

Background: Gefitinib treatment is limited by primary or acquired TKI resistance in NSCLC.
Results: 36245-PHF was identified and investigated in NSCLC, and its underlying mechanism of action was evaluated.
Conclusion: 36245-PHF inhibits proliferation and tumor growth of gefitinib-sensitive and -resistant NSCLC cells by targeting PI3K and Aurora A and B.
Significance:36245-PHF was identified as a novel therapeutic agent to overcome gefitinib-resistant NSCLC.
Keywords: Flavonoid, Lung Cancer, Protein Kinase, Receptor Tyrosine Kinase, Signal Transduction, Aurora Kinase, NSCLC, P13K, Gefitinib Resistance, Multikinase Inhibitor
Abstract
Non-small cell lung cancer (NSCLC) is the most lethal cancer, causing more than 150,000 deaths in the United States in 2013. The receptor tyrosine kinase inhibitors such as gefitinib are not perfect clinical therapeutic agents for NSCLC treatment due to primary or acquired tyrosine kinase inhibitor resistance. Herein, 3,6,2′,4′,5′-pentahydroxyflavone (36245-PHF) was identified as a multiple kinase inhibitor for NSCLC treatment based on the computational screening of a natural products database. 36245-PHF was shown to inhibit PI3K and Aurora A and B kinases and overcome gefitinib-resistant NSCLC growth. Our data clearly showed that 36245-PHF markedly inhibited anchorage-independent growth of gefitinib-resistant NSCLC cell lines and exerted a substantial chemotherapeutic effect following oral administration in a gefitinib-resistant NSCLC xenograft model. The evidence from three different subsequent methodological approaches, in vitro, ex vivo, and in vivo, all confirmed that 36245-PHF as a multiple protein kinase inhibitor. Overall, we identified 36245-PHF as a multiple protein kinase inhibitor and as a novel therapeutic agent to overcome gefitinib-resistant NSCLC growth, which could provide a new option for clinical NSCLC oral treatment.
Introduction
NSCLC4 is one of the most lethal cancers. Unfortunately, NSCLC has limited options for treatment currently, and the overall 5-year survival rate for NSCLC is only 18.2% in the United States (1). Tyrosine kinase inhibitors (TKIs) such as gefitinib have shown an immediate and dramatic clinical response in patients with NSCLC that harbor somatic mutations in the epidermal growth factor receptor (EGFR), including a deletion in exon 19 and an L858R mutation in exon 21 (2). Nonetheless, most patients who initially respond to TKIs eventually develop acquired resistance (3). Reports indicate that approximately one-half of the cases of TKI resistance are associated with the emergence of a secondary mutation of EGFR, T790M (4), whereas the activation of ErbB3/PI3K/Akt signaling mediated by mesenchymal-epithelial transition factor (MET) amplification accounts for ∼22% of acquired resistance (5). Moreover, other mechanisms, including HER2 (human epidermal growth factor receptor 2) amplification, K-ras (v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog) mutations and PTEN (phosphatase and tensin homolog) loss all have been reported to be associated with primary or acquired TKI resistance (6). Although resistance to TKI treatment in NSCLC has increased clinically, the treatment strategies to successfully overcome TKI resistance are still limited.
The PI3K signaling pathway plays a very important role in NSCLC as well as TKI resistance (7). The most common mechanisms of resistance to TKIs in patients such as the secondary T790M mutation of EGFR and mesenchymal-epithelial transition factor (MET) amplification promote cell proliferation and suppress apoptosis in cancer cells by continuously activating the PI3K/Akt signaling pathway (5, 8, 9). Approximately 4% of Japanese lung cancer patient samples were found to harbor a mutation in the PIK3CA gene, which codes for the p110α catalytic subunit of PI3K (10). Consequently, a large number of PI3K inhibitors are now in development and evaluation for clinical trials. Nonetheless, accumulating evidence suggests that combining PI3K inhibitors with other therapies might be more effective than PI3K inhibitors alone (7). Studies suggest that inhibition of both mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinases (ERKs) and the PI3K/Akt pathway might improve efficacy for cancer treatment (11).
Our previous studies have shown that the therapeutic potential of inhibitors targeting Aurora A and B for the treatment of gefitinib-resistant NSCLC is attractive (12, 13). These results indicated that another good strategy might be to combine inhibition of the Aurora kinase pathway with the PI3K/Akt pathway to treat gefitinib-resistant NSCLC. The Aurora family of serine/threonine kinases is another target in cancer therapeutics that plays a crucial role in mitosis by controlling chromatid segregation (14). In malignancy, alterations of Aurora kinase have been linked with genetic instability, including mitotic errors and chromosomal aneuploidy, which are highly associated with tumorigenesis (15). Aurora A and B are overexpressed in many tumor types and have been linked to poor patient prognosis in cancer, including NSCLC (16, 17).
