

Abstract
Three types of methyltransferases (MTases) generate 5-methylpyrimidine in nucleic acids, forming m5U in RNA, m5C in RNA and m5C in DNA. The DNA:m5C MTases have been extensively studied by crystallographic, biophysical, biochemical and computational methods. On the other hand, the sequence–structure–function relationships of RNA:m5C MTases remain obscure, as do the potential evolutionary relationships between the three types of 5-methylpyrimidine-generating enzymes. Sequence analyses and homology modeling of the yeast tRNA:m5C MTase Trm4p (also called Ncl1p) provided a structural and evolutionary platform for identification of catalytic residues and modeling of the architecture of the RNA:m5C MTase active site. The analysis led to the identification of two invariant residues that are important for Trm4p activity in addition to the conserved Cys residues in motif IV and motif VI that were previously found to be critical. The newly identified residues include a Lys residue in motif I and an Asp in motif IV. A conserved Gln found in motif X was found to be dispensable for MTase activity. Locations of essential residues in the model of Trm4p are in very good agreement with the X-ray structure of an RNA:m5C MTase homolog PH1374. Theoretical and experimental analyses revealed that RNA:m5C MTases share a number of features with either RNA:m5U MTases or DNA:m5C MTases, which suggested a tentative phylogenetic model of relationships between these three classes of 5-methylpyrimidine MTases. We infer that RNA:m5C MTases evolved from RNA:m5U MTases by acquiring an additional Cys residue in motif IV, which was adapted to function as the nucleophilic catalyst only later in DNA:m5C MTases, accompanied by loss of the original Cys from motif VI, transfer of a conserved carboxylate from motif IV to motif VI and sequence permutation.
INTRODUCTION
Methylation of nucleic acids is catalyzed by a large and diverse class of S-adenosyl-l-methionine (AdoMet)-dependent methyltransferases. The enzymes characterized to date include members of two unrelated superfamilies: ‘classical’ Rossmann-fold-like (1,2) and SPOUT (3). The relatively small SPOUT superfamily includes only a few characterized RNA-specific enzymes with 2′-O-ribose or guanosine-N1 modification specificity that will not be discussed further in this article. The Rossmann-fold superfamily (hereafter referred to as ‘MTases’) groups together enzymes acting on RNA, DNA, proteins, lipids and various small molecules. MTases have a catalytic domain with a common structural core and AdoMet-binding site. Shared motifs are usually detectable at the sequence level, but in some cases motifs that have diverged beyond recognition by sequence comparison can be identified by structural comparisons. The most common nomenclature involves motifs I–X, initially assigned to MTases that generate 5-methylcytosine (m5C) in DNA and which correspond to the key structural and functional elements associated with the cofactor-binding site (I–III), the catalytic pocket (X, IV, VI and VIII) and motifs implicated in preservation of the common fold (V and VII) (2,4,5). However, DNA and RNA MTases exhibit sequence permutation, resulting in a variable linear order of the conserved motifs (6,7).
DNA and RNA differ with respect to the number of observed modifications. Only three modified bases are typically found in DNA: m5C, N4-methylcytosine (m4C) and N6-methyladenine (m6A). Methylated nitrogens (m6A and m4C) occur primarily in Prokaryota, but m5C is found in organisms from all three Domains. Crystal structures have been determined for DNA MTases that generate each of these common modifications and a plethora of DNA-specific MTases have been cloned and characterized biochemically (reviewed in 8). In sharp contrast to the well-studied DNA MTase families, RNA MTases remain poorly characterized from the perspective of structure–function relationships. To date, crystal structures have been solved for several known or putative RNA MTases (reviewed in 9,10), but typically without a substrate, which limits the ability to correlate reaction mechanisms with active site architectures.
The RNA:m5C MTases are a fascinating group of RNA modification enzymes, for which some useful information has been obtained by separate structural, biochemical and evolutionary studies. Following cloning of the first representative, 16S rRNA:m5C967 MTase RsmB (previously called Sun or Fmu) from Escherichia coli by two groups (11,12), homologous sequences were identified and additional paralogous RNA:m5C MTase subfamilies were predicted (13). Among these putative m5C MTases, two eukaryotic proteins were characterized on the sequence–function level: a multisite-specific tRNA:m5C MTase Trm4p (14,15) and apparent rRNA MTase Nop2p (15–17). Despite the apparent size and wide distribution of this protein family, RsmB (Fmu) and Trm4p remain the only proteins with biochemically confirmed RNA m5C methyltransferase activity.
A particularly interesting aspect of RNA:m5C MTases is their relationship to two distinct classes of enzymes that generate 5-methylpyrimidine in nucleic acids, namely RNA:m5U MTases and DNA:m5C MTases. The enzymatic mechanism of DNA:m5C methylation has been extensively studied by crystallography, mutagenesis, biophysical methods and molecular dynamics simulations (18–25). Briefly, it involves an attack by the thiol of an invariant Cys residue from motif IV on the 6 position of the cytosine base to form a covalent complex, thereby activating the 5 position for methyl group transfer, which is followed by deprotonation and β-elimination to restore the free enzyme and release the methylated product (reviewed in 8,26). For RNA:m5U MTases an analogous mechanism has been proposed, albeit involving an unrelated Cys from motif VI (27,28).
Remarkably, RNA:m5C MTases possess counterparts of both DNA:m5C-like and RNA:m5U-like cysteine residues (13,15). Mutational analysis has suggested that in RNA:m5C methylation the RNA:m5U-like thiol acts in a classical fashion by forming a covalent link to carbon 6 of the pyrimidine base (29), while the DNA:m5C-like thiol assists breakdown of the covalent adduct (15). However, no other residues in the active site of RNA:m5C MTases have been studied. Therefore, the details of the catalytic mechanism remain obscure, as do the potential evolutionary relationships between the three types of 5-methylpyrimidine MTases. These enzymes could be the result of divergent evolution from a common ancestor, the product of progressive changes where one of the three mechanisms is an intermediate between the other two or the result of convergence where the MTase fold was independently adapted to perform three chemically similar reactions.
During the course of the work two crystal structures were solved for members of the RNA:m5C MTase family. PH1374 is a putative RNA:m5C MTase of unknown specificity from Pyrococcus horikoshii. A partial PH1374 structure is available from the Protein Data Bank (PDB) under accession number 1ixk, but as of January 2004, the work has not been published. The second structure was reported for RsmB from E.coli (30). It has been deposited in the PDB under accession numbers 1SQG and 1SQF, but not released prior to publication of this work. Neither of these structures was solved in the presence of a RNA substrate. Moreover, the limited functional analyses reported for RsmB did not include mutagenesis of the presumed active site, hence our knowledge of the residues required for catalysis of the RNA:m5C methylation reaction remains incomplete.
