

SUMMARY
MLKL is crucial for necroptosis, permeabilizing membranes through its N-terminal region upon phosphorylation of its kinase-like domain by RIP3. However, the mechanism underlying membrane permeabilization is unknown. The solution structure of the MLKL N-terminal region determined by NMR spectroscopy reveals a four-helix bundle with an additional helix at the top that is likely key for MLKL function, and a sixth, C-terminal helix that interacts with the top helix and with a poorly packed interface within the four-helix bundle. Fluorescence spectroscopy measurements indicate that much of the four-helix bundle inserts into membranes, but not the C-terminal helix. Moreover, we find that the four-helix bundle is sufficient to induce liposome leakage and that the C-terminal helix inhibits this activity. These results suggest that the four-helix bundle mediates membrane breakdown during necroptosis and that the sixth helix acts as a plug that prevents opening of the bundle and is released upon RIP3 phosphorylation.
INTRODUCTION
Cell death triggered by defined biochemical pathways plays critical roles in normal tissue development as well as in diverse pathological processes (Fiers et al., 1999). Apoptosis and necrosis constitute the two major forms of cell death caused by these pathways, and are characterized by distinct morphological and biochemical alterations. Apoptosis is a process of programmed cell death that results in structural changes such as nuclear membrane collapse, chromatin fragmentation, cell condensation and breakdown into small vesicles called apoptotic bodies, which are promptly eliminated by phagocytosis (Kerr et al., 1972). The molecular events leading to apoptosis have been exquisitely well characterized. A key step is the activation of cysteine proteases known as caspases (Thornberry and Lazebnik, 1998), which can be induced extrinsically by cytokines from the tumor necrosis factor (TNF) family and intrinsically by members of the Bcl-2 family of proteins such as Bax (Danial and Korsmeyer, 2004). These proteins can form oligomeric pores in membranes (Epand et al., 2002) and permeabilize mitochondria (Wei et al., 2001; Kuwana et al., 2002), causing the release into the cytosol of factors such as cytochrome c that are crucial for downstream events leading to caspase activation (Li et al., 1997).
Necrosis is characterized by cellular swelling, rupture of the plasma membrane and release of the cellular components to the extracellular space (Laster et al., 1988; Proskuryakov et al., 2003). While necrosis was initially considered a passive form of cell death resulting for instance from extreme cellular stress, it has become clear that necrosis can be induced in a regulated manner by certain physiological or pathophysiological stimuli (Proskuryakov et al., 2002), and viral or chemical inhibitors of caspases cause an increase in necrotic cell death (Vercammen et al., 1998; Cho et al., 2009). This form of programmed necrotic death, called necroptosis, is believed to provide a mechanism to defend against pathogens that suppress apoptosis (Vandenabeele et al., 2010; Vanlangenakker et al., 2012), and participates at least in part in diseases involving acute tissue damage such as terminal ileitis, systemic inflamatory response and ethanol-induced liver injury (Duprez et al., 2011; Gunther et al., 2011; Roychowdhury et al., 2013). Moreover, in a recent study we showed that necroptosis is activated in liver biopsy samples of patients with drug-induced live injury (Wang et al., 2014). Hence, understanding the molecular pathways underlying necroptosis can provide novel therapeutic opportunities for a wide variety of devastating diseases.
Necroptosis requires receptor-interacting kinase 1 (RIP1) (Holler et al., 2000; Degterev et al., 2008), which also plays an important role in TNF-induced apoptosis (Festjens et al., 2007; Wang et al., 2008). The key factor that switches TNF-induced cell death from apoptosis to necrosis is the RIP1-related kinase RIP3 (Cho et al., 2009; He et al., 2009; Zhang et al., 2009). During necroptosis, RIP3 interacts with RIP1 to recruit the downstream effector mixed-lineage kinase domain-like (MLKL) protein to form the necrosome, within which MLKL becomes phosphorylated at Threonine 357 and Serine 358 (Sun et al., 2012; Wang et al., 2014). The functional importance of MLKL was emphasized by the finding that a small molecule called necrosulfonamide (NSA) specifically blocks necroptosis downstream of RIP3 activation by covalently reacting with Cys86 of human MLKL (Sun et al., 2012).
To elucidate how MLKL causes necroptosis, we have investigated its biochemical and structural properties, and a molecular understanding of MLKL function has started to emerge from data that we present in our separate study (Wang et al., 2014) as well as from results reported by other groups during the course of our work. The crystal structure of mouse MLKL has shown that this protein comprises a C-terminal kinase-like domain that contains an unusual pseudoactive site near the RIP3 phosphorylation site and is tethered to an N-terminal four-helix bundle through a connecting α-helix, although the region harboring the residue homologous to C86 of human MLKL was not observable (Murphy et al., 2013). We and others found that MLKL oligomizes and translocates to membranes (Cai et al., 2014; Chen et al., 2014; Wang et al., 2014). However, while this event was proposed to mediate localization of MLKL to the plasma membrane and to induce Ca2+ influx (Cai et al., 2014) or Na+ influx, thus increasing osmotic pressure and eventually causing membrane rupture (Chen et al., 2014), we found that phosphorylated MLKL localizes not only to the plasma membrane but also to internal membranes, and that the MLKL N-terminal region binds directly to phospholipids and induces membrane permeabilization (Wang et al., 2014). These findings suggest that necrotic cell death results from disruption of multiple membranes by the MLKL N-terminal region upon phosphorylation of the MLKL kinase-like domain by RIP3. However, the molecular mechanism underlying membrane permeation by MLKL remains unclear.
The study presented here was designed to dissect how different structural elements of the MLKL N-terminal region contribute to MLKL function. We determined the solution structure of the MLKL N-terminal region determined by NMR spectroscopy and found a similar architecture to that observed in the crystal structure of MLKL, with a four-helix bundle bound to a C-terminal helix, but with an additional α-helix that includes C86 and also contacts the C-terminal helix at the top of the domain. Fluorescence experiments with the environment sensitive probe 7-nitrobenz-2-oxa-1,3-diazole (NBD) and liposome leakage assays reveal that the four-helix bundle is responsible for membrane insertion and membrane permeabilization, while the C-terminal helix inhibits this activity. Our results suggest a model whereby opening of the four-helix bundle at a site where two helices are poorly packed exposes the hydrophobic interior to enable membrane insertion, and the C-terminal helix acts as a plug that hinders such opening and couples allosteric changes caused by RIP3 phosphorylation on the kinase-like domain to activate membrane permeation by MLKL.