Based on the results of computational screening of a natural products database, we identified 36245-PHF, a flavonoid derivative (18), as a multiple protein kinase inhibitor for use in NSCLC therapy. We investigated the therapeutic effect in NSCLC in vitro and in vivo and evaluated its underlying mechanism of action.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
3,6,2′,4′,5′-pentahydroxyflavone (36245-PHF) (99%) was purchased from Indofine Chemical Co., Inc. (Hillsborough, NJ), and gefitinib was from AstraZeneca Pharmaceuticals (Wilmington, DE). All other compounds were obtained from Sigma-Aldrich. Fetal bovine serum (FBS) was from Gemini Bio-Products (West Sacramento, CA). Eagle's minimum essential medium, F-12K medium, RPMI 1640 medium, basal medium Eagle, gentamicin, penicillin, and l-glutamine were all from Invitrogen. CNBr-activated SepharoseTM 4B beads were from GE Healthcare Biosciences. The antibody against β-actin was from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), and the primary antibodies and kinase buffer were purchased from Cell Signaling Technology (Danvers, MA). All active kinases were from EMD Millipore Corporation (Billerica, MA).
Cell Culture
MRC-5, A549, H1299, H1650, and HCC827 human lung cell lines were obtained from American Type Culture Collection (ATCC; Manassas, VA). Gefitinib-resistant HCC827 (HCC827GR) cells were a gift from Dr. Pasi A. Jänne (Harvard Medical School, Boston, MA). All cells were cytogenetically tested and authenticated before freezing and were thawed and cultured with antibiotics at 37 °C in a CO2 incubator for a maximum of 10 passages. The MRC-5 cell line was cultured in Eagle's minimum essential medium/10% FBS. The A549 cell line was cultured in F-12K medium/10% FBS, and all other cell lines were cultured in RPMI 1640 medium/10% FBS.
MTS Assay
Cells (5 × 103 cells per well) for estimating compound cytotoxicity were seeded onto a 96-well plate and cultured overnight. Then cells were treated with different doses of compound and incubated for various times. The CellTiter96 AQueous MTS reagent (20 μl; Promega Corporation, Madison, WI) was added to each well, and then cells were incubated for 90 min at 37 °C. The optical density (OD) was measured at 490 nm using a plate reader.
Anchorage-independent Cell Growth Assay
A total of 8,000 cells was suspended between a layer of solidified basal medium Eagle/10% FBS/0.5% bottom agar with different concentrations of compound or vehicle and 1 ml basal medium Eagle/10% FBS/0.33% top agar with the same concentration of compound or vehicle in each well of six-well plates. After maintenance at 37 °C, 5% CO2 for 2 to 3 weeks, colonies were scored under a microscope using the Image-Pro Plus software (version 6.2) program (Media Cybernetics. Rockville, MD).
Western Blot
The protein concentration of samples was measured using a protein assay kit (Bio-Rad). Total proteins (20 to 100 μg) from each different sample were resolved by SDS-PAGE and then transferred onto polyvinylidene difluoride (PVDF) membranes (EMD Millipore Corp.) and blocked with 5% milk. Blots were probed with primary antibodies (1:1,000) overnight at 4 °C and followed by incubation with a horseradish peroxidase (HRP)-conjugated secondary antibody for hybridization. Protein bands were visualized with a chemiluminescence reagent (GE Healthcare Biosciences).
Kinase Profiler Screening
Kinase specificity screening was performed by Millipore's Kinase Profiler Service detailed online. The profiling was performed with 10 μm 36245-PHF and 10 μm ATP according to the manufacturer's instructions.
In Vitro Aurora Kinase Assay
Substrate (1 μg) and active Aurora kinase (100 μg) were mixed with different doses of 36245-PHF or hesperadin in a 20 μl reaction, which was conducted in 100 μm ATP and 1 × kinase buffer (Cell Signaling Technology) at 30 °C for 30 min. Reactions were stopped by boiling samples in 6 × SDS buffer, and proteins were analyzed by Western blot.
In Vitro PI3K Assay
The assay was conducted at 30 °C for 10 min with 100 ng of PI3K and different doses of 36245-PHF or LY294002. Each sample was mixed with 20 μl of 0.5 mg/ml phosphatidylinositol as substrate at room temperature for 5 min and then incubated an additional 10 min at 30 °C with reaction buffer (0.25 mm ATP containing 10 μCi [γ-32P]ATP, 10 mm Tris-HCl (pH 7.6), and 60 mm MgCl2). The reaction was stopped by adding 15 μl of 4 n HCl and 130 μl of chloroform:methanol (1:1). After vortexing and fractionation, the lower chloroform phase was spotted onto a silica gel plate (EMD Millipore Corp.) that was preheated at 110 °C for 1 h. The resulting radiolabeled spots were visualized by autoradiography.
Molecular Modeling
To find inhibitors against multiple targets, PI3K, ERK1/2, Aurora A and B, the x-ray crystal structures of PI3K (p110α), ERK1/2, and Aurora A and B were obtained from the Protein Data Bank (19). These structures were then prepared under the standard procedure of Protein Preparation Wizard in Schrödinger Suite 2013 (20). Hydrogen atoms were added consistent with a pH of 7, and all water molecules were removed. Finally, an ATP-binding site-based receptor grid of each kinase was generated for the screening study. The ZINC natural compound database was prepared with the program of LigPrep of Schrödinger 2013 using default parameters for screening. Screening was accomplished using the program Glide in Schrödinger Suite 2013 using default parameters under the standard precision mode and then followed by the extra precision mode. Herein, we could obtain the top scored representative list.