To learn more about the mechanism of RNA:m5C methylation and as an aid to resolution of the relationships between 5-methylpyrimidine MTases we have carried out extensive sequence analysis. The sequence comparison was followed by homology modeling of the yeast enzyme Trm4p, which has led to the identification of additional potential catalytic residues. Site-directed mutagenesis of three amino acids was carried out and the mutants were studied in vitro. The structural model was also used to rationalize the effect of site-directed mutants of Trm4p obtained previously (15). The results of the sequence analysis suggest that these findings can be extrapolated to other members of the RNA:m5C MTase family. Finally, the potential evolutionary relationships between different types of 5-methylpyrimidine MTases are discussed in the light of the available structural and biochemical data.
MATERIALS AND METHODS
Recombinant RNasin was obtained from Promega and the Talon affinity resin was purchased from Clontech. BL21-Codon Plus(DE3)-RIL cells and the QuikChange mutagenesis kits are products of Stratagene. Calf liver tRNA was a product of Boehringer Mannheim. Oligonucleotides were synthesized by Integrated DNA Technologies (www.idtdna.com). DNA sequence analysis was carried out by the Biochemistry Biotechnology Facility at the Indiana University School of Medicine, Indianapolis IN.
Cloning and site-directed mutagenesis of the NCL1/TRM4 gene
Amplification of the NCL1/TRM4 gene from yeast genomic DNA, cloning of the amplified product to generate pGEMNCL1 and transfer of the cloned gene into a modified version of the pET28b expression plasmid were previously described (15). Site-directed mutations were generated by the use of Stratagene’s QuikChange kit that utilizes two complementary oligonucleotides for each mutation. Table 1 shows the sequence of the positive strand oligonucleotide used for each mutation, the restriction site generated by the change and the pair of restriction sites used to move the altered region from the modified pGEMNCL1 plasmid to the pET28-based expression plasmid. The BglII–HindIII region of the D257A expression construct was sequenced in both directions using the primers previously reported for that purpose (15), but the SacI–BglII regions of the Q150A and K179M expression constructs were sequenced using a T7 promoter primer that initiates within the expression plasmid in the forward direction and the NCL1SEQR2 primer (GAAGAATTGGGCG TCATGGTT) in the reverse direction.
Table 1. Site-directed mutagenesis informaton.
| Mutant | Coding strand oligonucleotide (new restriction site underlined) | Restriction site formed | Sites used to move mutant fragment to expression plasmid |
|---|---|---|---|
| Q150A |
CCGTTGGTAATATCTCGAGAGCGGAAGCCGTTTCAATGATTCC |
XhoI |
SacI–BglII |
| D257A |
GACAGAATCCTGTGCGCAGTTCCATGTTCTGGTGATGG |
FspI |
BglII–HindIII |
| K179M | GTGTGCTGCTCCTGGATCCATGACTGCTCAATTAATCGAAGC | BamHI | SacI–BglII |
Expression and purification of Trm4p
BL21-Codon Plus(DE3)-RIL cells carrying TRM4 plasmids were grown in Luria Broth to an OD600 of 0.4, then Trm4p expression was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside for 2 h. The cell pellet from each 50 ml culture was suspended in 3 ml of extraction buffer (see below) containing 0.75 mg/ml lysozyme for 30 min at 24°C. Samples were then ice chilled, shaken with 200–300 µm glass beads for 30 s using a Mini Bead Beater at 3800 r.p.m. and centrifuged at 16 000 g for 10 min at 8°C. Trm4p was purified from the supernatant with 1 ml of Talon affinity resin using a combined batch–column procedure. Affinity resin equilibrated with extraction buffer (50 mM sodium phosphate pH 8, 300 mM NaCl and 5 mM benzamidine) was mixed for 20 min with the extract from one or two 50 ml cultures diluted to a volume of 10 ml, then sedimented by centrifugation at 700 g for 5 min. The resin was washed twice with 10 bed vol of wash buffer which was identical to extraction buffer except that the pH was 7.0. Washed resin was transferred to a minicolumn and eluted with pH 7 buffer containing 50 mM sodium phosphate, 300 mM NaCl and 150 mM imidazole. Protein concentrations were determined with the Coomassie Protein Assay reagent from Pierce using cytochrome C as the standard. Enzyme preparations were stored in Teflon capped glass vials that were flushed with N2 and the assays were done within 3 days of enzyme preparation. For samples to be phenol extracted, the concentration of NaCl in the purification buffers was reduced to 150 mM.
Enzyme assay
RNA substrate was extracted from a yeast Trm4 deletion strain (YPW17) (31) and fractionated essentially by the procedure of Knapp, but was not radiolabeled (32). RNA was phenol extracted twice from cells suspended in 1% SDS, 2 mM EDTA and 0.1 M sodium acetate at pH 5. The combined aqueous phases were extracted with chloroform:isoamyl alcohol (24:1) followed by RNA precipitation with ethanol. Total RNA was dissolved in TE (10 mM Tris, 1 mM EDTA pH 7.4) containing 300 mM NaCl and applied to DEAE–cellulose (Whatman DE23). Small RNA molecules were eluted with TE containing 1 M NaCl, alcohol precipitated and then dissolved in DEPC-treated water and stored at –20°C.
Assay mixtures contained either 0.5 or 1 µg affinity-purified Trm4p, 3 µg DEAE-purified yeast RNA and 0.20 µM [methyl-3H]AdoMet (1.9 µCi) in a total reaction volume of 120 µl. The reaction also contained 100 mM MOPS at pH 7.8, 100 mM ammonium acetate, 1 mM magnesium acetate, 5 mM dl-dithiothreitol and 6 U RNasin. Reactions were incubated at 30°C and 25 µl of the mixture was placed into 1 ml of 5% trichloroacetic acid (TCA) at each time point. Each sample was vacuum filtered through a 24 mm Whatman GF/C filter which was then washed five times with 2 ml of 5% TCA and twice with 2.5 ml of 95% ethanol to remove unincorporated radioactivity. The amount of radiolabel incorporated into RNA was determined by placing the filter in 5.5 ml of scintillation fluid and counting in a Beckman LS 5000CE scintillation counter.
Protein sequence analyses
PSI-BLAST (33) was used to search the non-redundant version of current sequence databases and the publicly available unfinished eukaryotic and prokaryotic genomic sequence databases at NCBI (Bethesda, MD) (http://www.ncbi.nlm.nih.gov) and IIMCB (Warsaw, Poland) (http://genesilico.pl/~blast). The EST (expressed sequence tag), STS (sequence-tagged site), HTG (high throughput genomic) and GSS (genome survey sequence) divisions of GenBank were searched with TBLASTN. Full-length sequences were assembled from fragments and the predicted splicing sites were verified in reciprocal BLAST searches against the local database comprising validated sequences of MTases.
