

Abstract
Background
Clinical heterogeneity in the development of levodopa-induced dyskinesias (LID) suggests endogenous factors play a significant role in determining their overall prevalence.
Objective
We hypothesised that single nucleotide polymorphisms (SNPs) in specific genes may result in a clinical phenotype conducive to an increased risk of LID.
Methods
We examined the influence of SNPs in the catechol-O-methyltransferase (COMT), monoamine oxidase A (MAO-A) and brain-derived neurotrophic factor (BDNF) genes on LID in a cohort of 285 pathologically confirmed Parkinson’s disease patients, using data from their complete disease course.
Results
Dyskinetic patients demonstrated younger age at disease onset (60.3 vs. 66.4 years, p < 0.0001), a longer disease duration (17.0 vs. 12.0 years, p < 0.0001) and a higher maximum daily levodopa equivalent dose (LED; 926.7 vs. 617.1 mg/day, p < 0.0001) than patients without dyskinesias. No individual SNP was found to influence prevalence or time to onset of dyskinesias, including after adjustment for known risk factors. We observed that patients carrying alleles conferring both high COMT activity and increased MAO-A mRNA expression received significantly higher maximum and mean daily LEDs than those with low enzyme activity/mRNA expression (max LED: 835 ± 445 vs. 508 ± 316 mg; p = 0.0056, mean LED: 601 ± 335 vs. 398 ± 260 mg; p = 0.025).
Conclusions
Individual SNPs in BDNF, COMT and MAO-A genes did not influence prevalence or time to onset of dyskinesias in this cohort. The possibility that combined COMT and MAO-A genotype is a significant factor in determining an individual’s lifetime levodopa exposure warrants further investigation.
Keywords: Parkinson’s disease, Levodopa-induced dyskinesias, Catechol-O-methyltransferase, Monoamine oxidase A, Brain-derived neurotrophic factor
Introduction
Levodopa-induced dyskinesias (LID) are a substantial barrier to effective symptomatic management of Parkinson’s disease (PD) and occur as a consequence of chronic levodopa (L-DOPA) treatment. Despite their high prevalence, therapeutic options are currently limited [1]. Epidemiological evidence suggests that the onset and severity of LID varies considerably, with some patients never developing them despite equivalent treatment regimens. This observation suggests endogenous factors play a role in individual susceptibility to LID, and despite established risk factors for developing LID including younger age at PD onset, higher L-DOPA dose, greater disease severity and longer disease duration [2], there has been little research to date regarding genetic susceptibility to LID.
In this study, we used a candidate gene approach to examine functional single nucleotide polymorphisms (SNPs) in which the cellular consequences of the polymorphism have been identified and are directly relevant to dyskinesias. Specifically, we focused on functional SNPs in genes encoding the catechol-O-methyltransferase (COMT) and monoamine oxidase A (MAO-A) enzymes, both of which catabolise dopamine and are central to the therapeutic response of L-DOPA. A valine to methionine substitution at codon 158 of the COMT gene produces a Met variant that catabolises dopamine up to four times slower than its Val counterpart [3]. Given the overlapping role of MAO with COMT, we anticipated that a similar finding might be observed for the synonymous substitution of T to G in exon 8 of the MAO-A gene, which promotes MAO-A mRNA expression [4].
Finally, a valine to methionine substitution at codon 66 of the brain-derived neurotrophic factor (BDNF) gene has been identified as having a putative role in influencing time to onset of dyskinesia in PD [5]. We hypothesised that these polymorphisms, individually or combined, may contribute to the risk of developing dyskinesias in PD.
Patients and Methods
Case Selection
We identified 285 pathologically confirmed PD cases from the Australian Brain Bank Network (ABBN), Australia and the Queen Square Brain Bank for Neurological Disorders (QSBB), UK with a history of L-DOPA usage and a disease duration of at least 5 years. Patients were excluded if they had confirmed monogenic PD, early symptom onset (≤40 years of age) or late symptom onset (≥80 years of age). Approval for the collection of brain tissue as well as retention of and access to clinical records was granted by the Human Research Ethics Committee of the University of Melbourne (ABBN) and the London Multi-Centre Research Ethics Committee (QSBB).
Genotyping
DNA was extracted using standard methods (QIAamp DNA Kit, Qiagen) and genotyping was performed via the SEQUENOM™ genotyping platform at the Australian Genome Research Facility for all Australian cases. Cases from the UK were genotyped for the Val158Met COMT polymorphism (dbSNP rs4680) and the T941G MAO-A polymorphism (rs6323) as per Spencer et al. [6]. The BDNF Val66Met polymorphism (rs6265) was genotyped as per Foltynie et al. [7].