Pulldown and ATP Competition Assays
36245-PHF-Sepharose 4B beads were prepared following the protocol provided by GE Healthcare Biosciences. A549 cell lysate (500 μg) or an active kinase with different concentrations of ATP was incubated with 36245-PHF-Sepharose 4B beads or Sepharose 4B beads only in reaction buffer (50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 5 mm EDTA, 1 mm DTT, 0.01% Nonidet P-40, 0.02 mm phenylmethysulfonyl fluoride, 1 × protease inhibitor mixture and 2 μg/ml bovine serum albumin) at 4 °C with gentle rocking overnight. The beads were then washed with buffer (50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 5 mm EDTA, 1 mm DTT, and 0.01% Nonidet P-40, and 0.02 mm phenylmethysulfonyl fluoride) 5 times. Binding results were analyzed by Western blotting.
Xenograft Mouse Model
Based on the guidelines approved by the University of Minnesota Institutional Animal Care and Use Committee, 6-week-old female athymic nude mice (The Jackson Laboratory, Bar Harbor, ME) were divided into eight groups (n = 10 per group). A549 or HCC827 human lung cancer cells (2 × 106/0.1 ml) were injected subcutaneously into the right flank of each mouse. Tumor and body weight measurements were performed twice a week, and tumor volume was calculated using the ellipsoid formula (length × width × height × 0.52). When tumors reached an average volume of 50 mm3, treatment with 36245-PHF, gefitinib, or vehicle control was initiated and repeated daily by oral gavage in dimethyl sulfoxide (5%), polyethylene glycol (PEG400; 5%), and Tween 80 (5%) in PBS.
Immunohistochemistry Staining
Animal tumor tissues were embedded in paraffin and subjected to immunohistochemistry staining. Tissues were deparaffinized and hydrated and then permeabilized in 0.5% Triton X-100/1 × PBS for 10 min. Immunohistochemical staining for Ki-67 (1:150), phosphorylated (p)-histone H3 (1:400) or p-Akt (1:50) was performed using the indirect avidin biotin-enhanced horseradish peroxidase method according to the manufacturer's instructions (Vector Laboratories, Burlingame, CA). After developing, all sections were observed by microscope (100×) and analyzed using Image-Pro Plus software (version 6.2) program (Media Cybernetics).
Statistical Analysis
Each experiment was performed three times independently, and all data are presented as mean values ± S.D. unless otherwise indicated. Statistically significant differences were determined using the Student's t test to compare data between two groups, or one-way analysis of variance and the Bonferroni's correction to compare data between three or more groups. A p value < 0.05 was considered statistically significant.
RESULTS
36245-PHF, a Potential Multikinase Inhibitor, Has Little Cytotoxicity
From the computational screening result, we found that 36245-PHF (Fig. 1A), a flavonoid derivative, can bind well with PI3K (p110α), ERK1/2, and Aurora A and B at the ATP binding site. Thus, 36245-PHF was selected for further study. We then investigated whether 36245-PHF exerted any cytotoxic effect on normal lung cells. MRC-5 cells were treated with different doses of 36245-PHF for 24 or 48 h and measured by MTS assay. The result showed that 36245-PHF had no cytotoxicity against MRC-5 cells at concentrations <100 μm (Fig. 1B).
FIGURE 1.
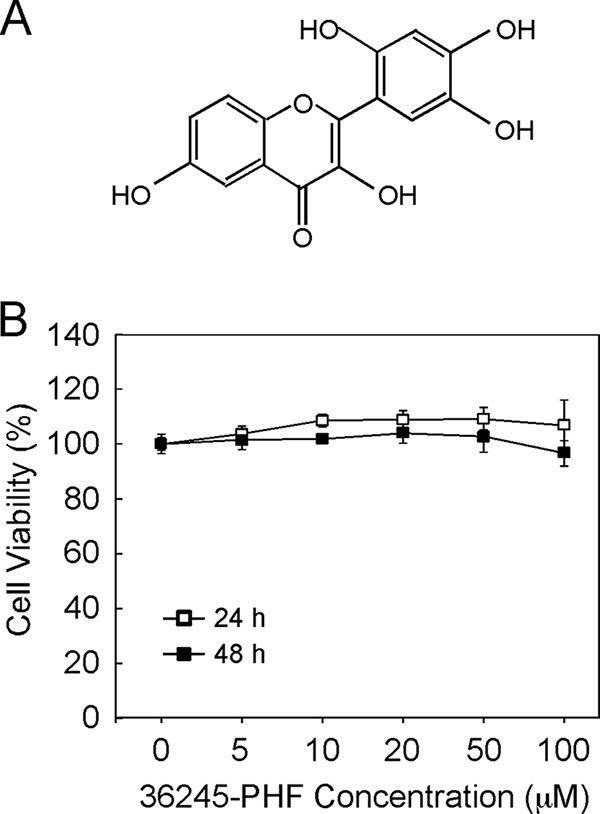
The structure and cytotoxicity of 36245-PHF. A, chemical structure of 36245-PHF. B, cytotoxicity of 36245-PHF was measured in normal lung cells by MTS assay. MRC-5 cells were treated with various concentrations of 36245-PHF for 24 or 48 h. Data are shown as means ± S.D. from triplicate experiments.