All sequences were subsequently realigned using the CLUSTAL X program (34) to the degapped profiles obtained from the multiple sequence alignments reported by PSI-BLAST. The evolutionary inference was carried out for the conserved blocks of the multiple sequence alignment (corresponding to the common MTase domain) with the MEGA package according to the neighbor-joining method (35). The sampling variance of the distance values was estimated from 1000 bootstrap resamplings of the alignment columns.
Protein structure prediction
Protein structure prediction was carried out via the GeneSilico Meta Server gateway (http://genesilico.pl/meta/) (36), using publicly available online services for fold recognition [SAM-T02 (37), 3DPSSM (38), mGENTHREADER (39), FFAS (40), and FUGUE (41)] and secondary structure prediction [PSIPRED (42), SSPRO (43), and PROF (44)]. Disordered regions were predicted with PONDR (45). Homology modeling was carried out according to the ‘FRankenstein’s monster’ approach (46). Briefly, the fold recognition alignments between the Trm4p sequence and various proteins with known structures obtained from the Meta Server were converted into a set of homology models using MODELLER (47). The sequence–structure fit in these models was evaluated using VERIFY3D (48) (window 5 amino acids). Polypeptide fragments with scores <0.1 were deleted and the remaining parts were merged to produce a ‘consensus’ model. Final adjustments have been made using fragments of known MTase structures. The consensus model was energy minimized to relieve steric clashes. The coordinates of the final model are freely available online at ftp://genesilico.pl/iamb/models/Trm4/.
RESULTS
Phylogenetic analysis of the RNA:m5C MTase family
The amino acid sequence of Saccharomyces cerevisiae Trm4p was used as a query in PSI-BLAST (33) searches of the NRDB database to identify homologous proteins (see Materials and Methods). Since we were interested in the identification of closely related enzymes with similar functions and not all MTases, a stringent cut-off of expectation (e) value of 10–15 was used to avoid expansion of the profile onto the whole AdoMet-dependent MTase superfamily. After convergence, the search was continued and the sequences with lower scores were arbitrarily included in subsequent iterations if they fulfilled two criteria: (i) conservation of two cysteine residues typical for RNA:m5C MTases (49); (ii) the reciprocal BLAST searches initiated with these sequences reported RNA:m5C MTases with the highest, significant scores. Preliminary phylogenetic analysis suggested a few groups, from very large to very small ones (typically bacterial and eukaryotic, respectively, reflecting the number of complete genomes available for the organisms from these two domains). Members of the under-represented eukaryotic lineages have been used as queries in TBLASTN searches of other databases (see Materials and Methods) to identify more putative RNA:m5C MTases for a robust phylogenetic analysis. Altogether, we identified 260 homologs of RNA:m5C MTases, compared to 52 sequences reported previously (13). All sequences obtained were pooled together and aligned with the PCMA program, followed by manual adjustments of the multiple sequence alignment to correct misplaced gaps. In the final multiple sequence alignment a common domain was identified (see Fig. 1 for the most conserved fragments) and used to generate a phylogenetic tree of the RNA:m5C MTase family (Fig. 2).
Figure 1.

Multiple sequence alignment of regions forming the active site in the RNA:m5C MTase family. Only representative members of the major subfamilies (see Fig. 2) are shown. The full alignment is available from the authors upon request. Conserved motifs are labeled according to the nomenclature described originally for DNA:m5C MTases (56). Identical residues are highlighted in black, conserved residues are highlighted in gray. Residues studied by site-directed mutagenesis in this work and earlier (15,17) are indicated by * and +, respectively.
Figure 2.

Unrooted phylogenetic tree of the RNA:m5C MTase family. Subfamilies are shown in different colors (Eukaryota, blue and cyan; Archaea, red; Bacteria, green) and labeled with the names of representative members. In eukaryotic lineages, the yeast members are shown in blue and human members in black. Orthologous groups are indicated by arcs.
Our analysis revealed more members of the RNA:m5C MTase family from higher eukaryotes than reported previously (13). Previous analyses identified only three eukaryotic lineages, corresponding to the yeast proteins Trm4p, Nop2p and Ynl022c. The alignment in Figure 1 shows the representative members of all major lineages of the RNA:m5C MTase family identified in this work, with special emphasis on seven human members. In conjunction with the phylogenetic tree (Fig. 2), we predict that new human RNA:m5C MTase candidates FLJ22609 and MGC22960 are most closely related to the Trm4p lineage (including the human Trm4p ortholog FLJ20303). We confirm that the human protein Nol1p is orthologous to the yeast protein Nop2p. However, our results suggest that WBSCR20, a predicted Nol1p ortholog that is one of several genes encoded within a chromosomal region implicated in the pathogenesis of Williams–Beuren syndrome (WBS) (50), is orthologous to Ynl022c rather than to Nol1p. We have also identified a lineage paralogous to Ynl022c/WBSCR20, which includes a human protein FLJ14001 and a few other proteins exclusively from metazoans. The topology of the tree reveals that the Ynl022c/WBSCR20/FLJ14001 lineage clusters together with the uncharacterized archaeal lineage previously denoted ‘subfamily IV’ (13) and with the bacterial RsmB lineage. That these lineages are orthologous is supported by the fact that the bacterial and archaeal proteins share the N-terminally fused NusB domain. Finally, our analysis revealed that the uncharacterized human protein FLJ23743 (NopD1p) is an ortholog of the archaeal lineage earlier denoted as ‘subfamily VI’ (13), since both these lineages share the unique feature of a PUA domain (51) inserted between motifs N1 and X.
The results of our phylogenetic analysis provide a convenient platform for experimental characterization of the putative human m5C MTases. For instance, human proteins FLJ22609 and MGC22960 with no orthologs in yeast could share a general specificity for tRNA with their close paralog Trm4p and be responsible for generation of higher Eukaryote-specific m5C modifications at positions 50 and 72 in tRNA. Likewise, that Ynl022c and WBSCR20 are predicted to be orthologous to RsmB suggests that they share its specificity for rRNA methylation. Interestingly, an RNA:m5C MTase homolog FLJ14001 seems to lack a few important residues of the active site identified in this work, which suggests that it may be inactive. Finally, the clustering of Nol1p and FLJ23743 with their sister archaeal lineages suggests that these subfamilies of enzymes may share specificity for an RNA:m5C modification present in Eukaryota and Archaea but absent from Bacteria.
