

Abstract
Wnt-targeted gene therapy has been proposed as a treatment for human colorectal cancer (CRC). The Cre-Lox system consists of methodology for enhancing targeted expression from tissue-specific or cancer-specific promoters. We analyzed the efficiency of Wnt-specific promoters as drivers of the Cre-mediated activity of a luciferase reporter gene or cell death effector gene in CRC cell lines in the presence and absence of two modulators of Wnt activity, sodium butyrate and lithium chloride. Butyrate is present in the colonic lumen after digestion of fiber-rich foods, whereas the colonic lumen is readily accessible to lithium chloride. In both SW620 and HCT-116 CRC cells, a physiologically relevant concentration of butyrate upregulated reporter and effector activity and altered the Wnt-specific expression pattern. Lithium chloride markedly enhanced Cre-Lox-mediated Wnt-specific reporter expression only in APC wild-type CRC cells. Possibilities for genetic modulation of the proposed CRC therapy included Wnt-specific expression of a floxed Lef1-VP16 fusion that enhanced Wnt-specific cell death and of a floxed dominant-negative Tcf4 that specifically downregulated endogenous Wnt activity. These findings demonstrated that the Cre-Lox system, in combination with pharmacological and genetic modulators, represents effective methodology for enhancing Wnt-targeted gene therapy.
INTRODUCTION
Wnt signaling is characterized by the stabilization of β-catenin, which complexed with the T-cell factor/ lymphocyte enhancer factor (Tcf-Lef) family of transcription factors upregulates the expression of genes whose promoters contain Tcf/Lef-binding sites (1–6). As a result of mutations in the adenomatous polyposis coli (APC) or β-catenin genes, the majority of human colorectal cancers (CRCs) exhibit constitutive upregulation of the Wnt transcriptional pathway, which has been implicated as a crucial step in the initiation and development of colonic tumorigenesis (7–9).
Gene therapy approaches for CRC (10) have utilized genes causing cell death (11–19), genes which encode for enzymes converting prodrugs to their active forms (20–23) or genes necessary for viral replication (24,25). Cancer-targeted gene therapy depends on differences in gene expression between malignant and normal cells; thus, the expression of a targeted gene must be both specific to abnormal cells and strong enough to induce cell death efficiently. Although some studies on Wnt-targeted therapeutic expression have been performed (10,18,22,24), a more systematic molecular examination of the factors that influence the effectiveness and specificity of Wnt-mediated gene therapy has not yet been performed.
The histone deacetylase inhibitor butyrate enhances colon cell differentiation and/or apoptosis (26–28) and, as demonstrated by us, modulates Wnt signaling in CRC cell lines in a cell-type- and promoter-dependent manner (29,30). Given that butyrate is normally found in the colonic lumen in high concentrations from bacterial fermentation of dietary fiber, the ability of butyrate to modulate Wnt signaling is an important consideration for Wnt-targeted therapeutic gene expression. In addition to butyrate, lithium enhances Wnt signaling (31) by the active β-catenin form (32). Thus, we have found that lithium chloride (LiCl) enhances Wnt-specific expression in HCT-116 CRC cells (APC+/+, β-catenin+/–), but not in SW620 CRC cells (APC+/–) (unpublished data). Thus lithium, commonly used for the treatment of psychiatric disorders, is another reagent whose ability to modulate Wnt activity may be of importance for Wnt-specific gene therapy approaches.
Other important factors that influence Wnt-targeted gene expression is the variability in levels of free β-catenin, β-catenin–Tcf complexes and, consequently, Wnt activity in CRC cell lines (29,30,33–35). Low passage colon cancer cell lines derived from CRC primary tumors have shown quite varied levels of intrinsic Wnt activity (36), suggesting that the above mentioned variations in established CRC cell lines represent an in vivo property of CRCs. Thus, in view of the variability in the levels of Wnt activity in CRC cells, Wnt-targeted gene therapy would appear to benefit significantly by the use of a system that enhances both expression levels and specificity. The Cre-Lox system represents such an approach to enhance expression from tissue- or cancer-specific promoters.
The overall aims of the present study were (i) to create a Wnt-specific Cre-Lox-mediated gene expression cell kill system for CRC cells and (ii) to evaluate the effects of pharmacological (butyrate, lithium) and genetic (Lef-VP16) modulators of this system. In the present report we describe comprehensive analyses of the effects of butyrate and lithium on the Cre-Lox system combined with a variety of Wnt-specific promoters to drive the expression of a floxed luciferase reporter or that of floxed FADD and diphtheria toxin A-chain cell death genes. In addition, FADD, which induces cell death via activation of caspase 8 as part of the Fas-mediated apoptotic pathway (37), has been previously proposed as an effector of Wnt activity in a direct TcfTK system (18). The diphtheria toxin A-chain, which induces cell death by ADP ribosylation of elongation factor 2 and irreversible inhibition of protein synthesis, has also been established as a potent effector gene in several gene therapy model systems (11–15). The ability of the Cre-Lox system to enhance expression from specific promoters (38–42) was examined. The rationale behind this approach was as follows: a stop cassette, flanked by Lox sites (‘floxed’), was placed between a strong promoter and the gene of interest (e.g. FADD or diphtheria toxin A-chain). In the absence of CRE recombinase, which has the ability to excise floxed sequences, the gene of interest is not expressed due to the presence of the retained stop cassette. However, in the presence of CRE recombinase, the stop cassette is excised, allowing for high levels of expression from a strong constitutively active promoter. Specificity is generated by having the expression of CRE recombinase under the transcriptional control of a tissue-specific and/or inducible promoter (e.g. a Wnt-specific promoter). Since low levels of CRE recombinase enzyme are sufficient to catalyze the excision of floxed DNA sequences, highly specific but relatively weak promoters driving CRE recombinase expression are adequate. The low levels of expression from such promoters are amplified by the enzymatic activity of CRE recombinase. In addition to the analysis of variable Wnt-specific promoters combined with the Cre-Lox system, we have explored the possibility of enhancing the specific cell kill genetically, as well as by using modulators of Wnt activity such as butyrate and LiCl.
Thus, using a floxed luciferase reporter, we demonstrate that Wnt-specific gene expression in SW620 and HCT-116 CRC cells is influenced by the promoter identity and the cell type. The activity is also significantly modulated by butyrate in APC mutant SW620 cells and by both butyrate and lithium in APC wild-type cells, such as HCT-116. Expression of Lef-VP16, a genetic modulator of Wnt activity (43), enhances Wnt-specific kill of CRC cells in the absence of butyrate. Finally, we demonstrate that a Cre-Lox-mediated system is capable of downregulating Wnt activity in SW620 cells in a Wnt-specific manner. Such an approach, combined with Wnt-mediated cell-death systems, has the potential to increase the efficacy of Wnt-targeted therapy of CRC. The implications of these approaches for possible Wnt-targeted genetic therapeutics are discussed.
MATERIALS AND METHODS
Plasmids
The following plasmids were kind gifts: pTOP/FOPFLASH, pTOP/FOPCAT and pTOP/FOPGlow from Dr Hans Clevers (University Hospital Utrecht, The Netherlands); pDN-Tcf4 and pCMV-APC from Dr Bert Vogelstein and Dr Ken Kinzler (Johns Hopkins Medical School, Baltimore, MD); Lef1-fusion constructs to VP16 from Dr Peter K. Vogt (Scripps Research Institute, La Jolla, CA); pCMV-CREM, pCMV-RFP/CREM, pSV-EGFPβ–Gal and pCMVe-βAc-STOP-Luc vectors from Dr Jeffrey E. Green (National Cancer Institute, Bethesda, MD); diphtheria toxin-A chain wild-type vector from Dr Ian Maxwell (University of Colorado, Denver, CO); TOP/FOP-CMV vectors from Dr M.C. Hung (M. D. Anderson Cancer Center, Houston, TX). pGL3Basic, pGL3Control and pRL-TK were obtained from the Promega Corporation (Madison, WI).
pTOP/FOPTKFLASH were constructed by ligating the small (BamHI-Klenow-XhoI) fragment containing the luciferase reporter of the pGL3Basic vector into the large SmaI-XhoI fragment of pTOPCAT and pFOPCAT, respectively. 0-CRE was constructed by ligating the XhoI-MscI fragment of pCMV-CREM, containing the modified CRE coding sequence and human β-globin intron, to the XbaI-Klenow-XhoI fragment of pGL3Basic. The BglI-Klenow-NheI promoter fragments from pTOP/FOPTK-FADD and pTOP/FOPFos-FADD (M. Bordonaro, D. L. Lazarova, R. Carbone and A. C. Sartorelli, submitted for publication) were ligated to the XhoI-Klenow-NheI fragment of 0-CRE to yield pTOP/FOPTK-CRE and pTOP/FOPFos-CRE, respectively. The Hind3-Klenow-XhoI fragments from pTOP/FOPGlow were ligated to the NheI-Klenow-XhoI fragment of 0-CRE to yield pTOP/FOPTATA-CRE. The TOP/FOP-CMV promoter plasmids were digested with BglII, Klenow treated and digested with XhoI, and the fragments containing the promoters were ligated to 0-CRE digested with NheI, Klenow treated and digested with XhoI to form pTOP/FOPCMV-CRE.
The ClaI-Klenow-Hind3 fragment of 0-FADD was ligated to the SalI-Klenow-Hind3 fragment of pCMVe-βAc-STOP-Luc, placing the FADD coding sequence and the SV40 late polyadenylation site downstream of the CMV-β-actin promoter and floxed stop cassette, creating the construct pSTOP-FADD. The coding sequence of VP16, fused to both the β-catenin-binding and DNA-binding domains of Lef1, was amplified from CMV-Lef-VP16 by PCR using primers containing NheI and XbaI sites, and the PCR product was digested with NheI, Klenow treated and digested with XbaI. This fragment was ligated to the Hind3-Klenow-SalI fragment of pCMVe-βAc-STOP-Luc and the XbaI-SalI fragment of pGL3 to form pSTOP-LefVP16. The Hind3-Klenow-SalI fragment of pCMVe-βAc-STOP-Luc, the PstI-Klenow-XbaI fragment of DT-WT (containing the wild-type diphtheria toxin A-chain gene) and the XbaI-SalI fragment of pGL3-Basic were ligated together to form pSTOP-DT. The Hind3-Klenow-SalI fragment of pCMVe-βAc-STOP-Luc, the BglI-Klenow-XbaI fragment of DN-Tcf4 and the XbaI-SalI fragment of pGL3 were ligated together to form pSTOP-DNTcf4. The Hind3-Klenow-NheI fragment of pCMVe-βAc-STOP-Luc, containing the pCMVe-βAc promoter and Stop cassette, was ligated to the XbaI-Klenow-NheI backbone fragment of pGL3, forming pSTOP-GL3. Constructed plasmids were analyzed by restriction digestion and DNA sequencing. All enzymes were from New England Biolabs (Beverly, MA), except where otherwise noted.