36245-PHF Effectively Suppresses Anchorage-independent Growth of Both Gefitinib-sensitive and -Resistant Lung Cancer Cells
Based on the previous data, we then determined whether 36245-PHF had any effect on anchorage-independent growth of human lung cancer cells. First, we examined the effects of 36245-PHF on HCC827, a gefitinib-sensitive NSCLC cell line. The results indicated that gefitinib strongly inhibited HCC827 growth as expected and 36245-PHF could also significantly inhibit HCC827 cell growth in soft agar at different concentrations (Fig. 2A). We also determined the effects of 36245-PHF on growth of four gefitinib-resistant NSCLC cell lines, including HCC827GR, A549, H1299, and H1650 cells. Results showed that 36245-PHF markedly suppresses colony formation of all four gefitinib-resistant NSCLC cell lines dose-dependently, whereas no effect of gefitinib was observed on any of these same cell lines (Fig. 2, B–E).
FIGURE 2.

36245-PHF suppresses anchorage-independent soft agar growth of both gefitinib-sensitive and -resistant NSCLC cell lines. A, the gefitinib-sensitive NSCLC cell line HCC827 was assayed for its ability to proliferate in the presence of 36245-PHF in soft agar. Four gefitinib-resistant NSCLC cell lines, including HCC827GR (B), A549 (C), H1299 (D), and H1650 (E) were also subjected to the soft agar assay. Multicellular colony formation was photographed at ×25 magnification (*, p < 0.05 versus untreated group).
36245-PHF Is a Multikinase Inhibitor in Vitro
To better understand the mechanisms and the activity of 36245-PHF, we profiled its in vitro kinase activity against 41 candidate kinases, which included the previously predicted targeted kinases. Kinase profiling was performed by Millipore's KinaseProfiler based on the protocols provided by Millipore. Data are presented as percentages of kinase activity remaining after treatment with 36245-PHF (Table 1). At a concentration of 10 μm, 36245-PHF inhibited the activity of Aurora B by nearly 80% and also suppressed the activity of Aurora A and PI3K (p110α/p85α) by ∼50% or more (Table 1). However, 36245-PHF, at the same concentration, had little effect on the activity of ERKs or MEK. We then conducted in vitro kinase assays to verify the kinase profiling results. As predicted, 36245-PHF strongly inhibited PI3K kinase activity (Fig. 3A). The findings also revealed that 36245-PHF substantially inhibited the kinase activity of Aurora A and B in a dose-dependent manner from 2.5 to 40 μm (Fig. 3, B and C). All of these results indicate that PI3K (p110α) and Aurora A and B, but not ERKs, are effective targets of 36245-PHF in vitro.
TABLE 1.
Kinase profiling of 36245-PHF
Kinase profiler screening was performed with 10 μm 36245-PHF and 10 μm ATP according to Millipore's protocol. Scores are represented as percent (%) of activity remaining after treatment. The letter h represents human.
| Kinase | Activity | Kinase | Activity |
|---|---|---|---|
| Aurora A (h) | 50 | MET (h) | 91 |
| Aurora B (h) | 23 | mTOR (h) | 97 |
| B-Raf (h) | 103 | p70S6K (h) | 72 |
| CDK1/cyclin B (h) | 103 | PDK1 (h) | 104 |
| CDK2/cyclin A (h) | 87 | Pim-1 (h) | 82 |
| Fyn (h) | 77 | PKBα (h) | 87 |
| GSK3β (h) | 74 | PKBβ (h) | 62 |
| IKKα (h) | 86 | PKCα (h) | 60 |
| IKKβ (h) | 85 | MEK1 (h) | 105 |
| JAK2 (h) | 109 | PKCδ (h) | 70 |
| JNK1α1 (h) | 103 | Ret (h) | 55 |
| JNK2α2 (h) | 98 | Rsk1 (h) | 96 |
| JNK3 (h) | 88 | Rsk2 (h) | 87 |
| KDR (h) | 79 | SAPK2a (h) | 107 |
| Lck (h) | 79 | Src (1–530) (h) | 78 |
| Lyn (h) | 93 | TGFBR1 (h) | 85 |
| MAPK1 (h) | 93 | PI3K (p110α/p85α) (h) | 45 |
| MAPK2 (h) | 76 | PI3K (p110β/p85α) (h) | 63 |
| MAPKAP-K2 (h) | 107 | PI3K (p110δ/p85α) (h) | 52 |
| MSK1 (h) | 101 | PI3K (p110γ) (h) | 87 |
| MSK2 (h) | 95 |
FIGURE 3.

36245-PHF inhibits multiple kinases in vitro. A, 36245-PHF inhibits PI3K kinase activity in vitro. Active kinase PI3K (p110α) and different doses of 36245-PHF or LY294002, a PI3K kinase inhibitor, were mixed with the substrate phosphatidylinositol and then incubated with a [γ-32P]ATP mixture. The resulting radiolabeled spots were visualized by autoradiography. 36245-PHF also inhibits Aurora A (B) or Aurora B (C) kinase activity in a dose-dependent manner. The substrate histone H3 and active kinase Aurora A or Aurora B were mixed with different doses of 36245-PHF or hesperadin, a reported Aurora kinase inhibitor. Reactions were analyzed by Western blot. Data are represented as means ± S.D. from three independent experiments (*, p < 0.05 versus kinase only). DMSO, dimethyl sulfoxide.