Clinical Data
A systematic review of patients’ medical records was performed by movement disorder specialists (K.B., H.L. and S.O’S.) and data including age at PD onset, disease duration, prevalence and time of onset of dyskinesia, and dopaminergic medication history were recorded. Levodopa equivalent dose (LED), which accounts for other antiparkinsonian drugs, was calculated as per Tomlinson et al. [8]. An approximation of the cumulative lifetime L-DOPA dose was estimated using previously published methods [9]. Mean daily LED was obtained using the calculated lifetime estimate and adjusting for the number of years of L-DOPA treatment.
Statistical Analysis
We constructed Kaplan-Meier survival curves, with the time-dependent variable set as the first recorded incidence of dyskinesias in dyskinetic patients, or disease duration for patients without dyskinesia. Time zero was set as age of PD onset, as this data was more reliably recorded than age at first L-DOPA administration. We used Cox proportional hazards regression and generalised linear modelling to examine the relationship between genotype and time to onset of LID, adjusting for established risk factors. As the distribution of LED was skewed, we used logarithmic transformation to normalise the distribution prior to linear modelling. Statistical analysis was performed using GraphPad Prism (version 5.0, GraphPad Software, San Diego, Calif., USA) or SPSS (version 20, IBM SPSS, New York, N.Y., USA).
Results
Patients in our combined cohort demonstrated typical PD demographics, with a mean age of onset of 63.0 ± 9.2 years, a mean disease duration of 14.8 ± 6.4 years and a mean maximum daily LED of 794.2 ± 431.6 mg/day. Dyskinesias were reported in 61.3% of patients. Dyskinetic patients demonstrated established risk factors for dyskinesias, including younger age of PD onset (60.3 vs. 66.4 years, p < 0.0001), a longer disease duration (17.0 vs. 12.0 years, p < 0.0001) and a higher maximum daily LED (926.7 vs. 617.1 mg/day, p < 0.0001) than patients without dyskinesias.
When patients were stratified according to COMT, MAO-A or BDNF genotype, no individual genotype was found to independently influence the prevalence or time to onset of dyskinesias (fig. 1). Individual genotypes were compared for each gene, and genotypes were pooled to examine the effect of homozygosity for either allele. No difference was observed for any clinical feature, including age at onset, disease duration and maximum or mean daily LED (table 1).
Fig. 1. Survival curves of time to onset of dyskinesia according to COMT (a), MAO-A (b) or BDNF (c) genotype.
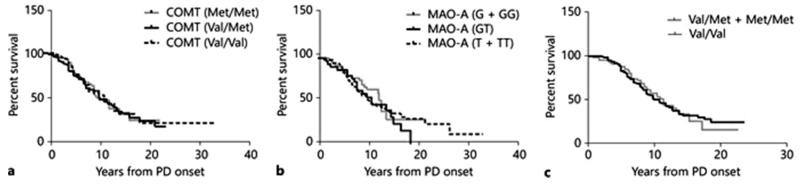
Kaplan-Meier curves demonstrating duration from PD onset to first reported incidence of LID in patients stratified according to genotype. For the BDNF Val66Met polymorphism, given the low prevalence of the variant Met allele in European populations, Met/Met homozygous patients were pooled with Val/Met heterozygotes for statistical analysis. As the MAO-A gene is X-linked, males and females were analysed both separately and with male hemizygotes pooled with female homozygotes to directly compare the effects of T or G allele carriage.
Table 1. Effect of COMT, MAO-A and BDNF genotype on incidence and time to onset of dyskinesias.
|
COMT genotype |
MAO-A genotype |
BDNF genotype |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Met/Met | Val/Met | Val/Val | P value | GG♀ + G♂ | GT♀ | TT♀ + T♂ | P value | Val/Met + Met/Met | Val/Val | P value | |
| Number of cases | 70 | 127 | 80 | n/a | 63 | 36 | 169 | n/a | 63+8 | 153 | n/a |
| Age at PD onset, years | 64.0±9.1 | 62.7±9.3 | 66.7±9.1 | 0.51 | 63.3±9.5 | 64.2±8.1 | 63.0±9.3 | 0.80 | 64.3±9.4 | 62.5±8.9 | 0.14 |
| Disease duration, years | 14.0±6.3 | 14.8±6.5 | 15.7±6.5 | 0.26 | 13.9±5.9 | 15.6±5.1 | 14.9±6.7 | 0.24 | 14.1±6.2 | 14.8±5.9 | 0.31 |
| Time to onset of dyskinesia, years | 8.7±4.9 | 7.9±4.9 | 8.4±4.2 | 0.67 | 8.0±4.3 | 9.2±5.6 | 8.0±4.6 | 0.58 | 8.1±4.2 | 7.7±3.9 | 0.62 |
| Dyskinesia reported, % | 58.5 | 59.6 | 63.9 | 0.78* | 55.4 | 75.0 | 56.2 | 0.12* | 62.0 | 62.0 | >0.90* |
| Mean daily LED, mg/day | 588±304 | 593±299 | 520±275 | 0.30 | 551±312 | 551±235 | 590±299 | 0.83 | 497±202 | 619±335 | 0.06 |
| Maximum daily LED, mg/day | 667±294 | 846±464 | 783±424 | 0.11 | 710±356 | 687±316 | 814±430 | 0.33 | 721±374 | 845±465 | 0.08 |
Values are mean ± SD. Statistical analysis was non-parametric Kruskal-Wallis or Mann-Whitney test.
p value from a χ2 (Fisher’s exact) test.
