

Abstract
Breast and ovarian cancer susceptibility genes BRCA1 and PALB2 have enigmatic roles in cellular growth and mammalian development. While these genes are essential for growth during early developmental programs, inactivation later in adulthood results in increased growth and formation of tumors, leading to their designation as tumor suppressors. We performed genome-wide analysis assessing their chromatin residence and gene expression responsiveness using high-throughput sequencing in breast epithelial cells. We found an intimate association between BRCA1 and PALB2 chromatin residence and genes displaying high transcriptional activity. Moreover, our experiments revealed a critical role for BRCA1 and, to a smaller degree, PALB2 in transcriptional responsiveness to NF-κB, a crucial mediator of growth and inflammatory response during development and cancer. Importantly, we also uncovered a vital role for BRCA1 and PALB2 in response to retinoic acid (RA), a growth inhibitory signal in breast cancer cells, which may constitute the basis for their tumor suppressor activity. Taken together, our results highlight an important role for these breast cancer proteins in the regulation of diverse growth regulatory pathways.
Keywords: breast cancer, chromatin, elongation, NF-κB, RNA polymerase
Introduction
BRCA1, BRCA2, and PALB2 correspond to genes whose mutations result in familial cases of breast and ovarian cancers (Castilla et al, 1994; Wooster et al, 1994; Erkko et al, 2007; Rahman et al, 2007). In addition, PALB2 and BRCA2 are found mutated in Fanconi anemia, a rare congenital syndrome defined by a bone marrow failure that can ultimately result in leukemia (Reid et al, 2007; Xia et al, 2007; D'Andrea, 2010). PALB2 mutations may also confer susceptibility to pancreatic cancer (Erkko et al, 2007; Rahman et al, 2007; Reid et al, 2007; Xia et al, 2007; Jones et al, 2009). The observation of DNA damage-induced nuclear foci containing BRCA1 and RAD51 propelled several laboratories to dissect the role of BRCA complexes in DNA recombination and repair (Scully et al, 1997b). Moreover, BRCA1 and BRCA2 defective cells are highly susceptible to DNA interstrand cross-links (Venkitaraman, 2009) and have been shown to play a critical role in the maintenance of genome stability, acting within the homologous recombination pathway (Moynahan et al, 1999, 2001). In particular, BRCA1 could be targeted to DNA double-strand breaks through the recently characterized BRCA1–RAP80–Abraxas complex (Sobhian et al, 2007). Independently, BRCA1 was also implicated in the regulation of transcription. Initial experiments suggested a role for BRCA1 as a co-activator of transcription (Chapman & Verma, 1996; Ouchi et al, 1998), perhaps through its association with RNA polymerase II (Scully et al, 1997a). Moreover, we previously identified a distinct BRCA1-containing complex that interacted with components of the SWI/SNF and exhibited chromatin remodeling activity (Bochar et al, 2000).
We had previously shown that the breast cancer susceptibility proteins BRCA1 and BRCA2 could be detected in nuclear fractions enriched for chromatin (Kumaraswamy & Shiekhattar, 2007) and a number of reports implicated BRCA1 in transcriptional regulation (Mullan et al, 2006; Rosen et al, 2006). Moreover, recombinant BRCA1 was shown to bind specific DNA structures, while BRCA1-containing complexes were reported to interact with specific DNA sequences in vitro (Paull et al, 2001; Cable et al, 2003). Additionally, PALB2 was suggested to bind DNA upon genetic lesions allowing for BRCA2 localization to DNA repair foci (Xia et al, 2006). Importantly, functional and biochemical analyses have found PALB2 to bridge the interaction between BRCA2 and BRCA1 proteins (Sy et al, 2009b; Zhang et al, 2009), and a chromatin association motif within PALB2 named ChAM was described as necessary and sufficient to mediate PALB2 chromatin association in both unperturbed and damaged cells (Bleuyard et al, 2012).
Here, we describe an unbiased functional genomics approach revealing an important role for BRCA1 and PALB2 in the transcription of RNAPII genes. We found BRCA1 and PALB2 associated with genes with high transcriptional activity in breast epithelial cells, MCF10A. Our functional analysis has uncovered a role for BRCA1 and PALB2 in transcriptional responsiveness to two critical growth regulating pathways, the NF-κB and retinoic acid (RA) signaling. The NF-κB family of transcription factors consists of five members: p65/RelA, c-Rel, RelB, p50/p105, and p52/p100 (Perkins, 2012). They reside in the cytoplasm as inactive homodimer or heterodimer associated with an inhibitory IκB family member. Numerous activation stimuli such as tumor necrosis factor α (TNF-α) and interleukin-1 (IL-1) trigger a phosphorylation-dependent degradation of IκB which sets the NF-κB dimer free to translocate to the nucleus and bind sequence-specific promoter elements. NF-κB recruits co-activators such as p300 and CBP to activate the transcription of genes involved in immune and inflammatory reactions, anti- and pro-apoptotic processes, and cell cycle regulation (Smale, 2012).
The retinoic acid pathway governs growth, cell differentiation, and tissue development in a variety of cell types and conditions, ranging from embryonic development to adult cell homeostasis. In mammals, RA mediates its effects through association with three different retinoic acid receptors (RAR alpha, beta, and gamma) (Amann et al, 2011). RARs are ligand-inducible transcriptional activators, which act in heterodimeric combination with the retinoid X receptors (RXRs) via recognition of specific RA-response elements (RAREs) (Amann et al, 2011). Retinoic acid was previously shown to have anti-proliferative effects in breast cancer cells (del Rincon et al, 2003; Hua et al, 2009) and might constitute an important tumor suppressor pathway regulated by the breast cancer proteins.
Results
Genome-wide occupancy of BRCA1 and PALB2
We sought to determine the genome-wide occupancy of the breast cancer susceptibility proteins in breast epithelial cells. We used polyclonal antibodies against BRCA1 and PALB2 to perform chromatin immunoprecipitation (ChIP) followed by high-throughput sequencing (ChIP-seq) in asynchronously growing MCF10A, a non-tumorigenic mammary epithelial cell line. Unbiased clustering of ChIP-seq reads revealed a compelling overlap between BRCA1 and PALB2 binding pattern across human RefSeq genes (Fig 1A). The highest co-occupied cluster (Fig 1A, represented by class I) comprises 373 genes, displaying greatest transcriptional activity as revealed by RNA sequencing (Fig 1, A and B, Supplementary Fig S1A, Supplementary Table S1). Such genes include ribosomal proteins, histones, growth regulators (FOS, JUN, MYC), and modulators of inflammation and stress (NFKBIA, IL-8, CXCL1, SOD2) (Fig 1, C and D and Supplementary Fig S1B). The average read profiles of the 373 target genes (Fig 1B) reflect the occupancy of BRCA1 and PALB2 over the entire gene body. Interestingly, while BRCA1 peaks at the transcription start site (TSS) and gradually decreases over the reading frame, PALB2 reads persist throughout the locus and peak at the 3′ untranslated region (3′ UTR) (Fig 1B). Additionally, a large number of transcriptionally active genes were occupied by BRCA1 and, to a smaller extent, by PALB2 on their TSS alone (represented by class II, Fig 1A, Supplementary Fig S1A, Supplementary Table S1).
Figure 1. Chromatin occupancy of BRCA1 and PALB2.
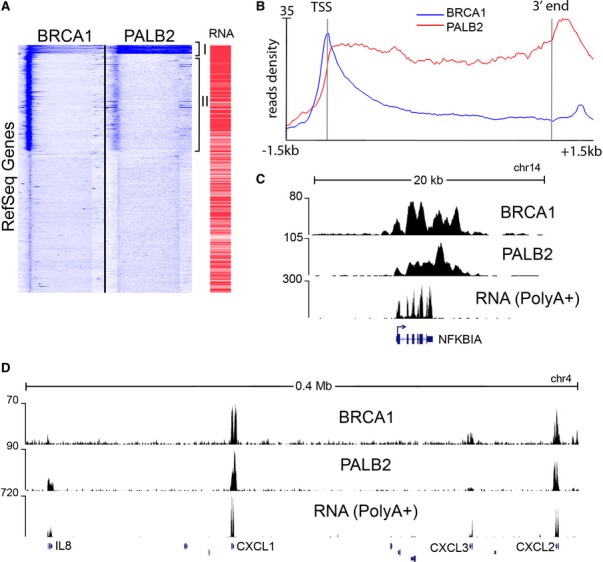
- Unbiased clustering of BRCA1 and PALB2 ChIP-seq data reveals that the breast cancer proteins co-occupy a set of genes. The heatmap representation shows the fraction of RefSeq coding genes incorporating the highest density of reads. The bracketed cluster on top contains 373 genes that are the most occupied by both proteins across the entire locus. A second larger cluster contains genes that are occupied by BRCA1 limited to their TSS, while they show minimal PALB2 binding. An additional column (RNA) shows the expression level of the transcripts as measured by FPKM (fragments per kilobase per million sequenced reads), indicating that the highest occupied genes are among the most expressed in MCF10A cells. Heatmaps are calculated over the whole gene body from the 5′ (left side of each column) to the 3′ (right side), with a further 1.5kb extension at both ends.