RESULTS
Residues 2-154 of MLKL form an autonomously folded module
Secondary structure predictions suggested that human MLKL contains a helical region from residues 1 to 178. In a separate study, we showed that a minimal fragment spanning this region (residues 2-178) binds to membranes containing polyacidic phospholipids such as phosphatidylinositol 4,5-bisphosphate (PIP2), a component of the plasma membrane, or cardiolipin, a component of the mitochondrial membranes (Wang et al., 2014). To further study the biochemical and structural properties of the N-terminal helical region of MLKL, we first tried to identify a minimal folded domain. For this purpose, we acquired 1H-15N heteronuclear single quantum coherence (HSQC) spectra of MLKL(2-178) and a shorter fragment ending at residue 154 [MLKL(2-154)] that could also be expressed in soluble form. 1H-15N HSQC spectra can be considered like protein fingerprints with one cross-peak for each non-proline residue in a 15N-labeled protein, and provide a powerful tool to assess the folding of the protein from the dispersion of the cross-peaks, as well as to study interactions with a target molecule(s) from the perturbations in the fingerprints caused by the target molecule (Rizo et al., 2012). The 1H-15N HSQC spectrum of MLKL(2-154) was of high quality and exhibited excellent dispersion (Figure 1, black contours), as expected for a monomeric, well-folded domain. The 1H-15N HSQC spectrum of MLKL(2-178) was similar, with well-resolved cross-peaks that were in the same positions observed for MLKL(2-154) but had broader line widths, and additional cross-peaks in the center of the spectrum (Figure 1, red contours). A few of the cross-peaks from residues 2-154 were shifted and/or strongly broadened in MLKL(2-178) but, based on the resonance assignments obtained for structure determination (see below), these cross-peaks correspond to residues that are located near residue 154 (Figure S1 available online). Hence, the perturbations in these cross-peaks can be attributed to proximity to the additional C-terminal residues in MLKL(2-178) and do not arise from overall conformational changes. These results show that MLKL(2-154) forms a monomeric autonomously folded domain in solution, that MLKL(2-178) has a higher tendency to aggregate, and that the additional C-terminal residues in MLKL(2-178) are largely unstructured in the context of this longer fragment.
Figure 1.
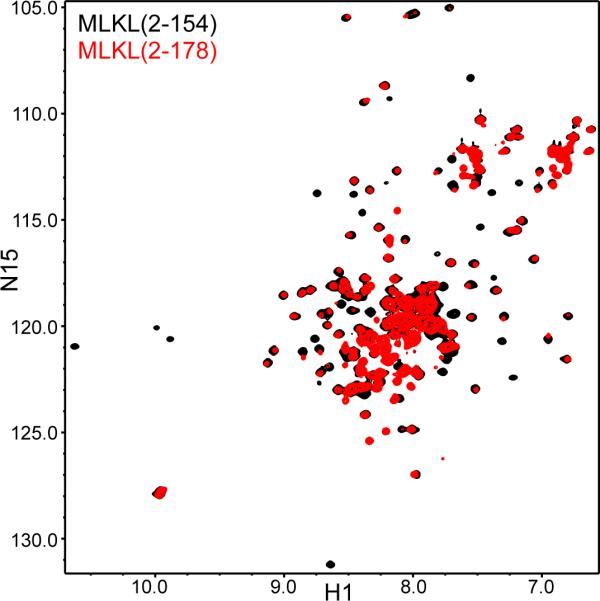
MLKL(2-154) forms an autonomously folded domain. The contour plots show superimposed 1H-15N HSQC spectra of MLKL(2-154) (black contours) and MLKL(2-178) (red contours.
MLKL(2-154) causes permeabilization of cardiolipin-containing membranes
To test whether MLKL(2-154) binds to membranes containing cardiolipin, as described for MLKL(2-178) (Wang et al., 2014), we performed co-floatation assays with liposomes containing 2%, 5% and 15% cardiolipin. Note that the larger percentages of cardiolipin correspond with those present in mitochondrial membranes (Getz et al., 1962). We observed increased binding of the MLKL(2-154) to liposomes with increasing amounts of cardiolipin and liposome binding was concomitant with the formation of MLKL(2-154) oligomers under the non-reducing conditions used in these experiments (Figure S2). This finding suggested that disulfide bond formation contributed to MLKL(2-154) oligomerization, although it may also arise from the confinement of the protein on the membranes.
To study how MLKL(2-154) binds to membranes, we attempted to use nanodiscs, which are self-assembled disc-like phospholipid bilayers surrounded by a membrane scaffolding protein. Because nanodiscs of relatively small sizes (e.g. 10-12 nm) can be obtained by using appropriate fragments of apolipoprotein A1 (Denisov et al., 2004), they provide a powerful tool to analyze proteins in membrane environments by NMR spectroscopy using transversed relaxation optimized (TROSY)-based experiments (Shenkarev et al., 2009; Brewer et al., 2011; Hagn et al., 2013). Intriguingly, addition of nanodiscs containing 15% cardiolipin did not cause any significant changes in the positions and intensities of the 1H-15N TROSY-HSQC cross-peaks of MLKL(2-154), whereas strong decreases in cross-peak intensities were observed upon addition of liposomes with the same phospholipid composition (compare black and red contours in the two-dimensional plots and the one-dimensional traces shown in Figures 2A,B). The latter result was expected because liposome binding should result in broadening of the cross-peaks beyond detection [the residual observable signals can be attributed to residual MLKL(2-154) that remains unbound because of membrane saturation]. Conversely, the absence of any effects of the nanodiscs on the 1H-15N TROSY-HSQC spectrum of MLKL(2-154) is surprising and indeed remarkable, since these spectra are very sensitive to even a small amount of binding to large species such as nanodiscs (Rizo et al., 2012), and shows that there is no detectable binding of MLKL(2-154) to nanodiscs even at the total lipid concentrations used in these experiments (1 mM).
Figure 2.

MLKL(2-154) binds to liposomes but not to nanodiscs. A,B. 1H-15N TROSY-HSQC spectra of MLKL(2-154) in the absence (black contours) and presence (red contours) of nanodiscs (A) or liposomes (B) containing 45% POPC, 30% POPE, 10% PI and 15% cardiolipin. The one-dimensional traces shown above the two-dimensional contour plots were taken at the 15N chemical shift indicated by the blue arrows and illustrate how the cross-peak intensities were not significantly affected by the nanodiscs but were strongly decreased by the liposomes.
The finding that MLKL(2-154) binds to liposomes but not to nanodiscs suggested that phospholipid binding does not reflect merely a surface interaction but rather involves insertion of at least part of MLKL(2-154) into the phospholipid bilayer, and such insertion is prevented in nanodiscs because a large number of lipids would need to displaced within the constraints imposed by the scaffolding protein to make room for MLKL(2-154). This effect against insertion into nanodiscs is expected to be compounded by formation of MLKL(2-154) oligomers [Figure S2; see also (Wang et al., 2014)]. These observations, and the fact that other protein domains that are highly soluble in solution but oligomerize within membranes can form membrane pores [e.g. (Mukherjee et al., 2014)], led us to hypothesize that MLKL may permeabilize membranes. Indeed, we found that MLKL causes liposome leakiness, that mutations mimicking phosphorylation by RIP3 enhance this activity, and that leakiness is caused by MLKL(2-178) but not by the MLKL kinase-like domain (Wang et al., 2014). In the context of the present study, we tested whether the smaller MLKL(2-154) fragment is sufficient to cause liposome leakiness using a sulforhodamine B de-quenching assay. We found that MLKL(2-154) induces liposome leakiness in a dose-dependent, saturable manner, with an EC50 of 140 nM under the conditions of our experiments (Figure 3). At concentrations on the 150 nM range, MLKL(2-154) did not cause an increase in vesicle size (Figure S3), indicating that leakiness does not arise from large membrane rearrangements leading to fusion. This result correlates with the finding that the pores induced in liposomes by MLKL under similar conditions are permeable mostly to molecules smaller than 10 kDa, although increasingly larger molecules permeate with high MLKL concentrations (Wang et al., 2014).
