

Abstract
Identification of the coding elements in the genome is a fundamental step to understanding the building blocks of living systems. Short peptides (< 100 aa) have emerged as important regulators of development and physiology, but their identification has been limited by their size. We have leveraged the periodicity of ribosome movement on the mRNA to define actively translated ORFs by ribosome footprinting. This approach identifies several hundred translated small ORFs in zebrafish and human. Computational prediction of small ORFs from codon conservation patterns corroborates and extends these findings and identifies conserved sequences in zebrafish and human, suggesting functional peptide products (micropeptides). These results identify micropeptide-encoding genes in vertebrates, providing an entry point to define their function in vivo.
Keywords: micropeptides, non-coding RNA, ribosome profiling, small ORFs, translation
See also: SM Cohen (May 2014)
Introduction
Analysis of the genome has identified many putative transcripts that lack the classical hallmark of eukaryotic protein-coding genes: a single, long, conserved coding sequence (CDS) encoding a protein of more than 100 amino acids (Carninci et al, 2005; Birney et al, 2007; Tautz, 2009; Ulitsky et al, 2011; Derrien et al, 2012; Pauli et al, 2012). However, many of these transcripts (including lincRNAs) (Khalil et al, 2009; Guttman et al, 2010; Ingolia et al, 2011) contain multiple putative small open reading frames (smORFs, ≤ 100 aa) that can potentially be translated and thus might have a coding function (Ingolia et al, 2011; Chew et al, 2013; Slavoff et al, 2013). Recent examples have revealed functional, protein-coding smORFs across various genomes in RNAs previously thought to be non-coding (Savard et al, 2006; Galindo et al, 2007; Kondo et al, 2007, 2010; Pueyo & Couso, 2008; Magny et al, 2013). mille-pattes and tarsal-less/polished-rice were found to encode several micropeptides required during development in Tribolium and Drosophila, respectively (Savard et al, 2006; Kondo et al, 2007; Pueyo & Couso, 2008). Similarly, the predicted non-coding pncr003:2L gene encodes two micropeptides, each smaller than 30 aa, that regulate cardiac contraction in Drosophila (Magny et al, 2013).
Comprehensive identification of smORFs has been challenging and has mainly relied on evolutionary conservation (Stark et al, 2007; Lin et al, 2011), known patterns of codon occurrence and mass spectrometry (Schwaid et al, 2013; Slavoff et al, 2013). However, these approaches can be limited by the size, abundance and amino acid composition of the polypeptide. Ribosome footprinting measures translation by direct quantification of mRNA fragments protected by the 80S ribosome (ribosome-protected fragments, RPFs) after nuclease digestion (Fig1A) (Wolin & Walter, 1988; Ingolia et al, 2009). Recent studies have used ribosome footprinting (Ingolia et al, 2009; Bazzini et al, 2012) to characterize the coding potential of different transcripts (Chew et al, 2013; Guttman et al, 2013) and identify translated protein-coding sequences (Ingolia et al, 2011; Brar et al, 2012; Michel et al, 2012; Stern-Ginossar et al, 2012; Chew et al, 2013; Crappe et al, 2013; Menschaert et al, 2013; Pauli et al, 2014). However, it has been questioned whether fragments recovered from ribosome profiling libraries always reflect a translating ribosome as opposed to regions protected by other RNA-binding proteins, or spurious binding to the ribosome (Guttman et al, 2013), and therefore, it is unclear how well these methods perform to identify individual smORFs. Unlike other interactions between mRNA, proteins, or scanning ribosomes, actively translating ribosomes have a unique property: the discrete movement along the message in three-nucleotide steps (phasing) (Ingolia et al, 2009; Guo et al, 2010; Michel et al, 2012), a feature that has been used to identify frame shifts and dually decoded regions in the genome (Michel et al, 2012). Since phased ribosome binding is a direct consequence of active translation, we reasoned that using phased binding as a criterion would reduce the noise in conventional ribosome profiling analyses and would allow us to identify smORFs undergoing translation in vivo.
Figure 1. Ribosome profiling in zebrafish.
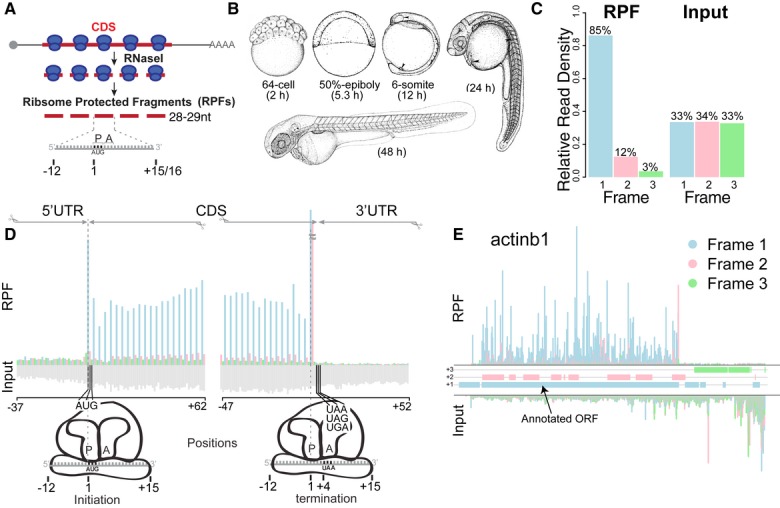
- Schematic representation of ribosome profiling: 28 to 29-nt-long ribosome-protected fragments (RPFs) are generated from nuclease digestion, where the P-site of the ribosome is in position 13.
- Developmental stages at which ribosome profiling was performed.
- Subcodon position of the ribosome footprints (position 13) for the RPF and input reads. Plot shows the proportion of RPFs or input reads aligned to the coding sequence of RefSeq genes at each position relative to the codon. Input reads were obtained after poly-(A) fractionation and random fragmentation of the naked RNA.
- RPFs and input reads mapped to a composite RefSeq transcript. RPFs mainly map to the CDS with a 3-nucleotide periodicity. RPF reads are colored as in (C) based on the position with respect to the frame of the CDS. Input reads map to both the UTRs and CDS (gray).
- Subcodon profile plot showing RPF and input reads aligned to actinb1. Reads are colored based on the frame (1, 2 or 3) position relative to the transcript (Michel et al, 2012). All putative ORFs (distal AUG-Stop) were also colored for each respective frame (blue, pink and green boxes). Note that most of the RPFs from the annotated ORF match the color of the box, consistent with a strong in-frame distribution of reads within individual transcripts.
Results
Ribosome footprinting in zebrafish with subcodon resolution
To define the coding potential of the transcriptome and identify translated smORFs, we analyzed the positional distribution of active ribosomes during zebrafish development. To this end, we generated high-depth ribosome footprinting with subcodon resolution across embryogenesis (at 2, 5, 12, 24 and 48 h post-fertilization, hpf) (Fig1B). This resulted in approximately 200 million mapped reads after filtering for ribosomal RNAs, tRNAs and snoRNAs (Supplementary Table S1). 95% of reads within RefSeq protein-coding genes overlapped the CDS. Because phasing of the ribosome footprints can vary with fragment size, we first analyzed the distribution of RPFs within a composite RefSeq transcript (Supplementary Fig S1). Metagene analysis of the reads mapping to the annotated CDS revealed that 84.6% of the 28 and 29 nt RPFs were in-frame relative to their 5′ ends (position 1 in the codon of the P-site, offset +12 nt), whereas the RNA input fragments did not present any bias in their distribution (Fig1C). The periodic distribution of RPFs observed along the CDS within each codon (Fig1D, Supplementary Fig S1) reflects the stepwise translocation of active ribosomes (Ingolia et al, 2009). We reasoned that this pattern should derive from a biased in-frame distribution of RPFs within each individual CDS (Fig1E). Thus, we hypothesized that this pattern could be used to define actively translated regions and distinguish them from background signal.
