

Abstract
Previous studies reported that chronic intermittent hypoxia (CIH) results in an imbalanced expression of hypoxia-inducible factor-α (HIF-α) isoforms and oxidative stress in rodents, which may be due either to the direct effect of CIH or indirectly via hitherto uncharacterized mechanism(s). As neural activity is a potent regulator of gene transcription, we hypothesized that carotid body (CB) neural activity contributes to CIH-induced HIF-α isoform expression and oxidative stress in the chemoreflex pathway. Experiments were performed on adult rats exposed to CIH for 10 days. Rats exposed to CIH exhibited: increased HIF-1α and decreased HIF-2α expression; increased NADPH oxidase 2 and decreased superoxide dismutase 2 expression; and oxidative stress in the nucleus tractus solitarius and rostral ventrolateral medulla as well as in the adrenal medulla (AM), a major end organ of the sympathetic nervous system. Selective ablation of the CB abolished these effects. In the AM, sympathetic activation by the CB chemoreflex mediates CIH-induced HIF-α isoform imbalance via muscarinic acetylcholine receptor-mediated Ca2+ influx, and the resultant activation of mammalian target of rapamycin pathway and calpain proteases. Rats exposed to CIH presented with hypertension, elevated sympathetic activity and increased circulating catecholamines. Selective ablation of either the CB (afferent pathway) or sympathetic innervation to the AM (efferent pathway) abolished these effects. These observations uncover CB neural activity-dependent regulation of HIF-α isoforms and the redox state by CIH in the central and peripheral nervous systems associated with the chemoreflex.
Key points
Rats exposed to chronic intermittent hypoxia (CIH) exhibited imbalanced expression of hypoxia-inducible factor (HIF)-α isoforms and oxidative stress in brainstem regions associated with the carotid body (CB) chemoreflex, and in the adrenal medulla, an end organ of the sympathetic nervous system.
Selective ablation of the CB abolished the effects of CIH on HIF-α isoform expression and oxidative stress.
In the adrenal medulla, chemoreflex-mediated sympathetic activation regulates HIF-α isoform expression via muscarinic acetylcholine receptor-mediated Ca2+ influx and the resultant activation of the mammalian target of rapamycin pathway and calpain proteases.
Thus, CB neural activity regulates HIF-α isoform expressions and redox state in the central and peripheral nervous system associated with the chemoreflex pathway under the setting of CIH.
Introduction
Carotid bodies (CBs) are the sensory organs for detecting changes in arterial blood O2 levels (Kumar & Prabhakar, 2012). Increased CB sensory nerve activity by hypoxia results in reflex activation of breathing and sympathetic nerve activity. Breathing disorders with recurrent apnoea manifest as chronic intermittent hypoxia (CIH). Rodents exposed to CIH exhibit augmented CB sensory responses to hypoxia (Peng & Prabhakar, 2004; Rey et al. 2004) and long-lasting increase in baseline CB neural activity known as sensory long-term facilitation (Peng et al. 2003, 2006). Sympathetic nerve activity and blood pressures (BPs) are elevated in CIH-exposed rodents (Fletcher et al. 1992; Kanagy et al. 2001; Zoccal et al. 2007; Silva & Schreihofer, 2011; Peng et al. 2012). Remarkably, ablation of carotid sinus nerves prevents CIH-induced hypertension (Fletcher et al. 1992; Lesske et al. 1997). These findings suggest that an augmented CB chemoreflex contribute to CIH-induced autonomic abnormalities, including the hypertension.
Emerging evidence suggests that oxidative stress from increased reactive oxygen species (ROS) signalling underlies a major cellular mechanism by which CIH mediates autonomic dysfunction (Prabhakar et al. 2007). Evidence includes that CIH-exposed rodents exhibit increased ROS levels in several tissues (Peng et al. 2003, 2006; Kumar et al. 2006), and antioxidant treatment moderates sympathetic activity and prevents hypertension (Kumar et al. 2006). Recent studies on cell cultures showed that CIH-induced ROS signalling is due to dysregulated expression of transcriptional activators, hypoxia-inducible factor (HIF)-1 and -2 (Prabhakar & Semenza, 2012). CIH increases HIF-1α and decreases HIF-2α protein levels, the O2-sensitive regulatory subunits of HIF-1 and HIF-2, respectively (Peng et al. 2006; Nanduri et al. 2009, 2013). CIH-induced oxidative stress involves HIF-1-mediated transcriptional upregulation of genes encoding pro-oxidant enzymes such as NADPH oxidase 2 (Nox2; Yuan et al. 2011). Downregulation of HIF-2α decreases transcription of genes encoding antioxidant enzymes such as superoxide dismutase 2 (Sod2) (Nanduri et al. 2009, 2013). Furthermore, mice with a partial deficiency of HIF-1α do not exhibit oxidative stress, sympathetic activation and/or hypertension when challenged with CIH (Peng et al. 2006), whereas mice with a partial deficiency of HIF-2α present with hypertension and oxidative stress similar to wild-type mice exposed to CIH (Peng et al. 2011; Yuan et al. 2013). Collectively, these studies on cell cultures and rodents demonstrate that CIH-induced oxidative stress stems from an imbalance in pro- and antioxidant enzymes due to the dysregulated HIF-α isoforms.
Remarkably, in intact rodents, CIH-induced HIF-α isoform imbalance and oxidative stress occur not only in the highly O2-sensitive CB but also in the adrenal medulla (AM), which is relatively insensitive to hypoxia in adults (Peng et al. 2006; Nanduri et al. 2009; Yuan et al. 2011). Consequently, a major outstanding question is whether altered HIF-α isoform expression and the oxidative stress in intact animals are direct consequence of CIH, or rather, indirect result of hitherto uncharacterized mechanism(s). Several lines of evidence suggest that neural activity is a potent regulator of gene transcription in the nervous system (Fields et al. 2005; Carulli et al. 2011; Ganguly & Poo, 2013). Given that CIH increases CB neural activity, and ablation of carotid sinus nerves, like antioxidant treatment, prevent CIH-induced autonomic dysfunction, we hypothesized that a heightened CB neural activity triggers CIH-induced HIF-α isoform imbalance and oxidative stress. This possibility was tested on adult rats exposed to CIH for 10 days. Our results demonstrate that selective ablation of the CB prevents CIH-induced dysregulation of HIF-α isoforms and oxidative stress in brainstem regions associated with the chemoreflex and in the AM. We further discovered that CB chemoreflex-mediated sympathetic activation regulates HIF-α isoform expression in the AM via muscarinic acetylcholine (ACh) receptor triggered Ca2+ influx, which activates the mammalian target of rapamycin (mTOR) and calpain proteases.
Methods
Experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of Chicago. Experiments were performed on adult, male Sprague–Dawley rats weighing between 250 and 300 g. Rats were killed with an overdose of anaesthesia (urethane 3 g kg−1 i.p.) at the termination of the experiment.
Procedures
Rats were anaesthetized with a ketamine/xylazine mixture (70/9 mg kg−1 i.p.). Carotid artery bifurcation was exposed, and the CBs were cryocoagulated with liquid nitrogen as previously described (Verna et al. 1975). Sham-operated rats served as controls. The adrenal gland and splanchnic nerve were exposed retroperitoneally and the branch of the splanchnic nerve innervating the adrenal gland was transected unilaterally, while leaving the sympathetic innervation to the contralateral AM intact. Four weeks of postoperative period was allowed for CB-ablated (CBA) rats for recovery of baroreceptor innervation. Rats that underwent adrenal sympathetic ablation (ASA) were allowed to recover from surgery for 1 week. During the postoperative period, rats were given buprenorphine, an analgesic, every 12 h (0.05 mg kg−1 s.c.).
Exposure to chronic intermittent hypoxia
Following surgical recovery, sham-operated, CBA and ASA rats were exposed to CIH. The protocols for CIH exposure were the same as described previously (Peng et al. 2003, 2006). Briefly, conscious rats were exposed to alternating cycles of 5% O2 for 15 s and 21% O2 for 5 min, between 09.00 and 17.00 h for 10 d. Blood O2 saturation levels were monitored during IH in conscious rats using a small animal pulse oximeter probe placed around the neck region (MouseQx Plus; Starr Life Sciences, Oakmont, PA, USA). Control experiments were performed on rats exposed to alternating cycles of room air instead of hypoxia. Within ∼16 h after terminating the 10th day of CIH, BP and heart rate (HR) were measured in conscious rats and tissue as well as blood samples were collected under anaesthesia (urethane 1.2 g kg−1 i.p.).
Measurements of blood pressure and heart rate in conscious rats
BP was monitored by the tail cuff method in conscious rats using a non-invasive BP system (IITC Life Science Inc., Woodland Hills, CA, USA) as described (Peng et al. 2006, 2011). HR was derived from the electrocardiograph signals and processed with a data acquisition system (PowerLab/8P; AD Instruments, Colorado Springs, CO, USA).
Measurements of phrenic and adrenal sympathetic nerve activities
Acute experiments were performed on rats anaesthetized with urethane (1.2 g kg−1 i.p.). After tracheal intubation, the femoral artery and vein were cannulated. BP was monitored via the femoral artery catheter. The rectal temperature was maintained at 37.5°C ± 1°C by means of a heating pad. The rats were mechanically ventilated (Harvard Apparatus, Holliston, MA, USA) with oxygen-enriched room air and treated with pancuronium bromide (2.5 mg kg−1 h−1 i.v.) to prevent spontaneous breathing. Bilateral vagotomy was performed in the mid-cervical region. The right phrenic nerve was isolated from a ventral approach at the level of C3–C4. The nerve was transected distally, desheathed and placed on bipolar platinum-iridium electrodes. Action potentials were filtered, amplified, rectified, integrated and stored in a computer via an A/D translation board (PowerLab/8P; AD Instruments) for further analysis. The branch of the left splanchnic nerve innervating the AM was isolated retroperitoneally, transected and desheathed. In the experiments involving assessing baroreflex function, the left splanchnic nerve was isolated and cut above the coeliac ganglion. The central cut ends of the nerves were placed on bipolar platinum-iridium electrodes for recording electrical activity as described previously (Peng et al. 2012). At the end of the experiment, hexamethonium (25 mg kg−1 i.v.) was administered to obtain ‘zero’ electrical activity.