Cell culture
SW620 and HCT-116 cells obtained from the American Type Culture Collection (Manassas, VA) and RKO cells obtained from Dr Michael Kastan (St Jude Children’s Hospital, Memphis, TN) were maintained in minimal essential medium (MEM) supplemented with 10% fetal bovine serum (FBS) at 37°C in the presence of 5% carbon dioxide.
Transfection and luciferase assays
For transfection for luciferase assays, 100 000 SW620, 60 000 HCT-116 or 35 000 RKO cells were plated into each well of a 24-well dish (Corning, Corning, NY). After 3 days, plasmid DNA was mixed with Gene Porter (Gene Therapy Systems, San Diego, CA) in MEM without serum at a ratio of 5 µl of GenePorter to 1 µg of DNA for 45 min and added to the wells (250 µl/well for 24-well plates). After 5 h, 250 µl of MEM with 20% FBS were added to each well and left overnight, after which the transfection mixture was removed and 1 ml of fresh MEM plus 10% FBS was added to each well. At this time, other reagents were added as indicated. Normalization with pRL-TK and treatment with sodium butyrate (Sigma, St Louis, MO) were accomplished as previously described (30). LiCl (Sigma) was added to a final concentration of 20 mM. Assays with pSTOP-FADD and pSTOP-DT-A were normalized by the protein concentration measured by the Bio-Rad protein assay reagent (Bio-Rad, Hercules, CA) at 595 nm. The Promega Luciferase system and Turner Luminometer (Promega) were used for luciferase assays, which were performed as described previously (30).
Transfection and GFP/annexin assays
Cells were plated 24 h prior to transfection at 1 × 106 cells per well in 24-well plates. The green fluorescent protein (GFP) expressing plasmid pTracer-EF/Bsd A EGFP (Invitrogen, Carlsbad, CA) was cotransfected with STOP-FADD and 0-CRE, TOPCMV-CRE or FOPCMV-CRE at a ratio of 0.6:0.2:0.2 µg/well, with 2 µl of Lipofectamine (Invitrogen) per well. Six hours post-transfection, cells were transferred to six-well plates. At 24 h post-transfection, cells were untreated or treated with 5 mM butyrate. The adherent cells were harvested 48 h later (72 h post-transfection) by trypsinization, pelleted at 1000 g and resuspended in 200 µl of 1× phosphate-buffered saline. The number of GFP-positive (viable transfected) cells was measured by flow cytometry. For each experiment, two controls were run for both untreated and butyrate-treated samples. The first control (negative) had no GFP vector (mock transfection); the second control (positive) had the GFP expression vector alone. Positive (GFP-only) control values were used to normalize for differences in transfection efficiency between treatments for each experiment. Flow cytometry was performed using a FACS Vantage flow cytometer (Becton-Dickinson Immunocytometry Systems, San Jose, CA). The cells were excited at 488 nm and the GFP fluorescence was observed through a 510/20 bandpass filter. In each group of samples, the percentage positivity was calculated using CellQuest Pro software by placing a marker to the immediate right of the background fluorescence peak in the mock transfected sample. Fifty thousand cells were observed in each sample in order to render small differences in GFP positivity meaningful.
To measure cell viability, transfections were conducted as described above for the GFP assay. Cells were untreated or treated with 5 mM butyrate for 24 h. Subsequently, 100 000 cells from the total cell population (floating as well as adherent cells) were collected as described above. The staining of the EGFP-expressing cells was performed with the Annexin V-PE apoptosis detection kit 1 (BD Pharmingen, San Diego, CA). The samples were analyzed with a FACS Vantage flow cytometer; cells were excited at 488 nm and the fluorescence of EGFP, Annexin V-PE and 7-amino-actinomycin D was collected through 510/20 BP, 575/26 BP and 660/20 BP filters, respectively. The data were analyzed with CellQuest Pro software.
Statistics
All p values were calculated using Student’s t test; p ≤ 0.05 was considered statistically significant.
RESULTS
Cre-Lox-controlled Wnt-specific luciferase reporter expression in SW620 cells
The Cre-Lox system was analyzed for its ability to enhance Wnt-specific cell death in CRC cells in combination with two effector genes, the FADD (Fas-associated, Death Domain) gene or the diphtheria toxin A-chain (DT-A) gene. The characteristics of a set of Wnt-specific promoters combined with the Cre-Lox system were initially analyzed. The Wnt-specific promoters (TOP) employed represented minimal promoters from different genes to which Tcf/Lef binding sites were attached. Each Wnt-specific promoter had a control promoter (FOP) in which wild-type Tcf/Lef sites were replaced by mutated non-functional sites. We used the floxed luciferase reporter pCMVe-βAc-STOP-Luc (henceforth called STOP-Luc) to determine the relative strength and specificity of the four sets of Wnt-specific promoters (TK, CMV, Fos and TATA box) employed. The expression of luciferase in this system modeled the expression of an effector gene such as FADD or DT-A; therefore, reporter data allowed a precise determination of the effects of pharmacological modulators of Wnt activity. In particular, we were interested in determining the effects of butyrate on Wnt-specific gene expression as mediated by the Cre-Lox system. The ability of butyrate, normally found in the colonic lumen, to modulate Wnt signaling could influence, either positively or negatively, the efficiency and specificity of Wnt-targeted expression of therapeutically relevant genes. The luciferase reporter assay provides a model for ascertaining such effects.
The ability of the TK-, Fos-, TATA- and CMV-CRE constructs (Fig. 1) to stimulate Wnt-specific expression from STOP-Luc in SW620 cells was evaluated in the presence and absence of 5 mM butyrate (Fig. 2). The concentration of CRE expression vectors used (50 pg/well) resulted in very low levels of luciferase activity with the promoterless 0-CRE construct. The 0-CRE vector was employed as a control for the non-specific expression of CRE recombinase and hence for the non-specific expression of the reporter/effector gene. The levels of luciferase activity induced by the wild-type (TOP) version of each CRE construct, which measured the strength of each wild-type promoter, were determined (Fig. 2A). TOPTK-CRE produced a level of luciferase activity ∼5-fold higher than that from TOPFos-CRE (p < 0.001), whereas TOPTATA-CRE led to luciferase expression 20 times lower than luciferase expression from TOPTK-CRE (p < 0.05) and approximately five times lower than that from TOPFos-CRE (p < 0.0001). The level of expression from the TOPCMV-CRE was ∼36% less than that from TOPTK-CRE (p < 0.05). Thus, the relative strength of the promoters was as follows: TOPTK > TOPCMV > TOPFos > TOPTATA.
Figure 1.

Cre-Lox constructs used to stimulate Wnt-specific expression. For each set, copies of either the wild-type (TOP) or mutant (FOP) Tcf-binding site, along with a minimal promoter or a TATA box, was inserted upstream of a modified CRE gene (41).
Figure 2.

Measurement of Wnt-specific expression by SW620 cells. (A, B and C) SW620 cells in a 24-well plate were cotransfected with 0.2 µg of pCMVe-βAc-STOP-Luc (STOP-Luc) and 50 pg of various CRE expression vectors per well, and untreated or treated with 5 mM sodium butyrate for 48 h. To normalize the luciferase values, the pRL-TK vector was cotransfected (30). (D, E and F) Alternatively, 500 pg (10× concentration) or 5 ng (100×) of TOP/FOPTATA-CRE or 500 pg (10×) of TOP/FOPCMV-CRE vectors were transfected along with STOP-Luc and pRL-TK vectors as described in Materials and Methods. (A) Luciferase activity from wild-type (TOP) vectors in the absence of butyrate (1× concentration of CRE vectors). (B) Luciferase activity, similar to (A), in the presence of 5 mM butyrate. (C) The ratio (Wnt specificity ratio) of wild-type (TOP) to mutant (FOP) luciferase expression (T/F) both in the absence and presence of butyrate. (D) Luciferase expression from 10× or 100× TOP vectors in the absence of butyrate. (E) Luciferase expression, similar to (D), in the presence of butyrate. (F) The ratio of TOP to FOP luciferase expression in the absence or presence of butyrate. The input plasmids for each experiment are listed at the top of each graph. Results are from three independent experiments, except for the 10× TOP/FOPTATA-CRE data, which were derived from four separate experiments. Bars, standard deviations.
In the absence of butyrate, the Wnt-specific CRE-mediated activation of STOP-Luc was demonstrated by the very low levels of expression driven by the mutant Wnt-specific promoter vectors (FOP-CRE) and the very high levels of expression stimulated by the wild-type Wnt-specific promoter vectors (TOP-CRE) (Fig. 2C). Thus the luciferase expression driven by TOPTK-CRE was 325-fold higher than that induced by the FOPTK-CRE (p < 0.0001). The luciferase expression stimulated by TOPFos-CRE was 107-fold higher than that caused by FOPFos-CRE (p < 0.001). The luciferase expression produced by TOPTATA-CRE was 137-fold higher than that induced by the mutant FOPTATA-CRE (p < 0.05), while that driven by TOPCMV-CRE was 711-fold higher than that stimulated by FOPCMV-CRE (p < 0.01).