ORFscore, a method to identify actively translated smORFs
Guided by this hypothesis, we developed a method (ORFscore) that quantifies the biased distribution of RPFs toward the first frame of a given CDS (Fig2). Given a putative ORF in the transcriptome (AUG to stop), we quantified the number of RPFs in each frame and determined whether RPFs were uniformly distributed or preferentially accumulated in one frame. We assigned a negative value to RPF distributions inconsistent with the frame of the ORF (Fig2A). To filter ORFs with single or few codons covered by reads, we calculated the proportion of codons with in-frame reads (coverage) (Fig2B). Next, we tested the predictive value of our methodology. Several lines of evidence suggest that our method identifies individual ORFs with coding potential. First, we analyzed all possible ORFs in annotated coding RefSeq transcripts. ORFscore was generally high across RefSeq CDS regions, with 85% of the expressed genes (> 1RPKM) having ORFscore ≥ 6.044 and coverage ≥ 10% (Fig3A, C). In contrast, scores for most ORFs in the 5′UTR, the 3′UTR or overlapping the annotated CDS out of frame fell below these levels, reflecting their lack of coding potential (Fig3A). Selecting the ORF with the highest ORFscore per transcript correctly identified the annotated CDS in 99% of the expressed coding transcripts, clearly distinguishing them from other possible ORFs in each transcript (P < 2.2e-16, Chi-squared test) (Fig3B).
Figure 2. Defining actively translated regions by ribosome profiling.
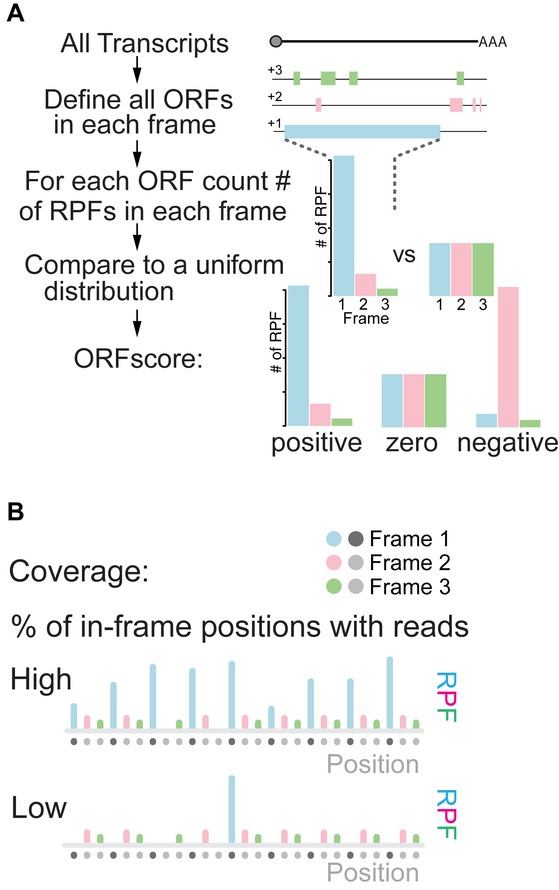
- Workflow to define the ORFscore: Top diagram represents a transcript, below solid bars represent all possible ORFs (Distal AUG-Stop) identified in each frame (+1, +2, +3). The RPF distribution in each frame is compared to an equally sized uniform distribution using a modified chi-squared statistic (see Materials and Methods). The resulting ORFscore is assigned a negative value when the distribution of RPFs is inconsistent with the frame of the CDS.
- Coverage is determined by measuring the proportion of in-frame CDS positions with ≥ 1 reads.
Figure 3. ORFscore discriminates translated from non-translated regions.
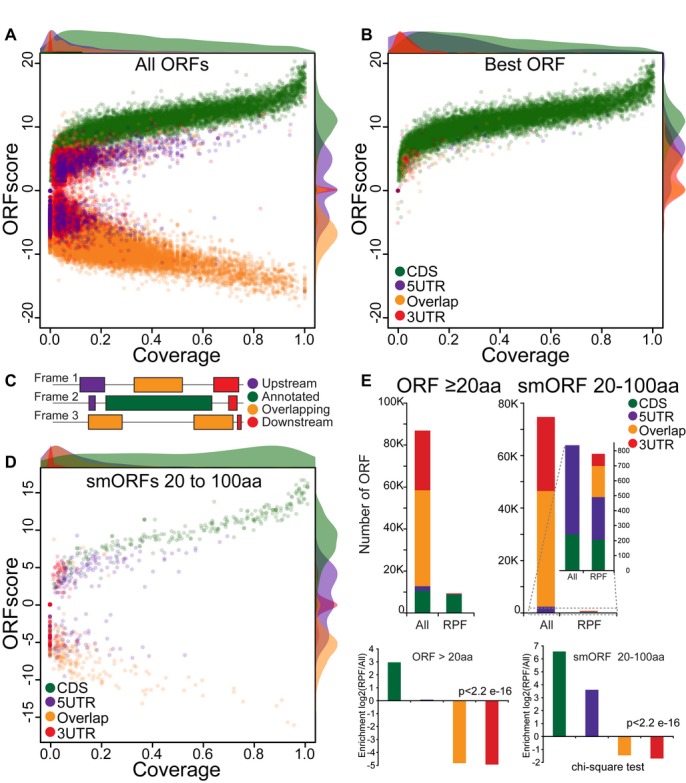
A–D Scatterplot of the ORFscore and coverage for all ORFs (A), the subset of ORFs with the highest ORFscore per transcript (B) and short (20–100 aa) annotated CDS (D). Relative density plots (scaled to the maximum value for each group) of the ORFscore and coverage are shown for each ORF type. Note the separation between annotated ORFs from the rest of the ORFs, even for short (20–100 aa) annotated CDSs. (C) Color code used to label different ORF types found in RefSeq protein-coding transcripts: annotated CDS (green), 5′UTR ORFs (purple), 3′UTR ORFs (red) and ORFs overlapping the annotated CDS (orange).
E Bar plots representing the number of ORFs identified on the basis of their ORFscore and coverage and defined as translated for each ORF type as in (C). Among all putative ORFs, the distribution of annotated ORFs was significantly different from the overall set (P = 2.2e-16, chi-squared test) with long and short CDS showing the highest fold-change enrichment in translated ORFs compared to other ORF types.
Annotated CDSs are usually longer than ORFs in the 5′- and 3′-UTRs (Supplementary Fig S2). To ensure that the correct identification of annotated CDSs is not simply due to this bias in size, we restricted the analysis to transcripts containing known coding regions ≤ 100 aa, using the same parameters (Fig3D). This analysis identified 86% (208 out of 241) of short annotated CDSs, distinguishing them from 74,669 other putative short ORFs in expressed RefSeq coding transcripts (20-to-100 aa, P < 2.2e-16, Chi-squared test) (Fig3E). Thus, combining frame bias and coverage provides a measure of coding potential that can be used to confidently identify small translated ORFs.
