

Abstract
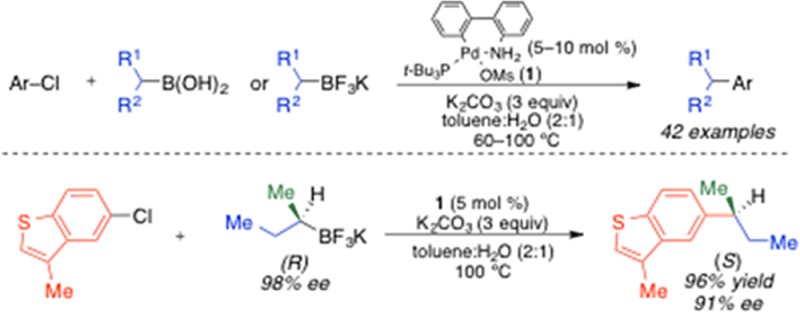
We report the development of a Pd-catalyzed process for the stereospecific cross-coupling of unactivated secondary alkylboron nucleophiles and aryl chlorides. This process tolerates the use of secondary alkylboronic acids and secondary alkyltrifluoroborates and occurs without significant isomerization of the alkyl nucelophile. Optically active secondary alkyltrifluoroborate reagents undergo cross-coupling reactions with stereospecific inversion of configuration using this method.
The development of transition metal-catalyzed carbon–carbon cross-coupling reactions has revolutionized the manner by which we approach the synthesis of complex organic molecules.1 Classical studies have focused mainly on the creation of C(sp2)–C(sp2) bonds for the construction of planar molecular topologies. More recently, C(sp3) nucleophiles and electrophiles have been investigated in cross-coupling reactions, which more readily allows the manipulation of the three-dimensional space of a molecule.2 The use of C(sp3) coupling partners in transition metal-catalyzed reactions is complicated by slow transmetalation and the propensity of alkyl ligands to undergo β-hydride elimination processes. These complications increase for secondary and tertiary alkyl coupling partners, which readily undergo isomerization and racemization via a β-hydride elimination/reinsertion sequence following transmetalation (Figure 1).2,3 A major focus of the research in our laboratory is the development of efficient metal-catalyzed reactions that broadly tolerate the use of secondary and tertiary alkyl nucleophiles.3,4 In particular, we are interested in the use of optically active alkyl nucleophiles that are configurationally stable and isolable. Such nucleophiles can, in principal, be employed as building blocks for the rapid and reliable construction of optically active drug libraries via stereospecific cross-coupling reactions. Recently, we reported a Pd-catalyzed Stille cross-coupling reaction that enabled the broad use of unactivated secondary alkylstannatranes.4d This was the first process that did not require the use of activated (containing a C(sp2) α-carbon, heteroatomic α-carbon, or strongly coordinating β-carbonyl group) alkylmetal nucleophiles to achieve stereospecific cross-coupling reactions.5 Building upon this work, we sought to extend the use of secondary alkyl nucleophiles to alkylboron nucleophiles, which are more configurationally stable and more easily prepared than their alkylstannatrane analogues.6 Herein, we report the first palladium-catalyzed process that permits the broad use of unactivated secondary alkylboron nuclophiles in cross-coupling reactions without concurrent isomerization. Secondary alkylboronic acids and secondary alkyltrifluoroborates both undergo cross-coupling reactions with aryl chlorides using this procedure. Importantly, when enantioenriched alkyltrifluoroborates are employed, cross-coupling products are obtained with highly enantiospecific inversion of absolute configuration.
Figure 1.
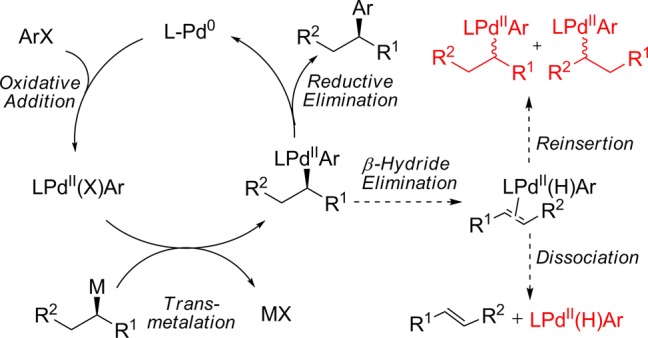
Catalytic cycle and possible side reactions in the Pd-catalyzed cross-coupling reaction of optically active nucleophiles and aryl electrophiles.