These expression patterns changed (Fig. 2B) when the transfected cells were exposed to 5 mM butyrate, a level that can be attained in the colonic lumen after digestion of dietary fiber (44). The level of expression from STOP-Luc cotransfected with promoterless 0-CRE remained low in the presence of butyrate. However, the level of expression induced by TOPTK-CRE was increased 1.6-fold (not statistically significant) by butyrate, but the level of luciferase activity from FOPTK-CRE was increased by ∼7-fold, resulting in a Wnt specificity ratio (wild-type/mutant ratio of luciferase activity) of 7.4 (p < 0.01) (Fig. 2C). Therefore, exposure to butyrate produced a slight increase in expression induced by the wild-type vector, but at the cost of greatly reduced Wnt specificity. Butyrate increased the level of expression induced by TOPFos-CRE by 7.3-fold (p < 0.02); however, butyrate also increased the luciferase expression induced by FOPFos-CRE. Thus, the Wnt specificity ratio for Fos was 29 (p < 0.01). Luciferase activity induced by TOPTATA-CRE was increased 1.8-fold (not statistically significant) in the presence of butyrate and was significantly lower than that induced by both wild-type TK-CRE (p < 0.01) and Fos-CRE (p < 0.005). While the activity induced by mutant (FOP) TcfTATA-CRE was also enhanced by butyrate, the level of STOP-Luc activation was extremely low; thus, the Wnt specificity ratio for the TATA-based clones was 29 (p < 0.002). For the CMV-CRE vectors, luciferase expression induced by TOPCMV-CRE was increased 2.4-fold (not statistically significant) by butyrate treatment, whereas expression caused by the FOPCMV-CRE was induced 22-fold (p < 0.02), resulting in a Wnt specificity ratio of 76 (p < 0.02). In the presence of butyrate the order of the Wnt specificity ratios for these promoters was CMV > Fos = TATA > TK, which can be contrasted with that observed in the absence of butyrate: CMV > TK > TATA > Fos.
Given the low background luciferase activity observed with FOPTATA-CRE and FOPCMV-CRE, we evaluated whether increasing the concentration of TATA-CRE and CMV-CRE constructs 10-fold would enhance expression without sacrificing specificity. The increased plasmid concentration resulted in higher levels of luciferase activity in both the presence and absence of butyrate (Fig. 2D and E). However, the Wnt specificity of 10× CMV-CRE was decreased by ∼10-fold (710 to 72) in the absence of butyrate and by ∼1.5-fold (76 to 50) in the presence of butyrate (Fig. 2F). The 10× concentration of the TATA-CRE vectors exhibited a high level of specificity in both the absence and presence of butyrate. Thus, in the absence of butyrate, the Wnt specificity ratio was 3096 (p < 0.001), >20-fold higher than that achieved at the lower concentration. In the presence of 5 mM butyrate, expression from the TATA-CRE vectors increased, with a Wnt specificity ratio of 280 (p < 0.001), almost 10-fold higher than at the 1× concentration. Given the high levels of Wnt-specific expression obtained from 10× TATA-CRE, we also transfected SW620 cells with a 100× concentration of the TATA-CRE constructs (Fig. 2D and E). At this concentration the low background expression from the promoterless 0-CRE resulted in markedly elevated luciferase activity, whereas the 100× FOPTATA-CRE still maintained low levels of luciferase activity. Thus, in the absence of butyrate the Wnt specificity ratio was 1220 (p < 0.02), whereas in the presence of butyrate the ratio was 680 (p < 0.001) (Fig. 2F). This latter value represented the highest Wnt specificity observed in butyrate-treated SW620 cells.
As a negative control for Wnt-specific expression in CRC cells, we determined the level of luciferase activity induced by TOP/FOP CMV-CRE and STOP-FADD in RKO CRC cells, which lack endogenous Wnt signaling (45). In both the presence and absence of butyrate, the ratio of luciferase activity induced by TOP versus FOP CMV-CRE was <2.0 (data not shown), which was several orders of magnitude less than that exhibited by SW620 cells (Fig. 2).
Wnt-specific Cre-Lox-mediated kill of SW620 cells
After establishing the characteristics of the Wnt-responsive promoters utilizing luciferase reporter assays, we evaluated the efficiency of the same promoters when driving the expression of the cell death effector gene FADD. Overexpressed FADD has been shown to induce apoptosis (37), and this property has been used to induce CRC-specific cell kill (18). To accomplish these measurements, SW620 cells were cotransfected with a combination of three vectors: the pGL3Control luciferase reporter, the construct [pSTOPFADD (Fig. 1)] containing the floxed FADD cDNA downstream of a CMV-β-actin promoter and the promoterless control vector (0-CRE), and the wild-type (TOP) or mutant (FOP) versions of the four sets of the different Wnt-responsive constructs (Fig. 1). Post-transfection, cells were treated with 5 mM butyrate for 48 h, and the remaining luciferase expression, indicative of cell survival (12,15), was compared with control. The concentration of CRE expression vectors was titrated to maximize the difference between cell kill resulting from expression of the wild-type vector and that of the mutant construct (data not shown).
In the absence of butyrate, less than half (44%, p < 0.05) of the luciferase expression remained after transfection with TOPTK-CRE (0.05 µg/well) (Fig. 3A). No background kill due to FOPTK-CRE was detectable. In addition, when a control vector (STOP-GL3; see Materials and Methods) containing only a stop cassette and no FADD cDNA was used in place of STOP-FADD, no decrease in luciferase activity after transfection with TK-CRE vectors was observed (data not shown). Therefore, the diminished luciferase activity obtained with cotransfection of STOP-FADD and CRE-expressing vectors was due to effector gene expression.
Figure 3.

Wnt-specific SW620 cell kill. SW620 cells in a 24-well plate were transfected with 0.05 µg of pGL3-Ctl, 0.5 µg of STOP-FADD and (A) 0.05 µg of TOP/FOPTK-CRE, (B) 0.0067 µg of TOP/FOPCMV-CRE, (C) 0.1 µg of TOP/FOPFos-CRE or (D) 0.15 µg of TOP/FOPTATA-CRE, with equivalent amounts of the 0-CRE control expression vector per well, and untreated or treated with 5 mM butyrate, and assayed for luciferase expression 48 h later. Luciferase readings were normalized to protein concentration determined for each sample. The input plasmids for each experiment are listed at the top of each graph. Results are from three independent experiments. Bars, standard deviations.
In the absence of butyrate, the relative luciferase activity resulting from transfection of TOPCMV-CRE (0.0067 µg/well) was 34% (p < 0.002) of the control, while FOPCMV-CRE resulted in a slightly reduced level of luciferase activity (82% of control); this latter difference was not statistically significant (Fig. 3B). Transfection of TOPFos-CRE (0.1 µg/well) resulted in a 50% decrease in luciferase activity and a 9% decline (not statistically significant) with the mutant FOPFos vector (Fig. 3C). Consistent with that observed with the reporter assays (Fig. 2), the TATA-box-based promoter required the highest concentration (0.15 µg/well) of the transfected plasmid to achieve cell kill; in addition, also consistent with the reporter assays, the FOPTATA-CRE exhibited markedly lower levels of background activity than that of promoterless 0-CRE. Thus, the decrease in luciferase activity obtained with TOPTATA-CRE was approximately the same as that obtained with the 0-CRE control; however, both exhibited lower activity than the mutant FOPTATA-CRE (Fig. 3D). Thus, luciferase activity after transfection with TOPTATA-CRE was 38% (p < 0.05) of that obtained following transfection of FOPTATA-CRE.
Consistent with the trend observed with the luciferase reporter assays (Fig. 2), treatment with a physiologically relevant concentration (5 mM) of butyrate significantly decreased the Wnt specificity of the STOP-FADD system with the TK and Fos minimal promoters. While relative luciferase expression resulting from TOPTK-CRE was reduced to 18.5% (p < 0.005) of the control, the relative luciferase expression resulting from FOPTK-CRE was reduced to 36.5% (p < 0.02) of the control. Transfection with FOPCMV-CRE followed by exposure to butyrate resulted in a level of luciferase activity (78%) similar to that observed in the absence of butyrate, which was not significantly different from the control. However, luciferase activity resulting from TOPCMV-CRE was further reduced by butyrate to 16% (p < 0.005) of control. Butyrate treatment resulted in a large increase in cell kill from both the TOP (21% of control, p < 0.02) and FOP (39% of control, p < 0.05) versions of Fos-CRE. For TOPTATA-CRE in the presence of butyrate, luciferase activity was decreased to 20% (p < 0.05) of that from FOPTATA-CRE.
Wnt-specific cell kill by CMV-CRE vectors determined by GFP-based assays
To confirm that the luciferase assay used in Figure 3 (and throughout the current study) measures the decrease in cell number resulting from expression of the cell-death-inducing FADD gene, we have performed additional experiments. We have focused on the STOP-FADD/CMV-CRE system in SW620 CRC cells, which produced the most efficient and specific cell kill measured by the luciferase assay (Fig. 3B). SW620 cells were cotransfected with the STOP-FADD and CRE expression vectors and with an expression vector for the green fluorescent protein (GFP), which acted as a marker for transfected cells. Flow cytometry was used to determine the number of GFP-positive adherent cells, comparing control (0-CRE promoterless vector), TOPCMV-CRE and FOP-CMV-CRE transfected cells. This assay is essentially equivalent to the β-galactosidase staining assay for cell survival (18); however, the GFP-based assay is more quantitative and objective, since the flow cytometer examines a large number of cells randomly selected from the total adherent cell population. In the absence of butyrate, transfection of SW620 cells with STOP-FADD and the wild-type TOPCMV-CRE vector resulted in a decrease in the number of adherent GFP-positive cells compared with that of the 0-CRE control (Fig. 4A); the number of GFP-positive cells was reduced by 69% after transfection with TOPCMV-CRE (p < 0.05), but reduced by only 26% after transfection with FOPCMV-CRE (difference compared with control not statistically significant). Thus, as measured by the GFP assay, there is a marked and statistically significant difference (2.4-fold, p < 0.03) between the cell kill induced by TOPCMV-CRE and that caused by FOPCMV-CRE. In the presence of butyrate, GFP positivity was reduced by 78% (p < 0.01) after transfection with TOPCMV-CRE, a 1.4-fold increase in cell kill compared with that observed in the absence of butyrate (p < 0.01). GFP positivity after transfection with FOPCMV-CRE in the presence of butyrate was reduced by only 22% (difference compared with control not statistically significant); this level of GFP positivity was statistically equivalent to that observed with FOPCMV-CRE in the absence of butyrate. The Wnt specificity ratio of relative cell kill by wild-type versus mutant TcfCMV-CRE in the presence of butyrate is 3.6 (p < 0.03). Thus, the results of the GFP assay, which examines the number of remaining transfected cells, matches almost exactly the findings determined by the luciferase assay (Fig. 3B) with respect to the level of cell kill, Wnt specificity and the ability of butyrate to enhance the kill while maintaining, or increasing, the specificity of cell kill.
Figure 4.