Identification of novel smORFs by ORFscore
To identify novel translated ORFs, we applied the ORFscore method to transcripts without defined coding sequences, including previously annotated long non-coding RNAs (Ulitsky et al, 2011; Pauli et al, 2012; Howe et al, 2013) and uncharacterized processed transcripts from Ensembl (Howe et al, 2013) (Fig4A). In this analysis, Ensembl-annotated smORFs were used as a positive control (Fig4A). Out of 2450 genes without previously defined coding sequences, many of which are thought to be non-coding, our analysis found experimentally supported coding ORFs in 303 genes. Of these, 214 (71%) encode smORFs between 20 and 100 aa long corresponding to 190 non-redundant smORF loci (Fig4C and E, Supplementary Fig S3, Supplementary Table S2 and Supplementary File S1) and 89 (29%) encode for proteins longer than 100 aa. The majority of defined smORFs do not share significant amino acid sequence homology with known proteins in zebrafish (Fig4D). An additional set of 53 non-redundant smORFs (Fig4E) was identified after relaxing the coverage requirements while maintaining requirement for phasing of the ribosomes through the ORFscore. In contrast, 959 expression-matched transcripts lacked evidence for coding ORFs, including the known non-coding RNAs cyrano and megamind (Ulitsky et al, 2011; Chew et al, 2013) (Fig4B, Supplementary Fig S4). Our analysis also provided experimental support for translation of 302 (52%) of the smORFs that were previously predicted by Ensembl and RefSeq (Howe et al, 2013) (Fig4E) and distinguished them from size-matched ORFs in the 3′UTR that were used as control for non-coding regions (Fig4A). Gene expression analysis revealed developmental regulation of mRNA levels for both smORF-containing and non-coding RNAs during embryogenesis (Fig4K, Supplementary Fig S4). As an independent analysis, we determined whether the polypeptide products from translated smORFs are detected by mass spectrometry (MS) (Supplementary Fig S5). We identified peptides for 98 annotated smORFs (˜32% out of 302) and 6 novel smORFs (˜3% out of 190) (Fig4F), including those encoded by ENSDART00000145781 and linc-brsk1 (Fig4I and J, Supplementary Fig S3). Identification of proteins by shotgun proteomics depends, among other factors, on protein and peptide lengths and abundances (Slavoff et al, 2013), which may explain why novel smORFs appear to be underrepresented in our recovered set, since they are shorter than previously annotated peptides (P = 1.6e-43, Wilcoxon test) (Fig4G). Translated smORFs are also present in canonical protein-coding transcripts. Using ORFscore, we identify 311 (5′UTR) and 93 (3′UTR) translated ORFs, of which 17 and 10, respectively, were also identified by mass spectrometry (Fig4H, Supplementary Fig S3). Future studies will be needed to further characterize the function of this large set of upstream and downstream ORFs, as they may regulate mRNA stability or translation of the main CDS (Barbosa et al, 2013). Taken together, these results reveal expression of several hundred smORFs present in transcripts with previously undefined coding sequences.
Figure 4. Identification of small coding ORFs (smORFs) in non-coding RNAs.

A Scatterplot of ORFscore and coverage for the ORF with highest ORFscore per transcript. Shown are annotated short ORF (20–100 aa) (green), annotated lincRNA and “processed transcripts” from Ensembl (orange), non-coding RNAs described by Ulitsky et al (2011) (set 1, dark blue) and by Pauli et al (2012) (set 2, light blue) and ORFs in annotated 3′UTR used as negative control (red). Note that several ORFs in non-coding annotated transcripts score at comparable levels to annotated CDSs. Inset shows the scatter plot for annotated smORFs and 3′UTR ORFs. Relative density plots (scaled to the maximum value for each group) of the ORFscore and coverage are shown for each ORF type.
B Subcodon profile plot showing a known non-coding RNA, cyrano, depleted of ribosome footprints.
C Stacked plot showing the proportion of genes in which a translated ORF was defined by ORFscore and 10% coverage (*, stringent) or only ORFscore (**, permissive) and transcripts with low ORFscore (undetermined). The number of transcripts in each fraction is indicated.
D Pie chart of BLASTp results against several organisms for the 241 newly defined translated regions, collapsed on amino acid sequence.
E Bar plot showing the number of unique novel smORFs and Ensembl-predicted smORFs (≤ 100 aa), defined by ORFscore and 10% coverage (*, stringent and predicted) or only ORFscore (**, permissive).
F Bar plot displaying the number of novel and Ensembl-predicted smORFs identified by tandem mass spectrometry (MS-MS).
G Box plot representing the size distribution of the ORFs defined by ORFscore and MS-MS.
H Bar plot showing the number of genes with translated ORFs in the 5′ or 3′ UTR defined by ORFscore or detected by MS-MS.
I, J Subcodon profile plots showing individual examples of identified smORFs: Ribosome profiling data show the translated ORF and fragmentation spectra identifying the encoded peptides.
K Heat-map showing dynamic expression of novel smORF-containing genes during zebrafish embryogenesis (n = 190).
Computational prediction of smORFs from codon conservation patterns
Translation by ribosomes per se does not imply that a smORF encodes a functional peptide. For example, the peptide could be unstable or translation could function to regulate transcript stability. Moreover, a fraction of smORFs may have been missed using ribosome footprinting if they were not sufficiently expressed in the stages analyzed. Alternatively, conservation analysis can reveal evolutionary pressure to maintain the amino acid sequence of functional peptides. Thus, as an independent and complementary approach, we developed a computational pipeline (micropeptide detection pipeline, micPDP) to search for smORFs and evaluate the evidence for negative selection on the encoded amino acid sequence from codon substitutions observed in whole-genome alignments. We filtered candidate alignments by coverage and reading frame conservation and then used phyloCSF (Lin et al, 2011) to score the coding potential from codon substitutions observed in whole-genome multiple alignments. We also used a simple parameter-free method (Ka/Ks) as control, which yields comparable results (Supplementary Fig S6). From published catalogs of zebrafish transcripts without annotated coding sequences, including lincRNAs (Ulitsky et al, 2011; Pauli et al, 2012; Howe et al, 2013), we evaluated 15,743 ORFs and from these predicted 63 conserved smORFs on 60 different transcripts (Fig5A). 23 of these were also found by ORFscore (P < 2e-22, Fisher's exact test) (Fig5B). Note that only 45 of the experimentally detected smORFs in zebrafish had sufficient sequence alignment and could be scored by phyloCSF. Experimental and computational scores are correlated in zebrafish (Fig5B and C), and smORFs score better by one method if they were detected by the other than if they were not (P < 3e-15, Mann–Whitney U-test, for both cases). Analysis of 33,961 human lincRNAs from Ensembl and RefSeq (Cabili et al, 2011; Derrien et al, 2012) yields comparable results, predicting 173 smORFs on 160 different transcripts (Fig5A and C). Using ORFscore to analyze previously published ribosome footprinting data in HeLa cells (Guo et al, 2010) (Supplementary Fig S6) defines 135 translated smORFs (118 unique loci) in human lincRNAs (Fig5D, Supplementary Table S2 and Supplementary File S2) and a small overlap of seven candidates out of 95,780 smORFs with the computational results (P < 6.3e-9, Fisher's exact test) (Fig5C and E). Taken together, we identify hundreds of translated smORFs in human and fish and define an overlapping set of smORFs encoding evolutionarily conserved peptides.