Structure prediction of Trm4p
In order to provide a structural platform for sequence–function studies of Trm4p, we carried out computational structure modeling (see the Materials and Methods). The protein fold recognition analysis revealed that the Trm4p sequence is compatible with the Rossmann-like MTase fold (9). All fold recognition algorithms reported various MTase structures with very high scores, in particular the only member of the RNA:m5C MTase family available in the PDB (putative archaeal MTase PH1374) (data not shown). For over 50 alternative sequence–structure alignments we have generated preliminary models and used their best scoring fragments to generate a hybrid model (46) (see also Materials and Methods for details). The N- and C-terminal extensions with no counterparts in the template structures were predicted to be at least partially disordered and therefore were not included in the final model. Only the structural core comprising residues 22–411 was homology modeled. The final model passed the quality test implemented in VERIFY3D (48). The model of the Trm4p–AdoMet–cytidine complex was constructed by transferring the ligand from the superimposed coordinates of the DNA:m5C MTase M.HhaI crystal structure (see Supplementary Material for images of the Trm4p model superimposed on the crystal structures of M.HhaI and PH1374).
Structure-based mutagenesis
Examination of the RNA m5C MTase model and the multiple sequence alignment revealed two highly conserved amino acids within the active site (Figs 1 and 3). These correspond to Trm4p residues Gln150 and Asp257, which could aid catalysis in addition to the pair of cysteines already implicated in catalysis (15). Gln150 is positioned behind the cytosine base relative to the conserved cysteines (Cys260 and Cys310) and is adjacent to the AdoMet cofactor (Fig. 3). Thus, Gln150 could help form substrate-binding sites or it could have a more direct catalytic role. We found that mutation of Gln150 to Ala had little or no effect on the initial enzymatic activity of the expressed enzyme (Fig. 4A), but the activity of the mutant enzyme declines more rapidly than that of wild-type Trm4p upon storage (data not shown). Therefore, the invariant Gln150 is not essential for catalysis, but its conversion to Ala appears to slightly alter Trm4p. Increased sensitivity to oxidation might result from a reduced affinity for tRNA (see below), which could make the active site cysteines more accessible to oxidizing agents.
Figure 3.

Homology model of Trm4p. The protein backbone is shown as a cartoon (sheets as blue arrows, helices as red spirals). Docked ligands (AdoMet and the target cytosine) and amino acid residues studied by mutagenesis are shown in the wireframe representation and labeled.
Figure 4.

Methyltransferase activity of altered Trm4p. Each panel compares the activity of wild-type Trm4p (open squares), a mutant form of Trm4 (solid triangles) and an equal volume of extract from cells not expressing Trm4p (open circles). (A) The activity of 0.5 µg Q150A-Trm4p and Trm4p are compared to a control extract containing 0.16 µg protein. (B) The activity of 1.0 µg D257A-Trm4p and Trm4p are compared to a control extract containing 0.27 µg protein. (C) The activity of 1.0 µg K179M-Trm4p and Trm4p are compared to a control extract containing 0.18 µg protein. The incorporation of radioactivity required the addition of RNA (data not shown). Plotted values are averages of triplicate assays.
In the model, Asp257 is located adjacent to the cytosine base and near the AdoMet cofactor. Conversion of Asp257 to Ala inactivates Trm4p, with no observed methyl transfer activity above the background level of control extract prepared from cells not expressing Trm4p (Fig. 4B). The levels of 3H incorporation into RNA indicate that the D257A mutant has less than 1% of the activity of the wild-type protein.
The presence of a potentially charged aspartic acid residue within the active site drew our attention to Lys179 of motif I, because the side chain of this conserved residue extends into the active site with the terminal amino group residing near Asp257. An isosteric (shape and size conservative) mutation of Lys179 to Met was made to eliminate the amino group. Like the Asp257 mutant, the K179M mutant is completely inactive (Fig. 4C). The strong effect of the D257A and K179M mutations combined with their possible juxtaposition in the active site indicates that an interaction between these residues may be critical for catalysis or proper conformation of the active site region.
In previous studies, ethidium bromide staining of SDS–PAGE gels containing Trm4 revealed that small RNA molecules co-purify with the wild-type protein, although they are not covalently attached as observed with a Cys260 mutant (15). To determine if the Trm4p mutants developed here retain the ability to bind RNA, affinity-purified enzyme preparations were phenol extracted and the isolated material was resolved in denaturing gels (Fig. 5). Bacterial RNAs the size of tRNAs co-purify with all three mutant forms of Trm4p reported above, although it is not known if the mutants retain identical tRNA affinity. In fact, a reduced amount of RNA seems to be associated with the Q150A mutant despite its normal level of activity. This finding suggests essentially normal folding for all of the mutants, although minor structural perturbations that could influence docking of the target base into the active site that could significantly affect enzyme activity cannot be excluded.
Figure 5.
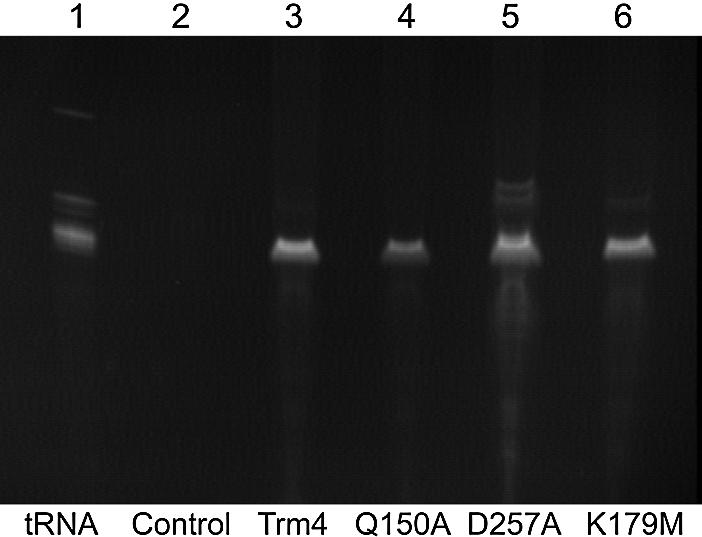
Evidence for tRNA binding by Trm4 mutants. Talon affinity purified Trm4p preparations and the affinity eluate from cells not expressing Trm4 were assayed for protein content then phenol extracted and ethanol precipitated. Isolated material was run on a 10% acrylamide gel containing 7 M urea and stained with ethidium bromide. The sample volumes were proportional to original concentration of expressed Trm4p. For the non-expression control (lane 2) the volume loaded equaled that of the largest sample containing expressed protein. Lane 1 contains 1 µg commercial calf liver tRNA preparation.
The locations of the three residues mutated in this study are in very good agreement with the X-ray structure of a RNA:m5C MTase homolog PH1374. They are all relatively near the AdoMet-binding site in our model and in the Fmu (RsmB) structure (30). Lys260 of Fmu corresponds to Trm4p Lys179 and the former has been proposed to be involved in cofactor binding, but to date we have not been able to obtain evidence as to the ability or inability of the D257A or K179M Trm4p mutants to bind the AdoMet cofactor (via electrostatic interactions and/or hydrogen bonds). Therefore, it cannot be excluded that the enzymatic defects in the Asp257 and Lys179 mutants are due to compromised cofactor binding, but the severity of the activity loss would also be consistent with a faulty catalytic mechanism.