Ex vivo carotid body sensory activity
The CB along with carotid bifurcation was harvested in rats anaesthetized with urethane (1.2 g kg−1
i.p.). CB sensory activity was recorded using an ex vivo preparation as described previously (Peng et al. 2003, 2006). Clearly identifiable action potentials (two to three active units) were recorded from one of the nerve bundles of the carotid sinus nerve with a suction electrode. Single units were selected based on the height and duration of the individual action potentials using a spike discrimination program (Spike Histogram Program, Power Laboratory; AD Instruments). The 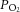 and
and 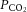 of the superfusion medium were determined by a blood gas analyser (ABL-5; Radiometer, Cleveland, OH, USA). CB sensory activity from single units was averaged during 3 min of baseline (
of the superfusion medium were determined by a blood gas analyser (ABL-5; Radiometer, Cleveland, OH, USA). CB sensory activity from single units was averaged during 3 min of baseline (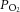 = 142 ± 4 mmHg) and during 3 min of hypoxic challenge (
= 142 ± 4 mmHg) and during 3 min of hypoxic challenge (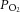 = 40 ± 3 mmHg) and expressed as impulses per second.
= 40 ± 3 mmHg) and expressed as impulses per second.
Blood pressure responses to acute intermittent hypoxia
In anaesthetized rats (urethane 1.2 g kg−1 i.p.), arterial BP was monitored via a catheter in the right femoral artery. The trachea was cannulated and the rats were allowed to breathe room air spontaneously. Ten cycles of intermittent hypoxia (15 s of 12% O2 alternating with 5 min of 21% O2) were delivered via a needle placed inside the tracheal cannula.
Isolation of tissues for analysis of hypoxia-inducible factor-α isoforms, mRNA and oxidative stress
Adrenal glands and brainstem were removed from anaesthetized rats (urethane 1.2 g kg−1 i.p.), frozen in liquid nitrogen and stored at –80°C for further analysis. Coronal brainstem sections (300 μm thick) were cut with a cryostat at –20°C, and rostral ventrolateral medulla (RVLM), nucleus tractus solitarius (nTS) and control brainstem regions were excised with a chilled micro-punch needle. The AM was dissected under ice-cold conditions.
Immunohistochemistry
Anaesthetized rats (urethane 1.2 g kg−1 i.p.) were perfused transcardially with heparinized phosphate-buffered saline (PBS) for 20 min followed by 4% paraformaldehyde in PBS. Brainstem, CBs and adrenal glands were harvested, post-fixed in 4% paraformaldehyde overnight, and cryoprotected in 30% sucrose/PBS at 4°C. Frozen tissues were serially sectioned at a thickness of 8 μm (CB and adrenal gland) or 20 μm (brainstem, coronal section) and stored at –80°C. CB and adrenal gland sections were treated with 20% normal goat serum and 0.2% Triton X-100 in PBS for 30 min (CB) or 2 h (adrenal gland). CB sections were incubated with a monoclonal antibody against tyrosine hydroxylase (1:2000; Sigma, St. Louis, MO, USA). AM sections were incubated with a polyclonal antibody against choline acetyl transferase (1:100; Millipore, Billerica, MA, USA). Sections were rinsed with PBS, stained with 4,6′-diamidino-2-phenylindole and visualized using a fluorescent microscope (Eclipse E600; Nikon, Melville, NY, USA). Brainstem sections were treated with 20% normal goat serum, 0.1% bovine serum albumin and 0.1% Triton X-100 in PBS for 30 min. Sections were incubated with antibodies against HIF-1α (1:1000; Novus Biologicals; Littleton, CO, USA; catalogue no. NB100-479) or HIF-2α (Novus Biologicals; catalogue no. NB100-132) and neuron-specific nuclear protein referred as NeuN (1:500; Millipore). After rinsing with PBS, sections were mounted, covered with coverslip and were viewed under a microscope.
Measurement of plasma adrenaline and noradrenaline levels
Blood samples were collected from anaesthetized rats (urethane 1.2 g kg−1 i.p.) by cardiac puncture. Plasma was separated and catecholamines (CAs) were extracted with acid-activated alumina and plasma noradrenaline and adrenaline levels were determined by high-performance liquid chromatography combined with electrochemical detection, using dihydroxybenzylamine as an internal standard (Kumar et al. 2006).
Measurement of catecholamine secretion from the AM
Experiments were performed on freshly harvested AM from anaesthetized rats (urethane 1.2 g kg−1
i.p.). Two hundred micrometre thick sections were cut. To stabilize the basal efflux, AM slices were incubated in Krebs–Ringer solution equilibrated with room air (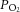 = 142 ± 5 mmHg) and then challenged with hypoxia (
= 142 ± 5 mmHg) and then challenged with hypoxia (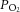 = 40 ± 3 mmHg) for 5 min. Noradrenaline and adrenaline levels in the medium were analysed by high-performance liquid chromatography–electrochemical detection system as previously described (Kumar et al. 2006). The concentrations of noradrenaline and adrenaline were calculated using standard curves constructed with known amounts of CAs. The minimum detection limits for noradrenaline and adrenaline were 50 and 40 fmol, respectively.
= 40 ± 3 mmHg) for 5 min. Noradrenaline and adrenaline levels in the medium were analysed by high-performance liquid chromatography–electrochemical detection system as previously described (Kumar et al. 2006). The concentrations of noradrenaline and adrenaline were calculated using standard curves constructed with known amounts of CAs. The minimum detection limits for noradrenaline and adrenaline were 50 and 40 fmol, respectively.
Reverse-transcriptase quantitative real-time polymerase chain reaction
Nox2 and Sod2 mRNA levels were analysed by RT-qPCR using SYBR Green and normalized to the 18S rRNA signal as described (Nanduri et al. 2009; Yuan et al. 2011). The following primers were used: Nox2: 5′-GTG GAG TGG TGT GTG AAT GC-3′ and 5′-TTT GGT GGA GGA TGT GAT GA-3′; Sod2: 5′-GGC CAA GGG AGA TGT TAC AGC-3′ and 5′-GGC CTG TGG TTC CTT GCA G-3′; 18S rRNA: 5′-CGC CGC TAG AGG TGA AAT TC-3′ and 5′-CGA ACC TCC GAC TTT CGT TCT-3′.
Cell cultures
Rat pheochromocytoma 12 (PC12) cells were plated on coverslips coated with collagen (Sigma), and cultured in DMEM supplemented with 10% (vol vol−1) horse serum, 5% (vol vol−1) fetal bovine serum, penicillin (100 U ml−1) and streptomycin (100 μg ml−1), under 20% O2 and 10% CO2 at 37°C. Cells were placed in antibiotic-free medium and serum starved for 16 h to avoid confounding effects of serum on protein expression. PC12 cells were challenged with repetitive application of muscarine (1, 3, 10, 50 μm; Sigma) or nicotine (3, 10, 50 μm; Sigma) for 5 min followed by 10 min of vehicle for a total of three cycles or were exposed to continuous application of agonists for 15 min. PC12 cells were pretreated with atropine (5 μm; Sigma), BAPTA (10 μm; Invitrogen, Grand Island, NY, USA), N-acetyl-leucine-leucine-methionine-aldehyde (Ac-LLM-CHO 10 μm; Calbiochem, San Diego, CA, USA) or rapamycin (100 nm; Alexis Biochemicals, San Diego, CA, USA) for 30 min before muscarine treatment. Cells were treated with cycloheximide (20 nm; Sigma) immediately after third repetitive application of muscarine.
Intracellular Ca2+ measurements
[Ca2+]i was monitored in PC12 cells using Fura-2. Background fluorescence was subtracted from signals. Image intensity at 340 nm was divided by 380 nm image intensity to obtain the ratiometric image. Ratios were converted to free [Ca2+]i using calibration curves constructed in vitro by adding Fura-2 (50 μm, free acid) to solutions containing known concentrations of Ca2+ (0–2 μm). The recording chamber was continually superfused with solution from gravity-fed reservoirs. After baseline [Ca2+]i levels were monitored, cells were treated with muscarine (10 μm) for 5 min followed by 10 min incubation in Hank's balanced salt solution (HBSS, Gibco®; Life Technologies, Grand Island, NY, USA) for three cycles. Control cells were exposed to HBSS only. To examine the effect of atropine, cells were pretreated with atropine (5 μm) for 30 min before muscarine or HBSS challenge.
Measurement of malondialdehyde levels and aconitase, NADPH oxidase, superoxide dismutase 2 and calpain enzyme activities
Malondialdehyde (MDA) levels were determined as described previously (Nanduri et al. 2009). Mitochondrial and cytosolic fractions were isolated from the AM by differential centrifugation. Aconitase, Nox, Sod2 and calpain activities were determined as described (Nanduri et al. 2009; Yuan et al. 2011). Protein levels were determined by Bradford assay kit (Bio-Rad, Hercules, CA, USA). Micro-punches of RVLM, nTS and a control brainstem region were pooled from two rats for measurements of mRNA levels and activities of pro- and antioxidant enzymes as well as MDA levels; whereas two AMs were pooled for measuring aconitase activity, pro- and antioxidant mRNA levels and enzyme activities.
