

 Isotopically labelled Ni57Fe models of the [NiFe] hydrogenase active site have been prepared and studied with nuclear resonant vibrational spectroscopy, enabling direct characterization of metal–metal bonding.
Isotopically labelled Ni57Fe models of the [NiFe] hydrogenase active site have been prepared and studied with nuclear resonant vibrational spectroscopy, enabling direct characterization of metal–metal bonding.
Abstract
A new route to iron carbonyls has enabled synthesis of 57Fe-labeled [NiFe] hydrogenase mimic (OC)3 57Fe(pdt)Ni(dppe). Its study by nuclear resonance vibrational spectroscopy revealed Ni–57Fe vibrations, as confirmed by calculations. The modes are absent for [(OC)3 57Fe(pdt)Ni(dppe)]+, which lacks Ni–57Fe bonding, underscoring the utility of the analyses in identifying metal–metal interactions.
Despite our extremely low atmospheric concentration of dihydrogen (∼1 ppm), this substrate is a key metabolite of many anaerobic bacteria.1 In such living systems can be found the most prevalent enzymes for hydrogen processing, the nickel–iron hydrogenases ([NiFe]–H2ases).1,2 These electrocatalysts specifically mediate the redox reaction H2 ⇌ 2H+ + 2e– at several hundred turnovers per second.3 Their heterobimetallic active sites exist in several states, some of which are summarized below (Fig. 1, left and centre).
Fig. 1. Key [NiFe]–H2ase states (left and centre) and two model complexes (right).

The active sites feature Ni bound to four cysteinato residues, two of which bridge to an Fe(CO)(CN)2 fragment. In the Ni–C state, Ni(iii)Fe(ii) centres bind a bridging hydride (H–), reductive elimination of which affords Ni–L.4 Thus, H+ is abstracted by a terminal cys ligand (Fig. 1, centre top), leaving a Ni(i)Fe(ii) core with a 2e– bond between the metals.4
The use of vibrational spectroscopy to study [NiFe]–H2ase is convenient in that its active site features chromophores easily identifiable by such techniques. Spectral analyses are often aided by comparison to data from synthetic models5 whose structures are well understood. Specificity for 57Fe-coupled modes is afforded by nuclear resonance vibrational spectroscopy (NRVS, vide infra). This has recently enabled observation of characteristic Fe–CN/Fe–CO bending and stretching modes in [NiFe]–H2ase (Ni–A and Ni–R) and the Fe subsite model [Fe(benzenedithiolato)(CN)2CO]2–.6 However, no NRVS studies have reported on metal–metal bonding, which is expected for low-valent clusters like Ni–L and its models.4,7
A near-complete [NiFe]–H2ase mimic is the Ni(ii)Fe(i) species [(OC)3Fe(pdt)Ni(dppe)]+ ([1]+, pdt2– = –S(CH2)3S–; dppe = 1,2-bis(diphenylphosphino)ethane), a model for Ni–L, albeit with metal oxidation states reversed (Fig. 1, right).8 This S = 1/2 model is prepared from (OC)3Fe(pdt)Ni(dppe) (1),9 itself the subject of density functional theory (DFT) and resonance Raman investigations.10 Disclosed here is methodology for 57Fe-labeled prototypes [(OC)3 57Fe(pdt)Ni(dppe)]0/+ ([1]0/+), enabling the study of metal–metal bonding with NRVS.
