

Abstract
Despite the prevalent studies of DNA/Chromatin related epigenetics, such as, histone modifications and DNA methylation, RNA epigenetics has not drawn deserved attention until a new affinity-based sequencing approach MeRIP-Seq was developed and applied to survey the global mRNA N6-methyladenosine (m6A) in mammalian cells. As a marriage of ChIP-Seq and RNA-Seq, MeRIP-Seq has the potential to study the transcriptome-wide distribution of various post-transcriptional RNA modifications.
We have previously developed an R/Bioconductor package ‘exomePeak’ for detecting RNA methylation sites under a specific experimental condition or the identifying the differential RNA methylation sites in a case control study from MeRIP-Seq data. Compared with other relatively well studied data types such as ChIP-Seq and RNA-Seq, the study of MeRIP-Seq data is still at very early stage, and existing protocols are not optimized for dealing with the intrinsic characteristic of MeRIP-Seq data. We therein provide here a detailed and easy-to-use protocol of using exomePeak R/Bioconductor package along with other software programs for analysis of MeRIP-Seq data, which covers raw reads alignment, RNA methylation site detection, motif discovery, differential RNA methylation analysis, and functional analysis. Particularly, the rationales behind each processing step as well as the specific method used, the best practice, and possible alternative strategies are briefly discussed.
Introduction
Despite the unprecedented advance in epigenetics studies of DNA methylation and histone modifications with next-generation sequencing (NGS), RNA epigenetics remains a largely uncharted territory [1] and has not benefitted as much from the advancement in sequencing technology until lately. A new powerful protocol MeRIP-Seq (independently named as ‘m6A-Seq’ [2, 3] and ‘MeRIP-Seq’[4]) was proposed in two recent studies on transcriptome-wide mRNA N6-methyladenosine (m6A) methylation [3, 4], where mRNA is fragmented before the immunoprecipitation with anti-m6A antibody, and the immunoprecipitated and input control fragments are then sequenced and aligned for reconstructing the m6A RNA methylome (See Figure 1).
Figure 1.

Illustration of MeRIP-Seq Technique
MeRIP-Seq (more comprehensively detailed in [2]) in theory enabled the transcriptome-wide unbiased study of a large number of known post-transcriptional RNA modifications [5] at a high resolution, provided that the corresponding antibody is available. As one of the primary application of next generation sequencing that targets the RNA modifications (see Table 1), the protocol is expected to gain increasing popularity in the near future.
Table 1.
MeRIP-Seq in the Large Picture of Next Generation Sequencing
| Analysis Types | DNA related | RNA related |
|---|---|---|
| Assembly | Genome Reconstruction | Transcriptome Reconstruction |
| Sequence | Single Nucleotide Polymorphism and Insertion and Deletion | RNA Editing |
| Quantity | Copy Number Variation | Differential Expression Analysis |
| Modification | DNA Methylation and Histone Modifications | MeRIP-Seq for Posttranscriptional RNA Modifications |
From a technological perspective, MeRIP-Seq can be considered a marriage of three relatively well-studied techniques: ChIP-Seq [6, 7], RNA-Seq [8, 9] and MeDIP-Seq [10, 11], yet it brings new computational challenges not addressed previously [12]. Next, we discuss briefly the best practice for MeRIP-Seq data analysis by drawing its connections with RNA-Seq, ChIP-Seq and MeDIP-Seq.
• Mapping and Filtering Short Reads
As MeRIP-Seq sequences mRNA indirectly from cDNA, spliced aligners that allow reads to span exon–exon junctions should be implemented. As in many other NGS based techniques, an important issue is how to best deal with the widespread repetitive elements [13] in a broad range of species (around 50% of the human genome) that can lead to multi-reads (reads that could be mapped to multiple genomic locations) and the mapping ambiguities in the alignment. Of the various existing strategies, the simplest yet very effective way is to exclude all the multi-reads completely from the analysis.