We considered that individual functional SNPs may be unlikely to exert a significantly large effect on dyskinesia in isolation, and that given the overlapping function of COMT and MAO-A, double homozygosity for both SNPs may exert a greater effect than homozygosity for either SNP individually. However, pooling genotypes demonstrated that patients with alleles conferring high COMT activity (Val/Val) and high MAO-A mRNA expression (T males; TT females) did not have a statistically significantly increased prevalence of dyskinesias (58.0 vs. 43.8%, χ2 = 0.62, d.f. = 1, p = 0.43) compared to those with low enzyme activity/mRNA expression. Interestingly, we observed that patients homozygous for high COMT activity and high MAO-A mRNA expression alleles received significantly higher maximum daily LED (835 ± 445 mg) than those homozygous for low activity/expression alleles (508 ± 316 mg; p = 0.0056), as well as a higher mean daily LED (601 ± 335 vs. 398 ± 260 mg; p = 0.0248). Generalised linear modelling confirmed the association between pooled COMT and MAO-A genotype and LED, with low activity/expression allele carriers receiving a mean maximum daily LED 43% lower (p = 0.003) than other genotype groups, after controlling for disease duration.
Discussion
In this study of pathologically confirmed PD, polymorphisms in COMT, MAO-A and BDNF genes, either individually or combined, had no influence on the prevalence or time to onset of LID (fig. 1). While COMT and BDNF have previously been implicated in the pathophysiology of dyskinesias, MAO-A is a novel candidate. COMT is primarily responsible for the breakdown of L-DOPA when DOPA decarboxylase inhibitors are co-administered with L-DOPA, blocking DOPA decarboxylase activity. The COMT rs4680 SNP has been associated with an increased risk of dyskinesias in a recent prospective study [10], although earlier cross-sectional studies failed to find a statistically significant effect [11].
MAO-A has a similar role to COMT in catabolising monoamines, and the T allele of the T941G MAO-A SNP, which confers increased mRNA expression, has been investigated as a risk factor for developing PD [12]. Despite this overlap in function, we found no evidence to suggest that this SNP individually influences dyskinesias. Similarly, met allele carriage of the BDNF Val66Met polymorphism has been associated with an earlier time to onset of dyskinesias [5]; however, our study found no association between BDNF genotype and prevalence or time to onset of dyskinesias, which may be a reflection of different study designs.
Our data suggest a putative relationship between combined COMT and MAO-A genotype and total lifetime L-DOPA exposure. Patients with alleles conferring high COMT activity and high MAO-A mRNA expression received significantly higher L-DOPA exposure than low activity/expression carriers. Although these data should be interpreted with caution given the small sample size of this subgroup of double homozygotes, we note that if we reduce our p value to p < 0.01 to correct for multiple comparisons, our data remain statistically significant.
In a clinical setting, patients with increased COMT enzyme activity and increased MAO-A expression will metabolise L-DOPA faster, which may result in increased wearing off, necessitating higher or more frequent L-DOPA doses over the day, resulting in the higher lifetime exposure to L-DOPA observed in our study. This finding warrants replication in a larger cohort, although it is difficult to increase the sample size of a post-mortem cohort without adding cases from additional tissue banks, introducing genetic diversity that can obscure results.
The present study has some important limitations. The data were collected retrospectively using clinical case notes with the implicit assumption that a reasonably consistent threshold would apply for specialists to report dyskinesias. While our patient demographics appear to represent a typical PD population on the basis of age of onset and disease duration, there is likely to be a selection bias in cases submitted for brain banking.
In summary, we find no evidence to suggest the SNPs in COMT, MAO-A or BDNF examined in this study influence the prevalence or time to onset of LID. Our data suggest that combined COMT and MAO-A genotype may influence L-DOPA use, but further studies in different populations are required to clarify this effect.