36245-PHF Binds Each Target Kinase at the ATP-binding Pocket
To determine whether 36245-PHF could bind with the potential target proteins, we conducted an in silico docking study. Our computer screening returned positive results, which indicated that 36245-PHF formed interactions within the ATP-binding pocket of PI3K (Fig. 4A), Aurora A (Fig. 4B), and Aurora B (Fig. 4C). To further validate these simulations, we conducted ATP competition assays with 36245-PHF-conjugated Sepharose 4B beads. The result showed that PI3K (p110α) was pulled down by 36245-PHF-conjugated Sepharose 4B beads but not by Sepharose 4B beads alone. Moreover, the binding ability of 36245-PHF with PI3K (p110α) was reduced in the presence of ATP (Fig. 4A), and similar results were obtained for the other two potential kinases (Fig. 4, B and C). Furthermore, we observed ex vivo binding between 36245-PHF and PI3K (p110α) and Aurora A or B in A549 cell lysates (Fig. 4, A–C). Taken together, our data indicated that 36245-PHF interacts with PI3K and Aurora A and B in the ATP-binding pocket.
FIGURE 4.

36245-PHF binds to target kinases at the ATP-binding pocket. Computer docking model of 36245-PHF and different target kinases, including PI3K (A) and Aurora A (B) and B (C). The binding was further confirmed by in vitro and ex vivo experiments. Ex vivo binding and in vitro ATP competitive binding of 36245-PHF and kinases were assessed by pulldown and ATP competition assays. The A549 cell lysate or the mixture of active kinase and different concentrations of ATP were incubated with 36245-PHF-Sepharose 4B beads or Sepharose 4B beads only. The protein expression was evaluated by Western blotting (WB).
36245-PHF Is Associated with Inhibition of PI3K/Akt and Aurora Kinase Signaling Pathways in NSCLC Cells
We examined the effect of 36245-PHF on the downstream molecules of PI3K/Akt in human NSCLC cells. 36245-PHF dose-dependent treatment of a gefitinib-sensitive cell line (HCC827) and three gefitinib-resistant NSCLC cell lines (HCC827GR, A549, and H1650) led to a substantial inhibition of phosphorylated Akt (Ser-473), p70S6K (Thr-389) and S6 (Ser-235/236), well known downstream molecules of PI3K (Fig. 5A). We also determined the effects of 36245-PHF on downstream Aurora A and B signaling in NSCLC cells (Fig. 5B). Results indicated that auto-phosphorylation of Aurora A at Thr-288 and of Aurora B at Thr-232 was significantly decreased in the presence of 36245-PHF in either gefitinib-sensitive or -resistant NSCLC cell lines. Furthermore, 36245-PHF substantially blocked the phosphorylation of histone H3 at Ser-10 in a dose-dependent manner. All of our data indicate that 36245-PHF suppresses the activation of PI3K/Akt and Aurora kinases signaling pathway by inhibiting the phosphorylation of their downstream molecules in both gefitinib-sensitive and -resistant NSCLC cells.
FIGURE 5.

The effect of 36245-PHF on signaling pathways on NSCLC cells. 36245-PHF inhibits the PI3K (A) and Aurora kinase (B) signaling pathways in both the gefitinib-sensitive NSCLC cell line (HCC827) and gefitinib-resistant NSCLC cell lines (HCC827GR, H1650, A549). Cells were treated with different concentrations of 36245-PHF for 24 h, dimethyl sulfoxide for 24 h, wortmannin (reported PI3K inhibitor) for 3 h or hesperadin (reported Aurora kinase inhibitor) for 2 h. Total proteins are shown as loading controls.
36245-PHF Suppresses Tumor Growth of Both Gefitinib-sensitive and -Resistant Xenografts
To determine the chemotherapeutic effect of 36245-PHF in vivo, we used an athymic nude xenograft mouse model of gefitinib-sensitive HCC827 cells and the gefitinib-resistant A549 cells. Data showed that gefitinib only inhibited tumor growth in the HCC827 xenograft model as expected, whereas 36245-PHF markedly reduced tumor size in both the HCC827 and A549 xenograft models dose-dependently (Fig. 6, A and B). The higher doses of 36245-PHF (200 mg/kg) treatment showed significantly more inhibition compared with the lower dose (40 mg/kg). Weights of all animals increased normally after daily treatment by oral gavage with 36245-PHF, gefitinib, or vehicle control (Fig. 6, A and B). Additionally, immunohistochemical analysis of 36245-PHF-treated A549 xenograft tumors was conducted to evaluate the expression level of phosphorylated Akt, histone H3, and Ki-67, a cell proliferation marker. Our results showed that Ki-67, phosphorylation of Akt and histone H3 were significantly suppressed in the 36245-PHF-treated group compared with the vehicle- or gefitinib-treated group (Fig. 6C). These results clearly indicated that 36245-PHF exerts a substantial chemotherapeutic effect to overcome gefitinib-resistant xenograft growth in mice through the suppression of PI3K and Aurora kinase activation.