Immunoblot assays
HIF-1α, HIF-2α and HIF-1β protein levels in lysates prepared from brainstem, AM and PC12 cells were analysed as described (Yuan et al. 2013). The following primary antibodies were used (at the indicated dilution): HIF-1α (1:500; Novus Biologicals; catalogue no. NB100-479), HIF-2α (1:1000; Novus Biological; catalogue no. NB100-132) and HIF-1β (1:1000; Novus Biologicals; catalogue no. NB100-124); phosphorylated mTOR (1:1000) and total mTOR (1:1000) from Cell Signaling (Billerica, MA, USA).
Data analysis
All data are presented as means ± s.e.m. Statistical significance was assessed by one-way ANOVA followed by Tukey's test and unpaired t test, when appropriate. P < 0.05 was considered significant.
Results
Selective ablation of the carotid body
CBs were harvested from anaesthetized sham-operated and CBA rats and were sectioned, stained for tyrosine hydroxylase, a marker of glomus cells, the major chemosensory cell type in the CB. In sham-operated rats, CBs exhibited many tyrosine hydroxylase-positive cells, whereas tyrosine hydroxylase staining was nearly absent in CBA rats, indicating that CB cryocoagulation results in the loss of O2-sensitive glomus cells (Fig. 1A).
Figure 1. CBA selectively impairs chemoreflex but not baroreflex function.
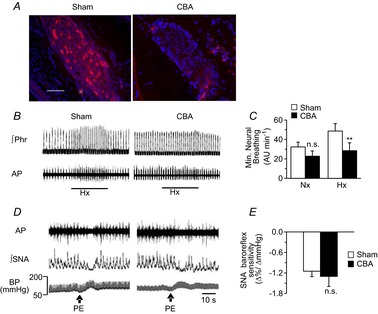
A, rats were subjected to sham surgery or CBA by cryocoagulation. Sections of the carotid body were stained with 4,6′-diamino-2-phenylindole (blue) to detect nuclei and antityrosine hydroxylase antibody (red) to detect type I glomus cells (scale bar, 20 μm). B, representative tracings of phrenic nerve activity during Hx (12% O2) in sham-operated and CBA rats are shown. The black bar indicates the duration of hypoxic challenge. C, minute neural breathing (in AU min–1; means ± s.e.m.) was determined during Nx and Hx in sham-operated (n = 6) and CBA (n = 6) rats. **P < 0.01; n.s., P > 0.05 compared to sham-operated rats. D, representative tracings of SNA responses to increased BP induced by PE (3 μg kg−1 i.v. at arrows) in sham-operated and CBA rats are shown. E, SNA baroreflex response (means ± s.e.m.) of sham-operated (n = 6) and CBA (n = 6) rats; n.s., P > 0.05. AP, action potential; BP, blood pressure; CBA, carotid body ablation; Hx, hypoxia; Nx, normoxia; PE, phenylephrine; ∫Phr, integrated phrenic nerve activity; ∫SNA, integrated splanchnic nerve activity. Note the absence of hypoxic ventilatory response in CBA rats with a fully preserved SNA baroreflex response.
To assess the impact of CBA on chemoreflex function, efferent phrenic nerve responses to hypoxia were determined in anaesthetized rats. Hypoxia (12% inspired O2) increased phrenic nerve activity in sham-operated rats, while this response was absent in CBA rats (Fig. 1B and C). Baroreflex function was determined by recording the splanchnic sympathetic nerve activity (SNA) in response to elevated BP induced by phenylephrine (3 μg kg−1 i.v.). SNA was inhibited in response to increased BP in both sham-operated and CBA rats (Fig. 1D and E). These results established that CBA by cryocoagulation selectively eliminated the CB chemoreflex while preserving the carotid baroreflex function.
Impact of carotid body ablation on chronic intermittent hypoxia-induced hypoxia-inducible factor-α isoform expression in brainstem neurons
Axons from the CB course through the carotid sinus nerve to the brainstem, where they synapse with neurons in the nTS and RVLM, from which the signal is transmitted to the sympathetic nervous system. The neural activity in nTS and RVLM is increased in CIH-exposed rats (Kline et al. 2007; Silva & Schreihofer, 2011; Costa-Silva et al. 2012). We determined the effects of CIH on HIF-α isoform expression in the nTS and RVLM.
Sham-operated and CBA rats were exposed to CIH for 10 days. An example illustrating ambient O2 levels and blood O2 saturation profile during 8 h of IH in a conscious sham-operated rat is shown in Fig. 2A. O2 saturation, on average, decreased from 97% to ∼80% with each episode of hypoxia. The O2 saturation profiles during IH exposure were comparable between sham-operated and CBA rats (P > 0.05).
Figure 2. CBA prevents CIH-induced changes in HIF-α isoform expression in brainstem regions.
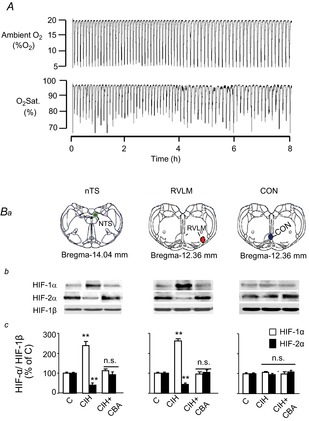
A, representative example of simultaneous measurements of ambient O2 and blood O2 saturation in a conscious sham-operated rat during exposure to 8 h of intermittent hypoxia. B, three groups of rats were studied: sham-operated and exposed to normoxia (C); sham-operated and exposed to CIH; and CIH-exposed CBA rats (CIH + CBA). Anatomical localization of nTS (green), RVLM (red) and a brainstem region that neither receives carotid body sensory input nor regulates sympathetic nerve activity (CON, blue) based on adult rat brain atlas (Paxinos & Watson, 2006) are shown (Ba). Representative immunoblots (Bb) and densitometric analysis (Bc) of HIF-1α, and HIF-2α in the micro-punches from nTS, RVLM and CON regions are shown. Changes in HIF-α isoforms were normalized to HIF-1β (HIF-α/HIF-1β) and presented as means ± s.e.m. relative to sham-operated, normoxia-exposed control rats. n = 6–7 rats each, **P < 0.01; n.s., P > 0.05. C, control; CBA, carotid body ablation; CIH, chronic intermittent hypoxia; HIF, hypoxia-inducible factor; nTS, nucleus tractus solitarious; RVLM, rostral ventrolateral medulla.
Following 10 days of CIH exposure, RVLM, nTS and an adjacent brainstem area unrelated to the CB chemoreflex were harvested by micro-punches (for coordinates see Fig. 2Ba). Immunoblot assays revealed increased HIF-1α and decreased HIF-2α protein levels in the nTS and RVLM of CIH-exposed sham-operated rats. CIH did not alter HIF-1α or HIF-2α levels in the control brainstem region that does not receive input from the CB (Fig. 2Bb and Bc). Immunohistochemical assays revealed increased HIF-1α and decreased HIF-2α-like immunoreactivity in nTS and RVLM neurons as evidenced by colocalization with NeuN, a neuronal marker (Fig. 3). The CIH-induced changes in HIF-α isoform expression in the nTS and RVLM were absent in CBA rats ( Figs 2B and 3).
Figure 3. CBA prevents CIH-induced changes in HIF-α isoform expression in nTS and RVLM neurons.
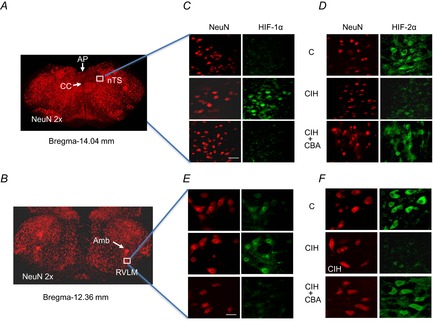
Three groups of rats were studied: sham-operated and exposed to normoxia (C); sham-operated and exposed to CIH; and CIH-exposed CBA rats (CIH + CBA). Brainstem sections containing either nTS or RVLM were obtained based on coordinates from adult rat brain atlas (Paxinos & Watson, 2006; nTS, Bregma-14.04 mm; RVLM, Bregma-12.36 mm). A and B, low magnification (2×) of sections stained with NeuN (red), a neuronal marker. C–F, high magnifications of sections containing nTS (C and D) and RVLM (E and F) double stained with either NeuN and HIF-1α or NeuN and HIF-2α are shown scale bar, 20 μm). Amb, nucleus ambiguus; AP, area postrema; CBA, carotid body ablation; CC, central canal; CIH, chronic intermittent hypoxia; HIF, hypoxia-inducible factor; nTS, nucleus tractus solitarious; RVLM, rostral ventrolateral medulla.
Effect of carotid body ablation on chronic intermittent hypoxia-induced changes in NADPH oxidase 2, superoxide dismutase-2 and redox state in the brainstem
HIF-1 and HIF-2 regulate genes encoding pro-oxidant and antioxidant enzymes, respectively (Scortegagna et al. 2003; Nanduri et al. 2009; Yuan et al. 2011). To assess the functional significance of changes in HIF-α isoforms by CIH, we first determined the expression of mRNAs encoding Nox2 and Sod2, which are the major pro-oxidant and antioxidant enzymes, respectively, along with their enzyme activities. In sham-operated, CIH-exposed rats, Nox2 mRNA levels and enzyme activity were increased in the nTS and RVLM, whereas those of Sod2 were decreased (Fig. 4A–D). In contrast, mRNA expressions and activities of Nox2 and Sod2 were unaltered in the control brainstem region (Fig. 4A–D). ROS inhibits aconitase enzyme activity in the cytosol and mitochondria, and serves as a measure of oxidative stress (Gardner, 2002). We found that the amount of protein from micro-punches of RVLM and nTS is insufficient to measure the aconitase activity in cytosolic and mitochondrial fractions. MDA levels represent oxidized lipids (Janero, 1990). Therefore, MDA levels were determined in brainstem micro-punch samples as an index of oxidative stress. MDA levels increased in the nTS and RVLM, in sham operated, CIH-exposed rats, indicating increased ROS generation (Fig. 4E). In CBA rats, CIH-induced changes in Nox2 and Sod2 mRNA levels and enzyme activity, as well as MDA levels, were absent in the nTS and RVLM (Fig. 4A–E).