Figure 3.
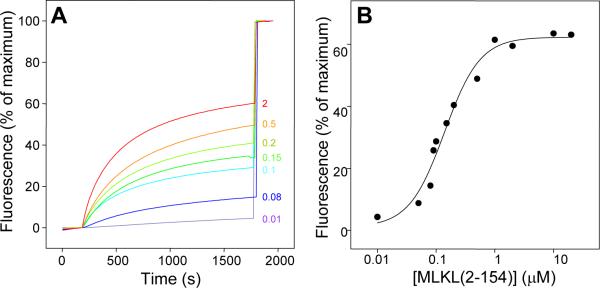
MLKL(2-154) causes membrane leakiness. A. Liposome leakage assays monitoring the de-quenching of sulforhodamine B fluorescence as a function of time in the presence of difference concentrations of MLKL(2-154) (color-coded and indicated on the right in μM concentrations). The protein was added after 200 s. The data were normalized with the fluorescence observed after adding detergent, and setting at zero the fluorescence right before protein addition. B. Plot of the fluorescence intensity observed after 1700 s (expressed as % of the maximum) as a function of MLKL(2-154) concentration. The curve shows the fit to a Hill equation, which yielded an EC50 of 140 nM and a Hill coefficient of 1.5.
Solution structure of MLKL(2-154)
Our results and the prediction that MLKL(2-154) contains multiple α-helices suggested that the mechanism of action of the MLKL N-terminal domain might resemble that of proteins from the Bcl-2 family of proteins such as Bax, which contain soluble helical domains that insert into membranes and permeabilize them (Epand et al., 2002; Wei et al., 2001; Kuwana et al., 2002). To investigate whether the architecture of MLKL(2-154) is similar to that of Bcl-2 proteins, we attempted to crystallize this MLKL fragment but very limited precipitation was observed in a variety of screens even at a protein concentration of 100 mg/ml, showing that the fragment is highly soluble. Hence, we determined the three-dimensional structure of MLKL(2-154) in solution using NMR spectroscopy. Final structures were calculated using 2,864 experimentally-derived restraints. The structural statistics are summarized in Table 1, and Figure 4A shows a backbone superposition of the 20 structures with the lowest NOE energies. The superposition shows that the structure is well defined throughout much of MLKL(2-154). At the outset, the availability of the crystal structure of mouse MLKL that was recently reported (Murphy et al., 2013) allowed us comparison of our solution structure of the isolated N-terminal domain with the structure of this domain in the context of the crystals of full-length MLKL. A sequence alignment of MLKL N-terminal domains from different species is shown in Figure S4 to facilitate assessment of the degree of evolutionary conservation of residues discussed below.
Table 1.
Structural statistics for the 20 simulated annealing structures of the MLKL N-terminal domain with the lowest NOE energies
| Average r.m.s. deviations from experimental restraints (2864 total) | ||
| NOE distance restraints (Å) | ||
| All | 2498 | 0.012 ± 0.0002 |
| Intraresidue | 324 | 0.010 ± 0.0011 |
| Sequential (|i-j|=1) | 475 | 0.009 ± 0.0008 |
| Short range (|i-j|=2-4) | 923 | 0.014 ± 0.0004 |
| Long range (|i-j|>4) | 776 | 0.012 ± 0.0004 |
| Hydrogen bonds (Å) | 134 | 0.008 ± 0.0010 |
| Dihedral angles (°) | 232 | 0.105 ± 0.017 |
| Average r.m.s deviations from idealized covalent geometry | ||
| Bonds (Å) | 0.0021 ± 0.0004 | |
| Angles (°) | 0.41 ± 0.007 | |
| Impropers (°) | 0.30 ± 0.006 | |
| Ramachandran plot statistics (%)1 | ||
| Residues in most favored regions | 86.1 | |
| Residues in additionally allowed regions | 12.2 | |
| Residues in generously allowed regions | 1.2 | |
| Residues in nonallowed regions | 0.5 | |
| Average r.m.s deviations of atomic coordinates (Å) | ||
| Backbone residues (2-150) | 0.48 | |
| Heavy-atom residues (2-150) | 1.31 | |
| Backbone secondary structure2 | 0.46 | |
| Heavy-atom secondary structure2 | 1.20 | |
Calculated using the program PROCHECK.
Six α-helices, residues 2-20, 25-47, 56-80, 82-91, 99-120, 133-150
Figure 4.

Structure of MLKL(2-154) in solution. A. Backbone superposition of the 20 NMR structures of MLKL(2-154) with the lowest NOE energies. The six α-helices are labeled H1-H6 and correspond to residues 2-20 (H1), 25-47 (H2), 56-80 (H3), 82-91 (H4), 99-120 (H5) and 133-150 (H6). N and C indicate the N- and C-termini, respectively. B. Ribbon diagram of the solution structure of MLKL(2-154). The helices that form the four-helix bundle are colored in blue, the helix in the top that contains C86 (helix H4) in yellow, and the C-terminal helix (H6) in red. The positions of selected residues are indicated to help identifying the locations of residues discussed in the text within the structure. C. Superposition of ribbon diagrams of the solution structure of MLKL(2-154) (blue) and the crystal structure of mouse MLKL (orange) (PDB accession code 4BTF) (Murphy et al., 2013). The atoms of the residue of the kinase-like domain of mouse MLKL that is phosphorylated by RIP3 (S345) are shown as spheres and colored in green. D. Close-up of the interface between the kinase-like domain and the N-terminal helical domain in the structural superposition shown in panel C but showing also the proteins as stick models. The red ellipses show areas where helix H4, which is observed in our solution structure but is not observed in the crystal structure, would have steric clashes with the kinase-like domain in the relative orientation between the two domains observed in the crystal structure. Selected residues involved in the clashes are labeled.
The solution structure of MLKL(2-154) revealed six α-helices that we will refer to as H1-H6 (Figures 4A,B). Helices H1, H2, H3 and H5 form a four-helix bundle that packs against the C-terminal helix (H6) at the top half of the domain (in the orientation of Figure 4B). This architecture is similar to that observed for the N-terminal domain in the crystal structure of mouse MLKL but, apart from some differences in the packing of the helices that are discussed below, a clear difference between the two structures is the presence of helix H4 at the top of the domain in our solution structure (Figures 4B,C). This helix contains C86, the cysteine that constitutes the target of NSA to block necroptosis (Sun et al., 2012), but no density is observable for the corresponding sequence in the crystal structure of mouse MLKL (Figure 4C) (Murphy et al., 2013). The structural superposition of Figure 4D shows that steric clashes between H4 and the kinase-like domain would occur if helix H4 were formed in the context of the crystal structure. These observations suggest that steric clashes prevent formation of helix H4 in the conformation observed in the crystals and that there is an intimate interplay between the orientation of the kinase-like domain and the structure of the N-terminal domain, particularly at the top of the domain.
The four-helix bundle of MLKL(2-154) mediates membrane insertion
To explore whether membrane insertion involves large changes in secondary structure, we acquired circular dichroism (CD) spectra of MLKL(2-154) in the absence and presence of liposomes (Figure S5A). The CD spectrum of free MLKL(2-154) was characteristic of a highly α-helical protein, as expected. Addition of lipsomes caused only small changes, indicating that the protein remains highly α-helical upon membrane insertion, but this finding does not exclude the posibility of large changes in tertiary structure.