• Fragment Length and Shifting Size
Currently, the most popular RNA sequencing protocol (unstranded and single-end sequencing) produces two shifted peaks on the ‘+’ and ‘−’ strands with a distance equal to the fragment length when using 5’ end position to denote the position of the reads (The distance is equal to ‘fragment length’ minus ‘read length’ when using ‘Pos’ in the SAM/BAM format to denote reads’ positions.). The bimodal pattern is naturally observed in MeRIP-Seq data [12]. To correctly predict the precise methylation sites, reads need to be shifted by half of the fragment length or extended to the full length towards the 3’ end. In case that the fragment length is unknown, it may be estimated from the bimodal pattern [14, 15] or the cross-strand correlation [16]. Noted that, different from MeRIP-Seq, the current standard RIP-Seq protocol takes advantages of strand-specific sequencing technique [17], so the reads from a transcript are mapped to ‘+’ or ‘−’ strand only, and the bimodal pattern is not observable [18].
• Peak Calling, Sequencing Bias and Control Sample
The detection of methylation sites has been mainly formulated as the peak detection problem in ChIP-Seq [19, 20]. Different from the mild sequencing bias in ChIP-Seq, which is mainly owing to nucleosome loss around the transcription starting sites, MeRIP-Seq suffers from the depletion at both 5’ and 3’ ends as a result of RNA fragmentation [9], considerable variations of expression levels for different genes, and most importantly, the positional bias on the locus of the same gene due to different isoform transcripts. Although ChIP-Seq peak calling can be conducted in the absence of a control sample by estimating the background from the neighborhood genomic regions, MeRIP-Seq peak calling requires the paired input control sample of fragmented RNAs before immunoprecipitation (input control sample) as opposed to an immunoglobulin G control sample (IgG control sample) as used in ChIP-Seq [6]. In MeDIP-Seq, of interests are the CpG islands, thus peak calling is usually unnecessary.
• Peak Annotation, Gene and Isoform Transcripts
The association between detected RNA methylation sites and the specific mRNA transcripts can be problematic due to the complexity of transcriptome. Recent study showed that with an average of 10 to 12 isoforms per gene, most genes tend to express multiple isoforms simultaneously [21]. Since the fragment length is 100bp for the current MeRIP-Seq protocol, isoform quantification can be difficult, not to mention the identification of sites on each individual isoform transcript. Nevertheless, an mRNA methylation site may be uniquely associated with a transcript when the site spans across the nearest exon(s) that uniquely belongs to that transcript. On the other hand, the association between peaks and genes is trivial except for the case of antisense RNA [22] Because identifying isoforms based on MeRIP-Seq is still not realistic, it should be prudent to report the association between gene and methylation sites instead of transcripts at the current stage.
• Differential Methylation
Differential analysis for MeRIP-Seq identifies differences in RNA methylome in a case-control study (e.g., normal and cancer). The RNA methylome is influenced by “methylation potential” [23], which is the ratio of S-adenosylmethionine (SAM, the universal methyl donor cosubstrate) and S-adnosylhomocysteine (SAH, the by-product of SAM that acts as competitive inhibitor). In this respect, it is comparable to the DNA methylation, where the percentage of methylated molecule (or Beta value as in bisulfite-Seq or DNA methylation microarray) is adopted to represent the degree of methylation. The differential methylation analysis is then equivalent to testing whether the percentage of modified molecules1 are the same under two experimental conditions in a case-control study. For affinity-based methods developed for DNA epigenetics (such as MeDIP-Seq and ChIP-Seq), since the total amount of DNA remains the same under two conditions after compensating for sequencing depth, the percentage of modified DNA molecule is linearly correlated with the absolute amount, and the difference is consistent regardless if the relative (percentage) or absolute amount is used. However, in MeRIP-Seq, due to the effect of transcriptional differential expression, it is possible that while the absolute amount of methylated RNA increases, the relative amount (percentage of methylated RNA) decreases (See Figure 2). Therein it is of crucial importance to untangle the transcriptional regulation (which directly changes the RNA abundance) and the enzymatic regulations of the RNA methylome by methylases and demethylases, which directly changes the percentage of methylated RNA molecules.