Figure 5. Computational identification of evolutionarily conserved smORFs (MicPDP).

A Number of smORFs detected within putative non-coding RNA transcripts in zebrafish and human.
B,C Scatterplot of ORFscore and phyloCSF score for 686 zebrafish and 45,079 human smORFs with sufficient alignment coverage. The predictions of the two methods have small but significant overlap (light blue dots; P < 2e-22 and P < 6.3e-9 respectively, Fisher's exact test), and zebrafish experimental and computational results are correlated (Spearman's ρ = 0.49, P < 4e-42).
D Scatterplot of ORFscore and coverage for 2,000 randomly selected human Ensembl-annotated coding ORFs (green), 2,000 ORFs in the 3′UTR and the set of coding ORFs from human lincRNAs as defined by ORFscore (blue, best ORFscore per unique genomic locus).
E Subcodon profile plot, showing a smORF in the human predicted non-coding RNA ENST00000426713 (LINC00116-002) that presented high phyloCSF score and ORFscore.
Discussion
Our analysis of the zebrafish transcriptome using ribosome profiling provides two key insights into the genome-wide expression of smORFs in vertebrates. First, smORFs are widely distributed and are translated from a large body of transcripts, many of which were thought to lack coding potential. We experimentally identified hundreds of translated smORF regions that encode small proteins (micropeptides), defining 190 smORFs, 311 ORFs in the 5′UTR and 93 in the 3′UTR, and validated a portion of these by mass spectrometry. Previous studies have used ribosome footprinting to define classes of transcripts within non-coding RNAs based on the pattern of ribosome footprints when compared to known coding genes (Chew et al, 2013; Guttman et al, 2013), but in most cases this classification does not define the translated ORF. In contrast to existing methods (Supplementary Fig S7) (Ingolia et al, 2011; Michel et al, 2012; Chew et al, 2013; Guttman et al, 2013), the ORFscore leverages the periodicity of high-quality ribosome-protected fragments to define small translated ORFs (Supplementary Fig S7) independent of the surrounding sequence context in zebrafish and humans (Supplementary Fig S7) (Guttman et al, 2013). While our method provides strong support for translation of individual ORFs using the parameters defined, we observe that relaxing the coverage cutoff recovers an additional set of ORFs that are defined with lower confidence but maintain strong phasing of the ribosome, suggesting active translation (Fig E; Supplementary Fig S6). Indeed, the Translated ORF Classifier designed by Chew et al (2013) identifies a fraction of these transcripts as coding, providing a complementary method to define the coding potential of RNA (Supplementary Fig S6). The ORFscore strongly depends on the phasing of the ribosomes, therefore overlapping ORFs that are translated on different reading frames (Michel et al, 2012) can be missed by this method depending on the region of overlap for these ORFs; thus, future refinement will be necessary to define overlapping translated ORFs in zebrafish (Supplementary Fig S3). Applying the ORFscore analysis to previously published ribosome footprinting data in human cell lines (Guo et al, 2010) provides evidence for translation of smORFs present in human RNAs previously classified as non-coding (Supplementary Table S2). The presence of these small translated regions does not rule out a direct function of the mRNA transcript independent of the encoded peptide. Translation of small regions in these transcripts may be necessary for RNA function through localization, folding or triggering non-sense-mediated decay (Medenbach et al, 2011; Chew et al, 2013; Guttman et al, 2013; Somers et al, 2013). Indeed, we have observed in lincRNAs a class of tiny ORFs (< 20 aa) supported by in-frame RPFs: For example, cyrano contains a 2aa ORF in zebrafish that displays high in-frame translation (Fig4B). Due to the small size of these ORFs, further work will be needed to characterize their functions in vivo. Our method also defines a subset of transcripts with no evidence of translation, supporting a non-coding function for these transcripts.
Second, independent of the ribosomal footprint analyses, we developed a computational pipeline (micPDP) that identified a set of micropeptides which are likely under natural selection by computationally analyzing codon conservation patterns in multiple species alignments of annotated human lincRNAs and fish transcripts without previously defined coding sequences, including lincRNAs. Half of the translated micropeptides analyzed by ribosome footprinting with sequence alignment across species (23 out of 45) present strong patterns of evolutionary conservation (Fig5B and C), supporting a functional role of these coding sequences. A small group of the identified smORFs (25%) have predicted homologs (e.g. RPL41) (Fig4D), complementing current genome annotations (Howe et al, 2013). We note that in human, the number of micropeptides with good conservation scores, but low ORFscore was much higher than in zebrafish. This could be explained by lower depth and phasing of the human ribosomal profiling data used or by micropeptides expressed specifically in developmental stages and not in the human cell line. Further, the larger number of species in the human genome alignment allowed us to score smORF coding potential more comprehensively than in zebrafish. Still, smORFs might have different codon usage and conservation patterns compared to canonical protein-coding genes and may therefore be only incompletely captured by our computational pipeline. Also, smORFs encoding for lineage-specific or fast-evolving peptides, or smORFs with purely regulatory function, will be missed since our comparative method is tailored toward the identification of smORFs with conservation of their encoded amino acid sequence over larger evolutionary distances. The conceptual differences between experimental and computational approaches lend strong support to jointly identified smORFs but translate into an inability to use one method to estimate false-positive or false-negative rate of the other. In sum, the computational results reported here reveal numerous smORFs in zebrafish and human and suggest that they have evolutionarily conserved sequence and function.
Central roles of small peptides have been known for decades based on the importance of neuropeptides, peptide hormones (such as secretin or insulin) and secreted signaling molecules (such as FGF1). However, the majority of these small peptides are encoded as large pre-proteins that undergo post-translational cleavage and modification. By contrast, similar functional small peptides such as ELABELA/toddler (Chng et al, 2013; Pauli et al, 2014) (also identified in our set of smORFs; Supplementary Fig S3) are directly translated from small ORFs. Indeed, Pauli et al (2014) recently reported the independent identification of more than 300 previously unannotated zebrafish proteins. Small proteins are also found in viral genomes, where transmembrane proteins (often shorter than 50 aa) have been shown to play vital roles in virus replication and virulence (Dimaio, 2014). It remains unclear how many of the peptides encoded by our in silico or in vivo identified smORFs are biologically relevant. However, ELABELA/toddler (Chng et al, 2013; Pauli et al, 2014), together with small peptides from other organisms and viruses (Savard et al, 2006; Galindo et al, 2007; Kondo et al, 2007, 2010; Pueyo & Couso, 2008; Magny et al, 2013), show that smORFs can play important roles across development and physiology. In sum, our identification of hundreds of translated smORFs significantly expands the set of micropeptide-encoding vertebrate genes providing an entry point to investigate their function in vivo.