- Read density profiles detail the binding pattern of BRCA1 and PALB2 across the 373 genes cluster. BRCA1 peaks at the 5′ of the gene further extending into the gene body, where its occupancy gradually decreases. PALB2 occupies the entire body of the gene, peaking at the 3′ end.
- A snapshot of NFKBIA locus (Hg18 UCSC Genome Browser). Genome-aligned tracks represent raw read numbers from ChIP-seq experiments. RNA-seq data from the poly-adenylated fraction of RNA are also shown, indicating that NFKBIA is highly transcribed.
- BRCA1 and PALB2 occupy 4 gene loci encoding for members of the chemokine family (for which the gene symbols are shown), within a 400-megabase region. Such profiles coincide with sustained mRNA transcription. The remaining 6 loci that are not targeted by the BRCA complex do not show evidence of transcription.
While BRCA1 and PALB2 also co-occupied several loci encoding for uridylate-rich small nuclear RNAs (U snRNAs) (Supplementary Fig S1C,D and Supplementary Table S1), BRCA1 alone was detected at several tRNAs as well as other RNAPIII genes (Supplementary Table S1), suggesting a broader role for BRCA1 in transcriptional regulation and DNA repair. Such diverse genomic localization by BRCA1 may represent the distribution of distinct BRCA1-containing complexes, including the BRCC and BRCA1/BACH1 complexes that are independent of PALB2 (Dong et al, 2003; Kumaraswamy & Shiekhattar, 2007). We validated the BRCA1 and PALB2 localization across multiple cell types, using a number of antibodies, and confirmed the specificity of their signal following the depletion of BRCA1 and PALB2 using RNA interference (Supplementary Fig S2A–E). Moreover, we performed an additional ChIP-seq using a monoclonal antibody against BRCA1, which displayed a similar pattern of occupancy for BRCA1 as that obtained using polyclonal antibodies (Fig 2A–C). Furthermore, we performed additional ChIP followed by real-time PCR using antibodies against Flag epitope in cell lines expressing stably integrated Flag-PALB2, which corroborated our results using anti-PALB2 antibodies (Supplementary Fig S2F).
Figure 2. Chromatin occupancy of BRCA1.
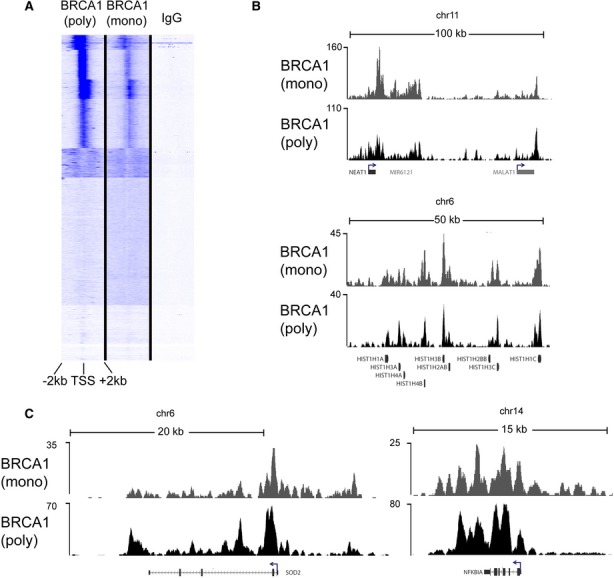
A BRCA1 occupancy, as determined by the polyclonal I-20 antibody (Fig 1), was compared to a mouse monoclonal antibody (D-9). Unbiased clustering reveals a strong correlation between the two independent ChIP-seq analyses of BRCA1 at all Human RefSeq Genes (Hg 18).
B,C Raw ChIP-seq data in MCF10A cells show a very similar pattern of chromatin binding at two large genomic regions encompassing the highly transcribed non-coding RNAs NEAT1 and MALAT1 (upper panel) and a cluster of histone genes on chr6 (lower panel). A closer look at individual genes (SOD2, NFKBIA) also reveals a very similar pattern.
Association of BRCA1 and PALB2 with elongating RNAPII
The chromatin residence of BRCA1 and PALB2, occupying the 5′-end and extending into the body of the transcriptionally active genes (Fig 1), suggested a functional association between BRCA1/PALB2 and the elongating form of RNAPII. Indeed, recent in vitro studies described the association of BRCA1 with the C-terminal domain (CTD) of RNAPII (Moisan et al, 2004), and the ubiquitination of elongating polymerase by the BRCA1/BARD1 heterodimer (Kleiman et al, 2005). To explore the association of BRCA1 and PALB2 with RNAPII in vivo, we compared our genome-wide occupancy of BRCA1 and PALB2 to an RNAPII ChIP-seq experiment that we previously performed in MCF10A cells using polyclonal antibodies (N-20), which recognize RPB1 independent of its phosphorylation status (Baillat et al, 2012). Analysis of 373 highly active genes revealed a similar pattern of occupancy for RNAPII and the breast cancer susceptibility proteins (Fig 3A). Interestingly, BRCA1 occupancy peaks with initiating form of RNAPII at the 5′-end of genes, while PALB2 occupancy resembles that of elongating RNAPII (Fig 3A).
Figure 3. The breast cancer genes mirror RNA polymerase II occupancy and are functionally linked with its elongating form.
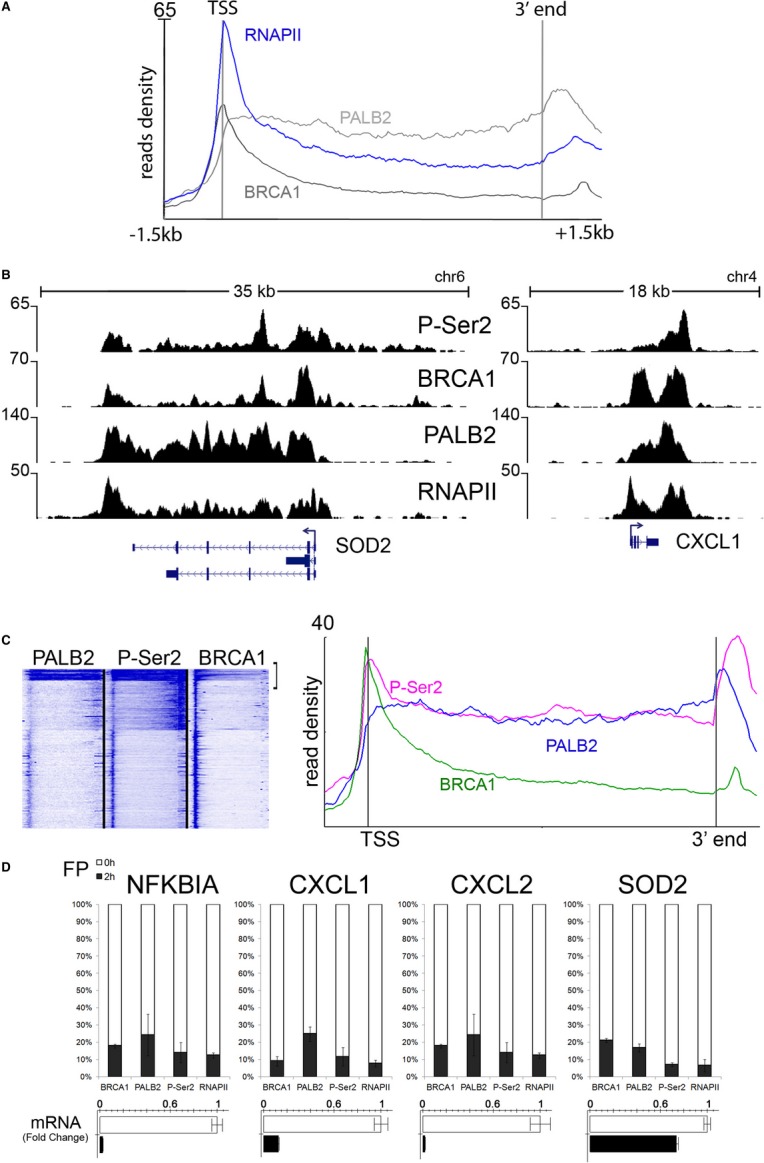
- The average profiles of RNAPII, BRCA1, and PALB2 across class I genes (n = 373) in MCF10A cells. The average read density of RNAPII peaks at the transcription start site and remains elevated across the entire gene body similar to the BRCA proteins.
- Genome-wide analysis of elongating RNAPII was performed in MCF10A cells using antibodies against phosphorylated Ser2 of the CTD (P-Ser2). Snapshots of aligned reads for P-Ser2, PALB2, BRCA1, and RNAPII show a compelling overlap between the four ChIP-seq datasets at two representative BRCA1/PALB2 target genes. Although spanning the entire gene body, P-Ser2 peaks beyond the 3′ end of the gene (multiple isoforms in the case of SOD2), similar to PALB2.
- Unbiased clustering of PALB2, BRCA1, and P-Ser2 reveals an intimate association between the genes implicated in familial breast cancer and the elongating form of RNA polymerase II on the left panel (see Supplementary Fig S3 for the whole cluster). The right panel depicts the average read profiles of the most actively transcribed genes (comprising approximately 300 genes).