Figure 4. Effect of CBA on CIH-induced changes in the redox state in brainstem regions.
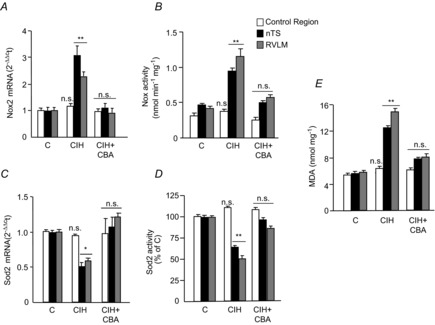
Nox2 mRNA levels (A), Nox enzyme activity (B), Sod2 mRNA levels (C), Sod2 enzyme activity (D) and MDA levels (E) were determined in nTS, RVLM and control region micro-punches from the following groups of rats: sham-operated and exposed to normoxia (C); sham-operated and exposed to CIH; and CIH-exposed CBA rats (CIH + CBA). Data in bar graphs are presented as means ± s.e.m., n = 6–7 rats per group; *P < 0.05; **P < 0.01; and n.s., P > 0.05. CBA, carotid body ablation; CIH, chronic intermittent hypoxia; HIF, hypoxia-inducible factor; MDA, malondialdehyde; Nox, NADPH oxidase; nTS, nucleus tractus solitarious; RVLM, rostral ventrolateral medulla; Sod, superoxide dismutase.
Chronic intermittent hypoxia-evoked hypoxia-inducible factor-α isoform expression in the end organ of the sympathetic nervous system
We next investigated whether the CB chemoreflex contribute to CIH-induced HIF-α isoform expression in the AM, a major end organ of the sympathetic nervous system. In the AM of sham-operated CIH-exposed rats, HIF-1α protein levels increased, whereas HIF-2α levels decreased (Fig. 5A). The changes in HIF-α isoforms were associated with upregulation of Nox2 mRNA and enzyme activity and downregulation of Sod2 mRNA and enzyme activity (Fig. 5B–E). Aconitase enzyme activity was determined as a measure of oxidative stress in the AM (Gardner, 2002) because of the availability of sufficient cytosolic and mitochondrial protein. Aconitase activity was decreased in both cytosolic and mitochondrial fractions of the AM from sham-operated CIH-exposed rats (Fig. 5F) indicative of oxidative stress. In the AMs of CBA rats, CIH-induced changes in HIF-α isoforms, Nox2, Sod2 and aconitase activity were absent (Fig. 5A–F).
Figure 5. Effects of CBA on CIH-induced changes in HIF-α isoform and redox state in the AM.
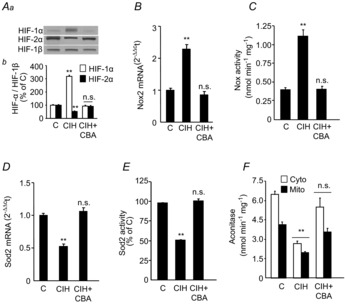
Three groups of rats were studied: sham-operated and exposed to normoxia (C); sham-operated and exposed to CIH; and CIH-exposed CBA rats (CIH + CBA). A, representative immunoblots (Aa) and densitometric analysis (Ab) of HIF-1α and HIF-2α in the AMs of sham-operated, normoxia-exposed control rats (C) presented as ratio of HIF-α/HIF-1β. B–F, Nox2 mRNA levels (B), Nox enzyme activity (C), Sod2 mRNA levels (D), Sod2 enzyme activity (E) and cytosolic and mitochondrial aconitase activities (F). Data in bar graphs are presented as means ± s.e.m., n = 6–7 rats per group. **P < 0.01; n.s., P > 0.05 compared to sham-operated, normoxia-exposed rats (C). CBA, carotid body ablation; CIH, chronic intermittent hypoxia; Cyto, cytosolic; HIF, hypoxia-inducible factor; Mito, mitochondrial; Nox, NADPH oxidase; Sod, superoxide dismutase.
Sympathetic activation mediates the effects of chronic intermittent hypoxia on the adrenal medulla
Sympathetic innervation to the AM is derived from a branch of the splanchnic nerve (Kesse et al. 1988). We hypothesized that sympathetic activation by CB chemoreflex mediates the effects of CIH in the AM. To test this possibility, we first recorded the adrenal sympathetic nerve activity (ASN) in rats exposed to CIH. Sham-operated, CIH-exposed rats exhibited elevated ASN activity under both normoxic and hypoxic conditions, but these responses were absent in CIH-exposed CBA rats (Fig. 6A and B).
Figure 6. CBA prevents CIH-induced ASN.
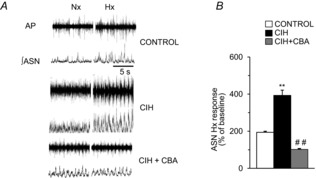
A, three groups of rats were studied: sham-operated and Nx-exposed (control); sham-operated and exposed to CIH; and CIH-exposed CBA rats (CIH + CBA). Representative tracings of ASN responses to Nx (21% O2) and Hx (12% O2). B, ASN response to Hx was quantified (means ± s.e.m., n = 6–7 rats each); **P < 0.01 compared to control and ##P < 0.01 compared to CIH-exposed rats. AP, action potential; ∫ASN, integrated adrenal sympathetic nerve activity; CBA, carotid body ablation; CIH, chronic intermittent hypoxia; Hx, hypoxia; Nx, normoxia.
To determine whether sympathetic activation contributes to CIH-induced transcriptional and redox changes in the AM, ASN was ablated unilaterally (ASA) leaving the contralateral ASN intact to serve as an internal control. After allowing for postoperative recovery, ASA was confirmed by the absence of choline acetyltransferase immunoreactive nerve fibres in the AM (Fig. 7A). CIH increased HIF-1α levels and decreased HIF-2α levels in the AM with intact ASN, leading to upregulation of Nox2 mRNA and enzyme activity and downregulation of Sod2 mRNA and enzyme activity (Fig. 7B–F). HIF-α isoform expression and Nox/Sod2 activity were unchanged in the ipsilateral AM that underwent ASA (Fig. 7B–F). ASA also prevented CIH-induced oxidative stress, as evidenced by the maintenance of cytosolic and mitochondrial aconitase activity in the AM (Fig. 7G).
Figure 7. ASA abolishes CIH-induced changes in HIF-α isoform and redox state in the AM.
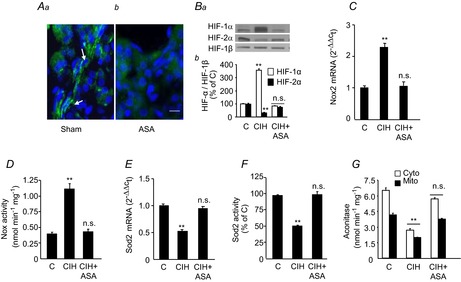
Three groups of rats were studied: sham-operated and exposed to normoxia (C); sham-operated and exposed to CIH; and unilateral ASA and exposed to CIH (CIH + ASA). A, immunofluorescence of choline acetyltransferase expression (stained in green) in sections from the AM of rats subjected to sham surgery (Aa) or ipsilateral ASA (Ab). B, representative immunoblots (Ba) and densitometric analysis (Bb) of HIF-1α and HIF-2α in the AM presented as HIF-α/HIF-1β relative to sham-operated, normoxia-exposed control rats (C). C–G, Nox2 mRNA levels (C), Nox enzyme activity (D), Sod2 mRNA levels (E), Sod2 enzyme activity (F) and cytosolic and mitochondrial aconitase activities (G) in the AM are shown. Data in bar graphs are presented as means ± s.e.m., n = 6–7 rats per group; **P < 0.01; n.s., P > 0.05 compared to vehicle-treated, normoxia-exposed rats (C). AM, adrenal medulla; ASA, adrenal sympathetic nerve ablation; CIH, chronic intermittent hypoxia; Cyto, cytosolic; HIF, hypoxia-inducible factor; Mito, mitochondrial; Nox, NADPH oxidase; Sod, superoxide dismutase.
Muscarinic acetylcholine receptors mediate the effects of sympathetic activation on the adrenal medulla
We then asked how sympathetic activation leads to transcriptional and redox changes in the AM of CIH-exposed rodents. The neurotransmitter ACh activates either nicotinic ACh (nAChR) and/or muscarinic ACh receptors (mAChR) on chromaffin cells in the AM. Previously, we showed that CIH downregulates nAChR expression in the AM (Souvannakitti et al. 2010) and that mAChR activation is sufficient to increase HIF-1α levels (Hirota et al. 2004). To test whether mAChRs contribute to dysregulation of HIF-α isoforms, rats were treated with atropine (10 mg kg−1 day−1 i.p.), a pan-mAChR blocker, daily during the 10 days of CIH exposure. Atropine-treated rats exhibited a small but significant increase in HR as compared to vehicle-treated rats exposed to CIH (Fig. 8A), demonstrating the efficacy of atropine in blocking mAChRs in the heart. However, atropine treatment had no effect on the CIH-induced augmented CB or ASN response to hypoxia (Fig. 8B–E), indicating that blockade of the mAChRs had no impact on these responses. However, atropine treatment prevented the CIH-induced imbalance in HIF-α isoforms, changes in pro-oxidant and antioxidant enzyme activities, and oxidative stress in the AM (Fig. 8F–I).
Figure 8. Effects of ATR on CIH-induced autonomic responses and functional changes in the AM.