Materials and Methods
RPF Library/Prep/SEQ
Ribosome-protected fragments: 50 embryos were collected for each time point and immediately frozen in liquid nitrogen. Each sample was lysed in 800 μl of Lysis buffer (1× Polysome Buffer, 1% Triton X-100, 1 mM DTT, 25 U/ml DNaseI and 100 μg/ml cycloheximide) according to manufacturer specifications (Epicentre, Artseq Ribosome Profiling Kit Mammalian, HRPHMR12126). The mix was clarified for 10 minutes (min.) at 10,000 g at 4°C. Three microliters of ARTseq Nuclease (Epicentre, HRPHMR12126) was added to 400 μl of the lysed supernatant and incubated at room temperature for 45 min with gentle mixing. Nuclease digestion was stopped with 15 μl of SUPERase·In™ RNase Inhibitor (Life Technologies, cat. no. AM2696) and chilled on ice. Ribosomes were purified by Sephacryl S400 spin column chromatography (GE Healthcare, cat. no. 27-5140-01) or sucrose cushion ultracentrifugation according to manufacturer specifications (Epicentre, Artseq Ribosome Profiling Kit). The 400 μl of treated lysate was divided into four S400 spin columns (1 min/735 g). One hundred microliters of 1× Polysome Buffer was added to the same columns and spin again (1 min/735 g). RNA from the 400 μl samples was extracted with Trizol, and rRNA was subtracted using RiboZero (Ribo-Zero™ Magnetic Kit (Human/Mouse/Rat)) (Epicentre; cat no. MRZH11124), following the manufacturer's protocol, omitting the 50°C incubation step. Ribosome-protected fragments (RPFs) were separated in a 15% Urea gel, and the region from 28-to-30 nucleotides was excised. RNA was eluted overnight in 300 mM NaOAc pH 5.5; 1 mM EDTA; 0.1 U/μl SUPERase In (Ambion #AM2694), followed by Ethanol precipitation.
RNA input
Total RNA was isolated from 400 μl of the 800 μl of clarified extract before ARTseq Nuclease treatment. Poly-A selection was done according to the manufacturer guidelines (Dynabeads mRNA Purification Kit, Cat no.610.06), and RNA was fragmented using the ARTseq Ribosome Profiling Kit Mammalian protocol.
Library preparation
Both RPF and RNA input fragments were cloned according to the ARTseq Ribosome Profiling Kit, Mammalian. The final PCR was carried out with an initial 15-s denaturation at 98°C, followed by 9-12 cycles of 15 s at 98°C, 5 s at 55°C and extension at 72°C for 10 s. Reactions were separated on a non-denaturing 8% polyacrylamide TBE gel, and DNA fragments of the correct size (113 + 28∼29 nt) were extracted and sequenced using an Illumina HiSeq 2000 sequencer.
Filtering and alignment of ribosome profiling reads
Base calling was performed using CASAVA-1.8.2. The Illumina TruSeq index adaptor sequence was then trimmed from raw reads by aligning its sequence, requiring 100% match of the first five base pairs and a minimum global alignment score of 60 (Matches: 5, Mismatches: −4, Gap opening: −7, Gap extension: −7, Cost-free ends gaps). Trimmed reads were then depleted of rRNA, tRNA, snRNA, snoRNA and misc_RNA from Ensembl and RepeatMasker annotations using strand-specific alignment performed with Bowtie2 v2.1.0 (Flicek et al, 2013) with default parameters. Filtered reads were finally aligned strand-specifically to the Zebrafish Zv9 (danRer7) genome assembly using Tophat v2.0.8 (Kim et al, 2013) with default parameters and the exon-junction coordinates from Ensembl r70. The same procedure was applied to the “input” reads.
The GEO GSE22004 filtered sequence set (Guo et al, 2010) was analyzed with the same procedure and the following modifications. The trimming required only five base pairs of the adaptor matching more than 80%. The filtering step used the tRNA from the Genomic tRNA database (Chan & Lowe, 2009) and Mt_rRNA, Mt_tRNA, rRNA, snoRNA, snRNA from Ensembl r73 non-coding RNAs. Sequencing reads were combined for the 12 and 32 h time points.
Defining transcript sets
To define annotated zebrafish transcripts, Ensembl transcripts (release 73) were downloaded from the Ensembl FTP repository and imported into R using the GenomicFeatures package. RefSeq transcripts were retrieved from UCSC and imported using the R GenomicFeatures package (Gentleman et al, 2004). To assemble a set of published lincRNA transcripts, supplemental data were downloaded from (Ulitsky et al, 2011) and (Pauli et al, 2012) and loaded into R using the RTrackLayer package. To define annotated human transcripts, the GenomicFeatures package was used to retrieve Ensembl (r73) transcript sets, and scaffolds were filtered out leaving transcripts on chromosomes 1–22, X and Y. Zebrafish transcript sequences were retrieved using genome assembly version danRer7/Zv9, and human sequences were retrieved from assembly version hg19. Each gene and each transcript were assigned a unique ID so that transcript variants could be analyzed individually.
Defining ORFs
Using the spliced version of each transcript, all possible stop codons in each of the three reading frames were defined. Each stop codon was paired with the most distal in-frame AUG codon without an intervening stop. Each region from most distal AUG to stop was thus defined as an ORF. ORFs were stored in both genomic and transcript-relative coordinates for further analyses. For position-related calculation of ORFscore and coverage of position 1, only reads within the ORF (defined as the region excluding the reads aligning to the start and stop (−1) codons) were counted.
Processing alignments/reads
Aligned reads were imported into R using the GenomicFeatures package. For downstream analysis, all sequencing experiment replicates were combined per time point. For “global” values, all time points were combined. RPF reads were filtered by size (28 & 29 nt). RNA expression was calculated in RPKM using corresponding input samples, using reads mapped to each transcript set as a total. For each transcript, all reads aligning to that transcript were mapped to transcript coordinates. For a given open reading frame, the position of each RPF read was designated as the +12 offset position of the read, corresponding to the P-site of the ribosome (Lawrence et al, 2009).
Calculating ORFscore
To calculate the ORFscore, reads were counted at each position within the ORF, excluding the first and last coding codons. To filter out putative artifactual peaks, the most abundant read position was masked if reads aligning to that position comprised more than 70% of the total reads in the ORF. This filter was determined empirically by applying a variable filter and minimizing 3′UTR ORFs that were misclassified as coding based on such peaks (Supplementary Fig S1). The ORFscore was then calculated as:
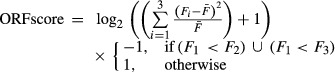 |
where Fn is the number of reads in reading frame n, 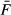 is the total number of reads across all three frames divided by 3.
is the total number of reads across all three frames divided by 3.
Calculating coverage
To calculate coverage in position 1, each position was considered covered if the P-site of at least one RPF read aligned to that position. For a given ORF, the coverage in position 1 is the resulting ratio of first frame positions covered versus all possible first frame positions in the ORF.
Calculating RRS
RRS (Ribosome Release Score) values for the 5 h time point were calculated to compare with ORFscore values. The RRS calculation and FindORF programs were provided by Guttman et al (2013). To calculate the RRS score of annotated CDSs, a raw bed file was provided to the calculation program; to calculate the scores of other ORFs, the FindORF program was used to generate an input file. For coding genes, the program was run using various 3′UTR options (using either the whole 3′UTR or the minimal 3′UTR until next ORF start), resulting in a slight improvement in RRS score of CDS ORFs when using the whole 3′UTR, but similar clustering of results with both methods.
Calculating translation efficiency
Translation efficiency (TE) was calculated as the base 2 logarithmic ratio of normalized RPFs over the normalized input for each ORF (Ingolia et al, 2011; Bazzini et al, 2012).
Metagene analysis
Metagene plots were generated using R, taking read sets and CDS boundaries in transcript coordinates as input. For each RefSeq transcript with an annotated CDS, reads were counted at each position within two windows (surrounding the start and stop codons). These counts were normalized per transcript by dividing each position's count by the sum of all reads in that window. These normalized counts were summed across all genes.