To gain further insights into how MLKL(2-154) inserts into membranes, we designed an approach that takes advantage of the high sensitivity of the fluorescent probe 7-nitrobenz-2-oxa-1,3-diazole (NBD) to its environment, exhibiting large increases when the probe is transferred from aqueous solution to a hydrophobic milieu (Crowley et al., 1993; Arac et al., 2006). The approach is also based on the finding that DTT impaired the liposome-leaking activity of MLKL(2-154) only to a small extent (Figure S5B), indicating that cysteines are not crucial for this activity. Hence, we created single cysteine versions of MLKL(2-154) by mutating the four native cysteines to serines, and then introducing cysteines in strategic positions of the different helices that are solvent exposed in the structure of MLKL(2-154). Below we will refer to these mutants naming only the residue that was mutated to cysteine (e.g. T9C for the mutant with all native cysteines mutated to serine and Thr9 mutated to cysteine). We also prepared one MLKL(2-154) mutant where we retained the native cysteine C86, which is located in helix H4, and mutated the other three native cysteines to serines (referred to as C86 mutant). All mutant proteins were labeled with NBD and their fluorescence spectra were recorded in the absence and presence of liposomes (Figure 5). The wavelengths of the fluorescence maxima observed, their intensities and the ratios between the maximum intensities in the absence and presence of liposomes (r) are listed in Table 2. All NBD-labeled proteins were able to induce liposomes leakiness albeit to different extents (Figure S5C,D). Thus, although the different activities in the leakiness assay may reflect differential effects of the mutations on the structure and/or oligmerization propensity of membrane-bound MLKL(2-154), all mutants remained functional to some degree.
Figure 5.

The MLKL four-helix bundle is responsible for membrane insertion. A-K. NBD fluorescence spectra of the indicated MLKL(2-154) mutants in the absence (black) and presence (red) of liposomes containing 15% cardiolipin. L. Ribbon diagram of the solution structure of MLKL(2-154) summarizing the NBD fluorescence results shown in panels A-K. The side chain of C86 and of residues that were replaced with cysteine and labeled with NBD are shown as spheres, labeled with the residue number and color coded: red indicates low NBD fluorescence in the absence of liposomes and strong increase in intensity upon addition of liposomes, orange indicates stronger than usual NBD fluorescence in the absence of liposomes (which increased further upon liposome addition), yellow indicates low NBD fluorescence in the absence of liposomes and a moderate increase upon liposome addition, and gray denotes low NBD fluorescence in the absence and presence of liposomes.
Table 2.
Fluorescence changes upon liposome binding for NBD labels placed at different positions of MLKL(2-154)
| Residue | λmax - lipidsa | Imax - lipidsb | λmax + lipidsa | Imax + lipidsb | rc |
|---|---|---|---|---|---|
| T9C | 548 | 0.32 | 537 | 1.34 | 4.2 |
| V13C | 547 | 0.24 | 532 | 1.20 | 5.0 |
| H33C | 546 | 0.31 | 534 | 1.39 | 4.5 |
| M44C | 551 | 1.10 | 536 | 1.56 | 1.4 |
| T60C | 548 | 0.29 | 539 | 1.05 | 3.6 |
| A67C | 557 | 0.72 | 536 | 1.22 | 1.7 |
| C86 | 542 | 1.42 | 535 | 1.85 | 1.3 |
| D100C | 551 | 0.24 | 533 | 1.05 | 4.4 |
| D107C | 550 | 0.26 | 536 | 1.11 | 4.3 |
| L114C | 552 | 0.24 | 539 | 0.61 | 2.5 |
| Q139C | 551 | 0.18 | 546 | 0.12 | 0.7 |
In nm. Estimated error ± 2 nm for the NBD labels with stronger fluorescence intensities and ± 3 nm for those with smaller intensities.
In arbitrary units. Values represent averages from at least five measurements. Standard deviations range from 3% to 7%.
The ratio r is defined as (Imax + lipids)/(Imax - lipids).
Six of the mutants (T9C, V13C, H33C, T60C, D100C and D107C) exhibited low NBD fluorescence intensities in the absence of liposomes and large intensity increases upon liposome binding (Figures 5A,B,C,E,H,I), with intensity ratio r ranging from 3.6 to 5.0 (Table 2; see residues colored in red in Figure 5L). This behavior is expected for NBD probes that are exposed to the aqueous millieu in monomeric soluble MLKL(2-154) but are transferred into a hydrophobic environment upon lipid binding, either because they are in direct contact with the acyl chains of the bilayers or because they become buried within new intramolecular or intermolecular helix-helix interfaces formed in the membrane-bound oligomer. The L114C mutant (colored in yellow in Figure 5L) also had a low NBD fluorescence intensity in the absence of lipids but underwent a considerably lower increase in intensity upon lipid binding (Figure 5J), indicating that the probe occupies a more interfacial location of the bilayer. Three of the mutants (M44C, A67C and C86) exhibited higher fluorescence intensities in the absence of lipids than expected for an aqueous environment (Figures 5D,F,G), suggesting that the NBD probe already experiences a somewhat hydrophobic environment in soluble MLKL(2-154) due to packing with neighboring side chains. Nevertheless, increases in NBD fluorescence intensities were also observed for these three mutants (residues colored in orange in Figure 5L). Therefore, the NBD probes in these mutants may also become in contact with the lipids or may be buried within the protein upon membrane binding. Finally, a low NBD fluorescence intensity was observed for the Q139C mutant in the absence and presence of liposomes (Figure 5K; residue colored in gray in Figure 5L)), showing that the probe remains an aqueous environment even upon binding of MLKL(2-154) to the lipids. While the individual results obtained for each mutant need to be interpreted with caution because of the potential effects that the mutations or NBD labeling might have on the structure of membrane bound MLKL(2-154), the overall NBD fluorescence data (summarized in Figure 5L) indicate that much of the four-helix bundle formed by helices H1, H2, H3 and H5 is involved in membrane insertion, whereas the C-terminal helix H6 is not.
Inhibition of membrane permeabilization activity by helix H6 and NSA
To further understand the results obtained in the NBD fluorescence experiments and the mechanism of membrane permeabilization by MLKL(2-154), we examined the packing between the α-helices in our solution structure. Interestingly, we found that the helical pairs H1-H2, H1-H3 and H3-H5 are well packed (Figures 6A-C), as is common in a well-folded domain, but helices H2 and H5 are not well packed against each other and are more distant from each other than is common in neighboring helices (compare Figure 6D with Figures 6A-C). This observation does not arise merely from a lack of NOE data to define the packing in this region of the domain, which can occur in structure determination by NMR spectroscopy (Lu et al., 2002), since the separation between helices H2 and H5 is very similar in our NMR structure of human MLKL(2-154) and the X-ray structure of mouse MLKL (Figure 6E), and the packing between these two helices is also less than optimal in the crystal structure (Figure S6A). We did observe some overt differences in the orientation of some conserved side chains such as W109 (W108 in mouse MLKL) in the interface between helices H2 and H5 (Figure 6E), and these differences do not arise from a poor definition of the NMR structure, since the orientation of the W109 side chain is defined by a large number of long-range NOEs (e.g. Figures S6B,C). We also observed some differences in the mode of packing of helix H6 against helix H2, and in the overall orientation of helix 6, when comparing the NMR and X-ray structures, despite the fact that the side chains in the interface are generally conserved (Figures 6F,G).