The groups of Molander and Dreher7a and of van der Hoogenband7b reported the first attempts to develop a Pd-catalyzed process that enabled the broad use of secondary alkylboron nucleophiles in cross-coupling reactions.8 Using bulky, electron-rich, phosphine ligands, they reported processes that permitted the cross-coupling of secondary carbocyclic boron reagents and aryl electrophiles. However, noncyclic secondary alkylboron nucleophiles underwent extensive isomerization using these methods. Inspired by these studies, we set fourth to develop a general process that could overcome these limitations, while also enabling the development of a stereospecific variant for the Suzuki coupling. While exploring the use of alternative alkylboron reagents in Suzuki reactions, we discovered that the isomerization of acyclic secondary alkylboronic acids could be overcome through modification of the conditions used by Molander and Dreher. In the cross-coupling of 4-chloroanisole and s-BuB(OH)2, isomerization was suppressed through use of a preformed Pt-Bu3 palladium precatalyst9 (1) and K2CO3 in a 2:1 mixture of toluene and water (Table 1). When Pd(dba)2 and P(t-Bu)3·HBF4 were used in place of the preformed palladium catalyst (entry 3), the reaction did not reach full conversion. The use of other trialkylphosphine ligands resulted in increased isomerization. When K2CO3 was replaced with Cs2CO3, the reaction failed completely (entry 4). By increasing the catalyst loading from 2 to 5 mol %, we observed enhanced selectivity for the branched product 2.
Table 1. Optimization of Reaction Conditions for the Pd-Catalyzed Cross-Coupling of s-BuB(OH)2 and 4-Chloroanisole.
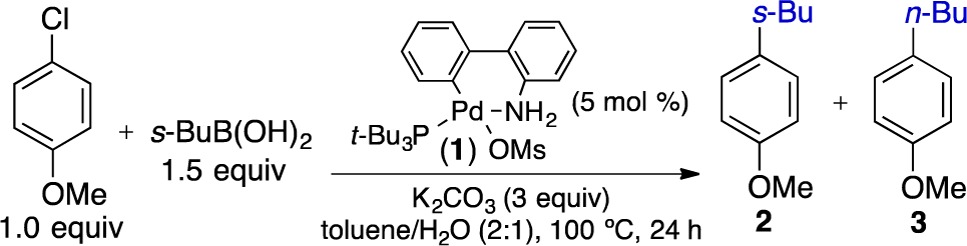
| entry | variations from above | yield (%)a | 2:3a |
|---|---|---|---|
| 1 | none | 98 | >200:1 |
| 2 | 2 mol % P(t-Bu)3 precatalyst | 98 | 36:1 |
| 3 | 2 mol % Pd(dba)2, 2 mol % P(t-Bu)3·HBF4 | 70 | 36:1 |
| 4 | Cs2CO3 (3 equiv) instead of K2CO3 | <5 | – |
| 5 | K3PO4-H2O (3 equiv) instead of K2CO3 | 94 | 69:1 |
| 6 | 5 mol % n-BuP(Ad)2 precatalyst | 89 | 4:1 |
| 7 | 5 mol % PhP(t-Bu)2 precatalyst | 94 | 32:1 |
| 8 | toluene/H2O(1:2) | 89 | 70:1 |
| 9 | THF instead of toluene | <5 | – |
| 10 | dioxane instead of toluene | <5 | – |
| 11 | methylcylcohexane instead of toluene | 97 | >200:1 |
Yields and selectivities determined by GC.
The generality of the cross-coupling reaction was evaluated using i-PrB(OH)2 (4) (no α-branching) and s-BuB(OH)2 (5) (containing α-branching) as nucleophiles alongside a variety of aryl chloride electrophiles (Table 2). Electron-rich and electron-deficient aryl chlorides provided the corresponding cross-coupling products in high yields. The presence of an ortho substitutent on the electrophile was well-tolerated in the reaction. Additionally, heterocyclic aryl chlorides could be employed in these reactions with moderate generality. For all substrates, a >50:1 ratio of retention to isomerization products was achieved. When heteroarenes with the chloride leaving group located directly on the heteroaryl ring (e.g., 3-chloropyridine) were employed, the reaction uniformly gave poor results. In our eyes, this constitutes the major scope limitation of the aryl chloride component. We were pleased to observe that the identical conditions employed for secondary alkylboronic acids could be used for i-PrBF3K (6) and s-BuBF3K (7). A similar substrate scope was also achieved using secondary alkyltrifluoroborates (Table 2). Since secondary alkyltrifluoroborates are significantly more stable than their secondary alkylboronic acid analogues,10 the ability to employ alkyltrifluoroborate reagents in this reaction greatly increases the overall utility of this process. When 4-bromoanisole was employed in a cross-coupling reaction with 6 using these conditions, the desired product was obtained in 60% yield. However, the use of aryl sulfonates resulted in the formation of <10% desired product. For all substrates, a >50:1 ratio of retention to isomerization products was again achieved.