Figure 2.

Comparison of the differential methylation in DNA and RNA In DNA related analysis, the background are considered the same in two experimental conditions, and the absolute of methylation is linearly correlated with the percentage of methylated molecules; However, in RNA, due to differential expression, there might be more copies of a specific RNA under one condition, thus the increase of absolute amount of methylation might not indicate stronger RNA enzymatic hypermethylation. While the differential analysis of ChIP-Seq and MeDIP-Seq doesn’t directly require the input control samples when a testing region is specified, differential RNA methylation analyisis does require the input control samples to estimate the total number of RNA molecules.
• Mechanism
The transcriptome-wide RNA methylation, or epitranscriptome, is simultaneously regulated transcriptionally and enzymatically (post-transcriptionally) (See Figure 3). While the transcriptional regulation in response to stimulus changes modulate directly the absolute amount of RNA causing the absolute amount of methylation changes coordinately, enzymatic regulation by methylases/demethylases changes directly the percentage of methylated molecule. In practice, the two layers of regulation contribute simultaneously to form the epitranscriptome. The identification of RNA differential methylation due to enzymatic regulation must compensate the changes in transcriptional level, making it fundamentally different from other affinity-based sequencing approaches used in DNA-templated epigenetic studies, such ChIP-Seq and MeDIP-Seq.
Figure 3. The regulation of RNA methylome.

In practice, the dynamics in epitranscriptome are a result of a joint effect of both transcriptional and enzymatic regulations. On the one hand, transcriptional regulation directly changes the amount of RNA molecules and leads to coordinated changes in the absolute amount of methylated molecules, leaving the relative amount unchanged. On the other hand, enzymatic regulation of the RNA methylome by ‘methylation potential’ changes directly the percentage of methylated molecules. For the above illustration, under the joint effects of transcriptional down-regulation and enzymatic hypermethylation, the absolute amount of methylated RNA stays unchanged.
• Molecular Structure and Motif finding
The sequence motifs, conjectured to have a biological significance, can be identified in both ChIP-Seq and MeRIP-Seq. The main computational difference is whether to search the reverse complement strand. While a DNA motif may appear on either strand of the two, RNA motif should appear only on the strand where the transcript is located, and thus the strand information should be kept at all times. When the strand information of the RNA fragment is lost in MeRIP-Seq unstranded library construction, it may still be derived from the information of transcripts to which the fragments (reads) are mapped.
• Functional analysis
The functional analysis of RNA methylation should still mainly rely on various gene annotations, such as, KEGG pathways, gene ontology (GO), TRANSFAC, etc. We may use many software programs to achieve this goal including DAVID [24], Ingenuity Pathway Analysis, GSEA [25], etc.
We have previously developed exomePeak [12], an open source R package for analyzing the MeRIP-Seq data. exomePeak addressed the aforementioned unique issues with MeRIP-Seq and was shown to be able to achieve improved performance than ChIP-Seq based algorithms. In this paper, we explain the detailed procedure of performing differential methylation analysis with exomePeak by using the MeRIP-Seq dataset that profiles transcriptome-wide methylation in mouse midbrain under wild type condition and FTO deficiency condition [26].
A case study: differential RNA methylation in mouse midbrain under FTO deficiency condition
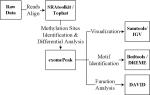
The exemplar MeRIP-Seq dataset (GEO GSE47217) measures the transcriptome-wide m6A profiles in mouse midbrain under wild type condition and FTO deficiency condition [26]. The software tools used for the analysis of the MeRIP-Seq dataset is summarized in Table 2, and this analysis also relies on Bash UNIX Shell and R system. This example starts from the raw data downloaded directly from GEO database and conducts reads alignment, RNA methylation site detection, differential analysis, RNA methylation site visualization, motif identification and functional annotation. The details of each step are provided in the next.