Figure 6.

Analysis of packing between helices in MLKL(2-154). A-D. Ribbon diagrams (left) and space filling models (right) of the four pairs of neighboring helices in the four-helix bundle. The helical pairs are oriented with their polar surface in front. The atoms of the side chains of basic residues are colored in blue and those of acidic side chains are colored in red. In other side chains and the backbone, carbon atoms are in green, oxygen atoms are in red, nitrogen atoms are in blue and sulfur atoms are in yellow. The helices are indicated at the bottom. Note the poor packing between helices H2 and H5 (D). E. Superposition of helices H2 and H5 from our solution structure of MLKL(2-154) (blue) and the crystal structure of MLKL (orange) (Murphy et al., 2013). Side chains are shown as stick models. The side chains of W109 of our solution structure and the homologous residue in mouse MLKL (W108) are labeled to emphasize their different orientations. F,G. Ribbon diagrams of helices H2 and H6 in our solution structure of MLKL(2-154) (F) and in the crystal structure of MLKL (G). Side chains are shown as stick models with oxygen atoms in red, nitrogen atoms in dark blue and carbon atoms in lighter blue (F) or orange (G). Residues in the interface between the two helices are labeled. Note that in H2 the residue numbers are the same for homologous residues in the two structures, whereas they are shifted by one residue in H6.
The picture that emerges from these observations is that the structure of MLKL(2-154) is somewhat unusual for a globular protein domain because some of the neighboring elements of secondary structure are not well packed. In particular, the interface between helices H2 and H5 appears to be malleable due to the poor packing and therefore offers a natural place where the structure of the four-helix bundle can ‘open’ and expose its hydrophobic interior to favor membrane insertion and oligomerization. Such oligomerization likely allows formation of pores lined by the abundant charges on the surface of the four-helix bundle, since it would be energetically unfavorable to bury such charged residues in the hydrophobic interior of the bilayer. In this model, the intramolecular interactions of helix H6 with helices H2 and H5 would stabilize the monomeric, water-soluble structure to hinder membrane insertion, and alteration of these interactions by phosphorylation of MLKL would play a key role in the mechanism of activation of its membrane permeabilization function.
To test our model, we prepared a shorter MLKL fragment spanning the four-helix bundle [MLKL(2-123)]. Although this fragment could be expressed efficiently in bacteria in a soluble and monomeric form (as judged by gel filtration), its 1H-15N HSQC spectrum exhibited broader and considerably fewer well-resolved cross-peaks than that of MLKL(2-154) (Figure S7A), and we also observed a tendency to degradation. These findings are consistent with the notion that the structure of MLKL(2-123) is not very stable. Moreover, we found that MLKL(2-123) is much more active than MLKL(2-154) in inducing liposome leakage (Figure 7A; compare red and black curves), strongly supporting the notion that the four-helix bundle is responsible for this activity. To further test our model, we prepared a double mutant of MLKL(2-154) where two basic residues of helix H2 (K26 and R30; see Figure 7B), which form salt bridges with acidic residues of helix H6 (Figure 6F) and hence are likely to stabilize the H2-H6 interface, were mutated to glutamate (K26E,R30E mutant). As expected, this mutation increased the activity of MLKL(2-154) in inducing liposome leakage considerably (Figure 7A, green curve), albeit not as much as truncation of the entire H6 helix. The specificity of this effect was shown by the observation that the liposome leakage activity was impaired by neutralizing K22 and K25, two basic residues that are at the top of the four-helix bundle, near K26 and R30 (Figure 7B), but do not participate in interactions with helix H6 (Figure 7A, blue curve). Co-floatation assays showed that MLKL(2-123) and the MLKL(2-154) K26E,R30E and K22Q,K25Q mutants bind to liposomes to similar extents that appeared to be somewhat lower than that of WT MLKL(2-154) (Figure S7B). Hence, while the impaired activity of the K22Q,K25Q mutant in the leakage assay may arise from partial disruption of lipid binding, consistent with the importance of polyacidic head groups for binding of MLKL to phospholipids (Wang et al., 2014), the increased leakage activities of MLKL(2-123) and the MLKL(2-154) K26E,R30E do not arise from enhanced lipid binding. We also tried to mutate helix 6 in MLKL(2-154) to disrupt its interaction with the four-helix bundle, but unfortunately all mutations attempted (D140K; E136K,D140K; E136Q,D140K; and E136Q,D140Q,E134Q) yielded proteins with a high tendency to degradation and/or aggregation.
Figure 7.
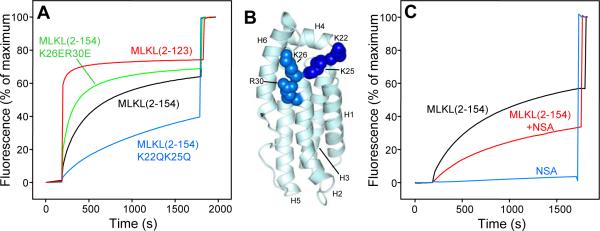
The MLKL four-helix bundle is sufficient to induce liposome leakiness and helix H6 inhibits this activity. A. Liposome leakage assays monitoring the de-quenching of sulforhodamine B fluorescence as a function of time in the presence of WT MLKL(2-154) (black curve), MLKL(2-154) K26E,R30E mutant (green curve), MLKL(2-154) K22Q,K25Q mutant (blue curve) and MLKL(2-123) (red). B. Ribbon diagram of MLKL(2-154) showing the side chains of the mutated residues as spheres (dark blue for K22 and K25; lighter blue for K26 and R30). C. Liposome leakage assays monitoring the de-quenching of sulforhodamine B fluorescence as a function of time in the presence of WT MLKL(2-154) (black curve), NSA (blue curve) and MLKL(2-154) that had been pre-incubated with NSA (red curve). In panels A and C, proteins and/or NSA were added after 200 s. The data were normalized with the fluorescence observed after adding detergent, and setting at zero the fluorescence right before protein or NSA addition.
In a final set of experiments, we tested whether NSA affects the liposome leakage activity of MLKL(2-154), since the inhibition of necroptosis by this small compound was key for the discovery of the functional importance of MLKL (Sun et al., 2012). Pre-incubation of MLKL(2-154) with NSA markedly inhibited its ability to induce liposome leakage, as we observed in experiments performed with full-length MLKL (Wang et al., 2014), and NSA by itself did not cause liposome leakage (Figure 7C). Hence, these leakage assays with the N-terminal helical domain of MLKL appear to recapitulate at least to some extent the inhibition of MLKL function caused by NSA in cells.