- Inhibition of elongation abrogates BRCA1 and PALB2 recruitment at target genes. MCF10A cells were treated with flavopiridol for 2 h and subjected to ChIP analysis at the 3′ end of a group of highly active genes. The stacked bars indicate the residual amount of BRCA1, PALB2, RNAPII, and P-Ser2 after flavopiridol treatment, relative to untreated cells (100%). Average of three IPs is shown. A dramatic decrease in elongating RNAPII is mirrored by the loss of over 70–80% of the BRCA1-PALB2 complex at chromatin. RT-PCR analysis of the mRNA levels after flavopiridol treatment is also shown.
To directly analyze the elongating form of RNAPII, we performed ChIP-seq using antibodies against the Ser2-phosphorylated form of RNAPII (P-Ser2). Genes targeted by BRCA1 and PALB2 display high levels of Ser2 phosphorylation (Fig 3B). Importantly, the average profile of elongating form of RNAPII is highly similar to that of PALB2 at highly active genes (Fig 3C), while the BRCA1 profile reflects its association with the promoter proximal RNAPII (Fig 1B–C). Indeed, unbiased clustering of genes occupied by PALB2 and elongating form of RNAPII revealed a strikingly similar profile at all RefSeq genes (Fig 3C and Supplementary Fig S3). To assess whether the association of BRCA1 and PALB2 with active genes requires the elongating form of RNAPII, we inhibited transcriptional elongation using flavopiridol, an inhibitor of RNAPII elongation (Chao & Price, 2001; Rahl et al, 2010). Treatment of MCF10A cells with flavopiridol resulted in concomitant decrease in transcriptional activity as well as the occupancy of BRCA1, PALB2, and elongating form of RNAPII at the 3′-end of all genes tested (Fig 3D). Since flavopiridol has been reported to also interact with duplex DNA and elicit a DNA damage response (Bible et al, 2000), we asked whether the treatment of cells with other DNA damage-inducing agents could alter occupancy of BRCA1, PALB2, and elongating form of RNAPII. Treatment of MCF10A cells with mitomycin C (MMC) led to a reduction in the occupancy of BRCA1, PALB2, and Ser-2 phosphorylated form of RNAPII concomitant with a corresponding reduction in transcription of a set of candidate BRCA target genes (Supplementary Fig S4A,B). While the treatment of MCF10 A cells with flavopiridol elicited a larger reduction in BRCA1 and PALB2 occupancy than that observed with MMC (compare Fig 3D and Supplementary Fig S4), it is likely that a component of the BRCA1 and PALB2 changes following flavopiridol treatment is due to the induction of the DNA damage response. Taken together, our results suggest a close functional association of BRCA1 and PALB2 with RNAPII and point to a role for PALB2 in some aspects of transcriptional elongation or 3′-end processing.
BRCA1 and PALB2 co-regulation of gene expression
To assess the functional impact of BRCA1 and PALB2 in gene expression, we depleted their levels by infecting MCF10A cells with lentiviral vectors containing small RNA hairpins (shRNAs) against BRCA1 and PALB2 and used non-targeting shRNAs as a control (Fig 4A). Following the depletion of ribosomal transcripts, total RNA was subjected to high-throughput sequencing. We measured changes in transcription for genes displaying highest levels of BRCA1 and PALB2 occupancy, since such changes should reflect the direct effect of their knock-down (Supplementary Table S2). Depletion of BRCA1 and PALB2 led to a concomitant reduction in a number of highly active genes including IL-8, CXCL1, and SOD2 loci (Fig 4B). Overall, depletion of BRCA1 or PALB2 led to the inhibition of transcription of nearly 1/3 of the highly active (class I) genes (101 and 92, respectively, out of a total 373 genes), with nearly half of the genes (56) displaying a co-regulation by both BRCA1 and PALB2 (Fig 4B, C, D). In contrast to genes whose transcription required BRCA1 and PALB2, a smaller set of genes were up-regulated and there was very little overlap between these genes following the depletion of each protein (Fig 4C). We validated these results across 5 independent experiments using real-time PCR (Fig 4D).
Figure 4. Transcriptional regulation by BRCA1 and PALB2.

- Depletion of BRCA1 and PALB2 in MCF10A cells with lentiviral-transduced shRNAs. While BRCA1 was directly detected using Western blot analysis, PALB2 was immunoprecipitated using anti-PALB2 antibodies in MCF10A (using the same antibodies employed for ChIP and ChIP-seq). Western blots were analyzed using antibodies to PALB2 and BRCA2.
- RNA-seq analysis of MCF10A cells after lentiviral-mediated depletion of BRCA1 and PALB2. Snapshots of IL-8, CXCL1, and SOD2 genes showing a significant down-regulation. Raw reads were aligned to the Hg18 version of the Human Genome Browser.
- Comparative analysis of adjusted FPKM at the 373 BRCA1 and PALB2 common target genes. Approximately 40% of genes occupied by the breast cancer proteins are regulated by either BRCA1 (141) or PALB2 (136) with a log2 (fold change) > 0.4. There is a large overlap (> 50%) between the group of genes down-regulated in both conditions (P < 0.01) as opposed to up-regulated genes, suggesting that BRCA1 and PALB2 mainly act as positive transcriptional regulators.
- Validation of the effect of BRCA1 and PALB2 in gene expression. A panel of 8 genes that are regulated by RNA-seq were validated by qPCR across 5 independent experiments. All genes are significantly regulated (P > 0.01). Knock-down efficiency of BRCA1 and PALB2 was also assessed.
BRCA1 and PALB2 regulate NF-κB-responsive genes
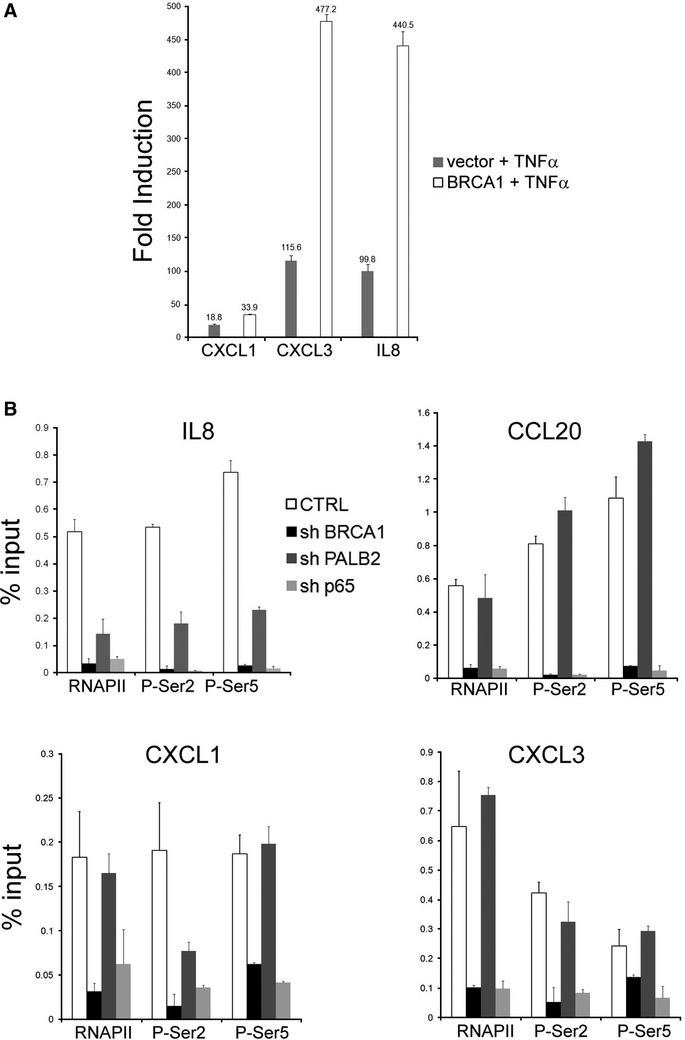
Since depletion of BRCA1/PALB2 led to a significant down-regulation of a subset of genes, we surmised that the breast cancer proteins may serve as co-activators for specific transcription pathways. We set out to test this hypothesis with an in silico analysis of BRCA1/PALB2 target genes. We searched for upstream regulators of genes whose expression required BRCA1 and PALB2 using a prediction algorithm based on a literature compiled dataset (IPA Upstream Regulator Analysis). The analysis on the group of 56 commonly regulated genes revealed NF-κB as the most significant transcriptional regulator (P < 4.31E-12) (Fig 5A). Additionally, motif analysis on the group of 373 BRCA1/PALB2 occupied genes revealed a strong enrichment in motifs bound by the REL homology domain (Supplementary Table S3). Indeed, 3 out of the 4 top human motifs correspond to NF-κB pathway (Supplementary Table S3). We depleted p65/RelA and confirmed the specific regulation of BRCA1 and PALB2 co-regulated genes by NF-κB in MCF10A cells (Fig 5B and C). Importantly, depletion of p65/RelA resulted in a concomitant reduction in PALB2, BRCA1, and RNAPII at NF-κB-responsive promoters, demonstrating the p65/RelA-mediated recruitment of BRCA1 and PALB2 (Fig 5D). Depletion of p65/RelA did not result in a significant decrease in BRCA1, PALB2, or RNAPII occupancy for genes not targeted by p65/RelA (Fig 5E).
Figure 5. BRCA1 and PALB2 regulate the expression of NF-κB-responsive genes.