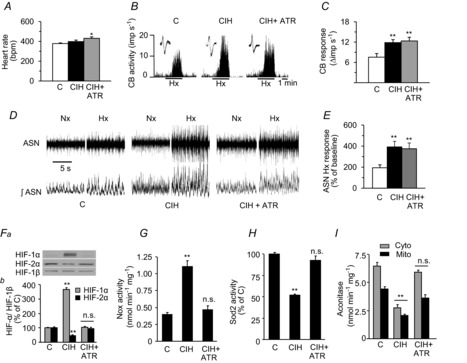
Three groups of rats were studied: vehicle-treated and exposed to Nx (C); vehicle-treated and exposed to CIH; and CIH-exposed rats ATR-treated (10 mg kg−1 day−1 i.p., CIH + ATR). A–E, effects of ATR on heart rate (A), Hx-induced increase in CB sensory activity (B and C), and ASN in Nx and Hx (D and E). F, representative immunoblots (Fa) and densitometric analysis (Fb) of HIF-1α and HIF-2α in the AM presented as a ratio of HIF-α/HIF-1β are shown. G–I, Nox, Sod2 and aconitase enzyme activities in the AM are shown. Data in bar graphs are presented as means ± s.e.m., n = 6–7 rats per group; *P < 0.05; **P < 0.01; and n.s., P > 0.05 compared to vehicle-treated, Nx-exposed rats (C). AM, adrenal medulla; ASN, adrenal sympathetic nerve activity; ATR, atropine; CB, carotid body; CIH, chronic intermittent hypoxia; Cyto, cytosolic; HIF, hypoxia-inducible factor; Hx, hypoxia; imp, impulse; Mito, mitochondrial; Nox, NADPH oxidase; Nx, normoxia; Sod, superoxide dismutase.
Mechanisms of hypoxia-inducible factor-α isoform regulation by muscarinic acetylcholine receptor signalling
Next, we investigated the mechanisms underlying HIF-α isoform regulation by mAChR signalling using PC12 cells, AM-derived tumour cells. As sympathetic nerve activity increases with each episode of intermittent hypoxia, we tested whether repetitive application of muscarine, a mAChR agonist, simulates CIH-induced HIF-α isoform expression. Repetitive application of muscarine increased HIF-1α while decreasing HIF-2α protein levels, recapitulating the effects of CIH on HIF-α isoform expression in the AM (Fig. 9A). Atropine prevented the effects of repetitive application of muscarine (Fig. 9B). In contrast, continuous applications of muscarine or repetitive or continuous applications of nicotine were ineffective in causing an imbalance in the HIF-α isoforms (Fig. 9C–E).
Figure 9. Effects of Mus and nicotine on HIF-α isoform expression in pheochromocytoma 12 cells.
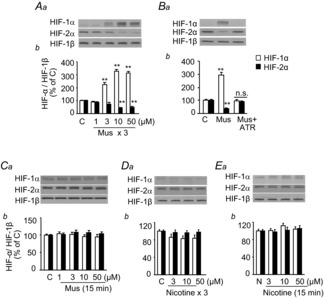
A, Mus, at indicated concentration, was applied to pheochromocytoma 12 cell cultures repetitively (Mus for 5 min followed by vehicle for 10 min, repeated for three cycles). Representative immunoblots (Aa) and densitometric analysis (Ab) of HIF-1α and HIF-2α are shown. B, effects of ATR on repetitive Mus-induced (Mus + ATR) changes in HIF-α isoform expression are shown. C–E, effects of continuous Mus treatment (C), repetitive (nicotine for 5 min followed by vehicle for 10 min, three cycles; D) and continuous nicotine treatment (for 15 min; E) on HIF-α isoform expression are shown in representative immunoblots (Ca–Ea) and densitometric analysis (Cb–Eb) of HIF-1α and HIF-2α. Changes in HIF-1α and HIF-2α are analysed as HIF-α/HIF-1β. Data are means ± s.e.m., n = 6–7 independent experiments. **P < 0.01; n.s., P > 0.05 compared to control cells (C). ATR, atropine; C, control; HIF, hypoxia-inducible factor; Mus, muscarine.
The increased HIF-1α expression induced by repetitive muscarine application persisted for as long as 90 min after termination of its application (Fig. 10A). Cycloheximide, an inhibitor of protein synthesis, prevented accumulation of HIF-1α (Fig. 10B), suggesting that mAChR activation increases HIF-1α protein synthesis. Previous studies have shown that Ca2+-dependent mTOR activation increases HIF-1α protein synthesis in PC12 cells (Yuan et al. 2008). Given that mAChR activation increases [Ca2+]i levels (Prakriya et al. 1996; Inoue et al. 2008; Harada et al. 2011), we hypothesized that a mAChR-mediated increase in [Ca2+]i activates mTOR to increase HIF-1α protein expression. Repetitive application of muscarine increased [Ca2+]i and mTOR activity (as evidenced by increased phosphorylation) and both responses were blocked by atropine (Fig. 10C and D). Muscarine-induced mTOR phosphorylation and increased HIF-1α protein expression were blocked by pretreatment with the calcium chelator BAPTA-AM or the mTOR inhibitor, rapamycin (Fig. 10D and E).
Figure 10. Signalling mechanisms associated with repetitive Mus-mediated regulation of HIF-α isoform in pheochromocytoma 12 cells.
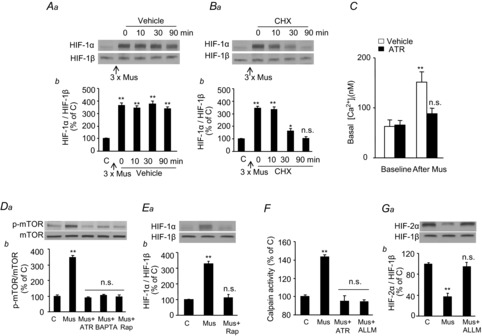
A and B, following repetitive Mus treatment (10 μm, three cycles), cells were treated with either vehicle (A) or CHX (20 nm; B) for up to 90 min. Representative immunoblots (Aa and Ba) and densitometric analysis (Ab and Bb) of HIF-1α presented as HIF-1α/HIF-1β relative to control (C) cells are shown. C, effect of ATR (5 μm) on repetitive Mus-induced changes in [Ca2+]i levels. D, representative immunoblots (Da) and densitometric analysis (Db) of p-mTOR protein levels presented as ratio of p-mTOR/mTOR in cells treated repetitively with Mus (10 μm, three cycles) in the absence or presence of either ATR (5 μm), calcium chelator (BAPTA, 10 μm), or Rap (100 nm) are shown. E, representative immunoblots (Ea) and densitometric analysis (Eb) of HIF-1α presented as HIF-1α/HIF-1β following repetitive Mus (10 μm, three cycles) treatment in the absence or presence of Rap (100 nm). F, calpain activity in cells treated repetitively with Mus (10 μm, three cycles) in the absence or presence of ATR (5 μm) or ALLM (10 μm) is shown. G, representative immunoblots (Ga) and densitometric analysis (Gb) of HIF-2α presented as HIF-2α/HIF-1β protein levels following repetitive Mus (10 μm, three cycles) treatment in the absence or presence of calpain inhibitor (ALLM, 10 μm). Data in bar graphs are presented as means ± s.e.m., n = 6–7 independent experiments; *P < 0.05; **P < 0.01; n.s., P > 0.05 compared to control cells (C). ALLM, N-acetyl-leucine-leucine-methionine-aldehyde; ATR, atropine; CHX, cycloheximide; HIF, hypoxia-inducible factor; mTOR, mammalian target of rapamycin; Mus, muscarine; p-mTOR, phosphorylated form of mTOR; Rap, rapamycin.
We next examined the mechanism underlying HIF-2α degradation by repetitive muscarine treatment. Recent studies have demonstrated that increases in [Ca2+]i in PC12 cells activate the Ca2+-dependent protease calpains, leading to degradation of HIF-2α (Nanduri et al. 2009, 2013). Repetitive muscarine application increased calpain activity in PC12 cells and this effect was blocked by either atropine or Ac-LLM-CHO, a calpain inhibitor (Fig. 10F). Ac-LLM-CHO also blocked muscarine-induced HIF-2α degradation (Fig. 10G).
The above in vitro results demonstrate that mTOR and calpain protease activities, which are induced by mAChR signalling, are critical mediators of HIF-α isoform dysregulation. To test whether this signalling pathway also contributes to CIH-induced HIF-α dysregulation in vivo in the AM, studies were performed on rats exposed to CIH. AMs from CIH-exposed rats exhibited increased levels of phosphorylated mTOR and calpain activity, and these effects were absent in CIH-exposed rats treated with either atropine or those subjected to CBA or ASA (Fig. 11A and B).
Figure 11. Effects of CIH on mTOR and calpain activation in the adrenal medulla.
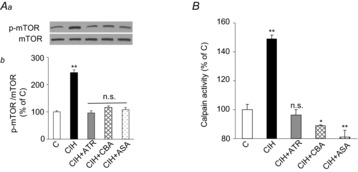
Five groups of rats were studied: vehicle-treated and exposed to normoxia (C); vehicle-treated and CIH-exposed (CIH); ATR-treated (10 mg kg−1 day−1 i.p.) and CIH-exposed (CIH + ATR); CIH-exposed CBA (CIH + CBA); and CIH-exposed bilateral ASA (CIH + ASA). Representative immunoblots (Aa) and densitometric analysis (Ab) of p-mTOR/total mTOR protein (A) and calpain activity (B). Data in bar graphs are presented as means ± s.e.m., n = 6–7 rats per group; *P < 0.05; **P < 0.01; n.s., P > 0.05. ASA, adrenal sympathetic nerve ablation; ATR, atropine; CBA, carotid body ablation; CIH, chronic intermittent hypoxia; mTOR, mammalian target of rapamycin; p-mTOR, phosphorylated mTOR.
