This disparity indicates that regulation of cancer cell proliferation at the primary site may be mainly contributed by LPA signaling in some cell lines or cancer types but only partially in others. Surprisingly, we found that Ki16425 treatment also affected tumor vascularity in RCC xenograft models while ATX-LPA signaling had no marked in vitro effect on endothelial cells. This may be due to LPA-mediated production of angiogenic cytokines such as IL-8 and GROα by the RCC cells.
The acquired resistance to anti-angiogenic therapies develops through a variety of poorly understood mechanisms and appears to be one of the greatest clinical questions in contemporary RCC translational science. The major difficulty in studying this phenomenon is the lack of sequential clinical tissues for comparative analyses. Using RCC xenograft models, we have demonstrated that ATX-LPA signaling is functionally implicated in the acquired resistance of RCC to sunitinib. Our data implicate LPA as a potent inducer of RCC proliferation both in vitro and in vivo, which may account for tumor regrowth, a major clinical observation in RCC patients while refractory to anti-angiogenic treatment. In addition, it has been noticed in several preclinical cancer models that tumors can adapt to anti-angiogenic therapies by developing into a more invasive and malignant phenotype [47, 48]. We found that LPA significantly enhances RCC invasiveness. Moreover, tumors may activate alternative signaling pathways to bypass VEGF blockade for reinitiation and persistence of tumor angiogenesis during acquired resistance. We demonstrated that LPA signaling mediates the restoration of tumor vasculature in RCC resistance against sunitinib, likely through production of tumor-derived specific cytokines such as IL-8. LPA stimulation has been previously reported to upregulate IL-8 and other cytokines [49]. Of note, IL-8 has been shown to preserve tumor angiogenesis in xenograft models of colon cancer [50] and to mediate the resistance of RCC to sunitinib [34]. Together, data from our study and others suggest that replenished ATX-LPA axis may functionally contribute to RCC tumorigenesis while escaping from sunitinib treatment.
In conclusion, we have proposed an involvement of ATX-LPA signaling pathway in renal cancer progression and especially in the acquired resistance of RCC to sunitinib through targeting the interactions of tumor cells and vasculature. Our findings indicate that combination therapy with both sunitinib and antagnoists of the ATX-LPA signaling cascade should be valuable in treatment of RCC and may shed additional light on the understanding of cancer biology and drug resistance in RCC.
Supplementary Material
Statement of translational relevance.
Targeted therapy for renal cell carcinoma (RCC) has made important strides in recent years. While the advent of antiangiogenic agents as the standard care for patients with metastatic RCC has resulted in improved survival rates, complete responses and disease cure are unfortunately rare due to angiogenic escape. A combination therapy of existing agents with new drugs against distinct signaling pathways which are critical for RCC biology or active in resistance will be of great clinical importance. This study demonstrated that endothelial autotaxin acts through lysophosphatidic acid signaling to promote renal tumorigenesis and is functionally involved in the acquired resistance of RCC to sunitinib, revealing for the first time that autotaxin-lysophosphatidic acid signaling axis regulates RCC pathogenesis and response to targeted therapy.
Acknowledgments
We thank Drs. Wadih Arap and Renata Pasqualini for assistance with the impedance-based cellular assay.
Financial Support: This work was supported by Erickson Kidney Cancer Research Fund, L.C. Biedenharn Jr. Kidney Cancer Research Fund, Beth Israel Deaconess Medical Center (5 P50 CA101942 08 2), and the Bosler Kidney Cancer Research Fund to CGW.
Footnotes
Conflicts of Interest: The authors declare no conflict of interest in regards to this manuscript.