FIGURE 6.

36245-PHF inhibits tumor growth in both gefitinib-sensitive and -resistant NSCLC xenograft mouse models. Treatment with 36245-PHF significantly inhibits tumor growth in the gefitinib-resistant A549 xenograft mouse model (A) or gefitinib-sensitive HCC827 xenograft mouse model (B) versus the vehicle- or gefitinib-treated group. For each xenograft mouse model, athymic nude mice were divided into four groups as described under “Experimental Procedures.” Mice (n = 10) were treated by daily oral gavage with vehicle (□), 150 mg/kg gefitinib (○), 40 mg/kg 36245-PHF (■), or 200 mg/kg 36245-PHF (●). All data are represented as means ± S.D., and significant differences were determined by factorial analysis of variance (**, p < 0.01 versus vehicle). Representative photographs show external appearance of tumors. C, immunohistochemical (IHC) staining of Ki-67, phosphorylated (p)-Akt, and p-histone H3 in tumors formed in mice treated with vehicle, 150 mg/kg gefitinib, or 200 mg/kg 36245-PHF. Representative photographs for each antibody and each group are shown. The integrated optical density (IOD) was evaluated using the Image-Pro Plus software (version 6.2) program. (**, p < 0.01 versus vehicle).
DISCUSSION
Each year, roughly 1.3 million new cases of NSCLC are diagnosed worldwide with only ∼25% being suitable for lobectomy with lymphadenectomy (21). Targeted therapy has emerged as an impressive approach for advanced NSCLC that relies on activated oncogenes in cancer. TKI treatment, one targeted therapeutic strategy, plays a major role in NSCLC treatment. However, according to the Iressa Pan-Asia Study and the literature, not all NSCLC patients respond to TKI treatment. In addition, the initial therapeutic effect of TKI treatment is eventually limited by acquired TKI resistance. Thus, our intent was to identify a novel multiple protein kinase inhibitor to overcome primary or acquired TKI resistance in NSCLC. Based on computational screening results, we identified a flavonoid derivative, 36245-PHF, as a potential multikinase inhibitor targeting PI3K, ERKs, and Aurora kinases. Studies with different cell and animal models showed that certain flavonoids exhibit effects almost equivalent to many Food and Drug Administration-approved anticancer agents and showed less or no cytotoxicity compared with regular anticancer agents (22).
In the present study, 36245-PHF markedly inhibited anchorage-independent growth of both gefitinib-sensitive and -resistant NSCLC cell lines (Fig. 2, A–E). The in vivo study results showed that 36245-PHF exerts a substantial chemotherapeutic effect against both gefitinib-sensitive and -resistant NSCLC xenograft athymic nude mouse models (Fig. 6, A and B). Previous studies indicated that when flavonoids were taken orally, the toxic effects might not be relevant in vivo (23). Our results indicated that consumption of 36245-PHF was well tolerated based on the observation that no significant body weight changes occurred even with the highest dose of 200 mg/kg of 36245-PHF administered by oral gavage (Fig. 6, A and B). To confirm the prediction of our computer docking and to investigate the underlying mechanism explaining how 36245-PHF could overcome gefitinib resistance of NSCLC cells, we conducted a kinase profiler screening assay with 41 candidate kinases. These data showed that PI3K and Aurora A and B, but not ERKs, are potential targets of 36245-PHF (Table 1). Three different approaches, in vitro (Fig. 3), ex vivo (Fig. 5), and in vivo (Fig. 6C), all confirmed that 36245-PHF functions as a multikinase inhibitor. All these results indicated that 36245-PHF overcomes gefitinib-resistant NSCLC cell growth by suppressing the over-activation of PI3K and Aurora A and B.
One of the 36245-PHF target kinases, PI3K, is essential for the activation and translocation of Akt, which links it to fundamental cellular functions such as cell growth, proliferation, differentiation, survival, and intracellular trafficking (24). More importantly, patients with acquired resistance to TKI often exhibit continuous activation of the PI3K/Akt pathway (5). Many researchers suggest that the PI3K/Akt pathway should be an effective target for oncological treatments. Several agents such as BZM120, PX-866 and SAR245408, which specifically target PI3K, are being developed as treatment for NSCLC (25–27). However, due to the presence of signaling feedback loops in cancer cells, not all of the specific PI3K inhibitors are effective for cancer therapeutics as a sole agent. The most effective strategy to overcome TKI resistance by targeting PI3K/Akt pathway might involve combining it with inhibition of other pathways (7). Another two target kinases, Aurora A and B, as indicated earlier, play a critical role in regulating diverse cell cycle events and regulate cell proliferation. However, abnormal activation or deregulation of Aurora A and B converts them into causes or drivers of cancer, including NSCLC (28, 29). Moreover, Aurora A has also been reported to promote gefitinib resistance by NF-κB pathway activation (30). Thus, the overexpression of these kinases in NSCLC makes them efficacious targets for 36245-PHF acting as a therapeutic agent. All of these findings suggest that inhibiting PI3K and Aurora kinases is reasonable for 36245-PHF to overcome gefitinib-resistant NSCLC cell growth.