DISCUSSION
Necroptosis has emerged as an important form of programmed cell death that is alternative to apoptosis and is implicated in multiple human diseases. While the molecular basis of apoptosis has been exquisitely well characterized, the mechanisms underlying necroptosis are just being discovered. It is now well established that RIP3 is key for commitment of cells to necroptosis (Cho et al., 2009; He et al., 2009; Zhang et al., 2009) and that MLKL constitutes a critical downstream target of RIP3 (Sun et al., 2012). In addition, MLKL was recently found to oligomerize, translocating to membranes (Cai et al., 2014; Chen et al., 2014; Wang et al., 2014), and our recent study showed that MLKL permeabilizes membranes through the N-terminal helical domain upon phosphorylation by RIP3 (Wang et al., 2014). Since a key characteristic of necroptosis is the breakdown of cellular membranes, membrane permeabilization by MLKL most likely constitutes the key event that is triggered by the signals that lead to necroptosis. It is thus crucial to understand how such permeabilization occurs. To address this question, we have determined the solution structure of the MLKL N-terminal region and have studied the molecular mechanism underlying the membrane permeabilization activity. Our results suggest a plug release mechanism whereby the four-helix bundle is responsible for membrane permeation and helix H6 serves as a plug to inhibit opening of the four-helix bundle and membrane insertion, providing at the same time a link to couple phosphorylation of the kinase-like domain to allosteric changes that activate the membrane permeation activity.
The finding that the MLKL N-terminal domain permeabilizes membranes and the prediction that this domain is formed by α-helices suggested a similarity with members of the Bcl-2 family of apoptotic proteins that are also helical and permeabilize membranes such Bax (Wei et al., 2001; Epand et al., 2002; Kuwana et al., 2002). However, our solution structure of MLKL(2-154) and the recently reported crystal structure of MLKL (Murphy et al., 2013) show that the soluble, monomeric form of the MLKL N-terminal is not structurally related to Bax, even though it has a similar size and is also formed by α-helices. Nevertheless, this finding does not completely rule out the possibility that the membrane permeabilization mechanisms of Bax and MLKL might be similar. In the case of Bax, this mechanism has been studied for many years but a consensus model has not yet emerged, which emphasizes the difficulties involved in elucidating how proteins form pores in membranes. Nevertheless, crystal structures of Bax have provided key insights into its activation mechanism, leading to a model whereby inhibitory intramolecular interactions within Bax must be released for its activation (Czabotar et al., 2013). This feature does resemble the mechanism for MLKL activation proposed here.
Our NBD fluorescence experiments (Figure 5) provided key data suggesting this mechanism. As mentioned above, the results obtained for each individual mutant need to be interpreted with caution because the mutations and NBD labeling could alter intramolecular and intermolecular interactions that stabilize the oligomeric pores formed by MLKL(2-154) in phospholipid bilayers. Indeed, the mutations caused a range of effects on the ability of MLKL(2-154) to induce liposome leakiness (Figure S5), which can arise from thermodynamic effects affecting the stability of the MLKL oligomers in the membranes and/or from kinetic effects caused by alteration of the rate of opening of the soluble, monomeric MLKL(2-154). Nevertheless, the overall data obtained from the NBD fluorescence experiments does provide a clear picture whereby much of the four-helix bundle is responsible for membrane insertion, whereas helix H6 does not insert into the membrane. Importantly, this conclusion can be rationalized naturally from examination of our solution structure of the MLKL N-terminal domain as well as from the MLKL crystal structure. Helices H1, H2, H3 and H5, which form the four-helix bundle, are very amphipathic, with hydrophobic surfaces buried inside the domain and highly charged surfaces exposed to the solvent in the monomeric soluble form (Figures 6A-D). Clearly, the charged surfaces cannot be inserted into the hydrophobic interior of a phospholipid bilayer, but the poorly packed interface between helices H2 and H5 provides a natural ‘weak’ point where the structure can open to expose the hydrophobic faces of the helices for insertion into the bilayer. Intramolecular binding of helix H6 to the groove between helices H2 and H5 is expected to stabilize the overall structure of MLKL(2-154) and thus hinder such opening of the structure. Further stabilization is likely provided by contacts of helix H6 with helix H4 at the top of the domain (Figure 4B), which include packing of Phe148 against C86 and other side chains of helix H4 (Figure S7C).
C86 is the residue of MLKL that reacts with the small molecule NSA, which blocks necroptosis (Sun et al., 2012). The finding that the liposome permeabilization activities of MLKL(2-154) (Figure 7C) and full-length MLKL (Wang et al., 2014) are inhibited by NSA indicate that the action of NSA in cells can be reproduced, at least to some extent, in the liposome leakage assays. The effect of NSA in these assays is less drastic that the impairment of necropotosis caused in cells (Sun et al., 2012), but it is likely that the ‘threshold’ for activation of MLKL in cells is considerably higher because of a lower cellular concentration of MLKL and/or because activation is hindered by interactions with other factors within the necrosome. This view is supported by the observation that, although mutations that mimic phosphorylation by RIP3 increase the liposome permeabilization activity of MLKL, the WT protein already has some activity in this assay, whereas phosphorylation of MLKL is required for necroptosis in cells (Wang et al., 2014). The inhibition caused by NSA in the liposome leakage assays suggests that the covalent modification of C86 disrupts intra or intermolecular interactions that are critical for formation of pores within the membranes. Hence, helix H6 likely acts as a plug that inhibits the membrane premeabilization activity not only by binding to the helix H2-helix H5 interface but also through its interactions with helix H4.
Our model is supported by the finding that a MLKL fragment containing the four-helix bundle but lacking helix H6 [MLKL(2-123)] is much more active in the liposome leakage assays than MLKL(2-154), and that mutation of two basic residues from helix H2 that contact helix H6 also enhances the activity of MLKL(2-154) in this assay (Figure 7A). Moreover, the MLKL four-helix bundle is sufficient to trigger necroptosis (Chen et al., 2014). The rest of the protein provides a natural mechanism to inhibit this activity and to couple the RIP3 signal to activation, thus leading to necroptosis. Helix H6 appears to be key for such coupling both because of its role as a plug that inhibits opening of the four-helix bundle and because it provides a connection to the kinase-like domain. The differences observed in the interactions between helices H2 and H6 in our solution structure and the crystal structure of MLKL (Figures 6 F,G) show that the interface between H6 and the four-helix bundle is malleable and hence can be altered through allosteric changes induced by phosphorylation of the kinase-like domain. The fact that helix H4 is observable in our solution structure of the isolated MLKL(2-154) fragment but not in the crystal structure of MLKL (Figure 4C) further shows that there are intimate contacts between the N-terminal domain and the kinase-like domain that can be remodeled upon phosphorylation.
Clearly, much research will be necessary to further test our model, including challenging high-resolution structural studies of the MLKL four-helix bundle in membranes, and it remains to be seen whether the mechanisms used by MLKL and Bax to permeabilize membranes are similar. Indeed, it would not be surprising if these mechanisms are fundamentally different given the lack of a clear structural similarity between their membrane permeabilizing domains, and the fact that there are diverse ways in which soluble proteins can insert into membranes to form pores [e.g. (Mukherjee et al., 2014)]. The work presented here provides an initial model and a framework to address these questions in the future.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
DNA constructs for bacterial expression of human MLKL (2-123), MLKL(2-154) and MLKL(2-178) were prepared from an original vector containing the full-length human MLKL sequence (Sun et al., 2012) and cloned into a pGEX-KG vector using standard recombinant DNA techniques. The different mutations of MLKL (2-154) were made by site-directed mutagenesis using the QuickChange site-directed mutagenesis kit (Stratagene). The proteins were expressed in bacteria (Escherichia Coli BL21) as GST fusion proteins, purified by affinity chromatography on glutathione-sepharose beads (GE Healthcare), cleaved with thrombin on beads, eluted with thrombin cleavage buffer containing 50 mM Tris·HCl (pH8.0)/150 mM NaCl/2.5 mM CaCl2, and further purified by size-exclusion chromatography on a S75 Superdex column (Amersham Biosciences, Pittsburgh, PA) in a buffer containing 20 mM HEPES pH 7.4, 100 mM NaCl. For NMR studies, uniform 15N-labeling or 15N- and 13C-labeling was achieved by growing bacteria in minimal medium containing 15NH4Cl or with 15NH4Cl and [13C6]glucose (CIL Inc.) as the sole nitrogen and carbon sources, respectively.