Acknowledgements
We would like to thank Catherine Smith, Monash University, for her assistance with statistical modelling and Linda Parsons, QSBB, for her assistance with tissue acquisition. We thank brain donors and their families, without whom this work would not have been possible. This work was funded by the Bethlehem Griffiths Research Foundation, the Brain Foundation, the Wellcome Trust (WTCCC2, grants 084747 and 083948) and Parkinson’s UK (K-0901). This work was partly undertaken at UCLH/UCL who received a proportion of funding from the Department of Health’s NIHR Biomedical Research Centres funding scheme. Support was also received from the Wellcome Trust/MRC Joint Call in Neurodegeneration Award (WT089698) to UCL, the MRC Unit in Dundee and the University of Sheffield. The ABBN is supported by Neuroscience Research Australia, the University of New South Wales and the National Health and Medical Research Council of Australia.
References
- 1.Fabbrini G, Brotchie JM, Grandas F, Nomoto M, Goetz CG. Levodopa-induced dyskinesias. Mov Disord. 2007;22:1379–1389. doi: 10.1002/mds.21475. [DOI] [PubMed] [Google Scholar]
- 2.Fahn S, Oakes D, Shoulson I, Kieburtz K, Rudolph A, Lang A, et al. Levodopa and the progression of Parkinson’s disease. N Engl J Med. 2004;351:2498–2508. doi: 10.1056/NEJMoa033447. [DOI] [PubMed] [Google Scholar]
- 3.Lachman HM, Papolos DF, Saito T, Yu YM, Szumlanski CL, Weinshilboum RM. Human catechol-O-methyltransferase pharmacogenetics: description of a functional polymorphism and its potential application to neuropsychiatric disorders. Pharmacogenetics. 1996;6:243–250. doi: 10.1097/00008571-199606000-00007. [DOI] [PubMed] [Google Scholar]
- 4.Pinsonneault JK, Papp AC, Sadee W. Allelic mRNA expression of X-linked monoamine oxidase a (MAOA) in human brain: dissection of epigenetic and genetic factors. Hum Mol Genet. 2006;15:2636–2649. doi: 10.1093/hmg/ddl192. [DOI] [PubMed] [Google Scholar]
- 5.Foltynie T, Cheeran B, Williams-Gray CH, Edwards MJ, Schneider SA, Weinberger D, et al. BDNF val66met influences time to onset of levodopa induced dyskinesia in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2009;80:141–144. doi: 10.1136/jnnp.2008.154294. [DOI] [PubMed] [Google Scholar]
- 6.UK Parkinson’s Disease Consortium. Wellcome Trust Case Control Consortium 2. Spencer CC, Plagnol V, Strange A, Gardner M, Paisan-Ruiz C, et al. Dissection of the genetics of Parkinson’s disease identifies an additional association 5′ of SNCA and multiple associated haplotypes at 17q21. Hum Mol Genet. 2011;20:345–353. doi: 10.1093/hmg/ddq469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Foltynie T, Lewis SG, Goldberg TE, Blackwell AD, Kolachana BS, Weinberger DR, et al. The BDNF Val66Met polymorphism has a gender specific influence on planning ability in Parkinson’s disease. J Neurol. 2005;252:833–838. doi: 10.1007/s00415-005-0756-5. [DOI] [PubMed] [Google Scholar]
- 8.Tomlinson CL, Stowe R, Patel S, Rick C, Gray R, Clarke CE. Systematic review of levodopa dose equivalency reporting in Parkinson’s disease. Mov Disord. 2010;25:2649–2653. doi: 10.1002/mds.23429. [DOI] [PubMed] [Google Scholar]
- 9.Parkkinen L, O’Sullivan SS, Kuoppamaki M, Collins C, Kallis C, Holton JL, et al. Does levodopa accelerate the pathologic process in Parkinson disease brain? Neurology. 2011;77:1420–1426. doi: 10.1212/WNL.0b013e318232ab4c. [DOI] [PubMed] [Google Scholar]
- 10.de Lau LM, Verbaan D, Marinus J, Heutink P, van Hilten JJ. Catechol-O-methyltransferase Val158Met and the risk of dyskinesias in Parkinson’s disease. Mov Disord. 2011;27:132–135. doi: 10.1002/mds.23805. [DOI] [PubMed] [Google Scholar]
- 11.Watanabe M, Harada S, Nakamura T, Ohkoshi N, Yoshizawa K, Hayashi A, et al. Association between catechol-O-methyltransferase gene polymorphisms and wearing-off and dyskinesia in Parkinson’s disease. Neuropsychobiology. 2003;48:190–193. doi: 10.1159/000074637. [DOI] [PubMed] [Google Scholar]
- 12.Williams-Gray C, Goris A, Foltynie T, Compston A, Sawcer S, Barker RA. No evidence for association between an MAOA functional polymorphism and susceptibility to Parkinson’s disease. J Neurol. 2009;256:132–133. doi: 10.1007/s00415-009-0899-x. [DOI] [PubMed] [Google Scholar]