Table 2.
Tools Used for MeRIP-Seq Differential RNA Methylation Analysis
| Step | Purpose | Tools | Flowchart |
|---|---|---|---|
| 1 | Raw data preprocessing and sequence alignment | SRAtoolkit / Tophat [27] |
|
| 2 | RNA methylation sites identification and differential analysis | exomePeak1 [12] | |
| 3 | Motif identification | Bedtools [28] / DREME [29] | |
| 4 | Methylation sites visualization | Samtools [30] / IGV [31] | |
| 5 | Function analysis | DAVID [24] |
The exomePeak package was initially developed as a MATLAB package [12] for RNA methylation site detection from MeRIP-Seq data. It has been recently extended with differential analysis capacity and implemented as an open source R/Bioconductor package.
Step 1 Download the raw data from GEO and aligned reads to reference genome. This step can be easily realized with Script 1 (bash script) provided in the next.
# Script 1
# !/bin/bash
# download the data from GEO
wget –r\
ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP/SRP023/SRP023108/ wget –r\
ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP/SRP023/SRP023107/ mkdir ./sra ./fastq ./tophat ./bam
find ./ -name '*.sra' -exec mv ./sra/
# conversion and alignment
# define function
sratool_and_tophat() {
fastq-dump ./sra/SRR“$1”.sra -O ./fastq # fastq-dump
tophat -o ./tophat/“$1” -G mm10_genes.gtf mm10_Bowtie2Index. /fastq/SRR“$1”.fastq # tophat mv ./tophat/“$1”/accepted_hits.bam ./bam/“$1”.bam
}
export -f sratool_and_tophat
# execution
for i in {866991..867002}
do
sratool_and_tophat $ {i}
done
After execution, the aligned bam files will be all saved under the “bam” folder of current working directory. Please note that:
It is important to check the quality of raw data (FASTQ files) using tools such as FastQC [32]. If necessary, the reads may be further trimmed to eliminate low quality regions. This reads trimming step is non-trivial and often conducted in sequencing facilities, so it will not be discussed in this protocol. Please refer to [33] for a comprehensive review. Specifically for MeRIP-Seq, it is still an open question what is the best trimming strategy for it. Since the fragment length in current MeRIP-Seq protocol is only 100 bp, compared with 250-500 bp fragment length used in transcriptome or genome de novo assemble, the reads quality in MeRIP-Seq is usually not the bottleneck.
For the reads alignment, please use spliced aligner and feed the aligner with known junctions.
Please make sure to select the matching genome assembly build (mm10_Bowtie2Index) and gene annotation file (mm10_genes.gtf), which may be downloaded from Illumina iGenomes[34].
This is the most time-consuming step. The speed of sequence alignment may be greatly improved through parallel computation, which can be easily realized by letting command “sratool_and_tophat” start in a new thread.
Step 2 Conduct RNA methylation site detection and differential methylation site detection with exomePeak. Script 2 (R script) will compare the two experimental conditions between wild type (untreated) and FTO knockout condition (treated) to report the differential RNA methylation sites due to enzymatic regulation.
# Script 2 # R script # Install exomePeak from Bioconductor source(“http://bioconductor.org/biocLite.R”) biocLite(“exomePeak”) # Define parameters and load library library(“exomePeak”) setwd(“./bam”) IP_BAM=c(“866997.bam”,“866999.bam”,“867001.bam”) INPUT_BAM=c(“866998.bam”,“867000.bam”,“867002.bam”) TREATED_IP_BAM=c(“866991.bam”,“866993.bam”,“866995.bam”) TREATED_INPUT_BAM=c(“866992.bam”,“866994.bam”,“866996.bam”) # comparison exomepeak(GENOME=“mm10”, IP_BAM=IP_BAM, INPUT_BAM=INPUT_BAM, TREATED_IP_BAM=TREATED_IP_BAM, TREATED_INPUT_BAM=TREATED_INPUT_BAM, EXPERIMENT_NAME=“FTO”)
Please note that:
ExomePeak may automatically download the required gene annotation from UCSC genome data, which is needed for transcriptome methylation site identification (pick calling). Please make sure the genome assembly selected (“mm10”) is consistent with previous steps.