GFP-based analysis of Wnt-specific cell kill in SW620 cells transfected with STOP-FADD and CMV-CRE vectors. (A) SW620 cells in a 24-well plate were transfected with 0.6 µg of GFP expression vector, 0.2 µg of STOP-FADD and 0.2 µg of 0-CRE or CMV-CRE vectors per well; 5 h post-transfection each well was transferred into a well of a 6-well plate. Twenty-four hours post-transfection, cells were untreated or treated with 5 mM butyrate and assayed 48 hours later. (B) SW620 cells were transfected and treated as above (for 24 h), and cells were analyzed with annexin and 7-amino-actinomycin D staining for cell viability, apoptosis and necrosis. The input plasmids for each experiment are listed at the top of each graph. The ratio of apoptotic and necrotic versus live cells [(A + N)/L] is shown. Results are from four independent experiments for the absence of butyrate, and three independent experiments for the presence of butyrate. Bars, standard deviations.
We have also determined the relative numbers of apoptotic and necrotic versus live cells in the total (floating as well as adherent) transfected cell population, after conducting transfections in the same manner as performed in Figure 4A. Samples were analyzed after 24 h to observe earlier stages of cell death. It is important to note that apoptosis is a dynamic process (viable cells to apoptosis to necrosis), while the main assay utilized in this study, i.e. luciferase confirmed by GFP cell count (Fig. 4A), determines the final level of remaining transfected cells. Thus small differences in the rate of apoptosis at any given time during the transfection results in a large difference in the final number of surviving cells. As expected, the ratio of apoptotic and necrotic versus live cells [(A + N)/L)] was highest after transfection with the wild-type TOPCMV-CRE vector, which has been shown to result in the largest decrease in luciferase expression (Fig. 3B) and cell number (Fig. 4A). The (A + N)/L ratio with TOPCMV-CRE transfection was 3-fold higher (p < 0.002) than that observed with the promoterless control, while the ratio after transfection with the mutant FOPCMV-CRE vector was similar to that observed with control (difference not statistically significant). The same patterns were observed after treatment with butyrate (Fig. 4B); the (A + N)/L ratio after transfection with TOPCMV-CRE was 3.3-fold higher than control (p < 0.001), while the ratios from the FOPCMV-CRE and control vectors were statistically equivalent.
The data presented in Figure 4 demonstrate that the luciferase assay measures the increase in cell death resulting from Wnt-specific expression of the effector. Thus, we used the sensitive and high-throughput luciferase assay to further examine aspects of Wnt-targeted expression in CRC cells.
Cre-Lox-mediated SW620 cell kill induced by DT-A expression
To determine whether a DT-A effector would be as effective and specific as the FADD effector in downregulating luciferase activity, SW620 cells were cotransfected with the CMV-CRE expression vectors (0.005 µg/well), the pGL3 control luciferase reporter and a new construct containing a floxed DT-A gene fragment [STOP-DT (Fig. 1)]; these cells were treated with 5 mM sodium butyrate for 48 h. For these experiments, the CMV-CRE constructs were used, since this set of expression vectors exhibited high efficacy and specificity in both untreated and butyrate-treated STOP-FADD transfected SW620 cells (Fig. 3).
In the absence of butyrate, the efficiency and Wnt specificity of SW620 cell kill was similar to that achieved with the FADD effector (Fig. 5A). Luciferase expression after transfection with TOPCMV-CRE and STOP-DT was reduced to 32% (p < 0.02) of the control, while FOPCMV-CRE resulted in a statistically non-significant 13% decline in the relative luciferase activity. In the presence of butyrate, luciferase activity in the presence of the TOPCMV-CRE vector was reduced to 20% (p < 0.01) of the control and the background activity of FOPCMV-CRE increased, resulting in luciferase activity being reduced to 61% of control (not statistically significant).
Figure 5.

SW620 cell kill utilizing the diphtheria toxin A-chain (DT-A) effector. SW620 cells in a 24-well plate were transfected with 0.05 µg of pGL3-Ctl and (A) 0.5 µg of STOP-DT and 0.0067 µg of TOP/FOPCMV-CRE expression vectors per well, or (B) 0.15 µg of STOP-DT and 0.03 µg of TOP/FOPCMV-CRE expression vectors per well, and untreated or treated with 5 mM butyrate, and assayed for luciferase expression 48 h later. Luciferase readings were normalized to protein concentration determined for each sample. The input plasmids for each experiment are listed at the top of each graph. Results are from three independent experiments. Bars, standard deviations.
Given the potential potency of the DT-A effector, we attempted to further optimize the effector and CRE expression vector concentrations. The concentration of STOP-DT was lowered to 0.15 µg/well, while the concentration of the CRE expression vectors was increased to 0.03 µg/well. At these concentrations, in the absence of butyrate, luciferase expression in the presence of TOPCMV-CRE was reduced to 21% (p < 0.02) of control, while FOPCMV-CRE resulted in a statistically non-significant 25% decline in relative luciferase activity (Fig. 5B). In the presence of butyrate, cotransfection with TOPCMV-CRE further reduced the luciferase activity by approximately two-thirds (p < 0.001), to 7.5% of control, whereas cotransfection of FOPCMV-CRE and STOP-DT butyrate further reduced luciferase activity by approximately one-third (p < 0.002), to 51% of the control. Thus, compared with STOP-FADD, the STOP-DT vector resulted in enhanced cell kill, but in the presence of butyrate non-specific kill was markedly higher with the DT-A effector compared with the FADD effector. Therefore, in view of its greater specificity in the presence of butyrate treatment, we have utilized the FADD gene as the effector gene for the remainder of these studies.
Cre-Lox-controlled Wnt-specific luciferase reporter expression in HCT-116 cells
We have previously shown that HCT-116 CRC cells (APC+/+, β-catenin+/–) differ from SW620 CRC cells (APC+/–) in their endogenous levels of Wnt activity, modulation of Wnt activity by butyrate and specific response of different Wnt-specific promoters to butyrate treatment (29,30,35). Compared with HCT-116 cells, SW620 cells exhibit higher levels of free β-catenin and β-catenin–Tcf transcriptional complexes (33,34), as well as higher levels of Wnt signaling both in the presence and absence of butyrate (29,30,35). The HCT-116 cell line, on the other hand, exhibits higher levels of non-Wnt specific transcriptional activity from certain minimal promoters. For example, luciferase reporter assays have shown that the minimal TK promoter exhibits an ∼50-fold higher level of activity in HCT-116 cells than in SW620 cells (30). Therefore, we evaluated Wnt-specific Cre-Lox-mediated luciferase expression in HCT-116 cells as an example of a Wnt-positive CRC cell line that exhibits low levels of Wnt activity and high levels of background promoter activity, thereby being a considerable challenge for Wnt-targeted gene expression.
In the absence of butyrate, transfection of the four sets of Tcf-CRE vectors into HCT-116 cells resulted in lower levels of luciferase reporter activity than observed in SW620 cells (Fig. 6A). The TOPTK-CRE construct produced luciferase activity that was ∼5-fold lower than that observed in SW620 cells. However, the activity of the FOPTK-CRE construct was very high in HCT-116 cells, and thus the Wnt specificity ratio of 2.0 was not statistically significant. TOPFos-CRE generated luciferase activity ∼2.5-fold lower than that observed in SW620 cells, with a Wnt specificity ratio of 6.9 (p < 0.03). TOPTATA-CRE gave very low levels of luciferase activity that were only 2.2-fold higher than the promoterless control and >5-fold lower than that observed in SW620 cells. Although the Wnt specificity ratio for this promoter was 5.5, owing to variability associated with the low luciferase readings this ratio was not statistically significant. In the absence of butyrate, the CMV-CRE constructs exhibited both the highest levels of luciferase activity and the greatest Wnt specificity ratio of 10 (p < 0.03). The order of the Wnt specificity ratios for these promoters in HCT-116 cells was as follows: CMV > Fos >TATA > TK (Fig. 6C).
Figure 6.

Wnt-specific expression by HCT-116 cells. HCT-116 cells in a 24-well plate were transfected with 50 pg each of the CRE expression vectors with STOP-Luc and pRL-TK, and treated and assayed as described in Figure 2. (A) Luciferase activity from the TOP vectors in the absence of butyrate or lithium. (B) Luciferase expression from the TOP vectors in the presence of 5 mM butyrate. (C) The Wnt specificity ratio (T/F) in the absence or presence of butyrate. (D) Luciferase expression from TOPCMV-CRE in the presence of 20 mM LiCl or 20 mM LiCl plus 5 mM butyrate combined. (E) The Wnt specificity ratio in the presence of LiCl or LiCl plus butyrate. The input plasmids for each experiment are listed at the top of each graph. Results are from three independent experiments. Bars, standard deviations.
Treatment of HCT-116 cells with butyrate enhanced the ability of all of the promoters to express CRE and to induce luciferase activity (Fig. 6B), whereas at the same time the Wnt specificity ratios were altered. Thus, butyrate treatment resulted in an 11-fold induction (p < 0.005) of luciferase activity by TOPTK-CRE; however, the Wnt specificity ratio was further reduced to 1.8 (p < 0.05). Luciferase activity resulting from TOPFos-CRE was increased 29-fold (p < 0.003) by butyrate and the Wnt specificity ratio was slightly increased to 8.2 (p < 0.005). The TATA-CRE vectors exhibited the highest Wnt specificity ratio of 13 (p < 0.001) in the presence of butyrate, with butyrate inducing a 37-fold (p < 0.001) increase in luciferase activity from TOPTATA-CRE. Butyrate also increased by 9.2-fold (p < 0.005) the induction of luciferase activity from TOPCMV-CRE, resulting in a Wnt specificity ratio of 6.9 (p < 0.01). Thus, in the presence of butyrate, the order of the Wnt specificity ratios in HCT-116 cells was as follows: TATA > Fos > CMV > TK (Fig. 6C).
LiCl enhanced Wnt-specific Cre-Lox-mediated gene expression in HCT-116 cells
Compared with SW620 cells, the overall levels of Cre-Lox-mediated luciferase activity, as well as of Wnt specificity, were lower in HCT-116 cells. Therefore, we ascertained whether LiCl would improve the levels and specificity of Wnt-mediated expression in HCT-116 cells, since LiCl is known to enhance Wnt activity by increasing levels of nuclear active β-catenin (32). While the APC mutant SW620 cells were unaffected by lithium treatment, because the APC mutation had already allowed for the maximum possible level of β-catenin stabilization (unpublished data), Wnt activity in APC wild-type HCT-116 cells was markedly enhanced by 20 mM LiCl (unpublished data). In these experiments, we evaluated the activity of the CMV-CRE system, which exhibited the greatest efficiency coupled to specificity in HCT-116 cells (Fig. 6A–C).