- Bioinformatic analysis of the upstream regulators of BRCA1- and PALB2-regulated genes uncovered NF-κB as the foremost significant transcription factor involved. All NF-κB target genes that are regulated by the BRCA complex are depicted in the diagram. The analysis and the diagram were obtained using Ingenuity IPA Upstream Regulator Analysis.
- Validation of p65/RelA depletion after lentiviral shRNAs infection. BRCA1 and PALB2 protein levels are not affected.
- Depletion of p65/RelA in MCF10A cells with two distinct shRNA constructs nearly abrogates the expression of IL-8 and greatly impairs CXCL1 and NFKBIA transcription, as opposed to a non-targeting hairpin (CTRL), while BRCA1 and PALB2 mRNA levels are not affected. FOS and HOXA1 were used as control genes not targeted by NF-κB. Samples were collected 72 h post-infection. The average values of three independent experiments are shown (P < 0.0005 for IL-8, NFKBIA, and CXCL1 regulation; non-significant for FOS and HOXA1).
- ChIP analysis upon p65 depletion at 72 h post-infection reveals a dramatic change in BRCA1, PALB2, and RNAPII recruitment at the transcription start site of IL-8, CXCL1, and NFKBIA genes. p65/RelA occupancy was also assessed.
- ChIP analysis upon p65 depletion reveals minimal change in BRCA1, PALB2, and RNAPII recruitment at the transcription start site of FOS and HOXA1, whose expression is not affected by p65 depletion.
We next examined the role of BRCA1 and PALB2 in TNF-α responsiveness in the breast cancer cell line MCF7. Treatment of MCF7 cells with 10 ng/ml TNF-α for 1 h resulted in increased occupancy of p65/RelA, BRCA1, and PALB2 at the promoter of candidate NF-κB target genes (Fig 6A). Moreover, in contrast to requirement for p65/RelA in the recruitment of BRCA1 and PALB2 (Fig 5D), depletion of BRCA1 or PALB2 did not affect the occupancy of p65/RelA (Fig 6B,C; Supplementary Fig S5A). Next we performed gene expression analysis using microarrays to determine the genome-wide responsiveness of MCF7 cells following the treatment with TNF-α. Overall, 33 genes (Supplementary Table S4) displayed a significant activation following the treatment of MCF7 cells with TNF-α (log2(fold change)>0.4, P < 0.01). We validated the response to TNF-α using real-time PCR for eight genes in three independent experiments. Additionally, we examined CXCL1, CXCL3, NFKBIA, and SOD2 (which while responsive to TNF-α using real-time PCR were not reliably detected on the microarray). While depletion of p65/RelA resulted in the loss of TNF-α responsiveness in 31 of 33 genes represented on the array and all four additional genes examined (35 of 37 genes examined), BRCA1 depletion led to a diminished responsiveness to TNF-α for a subset of genes (18 out of 37; Fig 6D). A smaller group of genes (10 out of 37) displayed a significant decrease in their response following PALB2 depletion (Fig 6E and Supplementary Table S4). A large number of TNF-α-responsive genes (24 out of 37) showed a significant decrease in their basal activity following the depletion of p65/RelA, BRCA1, or PALB2 (Fig 6D,E and Supplementary Table S4, P < 0.05). This is also indicated as a heat map for all 33 responsive genes on the array prior and following the stimulation with TNF-α (Supplementary Fig S6). Indeed, due to diminished changes in basal activity, the fold induction of most TNF-α-responsive genes was not significantly affected (Supplementary Fig S7). Moreover, due to decreased basal activity following the depletion of BRCA1 and PALB2, some genes displayed increased fold induction following TNF-α stimulation (Supplementary Fig S7).
Figure 6. BRCA1 and PALB2 affect TNF-α-mediated response at NF-κB target genes.

- ChIP analysis of TNF-α-stimulated MCF7 cells shows a substantial recruitment of p65/RelA, BRCA1, and PALB2 at a set of target genes. Cells were analyzed after 1 h of treatment.
- Depletion of BRCA1, PALB2, and p65 in MCF7 cells with lentiviral-transduced shRNAs. Due to PALB2 antibodies poorly performing in a total extract, we immunoprecipitated PALB2 before testing its depletion by immunoblot.
- Impact of BRCA1, PALB2, and p65/RelA depletion on p65/RelA recruitment upon TNF-α stimulation in MCF7 cells. ChIP analysis was performed after 1 h stimulation with TNF-α and reveals that neither BRCA1 nor PALB2 depletion affects p65/RelA binding to the TSS of its target genes.
- Transcriptional effects of TNF-α at selected genes in MCF7 cells. Depletion of BRCA1 but not PALB2 significantly impairs activation in all genes tested (P < 0.02) with the exception of NFKBIA. Most genes display attenuation of basal transcription following BRCA1, PALB2, and p65 depletion.
- Transcriptional effects of TNF-α at selected genes in MCF7 cells. Depletion of BRCA1 and PALB2 impairs gene activation mediated by NF-κB at IL-8, PHLDA1, BIRC3 (as resulted from the microarray analysis), and SOD2 (uncovered as a target gene in MCF10A cells). SOD2, IL-8, and BIRC3 also show a pronounced attenuation of basal transcription. All four genes displayed a significant attenuation of their responsiveness following the depletion of BRCA1 and PALB2 in three independent experiments (P < 0.05).
It is also important to note that infection of MCF10A or MCF7 cells with viral vectors used in our depletion studies may activate a component of the NF-κB pathway, which may contribute to the basal NF-κB responsiveness in MCF10A and MCF7 cells. Moreover, while depletion of BRCA1 and PALB2 did not elicit a change in expression of some TNF-α-responsive genes such as IRF, BCL3, or MAP3K8, we could still detect increased recruitment of BRCA1 and PALB2 on these genes following TNF-α induction (Supplementary Fig S5B), suggestive of additional redundant pathways for TNF-α responsiveness of these genes.
Next we asked whether BRCA1 mutant cell line HCC1937 displayed aberrant responsiveness to TNF-α. HCC1937 cells synthesize a truncated BRCA1 protein that is a product of a disease-producing mutant allele (5382insC) and no wild-type protein (Chen et al, 1998; Tomlinson et al, 1998). We used HCC1937 cells that were reconstituted with either wild-type BRCA1 construct or vector alone (Scully et al, 1999). We observed a robust enhancement of HCC1937 cells reconstituted with wild-type BRCA1 compared to the parental lines expressing an empty vector, consistent with the role for BRCA1 in TNF-α (Fig 7A). To gain further insight into the mechanism by which BRCA1 and PALB2 mediate responsiveness to TNF-α, we asked whether the depletion of BRCA1 or PALB2 decreases the recruitment of total RNAPII, its serine 2 or serine 5 phosphorylated forms. As shown earlier, while the depletion of BRCA1 or PALB2 did not affect the association of p65/RelA with TNF-α-responsive genes (Fig 6C), recruitment of RNAPII and its phosphorylated forms was substantially diminished following BRCA1 or PALB2 depletion in genes that displayed corresponding changes in their transcription (Fig 7B). These results indicate that while BRCA1 and PALB2 occupy the body of transcriptionally active genes and may fulfill roles in productive elongation (Fig 3), their depletion leads to substantial reduction in the recruitment of RNAPII to the TNF-α-responsive genes. Taken together, depletion of p65/RelA had a greater effect on TNF-α responsiveness than that of BRCA1 or PALB2 depletion, reflecting the fact that there may be other NF-κB co-activators and that BRCA1/PALB2 may only confer a component of the TNF-α response.
Figure 7. Requirement of BRCA1 for TNF-α activation.
- Response to TNF-α was investigated in HCC1937 cells, which carry one defective allele of BRCA1. Cells stably transfected with the empty vector or stably reconstituted with wild-type BRCA1 were stimulated with TNF-α for 1 h. Fold induction relative to t = 0 h for each cell line was calculated. Data are normalized to GUSB expression.
- Analysis of RNAPII and its phosphorylation status in MCF7 cells depleted of BRCA1, PALB2, or p65. qChIP was performed after TNF-α stimulation and reveals a substantial impairment of RNAPII recruitment after PALB2 and BRCA1 depletion, consistent with the effect on mRNA (Fig 6E). BRCA1 also impairs CXCL1, CXCL3, and CCL20 activation (Fig 6D), which reflects in a severe reduction of RNAPII recruitment.