DISCUSSION
The role of conserved residues in the active site of RNA:m5C MTases
Gln150 is located in motif X and superimposes well on the chemically similar invariant Asn residue of DNA:m5C MTases (Asn304 in M.HhaI; data not shown). The crystallographic analysis has suggested that Asn304 of M.HhaI is involved in binding of the target cysteine, flipped into the enzyme active site (52). The role of this residue, however, has not been studied by mutagenesis. Our analysis has shown that Gln150 of RNA:m5C MTase Trm4p is not essential for enzyme activity. Interestingly, this otherwise conserved Gln is substituted by hydrophobic residues in the two newly identified human paralogs of Trm4 (Fig. 4). It will be interesting to determine if the homologous Asn residue is required for catalysis in DNA:m5C MTases.
Lys179 is located in motif I, which is traditionally regarded as a part of the AdoMet-binding site rather than a part of the active site. However, it maps not to the loop shown to bind AdoMet in many crystal structures (reviewed in 2), but to the solvent-exposed face of the subsequent α-helix. According to the crystal structure of PH1374 and the homology modeled structure of Trm4p, the side chain of Lys179 points towards the active site rather than the cofactor-binding site. It will be of interest to study the role of this residue using high resolution biophysical and/or structural methods, as it may be the first catalytic residue of MTases located in such an unusual position. Alternatively, Lys179 may be an atypical component of the cofactor-binding site or be involved in substrate recognition and binding. While the severe loss of activity associated with the K179M mutation seems more characteristic of the loss of a catalytic residue than a ligand-binding residue, its position within the active site does not make these two potential roles mutually exclusive.
Asp257 is located in motif IV, which in RNA:m5C MTases includes the invariant, essential Cys260 residue and typically assumes the ‘DAPC’ pattern. Residues 1 and 4 of motif IV often contribute to the catalytic activity and reaction specificity of various nucleic acid MTase families owing to key interactions of the respective functional groups with the targeted base (reviewed in 2). Specifically, Asp257 of Trm4p is homologous to the Asp residue conserved in many ‘amino-MTases’, i.e. RNA and DNA MTases that methylate exocyclic amino groups in adenine (DPPY in members of the α-class of DNA:m6A MTases or DPPW in mRNA:m6A MTases), guanine (DPPF in rRNA:m2G MTases RsmC/RsmD) and cytosine (DPPH in DNA:m4C MTase M.NgoMXV) (reviewed in 53). It is noteworthy that the consensus sequence signature of amino-MTases is rather loosely defined as (S/D/N)-(I/P)-P-(Y/F/W/H), but it is neither specific nor unique to these enzymes, as there are known cases of amino-MTases with a different motif IV (54) and MTases acting on proteins rather than DNA, which exhibit the ‘NPPY’ tetrapeptide in motif IV (9). A more general (D/N/S)-X-P-X pattern (where X is any amino acid) is shared by the majority of nucleic acid MTases as well as some MTases acting on other substrates (2) and may be regarded as an ancestral form of one of the largest sub-groups of MTases of the class I fold (51; J.M.Bujnicki, E.V.Koonin and Aravind, manuscript in preparation). An Asp residue is also conserved in the position 1 of motif IV in RNA:m5U MTases, which typically assume the ‘DPPR’ form (sometimes substituted with ‘NPPR’) (49; our unpublished observations). On the other hand, all known DNA:m5C MTases possess the side chain-less Gly residue rather than Asp in position 1 of motif IV (typically GPPC) (55). Due to its conservation, structural location and functional importance, we hypothesize that Asp257 aids positioning of the cytosine base in the active site and may be the RNA MTase functional equivalent of the Glu found in motif VI (typically ENV) of the DNA m5C MTases (Glu119 in M.HhaI).
Previous mutation of the motif IV Asp residue in Nop2p (Asp421), a putative rRNA m5C MTase from yeast, failed to reveal any significance for the residue. The altered Nop2p supported yeast viability (16) but the effect of the mutation on enzyme activity was not determined due to lack of an in vitro assay for Nop2p (15,17). Although Nop2p is essential in yeast, later studies strongly indicate that RNA methylation is not its critical function, since a mutant with both active site cysteines replaced by alanine also supports yeast viability (15). Based on the Trm4p Asp257 mutant, we predict that the Nop2p Asp421 mutant will be found to be enzymatically inactive.
The essential residues Lys in motif I and Asp in motif IV are nearly invariant in the RNA:m5C MTase family and are missing only in the human protein FLJ14001 (Fig. 1) and its orthologs from other higher animals (data not shown). This suggests that the MTase activity of FLJ14001 may be compromised, even though it possesses the two catalytic Cys residues.
Cytosine methylation in DNA requires a single active site Cys, but biochemical experiments with Trm4p and Nop2p indicated that RNA m5C MTases utilize two active site Cys residues in the methylation of RNA (15). This concept is supported by the structural modeling reported here and by the structures of PH1374 and RsmB (30), all of which place the conserved Cys residues adjacent to each other within the active site. The Cys residue from motif VI of RsmB (corresponding to Trm4 Cys310) was previously shown to be required for the formation of a covalent intermediate (29). In DNA m5C MTases, the covalent intermediate is formed by the Cys found in motif IV. Mutation of the motif IV Cys to Ala or Ser in Nop2p causes yeast lethality (17), but similar experiments with Trm4p revealed that mutation of the motif IV Cys to Ala prevented release of RNA from Trm4p (15). This observation combined with the active site location of Cys260 suggests that the Cys in motif IV of RNA m5C MTases acts as a base to aid the proton extraction and β-elimination required for RNA release. Therefore, the conserved motif IV Cys residues in DNA and RNA m5C MTases are utilized in different ways during catalysis.
Relationships between 5-methylpyrimidine MTases
RNA:m5U MTases and RNA:m5C MTases share three characteristic features: (i) they act on RNA; (ii) they utilize the invariant Cys residue in motif VI as the catalytic nucleophile; (iii) they possess a conserved Asp residue in motif IV (like many other MTases acting on RNA) (51). RNA:m5C MTases and DNA:m5C MTases also share three characteristic features: (i) they methylate cytosine; (ii) they possess an invariant Cys residue in motif IV; (iii) they possess a conserved amide side chain (Gln or Asn, respectively) in motif X, which could make contact with the target base. RNA:m5U MTases and DNA:m5C MTases share none of the above-mentioned features: they act on different nucleic acids, methylate different bases, their invariant cysteine residues are located in different motifs and they do not share any other particularly similar residues known or predicted to interact with the target base (Table 2).