Treatment with LiCl alone resulted in a marked 6.2-fold (p < 0.005) upregulation of luciferase activity induced by TOPCMV-CRE and an increase in Wnt specificity to 24, compared with 10 with untreated cells (Fig. 6D and E). Cotreatment with lithium and butyrate enhanced expression more than with either reagent alone. Thus, in the presence of both reagents, luciferase activity induced by TOPCMV-CRE was 3.4-fold higher (p < 0.003) than in the presence of butyrate alone, and 5-fold higher (p < 0.002) than in the presence of lithium alone (Fig. 6D). The Wnt specificity ratio in the presence of both reagents was 14, lower than that of lithium alone, but approximately twice that observed in the presence of butyrate alone (Fig. 6E).
Wnt-specific Cre-lox-mediated cell kill of HCT-116 cells
To determine whether lithium and butyrate would also improve the efficacy of Cre-Lox-mediated kill of HCT-116 cells, we analyzed the effects of CMV-CRE vectors coupled to the FADD effector. Unlike that observed with SW620 cells (Fig. 3), no Wnt-specific kill was obtained with untreated HCT-116 cells (Fig. 7). Luciferase activity resulting from the transfection of TOPCMV-CRE was 82% of control, while luciferase activity resulting from transfection of FOPCMV-CRE was 90% of control. Neither decline in luciferase activity was statistically significant. These findings are consistent with weaker levels of expression of CRE in HCT-116 cells compared with SW620 cells (Figs 2 and 6). In the presence of 5 mM butyrate, luciferase activity in TOPCMV-CRE transfected cells was only 25% (p < 0.02) of control; however, there was also a large decline (49%) in luciferase activity in mutant FOPCMV-CRE transfected cells, indicative of substantial background kill. Surprisingly, treatment with 20 mM LiCl did not enhance cell kill, with levels of luciferase activity after transfection with TOP or FOP CMV-CRE being 87 and 95% of control, respectively (neither change was statistically significant). However, the combination of both butyrate and lithium resulted in an optimal level of cell kill and Wnt specificity. In the presence of both agents, luciferase activity of TOPCMV-CRE transfected cells was 31% (p < 0.002) of control, an ∼2.5-fold more efficient cell kill (p < 0.01) than that occurring in the absence of either agent. The background kill produced by FOPCMV-CRE resulted in luciferase activity that was 67% of control. This Wnt specificity ratio of 2.2 was statistically significant (p < 0.05), while for the other three treatments (no reagent, butyrate alone and lithium alone), the Wnt specificity ratio was not statistically significant. Therefore, cotreatment of HCT-116 cells with butyrate and lithium yielded the most effective results in conjunction with the CMV-CRE/STOP-FADD system.
Figure 7.
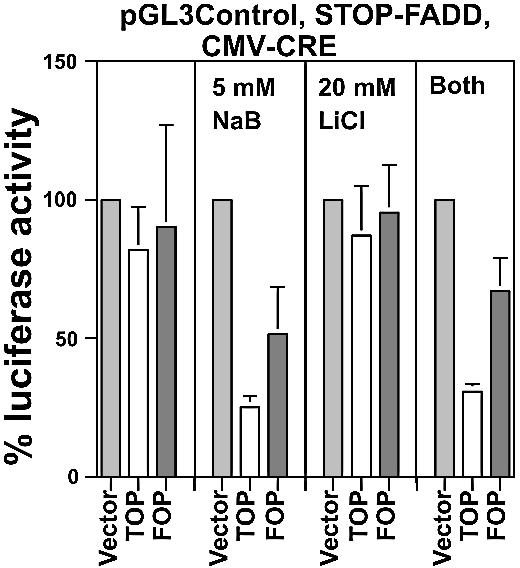
Wnt-specific HCT-116 cell kill. HCT-116 cells in a 24-well plate were transfected with 0.05 µg of pGL3-Ctl, 0.5 µg of STOP-DT and 0.0067 µg of TOP/FOPCMV-CRE expression vectors per well, and untreated or treated with 20 mM LiCl and 5 mM butyrate, and assayed for luciferase expression 48 h later. Luciferase readings were normalized to protein concentration determined for each sample. The input plasmids for the experiment are listed at the top of the graph. Results are from three independent experiments. Bars, standard deviations.
Lef1-VP16 fusion vector enhanced Wnt-specific cell kill
To increase the efficiency of the CRE/STOP-FADD system, we created the STOP-LefVP16 vector, in which expression of Lef1-VP16, which enhances transcription from Tcf/Lef-site containing promoters (43), is dependent upon the activity of CRE-recombinase, in a manner analogous to the STOP-FADD vector. Transfection of this vector along with Wnt-responsive CRE expression vectors would result in Wnt-specific expression of Lef-VP16, which in turn would enhance CRE expression from the TOP-CRE vectors. This enhanced CRE expression would boost the expression of cotransfected floxed genes in the cell (e.g. FADD, Lef-VP16). Therefore, cotransfection with STOP-LefVP16 is a genetic method of upregulating Wnt-specific gene expression and, in conjunction with STOP-FADD, of increasing Wnt-targeted cell death.
A pilot experiment was performed to determine whether cotransfection of STOP-LefVP16 with Wnt-responsive CRE expression vectors enhanced Wnt activity in a Wnt-specific manner (Fig. 8A). To measure changes in Wnt activity the TOPFLASH and FOPFLASH luciferase reporters were utilized, since the ratio of their activities (TOP/FOP) measures specific Wnt activity (5,29,30). Either TOPFLASH or FOPFLASH were cotransfected with STOP-LefVP16 and one of the following CRE expression vectors, promoterless 0-CRE (negative control), CMV-CRE (positive control), wild-type TOPTK-CRE or mutant FOPTKCRE, and the TOP/FOP ratio of luciferase activity from each combination of transfected vectors was compared. The TOP/FOP ratio resulting from mutant FOPTK-CRE was approximately the same as that of the promoterless negative control 0-CRE, whereas the TOP/FOP ratio of TOPTK-CRE (5-fold higher than 0-CRE) and CMV-CRE (5.5-fold higher than 0-CRE) were similar. These findings demonstrated that STOP-LefVP16, when activated by a TOP-CRE vector, can further upregulate Wnt activity; however, a FOP-CRE vector cannot activate STOP-LefVP16, ensuring specificity.
Figure 8.

Enhanced Wnt-specific expression produced by STOP-LefVP16. (A) SW620 cells in a 24-well plate were transfected with 0.1 µg of TOPFLASH or FOPFLASH, 0.2 µg of STOP-LefVP16, 0.005 µg of 0-CRE, TOPTK-CRE or FOPTK-CRE, and normalized with pRL-TK as described (30). (B) SW620 cells in a 24-well plate were transfected with 0.05 µg of pGL3-Ctl, 0.5 µg of STOP-FADD, 0.25 µg of STOP-LefVP16 and 0.05 µg of TOP/FOPCMV-CRE expression vectors per well, and untreated or treated with 20 mM LiCl and 5 mM butyrate, and assayed for luciferase expression 48 h later. (C) SW620 and (D) HCT-116 cells in a 24-well plate were transfected with 0.05 µg of pGL3-Ctl, 0.5 µg of STOP-FADD, 0.25 µg of STOP-LefVP16 and 0.0067 µg of TOP/FOPCMV-CRE expression vectors per well, and untreated or treated with 5 mM butyrate, and assayed for luciferase expression 48 h later. (B, C and D) Luciferase readings were normalized to protein concentration determined for each sample. (A) Results are from one experiment in triplicate. (B–D) The input plasmids for each experiment are listed at the top of each graph. Results are from three independent experiments. Bars, standard deviations.
The ability of STOP-LefVP16 to upregulate Wnt-specific kill of SW620 cells was analyzed with TOP/FOPTK-CRE and STOP-FADD (Fig. 8B). In the absence of butyrate, the cotransfection of TOPTK-CRE, STOP-FADD and STOP-LefVP16 reduced luciferase activity to 24% (p < 0.001) of control, which is almost 2-fold lower (p = 0.5) than that observed in the absence of STOP-LefVP16 (Fig. 2A). Luciferase activity from the cotransfection of FOPTK-CRE, STOP-FADD and STOP-LefVP16 was 91% of control, statistically equivalent to that observed in the absence of STOP-LefVP16 (Fig. 3A). In the presence of butyrate, TOPTK-CRE, STOP-FADD and STOP-LefVP16 reduced luciferase activity to 13% (p < 0.05) of control; FOPTK-CRE and the two STOP vectors reduced luciferase activity to 28% of control, a value statistically equivalent to that observed with TOPTK-CRE. This low Wnt specificity in the presence of butyrate was characteristic of the TK-CRE constructs and was also observed in the absence of STOP-LefVP16 (Fig. 3A).
The ability of STOP-LefVP16 to upregulate Wnt-specific kill of SW620 cells was analyzed with TOP/FOPCMV-CRE (Fig. 8C). In the absence of butyrate, TOPCMV-CRE, STOP-FADD and STOP-LefVP16 reduced luciferase activity to 15% of control (p < 0.005), which was >2-fold lower (p < 0.01) than that observed in the absence of STOP-LefVP16 (Fig. 3B). Luciferase activity expressed from FOPCMV-CRE and the two STOP vectors was 73% of control, statistically equivalent to that observed in the absence of STOP-LefVP16 (Fig. 3B). In the presence of butyrate, TOPCMV-CRE, STOP-FADD and STOP-LefVP16 together reduced luciferase activity to 11% (p < 0.003) of control; FOPCMV-CRE and the two STOP vectors reduced luciferase activity to 58% of control. These levels of luciferase activity were statistically equivalent to that observed in the absence of STOP-LefVP16 (Fig. 3B).
The ability of STOP-LefVP16 to upregulate Wnt-specific kill of HCT-116 cells with TOP/FOPCMV-CRE was also determined and the results are shown in Figure 8D. In the absence of butyrate, TOPCMV-CRE, STOP-FADD and STOP-LefVP16 reduced luciferase activity to 37% (p < 0.005) of control, which is >2-fold lower (p < 0.03) than that observed in the absence of STOP-LefVP16 (Fig. 7). Luciferase activity from FOPCMV-CRE and the two STOP vectors was 57% of control, statistically equivalent to that observed in the absence of STOP-LefVP16 (Fig. 7). In the presence of butyrate, TOPCMV-CRE, STOP-FADD and STOP-LefVP16 together reduced luciferase activity to 17% (p < 0.002) of control; this result represents a statistically significant decrease in luciferase activity (p < 0.03) compared with the absence of STOP-LefVP16 (Fig. 7). FOPCMV-CRE and the two STOP vectors reduced luciferase activity to 49% of control, statistically equivalent to that observed in the absence of STOP-LefVP16 (Fig. 7).