Alternative pathways, which evade or bypass the inhibition of EGF receptor signaling, enable multitarget agents to overcome TKI resistance by the simultaneous attack of multiple targets (6). As a multiple protein kinase inhibitor, 36245-PHF inhibits cell proliferation and tumor growth of gefitinib-resistant NSCLC cells both in vitro and in vivo by simultaneously targeting PI3K and Aurora A and B.
Numerous and compelling data from laboratory and clinical investigations demonstrated that flavonoids exert excellent effects in cancer chemotherapy and chemoprevention acting through anti-angiogenesis, anti-migration, carcinogen inactivation, cell cycle arrest, antiproliferation, induction of apoptosis, and inhibition of chemoresistance. Accordingly, 36245-PHF might exert its effects through other anti-tumor mechanisms in addition to antiproliferation and tumor growth inhibition by targeting PI3K and Aurora kinases. Further studies of this chemical on its antitumor efficacy in other cancer types in addition to NSCLC and additional targets will yield more information regarding this interesting chemical. To our knowledge, this is the first study to investigate the therapeutic effect of 36245-PHF in gefitinib-resistant NSCLC, and to evaluate the underlying mechanism. In summary, the present study identified 36245-PHF as a novel therapeutic agent to overcome gefitinib-resistant NSCLC cell growth and could provide a new option for clinical oral treatment of NSCLC.
This work was supported by The Hormel Foundation, United Soybean Board, and National Institutes of Health Grants CA1661011, CA172457, R37 C081064, CA027502, and ES016548.
- NSCLC
- non-small cell lung cancer
- 36245-PHF
- 3,6,2′,4′,5′-pentahydroxyflavone
- TKI
- tyrosine kinase inhibitor.
REFERENCES
- 1. DeSantis C. E., Lin C. C., Mariotto A. B., Siegel R. L., Stein K. D., Kramer J. L., Alteri R., Robbins A. S., Jemal A. (2014) Cancer treatment and survivorship statistics, 2014. CA Cancer J. Clin. 64, 252–271 [DOI] [PubMed] [Google Scholar]
- 2. Lynch T. J., Bell D. W., Sordella R., Gurubhagavatula S., Okimoto R. A., Brannigan B. W., Harris P. L., Haserlat S. M., Supko J. G., Haluska F. G., Louis D. N., Christiani D. C., Settleman J., Haber D. A. (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 350, 2129–2139 [DOI] [PubMed] [Google Scholar]
- 3. Jackman D., Pao W., Riely G. J., Engelman J. A., Kris M. G., Janne P. A., Lynch T., Johnson B. E., Miller V. A. (2010) Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J. Clin. Oncol. 28, 357–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pao W., Miller V. A., Politi K. A., Riely G. J., Somwar R., Zakowski M. F., Kris M. G., Varmus H. (2005) Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2, e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Engelman J. A., Zejnullahu K., Mitsudomi T., Song Y., Hyland C., Park J. O., Lindeman N., Gale C. M., Zhao X., Christensen J., Kosaka T., Holmes A. J., Rogers A. M., Cappuzzo F., Mok T., Lee C., Johnson B. E., Cantley L. C., Jänne P. A. (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316, 1039–1043 [DOI] [PubMed] [Google Scholar]
- 6. Wheeler D. L., Dunn E. F., Harari P. M. (2010) Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nat. Rev. Clin. Oncol. 7, 493–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Engelman J. A. (2009) Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat. Rev. Cancer 9, 550–562 [DOI] [PubMed] [Google Scholar]
- 8. Engelman J. A., Mukohara T., Zejnullahu K., Lifshits E., Borras A. M., Gale C. M., Naumov G. N., Yeap B. Y., Jarrell E., Sun J., Tracy S., Zhao X., Heymach J. V., Johnson B. E., Cantley L. C., Janne P. A. (2006) Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J. Clin. Invest. 116, 2695–2706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Turke A. B., Zejnullahu K., Wu Y. L., Song Y., Dias-Santagata D., Lifshits E., Toschi L., Rogers A., Mok T., Sequist L., Lindeman N. I., Murphy C., Akhavanfard S., Yeap B. Y., Xiao Y., Capelletti M., Iafrate A. J., Lee C., Christensen J. G., Engelman J. A., Janne P. A. (2010) Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 17, 77–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kawano O., Sasaki H., Endo K., Suzuki E., Haneda H., Yukiue H., Kobayashi Y., Yano M., Fujii Y. (2006) PIK3CA mutation status in Japanese lung cancer patients. Lung Cancer 54, 209–215 [DOI] [PubMed] [Google Scholar]
- 11. Li H., Schmid-Bindert G., Wang D., Zhao Y., Yang X., Su B., Zhou C. (2011) Blocking the PI3K/AKT and MEK/ERK signaling pathways can overcome gefitinib-resistance in non-small cell lung cancer cell lines. Adv. Med. Sci. 56, 275–284 [DOI] [PubMed] [Google Scholar]