NMR spectroscopy
NMR spectra for structure determination were acquired on INOVA600 or INOVA800 spectrometers at 25 °C using 1.5 mM samples of uniformly 15N-labeled or 15N,13C-labeled MLKL(2-154) in buffer containing 20 mM HEPES pH7.0, 100 mM NaCl, 2 mM TCEP, with 5% D2O. Chemical shift assignments, NOEs, and amide protection data for structural determination were obtained from a series of gradient enhanced two-dimensional (2D) and three-dimensional (3D) experiments. These experiments included: 3D 15N-edited total correlated spectroscopy (TOCSY)-HSQC, 15N,13C-edited NOESY-HSQC, HNCO, HNCACB, CBCA(CO)NH, H(C)(CO)NH-TOCSY, (H)C(CO)NH-TOCSY, and HCCH-TOCSY spectra for backbone and non-aromatic side chain assignments; 2D aromatic constant time (CT) 1H−13C−HSQC, HBCBCGCDHD, HBCBCGCDCEHE, DQF-COSY, TOCSY and NOESY spectra for aromatic side chain assignments (Zhang et al., 1994; Kay et al., 1994; Muhandiram and Kay, 1994; Kay, 1993; Kay et al., 1993). For 2D DQF-COSY, TCOSY and NOESY experiments, D2O was used as the solvent. The mixing time in NOESY experiments was 100 ms. Protection of amide protons from the solvent was identified from the intensities of exchange cross-peaks with the water resonance in 3D 1H−15N NOESY-HSQC and TOCSY-HSQC experiments. Stereospecific assignments of leucine and valine methyl groups were obtained from a 1H−13C CT-HSQC spectrum acquired on a 1.5 mM sample of 10% 13C-labeled MLKL(2-154). All data were processed with NMRPipe (Delaglio et al., 1995) and analyzed with NMRView (Johnson and Blevins, 1994). The resonance assignments have been deposited in the BioMagResBank with accession number 25135.
Structure calculations
Distance restraints were derived from NOE cross-peak intensities. Restraints of 1.8–2.8 Å, 1.8–3.5 Å, 1.8–5.0 Å and 1.8–6.0 Å were assigned to cross-peak intensities classified as strong, medium, weak, and very weak, respectively, with appropriate pseudoatom corrections using r−6 averaging. Phi and psi torsions angle restraints were generated with the program TALOS (Cornilescu et al., 1999). The restraints were centered at the values predicted by TALOS ± the maximum of 22.5° or 1.5 times the standard deviation observed in the TALOS database matches. The hydrogen bond restraints were set 1.3–2.5 Å for the H/O distances and 2.3–3.5 Å for the N/O distances. Structures of MLKL(2-154) were initially calculated and refined using torsion angle simulated annealing with the program CNS (Brunger et al., 1998). A total of 4,000 structures were calculated with the final set of restraints, and the 20 structures with the lowest NOE energy were selected. The coordinates of the 20 structures have been deposited in the Protein Data Bank (PDB) with accession number 2msv.
Dye leakage assays
Liposomes with lipid composition of 45% POPC, 30% POPE, 10% PI and 15% cardiolipin and with 40 mM sulforhodamine B encapsulated were prepared basically as described (Chen et al., 2006). Briefly, lipid mixtures dissolved in CHCL3 were dried in a glass tube with nitrogen gas and further dried in a vacuum overnight. The lipid films were first re-suspended in 400 μl buffer containing 20 mM HEPES pH7.4, 100 mM NaCl, 40 mM sulforhodamine B by vortexing at least 5 minutes, and then frozen and thawed five times. The final lipid concentration was 5 mM. Liposomes were formed by extruding the re-suspended lipid films through a 80 nm polycarbonate filter with an Avanti extruder for at least 29 times. Liposomes and the free sulforhodamine B were then separated using a PD10 column. For leakage assays, liposomes containing 150 μM lipids were incubated without or with 2 μM WT or mutant MLKL fragments as indicated in the figure legends, except for the titration experiments of Figure 3 where different concentrations of WT MLKL(2-154) were used. For the experiments to test the effect of NSA on the liposome leakage activity of MLKL(2-154), 100 μM 15N-labeled MLKL(2-154) was first incubated with 1 mM NSA at room temperature for 2 hours in a buffer containing 20 mM HEPES pH 7.4, 100 mM NaCl, and 200 μM TCEP. The completeness of the reaction with NSA was confirmed by monitoring the 1H-15N HSQC spectra of the 15N-labeled MLKL(2-154), which revealed shifts and/or broadening beyond detection of cross-peaks from residues near C86, as expected from the finding that this cysteine residue is the target of NSA (Wang et al., 2014) . Dye leakage of the liposomes was measured by monitoring the fluorescence increase at 587 nm (excitation at 565 nm) with a PTI spectrofluorometer. The distribution of liposome sizes was analyzed by DLS as described (Arac et al., 2006).