We hypothesized that cotransfection with STOP-LefVP16 would ensure adequate cell kill even in the presence of suboptimal levels of the CRE expression vector, a property potentially useful in gene therapy. Therefore, we utilized a 10-fold lesser concentration of the TK-CRE expression vectors, i.e. the same concentration used in Figure 8A, in both the absence (Fig. 9A) and the presence (Fig. 9B) of cotransfected STOP-LefVP16. In the absence of Lef-VP16 a 10-fold dilution of TOPTK-CRE resulted in a low level of cell kill in the absence of butyrate. In the presence of cotransfected STOP-LefVP16, the more dilute concentration of TOPTK-CRE reduced luciferase activity to 33% (p < 0.03) of control, a 2-fold enhancement of cell kill compared with that observed in the absence of STOP-LefVP16 (Fig. 9A). Thus, STOP-LefVP16 markedly enhanced effector activity resulting from 10-fold lower levels of CRE vectors. In the presence of butyrate and STOP-LefVP16, dilute TOPTK-CRE and FOPTK-CRE reduced luciferase activity to low levels statistically equivalent to that observed in the absence of Lef-VP16.
Figure 9.

Enhanced effects of diluted CRE expression produced by STOP-LefVP16. (A) SW620 cells in a 24-well plate were transfected with 0.05 µg of pGL3-Ctl, 0.5 µg of STOP-FADD and 0.005 µg of TOP/FOPTK-CRE, with equivalent amounts of the 0-CRE control, expression vectors per well, and untreated or treated with 5 mM butyrate for 48 h. (B) SW620 cells were transfected as in (A), except that 0.25 µg of STOP-LefVP16 was cotransfected. The input plasmids for each experiment are listed at the top of each graph. Results are from three independent experiments. Bars, standard deviations.
Wnt-specific downregulation of Wnt activity in SW620 cells
To determine if the Cre-Lox system would allow for alternative therapeutic approaches for Wnt-positive CRC other than those that lead to direct cell kill, we considered the possibility of downregulating the Wnt activity that has been related to high cell proliferation in CRC cells. Dominant-negative Tcf4 (DN-Tcf4) is an N-terminal deletion of Tcf4 that retains the ability to bind to DNA but cannot bind β-catenin (5). Thus, DN-Tcf4 acts as a dominant-negative analog of Tcf4, inhibiting Wnt signaling by blocking the access of transcriptionally active β-catenin–Tcf4 complexes to DNA binding sites. Through this mechanism, DN-Tcf4 downregulates CRC cell proliferation by inhibiting the expression of Wnt target genes such as cyclin D1 (46). Therefore, we determined whether activation of the expression of DN-Tcf4 by constitutive Wnt activity in CRC cells leads to Wnt-specific inhibition of Wnt activity.
This was accomplished by constructing a floxed DN-Tcf4 (STOP-DNTcf4) plasmid, similar in structure to the floxed FADD and DT-A vectors described in Figure 1. The floxed DN-Tcf4 vector was transfected simultaneously with the TOPFLASH or FOPFLASH luciferase reporters, and with the promoterless 0-CRE, TOPCMV-CRE or FOPCMV-CRE vectors. The ratio of luciferase activity of TOPFLASH to that of FOPFLASH (TOP/FOP) is a measure of Wnt activity. The combination of TOPCMV-CRE with STOP-DNTcf4 resulted in a 63% (p < 0.001) decline in Wnt activity compared with that observed with the combination of 0-CRE and STOP-DNTcf4 (Fig. 10). In contrast, background activity measured with the combination of FOPCMV-CRE with STOP-DNTcf4 resulted in only a 15% decline in Wnt activity, which was not statistically significant, compared with that observed with the 0-CRE control. Thus, it was possible to inhibit Wnt activity in a manner dependent on the initial presence of constitutively active Wnt signaling.
Figure 10.
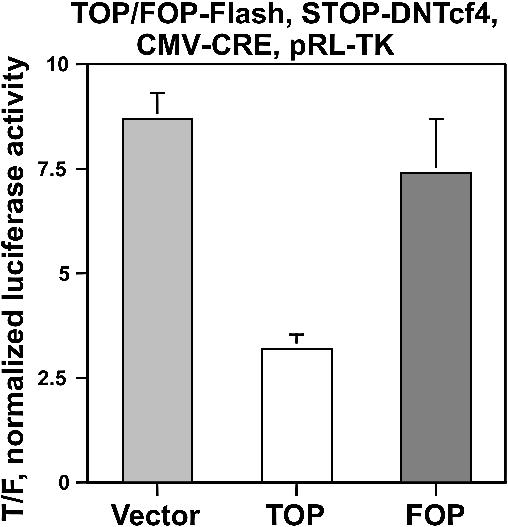
Wnt-specific downregulation of Wnt activity. SW620 cells in a 24-well plate were transfected with 0.1 µg of pTOPFLASH or pFOPFLASH, 0.3 µg of STOP-DNTcf4 and 0.004 µg of TOP/FOPCMV-CRE expression vectors per well, and assayed for luciferase expression 48 h later. Luciferase readings were normalized to cotransfected pRL-TK (30). The input plasmids for each experiment are listed at the top of each graph. Results are from three independent experiments. Bars, standard deviations.
DISCUSSION
The Wnt signaling pathway is constitutively activated in most CRCs and therefore has been viewed as a target for the Wnt-specific expression of therapeutic genes (18,22,24,25). A Wnt-targeted gene therapy system can be of particular practical use in the treatment of extensive CRC tumors as an adjunct or alternative to surgery, particularly as an alternative to total colonectomy, in the treatment of CRC metastases (the SW620 cell line is derived from a lymph node metastasis) or in the treatment of patients with familial adenomatous polyposis coli (FAP) as an alternative to prophylactic surgery. Some in vitro and in vivo success using Wnt-targeted systems has been reported (10,18,22); however, no systematic examination of Wnt-targeted expression, encompassing promoter identity, cell type and methods of enhancing efficacy, has been reported. The present study is the first comprehensive report that couples the specificity of Wnt-targeted expression with the Cre-Lox system (Fig. 11).
Figure 11.

Schematic summary of Wnt-targeted Cre-Lox expression. Wnt activity stimulates a Tcf-minimal promoter resulting in the expression of CRE recombinase. The activity of CRE excises the STOP cassette, allowing expression of a floxed gene, which in the case of the present study can be luciferase (reporter), FADD, DT-A, Lef-VP16 or DN-Tcf4 (effectors). Expression of FADD or DT-A causes cell death, while Lef-VP16 and DN-Tcf4 modulate Wnt activity positively or negatively, respectively. Wnt activity can also be modulated by butyrate or lithium, with effects on Cre-Lox-mediated expression.
We describe cell-type differences resulting in varying levels of expression from different Wnt-specific promoters, as well as differences in the modulation of this expression by pharmacological and genetic agents. Perhaps the most important factor heretofore ignored in considering Wnt-targeted gene therapy is the presence of butyrate in the colonic lumen. Butyrate can be directly introduced into the colonic lumen or given orally to patients as tributyrin (47). Butyrate is also present at significant physiological concentrations after consumption of fiber-rich foods (44). Butyrate is known to significantly modulate the Wnt signaling pathway in a promoter- and cell-type-specific manner at concentrations within the range normally found in the colonic lumen (29,30). Therefore, both natural and exogenously added butyrate can influence Wnt-targeted therapeutic gene expression, and its influence on the expression of genes of interest must be considered.
In the present study, we demonstrate that butyrate enhances CRE-driven luciferase; however, it decreases the Wnt specificity of gene expression (Fig. 2). Both of these effects can have significant consequences for Wnt-targeted gene therapy. In SW620 cells, the CMV-CRE vector was the most efficient at the lowest concentration of transfected plasmid, combining high Wnt specificity with high expression in both the presence and absence of butyrate. We established that the presence of butyrate alters the levels of expression and specificity of different Wnt-sensitive promoters. Thus, in butyrate-treated SW620 cells, the Fos-CRE vector has higher expression and specificity than the TK-CRE vector, while in the absence of butyrate the TK-CRE vector is the more effective. TATA-CRE vectors, however, exhibited high levels of Wnt specificity in both the presence and absence of physiological concentrations of butyrate. These same differences in activity were observed when the FADD gene was used as an effector (Fig. 3). Therefore, Wnt-specific kill of SW620 CRC cells differs with respect to promoter type and is significantly influenced by treatment with butyrate.
HCT-116 cells exhibited similarities to SW620 cells in that butyrate treatment had a marked effect on Wnt-specific Cre-Lox-mediated gene expression (Fig. 6) and cell kill (Fig. 7), and that varying promoter efficiencies existed even in the absence of butyrate. Therefore, both SW620 and HCT-116 cells exhibit marked and statistically significant changes in the efficiency and specificity of Wnt-targeted expression and effector activity after treatment with physiologically relevant levels of butyrate. In this regard, the CMV-CRE vectors appeared most specific and efficient, even in the presence of butyrate (Figs 3 and 7). However, marked differences were evident between these two cell lines with respect to promoter activity, Wnt specificity and butyrate responsiveness. For example, the Wnt specificity of the TK promoter was better in SW620 cells than in HCT-116 cells, while that of the Fos promoter was better in HCT-116 cells than in SW620 cells (Figs 2 and 6). To improve the performance of the Wnt-mediated gene expression system in HCT-116 cells, we attempted to increase Wnt levels in these cells by treatment with LiCl. Lithium has long been used for the treatment of human psychiatric disorders and is known to stimulate Wnt signaling in certain cell lines (31); therefore, it was reasonable to view lithium treatment as a means of enhancing the efficacy of Wnt-targeted expression and effector activity. Surprisingly, the upregulation of Wnt-specific Cre-Lox-mediated expression produced by lithium, as assayed by the luciferase reporter (Fig. 6D), did not translate into enhanced HCT-116 cell kill. A specific threshold of CRE expression necessary to activate floxed FADD sufficiently might well be responsible for this phenomenon, and this threshold was not reached with LiCl treatment of HCT-116 cells. In addition, a fundamental difference exists in the dynamics of a luciferase reporter system compared with a cell kill effector expression model. In the case of luciferase reporter assays, the activity of the reporter generally correlates with the level of expression of the reporter gene; thus, the more luciferase produced, the higher is the luciferase activity within the confines of its linear range of detection. Alternatively, once a level of FADD or DT-A sufficient to kill a given cell with near 100% certainty is reached, further enhanced expression of that gene is superfluous. In the case of such an effector, a rough correlation between expression and effect would exist only within a relatively narrow range of suboptimal effector expression. At the lower end of the range effector levels are too low to result in any detectable cell kill, and at the upper end of the range the probability of cell kill reaches ∼100%; therefore, a graph of concentration versus probability of kill levels off. It is possible that these differences in reporter versus effector dynamics are responsible for the observed differences between the effects of LiCl on luciferase compared with its effects on FADD activity in the HCT-116 cell line.