Defining coding ORFs
Translated ORFs were defined using the following parameters, in order to capture 85% of non-genome duplicated RefSeq annotated coding genes (N = 9,559): minimum size of 20 aa, ORFscore > 6.044, coverage of position 1 > 10% and expression greater than 1 RPKM at any time point. The less stringent criteria to define the relaxed set of translated ORFs used only ORFscore > 6.044.
Defining alternative (altORFs)
To define altORFs in 5′ and 3′ UTR, ORFs were excluded for which the genomic position of the start or stop codon overlapped any coding regions annotated by RefSeq or Ensembl. To define translated ORFs, see parameters above.
Defining Non-coding RNA
To define all the genes with no coding region (non-coding RNA), several filters were imposed to the transcript set described above. First, transcripts overlapping a coding exon were excluded. Second, all the transcripts from a genes with at least one ORF defined as coding (strict or relaxed) were excluded. Third, transcripts with coding potential predicted by Chew et al, 2013 were removed. Fourth, genes with RNA level lower than the first quartile of the genes encoding for defined smORFs (20–100 aa) were discarded. Finally, genes with any ORF that could encode for a protein larger than 100 aa were also excluded.
Defining translated small ORFs (smORFs)
Annotated smORFs were defined as all protein-coding ORFs annotated in RefSeq and Ensembl sized 20–100 aa that passed the same ORFscore and position 1 coverage thresholds (6.044, 10%) mentioned previously. To define smORFs, these thresholds were applied to “processed transcripts” and lincRNAs from Ensembl in addition to the lincRNAs previously described in (Ulitsky et al, 2011) and (Pauli et al, 2012). Any ORFs that overlapped an annotated coding exon were excluded. smORFs residing in the same gene as an ORF > 100 aa defined as coding were also excluded. To avoid multiply counting isoforms/variants that encode for the same peptide, only one of these transcripts was selected when more than one ORF shared the same genomic start and stop coordinates and coding length. For final smORF counts, only one transcript per gene was counted.
Embryo preparation for MSMS detection
About 2,500 embryos were collected at 5 h and deyolked. Groups of 100 embryos were transferred from an agar coated dish to a 1.5-ml tube filled with 1 ml of deyolking buffer (55 mM NaCl, 1.8 mM KCl, 1.25 mM NaHCO3), the yolk sac was disrupted by pipetting with a 200 μl narrow tip, and the embryos were shaken for 5 min at 1,100 rpm to dissolve the yolk (Thermomixer, Eppendorf) at 4°C. Cells were pelleted at 300 g for 30 s and the supernatant was discarded. Subsequently, two additional washes were performed with 1 ml of wash buffer (110 mM NaCl, 3.5 mM KCl, 2.7 CaCl2, 10 mM Tris/Cl pH 8.5) shaking for 2 min at 1100 rpm and centrifugation. Cell pellets were frozen in liquid nitrogen.
MS sample preparation
To optimize detection of small proteins, unfractionated samples and small-protein-enriched fractions were prepared from embryo extracts (Supplementary Fig S5). Fractions were either digested or not using different enzymes (LysC, trypsin or GluC) to increase sequence coverage. Unfractionated samples were prepared following two different protocols: filter-aided sample preparation (FASP) (Wisniewski et al, 2009) and in-solution protein digestion. For FASP, approximately 300 frozen cells were boiled at 95°C in 5% (w/v) SDS, 100 mM DTT in 100 mM Tris/Cl pH 7.6 for 15 min and then submitted to FASP protocol as previously described (Frohlich et al, 2013). For this fraction only, trypsin (Promega) digestion and 4 h chromatographic runs were used.
For in-solution digestion, approximately 700 frozen cells were boiled at 95°C in lysis buffer 100 mM Tris/Cl (300 μl, pH 7.6). After cooling to room temperature, urea powder (160 mg) was directly added to the boiled embryos solution and cells were lysed using a Dounce homogenizer on ice. Samples were prepared for standard in-solution digestion protocol using LysC (Wako), trypsin (Promega) and GluC (Promega) according to the manufacturers' recommendations.
The small-protein-enriched fractions were prepared from the rest of the embryos (˜1,500) by acid extraction (Oyama et al, 2004). Briefly, frozen cells were boiled in water (600 μl) at 95°C for 15 min. After cooling to room temperature, cells were lysed in 1 M acetic acid using a Dounce homogenizer on ice. Precipitates and cell debris were pelleted by centrifugation at 17,000 g for 10 min. The extracted peptides were adjusted to pH 8 with NH4OH and then subjected to standard reduction/alkylation steps. Peptides were incubated for 30 min with DTT (1 mM) at room temperature and alkylated with iodoacetamide (55 mM) in the dark for 20 min. Peptides were submitted to size exclusion centrifugation using a 10-kDa filter unit (Millipore) at 14,000 g for 10 min. Resulting fractions “< 10 kDa” and “> 10 kDa” were dried in a Speedvac and resolubilized in NH4HCO3 (100 μl). One-fourth of each fraction was kept for direct MS analysis without further treatment and the remainder of the fractions was submitted to three proteolytic digestions using LysC (Wako), trypsin (Promega) and GluC (Promega) according to the manufacturers' recommendations.
Each peptide fraction was acidified by trifluoroacidic acid (TFA) and cleared of precipitates by centrifugation at 17,000 g for 5 min. Two micrograms of peptides was desalted following the protocol for StageTip purification (47). Prior to MS injection, samples were eluted with 70 μl buffer B (80% ACN, 0.1% formic acid in H2O) and reduced to a final volume of 5 μl in a Speedvac.
Chromatography and mass spectrometry
Each peptide fraction was separated by reversed-phase chromatography on a Thermo Easy nLC 1000 system connected to a Q Exactive mass spectrometer (Thermo) through a nano-electrospray ion source. Peptides were separated on 50-cm columns (New Objective) with an inner diameter of 75 μm packed in house with 1.9 μm C18 resin (Dr. Maisch GmbH). For FASP samples, 265-min chromatographic runs were used and peptides were eluted with a linear gradient of acetonitrile from 5 to 30% in 0.1% formic acid for 240 min at a constant flow rate of 250 nl/min For the rest of the samples, 120-min chromatographic runs were used and peptides were eluted with a linear gradient of acetonitrile from 5 to 30% in 0.1% formic acid for 95 min at a constant flow rate of 250 nl/min. The column temperature was kept at 45°C. Eluted peptides were directly electrosprayed into the mass spectrometer. Mass spectra were acquired on the Q Exactive in a data-dependent mode to automatically switch between full scan MS and up to 5 (10 for FASP samples) data-dependent MS/MS scans. The maximum injection time for full scans was 10 ms (20 ms for FASP samples) with a target value of 3,000,000 at a resolution of 70,000 at m/z = 200. The five or ten most intense multiple charged ions (z ≥ 2) from the survey scan were selected with an isolation width of 2Th (3Th for FASP samples) and fragmented with higher energy collision dissociation (HCD) with normalized collision energies of 25. Target values for MS/MS were set to 10,000 with a maximum injection time of 120 ms at a resolution of 17,500 at m/z = 200. To avoid repetitive sequencing, the dynamic exclusion of sequenced peptides was set to 45 s for 265 min runs and to 35 s for 120 min runs.