- 12. Xie H., Lee M. H., Zhu F., Reddy K., Peng C., Li Y., Lim do Y., Kim D. J., Li X., Kang S., Li H., Ma W., Lubet R. A., Ding J., Bode A. M., Dong Z. (2013) Identification of an Aurora kinase inhibitor specific for the Aurora B isoform. Cancer Res. 73, 716–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li X., Li H., Li S., Zhu F., Kim D. J., Xie H., Li Y., Nadas J., Oi N., Zykova T. A., Yu D. H., Lee M. H., Kim M. O., Wang L., Ma W., Lubet R. A., Bode A. M., Dong Z. (2012) Ceftriaxone, an FDA-approved cephalosporin antibiotic, suppresses lung cancer growth by targeting Aurora B. Carcinogenesis 33, 2548–2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carvajal R. D., Tse A., Schwartz G. K. (2006) Aurora kinases: new targets for cancer therapy. Clin. Cancer Res. 12, 6869–6875 [DOI] [PubMed] [Google Scholar]
- 15. Dar A. A., Goff L. W., Majid S., Berlin J., El-Rifai W. (2010) Aurora kinase inhibitors–rising stars in cancer therapeutics? Mol. Cancer Ther. 9, 268–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ogawa E., Takenaka K., Katakura H., Adachi M., Otake Y., Toda Y., Kotani H., Manabe T., Wada H., Tanaka F. (2008) Perimembrane Aurora-A expression is a significant prognostic factor in correlation with proliferative activity in non-small-cell lung cancer (NSCLC). Ann. Surg. Oncol. 15, 547–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vischioni B., Oudejans J. J., Vos W., Rodriguez J. A., Giaccone G. (2006) Frequent overexpression of aurora B kinase, a novel drug target, in non-small cell lung carcinoma patients. Mol. Cancer Ther. 5, 2905–2913 [DOI] [PubMed] [Google Scholar]
- 18. Liu M. M., Zhou L., He P. L., Zhang Y. N., Zhou J. Y., Shen Q., Chen X. W., Zuo J. P., Li W., Ye D. Y. (2012) Discovery of flavonoid derivatives as anti-HCV agents via pharmacophore search combining molecular docking strategy. Eur. J. Med. Chem. 52, 33–43 [DOI] [PubMed] [Google Scholar]
- 19. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E. (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schrödinger LLC. (2013) Schrödinger Suite 2013, Schrödinger, LLC, New York, NY [Google Scholar]
- 21. Reck M., Heigener D. F., Mok T., Soria J. C., Rabe K. F. (2013) Management of non-small-cell lung cancer: recent developments. Lancet 382, 709–719 [DOI] [PubMed] [Google Scholar]
- 22. Kamei H., Koide T., Kojimam T., Hasegawa M., Terabe K., Umeda T., Hashimoto Y. (1996) Flavonoid-mediated tumor growth suppression demonstrated by in vivo study. Cancer Biother. Radiopharm. 11, 193–196 [DOI] [PubMed] [Google Scholar]
- 23. Calderón-Montaño J. M., Burgos-Morón E., Pérez-Guerrero C., López-Lázaro M. (2011) A review on the dietary flavonoid kaempferol. Mini Rev. Med. Chem. 11, 298–344 [DOI] [PubMed] [Google Scholar]
- 24. Osaki M., Oshimura M., Ito H. (2004) PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis 9, 667–676 [DOI] [PubMed] [Google Scholar]
- 25. Bendell J. C., Rodon J., Burris H. A., de Jonge M., Verweij J., Birle D., Demanse D., De Buck S. S., Ru Q. C., Peters M., Goldbrunner M., Baselga J. (2012) Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 30, 282–290 [DOI] [PubMed] [Google Scholar]
- 26. Hong D. S., Bowles D. W., Falchook G. S., Messersmith W. A., George G. C., O'Bryant C. L., Vo A. C., Klucher K., Herbst R. S., Eckhardt S. G., Peterson S., Hausman D. F., Kurzrock R., Jimeno A. (2012) A multicenter phase I trial of PX-866, an oral irreversible phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 18, 4173–4182 [DOI] [PubMed] [Google Scholar]
- 27. Shapiro G. I., Rodon J., Bedell C., Kwak E. L., Baselga J., Braña I., Pandya S. S., Scheffold C., Laird A. D., Nguyen L. T., Xu Y., Egile C., Edelman G. (2014) Phase I safety, pharmacokinetic, and pharmacodynamic study of SAR245408 (XL147), an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 20, 233–245 [DOI] [PubMed] [Google Scholar]
- 28. Lo Iacono M., Monica V., Saviozzi S., Ceppi P., Bracco E., Papotti M., Scagliotti G. V. (2011) Aurora Kinase A expression is associated with lung cancer histological-subtypes and with tumor de-differentiation. J. Transl. Med. 9, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smith S. L., Bowers N. L., Betticher D. C., Gautschi O., Ratschiller D., Hoban P. R., Booton R., Santibáñez-Koref M. F., Heighway J. (2005) Overexpression of aurora B kinase (AURKB) in primary non-small cell lung carcinoma is frequent, generally driven from one allele, and correlates with the level of genetic instability. Br. J. Cancer 93, 719–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu C. C., Yu C. T., Chang G. C., Lai J. M., Hsu S. L. (2011) Aurora-A promotes gefitinib resistance via a NF-κB signaling pathway in p53 knockdown lung cancer cells. Biochem. Biophys. Res. Commun. 405, 168–172 [DOI] [PubMed] [Google Scholar]