The differences between HCT-116 and SW620 cells demonstrate that cell-specific effects on promoter activity and responsiveness to butyrate can influence Wnt-specific Cre-Lox-mediated expression. Therefore, what is most effective in one CRC subtype may not be best in another. The type of Wnt-activating mutation present, which affects the responsiveness to lithium, the endogenous levels of Wnt activity (18,29,30,33–35) and the further modulation of Wnt activity by butyrate present in the colonic lumen or added by oral treatment with tributyrin (47), are all examples of cell-specific effects that need to be considered in the design and application of Wnt-targeted therapeutics. HCT-116 and SW620 CRC cells may represent types of CRCs which would exhibit different responses to Wnt-targeted gene therapy, particularly in the presence of physiological concentrations of butyrate. Tissue biopsies of CRCs or of colonic tissue of patients with FAP might well be evaluated for levels of nuclear β-catenin via immunocytochemistry and/or the relative levels of β-catenin–Tcf complexes could be ascertained, both of which could suggest the potential level of Wnt activity in CRC cells. Establishing the type of Wnt-activating mutation, if any, would be useful, since APC wild-type Wnt-positive tumors or adenomas, exemplified by HCT-116 cells, may be specifically sensitive to lithium-induced modulation of Wnt-targeted effector expression. In addition, the malignant cells of CRCs may differ in their levels of non-Wnt transcription factors, thereby exhibiting differential levels of expression due to background activity from minimal promoters. Finally, different CRC cells may differ in their sensitivity to the effector gene used.
We explored the possibility of genetically modulating Wnt-targeted CRCs utilizing the Lef-VP16 fusion protein. Figures 8 and 9 demonstrate that cotransfection with STOP-LefVP16 enhances Wnt-specific expression (Fig. 8A) and cell kill (Figs 8B–D and 9) in the absence of butyrate. However, in the presence of butyrate, addition of Lef-VP16 does not result in further enhancement of Wnt-specific cell kill, most likely because the system has already reached its maximal efficiency. Figure 9 demonstrates that the LefVP16 fusion can effectively enhance Wnt-specific cell kill in the presence of suboptimal concentrations of the CRE expression vector. Therefore, a STOP-LefVP16 construct could be incorporated as part of a transfected/infected suite of Wnt-targeted genes to specifically enhance the cell kill of Wnt-activated cancer cells. The STOP-LefVP16 vector methodology in a more general sense can be used to enhance activity from any targeted Cre-Lox system. If, for example, it is known that transcription factor ‘X’ binds to a promoter ‘Y’ that is used to drive CRE expression, then X can be fused to the transactivation domain of VP16 and inserted into a floxed vector. CRE expression from promoter Y would induce X-VP16 fusion activity, which would feed back to further stimulate Y-promoter activity, resulting in enhanced CRE expression and enhanced downstream activity of any other cotransfected or co-infected floxed genes.
The Wnt activity downregulatory system designed by us (Fig. 10) demonstrates that the Cre-Lox system can be utilized to drive various therapeutic methodologies. The Wnt-driven downregulation of Wnt signaling is novel and, as established in this report, is successful in vitro. Downregulation of Wnt activity has been shown to increase levels of drug-induced apoptosis (48) and to inhibit proliferation (46). Therefore, Wnt-dependent downregulation of Wnt activity could prime CRC cells to be effectively killed by other therapeutic approaches, including those used in the present study. The self-downregulation method can also be more generally applied to any situation in which a dominant-negative protein inhibits a signaling pathway essential for cell growth and/or survival
Although the current study is of a model system utilized to investigate fundamental aspects of Wnt-targeted Cre-Lox-mediated gene expression, one can speculate on how such a multicomponent system could be applied in vivo to the clinical and/or preclinical situation. One methodology for introducing genes for therapeutic applications is by viral infection. We note that a multicomponent Cre-Lox/prodrug expression system was successfully used in mouse models of gastric cancer and CRC, utilizing multiple infections with adenoviruses containing the Cre and Lox components of the system; significant beneficial effects measured as the reduction of tumor burden, prevention of metastasis and enhanced survival were observed (39,42,49). The same principle of multiple applications of the components of the expression system can be used for the introduction of these components by the application of plasmids, either as naked DNA or complexed with cationic lipids. Kaczmarczyk and Green (41) demonstrated the feasibility of placing both the Cre and Lox components of the system in the same vector, which would greatly simplify the clinical use of a Cre-Lox approach similar to that presented in this report. Thus, the Wnt-targeted Cre-Lox system described in the present study can be tested in vivo, utilizing current methodologies and experimental models. In summary, our report suggests that a Wnt-targeted Cre-Lox system has potential in the treatment of CRC, and that considerations such as the action of butyrate on Wnt signaling are important to maximize the effectiveness of such a system.
Acknowledgments
ACKNOWLEDGEMENTS
We wish to thank all the investigators who provided us with required reagents; their contributions are listed in Materials and Methods. We thank Dr Stanislaw Kaczmarczyk for helpful discussions on Cre-Lox plasmid cloning and on the reporter assay titrations. We also thank Dr Philip Penketh for assistance in formatting the figures. This research was supported in part by a grant from the American Institute for Cancer Research, and by NCI Core Grant CA-16359 to the Yale Cancer Center.
REFERENCES
- 1.Su L.-K., Vogelstein,B. and Kinzler,K. (1993) Association of the APC gene product with β-catenin. Science, 262, 1731–1734. [DOI] [PubMed] [Google Scholar]
- 2.Munimetsu S., Albert,I., Souza,B., Rubinfeld,B. and Polakis,P. (1995) Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc. Natl Acad. Sci. USA, 92, 3046–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Molenaar M., van de Wetering,M., Oosterwegel,M., Peterson-Maduro,J., Godsave,S., Korinek,V., Roose,J., Destree,O. and Clevers,H. (1996) XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell, 86, 391–399. [DOI] [PubMed] [Google Scholar]
- 4.Behrens J., Von Kries,J.P., Kuhl,M., Bruhn,L., Wedlich,D., Grosschedl,R. and Birchmeier,W. (1996) Functional interaction of β-catenin with the transcriptional factor LEF-1. Nature, 382, 638–642. [DOI] [PubMed] [Google Scholar]
- 5.Korinek V., Barker,N., Morin,P.J., van Wichen,D., de Weger,R., Kinzler,K.W., Vogelstein,B. and Clevers,H. (1997) Constitutive transcriptional activation by a beta-catenin–Tcf complex in APC –/– colon carcinoma. Science, 275, 1784–1787. [DOI] [PubMed] [Google Scholar]
- 6.Morin P.J., Sparks,A.B., Korinek,V., Barker,N., Clevers,H., Vogelstein,B. and Kinzler,K.W. (1997) Activation of beta-catenin–Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science, 275, 1787–1790. [DOI] [PubMed] [Google Scholar]
- 7.Kinzler K.W. and Vogelstein,B. (1996) Lessons from hereditary colorectal cancer. Cell, 87, 159–170. [DOI] [PubMed] [Google Scholar]
- 8.Roose J. and Clevers,H. (1999) Tcf transcription factors: molecular switches in carcinogenesis. Biochim. Biophys. Acta, 1424, M23–M27. [DOI] [PubMed] [Google Scholar]
- 9.Miyaki M., Iijima,T., Kimura,J., Yasuno,M., Mori,T., Hayashi,Y., Koike,M., Shitara,N., Iwama,T. and Kuroki,T. (1999) Frequent mutations of β-catenin and APC genes in primary colorectal tumors from patients with hereditary nonpolyposis colorectal cancer. Cancer Res., 59, 4506–4509. [PubMed] [Google Scholar]
- 10.Westphal E.M. and von Melchner,H. (2002) Gene therapy approaches for the selective killing of cancer cells. Curr. Pharm. Des., 8, 1683–1694. [DOI] [PubMed] [Google Scholar]
- 11.Maxwell I.H., Maxwell,F. and Glode L.M. (1986) Regulated expression of a diphtheria toxin A-chain transfected into human cells: possible strategy for inducing cancer cell suicide. Cancer Res., 46, 4660–4664. [PubMed] [Google Scholar]
- 12.Harrison G.S., Maxwell,F., Long,C.J., Rosen,C.A., Glode,L.M. and Maxwell I.H. (1991) Activation of a diphtheria toxin A gene by expression of human immunodeficiency virus-1 Tat and Rex proteins in transfected cells. Hum. Gene Ther., 2, 53–60. [DOI] [PubMed] [Google Scholar]
- 13.Maxwell I.H., Glode,L.M. and Maxwell F. (1991) Expression of the diphtheria toxin A-chain coding sequence under the control of promoters and enhancers from immunoglobulin genes as a means of directing toxicity to B-lymphoid cells. Cancer Res., 51, 4299–4304. [PubMed] [Google Scholar]