The Ni(i)Fe(i) complex 1 is usually accessed by interaction of (pdt)Ni(dppe) with an Fe carbonyl such as Fe2(CO)9 or Fe(CO)4I2.11–14 The precursor to these, Fe(CO)5, is not conveniently prepared from elemental Fe, a factor that necessitated a new route adaptable to 57Fe incorporation. Thus, metallic Fe was converted to the organo-soluble FeI2 source Fe2I4(iPrOH)4,15 which, upon combination with (pdt)Ni(dppe), gave the known diiodide I2Fe(pdt)Ni(dppe).14 While the diiodide does not bind CO in CH2Cl2, when treated with AgBF4 it converts to the putative electrophile ‘[IFe(pdt)Ni(dppe)]+’ (or perhaps its dimer), which undergoes carbonylation to afford [(OC)3FeI(pdt)Ni(dppe)]+ ([1I]+).14 Reduction with CoCp2 gave 1 in yields comparable to the (pdt)Ni(dppe)/Fe2(CO)9 route.11
In adapting the synthesis to the Ni57Fe target, elemental 57Fe was oxidized to 57Fe2I4(iPrOH)4,12 which was converted into violet [(OC)3 57FeI(pdt)Ni(dppe)]+ (ESI-MS: m/z 829.5 [1′I]+ vs. 828.9 [1I]+), with reduction affording green (OC)3 57Fe(pdt)Ni(dppe) (1′) in 35% yield (Scheme 1).
Scheme 1.

While ν CO energies and analytical and 31P{1H} NMR data for 1′ are virtually identical to those of natural abundance 1, LI-FDI-MS (liquid introduction field desorption ionization mass spectrometry) analyses are telling. Soft ionization of non-polar 1′ and 1 allowed for the detection of parent cations at m/z 702.9 and 701.9, respectively (Fig. S4, ESI†). Oxidation of 1′ with FcBF4 afforded mixed-valent salt [1′]BF4, whose EPR signal is broadened relative to that of [1]BF4 due to hyperfine interactions.
Investigations into Ni–Fe bonding in the new 57Fe-labeled variants of the reduced and oxidized complexes (respectively 1′ and [1′]+) were undertaken using NRVS. This technique, enabled by the development of third generation synchrotron sources, insertion devices and advanced X-ray optics,16–18 involves scanning an extremely monochromatic (e.g. 0.8 meV) X-ray beam through a nuclear resonance of a Mössbauer-active isotope (e.g. 14.4 keV for 57Fe). Subsequent relaxation causes the generation/annihilation of phonons, the detection of which reveals all modes in which the 57Fe nucleus moves along the direction of the incident X-ray. NRVS has several advantages over traditional IR and resonance Raman spectroscopies,19 not least in terms of element and isotope specificity and absence of the optical selection rules, which have allowed for the resolution of ν Fe–X (X = S, P, Cl, CO, CN, NO) vibrations in complicated systems.6,13,19,20 This relatively new but powerful technique in inorganic and biological iron chemistry is applied here to [1′]0/+. The analysed spectra in terms of 57Fe partial vibrational density of states (PVDOS) are given in Fig. 2.
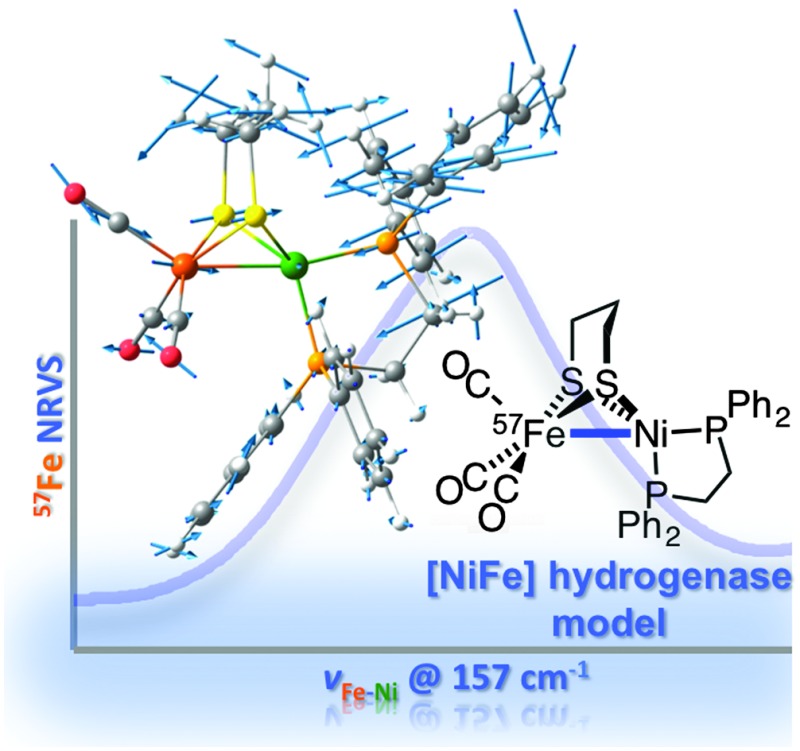
Fig. 2. Observed NRVS spectra for [1′]+ (thick red lines, (a) and (b)) and 1′ (thick blue lines, (a) and (c)) vs. DFT calculated 57Fe PVDOS spectra for [1′]+ (thin red line, (b)) and 1′ (thin blue line, (c)). Calculated Fe–Ni KED (green) is given in (b) for [1′]+, and in (c) for 1′. Key bands observed for 1′ are labelled in (a), with DFT counterparts in (c) indicated by vertical lines. Modes giving rise to bands with significant Fe–Ni character are marked (*) and shown in Fig. 3 and Fig. S10 (ESI†). For 0–650 cm–1 spectra, see Fig. S9 (ESI†).