ExomePeak outputs the differential methylation sites in BED and XLS formats. Specifically a new directory “exomePeak_output” will be generated with consistently differentially methylated sites saved in “con_sig_diff_peak.xls”. For this exemplar dataset, there are 9 hypomethylation sites (diff.log2.fc<0) and 1597 hypermethylation sites (diff.log2.fc>0). The dominant hypermethylation (99.44%) after FTO knockout is consistent with the fact that FTO is a known m6A demethylase [35].
Step 3 Visualization of the detected RNA methylation sites and aligned bam files. For consistently differential methylated sites, exomePeak automatically generates a BED file “sig_diff_peak.bed” that can be visualized in IGV browser. To visualize the generated bam files, multi-reads (reads that can be mapped to multiple locations) and local anomaly are be removed using Samtools [36] before generating a viewable TDF format using igvtools (part of IGV). We use Script 3 (bash script) to implement this task.
# Script 3
# Bash script
# generate viewable format from bam file
samtools_and_igvtools() {
samtools view -F 516 -q 30 -b ./bam/“$1”.bam | samtools sort - ./bam/“$1”_inter # filter reads
samtools rmdup -s ./bam/“$1”_inter.bam ./bam/“$1”_filtered.bam # remove duplicated reads
igvtools count -z 5 -w 10 -e 0 ./bam/“$1”_filtered.bam ./tdf/“$1”.tdf mm10 # generate TDF
}
export -f samtools_and_igvtools
# execution
mkdir ./tdf
for i in {866991..867002}
do
samtools_and_igvtools $ {i}
done
Make sure the genome matches previous setting. The generated TDF files together with BED file “sig_diff_peak.bed” can then be visualized together using IGV [31] browser (Figure 4)
Figure 4. A differential methylation site shown in IGV browser.

Compared with the INPUT samples, Hps1 is slightly down-regulated under FTO Knockout condition; when comparing the IP samples, the absolute amount of methylated RNA fragments actually increases. Together, the figure shows an RNA m6A hypermethylation site on the 3’UTR of Hps1 after FTO Knockout.
Step 4 Motif finding using DREME and bedtools. Besides differential methylation sites, exomePeak also reports all the detected RNA methylation sites in BED format “diff_peak.bed”, based on which the motifs of RNA methylation sites can be detected. Specifically, the stranded methylated RNA fragments can be extracted from bedtools with script 4 (bash script).
# Script 4 # Bash script bedtools getfasta -s -fi mm10.fa -bed diff_peak.bed -split -fo methylated.fa
Make sure to use the whole genome fasta (mm10.fa) consistent with previous steps. The generated “methylated.fa” can then be uploaded to MEME-ChIP [37]: http://meme.nbcr.net/meme/cgi-bin/meme-chip.cgi for strand-specific (scan given strand only) motif discovery. The RRACH motif of m6A [38, 39] (or the consensus sequence of m6A sites) can be corrected identified (p-value 4.1e-200) from the peaks called by the exomePeak R/Bioconductor package, indicating the specificity of m6A-targeted antibody. The motif occurrence is reported centrally enriched (p-value 8.3e-3) by CentriMo [40] (Figure 5), indicates it is the consensus sequence targeted by RNA-binding domains (RBDs) of RNA methyltransferases, such as, METTL3 and METTL14 [41], in post-transcriptional regulation process.
Figure 5.