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors’ Contributions
Conception and design: SC Su, X. Lin, C.G. Wood
Development of methodology: SC Su, XY Zhang, T. Maity
Acquisition of data: SC Su, XY Zhang, T. Maity, P.A. Kenney, M.M. Merrill, K.N. Babaian
Analysis and interpretation of data: SC Su, XY Zhang, T. Maity, X Hu, SF Yang
Writing, review, and/or revision of the manuscript: SC Su, X. Lin, C.G. Wood
Study supervision: X. Lin, C.G. Wood
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010 Sep-Oct;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Hollingsworth JM, Miller DC, Daignault S, Hollenbeck BK. Rising incidence of small renal masses: a need to reassess treatment effect. J Natl Cancer Inst. 2006 Sep 20;98(18):1331–4. doi: 10.1093/jnci/djj362. [DOI] [PubMed] [Google Scholar]
- 3.Biswas S, Eisen T. Immunotherapeutic strategies in kidney cancer–when TKIs are not enough. Nat Rev Clin Oncol. 2009 Aug;6(8):478–87. doi: 10.1038/nrclinonc.2009.91. [DOI] [PubMed] [Google Scholar]
- 4.Rini BI, Atkins MB. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol. 2009 Oct;10(10):992–1000. doi: 10.1016/S1470-2045(09)70240-2. [DOI] [PubMed] [Google Scholar]
- 5.Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007 Jan 11;356(2):115–24. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 6.Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Oudard S, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009 Aug 1;27(22):3584–90. doi: 10.1200/JCO.2008.20.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stracke ML, Krutzsch HC, Unsworth EJ, Arestad A, Cioce V, Schiffmann E, et al. Identification, purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. The Journal of biological chemistry. 1992 Feb 5;267(4):2524–9. [PubMed] [Google Scholar]
- 8.Nam SW, Clair T, Kim YS, McMarlin A, Schiffmann E, Liotta LA, et al. Autotaxin (NPP-2), a metastasis-enhancing motogen, is an angiogenic factor. Cancer Res. 2001 Sep 15;61(18):6938–44. [PubMed] [Google Scholar]
- 9.van Meeteren LA, Ruurs P, Stortelers C, Bouwman P, van Rooijen MA, Pradere JP, et al. Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Mol Cell Biol. 2006 Jul;26(13):5015–22. doi: 10.1128/MCB.02419-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003 Aug;3(8):582–91. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 11.Choi JW, Herr DR, Noguchi K, Yung YC, Lee CW, Mutoh T, et al. LPA receptors: subtypes and biological actions. Annual review of pharmacology and toxicology. 2010;50:157–86. doi: 10.1146/annurev.pharmtox.010909.105753. [DOI] [PubMed] [Google Scholar]
- 12.Liu S, Murph M, Panupinthu N, Mills GB. ATX-LPA receptor axis in inflammation and cancer. Cell Cycle. 2009 Nov 15;8(22):3695–701. doi: 10.4161/cc.8.22.9937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu S, Umezu-Goto M, Murph M, Lu Y, Liu W, Zhang F, et al. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell. 2009 Jun 2;15(6):539–50. doi: 10.1016/j.ccr.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin S, Wang D, Iyer S, Ghaleb AM, Shim H, Yang VW, et al. The absence of LPA2 attenuates tumor formation in an experimental model of colitis-associated cancer. Gastroenterology. 2009 May;136(5):1711–20. doi: 10.1053/j.gastro.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang M, Zhong WW, Srivastava N, Slavin A, Yang J, Hoey T, et al. G protein-coupled lysophosphatidic acid receptors stimulate proliferation of colon cancer cells through the {beta}-catenin pathway. Proc Natl Acad Sci U S A. 2005 Apr 26;102(17):6027–32. doi: 10.1073/pnas.0501535102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boucharaba A, Serre CM, Gres S, Saulnier-Blache JS, Bordet JC, Guglielmi J, et al. Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J Clin Invest. 2004 Dec;114(12):1714–25. doi: 10.1172/JCI22123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boucharaba A, Serre CM, Guglielmi J, Bordet JC, Clezardin P, Peyruchaud O. The type 1 lysophosphatidic acid receptor is a target for therapy in bone metastases. Proc Natl Acad Sci U S A. 2006 Jun 20;103(25):9643–8. doi: 10.1073/pnas.0600979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fulkerson Z, Wu T, Sunkara M, Kooi CV, Morris AJ, Smyth SS. Binding of autotaxin to integrins localizes lysophosphatidic acid production to platelets and mammalian cells. The Journal of biological chemistry. 