An intact carotid body chemosensory reflex is required for chronic intermittent hypoxia-induced hypertension
Previous studies have shown that elevated ROS signalling in CIH-exposed rats augments hypoxia-induced CA secretion from the AM (Kumar et al. 2006; Kuri et al. 2007). As CBA (removal of CB afferent input) and ASA (removal of efferent sympathetic output) prevented CIH-induced oxidative stress in the AM, we examined whether they also maintain normal CA secretion. Hypoxia elicited enhanced noradrenaline and adrenaline effluxes from the AM in rats exposed to CIH, whereas CBA or ASA prevented this augmented secretory response (Fig. 12A). CIH-exposed rats exhibited elevated plasma noradrenaline and adrenaline levels (Fig. 12B) and increased the basal BP (Fig. 12C). CBA or ASA prevented increases in plasma CA levels and BP (Fig. 12B and C).
Figure 12. Carotid body chemoreflex mediates CIH-induced changes in CA secretion from adrenal medulla, plasma CA and blood pressure.
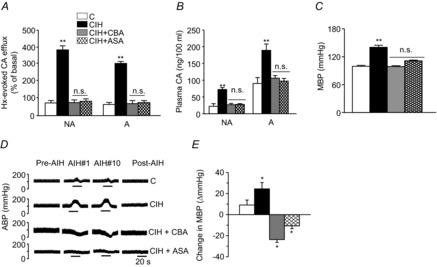
Four groups of rats were studied: sham-operated and normoxia-exposed (C); sham-operated and CIH-exposed (CIH); carotid body ablated (CBA) and CIH exposed (CBA + CIH); and bilateral ASA and CIH-exposed (CIH + ASA). A, Hx (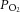 = 40 ± 3 mmHg, 5 min)-evoked NA and A secretion from adrenal medulla slices is presented as the percentage of basal release. B and C, plasma CA levels of NA and A (B) and MBP (C) are shown. D and E, effect of AIH (15 s at 12% O2, followed by 5 min at 21% O2) on ABP. Representative changes in ABP during pre-AIH, first and 10th AIH, and post-AIH periods (D) and MBP (E) are shown. Data in bar graphs are presented as means ± s.e.m., n = 6–7 rats; *P < 0.05; ***P < 0.001; n.s., P > 0.05. A, adrenaline; ABP, arterial blood pressure; AIH, acute intermittent hypoxia; ASA, adrenal sympathetic nerve ablation; C, control; CA, catecholamine; CBA, carotid body ablation; CIH, chronic intermittent hypoxia; Hx, hypoxia; MBP, mean blood pressure; NA, noradrenaline.
= 40 ± 3 mmHg, 5 min)-evoked NA and A secretion from adrenal medulla slices is presented as the percentage of basal release. B and C, plasma CA levels of NA and A (B) and MBP (C) are shown. D and E, effect of AIH (15 s at 12% O2, followed by 5 min at 21% O2) on ABP. Representative changes in ABP during pre-AIH, first and 10th AIH, and post-AIH periods (D) and MBP (E) are shown. Data in bar graphs are presented as means ± s.e.m., n = 6–7 rats; *P < 0.05; ***P < 0.001; n.s., P > 0.05. A, adrenaline; ABP, arterial blood pressure; AIH, acute intermittent hypoxia; ASA, adrenal sympathetic nerve ablation; C, control; CA, catecholamine; CBA, carotid body ablation; CIH, chronic intermittent hypoxia; Hx, hypoxia; MBP, mean blood pressure; NA, noradrenaline.
Patients with recurrent apnoea exhibit pronounced increases in BP with each episode of apnoea (Stoohs & Guilleminault, 1992; Imadojemu et al. 2002). We simulated recurrent apnoea by subjecting control or CIH-pre-exposed rats to acute intermittent hypoxia (AIH; 12% O2 for 15 s and then room air for 5 min, repeated for 10 cycles). Each AIH cycle induced a modest elevation in BP in control rats, but produced a two- to three-fold exaggerated increase in BP in CIH-exposed sham-operated rats (Fig. 12D and E). Either CBA or ASA prevented AIH-induced elevations in BP in CIH-exposed rats (Fig. 12D and E).
Discussion
The present results demonstrate that CIH leads to an imbalanced expression of HIF-α isoforms in the central and peripheral components of the CB chemoreflex, including neurons in the nTS, and RVLM, as well as in the AM. Remarkably, selective blockade of the CB sensory input abolished CIH-induced changes in HIF-α isoform expression. As the carotid baroreflex and aortic bodies are intact in CBA rats, the absence of CIH-induced changes in HIF-α isoforms can be attributed entirely to the lack of sensory input from the CB. It is unlikely that elevated BP itself influences HIF-α expression, as isoform expression was unaltered in brainstem areas that do not receive CB sensory input despite CIH-induced hypertension (Fig. 2B). Although we found changes in HIF-α isoforms in neurons of the nTS and RVLM, it remains to be investigated whether similar changes also occur in glia in these regions. None the less, our results delineate a previously uncharacterized CB neural activity-dependent regulation of HIF-α isoform expression in specific neuroendocrine tissues.
CIH-exposed rats exhibited oxidative stress in the nTS, RVLM and AM, a finding consistent with previous reports (Kumar et al. 2006; Peng et al. 2006; Kuri et al. 2007; Yuan et al. 2008; Raghuraman et al. 2009). The increased oxidative stress was associated with imbalanced expression of the Nox2 and Sod2 genes, which encode major pro-oxidant and antioxidant enzymes, respectively, which are known to be transcriptionally activated by HIF-1 and HIF-2, respectively (Scortegagna et al. 2003; Nanduri et al. 2009, 2013; Yuan et al. 2011). CBA prevented the CIH-induced imbalance in HIF-α isoforms, which otherwise results in dysregulation of Nox2 and Sod2 expression, and oxidative stress. These results demonstrate CB activity-dependent regulation of HIF-α isoforms alters the redox state in the central and peripheral nervous system during CIH.
Our results also illustrate how CB activity evokes changes in HIF-α isoforms and oxidative stress in the AM, an end organ of the sympathetic nervous system. The following findings suggest that mAChR activation is the primary mechanism by which CB activity triggers cellular and molecular changes in the AM: (a) systemic chronic administration of atropine abolished changes in HIF-α isoforms and the redox state of the AM without affecting CIH-induced CB hypersensitivity or sympathetic excitation, and (b) similar to CIH, repetitive application of muscarine upregulated HIF-1α and downregulated HIF-2α in PC12 cells, effects that were blocked by atropine. Our results further showed that mAChR-mediated increases in [Ca2+]i concomitantly activated the mTOR pathway and calpain proteases, leading to an increase in HIF-1α and decrease in HIF-2α levels, respectively. The observed increases in [Ca2+]i may involve mobilization of intracellular stores (Prakriya et al. 1996) or extracellular Ca2+ influx via inhibition of TASK-1 channels by mAChRs (Inoue et al. 2008; Harada et al. 2011). Which of the five mAChR isoforms (i.e. M1–M5) thus far identified (Caulfield & Birdsall, 1998) contribute to the effects of CIH remains to be investigated. A recent study reported that xanthine oxidase also mediates HIF-2α degradation by activating calpain in PC12 cells and in the AM (Nanduri et al. 2013). Whether xanthine oxidase also plays a role in mAChR-induced calapin activation requires further investigation. An AM-like detailed analysis of signalling pathways mediating CB activity-dependent HIF-α isoform expression could not be performed in nTS and RVLM neurons due to complex regulation of neurotransmission by multiple neurotransmitters and neuron–glial interactions in these areas.
Cell cultures exposed to CIH respond to molecular changes similar to those seen in intact rats and the effects of CIH on cell cultures were attributed to direct effects of hypoxia and re-oxygenation (Yuan et al. 2008; Nanduri et al. 2009). One may therefore ask why the CB neural activity is required to induce similar molecular changes in intact rats. We found only a modest decrease in arterial blood O2 saturation (from 97% to 80%) in rats during IH exposure (Fig. 2A). Under normoxia, while the arterial blood 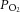 is ∼100 mmHg, tissue
is ∼100 mmHg, tissue 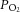 levels are much lower and range between 30 and 60 mmHg (Carreau et al. 2011). Given these low basal tissue
levels are much lower and range between 30 and 60 mmHg (Carreau et al. 2011). Given these low basal tissue 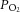 levels, modest fluctuations in arterial
levels, modest fluctuations in arterial 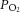 caused by IH may not be sufficient to induce changes in HIF-α isoform expression or oxidative stress in intact animals. The CB, on the other hand, has the highest blood flow per tissue weight of any organ in the body (Kumar & Prabhakar, 2012). Because of its high blood flow, and exquisite sensitivity to hypoxia, the CB is uniquely suited to sense and respond to a modest drop in
caused by IH may not be sufficient to induce changes in HIF-α isoform expression or oxidative stress in intact animals. The CB, on the other hand, has the highest blood flow per tissue weight of any organ in the body (Kumar & Prabhakar, 2012). Because of its high blood flow, and exquisite sensitivity to hypoxia, the CB is uniquely suited to sense and respond to a modest drop in 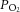 such as that occurs during transient episodes of hypoxia. Therefore, it is conceivable that CIH exerts a direct effect on the CB resulting in augmented neural activity (Peng & Prabhakar, 2004; Rey et al. 2004). The ensuing CB neural activity then triggers activity-dependent dysregulation of HIF-α isoforms and redox changes in the central and peripheral components of the chemoreflex, as delineated in the present study.
such as that occurs during transient episodes of hypoxia. Therefore, it is conceivable that CIH exerts a direct effect on the CB resulting in augmented neural activity (Peng & Prabhakar, 2004; Rey et al. 2004). The ensuing CB neural activity then triggers activity-dependent dysregulation of HIF-α isoforms and redox changes in the central and peripheral components of the chemoreflex, as delineated in the present study.