BRCA1 and PALB2 are critical for responsiveness to retinoic acid in breast cancer cells
Since BRCA1 and PALB2 proteins occupy a large number of transcriptionally active genes, we envisage that they may play a broader role in stimulus-dependent transcriptional activation. To test this contention, we examined three well-characterized signaling pathways, implicated in breast cancer: p53, epidermal growth factor (EGF), and retinoic acid (RA). We did not observe a significant reduction in transcriptional responsiveness following the depletion of BRCA1 or PALB2 in a set of p53- or EGF-responsive genes (Supplementary Fig S8A,B). However, analysis of the RA signaling, which was previously shown to stimulate cellular differentiation and display anti-proliferative effects in breast cancer cells (Donato & Noy, 2005; Hua et al, 2009), revealed a compelling requirement for BRCA1 and PALB2. Treatment of MCF7 cells with RA led to a concomitant increase in the recruitment of RNAPII, BRCA1, and PALB2 on HOXA1 and HOXA2 genes supporting a direct function for these breast cancer susceptibility proteins in RA responsiveness (Fig 8A,B). Importantly, BRCA1 and PALB2 are not only recruited at the TSS following stimulation with RA but also accumulate at the 3′ end of genes (Fig 8A,B). We next turned to examining the functional impact of BRCA1 and PALB2 on RA responsiveness. While treatment with RA induced a ˜50- and ˜20-fold activation of HOXA1 and HOXA2 genes, respectively, depletion of PALB2 or BRCA1 reduced the RA-induced activation to tenfold for each gene (Fig 8C). Importantly, depletion of retinoic acid receptor (RARα) diminished the recruitment of BRCA1, PALB2, and RNAPII following RA treatment concomitant with decreased RA responsiveness (Fig 8D and Supplementary Fig S9A). These results point to a role for RARα in the recruitment of BRCA proteins similar to that for the p65/RelA protein.
Figure 8. BRCA1 and PALB2 are required for RA-mediated activation of HOX genes.

A, B Recruitment of BRCA1, PALB2, and RNAPII at the transcription start site and the 3′ end of HOXA1 and HOXA2 genes upon stimulation with 10 μM RA. Wild-type MCF7 cells were collected at 0, 6 and 24 h following stimulation and cross-linked in formaldehyde for ChIP analysis. The breast cancer proteins are loaded on chromatin at HOX loci over time, peaking at 24 h, with recruitment kinetics similar to RNAPII.
C BRCA1 and PALB2 are required for proper activation of HOX genes. MCF7 cells were infected with lentiviral shRNAs against PALB2 and BRCA1. Stably transduced cells were stimulated with retinoic acid for 24 h (RA +). The expression analysis of HOXA1 and HOXA2 expression was normalized against GUSB expression and is reported as a fold increase over unstimulated cells (RA −) infected with a non-targeting construct (CTRL). HOXA1 activation is dampened by over 70% upon depletion of the breast cancer proteins (P < 0.001 over three independent experiments); HOXA2 activation is also significantly reduced (P < 0.02) after silencing of PALB2 (50%) and BRCA1 (65%).
D Recruitment of BRCA1 and PALB2 at HOX genes is RARα dependent. Chromatin occupancy of RNAPII, BRCA1, and PALB2 was analyzed upon 6 h of RA stimulation in MCF7 cells infected with lentiviral shRNAs against RARA. qChIP data reveal a defective recruitment of BRCA1 and PALB2, along with RNA polymerase, at HOXA1 and HOXA2 proximal promoters. RNA levels of HOXA1, HOXA2, and RARA are shown in Supplementary Fig S7A.
To assess the breadth of action of PALB2 and BRCA1 in RA responsiveness, we treated MCF7 cells with RA and performed a gene expression profiling using microarrays. Importantly, depletion of BRCA1 and PALB2 in three independent experiments blunted the responsiveness of nearly all RA-responsive genes (248 genes, Supplementary Table S5), underscoring the importance of these proteins in RA signaling (Fig 9A). Besides genes with known roles in RA-mediated apoptosis signaling, many genes involved in oxidative stress and interferon signaling lost their responsiveness to RA following the depletion of BRCA1 and PALB2 (Fig 9B).
Figure 9. BRCA1 and PALB2 are required for RA-mediated gene activation genome-wide.

- Global RA transcriptional response is diminished following BRCA1 and PALB2 depletion. The induction of RA top-responding genes at 24 h has been analyzed in MCF7 cells using gene expression arrays. The heatmap representation covers 248 microarray probes up-regulated by retinoic acid in normal conditions (CTRL shRNAs, expression fold change of t = 24 h over t = 0 h, log2(fold change) > 0.4). The color scale represents the modified log2 ratio (“sweep” function R, scaled by row) between the induced (24 h) and the basal state (0 h) of every shRNA condition. The color variation accounts for the difference in induction across the three conditions (Red = augmented induction, Green = decreased induction). Results from three independent experiments are shown.
- The table lists some of the most significant pathways regulated by retinoic acid (RA) in MCF-7 cells, including RA-mediated apoptosis and NRF2-dependent oxidative stress response (data were obtained using Ingenuity canonical pathways analysis).
- PALB2 and BRCA1 depletion attenuates RA growth suppression. MCF7 cells stably transfected with shRNAs against BRCA1, PALB2, and CTRL were plated at subconfluent density and treated with 10 μM RA for 5 days. Data are reported as percent of viable cells compared to the untreated control. There is a significant difference between CTRL and BRCA1/PALB2-depleted cells at day 4 and day 5 (P < 0.05).
RA induces growth suppression in a wide range of adult cells, including breast cancer cells (del Rincon et al, 2003; Hua et al, 2009). We tested the hypothesis that depletion of BRCA1 and PALB2, which resulted in impaired transcriptional response to RA, may also diminish the RA-mediated growth suppression using MCF7 cells. Indeed, depletion of BRCA1 or PALB2 resulted in a significant decrease in the RA-mediated growth suppression (Fig 9C and Supplementary Fig S9B), consistent with their roles in transcriptional co-activation of RA-responsive genes.
Discussion
The novelty of this work lies in the following. First, it demonstrates the genomic occupancy of BRCA1 and PALB2 using high-resolution ChIP sequencing. Previous studies had suggested the occupancy of these proteins on chromatin; however, the comprehensive high-resolution analysis of the association of these proteins with chromatin had not been performed. Second, it shows that BRCA1 and PALB2 co-occupy a large number of protein-coding genes and its localization to a large extent tracks with that of RNAPII. Third, it provides evidence for functional association of BRCA1 and PALB2 with the elongating form of RNAPII, consistent with their occupancy on the body of the protein-coding genes. Fourth, it shows a requirement for BRCA1 and PALB2 in p65/RelA-mediated NF-κB signaling. Fifth, it provides support for the BRCA1 and PALB2 proteins as co-activators in RA signaling, through a functional genome-wide analysis.
Analysis of ChIP-seq data revealed that BRCA1 occupies a large number of protein-coding genes. Importantly, BRCA1 was also associated with nearly all RNA polymerase III (RNAPIII) genes, suggesting a role for this protein in the regulation of RNAPIII genes (Veras et al, 2009). Indeed, BRCA1 displayed a greater occupancy genome-wide than that observed with PALB2. Since BRCA1 is a component of number of multiprotein complexes including the ubiquitin hydrolase complex BRCC and BRCA1/BACH1 complexes (Dong et al, 2003; Kumaraswamy & Shiekhattar, 2007), such diverse genomic localization may represent the distribution of distinct BRCA1-containing complexes. This study focused on the functional analysis of genomic sites that displayed a concomitant chromatin residence for BRCA1 and PALB2. Importantly, our functional analysis revealed that the NF-κB signaling pathway targets a large number of highly active genes whose expression is co-regulated by BRCA1 and PALB2. Indeed, we demonstrate that BRCA1 and PALB2 are recruited to such promoters in a p65/RelA-dependent manner and that the loss of p65/RelA at target genes resulted in diminished BRCA1 and PALB2 occupancy. However, while we observed a great reduction in basal activity of NF-κB-responsive genes following the depletion of BRCA1 and PALB2, the fold induction of these genes following TNF-α stimulation was not significantly affected. This effect was predominantly due to a decreased transcriptional activity of these genes following BRCA1 and PALB2 depletion prior to their treatment with TNF-α. Since NF-κB subunits have been shown to induce senescence and promote DNA repair (Wang et al, 2009; Chien et al, 2011; Jing et al, 2011), the loss of ongoing NF-κB signaling following the inactivation of BRCA1 or PALB2 could contribute to increased genomic instability associated with breast and ovarian cancers.
These studies are also consistent with recent findings that NF-κB functions as a critical pathway in BRCA1-induced chemoresistance (Harte et al, 2013). Moreover, we analyzed a published gene expression dataset of transformed mammary epithelial cells derived from individuals with BRCA1 mutations and we found that these cells bear a significantly down-regulation of the NF-κB target genes as compared to transformed mammary cells from BRCA1 wild-type donors (Proia et al, 2011) (Supplementary Table S6). Furthermore, while these results are consistent with a previous report indicating the association of p65/RelA with the N-terminus of BRCA1 (Benezra et al, 2003), we were unable to find a physical association between p65/RelA and BRCA1 in our model system. It is likely that such BRCA1 and PALB2 interaction with p65/RelA may take place once these proteins are in association with DNA or chromatin. Furthermore, other BRCA1-interacting partners such as RNA helicase A or CtIP may promote such physical interaction (Tetsuka et al, 2004; Volcic et al, 2012).