2011 Oct 7;286(40):34654–63. doi: 10.1074/jbc.M111.276725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanda H, Newton R, Klein R, Morita Y, Gunn MD, Rosen SD. Autotaxin, an ectoenzyme that produces lysophosphatidic acid, promotes the entry of lymphocytes into secondary lymphoid organs. Nat Immunol. 2008 Apr;9(4):415–23. doi: 10.1038/ni1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dennis J, White MA, Forrest AD, Yuelling LM, Nogaroli L, Afshari FS, et al. Phosphodiesterase-Ialpha/autotaxin’s MORFO domain regulates oligodendroglial process network formation and focal adhesion organization. Molecular and cellular neurosciences. 2008 Feb;37(2):412–24. doi: 10.1016/j.mcn.2007.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Beijnum JR, Rousch M, Castermans K, van der Linden E, Griffioen AW. Isolation of endothelial cells from fresh tissues. Nat Protoc. 2008;3(6):1085–91. doi: 10.1038/nprot.2008.71. [DOI] [PubMed] [Google Scholar]
- 22.Karam JA, Zhang XY, Tamboli P, Margulis V, Wang H, Abel EJ, et al. Development and characterization of clinically relevant tumor models from patients with renal cell carcinoma. Eur Urol. 2011 Apr;59(4):619–28. doi: 10.1016/j.eururo.2010.11.043. [DOI] [PubMed] [Google Scholar]
- 23.Masuda A, Nakamura K, Izutsu K, Igarashi K, Ohkawa R, Jona M, et al. Serum autotaxin measurement in haematological malignancies: a promising marker for follicular lymphoma. Br J Haematol. 2008 Oct;143(1):60–70. doi: 10.1111/j.1365-2141.2008.07325.x. [DOI] [PubMed] [Google Scholar]
- 24.Nakagawa H, Ikeda H, Nakamura K, Ohkawa R, Masuzaki R, Tateishi R, et al. Autotaxin as a novel serum marker of liver fibrosis. Clin Chim Acta. 2011 Jun 11;412(13–14):1201–6. doi: 10.1016/j.cca.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 25.Guan N, Deng J, Li T, Xu X, Irelan JT, Wang MW. Label-free monitoring of T cell activation by the impedance-based xCELLigence system. Molecular bioSystems. 2013 Apr 2;9(5):1035–43. doi: 10.1039/c3mb25421f. [DOI] [PubMed] [Google Scholar]
- 26.McGuinness R. Impedance-based cellular assay technologies: recent advances, future promise. Curr Opin Pharmacol. 2007 Oct;7(5):535–40. doi: 10.1016/j.coph.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 27.Croset M, Brossard N, Polette A, Lagarde M. Characterization of plasma unsaturated lysophosphatidylcholines in human and rat. The Biochemical journal. 2000 Jan 1;345(Pt 1):61–7. [PMC free article] [PubMed] [Google Scholar]
- 28.Fisher KE, Pop A, Koh W, Anthis NJ, Saunders WB, Davis GE. Tumor cell invasion of collagen matrices requires coordinate lipid agonist-induced G-protein and membrane-type matrix metalloproteinase-1-dependent signaling. Mol Cancer. 2006;5:69. doi: 10.1186/1476-4598-5-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang G, Xu Y, Fujiwara Y, Tsukahara T, Tsukahara R, Gajewiak J, et al. Alpha-substituted phosphonate analogues of lysophosphatidic acid (LPA) selectively inhibit production and action of LPA. ChemMedChem. 2007 May;2(5):679–90. doi: 10.1002/cmdc.200600280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tigyi G. Aiming drug discovery at lysophosphatidic acid targets. Br J Pharmacol. 2010 Sep;161(2):241–70. doi: 10.1111/j.1476-5381.2010.00815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohta H, Sato K, Murata N, Damirin A, Malchinkhuu E, Kon J, et al. Ki16425, a subtype-selective antagonist for EDG-family lysophosphatidic acid receptors. Mol Pharmacol. 2003 Oct;64(4):994–1005. doi: 10.1124/mol.64.4.994. [DOI] [PubMed] [Google Scholar]
- 32.Murph M, Mills GB. Targeting the lipids LPA and S1P and their signalling pathways to inhibit tumour progression. Expert Rev Mol Med. 2007;9(28):1–18. doi: 10.1017/S1462399407000476. [DOI] [PubMed] [Google Scholar]
- 33.Tamaskar I, Dhillon J, Pili R. Resistance to angiogenesis inhibitors in renal cell carcinoma. Clinical advances in hematology & oncology : H&O. 2011 Feb;9(2):101–10. [PubMed] [Google Scholar]