the identified RRACH motif of m6A and its distribution
Step 5 Functional analyses of FTO target genes using DAVID. The consistently differentially methylated sites and genes are shown in “con_sig_diff_peak.xls”. As FTO is a known m6A demethylase, its knockout should lead to the hypermethylation of its targets. We extracted the Entrez gene ID (4th column) associated with hypermethylation sites whose diff.log2.fc is larger than 0, and analyzed them using the DAVID functional annotation tool [24]: http://david.abcc.ncifcrf.gov/summary.jsp. Result indicates FTO targets are associated with synapse (p-value 4.77e-20), alternative splicing (p-value 4.72e-33), neuron projection (p-value 1.13e-16), and ion binding (p-value 1.19e-12), etc. (See Figure 6), consistent with previous studies [26].
Figure 6.

Functions that are enriched with FTO target genes
Summary
We proposed here a detailed protocol for differential analysis of RNA methylation MeRIP-Seq data set from two conditions to unveil the enzymatic regulation of RNA methylome by methyltransferases and demethylases, which is independent from transcriptional regulation. The inputs, tools and outputs are summarized in
Discussion
RNA epigenetics represents a novel mechanism that post-transcriptionally modifies RNA nucleotides, and embraces great potentials in physiological and pathological research. Different from the stable chemical structure of the DNA molecule, copy of RNA molecules are being synthesized and degraded, and thus the enzymatic regulation of RNA methylome must maintained in a more dynamic manner compared with DNA methylation, which is only reprogrammed during major event of the cell.
As one of the major progress in sequencing technique, MeRIP-Seq embraces enormous computational potentials that are yet addressed. The open source R-package “exomePeak” we developed is capable of detecting RNA methylation sites and performing RNA differential methylation. Mining this data, and especially by integrating additional layers from other omic data types, should enable us to address various important questions, such as: What are the functions of different post-transcriptional RNA modifications? Are different RNA modifications combined in a specific manner? As one of the fundamental mechanisms that exist in all three kingdoms of life with so many open questions, RNA epigenetics and the MeRIP-Seq techniques will certainly draw increasing attention in the next decade.
We provided here a detailed protocol for processing MeRIP-Seq data with exomePeak R/Bioconductor package [12]. Compared with previous protocol [2], this protocol for the first time addresses the comparison of RNA methylome between two experimental conditions, which is a key issue in RNA methylation research. exomePeak also improves in various other aspects, including reads alignment with spliced aligner, RNA methylation site detection with splicing-aware peak caller, strand-specific motif finding, and multiple biological replicates support, and also provided detailed data processing techniques, such as local anomaly and PCR artifacts removal using Samtools, etc. However, this protocol doesn't cover the technical details of generating MeRIP-Seq dataset, which has been previously address in [2].
Table 3.
Summary of the Protocol
| Category | Content |
|---|---|
| Inputs | MeRIP-Seq dataset [2] from 2 conditions with both the immunoprecipitated sample and input control sample, ideally with biological replicates. |
| Outputs | 1.The RNA methylation sites that are differentially methylated by RNA methyltransferases and demethylases between the 2 conditions, in BED, XLS and Rdata formats. |
| 2. The genes, biological functions and motifs that are associated with differential RNA methylation sites | |
| Tools | SRAtoolkit, Tophat [27], exomePeak [12], Bedtools [28], DREME [29], Samtools [30], IGV [31] and DAVID [24] |
| Database | Transcriptome information can be retrieved from UCSC with exomePeak package, or provided as a GTF file or TranscriptDb object. |
Acknowledgment
National Natural Science Foundation of China (No.61170134) to SZ; National Natural Science Foundation of China (No.81373469) to ZL; National Institutes of Health (NIH-NCIP30CA54174) to YC; National Science Foundation (CCF-0546345) to YH; Qatar National Research Fund (09-874-3-235) to YC and YH; National Natural Science Foundation of China (61201408) and the Fundamental Research Funds for the Central Universities (2014QNA84) to HL; Jiangsu Natural Science Foundation (SBK2014041258) to JM; China Postdoctoral Science Foundation (2012M511816) and the Fundamental Research Funds for the Central Universities (2014QNB47) to LZ; We thank computational support from the UTSA Computational System Biology Core, funded by the National Institute on Minority Health and Health Disparities (G12MD007591) from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The exomePeak R/Bioconductor package is freely available from Bioconductor: http://www.bioconductor.org/packages/release/bioc/html/exomePeak.html
The same percentage of methylated molecule under two experimental conditions may be approximated to the same fold enrichment in the IP sample compared with the input control sample for affinity based approaches, such as, ChIP-Seq, MeDIP-Seq and MeRIP-Seq.