Table 2. Similarities and differences between nucleic acid NH2-MTases and three pyrimidine-C5 MTase families.
| MTase family | Substrate | Asp in motif IV? | Glu in motif VI? | Nucleophile Cys? | Cys in motif VI? | Cys in motif IV? | Position of motif X |
|---|---|---|---|---|---|---|---|
| NH2-MTases |
RNA/DNA |
Yes |
No |
No |
No |
No |
variable |
| RNA:m5U |
RNA |
Yes |
No |
Yes |
Yes |
No |
N-terminal |
| RNA:m5C |
RNA |
Yes |
No |
Yes |
Yes |
Yes |
N-terminal |
| DNA:m5C | DNA | No | Yes | Yes | No | Yes | C-terminal |
Combination of the biochemical data with bioinformatic analyses (including homology modeling and comparison of models with experimentally solved structures) and phylogenetic studies (sequence analysis and identification of characters shared between distinct enzyme families) allows us to infer a possible scenario of evolution for the large class of pyrimidine-C5 MTases. The ‘mosaic’ similarity of RNA:m5C MTases to RNA:m5U MTases and DNA:m5C MTases (sharing features with both of the latter families) strongly suggests that RNA:m5C MTases may be the evolutionary intermediate. If this were the case, the question arises as to the direction of evolution, i.e. whether RNA:m5U MTases or DNA:m5C MTases are the best candidate for the ‘ancestral’ form and which of them may be regarded as the latest evolutionary development. A few observations support the scenario in which RNA:m5U MTases may be ancestral: (i) motif IV of RNA:m5U MTases conforms to the ancestral ‘DXPX’ pattern shared by the majority of nucleic acid MTases, while motif IV of DNA:m5C MTases strongly deviates from this pattern and may be considered as derived; (ii) in searches of sequence databases, RNA:m5U MTases show significant similarity to many other nucleic acid MTase families, including amino-MTases, while DNA:m5C MTases appear rather ‘isolated’, which is most likely due to strong divergence and a relatively large phylogenetic distance from the common ancestor; (iii) RNA:m5U MTases exhibit the most typical (ancestral) order of sequence motifs shared by all MTases acting on substrates other than nucleic acids (from the N-terminus, X, followed by I–VIII), while most of DNA:m5C MTases exhibit a unique circular permutation, in which motif X is transferred to the C-terminus (6), which again may be considered as derived rather than ancestral. Moreover, it is believed that RNA is more ancient than DNA, thus RNA:m5U MTases (as well as RNA:m5C MTases) could have evolved in the hypothetical ‘ribonucleoprotein world’, with DNA:m5C MTases having emerged once DNA took over as the principal carrier of cellular genetic information. The tentative scenario of relationships between the three pyrimidine C-5 MTases is shown in Figure 6. Alternative scenarios, including origin of the RNA MTases from DNA MTases or convergent evolution (independent origin of the major pyrimidine C-5 MTase families) cannot be completely excluded, but we find them rather unlikely.
Figure 6.

Evolutionary model of relationships between pyrimidine-C5 MTases. The model of major functional innovations in the evolution of pyrimidine-C5 methylation. Key residues in motifs IV and VI are indicated. Thin arrows indicate the sites of covalent bond formation between the Cys residues and the C6 atom of the target base or between the CH3 group of AdoMet and the C5 atom. Thick lines indicate the hypothetical direction of evolution.
We propose that RNA:m5U MTases are the most ancient of pyrimidine C-5 MTases, as they exhibit most of the primitive/ancestral features typical of most of the RNA Mtases, as well as many MTases acting on substrates other than nucleic acids (the common order of sequence motifs and the Asp residue in motif IV being most notable). They were probably the first to evolve the ability to catalyze the chemically complex reaction of pyrimidine-C5 methylation, involving an attack of the thiol of a Cys residue from motif VI on the 6 position of uracil to form a covalent complex, thereby activating the 5 position for methyl group transfer from AdoMet, to be followed by deprotonation and β-elimination to restore the free enzyme and release the methylated product (reviewed in 8,26). RNA:m5C MTases might have evolved very early from RNA:m5U MTases by developing a second Cys residue in motif IV to aid in breakdown of the covalent adduct. Alternatively, these two RNA MTase families might have evolved from a common ancestor that methylated both types of pyrimidine bases. The new ‘auxiliary’ Cys residue in motif IV becomes the principal catalyst in DNA:m5C MTases, which have lost the original Cys from motif VI. This transition has probably prompted ‘migration’ of a conserved essential carboxylate in the opposite direction, i.e. loss of the primitive Asp from motif IV and acquisition of Glu in motif VI. Finally, adaptation to recognition of DNA involved loss of the RNA-binding domain from the N-terminus and insertion of a DNA-binding domain after motif VIII. We speculate that the requirement for stabilization of the substrate-binding domain of DNA:m5C MTases in the new location prompted sequence permutation that transferred motif X from the N-terminus to the C-terminus; in this way, both termini of the DNA-binding domain are attached to the catalytic domain, as observed in the crystal structures of M.HhaI and M.HaeIII (reviewed in 8). It is also possible that replacement of the substrate-binding domain in the N-terminus (just before motif X in RNA:m5U and RNA:m5C MTases) was followed by its transfer together with motif X to the C-terminus in DNA:m5C MTases. This scenario can be validated and refined in the future as more sequences of pyrimidine-C5 MTases (especially from their early branching subfamilies) become available and especially as more experimental data on the function of representative members of all major subfamilies are obtained. We hope that our analysis will guide further comparative investigations of residues conserved between the RNA:m5C MTases and either the RNA:m5U or DNA:m5C MTases, as such studies have the potential to provide insight into the origin of m5C methylation, the most important type of DNA modification in Eukaryota, including humans.
CONCLUSIONS
To learn more about the mechanism of RNA:m5C methylation and as an aid to the resolution of the relationships between 5-methylpyrimidine MTases we have carried out extensive bioinformatics analysis followed by mutagenesis of three potential catalytic residues in the yeast tRNA:m5C MTase Trm4p. In addition to the previously reported Cys residues in motifs IV and VI (15), we have found two additional residues that are essential for enzyme activity: Lys in motif I and Asp in motif IV. Based on these analyses, we propose a structural model of the enzyme–substrate complex and a mechanistic role for the motif IV Asp. We have found that RNA:m5C MTases share a number of features with RNA:m5U MTases or DNA:m5C MTases, which prompted us to suggest a tentative phylogenetic model of origin of the three classes of 5-methylpyrimidine MTases, in which DNA:m5C MTases arose from RNA:m5C MTases following ‘switching’ of the role of the nucleophilic catalyst from the now lost Cys in motif VI to Cys in motif IV and a number of other evolutionary innovations, such as sequence permutation.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
J.M.B. thanks Henri Grosjean for critical reading of the manuscript and for useful comments and stimulating discussions. Technical assistance for the production of Trm4p mutants was provided by Michelle King. Funding for portions of this work was provided by a Biomedical Research Grant from the Indiana University School of Medicine. J.M.B. is supported by the EMBO/HHMI Young Investigator Programme award, by the Polish State Committee for Scientific Research (grant PBZ-KBN-088/P04/2003) and by a fellowship from the Foundation for Polish Science.