- 34.Huang D, Ding Y, Zhou M, Rini BI, Petillo D, Qian CN, et al. Interleukin-8 mediates resistance to antiangiogenic agent sunitinib in renal cell carcinoma. Cancer research. 2010 Feb 1;70(3):1063–71. doi: 10.1158/0008-5472.CAN-09-3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang D, Ding Y, Li Y, Luo WM, Zhang ZF, Snider J, et al. Sunitinib acts primarily on tumor endothelium rather than tumor cells to inhibit the growth of renal cell carcinoma. Cancer research. 2010 Feb 1;70(3):1053–62. doi: 10.1158/0008-5472.CAN-09-3722. [DOI] [PubMed] [Google Scholar]
- 36.Furuya M, Nishiyama M, Kimura S, Suyama T, Naya Y, Ito H, et al. Expression of regulator of G protein signalling protein 5 (RGS5) in the tumour vasculature of human renal cell carcinoma. The Journal of pathology. 2004 May;203(1):551–8. doi: 10.1002/path.1543. [DOI] [PubMed] [Google Scholar]
- 37.Kehlen A, Englert N, Seifert A, Klonisch T, Dralle H, Langner J, et al. Expression, regulation and function of autotaxin in thyroid carcinomas. International journal of cancer Journal international du cancer. 2004 May 10;109(6):833–8. doi: 10.1002/ijc.20022. [DOI] [PubMed] [Google Scholar]
- 38.Black EJ, Clair T, Delrow J, Neiman P, Gillespie DA. Microarray analysis identifies Autotaxin, a tumour cell motility and angiogenic factor with lysophospholipase D activity, as a specific target of cell transformation by v-Jun. Oncogene. 2004 Mar 25;23(13):2357–66. doi: 10.1038/sj.onc.1207377. [DOI] [PubMed] [Google Scholar]
- 39.Ptaszynska MM, Pendrak ML, Stracke ML, Roberts DD. Autotaxin signaling via lysophosphatidic acid receptors contributes to vascular endothelial growth factor-induced endothelial cell migration. Molecular cancer research : MCR. 2010 Mar;8(3):309–21. doi: 10.1158/1541-7786.MCR-09-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005 Oct;8(4):299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 41.Welti JC, Gourlaouen M, Powles T, Kudahetti SC, Wilson P, Berney DM, et al. Fibroblast growth factor 2 regulates endothelial cell sensitivity to sunitinib. Oncogene. 2011 Mar 10;30(10):1183–93. doi: 10.1038/onc.2010.503. [DOI] [PubMed] [Google Scholar]
- 42.Nakanaga K, Hama K, Aoki J. Autotaxin–an LPA producing enzyme with diverse functions. Journal of biochemistry. 2010 Jul;148(1):13–24. doi: 10.1093/jb/mvq052. [DOI] [PubMed] [Google Scholar]
- 43.Houben AJ, Moolenaar WH. Autotaxin and LPA receptor signaling in cancer. Cancer Metastasis Rev. 2011 Dec;30(3–4):557–65. doi: 10.1007/s10555-011-9319-7. [DOI] [PubMed] [Google Scholar]
- 44.Stassar MJ, Devitt G, Brosius M, Rinnab L, Prang J, Schradin T, et al. Identification of human renal cell carcinoma associated genes by suppression subtractive hybridization. Br J Cancer. 2001 Nov 2;85(9):1372–82. doi: 10.1054/bjoc.2001.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakasaki T, Tanaka T, Okudaira S, Hirosawa M, Umemoto E, Otani K, et al. Involvement of the lysophosphatidic acid-generating enzyme autotaxin in lymphocyte-endothelial cell interactions. Am J Pathol. 2008 Nov;173(5):1566–76. doi: 10.2353/ajpath.2008.071153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marshall JC, Collins JW, Nakayama J, Horak CE, Liewehr DJ, Steinberg SM, et al. Effect of inhibition of the lysophosphatidic acid receptor 1 on metastasis and metastatic dormancy in breast cancer. Journal of the National Cancer Institute. 2012 Sep 5;104(17):1306–19. doi: 10.1093/jnci/djs319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer cell. 2009 Mar 3;15(3):220–31. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Du R, Petritsch C, Lu K, Liu P, Haller A, Ganss R, et al. Matrix metalloproteinase-2 regulates vascular patterning and growth affecting tumor cell survival and invasion in GBM. Neuro Oncol. 2008 Jun;10(3):254–64. doi: 10.1215/15228517-2008-001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fang X, Yu S, Bast RC, Liu S, Xu HJ, Hu SX, et al. Mechanisms for lysophosphatidic acid-induced cytokine production in ovarian cancer cells. The Journal of biological chemistry. 2004 Mar 5;279(10):9653–61. doi: 10.1074/jbc.M306662200. [DOI] [PubMed] [Google Scholar]
- 50.Mizukami Y, Jo WS, Duerr EM, Gala M, Li J, Zhang X, et al. Induction of interleukin-8 preserves the angiogenic response in HIF-1alpha-deficient colon cancer cells. Nat Med. 2005 Sep;11(9):992–7. doi: 10.1038/nm1294. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.