MS data analysis
MS and MS/MS spectra were analyzed using MaxQuant (version 1.4.0.5), utilizing its integrated ANDROMEDA search algorithms (Cox et al, 2011). Scoring of peptides for identification was carried out with an initial allowed mass deviation of the precursor ion of up to 4.5 ppm for the search for peptides with a minimum length of five amino acids. The allowed fragment mass deviation was 20 ppm. The false discovery rate (FDR) was set to 0.01 for proteins and peptides. Peak lists were searched against manually curated databases corresponding to all annotated RefSeq proteins as well as all smORF and altORF putative peptides encoded from all isoforms predicted by ribosome profiling combined with 262 common contaminants. Maximum missed cleavages were set to 4. The search included carbamidomethylation of cysteine as fixed modification, methionine oxidation, N-terminal acetylation and phosphorylation as variable modifications.
ORF similarity analysis
To analyze the similarity of newly detected smORFs to existing proteins, the amino acid sequences of all smORFs defined as coding by ORFscore and coverage were collapsed and input to NCBI BLASTp with start (Met) and stop codon not included. Default parameters were used, with species restricted to a number of model and studied organisms: Danio rerio, Homo sapiens, Mus musculus, Xenopus tropicalis, Drosophila melanogaster, Rattus norvegicus, Strongylocentrotus purpuratus, Caenorhabditis elegans and Arabidopsis thaliana. Results were manually curated and classified into one of the following categories:
High similarity: 90% or more of the smORF aligns with score > 50 to known protein.
High similarity; predicted: 90% or more of the smORF aligns with score > 50 to predicted protein.
Partial similarity: < 90% of the smORF aligns with score > 50 to known protein.
Partial similarity: predicted: < 90% of the smORF aligns with score > 50 to predicted protein.
Non-significant: no part aligns with an alignment score > 50, but some hits are returned.
None: no hits.
Analysis of smORF coding signatures across species
Computational prediction of conserved smORFs was based on published catalogs of lincRNAs together with whole-genome alignments from the UCSC browser (human: alignment of 45 vertebrates to hg19; zebrafish: alignment of seven vertebrates to danRer7). lincRNAs for human were taken from Gencode v18 (Derrien et al, 2012) and from Cabili and colleagues (Cabili et al, 2011). For zebrafish, lincRNAs from Ensembl (v73), as well as catalogs from Ulitsky et al, (2011) and Pauli et al, (2012), were used. The spliced sequence of lincRNAs was then scanned for the longest putative open reading frame longer than 63 nt (≥ 20 aa), with a canonical ATG start codon closest to the next upstream stop. ORFs in human and zebrafish which overlapped (sense or antisense) with coding exons from annotated genes from Refseq (downloaded Nov. 14, 2013) or Ensembl (v73) were filtered out. For each smORF, the corresponding multiple alignment block (“stitched”) was then extracted from the whole-genome alignment. Sequences from species where less than 50% could be aligned, or with frameshift inducing insertions or deletions, were discarded, using an index for nucleotide insertions prepared from the original unstitched alignment blocks. Only smORFs where at least 50% of species with enough alignable sequence have no frameshift inducing indels were considered. These species were then used to calculate the phyloCSF score using the omega test (with –strategy = omega) (Pauli et al, 2012). A cutoff of 50 on the phyloCSF score was used (Pauli et al, 2012; Guttman et al, 2013). For ORF counts used in calculations, ORFs were filtered by unique genomic location.
As a control, the coding potential of smORFs was scored with a method that had not been previously used to annotate lincRNAs. A simple parameter-free empirical Ka/Ks calculation was implemented: Ka was defined as the number of non-synonymous mutations per non-synonymous site (adding pseudocounts equal to the fraction of non-synonymous sites in the sequence to numerator and denominator), and Ks similarly as the number of synonymous mutations per synonymous site (where pseudocounts equal to the fraction of synonymous sites are added). Ka/Ks ratios for pairs of species were summarized by taking the median. For significance estimation, the columns of the multiple species alignment of each putative smORF were permuted 1000 times. For each shuffled smORF, median Ka/Ks values were calculated as before. The empirical P-value was then defined as the percentage of shuffles with same or better median Ka/Ks value than the original smORF. Only smORFs with median Ka/Ks < 1 and P-value smaller than 0.05 were kept.
Data deposition
Sequencing data are deposited in GEO with accession number GSE53693.
Acknowledgments
We thank J. Weissman, N. Ingolia, M. Guttman and S. Kuersten for reagents, scripts and discussion, D. Cifuentes, C. Takacs and all the members of the Giraldez laboratory for intellectual and technical support and M. Hammarlund and A.F. Schier for comments on the manuscript. NR and SDM thank F. Payre for initial discussions and support as well as all members of the Rajewsky laboratory for stimulating feedback. This work was supported by T32GM007499 (TGJ), Pew Fellows Program in Biomedical Sciences (AAB), the Helmholtz Alliance on Systems Biology (MDC Systems Biology Network) and MDC funding (SDM), the Delbrück Fellows program of the MDC (BO), the Swiss National Science Foundation (CEV), R01GM081602-06 (AJG), R01GM103789-01 (AJG), R01HD074078-02 (AJG), F32HD071697-02 (MTL), R01GM095982 (TCW), R01GM097194 (TCW), the Pew Scholars Program in the Biomedical Sciences (AJG), the March of Dimes (AJG) and the Yale Scholars Program (AJG, TCW).
Author contributions
AAB and AJG conceived the project and together with TGJ and ESF designed and performed the experiments and data analysis. AAB, TGJ and AJG with input from MTL developed the ORFscore to define micropeptides using ribosome footprinting. TGJ and CEV analyzed the human data. AAB, TGJ and AJG wrote the paper with input from the other authors. AAB, RC and TCW designed and performed the MS experiments. SDM, BO and NR independently conceived, designed and implemented the computational micropeptide detection pipeline (micPDP).