References
- 1.He C. Grand challenge commentary: RNA epigenetics? Nat Chem Biol. 2010 Dec;6:863–5. doi: 10.1038/nchembio.482. [DOI] [PubMed] [Google Scholar]
- 2.Dominissini D, Moshitch-Moshkovitz S, Salmon-Divon M, Amariglio N, Rechavi G. Transcriptome-wide mapping of N(6)-methyladenosine by m(6)A-seq based on immunocapturing and massively parallel sequencing. Nat Protoc. 2013 Jan;8:176–89. doi: 10.1038/nprot.2012.148. [DOI] [PubMed] [Google Scholar]
- 3.Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, Sorek R, Rechavi G. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012 May 10;485:201–6. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 4.Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012 Jun 22;149:1635–46. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Machnicka MA, Milanowska K, Osman Oglou O, Purta E, Kurkowska M, Olchowik A, Januszewski W, Kalinowski S, Dunin-Horkawicz S, Rother KM, Helm M, Bujnicki JM, Grosjean H. MODOMICS: a database of RNA modification pathways--2013 update. Nucleic Acids Res. 2013 Jan;41:D262–7. doi: 10.1093/nar/gks1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kidder BL, Hu G, Zhao K. ChIP-Seq: technical considerations for obtaining high-quality data. Nat Immunol. 2011 Oct;12:918–22. doi: 10.1038/ni.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009 Oct;10:669–80. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garber M, Grabherr MG, Guttman M, Trapnell C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Meth. 2011;8:469–477. doi: 10.1038/nmeth.1613. [DOI] [PubMed] [Google Scholar]
- 9.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009 Jan;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bock C. Analysing and interpreting DNA methylation data. Nat Rev Genet. 2012 Oct;13:705–19. doi: 10.1038/nrg3273. [DOI] [PubMed] [Google Scholar]
- 11.Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet. 2010 Mar;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- 12.Meng J, Cui X, Rao MK, Chen Y, Huang Y. Exome-based analysis for RNA epigenome sequencing data. Bioinformatics. 2013 Jun;2915:1565–1567. doi: 10.1093/bioinformatics/btt171. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 2012;13:36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng JX, Liu T, Qin B, Zhang Y, Liu XS. Identifying ChIP-seq enrichment using MACS. Nature Protocols. 2012 Sep;7:1728–1740. doi: 10.1038/nprot.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kharchenko PV, Tolstorukov MY, Park PJ. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol. 2008 Dec;26:1351–9. doi: 10.1038/nbt.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levin JZ, Yassour M, Adiconis X, Nusbaum C, Thompson DA, Friedman N, Gnirke A, Regev A. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat Methods. 2010 Sep;7:709–15. doi: 10.1038/nmeth.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y, Zhao DY, Greenblatt JF, Zhang Z. RIPSeeker: a statistical package for identifying protein-associated transcripts from RIP-seq experiments. Nucleic Acids Research. 2013 Apr;411:e94. doi: 10.1093/nar/gkt142. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Micsinai M, Parisi F, Strino F, Asp P, Dynlacht BD, Kluger Y. Picking ChIP-seq peak detectors for analyzing chromatin modification experiments. Nucleic Acids Research. 