CB activity-dependent dysregulation of HIF-α isoforms and the ensuing oxidative stress was associated with augmented hypoxia-induced CA secretion from the AM. As CBA or ASA prevented HIF-α isoform imbalance, oxidative stress and excessive CA secretion, it seems possible that the AM acquires hypoxic hypersensitivity indirectly from the CB rather than directly from CIH. Emerging evidence suggests that T-type Ca2+ and KATP channels are key regulators of hypoxic sensitivity and CA secretion from AM. As expression of these channels is transcriptionally regulated by HIF-α isoforms (Prabhakar et al. 2012; Salman et al. 2014), they might contribute to CA hypersecretion by hypoxia from AM after CIH exposure.
It has been long thought that CIH increases circulating CA levels due to augmented global sympathetic activity (Fletcher et al. 1992). However, we found that either blockade of CB sensory input by CBA or sympathetic input to the AM by ASA alone was sufficient to prevent elevation in plasma CA levels in CIH-exposed rats. These findings suggest that CB-enhanced AM CA secretion, rather than overall sympathetic activation, is the primary source of elevated plasma CA levels during CIH. In addition to elevated basal BP, CIH-exposed rats exhibited marked increases in BP with each episode of AIH, a classic phenotype of patients with obstructive sleep apnoea (Stoohs & Guilleminault, 1992; Imadojemu et al. 2002). Originally, these periodic elevations in BP were attributed to augmented sympathetic activity affecting vascular tone (Imadojemu et al. 2002). However, either CBA or ASA was sufficient to prevent the abnormal increases in BP, suggesting that changes in BP during AIH are instead due to enhanced CA secretion from the AM. Acute atropine treatment is insufficient to prevent episodic elevation of BP in humans during sleep apnoea (Gulleminault et al. 1977; van den Aardweg & Karemaker, 1992). However, while our study demonstrated a potential link between CB-induced HIF-α isoform imbalance, oxidative stress and BP changes, further studies with tissue selective ablation of HIF-α isoforms in the chemoreflex pathway are necessary to establish a direct role of HIF-induced oxidative stress in CIH-evoked hypertension. Whether CB chemoreflex-mediated sympathetic activation facilitates pathologic HIF-α isoform dysregulation and oxidative stress in other sympathetic end organs remains an important question that requires further investigation.
Translational significance
Recurrent apnoea with CIH is a major respiratory disorder affecting several million adults (Young et al. 1993, 2008), and there is a strong correlation between the severity of apnoea and subsequent development of hypertension (Peppard et al. 2000). A growing body of evidence suggests that augmented sympathetic activity resulting from heightened CB chemoreflex is a major contributor to the hypertension in patients with obstructive sleep apnoea (Narkiewicz et al. 1999; Kara et al. 2003). The present study outlines that heightened CB neural activity mediates CIH-induced HIF-α isoform dysregulation and oxidative stress in components of the CB chemoreflex pathway, including the AM. The resulting oxidative stress probably leads to elevated CA secretion and thus, hypertension. In addition to sleep-disordered breathing, an augmented CB chemoreflex has been implicated in the sequelae of a hyperactive sympathetic nervous system, such as neurogenic hypertension (Abdala et al. 2012) and congestive heart failure (Schultz & Li, 2007). Whether the CB chemoreflex regulates HIF-α isoforms and oxidative stress in such pathologies is an important question that remains to be investigated.
Acknowledgments
None declared.
Glossary
- Ac-LLM-CHO
N-acetyl-leucine-leucine-methionine-aldehyde
- AChR
acetylcholine receptor
- AIH
acute intermittent hypoxia
- AM
adrenal medulla
- ASA
adrenal sympathetic ablation
- BAPTA
1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- BP
blood pressure
- CA
catecholamines
- CB
carotid body
- CBA
carotid body ablation
- CIH
chronic intermittent hypoxia
- HIF-α
hypoxia-inducible factor alpha
- HR
heart rate
- mAChR
muscarinic ACh receptor
- MDA
malondialdehyde
- mTOR
mammalian target of rapamycin
- nAChR
nicotinic ACh receptor
- Nox2
NADPH oxidase 2
- nTS
nucleus tractus solitarious
- PC12
pheochromocytoma 12
- ROS
reactive oxygen species
- RVLM
rostral ventrolateral medulla
- SNA
splanchnic sympathetic nerve activity
- Sod2
superoxide dismutase 2
Additional information
Competing interests
The authors declare no conflict of interest.
Author contributions
N.R.P. conceived the study and N.R.P. and G.K. designed the experiments. Y-J.P., G.Y., S.K., J.N., V.V.M., V.D.R., C.V., G.K. and N.R.P. were all responsible for the collection, analysis and interpretation of the data. N.R.P., G.K. and G.L.S. drafted the article. All authors approved the final version of the manuscript.
Funding
This research was supported by National Institutes of Health, Heart, Lung and Blood Institute, Grant PO1-HL-90554.
References
- Abdala AP, McBryde FD, Marina N, Hendy EB, Engelman ZJ, Fudim M, Sobotka PA, Gourine AV, Paton JF. Hypertension is critically dependent on the carotid body input in the spontaneously hypertensive rat. J Physiol. 2012;590:4269–4277. doi: 10.1113/jphysiol.2012.237800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreau A, El Hafny-Rahbi B, Matejuk A, Grillon C, Kieda C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med. 2011;15:1239–1253. doi: 10.1111/j.1582-4934.2011.01258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carulli D, Foscarin S, Rossi F. Activity-dependent plasticity and gene expression modifications in the adult CNS. Front Mol Neurosci. 2011;4:50. doi: 10.3389/fnmol.2011.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulfield MP, Birdsall NJ. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol Rev. 1998;50:279–290. [PubMed] [Google Scholar]
- Costa-Silva JH, Zoccal DB, Machado BH. Chronic intermittent hypoxia alters glutamatergic control of sympathetic and respiratory activities in the commissural NTS of rats. Am J Physiol Regul Integr Comp Physiol. 2012;302:R785–R793. doi: 10.1152/ajpregu.00363.2011. [DOI] [PubMed] [Google Scholar]
- Fields RD, Lee PR, Cohen JE. Temporal integration of intracellular Ca2+ signaling networks in regulating gene expression by action potentials. Cell Calcium. 2005;37:433–442. doi: 10.1016/j.ceca.2005.01.011. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Lesske J, Behm R, Miller CC, 3rd, Stauss H, Unger T. Carotid chemoreceptors, systemic blood pressure, and chronic episodic hypoxia mimicking sleep apnea. J Appl Physiol. 1992;72:1978–1984. doi: 10.1152/jappl.1992.72.5.1978. [DOI] [PubMed] [Google Scholar]
- Ganguly K, Poo MM. Activity-dependent neural plasticity from bench to bedside. Neuron. 2013;80:729–741. doi: 10.1016/j.neuron.2013.10.028. [DOI] [PubMed] [Google Scholar]
- Gardner PR. Aconitase: sensitive target and measure of superoxide. Methods Enzymol. 2002;349:9–23. doi: 10.1016/s0076-6879(02)49317-2. [DOI] [PubMed] [Google Scholar]
- Gulleminault C, Lehrman AT, Forno L, Dement WC. Sleep apnoea syndrome: states of sleep and autonomic dysfunction. J Neurol Neurosurg Psychiatry. 1977;40:718–725. doi: 10.1136/jnnp.40.7.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada K, Matsuoka H, Sata T, Warashina A, Inoue M. Identification and role of muscarinic receptor subtypes expressed in rat adrenal medullary cells. J Pharmacol Sci. 2011;117:253–264. doi: 10.1254/jphs.11125fp. [DOI] [PubMed] [Google Scholar]
- Hirota K, Fukuda R, Takabuchi S, Kizaka-Kondoh S, Adachi T, Fukuda K, Semenza GL. Induction of hypoxia-inducible factor 1 activity by muscarinic acetylcholine receptor signaling. J Biol Chem. 2004;279:41521–41528. doi: 10.1074/jbc.M405164200. [DOI] [PubMed] [Google Scholar]
- Imadojemu VA, Gleeson K, Gray KS, Sinoway LI, Leuenberger UA. Obstructive apnea during sleep is associated with peripheral vasoconstriction. Am J Respir Crit Care Med. 2002;165:61–66. doi: 10.1164/ajrccm.165.1.2009062. [DOI] [PubMed] [Google Scholar]
- Inoue M, Harada K, Matsuoka H, Sata T, Warashina A. Inhibition of TASK1-like channels by muscarinic receptor stimulation in rat adrenal medullary cells. J Neurochem. 2008;106:1804–1814. doi: 10.1111/j.1471-4159.2008.05521.x. [DOI] [PubMed] [Google Scholar]
- Janero DR. Malondialdehyde and thiobarbituric acid-reactivity as diagnostic indices of lipid peroxidation and peroxidative tissue injury. Free Radic Biol Med. 1990;9:515–540. doi: 10.1016/0891-5849(90)90131-2. [DOI] [PubMed] [Google Scholar]
- Kanagy NL, Walker BR, Nelin LD. Role of endothelin in intermittent hypoxia-induced hypertension. Hypertension. 2001;37:511–515. doi: 10.1161/01.hyp.37.2.511. [DOI] [PubMed] [Google Scholar]
- Kara T, Narkiewicz K, Somers VK. Chemoreflexes – physiology and clinical implications. Acta Physiol Scand. 2003;177:377–384. doi: 10.1046/j.1365-201X.2003.01083.x. [DOI] [PubMed] [Google Scholar]