REFERENCES
- 1.Bujnicki J.M. (1999) Comparison of protein structures reveals monophyletic origin of the AdoMet-dependent methyltransferase family and mechanistic convergence rather than recent differentiation of N4-cytosine and N6-adenine DNA methylation. In Silico Biol., 1, 1–8. [PubMed] [Google Scholar]
- 2.Fauman E.B., Blumenthal,R.M. and Cheng,X. (1999) Structure and evolution of AdoMet-dependent methyltransferases. In Cheng,X. and Blumenthal,R.M. (eds.), S-Adenosylmethionine-dependent Methyltransferases: Structures and Functions. World Scientific, Singapore, pp. 1–38. [Google Scholar]
- 3.Anantharaman V., Koonin,E.V. and Aravind,L. (2002) SPOUT: a class of methyltransferases that includes SpoU and TrmD RNA methylase superfamilies and novel superfamilies of predicted prokaryotic RNA methylases. J. Mol. Microbiol. Biotechnol., 4, 71–75. [PubMed] [Google Scholar]
- 4.Posfai J., Bhagwat,A.S., Posfai,G. and Roberts,R.J. (1989) Predictive motifs derived from cytosine methyltransferases. Nucleic Acids Res., 17, 2421–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar S., Cheng,X., Klimasauskas,S., Mi,S., Posfai,J., Roberts,R.J. and Wilson,G.G. (1994) The DNA (cytosine-5) methyltransferases. Nucleic Acids Res., 22, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malone T., Blumenthal,R.M. and Cheng,X. (1995) Structure-guided analysis reveals nine sequence motifs conserved among DNA amino-methyltransferases and suggests a catalytic mechanism for these enzymes. J. Mol. Biol., 253, 618–632. [DOI] [PubMed] [Google Scholar]
- 7.Bujnicki J.M., Feder,M., Radlinska,M. and Blumenthal,R.M. (2002) Structure prediction and phylogenetic analysis of a functionally diverse family of proteins homologous to the MT-A70 subunit of the human mRNA:m6A methyltransferase. J. Mol. Evol., 55, 431–444. [DOI] [PubMed] [Google Scholar]
- 8.Cheng X. and Roberts,R.J. (2001) AdoMet-dependent methylation, DNA methyltransferases and base flipping. Nucleic Acids Res., 29, 3784–3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schubert H.L., Blumenthal,R.M. and Cheng,X. (2003) Many paths to methyltransfer: a chronicle of convergence. Trends Biochem. Sci., 28, 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bujnicki J.M., Droogmans,L., Grosjean,H., Purushothaman,S.K. and Lapeyre,B. (2004) Bioinformatics-guided identification and experimental characterization of novel RNA methyltransferases. In Bujnicki,J.M. (ed.), Practical Bioinformatics. Springer-Verlag, Heidelberg. [Google Scholar]
- 11.Tscherne J.S., Nurse,K., Popienick,P., Michel,H., Sochacki,M. and Ofengand,J. (1999) Purification, cloning and characterization of the 16S RNA m5C967 methyltransferase from Escherichia coli. Biochemistry, 38, 1884–1892. [DOI] [PubMed] [Google Scholar]
- 12.Gu X.R., Gustafsson,C., Ku,J., Yu,M. and Santi,D.V. (1999) Identification of the 16S rRNA m5C967 methyltransferase from Escherichia coli. Biochemistry, 38, 4053–4057. [DOI] [PubMed] [Google Scholar]
- 13.Reid R., Greene,P. and Santi,D.V. (1999) Exposition of a family of RNA m(5)C methyltransferases from searching genomic and proteomic sequences. Nucleic Acids Res., 27, 3138–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Motorin Y. and Grosjean,H. (1999) Multisite-specific tRNA:m5C-methyltransferase (Trm4) in yeast Saccharomyces cerevisiae: identification of the gene and substrate specificity of the enzyme. RNA, 5, 1105–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.King M.Y. and Redman,K.L. (2002) RNA methyltransferases utilize two cysteine residues in the formation of 5-methylcytosine. Biochemistry, 41, 11218–11225. [DOI] [PubMed] [Google Scholar]
- 16.de Beus E., Brockenbrough,J.S., Hong,B. and Aris,J.P. (1994) Yeast NOP2 encodes an essential nucleolar protein with homology to a human proliferation marker. J. Cell Biol., 127, 1799–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.King M., Ton,D. and Redman,K.L. (1999) A conserved motif in the yeast nucleolar protein Nop2p contains an essential cysteine residue. Biochem. J., 337, 29–35. [PMC free article] [PubMed] [Google Scholar]
- 18.Wyszynski M.W., Gabbara,S. and Bhagwat,A.S. (1992) Substitutions of a cysteine conserved among DNA cytosine methylases result in a variety of phenotypes. Nucleic Acids Res., 20, 319–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mi S. and Roberts,R.J. (1993) The DNA binding affinity of HhaI methylase is increased by a single amino acid substitution in the catalytic center. Nucleic Acids Res., 21, 2459–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gabbara S., Sheluho,D. and Bhagwat,A.S. (1995) Cytosine methyltransferase from Escherichia coli in which active site cysteine is replaced with serine is partially active. Biochemistry, 34, 8914–8923. [DOI] [PubMed] [Google Scholar]
- 21.O’Gara M., Klimasauskas,S., Roberts,R.J. and Cheng,X. (1996) Enzymatic C5-cytosine methylation of DNA: mechanistic implications of new crystal structures for HhaI methyltransferase-DNA-AdoHcy complexes. J. Mol. Biol., 261, 634–645. [DOI] [PubMed] [Google Scholar]
- 22.Klimasauskas S., Szyperski,T., Serva,S. and Wuthrich,K. (1998) Dynamic modes of the flipped-out cytosine during HhaI methyltransferase–DNA interactions in solution. EMBO J., 17, 317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Serva S., Weinhold,E., Roberts,R.J. and Klimasauskas,S. (1998) Chemical display of thymine residues flipped out by DNA methyltransferases. Nucleic Acids Res., 26, 3473–3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lau E.Y. and Bruice,T.C. (1999) Active site dynamics of the HhaI methyltransferase: insights from computer simulation. J. Mol. Biol., 293, 9–18. [DOI] [PubMed] [Google Scholar]
- 25.Huang N., Banavali,N.K. and MacKerell,A.D.,Jr (2003) Protein-facilitated base flipping in DNA by cytosine-5 methyltransferase. Proc. Natl Acad. Sci. USA, 100, 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ivanetich K.M. and Santi,D.V. (1992) 5,6-Dihydropyrimidine adducts in the reactions and interactions of pyrimidines with proteins. Prog. Nucleic Acid Res. Mol. Biol., 42, 127–156. [DOI] [PubMed] [Google Scholar]