To extend our analysis of BRCA1 and PALB2 in transcriptional responsiveness, we assessed their role in RA signaling which was shown to inhibit the proliferation of a variety of breast cancer cell lines (Liu et al, 1998; Donato & Noy, 2005; Hua et al, 2009). Similar to their roles in p65/RelA signaling, BRCA1 and PALB2 are recruited to the RA-responsive promoters following the treatment of breast cancer cells with RA and play an important role in transcriptional activation. We determined a requirement for BRCA1 and PALB2 in RA signaling not only for such classically responsive genes such as the HOX cluster but also for nearly all genes whose activation required RA treatment. Importantly, depletion of BRCA1 or PALB2 diminished the RA-mediated growth suppression of breast cancer cells. Taken together, these results identify a role for BRCA1 and PALB2 in transcriptional responsiveness for at least two important signaling pathways, NF-κB and RA, whose activity is critical in developmental regulation and oncogenesis. Such a contention is consistent with recent studies showing that BRCA1 haploinsufficiency drives lineage differentiation defects (Liu et al, 2008; Proia et al, 2011), explaining the recurrence of a very specific molecular and cellular phenotype in BRCA1-defective tumors. The requirement of BRCA1 in the transcriptional response to a differentiation stimulus, such as RA, suggests that the co-activator function of BRCA1 might be at the center of a regulatory network that is critical for the development of mammary epithelial cells. In the light of the breadth of their genomic distribution, we envision that BRCA1 and PALB2 may play a more general role in transcriptional regulation responding to diverse sets of growth regulatory signals.
The chromatin residence of BRCA1 and PALB2, which extends into the body of the transcriptionally active genes, suggests a functional role for these proteins in regulating transcriptional elongation. Indeed, previous reports uncovered an association between PALB2 and the chromatin regulator MRG15 (Sy et al, 2009a). MRG15 by virtue of its chromodomain was shown to interact with di- and trimethylated histone H3 lysine 36, a chromatin mark highly correlated with transcriptional elongation (Zhang et al, 2006). Therefore, it is likely that the distinct enrichment of PALB2 at the 3′-end of the transcriptionally active genes is reflective of its association with MRG15. Indeed, a functional role for BRCA1 and PALB2 in transcriptional elongation is consistent with the loss of these proteins at the 3′-end of transcriptionally active genes following the treatment of MCF10A cells with the transcriptional elongation inhibitor flavopiridol, concomitant with the decreased levels of elongating form of RNAPII. However, since flavopiridol could elicit the DNA damage response pathway, the loss of BRCA1 and PALB2 occupancy at target sites following the treatment of cells with this drug may reflect a re-localization of these proteins to DNA damage sites.
Whether BRCA1 and PALB2 are required for transcriptional responsiveness to other transcriptional activators is not known. Our preliminary analysis indicated that BRCA1 and PALB2 do not play a significant role in a set of candidate genes tested for their responsiveness to either EGF signaling or transcriptional activation following the induction of p53 pathway by nutlin-3. Therefore, based on our current data, we envision that mutations in BRCA1 or PALB2 will render affected cells incapable of responding to certain incoming stimuli. It is quite likely that breast and ovarian cancer susceptibility genes may play a broader role as guardian of the genome and their deregulation may impact a much broader spectrum of cancers than those of hereditary breast, ovarian, and pancreatic cancers.
Materials and Methods
Cell culture
Breast epithelial MCF10A cells were cultivated in serum-free DMEM/F12 1:1 (Invitrogen) and supplemented with the following: 2 mM L-glutamine, 50 ng/ml cholera toxin, 10 μg/ml bovine insulin, 500 ng/ml hydrocortisone, 10 ng/ml of recombinant EGF, and 50 μg/ml bovine pituitary extract. HeLa and MCF7 cells were grown in high-glucose DMEM, supplemented with 2 mM L-glutamine and 10% fetal bovine serum.
Antibodies
All BRCA1 antibodies were obtained from Santa Cruz Biotechnology: rabbit polyclonal I-20 was used for ChIP-seq and validation by qChIP, polyclonal D-20 was also used in qChIP validation, mouse monoclonal D-9 was used for ChIP-seq, qChIP validation and the following ChIP assays, this latter antibody was also used in immunoblot. Rabbit anti-PALB2 antibodies (Bethyl) and anti-p65 antibodies (Santa Cruz) were used for either ChIP or immunoblot. Western blot analysis of γ-tubulin and CBP80 was carried out using mouse monoclonal antibodies from Santa Cruz. Antibodies against RNAPII were obtained from Bethyl (against phosphorylated Ser2 of the CTD) and from Santa Cruz (N-20, recognizing all forms of RNAPII).
ChIP-seq
25–30 × 106 asynchronously growing MCF10A cells were cross-linked with 1% formaldehyde for 10 min at room temperature, harvested, and washed twice with 1x PBS. The pellet was resuspended in ChIP lysis buffer (150 mM NaCl, 1% Triton-X 100, 0.7% SDS, 500 μM DTT, 10 mM Tris–HCl, 5 mM EDTA) and chromatin was sheared to an average length of 200–400 bp, using a Bioruptor sonication device (20 min with 30-second intervals). The chromatin lysate was diluted with SDS-free ChIP lysis buffer and aliquoted into single IPs of 2.5 × 106 cells each. A specific antibody or a total rabbit IgG control was added to the lysate along with Protein A magnetic beads (Invitrogen) and incubated at 4°C overnight. On day 2, beads were washed twice with each of the following buffers: mixed micelle buffer (150 mM NaCl, 1% Triton-X 100, 0.2% SDS, 20 mM Tris–HCl, 5 mM EDTA, 65% sucrose), buffer 500 (500 mM NaCl,% Triton-X 100, 0.1% Na deoxycholate, 25 mM HEPES, 10 mM Tris–HCl, 1 mM EDTA), LiCl/detergent wash (250 mM LiCl, 0.5% Na deoxycholate, 0.5% NP-40, 10 mM Tris–HCl, 1 mM EDTA), and a final wash was performed with 1× TE. Finally, beads were resuspended in 1× TE containing 1% SDS and incubated at 65°C for 10 min to elute immunocomplexes. Elution was repeated twice, and the samples were further incubated overnight at 65°C to reverse cross-linking, along with the untreated input (2.5% of the starting material). After treatment with 0.5 mg/ml proteinase K for 3 h, DNA was purified with Wizard SV Gel and PCR Clean-up system (Promega) according to the manufacturer's protocol and eluted in nuclease-free water. Eluates from different IPs were pooled together and concentrated by speed-vac. DNA concentration was assessed with Quant-it PicoGreen dsDNA kit (Invitrogen) and 5–10 ng was used to generate the sequencing libraries. DNA fragments of ˜150- to 400-bp range were isolated by agarose gel purification, ligated to primers, and then subject to Solexa sequencing using the manufacturer's recommendations (Illumina, Inc.).
ChIP-seq and RNA-seq data analysis
All RNA-seq and ChIP-seq raw and processed files are available at GEO (accession number GSE45715). ChIP-seq and RNA-seq data were obtained using an Illumina Genome Analyzer II. ChIP-seq 36-bp reads were aligned to the human genome hg18 using ELAND. For further bioinformatic analysis, we selected reads that uniquely aligned to the genome. In addition, multiple reads with an identical start site and mapping to the same strand were considered as experimental artifacts; therefore, they were counted only once. Snapshots of raw ChIP-seq data presented throughout the figures were obtained as follows: BigWiggle files for every ChIP-Seq were generated using Bed Tools and the utility bedGraphToBigWig (http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/); these tracks were then uploaded into the UCSC Genome Browser.
RNA-seq data for wild-type MCF10A (36-bp single reads) were obtained using the Epicentre (Illumina) mRNA-seq kit, generating a polyadenylated transcript library. MCF10A cells expressing shRNAs (PALB2, BRCA1, and control) were collected after 5 days from infection; RNA was depleted of ribosomal RNA using RiboZero (Epicentre) and processed with the ScriptSeqv2 kit along with ScriptSeq Index PCR primers (Epicentre) to generate a strand-specific library of total RNA. 72-bp single reads were obtained using an Illumina Genome Analyzer II.
Sequencing reads were aligned to the human genome hg18 using TopHat (Trapnell et al, 2009) that takes into account reads coming from splicing junctions (parameters were set to default). The expression level of all RefSeq transcripts was evaluated using Cufflinks (Trapnell et al, 2010), and a FPKM (fragments per kilobase of transcript per million fragments mapped) was calculated for each transcript (the parameters were left to default and hg18 RefSeq GTF table was used to define the transcripts). Differences in gene expression levels between the CRTL and PALB2 and BRCA1 depleted samples were assessed by Cuffdiff and calculated as log2 (fold change).
Clustering and Heatmap analysis
ChIP-seq data were subjected to unbiased clustering, with respect to a list of unique RefSeq genes or U snRNA genes, using the seqMINER 1.3.2 platform (Ye et al, 2011). We used Kmeans linear as the method of clustering, with the following parameters: left and right extension = 1.5 kb, internal bins (with respect to the gene body) = 160, number of cluster = 6. seqMINER was also used to generate the heatmaps and the average profiles of read density for the different clusters.
Clustering and heatmap analysis of Illumina bead array data was performed using the beadarray library in R and the heatmap.2 function of gplots.
qChIP
ChIP was performed in HeLa, MCF7, and MCF10A as previously described (Baillat et al, 2012). ChIP eluates from the specific antibodies, control IgG, and input were assayed by real-time quantitative PCR in a 20-μl reaction mixture with the following: 0.4 μM of each primer, 10 μl of iQ SYBR Green Supermix (BioRAD), and 5 μl of template DNA (corresponding to 1/40 of the elution material). Thermal cycling parameters were: 3 min at 95°C, followed by 40 cycles of 10 s at 95°C, and 30 s at 60°C. The strength of ChIP signal was calculated as the amount of immunoprecipitated DNA relative to that present in the input chromatin (% of input).