- Kesse WK, Parker TL, Coupland RE. The innervation of the adrenal gland. I. The source of pre- and postganglionic nerve fibres to the rat adrenal gland. J Anat. 1988;157:33–41. [PMC free article] [PubMed] [Google Scholar]
- Kline DD, Ramirez-Navarro A, Kunze DL. Adaptive depression in synaptic transmission in the nucleus of the solitary tract after in vivo chronic intermittent hypoxia: evidence for homeostatic plasticity. J Neurosci. 2007;27:4663–4673. doi: 10.1523/JNEUROSCI.4946-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar GK, Rai V, Sharma SD, Ramakrishnan DP, Peng YJ, Souvannakitti D, Prabhakar NR. Chronic intermittent hypoxia induces hypoxia-evoked catecholamine efflux in adult rat adrenal medulla via oxidative stress. J Physiol. 2006;575:229–239. doi: 10.1113/jphysiol.2006.112524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Prabhakar NR. Peripheral chemoreceptors: function and plasticity of the carotid body. Compr Physiol. 2012;2:141–219. doi: 10.1002/cphy.c100069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuri BA, Khan SA, Chan SA, Prabhakar NR, Smith CB. Increased secretory capacity of mouse adrenal chromaffin cells by chronic intermittent hypoxia: involvement of protein kinase C. J Physiol. 2007;584:313–319. doi: 10.1113/jphysiol.2007.140624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesske J, Fletcher EC, Bao G, Unger T. Hypertension caused by chronic intermittent hypoxia – influence of chemoreceptors and sympathetic nervous system. J Hypertens. 1997;15:1593–1603. doi: 10.1097/00004872-199715120-00060. [DOI] [PubMed] [Google Scholar]
- Nanduri J, Wang N, Yuan G, Khan SA, Souvannakitti D, Peng YJ, Kumar GK, Garcia JA, Prabhakar NR. Intermittent hypoxia degrades HIF-2alpha via calpains resulting in oxidative stress: implications for recurrent apnea-induced morbidities. Proc Natl Acad Sci U S A. 2009;106:1199–1204. doi: 10.1073/pnas.0811018106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanduri J, Vaddi DR, Khan SA, Wang N, Makerenko V, Prabhakar NR. Xanthine oxidase mediates hypoxia-inducible factor-2alpha degradation by intermittent hypoxia. PLoS One. 2013;8:e75838. doi: 10.1371/journal.pone.0075838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkiewicz K, van de Borne PJ, Pesek CA, Dyken ME, Montano N, Somers VK. Selective potentiation of peripheral chemoreflex sensitivity in obstructive sleep apnea. Circulation. 1999;99:1183–1189. doi: 10.1161/01.cir.99.9.1183. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 6th edn. Burlington, MA, USA: Academic Press; 2006. [Google Scholar]
- Peng YJ, Prabhakar NR. Effect of two paradigms of chronic intermittent hypoxia on carotid body sensory activity. J Appl Physiol. 2004;96:1236–1242. doi: 10.1152/japplphysiol.00820.2003. discussion 1196. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proc Natl Acad Sci U S A. 2003;100:10073–10078. doi: 10.1073/pnas.1734109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Yuan G, Ramakrishnan D, Sharma SD, Bosch-Marce M, Kumar GK, Semenza GL, Prabhakar NR. Heterozygous HIF-1alpha deficiency impairs carotid body-mediated systemic responses and reactive oxygen species generation in mice exposed to intermittent hypoxia. J Physiol. 2006;577:705–716. doi: 10.1113/jphysiol.2006.114033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Nanduri J, Khan SA, Yuan G, Wang N, Kinsman B, Vaddi DR, Kumar GK, Garcia JA, Semenza GL, Prabhakar NR. Hypoxia-inducible factor 2alpha (HIF-2alpha) heterozygous-null mice exhibit exaggerated carotid body sensitivity to hypoxia, breathing instability, and hypertension. Proc Natl Acad Sci U S A. 2011;108:3065–3070. doi: 10.1073/pnas.1100064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Nanduri J, Zhang X, Wang N, Raghuraman G, Seagard J, Kumar GK, Prabhakar NR. Endothelin-1 mediates attenuated carotid baroreceptor activity by intermittent hypoxia. J Appl Physiol. 2012;112:187–196. doi: 10.1152/japplphysiol.00529.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppard PE, Young T, Palta M, Skatrud J. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med. 2000;342:1378–1384. doi: 10.1056/NEJM200005113421901. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR, Semenza GL. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol Rev. 2012;92:967–1003. doi: 10.1152/physrev.00030.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar NR, Kumar GK, Nanduri J, Semenza GL. ROS signaling in systemic and cellular responses to chronic intermittent hypoxia. Antioxid Redox Signal. 2007;9:1397–1403. doi: 10.1089/ars.2007.1732. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR, Kumar GK, Peng YJ. Sympatho-adrenal activation by chronic intermittent hypoxia. J Appl Physiol (1985) 2012;113:1304–1310. doi: 10.1152/japplphysiol.00444.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M, Solaro CR, Lingle CJ. [Ca2+]i elevations detected by BK channels during Ca2+ influx and muscarine-mediated release of Ca2+ from intracellular stores in rat chromaffin cells. J Neurosci. 1996;16:4344–4359. doi: 10.1523/JNEUROSCI.16-14-04344.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuraman G, Rai V, Peng YJ, Prabhakar NR, Kumar GK. Pattern-specific sustained activation of tyrosine hydroxylase by intermittent hypoxia: role of reactive oxygen species-dependent downregulation of protein phosphatase 2A and upregulation of protein kinases. Antioxid Redox Signal. 2009;11:1777–1789. doi: 10.1089/ars.2008.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey S, Del Rio R, Alcayaga J, Iturriaga R. Chronic intermittent hypoxia enhances cat chemosensory and ventilatory responses to hypoxia. J Physiol. 2004;560:577–586. doi: 10.1113/jphysiol.2004.072033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salman S, Buttigieg J, Nurse CA. Ontogeny of O2 and CO2//H+ chemosensitivity in adrenal chromaffin cells: role of innervation. J Exp Biol. 2014;217:673–681. doi: 10.1242/jeb.086165. [DOI] [PubMed] [Google Scholar]
- Schultz HD, Li YL. Carotid body function in heart failure. Respir Physiol Neurobiol. 2007;157:171–185. doi: 10.1016/j.resp.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scortegagna M, Ding K, Oktay Y, Gaur A, Thurmond F, Yan LJ, Marck BT, Matsumoto AM, Shelton JM, Richardson JA, Bennett MJ, Garcia JA. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1-/- mice. Nat Genet. 2003;35:331–340. doi: 10.1038/ng1266. [DOI] [PubMed] [Google Scholar]
- Silva AQ, Schreihofer AM. Altered sympathetic reflexes and vascular reactivity in rats after exposure to chronic intermittent hypoxia. J Physiol. 2011;589:1463–1476. doi: 10.1113/jphysiol.2010.200691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souvannakitti D, Kuri B, Yuan G, Pawar A, Kumar GK, Smith C, Fox AP, Prabhakar NR. Neonatal intermittent hypoxia impairs neuronal nicotinic receptor expression and function in adrenal chromaffin cells. Am J Physiol Cell Physiol. 2010;299:C381–C388. doi: 10.1152/ajpcell.00530.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoohs R, Guilleminault C. Cardiovascular changes associated with obstructive sleep apnea syndrome. J Appl Physiol (1985) 1992;72:583–589. doi: 10.1152/jappl.1992.72.2.583. [DOI] [PubMed] [Google Scholar]
- van den Aardweg JG, Karemaker JM. Repetitive apneas induce periodic hypertension in normal subjects through hypoxia. J Appl Physiol (1985) 1992;72:821–827. doi: 10.1152/jappl.1992.72.3.821. [DOI] [PubMed] [Google Scholar]
- Verna A, Roumy M, Leitner LM. Loss of chemoreceptive properties of the rabbit carotid body after destruction of the glomus cells. Brain Res. 1975;100:13–23. doi: 10.1016/0006-8993(75)90239-5. [DOI] [PubMed] [Google Scholar]
- Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med. 1993;328:1230–1235. doi: 10.1056/NEJM199304293281704. [DOI] [PubMed] [Google Scholar]
- Young T, Finn L, Peppard PE, Szklo-Coxe M, Austin D, Nieto FJ, Stubbs R, Hla KM. Sleep disordered breathing and mortality: eighteen-year follow-up of the Wisconsin sleep cohort. Sleep. 2008;31:1071–1078. [PMC free article] [PubMed] [Google Scholar]
- Yuan G, Nanduri J, Khan S, Semenza GL, Prabhakar NR. Induction of HIF-1alpha expression by intermittent hypoxia: involvement of NADPH oxidase, Ca2+ signaling, prolyl hydroxylases, and mTOR. J Cell Physiol. 2008;217:674–685. doi: 10.1002/jcp.21537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan G, Khan SA, Luo W, Nanduri J, Semenza GL, Prabhakar NR. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J Cell Physiol. 2011;226:2925–2933. doi: 10.1002/jcp.22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan G, Peng YJ, Reddy VD, Makarenko VV, Nanduri J, Khan SA, Garcia JA, Kumar GK, Semenza GL, Prabhakar NR. Mutual antagonism between hypoxia-inducible factors 1alpha and 2alpha regulates oxygen sensing and cardio-respiratory homeostasis. Proc Natl Acad Sci U S A. 2013;110:E1788–1796. doi: 10.1073/pnas.1305961110. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zoccal DB, Bonagamba LG, Oliveira FR, Antunes-Rodrigues J, Machado BH. Increased sympathetic activity in rats submitted to chronic intermittent hypoxia. Exp Physiol. 2007;92:79–85. doi: 10.1113/expphysiol.2006.035501. [DOI] [PubMed] [Google Scholar]