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information for this article is available online: http://emboj.embopress.org
References
- Barbosa C, Peixeiro I, Romao L. Gene expression regulation by upstream open reading frames and human disease. PLoS Genet. 2013;9:e1003529. doi: 10.1371/journal.pgen.1003529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzini AA, Lee MT, Giraldez AJ. Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science. 2012;336:233–237. doi: 10.1126/science.1215704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, Kuehn MS, Taylor CM, Neph S, Koch CM, Asthana S, Malhotra A, Adzhubei I, Greenbaum JA, Andrews RM, Flicek P, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, Weissman JS. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science. 2012;335:552–557. doi: 10.1126/science.1215110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–1927. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, Kodzius R, Shimokawa K, Bajic VB, Brenner SE, Batalov S, Forrest AR, Zavolan M, Davis MJ, Wilming LG, Aidinis V, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- Chan PP, Lowe TM. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 2009;37:D93–D97. doi: 10.1093/nar/gkn787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew GL, Pauli A, Rinn JL, Regev A, Schier AF, Valen E. Ribosome profiling reveals resemblance between long non-coding RNAs and 5′ leaders of coding RNAs. Development. 2013;140:2828–2834. doi: 10.1242/dev.098343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chng SC, Ho L, Tian J, Reversade B. ELABELA: a hormone essential for heart development signals via the apelin receptor. Dev Cell. 2013;27:672–680. doi: 10.1016/j.devcel.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- Crappe J, Van Criekinge W, Trooskens G, Hayakawa E, Luyten W, Baggerman G, Menschaert G. Combining in silico prediction and ribosome profiling in a genome-wide search for novel putatively coding sORFs. BMC Genomics. 2013;14:648. doi: 10.1186/1471-2164-14-648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, Lagarde J, Veeravalli L, Ruan X, Ruan Y, Lassmann T, Carninci P, Brown JB, Lipovich L, Gonzalez JM, Thomas M, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimaio D. Viral miniproteins. Annu Rev Microbiol. 2014;68 doi: 10.1146/annurev-micro-091313-103727. . in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S, Gil L, García-Girón C, Gordon L, Hourlier T, Hunt S, Juettemann T, Kähäri AK, Keenan S, Komorowska M, et al. Ensembl 2013. Nucleic Acids Res. 2013;41:D48–D55. doi: 10.1093/nar/gks1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohlich F, Christiano R, Walther TC. Native SILAC: metabolic labeling of proteins in prototroph microorganisms based on lysine synthesis regulation. Mol Cell Proteomics. 2013;12:1995–2005. doi: 10.1074/mcp.M112.025742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galindo MI, Pueyo JI, Fouix S, Bishop SA, Couso JP. Peptides encoded by short ORFs control development and define a new eukaryotic gene family. PLoS Biol. 2007;5:e106. doi: 10.1371/journal.pbio.0050106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, Fan L, Koziol MJ, Gnirke A, Nusbaum C, Rinn JL, Lander ES, Regev A. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol. 2010;28:503–510. doi: 10.1038/nbt.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Russell P, Ingolia NT, Weissman JS, Lander ES. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell. 2013;154:240–251. doi: 10.1016/j.cell.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, Collins JE, Humphray S, McLaren K, Matthews L, McLaren S, Sealy I, Caccamo M, Churcher C, Scott C, Barrett JC, Koch R, Rauch GJ, White S, Chow W, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013;496:498–503. doi: 10.1038/nature12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, Regev A, Lander ES, Rinn JL. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci USA. 2009;106:11667–11672. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T, Hashimoto Y, Kato K, Inagaki S, Hayashi S, Kageyama Y. Small peptide regulators of actin-based cell morphogenesis encoded by a polycistronic mRNA. Nat Cell Biol. 2007;9:660–665. doi: 10.1038/ncb1595. [DOI] [PubMed] [Google Scholar]
- Kondo T, Plaza S, Zanet J, Benrabah E, Valenti P, Hashimoto Y, Kobayashi S, Payre F, Kageyama Y. Small peptides switch the transcriptional activity of Shavenbaby during Drosophila embryogenesis. Science. 2010;329:336–339. doi: 10.1126/science.1188158. [DOI] [PubMed] [Google Scholar]
- Lawrence M, Gentleman R, Carey V. rtracklayer: an R package for interfacing with genome browsers. Bioinformatics. 2009;25:1841–1842. doi: 10.1093/bioinformatics/btp328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MF, Jungreis I, Kellis M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 2011;27:i275–i282. doi: 10.1093/bioinformatics/btr209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magny EG, Pueyo JI, Pearl FM, Cespedes MA, Niven JE, Bishop SA, Couso JP. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science. 2013;341:1116–1120. doi: 10.1126/science.1238802. [DOI] [PubMed] [Google Scholar]
- Medenbach J, Seiler M, Hentze MW. Translational control via protein-regulated upstream open reading frames. Cell. 2011;145:902–913. doi: 10.1016/j.cell.2011.05.005. [DOI] [PubMed] [Google Scholar]
- Menschaert G, Van Criekinge W, Notelaers T, Koch A, Crappe J, Gevaert K, Van Damme P. Deep proteome coverage based on ribosome profiling aids mass spectrometry-based protein and peptide discovery and provides evidence of alternative translation products and near-cognate translation initiation events. Mol Cell Proteomics. 2013;12:1780–1790. doi: 10.1074/mcp.M113.027540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AM, Choudhury KR, Firth AE, Ingolia NT, Atkins JF, Baranov PV. Observation of dually decoded regions of the human genome using ribosome profiling data. Genome Res. 2012;22:2219–2229. doi: 10.1101/gr.133249.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyama M, Itagaki C, Hata H, Suzuki Y, Izumi T, Natsume T, Isobe T, Sugano S. Analysis of small human proteins reveals the translation of upstream open reading frames of mRNAs. Genome Res. 2004;14:2048–2052. doi: 10.1101/gr.2384604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli A, Valen E, Lin MF, Garber M, Vastenhouw NL, Levin JZ, Fan L, Sandelin A, Rinn JL, Regev A, Schier AF. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 2012;22:577–591. doi: 10.1101/gr.133009.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli A, Norris ML, Valen E, Chew GL, Gagnon JA, Zimmerman S, Mitchell A, Ma J, Dubrulle J, Reyon D, Tsai SQ, Joung JK, Saghatelian A, Schier AF. Toddler: an embryonic signal that promotes cell movement via apelin receptors. Science. 2014;343:746. doi: 10.1126/science.1248636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pueyo JI, Couso JP. The 11-aminoacid long tarsal-less peptides trigger a cell signal in Drosophila leg development. Dev Biol. 2008;324:192–201. doi: 10.1016/j.ydbio.2008.08.025. [DOI] [PubMed] [Google Scholar]
- Savard J, Marques-Souza H, Aranda M, Tautz D. A segmentation gene in tribolium produces a polycistronic mRNA that codes for multiple conserved peptides. Cell. 2006;126:559–569. doi: 10.1016/j.cell.2006.05.053. [DOI] [PubMed] [Google Scholar]
- Schwaid AG, Shannon DA, Ma J, Slavoff SA, Levin JZ, Weerapana E, Saghatelian A. Chemoproteomic discovery of cysteine-containing human short open reading frames. J Am Chem Soc. 2013;135:16750–16753. doi: 10.1021/ja406606j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavoff SA, Mitchell AJ, Schwaid AG, Cabili MN, Ma J, Levin JZ, Karger AD, Budnik BA, Rinn JL, Saghatelian A. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat Chem Biol. 2013;9:59–64. doi: 10.1038/nchembio.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somers J, Poyry T, Willis AE. A perspective on mammalian upstream open reading frame function. Int J Biochem Cell Biol. 2013;45:1690–1700. doi: 10.1016/j.biocel.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark A, Lin MF, Kheradpour P, Pedersen JS, Parts L, Carlson JW, Crosby MA, Rasmussen MD, Roy S, Deoras AN, Ruby JG, Brennecke J, Hodges E, Hinrichs AS, Caspi A, Paten B, Park SW, Han MV, Maeder ML, Polansky BJ, et al. Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature. 2007;450:219–232. doi: 10.1038/nature06340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern-Ginossar N, Weisburd B, Michalski A, Le VT, Hein MY, Huang SX, Ma M, Shen B, Qian SB, Hengel H, Mann M, Ingolia NT, Weissman JS. Decoding human cytomegalovirus. Science. 2012;338:1088–1093. doi: 10.1126/science.1227919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz D. Polycistronic peptide coding genes in eukaryotes–how widespread are they? Brief Funct Genomic Proteomic. 2009;8:68–74. doi: 10.1093/bfgp/eln054. [DOI] [PubMed] [Google Scholar]
- Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 2011;147:1537–1550. doi: 10.1016/j.cell.2011.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Wolin SL, Walter P. Ribosome pausing and stacking during translation of a eukaryotic mRNA. EMBO J. 1988;7:3559–3569. doi: 10.1002/j.1460-2075.1988.tb03233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.