Supplementary Material
ACKNOWLEDGMENTS
The DD2 console of one of the Agilent 600 MHz NMR spectrometers used for the research presented here was purchased with shared instrumentation grant S10RR026461 from the NIH (to Michael K. Rosen). This work was supported by grant I-1304 from the Welch Foundation (to JR) and NIH grant NS040944 (to JR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Arac D, Chen X, Khant HA, Ubach J, Ludtke SJ, Kikkawa M, Johnson AE, Chiu W, Sudhof TC, Rizo J. Close membrane-membrane proximity induced by Ca(2+)-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat. Struct. Mol. Biol. 2006;13:209–217. doi: 10.1038/nsmb1056. [DOI] [PubMed] [Google Scholar]
- Brewer KD, Li W, Horne BE, Rizo J. Reluctance to membrane binding enables accessibility of the synaptobrevin SNARE motif for SNARE complex formation. Proc. Natl. Acad. Sci. U. S. A. 2011;108:12723–12728. doi: 10.1073/pnas.1105128108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D. Biol. Crystallogr. 1998;54(Pt 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014;16:55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Arac D, Wang TM, Gilpin CJ, Zimmerberg J, Rizo J. SNARE-Mediated Lipid Mixing Depends on the Physical State of the Vesicles. Biophys. J. 2006;90:2062–2074. doi: 10.1529/biophysj.105.071415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Li W, Ren J, Huang D, He WT, Song Y, Yang C, Li W, Zheng X, Chen P, Han J. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24:105–121. doi: 10.1038/cr.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- Crowley KS, Reinhart GD, Johnson AE. The signal sequence moves through a ribosomal tunnel into a noncytoplasmic aqueous environment at the ER membrane early in translocation. Cell. 1993;73:1101–1115. doi: 10.1016/0092-8674(93)90640-c. [DOI] [PubMed] [Google Scholar]
- Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ, Huang DC, Kluck RM, Adams JM, Colman PM. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell. 2013;152:519–531. doi: 10.1016/j.cell.2012.12.031. [DOI] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. Nmrpipe - A Multidimensional Spectral Processing System Based on Unix Pipes. Journal of Biomolecular Nmr. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Denisov IG, Grinkova YV, Lazarides AA, Sligar SG. Directed self-assembly of monodisperse phospholipid bilayer Nanodiscs with controlled size. J. Am. Chem. Soc. 2004;126:3477–3487. doi: 10.1021/ja0393574. [DOI] [PubMed] [Google Scholar]
- Duprez L, Takahashi N, Van HF, Vandendriessche B, Goossens V, Vanden BT, Declercq W, Libert C, Cauwels A, Vandenabeele P. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35:908–918. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- Epand RF, Martinou JC, Montessuit S, Epand RM, Yip CM. Direct evidence for membrane pore formation by the apoptotic protein Bax. Biochem. Biophys. Res. Commun. 2002;298:744–749. doi: 10.1016/s0006-291x(02)02544-5. [DOI] [PubMed] [Google Scholar]
- Festjens N, Vanden BT, Cornelis S, Vandenabeele P. RIP1, a kinase on the crossroads of a cell's decision to live or die. Cell Death. Differ. 2007;14:400–410. doi: 10.1038/sj.cdd.4402085. [DOI] [PubMed] [Google Scholar]
- Fiers W, Beyaert R, Declercq W, Vandenabeele P. More than one way to die: apoptosis, necrosis and reactive oxygen damage. Oncogene. 1999;18:7719–7730. doi: 10.1038/sj.onc.1203249. [DOI] [PubMed] [Google Scholar]
- Getz GS, Bartley W, STIRPE F, Notton BM, RENSHAW A. The lipid composition of rat-liver mitochondria, fluffy layer and microsomes. Biochem. J. 1962;83:181–191. doi: 10.1042/bj0830181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath MF, Becker C. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477:335–339. doi: 10.1038/nature10400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagn F, Etzkorn M, Raschle T, Wagner G. Optimized phospholipid bilayer nanodiscs facilitate high-resolution structure determination of membrane proteins. J. Am. Chem. Soc. 2013;135:1919–1925. doi: 10.1021/ja310901f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000;1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- Johnson BA, Blevins RA. Nmr View - A Computer-Program for the Visualization and Analysis of Nmr Data. Journal of Biomolecular Nmr. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- Kay LE. Pulsed-Field Gradient-Enhanced 3-Dimensional Nmr Experiment for Correlating C-13-Alpha-Beta, C-13′, and H-1-Alpha Chemical-Shifts in Uniformly C-13-Labeled Proteins Dissolved in H2O. Journal of the American Chemical Society. 1993;115:2055–2057. [Google Scholar]
- Kay LE, Xu GY, Singer AU, Muhandiram DR, Formankay JD. A Gradient-Enhanced Hcch Tocsy Experiment for Recording Side-Chain H-1 and C-13 Correlations in H2O Samples of Proteins. Journal of Magnetic Resonance Series B. 1993;101:333–337. [Google Scholar]
- Kay LE, Xu GY, Yamazaki T. Enhanced-Sensitivity Triple-Resonance Spectroscopy with Minimal H2O Saturation. Journal of Magnetic Resonance Series A. 1994;109:129–133. [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- Laster SM, Wood JG, Gooding LR. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J. Immunol. 1988;141:2629–2634. [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Lu J, Garcia J, Dulubova I, Sudhof TC, Rizo J. Solution structure of the Vam7p PX domain. Biochemistry. 2002;41:5956–5962. doi: 10.1021/bi020050b. [DOI] [PubMed] [Google Scholar]
- Muhandiram DR, Kay LE. Gradient-Enhanced Triple-Resonance 3-Dimensional Nmr Experiments with Improved Sensitivity. Journal of Magnetic Resonance Series B. 1994;103:203–216. [Google Scholar]
- Mukherjee S, Zheng H, Derebe MG, Callenberg KM, Partch CL, Rollins D, Propheter DC, Rizo J, Grabe M, Jiang QX, Hooper LV. Antibacterial membrane attack by a pore-forming intestinal C-type lectin. Nature. 2014;505:103–107. doi: 10.1038/nature12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, varez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, Young SN, Varghese LN, Tannahill GM, Hatchell EC, Majewski IJ, Okamoto T, Dobson RC, Hilton DJ, Babon JJ, Nicola NA, Strasser A, Silke J, Alexander WS. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39:443–453. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- Proskuryakov SY, Gabai VL, Konoplyannikov AG. Necrosis is an active and controlled form of programmed cell death. Biochemistry (Mosc. ) 2002;67:387–408. doi: 10.1023/a:1015289521275. [DOI] [PubMed] [Google Scholar]
- Proskuryakov SY, Konoplyannikov AG, Gabai VL. Necrosis: a specific form of programmed cell death? Exp. Cell Res. 2003;283:1–16. doi: 10.1016/s0014-4827(02)00027-7. [DOI] [PubMed] [Google Scholar]
- Rizo J, Rosen MK, Gardner KH. Enlightening molecular mechanisms through study of protein interactions. J. Mol. Cell Biol. 2012;4:270–283. doi: 10.1093/jmcb/mjs036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roychowdhury S, McMullen MR, Pisano SG, Liu X, Nagy LE. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology. 2013;57:1773–1783. doi: 10.1002/hep.26200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenkarev ZO, Lyukmanova EN, Solozhenkin OI, Gagnidze IE, Nekrasova OV, Chupin VV, Tagaev AA, Yakimenko ZA, Ovchinnikova TV, Kirpichnikov MP, Arseniev AS. Lipid-protein nanodiscs: possible application in high-resolution NMR investigations of membrane proteins and membrane-active peptides. Biochemistry. 2009;74:756–765. doi: 10.1134/s0006297909070086. [DOI] [PubMed] [Google Scholar]
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- Vandenabeele P, Galluzzi L, Vanden BT, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- Vanlangenakker N, Vanden BT, Vandenabeele P. Many stimuli pull the necrotic trigger, an overview. Cell Death. Differ. 2012;19:75–86. doi: 10.1038/cdd.2011.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercammen D, Beyaert R, Denecker G, Goossens V, Van LG, Declercq W, Grooten J, Fiers W, Vandenabeele P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 1998;187:1477–1485. doi: 10.1084/jem.187.9.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133:693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- Wang H, Sun L, Su L, Rizo J, Liu L, Wang L-F, Wang F-S, Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell. 2014;54:133–146. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- Zhang O, Kay LE, Olivier JP, Forman-Kay JD. Backbone 1H and 15N resonance assignments of the N-terminal SH3 domain of drk in folded and unfolded states using enhanced-sensitivity pulsed field gradient NMR techniques. J. Biomol. NMR. 1994;4:845–858. doi: 10.1007/BF00398413. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.