Intense bands assigned to ν Fe–CO and δ Fe–CO modes were observed at 440–630 cm–1, with full-range NRVS spectra and Fe–C(O) kinetic energy distribution (KED) diagrams presented in Fig. S9 (ESI†). Similar signals were found for [NiFe]–H2ase.6 It was expected that ν Fe–S, δ Fe–S and ν Fe–Ni bands, if observable, would lie at low energies (≤400 cm–1). Upon comparing data for 1′ and [1′]+ (Fig. 2a), a sharp and prominent NRVS peak at 158 cm–1 for 1′ was noticed. This band, absent from the spectra of [1′]+, was tentatively ascribed to vibration of the Ni–Fe bond, such interactions not being significant in [1′]+. Differences in NRVS data of 1′ and [1′]+ were less marked in other spectral regions, although peaks for [1′]+ were broader.
The assignment of vibrational bands was elaborated using DFT calculations on [1′]0/+ as detailed in the ESI;† simulated NRVS (57Fe PVDOS) and Fe–Ni KED diagrams for [1′]+ and 1′ are also presented in Fig. 2b and c, respectively. Band positions and intensities in the calculated and observed spectra are largely in agreement (particularly in Fig. 2c; see also Fig. S9, ESI†). In the case of [1′]+, some differences are assigned to impurities (1′ and/or [1′H]+). The band for 1′ at 158 cm–1, calculated by DFT at 157 cm–1, indeed involves stretching of the Fe–Ni bond symmetrical to a Ni–P1 stretch (see Fig. 3, and the ESI† for animations). Such a vibrational coupling in the Fe–Ni–P1 triad implicates a strong Fe–Ni interaction, and it has no complement in the normal modes pool calculated for [1]+.
Fig. 3. Scaled arrow depiction of nuclear displacements for the normal mode calculated for 1′ at 157 cm–1 (a symmetric Fe–Ni–P1 stretch, see corresponding 57Fe PVDOS band in Fig. 2c). Key [1′]0/+ modes are animated in the ESI.† .