Lentiviral infection
pSiCo PGKpuro constructs (Ventura et al, 2004) against PALB2, BRCA1, p65/RelA, and a non-targeting control (see Supplemental Material for sequences) were transfected in 293T cells using Metafectene Pro (Biontex) as a carrier. Two different shRNAs were pooled and utilized for to produce viral supernatant for PALB2, BRCA1, and p65/RelA, unless specified otherwise. The supernatant was collected after 24 and 48 h and used to infect MCF10A and MCF7 cells. After 24 h, cells were selected with 2.5 μg/mL puromycin for 48–72 h.
Flavopiridol treatment
Exponentially growing MCF10A cells (70–80% confluency) were treated with flavopiridol at a final concentration of 2 μM; treated and untreated cells were collected after 2 h and subjected to ChIP analysis.
Growth suppression curve
MCF-7 cells were infected with lentiviral shRNAs against PALB2, BRCA1, and a non-targeting control. After 3 days of puromycin selection, cells were plated at 4 × 104 cells/well in 12-well plates in DMEM/F12 medium+5% FBS. Cells were incubated with 10 μM RA for 5 days and counted using a hemocytometer on days 3, 4, and 5. Each data point represents the percentage of viable cells with 10 μM RA compared to untreated cells. Data are average of triplicate wells ± standard deviation.
Gene expression arrays
MCF7 cells were infected with different shRNA constructs, stimulated after selection, and 400 ng of total RNA was amplified according to Illumina protocols and hybridized to Illumina HumanHT-12 v4 Expression BeadChip. Three biological replicates were analyzed for each condition. Data were processed using the beadarray library in R. Raw data were transformed to log2 and normalized by quantile normalization; fold changes and statistics were calculated using the LIMMA library in R (Linear Models for Microarray Data). Heatmaps have been created using GPLOT library with default parameters.
qRT-PCR
Total RNA was extracted using Trizol Reagent (Invitrogen), treated 15 min with 10 units of RNase-free DNase I (NEB), and cleaned up on RNeasy columns (Qiagen). cDNAs were synthesized from 2 μg of total RNA using the RevertAid first strand synthesis kit (Fermentas) with random primers. qPCR was performed as already described for ChIP samples, using 15 ng of cDNA. Each sample was run in triplicate. The mean value of the replicates for each sample was calculated and expressed as cycle threshold (CT, cycle number at which each PCR reaction reaches a predetermined fluorescence threshold, set within the linear range of all reactions). The amount of gene expression was then calculated as the difference (ΔCT) between the CT value of the sample for the target gene and the mean CT value of that sample for the endogenous control (GUSB).
Acknowledgments
All genome-wide data presented in the paper are available at GEO (GSE45715). We thank Ralph Scully for providing the HCC1937 cell lines. We thank members of the Wistar Institute Genomics facility for processing of ChIP-seq samples and Expression Arrays; we also thank the Bioinformatics Core Unit for the analysis of Illumina BeadArrays. This work was supported by Grant R01-CA 090758 (R.S.), PA Breast and Cervical Cancer Research Initiative to RS, P30 CA 010815 from the National Institute of Health. A.G. was supported by an American-Italian Cancer Foundation Post-Doctoral Research Fellowship.
Author contributions
The study was conceived by RS and AG. AG performed most of the experiments. DB performed some of the knock-down experiments and generated reagents. MC performed the bioinformatics analysis of ChIP-seq and RNA-seq data. RS and AG wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Amann PM, Eichmuller SB, Schmidt J, Bazhin AV. Regulation of gene expression by retinoids. Curr Med Chem. 2011;18:1405–1412. doi: 10.2174/092986711795029618. [DOI] [PubMed] [Google Scholar]
- Baillat D, Gardini A, Cesaroni M, Shiekhattar R. Requirement for SNAPC1 in transcriptional responsiveness to diverse extracellular signals. Mol Cell Biol. 2012;32:4642–4650. doi: 10.1128/MCB.00906-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benezra M, Chevallier N, Morrison DJ, MacLachlan TK, El-Deiry WS, Licht JD. BRCA1 augments transcription by the NF-kappaB transcription factor by binding to the Rel domain of the p65/RelA subunit. J Biol Chem. 2003;278:26333–26341. doi: 10.1074/jbc.M303076200. [DOI] [PubMed] [Google Scholar]
- Bible KC, Bible RH, Jr, Kottke TJ, Svingen PA, Xu K, Pang YP, Hajdu E, Kaufmann SH. Flavopiridol binds to duplex DNA. Cancer Res. 2000;60:2419–2428. [PubMed] [Google Scholar]
- Bleuyard JY, Buisson R, Masson JY, Esashi F. ChAM, a novel motif that mediates PALB2 intrinsic chromatin binding and facilitates DNA repair. EMBO Rep. 2012;13:135–141. doi: 10.1038/embor.2011.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochar DA, Wang L, Beniya H, Kinev A, Xue Y, Lane WS, Wang W, Kashanchi F, Shiekhattar R. BRCA1 is associated with a human SWI/SNF-related complex: linking chromatin remodeling to breast cancer. Cell. 2000;102:257–265. doi: 10.1016/s0092-8674(00)00030-1. [DOI] [PubMed] [Google Scholar]
- Cable PL, Wilson CA, Calzone FJ, Rauscher FJ, 3rd, Scully R, Livingston DM, Li L, Blackwell CB, Futreal PA, Afshari CA. Novel consensus DNA-binding sequence for BRCA1 protein complexes. Mol Carcinog. 2003;38:85–96. doi: 10.1002/mc.10148. [DOI] [PubMed] [Google Scholar]
- Castilla LH, Couch FJ, Erdos MR, Hoskins KF, Calzone K, Garber JE, Boyd J, Lubin MB, Deshano ML, Brody LC, Collins FS, Weber BL. Mutations in the BRCA1 gene in families with early-onset breast and ovarian cancer. Nat Genet. 1994;8:387–391. doi: 10.1038/ng1294-387. [DOI] [PubMed] [Google Scholar]
- Chao SH, Price DH. Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J Biol Chem. 2001;276:31793–31799. doi: 10.1074/jbc.M102306200. [DOI] [PubMed] [Google Scholar]
- Chapman MS, Verma IM. Transcriptional activation by BRCA1. Nature. 1996;382:678–679. doi: 10.1038/382678a0. [DOI] [PubMed] [Google Scholar]
- Chen J, Silver DP, Walpita D, Cantor SB, Gazdar AF, Tomlinson G, Couch FJ, Weber BL, Ashley T, Livingston DM, Scully R. Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells. Mol Cell. 1998;2:317–328. doi: 10.1016/s1097-2765(00)80276-2. [DOI] [PubMed] [Google Scholar]
- Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, Premsrirut P, Luo W, Chicas A, Lee CS, Kogan SC, Lowe SW. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25:2125–2136. doi: 10.1101/gad.17276711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Andrea AD. Susceptibility pathways in Fanconi's anemia and breast cancer. N Engl J Med. 2010;362:1909–1919. doi: 10.1056/NEJMra0809889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato LJ, Noy N. Suppression of mammary carcinoma growth by retinoic acid: proapoptotic genes are targets for retinoic acid receptor and cellular retinoic acid-binding protein II signaling. Cancer Res. 2005;65:8193–8199. doi: 10.1158/0008-5472.CAN-05-1177. [DOI] [PubMed] [Google Scholar]
- Dong Y, Hakimi MA, Chen X, Kumaraswamy E, Cooch NS, Godwin AK, Shiekhattar R. Regulation of BRCC, a holoenzyme complex containing BRCA1 and BRCA2, by a signalosome-like subunit and its role in DNA repair. Mol Cell. 2003;12:1087–1099. doi: 10.1016/s1097-2765(03)00424-6. [DOI] [PubMed] [Google Scholar]
- Erkko H, Xia B, Nikkilae J, Schleutker J, Syrjaekoski K, Mannermaa A, Kallioniemi A, Pylkas K, Karppinen SM, Rapakko K, Miron A, Sheng Q, Li GL, Mattila H, Bell DW, Haber DA, Grip M, Reiman M, Jukkola-Vuorinen A, Mustonen A, et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature. 2007;446:316–319. doi: 10.1038/nature05609. [DOI] [PubMed] [Google Scholar]