2012;40:e70–e70. doi: 10.1093/nar/gks048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilbanks EG, Facciotti MT. Evaluation of algorithm performance in ChIP-seq peak detection. PLoS One. 2010;5:e11471. doi: 10.1371/journal.pone.0011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, Xue C, Marinov GK, Khatun J, Williams BA, Zaleski C, Rozowsky J, Roder M, Kokocinski F, Abdelhamid RF, Alioto T, Antoshechkin I, Baer MT, Bar NS, Batut P, Bell K, Bell I, Chakrabortty S, Chen X, Chrast J, Curado J, Derrien T, Drenkow J, Dumais E, Dumais J, Duttagupta R, Falconnet E, Fastuca M, Fejes-Toth K, Ferreira P, Foissac S, Fullwood MJ, Gao H, Gonzalez D, Gordon A, Gunawardena H, Howald C, Jha S, Johnson R, Kapranov P, King B, Kingswood C, Luo OJ, Park E, Persaud K, Preall JB, Ribeca P, Risk B, Robyr D, Sammeth M, Schaffer L, See LH, Shahab A, Skancke J, Suzuki AM, Takahashi H, Tilgner H, Trout D, Walters N, Wang H, Wrobel J, Yu Y, Ruan X, Hayashizaki Y, Harrow J, Gerstein M, Hubbard T, Reymond A, Antonarakis SE, Hannon G, Giddings MC, Ruan Y, Wold B, Carninci P, Guigo R, Gingeras TR. Landscape of transcription in human cells. Nature. 2012 Sep 6;489:101–8. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mizuno T, Chou MY, Inouye M. A unique mechanism regulating gene expression: translational inhibition by a complementary RNA transcript (micRNA) Proceedings of the National Academy of Sciences. 1984 Apr;811:1966–1970. doi: 10.1073/pnas.81.7.1966. 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carmel R, Jacobsen DW. Homocysteine in health and disease. Cambridge University Press; 2001. [Google Scholar]
- 24.Da Wei Huang BTS, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols. 2008;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 25.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005 Oct 25;102:15545–15550. doi: 10.1073/pnas.0506580102. 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hess ME, Hess S, Meyer KD, Verhagen LA, Koch L, Bronneke HS, Dietrich MO, Jordan SD, Saletore Y, Elemento O, Belgardt BF, Franz T, Horvath TL, Ruther U, Jaffrey SR, Kloppenburg P, Bruning JC. The fat mass and obesity associated gene (Fto) regulates activity of the dopaminergic midbrain circuitry. Nat Neurosci. 2013 Aug;16:1042–8. doi: 10.1038/nn.3449. [DOI] [PubMed] [Google Scholar]
- 27.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013 Apr 25;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bailey TL. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics. 2011;27:1653–1659. doi: 10.1093/bioinformatics/btr261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009 Aug 15;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011 Jan;29:24–6. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andrews S. FastQC: A quality control tool for high throughput sequence data. Reference Source. 2010 [Google Scholar]
- 33.Del Fabbro C, Scalabrin S, Morgante M, Giorgi FM. An Extensive Evaluation of Read Trimming Effects on Illumina NGS Data Analysis. PLoS One. 2013;8:e85024. doi: 10.1371/journal.pone.0085024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Illumina iGenomes. Available: https://support.illumina.com/sequencing/sequencing_software/igenome.ilmn.
- 35.Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang Y-G. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nature chemical biology. 2011;7:885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Machanick P, Bailey TL. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics. 2011 Jun 15;27:1696–1697. doi: 10.1093/bioinformatics/btr189. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schibler U, Kelley DE, Perry RP. Comparison of methylated sequences in messenger RNA and heterogeneous nuclear RNA from mouse L cells. J Mol Biol. 1977 Oct 5;115:695–714. doi: 10.1016/0022-2836(77)90110-3. [DOI] [PubMed] [Google Scholar]
- 39.Harper JE, Miceli SM, Roberts RJ, Manley JL. Sequence specificity of the human mRNA N6-adenosine methylase in vitro. Nucleic Acids Res. 1990 Oct 11;18:5735–41. doi: 10.1093/nar/18.19.5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bailey TL, Machanick P. Inferring direct DNA binding from ChIP-seq. Nucleic Acids Research. 2012 May 18; doi: 10.1093/nar/gks433. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, Dai Q, Chen W, He C. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–95. doi: 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]