- 27.Kealey J.T. and Santi,D.V. (1991) Identification of the catalytic nucleophile of tRNA (m5U54)methyltransferase. Biochemistry, 30, 9724–9728. [DOI] [PubMed] [Google Scholar]
- 28.Kealey J.T., Gu,X. and Santi,D.V. (1994) Enzymatic mechanism of tRNA (m5U54)methyltransferase. Biochimie, 76, 1133–1142. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y. and Santi,D.V. (2000) m5C RNA and m5C DNA methyl transferases use different cysteine residues as catalysts. Proc. Natl Acad. Sci. USA, 97, 8263–8265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Foster P.G., Nunes,C.R., Greene,P., Moustakas,D. and Stroud,R.M. (2003) The first structure of an RNA m5C methyltransferase, Fmu, provides insight into catalytic mechanism and specific binding of RNA substrate. Structure (Camb.), 11, 1609–1620. [DOI] [PubMed] [Google Scholar]
- 31.Wu P., Brockenbrough,J.S., Paddy,M.R. and Aris,J.P. (1998) NCL1, a novel gene for a non-essential nuclear protein in Saccharomyces cerevisiae. Gene, 220, 109–117. [DOI] [PubMed] [Google Scholar]
- 32.Knapp G. (1989) Preparation of yeast transfer RNA precursors in vivo. Methods Enzymol., 180, 110–117. [DOI] [PubMed] [Google Scholar]
- 33.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thompson J.D., Gibson,T.J., Plewniak,F., Jeanmougin,F. and Higgins,D.G. (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res., 25, 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saitou N. and Nei,M. (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol., 4, 406–425. [DOI] [PubMed] [Google Scholar]
- 36.Kurowski M.A. and Bujnicki,J.M. (2003) GeneSilico protein structure prediction meta-server. Nucleic Acids Res., 31, 3305–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karplus K., Karchin,R., Barrett,C., Tu,S., Cline,M., Diekhans,M., Grate,L., Casper,J. and Hughey,R. (2001) What is the value added by human intervention in protein structure prediction? Proteins, 45 (Suppl. 5), 86–91. [DOI] [PubMed] [Google Scholar]
- 38.Kelley L.A., McCallum,C.M. and Sternberg,M.J. (2000) Enhanced genome annotation using structural profiles in the program 3D-PSSM. J. Mol. Biol., 299, 501–522. [DOI] [PubMed] [Google Scholar]
- 39.Jones D.T. (1999) GenTHREADER: an efficient and reliable protein fold recognition method for genomic sequences. J. Mol. Biol., 287, 797–815. [DOI] [PubMed] [Google Scholar]
- 40.Rychlewski L., Jaroszewski,L., Li,W. and Godzik,A. (2000) Comparison of sequence profiles. Strategies for structural predictions using sequence information. Protein Sci., 9, 232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi J., Blundell,T.L. and Mizuguchi,K. (2001) Fugue: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol., 310, 243–257. [DOI] [PubMed] [Google Scholar]
- 42.McGuffin L.J., Bryson,K. and Jones,D.T. (2000) The PSIPRED protein structure prediction server. Bioinformatics, 16, 404–405. [DOI] [PubMed] [Google Scholar]
- 43.Pollastri G., Przybylski,D., Rost,B. and Baldi,P. (2002) Improving the prediction of protein secondary structure in three and eight classes using recurrent neural networks and profiles. Proteins, 47, 228–235. [DOI] [PubMed] [Google Scholar]
- 44.Ouali M. and King,R.D. (2000) Cascaded multiple classifiers for secondary structure prediction. Protein Sci., 9, 1162–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garner E., Romero,P., Dunker,A.K., Brown,C. and Obradovic,Z. (1999) Predicting binding regions within disordered proteins. Genome Inf. Ser. Workshop Genome Inf., 10, 41–50. [PubMed] [Google Scholar]
- 46.Kosinski J., Cymerman,I.A., Feder,M., Kurowski,M.A., Sasin,J.M. and Bujnicki,J.M. (2003) A ‘Frankenstein’s monster’ approach to comparative modeling: merging the finest fragments of fold-recognition models and iterative model refinement aided by 3D structure evaluation. Proteins, 53 (Suppl.), 369–379. [DOI] [PubMed] [Google Scholar]
- 47.Sali A. and Blundell,T.L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol., 234, 779–815. [DOI] [PubMed] [Google Scholar]
- 48.Luthy R., Bowie,J.U. and Eisenberg,D. (1992) Assessment of protein models with three-dimensional profiles. Nature, 356, 83–85. [DOI] [PubMed] [Google Scholar]
- 49.Gustafsson C., Reid,R., Greene,P.J. and Santi,D.V. (1996) Identification of new RNA modifying enzymes by iterative genome search using known modifying enzymes as probes. Nucleic Acids Res., 24, 3756–3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Doll A. and Grzeschik,K.H. (2001) Characterization of two novel genes, WBSCR20 and WBSCR22, deleted in Williams-Beuren syndrome. Cytogenet. Cell Genet., 95, 20–27. [DOI] [PubMed] [Google Scholar]
- 51.Anantharaman V., Koonin,E.V. and Aravind,L. (2002) Comparative genomics and evolution of proteins involved in RNA metabolism. Nucleic Acids Res., 30, 1427–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klimasauskas S., Kumar,S., Roberts,R.J. and Cheng,X. (1994) HhaI methyltransferase flips its target base out of the DNA helix. Cell, 76, 357–369. [DOI] [PubMed] [Google Scholar]
- 53.Bujnicki J.M. (2000) Phylogenomic analysis of 16S rRNA:(guanine-N2) methyltransferases suggests new family members and reveals highly conserved motifs and a domain structure similar to other nucleic acid amino-methyltransferases. FASEB J., 14, 2365–2368. [DOI] [PubMed] [Google Scholar]
- 54.Bujnicki J.M., Leach,R.A., Debski,J. and Rychlewski,L. (2002) Bioinformatic analyses of the tRNA:(guanine 26, N2,N2)-dimethyltransferase (Trm1) family. J. Mol. Microbiol. Biotechnol., 4, 405–415. [PubMed] [Google Scholar]
- 55.Bujnicki J.M. and Radlinska,M. (1999) Molecular phylogenetics of DNA 5mC-methyltransferases. Acta Microbiol. Pol., 48, 19–33. [PubMed] [Google Scholar]
- 56.Posfai J., Bhagwat,A.S. and Roberts,R.J. (1988) Sequence motifs specific for cytosine methyltransferases. Gene, 74, 261–265. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}