- Harte MT, Gorski JJ, Savage KI, Purcell JW, Barros EM, Burn PM, McFarlane C, Mullan PB, Kennedy RD, Perkins ND, Harkin DP. NF-kappaB is a critical mediator of BRCA1-induced chemoresistance. Oncogene. 2013;33:713–723. doi: 10.1038/onc.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua S, Kittler R, White KP. Genomic antagonism between retinoic acid and estrogen signaling in breast cancer. Cell. 2009;137:1259–1271. doi: 10.1016/j.cell.2009.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing H, Kase J, Dorr JR, Milanovic M, Lenze D, Grau M, Beuster G, Ji S, Reimann M, Lenz P, Hummel M, Dorken B, Lenz G, Scheidereit C, Schmitt CA, Lee S. Opposing roles of NF-kappaB in anti-cancer treatment outcome unveiled by cross-species investigations. Genes Dev. 2011;25:2137–2146. doi: 10.1101/gad.17620611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, Lin JC, Palmisano E, Brune K, Jaffee EM, Iacobuzio-Donahue CA, Maitra A, Parmigiani G, Kern SE, Velculescu VE, Kinzler KW, Vogelstein B, Eshleman JR, Goggins M, Klein AP. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324:217. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiman FE, Wu-Baer F, Fonseca D, Kaneko S, Baer R, Manley JL. BRCA1/BARD1 inhibition of mRNA 3' processing involves targeted degradation of RNA polymerase II. Genes Dev. 2005;19:1227–1237. doi: 10.1101/gad.1309505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaraswamy E, Shiekhattar R. Activation of BRCA1/BRCA2-associated helicase BACH1 is required for timely progression through S phase. Mol Cell Biol. 2007;27:6733–6741. doi: 10.1128/MCB.00961-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Takayama S, Zheng Y, Froesch B, Chen GQ, Zhang X, Reed JC, Zhang XK. Interaction of BAG-1 with retinoic acid receptor and its inhibition of retinoic acid-induced apoptosis in cancer cells. J Biol Chem. 1998;273:16985–16992. doi: 10.1074/jbc.273.27.16985. [DOI] [PubMed] [Google Scholar]
- Liu S, Ginestier C, Charafe-Jauffret E, Foco H, Kleer CG, Merajver SD, Dontu G, Wicha MS. BRCA1 regulates human mammary stem/progenitor cell fate. Proc Natl Acad Sci U S A. 2008;105:1680–1685. doi: 10.1073/pnas.0711613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moisan A, Larochelle C, Guillemette B, Gaudreau L. BRCA1 can modulate RNA polymerase II carboxy-terminal domain phosphorylation levels. Mol Cell Biol. 2004;24:6947–6956. doi: 10.1128/MCB.24.16.6947-6956.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7:263–272. doi: 10.1016/s1097-2765(01)00174-5. [DOI] [PubMed] [Google Scholar]
- Mullan PB, Quinn JE, Harkin DP. The role of BRCA1 in transcriptional regulation and cell cycle control. Oncogene. 2006;25:5854–5863. doi: 10.1038/sj.onc.1209872. [DOI] [PubMed] [Google Scholar]
- Ouchi T, Monteiro AN, August A, Aaronson SA, Hanafusa H. BRCA1 regulates p53-dependent gene expression. Proc Natl Acad Sci U S A. 1998;95:2302–2306. doi: 10.1073/pnas.95.5.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Cortez D, Bowers B, Elledge SJ, Gellert M. Direct DNA binding by Brca1. Proc Natl Acad Sci USA. 2001;98:6086–6091. doi: 10.1073/pnas.111125998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins ND. The diverse and complex roles of NF-kappaB subunits in cancer. Nat Rev Cancer. 2012;12:121–132. doi: 10.1038/nrc3204. [DOI] [PubMed] [Google Scholar]
- Proia TA, Keller PJ, Gupta PB, Klebba I, Jones AD, Sedic M, Gilmore H, Tung N, Naber SP, Schnitt S, Lander ES, Kuperwasser C. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell. 2011;8:149–163. doi: 10.1016/j.stem.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA. c-Myc regulates transcriptional pause release. Cell. 2010;141:432–445. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, Reid S, Spanova K, Barfoot R, Chagtai T, Jayatilake H, McGuffog L, Hanks S, Evans DG, Eccles D, Easton DF, Stratton MR, Collabo BCS. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39:165–167. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, Neveling K, Kelly P, Seal S, Freund M, Wurm M, Batish SD, Lach FP, Yetgin S, Neitzel H, Ariffin H, Tischkowitz M, Mathew CG, Auerbach AD, Rahman N. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. 2007;39:162–164. doi: 10.1038/ng1947. [DOI] [PubMed] [Google Scholar]
- del Rincon SV, Rousseau C, Samanta R, Miller WH., Jr Retinoic acid-induced growth arrest of MCF-7 cells involves the selective regulation of the IRS-1/PI 3-kinase/AKT pathway. Oncogene. 2003;22:3353–3360. doi: 10.1038/sj.onc.1206485. [DOI] [PubMed] [Google Scholar]
- Rosen EM, Fan S, Ma Y. BRCA1 regulation of transcription. Cancer Lett. 2006;236:175–185. doi: 10.1016/j.canlet.2005.04.037. [DOI] [PubMed] [Google Scholar]
- Scully R, Anderson SF, Chao DM, Wei W, Ye L, Young RA, Livingston DM, Parvin JD. BRCA1 is a component of the RNA polymerase II holoenzyme. Proc Natl Acad Sci U S A. 1997a;94:5605–5610. doi: 10.1073/pnas.94.11.5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston DM. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997b;88:265–275. doi: 10.1016/s0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- Scully R, Ganesan S, Vlasakova K, Chen J, Socolovsky M, Livingston DM. Genetic analysis of BRCA1 function in a defined tumor cell line. Mol Cell. 1999;4:1093–1099. doi: 10.1016/s1097-2765(00)80238-5. [DOI] [PubMed] [Google Scholar]
- Smale ST. Dimer-specific regulatory mechanisms within the NF-kappaB family of transcription factors. Immunol Rev. 2012;246:193–204. doi: 10.1111/j.1600-065X.2011.01091.x. [DOI] [PubMed] [Google Scholar]
- Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007;316:1198–1202. doi: 10.1126/science.1139516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sy SM, Huen MS, Chen J. MRG15 is a novel PALB2-interacting factor involved in homologous recombination. J Biol Chem. 2009a;284:21127–21131. doi: 10.1074/jbc.C109.023937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci U S A. 2009b;106:7155–7160. doi: 10.1073/pnas.0811159106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetsuka T, Uranishi H, Sanda T, Asamitsu K, Yang JP, Wong-Staal F, Okamoto T. RNA helicase A interacts with nuclear factor kappaB p65 and functions as a transcriptional coactivator. Eur J Biochem. 2004;271:3741–3751. doi: 10.1111/j.1432-1033.2004.04314.x. [DOI] [PubMed] [Google Scholar]
- Tomlinson GE, Chen TT, Stastny VA, Virmani AK, Spillman MA, Tonk V, Blum JL, Schneider NR, Wistuba II, Shay JW, Minna JD, Gazdar AF. Characterization of a breast cancer cell line derived from a germ-line BRCA1 mutation carrier. Cancer Res. 1998;58:3237–3242. [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkitaraman AR. Linking the cellular functions of BRCA genes to cancer pathogenesis and treatment. Annu Rev Pathol. 2009;4:461–487. doi: 10.1146/annurev.pathol.3.121806.151422. [DOI] [PubMed] [Google Scholar]
- Ventura A, Meissner A, Dillon CP, McManus M, Sharp PA, Van Parijs L, Jaenisch R, Jacks T. Cre-lox-regulated conditional RNA interference from transgenes. Proc Natl Acad Sci USA. 2004;101:10380–10385. doi: 10.1073/pnas.0403954101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veras I, Rosen EM, Schramm L. Inhibition of RNA polymerase III transcription by BRCA1. J Mol Biol. 2009;387:523–531. doi: 10.1016/j.jmb.2009.02.008. [DOI] [PubMed] [Google Scholar]
- Volcic M, Karl S, Baumann B, Salles D, Daniel P, Fulda S, Wiesmuller L. NF-kappaB regulates DNA double-strand break repair in conjunction with BRCA1-CtIP complexes. Nucleic Acids Res. 2012;40:181–195. doi: 10.1093/nar/gkr687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Jacob NK, Ladner KJ, Beg A, Perko JD, Tanner SM, Liyanarachchi S, Fishel R, Guttridge DC. RelA/p65 functions to maintain cellular senescence by regulating genomic stability and DNA repair. EMBO Rep. 2009;10:1272–1278. doi: 10.1038/embor.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooster R, Neuhausen SL, Mangion J, Quirk Y, Ford D, Collins N, Nguyen K, Seal S, Tran T, Averill D, et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science. 1994;265:2088–2090. doi: 10.1126/science.8091231. [DOI] [PubMed] [Google Scholar]
- Xia B, Dorsman JC, Ameziane N, de Vries Y, Rooimans MA, Sheng Q, Pals G, Errami A, Gluckman E, Llera J, Wang W, Livingston DM, Joenje H, de Winter JP. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat Genet. 2007;39:159–161. doi: 10.1038/ng1942. [DOI] [PubMed] [Google Scholar]
- Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ, Livingston DM. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell. 2006;22:719–729. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Ye T, Krebs AR, Choukrallah MA, Keime C, Plewniak F, Davidson I, Tora L. seqMINER: an integrated ChIP-seq data interpretation platform. Nucleic Acids Res. 2011;39:e35. doi: 10.1093/nar/gkq1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, Yu X. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol. 2009;19:524–529. doi: 10.1016/j.cub.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Du J, Sun B, Dong X, Xu G, Zhou J, Huang Q, Liu Q, Hao Q, Ding J. Structure of human MRG15 chromo domain and its binding to Lys36-methylated histone H3. Nucleic Acids Res. 2006;34:6621–6628. doi: 10.1093/nar/gkl989. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.