- 14.Robinson D.F. and Maxwell,I.H. (1995) Suppression of single and double nonsense mutations introduced into the diphtheria toxin A-chain: a potential binary system for toxin gene therapy. Hum. Gene Ther., 6, 137–143. [DOI] [PubMed] [Google Scholar]
- 15.Lidor Y.J., Lee,W.E., Nilson,J.H., Maxwell I.H., Su,L.J., Brand,E. and Glode,L.M. (1997) In vitro expression of the diphtheria toxin A-chain gene under the control of human gonadotropin gene promoters as a means of directing toxicity to ovarian cancer cell lines. Am. J. Obstet. Gynecol., 177, 579–585. [DOI] [PubMed] [Google Scholar]
- 16.Cao G.W., Qi,Z.T., Pan,X., Zhang,X.Q., Miao,X.H., Feng,Y., Lu,X.H., Kuriyama,S. and Du,P. (1998) Gene therapy for human colorectal carcinoma using CEA promoter controlled bacterial ADP-ribosylating toxin genes human CEA:PEA & DTA gene transfer. World J. Gastoenterol., 4, 388–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kondo S., Ishizaka,Y., Okada,T., Kondo,Y., Hitomi,M., Tanaka,Y., Haqqi,T., Barnett,G.H. and Barna,B.P. (1998) FADD gene therapy for malignant gliomas in vitro and in vivo. Hum. Gene Ther., 20, 1599–1608. [DOI] [PubMed] [Google Scholar]
- 18.Chen R.H. and McCormick,F. (2001) Selective targeting to the hyperactive beta-catenin/T-cell factor pathway in colon cancer cells. Cancer Res., 61, 4445–4449. [PubMed] [Google Scholar]
- 19.Qiao J., Doubrovin,M., Sauter,B.V., Huang,Y., Guo,Z.S., Balatoni,J., Akhurst,T., Blasberg,R.G., Tjuvajev,J.G., Chen,S.H. and Woo,S.L. (2002) Tumor-specific transcriptional targeting of suicide gene therapy. Gene Ther., 9, 168–175. [DOI] [PubMed] [Google Scholar]
- 20.Richards C.A., Austin,E.A. and Huber,B.E. (1995) Transcriptional regulatory sequences of carcinoembryonic antigen: identification and use with cytosine deaminase for tumor-specific gene therapy. Hum. Gene Ther., 6, 881–893. [DOI] [PubMed] [Google Scholar]
- 21.Ge K., Jiang,Q., Xu,D.H., Zheng,Z.C. and Liu,X.Y. (1998) Experimental treatment for human colorectal carcinoma by tissue type specific expression of herpes simplex virus thymidine kinase gene. Shi Yan Sheng Wu Xue Bao, 31, 259–264. [PubMed] [Google Scholar]
- 22.Kwong K.Y., Zou,Y., Day,C.-P. and Hung,M.C. (2002) The suppression of colon cancer cell growth in nude mice by targeting β-catenin/TCF pathway. Oncogene, 21, 8340–8346. [DOI] [PubMed] [Google Scholar]
- 23.Nyati M.K., Sreekumar,A., Li,S., Zhang,M., Rynkiwicz,S.D., Chinnaiyan,A.M., Rehemtulla,A. and Lawrence,T.S. (2002) High and selective expression of yeast cytosine deaminase under a carcinoembryonic antigen promoter-enhancer. Cancer Res., 62, 2337–2342. [PubMed] [Google Scholar]
- 24.Fuerer C. and Iggo R. (2002) Adenoviruses with Tcf binding sites in multiple early promoters show enhanced selectivity for tumour cells with constitutive activation of the wnt signaling pathway. Gene Ther., 9, 270–281. [DOI] [PubMed] [Google Scholar]
- 25.Malerba M., Daeffler,L., Rommelaere,J. and Iggo,R.D. (2002) Replicating parvoviruses that target colon cancer cells. J. Virol., 77, 6638–6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morita A., Tsao,D. and Kim,Y.S. (1982) Effect of sodium butyrate on alkaline phosphatase in HRT-18, a human rectal cancer cell line. Cancer Res., 42, 4540–4545. [PubMed] [Google Scholar]
- 27.Hague A., Manning,A.M., Hanlon,K.A., Huschtscha,L.I., Hart,D. and Paraskeva,C. (1993) Sodium butyrate induces apoptosis in human colonic tumour cell lines in a p53-independent pathway: implications for the possible role of dietary fibre in the prevention of large-bowel cancer. Int. J. Cancer, 55, 498–505. [DOI] [PubMed] [Google Scholar]
- 28.Heerdt B.G., Houston. M.A. and Augenlicht,L.H. (1994) Potentiation by specific short-chain fatty acids of differentiation and apoptosis in human colonic carcinoma cell lines. Cancer Res., 58, 3288–3294. [PubMed] [Google Scholar]
- 29.Bordonaro M., Mariadason,J.M., Aslam,F., Heerdt,B.G. and Augenlicht,L.H. (1999) Butyrate-induced apoptotic cascade in colonic carcinoma cells: modulation of the beta-catenin–Tcf pathway and concordance with effects of sulindac and trichostatin A but not curcumin. Cell Growth Differ., 10, 713–720. [PubMed] [Google Scholar]
- 30.Bordonaro M., Lazarova,D.L., Augenlicht,L.H. and Sartorelli,A.C. (2002) Cell type- and promoter-dependent modulation of Wnt signaling pathway by sodium butyrate. Int. J. Cancer, 97, 42–51. [DOI] [PubMed] [Google Scholar]
- 31.Hedgepeth C.M., Conrad,L.J., Zhang,J. Huang,H.C., Lee,V.M. and Kelin,P.S. (1997) Activation of the Wnt signaling pathway: a molecular mechanism for lithium action. Dev. Biol., 85, 82–91. [DOI] [PubMed] [Google Scholar]
- 32.Staal F.J., Noort Mv M., Strous,G.J. and Clevers,H.C. (2002) Wnt signals are transmitted through N-terminally dephosphorylated beta-catenin. EMBO Rep., 3, 65–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crawford H.C., Fingleton,B.M., Rudolph-Owen,L.A., Goss,K.J., Rubinfeld,B., Polakis,P. and Matrisian,L.M. (1999) The metalloproteinase matrilysin is a target of beta-catenin transactivation in intestinal tumors. Oncogene, 18, 2883–2891. [DOI] [PubMed] [Google Scholar]
- 34.Stewart D.B. and Nelson,W.J. (1997) Identification of four distinct pools of catenins in mammalian cells and transformation-dependent changes in catenin distributions among these pools. J. Biol. Chem., 272, 29652–29662. [DOI] [PubMed] [Google Scholar]
- 35.Lazarova D.L., Bordonaro,M., Carbone,R. and Sartorelli,A.C. (2004) Linear relationship between Wnt activity levels and apoptosis in colorectal carcinoma cells exposed to butyrate. Int. J. Cancer, in press. [DOI] [PubMed] [Google Scholar]
- 36.Vecsey-Semjen B., Becker,K.-F., Sinski,A., Blennow,E., Vietor,I., Zatloukal,K., Beug,H., Wagner,E. and Huber,L.A. (2002) Novel colon cancer cell lines leading to better understanding of the diversity of respective primary cancers. Oncogene, 21, 4646–4662. [DOI] [PubMed] [Google Scholar]
- 37.Chinnaiyan A.M., O’Rourke,K., Tewari,M. and Dixit,V.M. (1995) FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell, 81, 505–512. [DOI] [PubMed] [Google Scholar]
- 38.Kijima T., Osaki,T., Nishino,K., Kumagai,T., Funakoshi,T., Goto,H., Tachibana,I., Tanio,Y. and Kishimoto,T. (1999) Application of Cre recombinase/loxP system further enhances antitumor effects in cell type-specific gene therapy against carcinoembryonic antigen-producing cancer. Cancer Res., 59, 4906–4911. [PubMed] [Google Scholar]
- 39.Ueda K., Iwahashi,M., Nakamori,M., Nakamura,M., Yamaue,H. and Tanimura,H. (2000) Enhanced selective gene expression by adenovirus vector using Cre/loxP regulation system for human carcinoembryonic antigen-producing carcinoma. Oncology, 59, 255–265. [DOI] [PubMed] [Google Scholar]
- 40.Goto H., Osaki,T., Kijima,T., Nishino,K., Kumagai,T., Funakoshi, T,. Kimura,H., Takeda,Y., Yoneda,T., Tachinana,I. and Hauashi,S. (2001) Gene therapy utilizing the Cre/loxP system selectively suppresses tumor growth of disseminated carcinoembryonic antigen-producing cancer cells. Int. J. Cancer., 94, 414–419. [DOI] [PubMed] [Google Scholar]
- 41.Kaczmarczyk S.J. and Green,J.E. (2001) A single vector containing modified cre recombinase and LOX recombination sequences for inducible tissue-specific amplification of gene expression. Nucleic Acids Res., 29, e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ueda K., Iwahashi,M., Nakamori,M., Nakamura,M., Yamaue,H. and Tanimura H. (2001) Carcinoembryonic antigen-specific suicide gene therapy of cytosine deaminase/5-fluorocytosine enhanced by the cre-loxP system in the orthotopic gastric carcinoma model. Cancer Res., 61, 6158–6162. [PubMed] [Google Scholar]
- 43.Aoki M., Hecht,A., Kruse,U., Kemler,R. and Vogt,P.K. (1999) Nuclear endpoint of Wnt signaling: neoplastic transformation induced by transactivating lymphoid-enhancing factor 1. Proc. Natl Acad. Sci. USA, 96, 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cummings J.H., Pomare,E.W., Branch,W.J., Naylor,C.P. and Macfarlane,G.T. (1987) Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut, 28, 1221–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.da Costa L.T., He,T.C., Yu,J., Sparks,A.B., Morin,P.J., Polyak,K., Laken,S., Vogelstein,B. and Kinzler K.W. (1999) CDX2 is mutated in a colorectal cancer with normal APC/beta-catenin signaling. Oncogene, 18, 5010–5014. [DOI] [PubMed] [Google Scholar]
- 46.Tetsu O. and McCormick,F. (1999) β-Catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature, 398, 422–426. [DOI] [PubMed] [Google Scholar]
- 47.Schroder C., Eckert,K. and Maurer,H.R. (1998) Tributyrin induces growth inhibitory and differentiating effects on HT-29 colon cancer cells in vitro. Int. J. Oncol., 13, 1335–1340. [DOI] [PubMed] [Google Scholar]
- 48.Chen S., Guttridge,D.C., You,Z., Zhang,Z., Fribley,A., Mayo,M.W., Kitajewski,J. and Wang,C.Y. (2001) Wnt-1 signaling inhibits apoptosis by activating beta-catenin/T cell factor-mediated transcription. J. Cell Biol., 152, 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ueda K., Iwahashi,M., Nakamori,M., Nakamura,M., Matsuura,I., Ojima,T. and Yamaue,H. (2003) Improvement of carcinoembryonic antigen-specific prodrug gene therapy for experimental colon cancer. Surgery, 133, 309–317. [DOI] [PubMed] [Google Scholar]