While very prominent in NRVS (owing to the large 57Fe displacement), the 157 cm–1 band is weaker (∼5%) in the Ni–Fe KED diagram, which reflects relative motion of Fe and Ni. Other Ni–Fe stretches, such as those calculated at 266, 311, and 386 cm–1 (see in Fig. 2c), are considerably stronger (representing 13%, 9%, and 11% total Ni–Fe KED, respectively). Yet only the first two modes can be associated with bands observed at 262 and 303 cm–1, while the last one has vanishing NRVS intensity (Fig. 2a). The modes at 266, 311, and 386 cm–1 have lower 57Fe PVDOS intensity as they involve displacement mostly of Ni (rather than Fe), this movement being evident from the DFT results (Fig. S10, ESI†). Analysis of Fe–Ni vibrations is further complicated by vibrational coupling to C/P/S atoms, in particular the bridging S donors. Notably, mixed ν Fe–Ni modes in the 220–360 cm–1 region involving up to 14% contribution from the Fe–Ni stretch were also reported for 1.10
Structural (X-ray diffraction) and DFT studies on unlabeled [1]0/+ have suggested that the reduced Ni(i)Fe(i) species may feature Ni–Fe bonding,9 while the oxidized Ni(ii)Fe(i) does not.8 This was confirmed by re-analysing bonding in [1]0/+ using ELF,21 ELI-D,22 and QTAIM23 electron density-based methods (Fig. 4; Fig. S11 and S12, ESI†), all of which indicated a Ni–Fe bond in 1. Notably, the ELF/ELI-D bond attractor and the bond critical point found by QTAIM are shifted from the Ni–Fe vector away from the bridging S atoms, such that Fe(i) is (pseudo)octahedral. Bonding involves overlap of two singly-occupied d(z 2) orbitals and is absent when Ni is oxidized, with [1]+ featuring a Fe-localized d(z 2) SOMO. In contrast, Ni–L has a SOMO with Ni d(z 2) and d(x 2 – y 2) character,4 and a 2e– Ni→Fe dative bond (optimized Fe–Ni distance: 2.47 Å).4 While [1]+ and Ni–L have the same electron count for the [NiFe]3+ site, only one has Ni–Fe bonding. A key distinction is Ni geometry, which is planar in [1]+ but SF4-like in Ni–L.
Fig. 4. Electron localization function (ELF) analysis of the Ni–Fe bonding in [1]0/+ (top/bottom). Ni–Fe bond attractor position for [1]0 is indicated by the localized area in green (center-top), absent for [1]+. See Fig. S11 and S12 (ESI†) for alternative bonding representations.

Since the Fe(i) centre in [1]+ is electron poor (average ν CO = 2010 cm–1)8 it cannot supply 2e– for a Fe→Ni coordinate bond. Another possibility, a covalent Ni–Fe bond, is unlikely due to the low donicity of Ni(ii). The metal centres in [1]+ are distant (our DFT result: 2.80 Å), while those in electron-rich 1 (average ν CO = 1977 cm–1) are sufficiently proximal (experimental: 2.47 Å,9 DFT: 2.46 Å) for covalent Fe–Ni bonding (see Table S2 and Fig. S3, ESI† for structural and IR details).
Obtaining direct evidence of metal–metal bonding in molecular systems is nontrivial, and distances do not guarantee presence/absence of bonding. For example, EXAFS studies reveal similar Fe–Ni distances for Ni–L and Ni–C,24 despite Ni not being bonded to Fe in the latter. This highlights that (i) [NiFe]–H2ase active site ligation is inflexible (relative to [1]0/+) and that (ii) EXAFS/XRD studies typically only afford nuclear positions via core electron density, with bonding electron density between metals being poorly resolved.25 In rare cases when very high quality single crystal data are obtained, multipole analysis, followed by topological analysis of static electron density (versus inspection of difference maps) can give insights into metal–metal bonding, as exemplified by (re)investigation of the archetypal Mn2(CO)10.26
Metal–metal bonding is common in organoiron chemistry, but it also plays a role in the reduced states of some metalloenzymes, stabilizing low-valent metal centres poised for substrate activation.7 In addition to [NiFe]–H2ase, Ni–Fe interactions are also proposed for carbon monoxide dehydrogenase (CODH),7,27 with Ni–Ni and Fe–Fe bonds being present in acetyl-CoA synthetase (ACS)7 and [FeFe]–H2ase,28 respectively. NRVS is demonstrably effective in unambiguous identification of low energy 57Fe-coupled modes.13,20,29 Compared to IR and resonance Raman, it avoids interference from solvent and ‘fingerprint’ bands, enabling identification of Fe-coupled vibrations, such as the 158 cm–1 ν Fe–Ni mode here. The use of NRVS to probe iron–metal modes in everything from small molecules to iron enzymes is anticipated to provide a wealth of information on these catalysts.
While primordial routes to iron carbonyls have been reported (e.g. the one-pot preparation of (CO)3Fe(pdt)Fe(CO)3 from FeCl2),30 they are typically limited in scope and reproducibility. In contrast, the Fe2I4(iPrOH)4/CO/CoCp2 strategy will likely be generalizable and afford iron carbonyls of relevance to H2ases and organoiron chemistry in general. With the isolation of 57Fe-labeled [NiFe]–H2ase mimics [1′]0/+, the element and isotope selectivity of NRVS was exploited to obtain the first vibrational spectroscopic evidence of Fe–Ni interactions. These results serve as an important benchmark, opening the door to work probing metal–metal bonding in redox-active H2ase enzymes as well as related enzymatic and model systems.
Thanks are given to Drs Mark J. Nilges and Haijun Yao for assistance with EPR and LI-FDI-MS, respectively. Financial support was provided by the National Institutes of Health (GM061153-10 to T.B.R. and GM-65440 to S.P.C.), U.S. Department of Energy Office of Biological and Environmental Research (DOE OBER) (S.P.C.), and the ‘Unifying Concepts in Catalysis' initiative of the German Research Council (V.P., F.M., and M.K.). NRVS experiments performed at SPring-8 BL09XU were funded by JASRI (beamtime proposal 2013A0032).
Footnotes
References
- Vignais P. M., Billoud B. Chem. Rev. 2007;107:4206. doi: 10.1021/cr050196r. [DOI] [PubMed] [Google Scholar]
- Ogata H., Lubitz W., Higuchi Y. Dalton Trans. 2009:7577. doi: 10.1039/b903840j. [DOI] [PubMed] [Google Scholar]
- Frey M. ChemBioChem. 2002;3:153. doi: 10.1002/1439-7633(20020301)3:2/3<153::AID-CBIC153>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Kampa M., Pandelia M.-E., Lubitz W., van Gastel M., Neese F. J. Am. Chem. Soc. 2013;135:3915. doi: 10.1021/ja3115899. [DOI] [PubMed] [Google Scholar]
- Kaur-Ghumaan S., Stein M. Dalton Trans. 2014;43:9392. doi: 10.1039/c4dt00539b. [DOI] [PubMed] [Google Scholar]
- Kamali S., Wang H., Mitra D., Ogata H., Lubitz W., Manor B. C., Rauchfuss T. B., Byrne D., Bonnefoy V., Jenney F. E., Adams M. W. W., Yoda Y., Alp E., Zhao J., Cramer S. P. Angew. Chem., Int. Ed. 2013;52:724. doi: 10.1002/anie.201204616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl P. A. J. Inorg. Biochem. 2012;106:172. doi: 10.1016/j.jinorgbio.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilter D., Nilges M. J., Chakrabarti M., Lindahl P. A., Rauchfuss T. B., Stein M. Inorg. Chem. 2012;51:2338. doi: 10.1021/ic202329y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W., Marr W. A. C., Wang Q., Neese F., Spencer D. J. E., Blake A. J., Cooke P. A., Wilson C., Schröder M., Huynh M. T., Schilter D., Hammes-Schiffer S., Rauchfuss T. B. Proc. Natl. Acad. Sci. U. S. A. J. Am. Chem. Soc. 2005;2014;102136:18280. 12385. doi: 10.1073/pnas.0505779102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafaat H. S., Weber K., Petrenko T., Neese F., Lubitz W. Inorg. Chem. 2012;51:11787. doi: 10.1021/ic3017276. [DOI] [PubMed] [Google Scholar]
- Barton B. E., Rauchfuss T. B. J. Am. Chem. Soc. 2010;132:14877. doi: 10.1021/ja105312p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll M. E., Chen J., Gray D. E., Lansing J. C., Rauchfuss T. B., Schilter D., Volkers P. I., Wilson S. R. Organometallics. 2014;33:858. doi: 10.1021/om400752a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y., Wang H., Xiao Y., Vogt S., Thauer R. K., Shima S., Volkers P. I., Rauchfuss T. B., Pelmenschikov V., Case D. A., Alp E., Sturhahn W., Yoda Y., Cramer S. P. Inorg. Chem. 2008;47:3969. doi: 10.1021/ic701251j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilter D., Rauchfuss T. B. Dalton Trans. 2012;41:13324. doi: 10.1039/c2dt31895d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes G. G., Bottini R. C. R., Reis D. M., Camargo P. H. C., Evans D. J., Hitchcock P. B., Leigh G. J., Sá E. L., Soares J. F. Inorg. Chim. Acta. 2004;357:1219. [Google Scholar]
- Seto M., Yoda Y., Kikuta S., Zhang X. W., Ando M. Phys. Rev. Lett. 1995;74:3828. doi: 10.1103/PhysRevLett.74.3828. [DOI] [PubMed] [Google Scholar]
- Sturhahn W., Toellner T. S., Alp E. E., Zhang X., Ando M., Yoda Y., Kikuta S., Seto M., Kimball C. W., Dabrowski B. Phys. Rev. Lett. 1995;74:3832. doi: 10.1103/PhysRevLett.74.3832. [DOI] [PubMed] [Google Scholar]
- Yoda Y., Imai Y., Kobayashi H., Goto S., Takeshita K., Seto M. Hyperfine Interact. 2012;206:83. [Google Scholar]
- Smith M. C., Xiao Y., Wang H., George S. J., Coucouvanis D., Koutmos M., Sturhahn W., Alp E. E., Zhao J., Cramer S. P. Inorg. Chem. 2005;44:5562. doi: 10.1021/ic0482584. [DOI] [PubMed] [Google Scholar]
- Tonzetich Z. J., Wang H., Mitra D., Tinberg C. E., Do L. H., Jenney, Jr. F. E., Adams M. W. W., Cramer S. P., Lippard S. J. J. Am. Chem. Soc. 2010;132:6914. doi: 10.1021/ja101002f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becke A. D., Edgecombe K. E., Savin A., Jepsen O., Flad J., Andersen O. K., Preuss H., Vonschnering H. G., Kohout M., Savin A. J. Chem. Phys. Angew. Chem., Int. Ed. Int. J. Quantum Chem. 1990;1992;1996;923160:5397. 187, 875. [Google Scholar]
- Kohout M., Kohout M., Pernal K., Wagner F. R., Grin Y. Int. J. Quantum Chem. Theor. Chem. Acc. 2004;2004;97112:651. 453. [Google Scholar]
- Bader R. F. W., Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990. [Google Scholar]
- Whitehead J. P., Gurbiel R. J., Bagyinka C., Hoffman B. M., Maroney M. J. J. Am. Chem. Soc. 1993;115:5629. [Google Scholar]
- Koritsanszky T. S., Coppens P. Chem. Rev. 2001;101:1583. doi: 10.1021/cr990112c. [DOI] [PubMed] [Google Scholar]
- Bianchi R., Gervasio G., Marabello D. Inorg. Chem. 2000;39:2360. doi: 10.1021/ic991316e. [DOI] [PubMed] [Google Scholar]
- Jeoung J.-H., Dobbek H. J. Am. Chem. Soc. 2009;131:9922. doi: 10.1021/ja9046476. [DOI] [PubMed] [Google Scholar]
- Nicolet Y., de Lacey A. L., Vernède X., Fernandez V. M., Hatchikian E. C., Fontecilla-Camps J. C. J. Am. Chem. Soc. 2001;123:1596. doi: 10.1021/ja0020963. [DOI] [PubMed] [Google Scholar]
- Cramer S. P., Xiao Y., Wang H., Guo Y., Smith M. C. Hyperfine Interact. 2006;170:47. [Google Scholar]
- Volkers P. I., Boyke C. A., Chen J., Rauchfuss T. B., Whaley C. M., Wilson S. R., Yao H. Inorg. Chem. 2008;47:7002. doi: 10.1021/ic800601k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.