Table 2. Pd-Catalyzed Cross-Coupling Reactions of Secondary Alkylboronic Acids and Aryl Chloridesa.
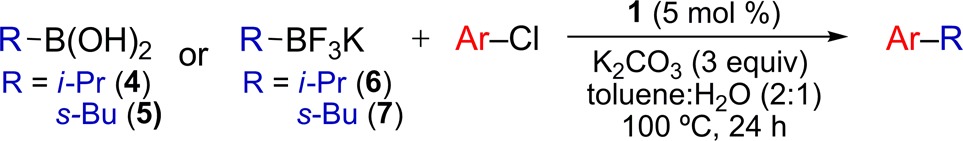
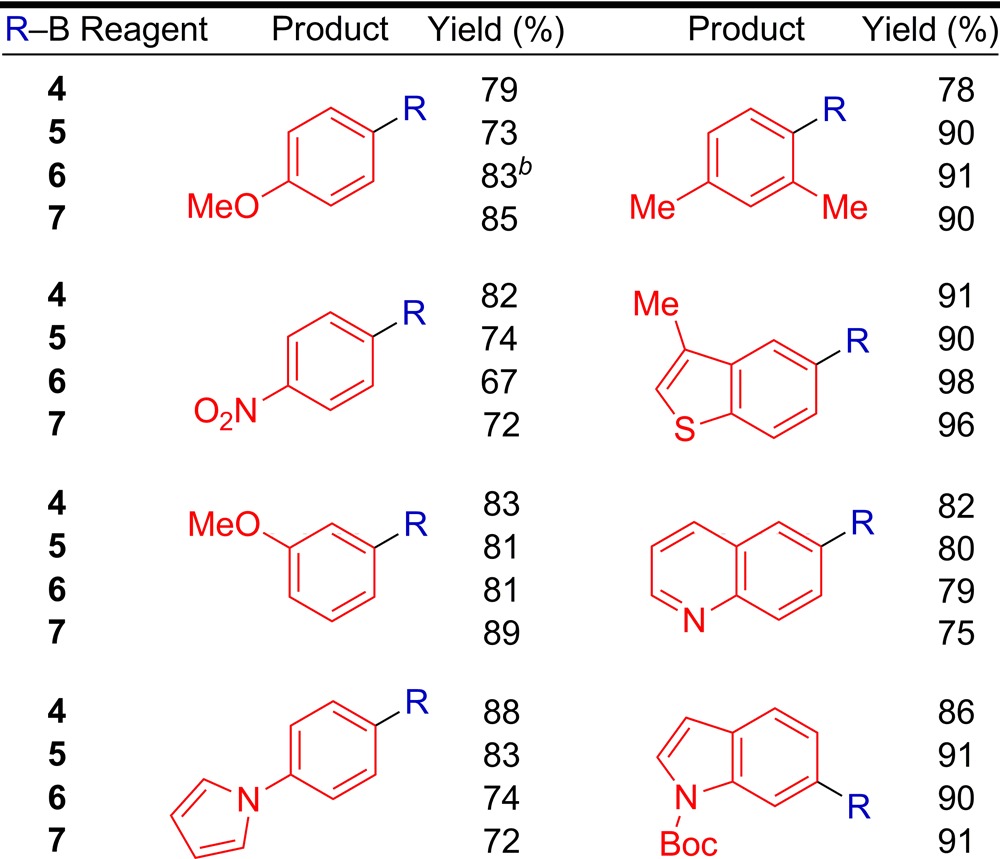
ArCl (0.5 mmol), RB(OH)2 (0.75 mmol), 100 °C, 24 h; average isolated yield of 2 runs; >50:1 ratio of retention to isomerization.
60% yield using 4-bromoanisole.
Because of the greater stability of alkyltrifluoroboronate reagents in comparison to alkylboronic acids, we investigated the extension of the nucleophilic coupling partner using secondary alkyltrifluoroborates (Table 3). The cross-coupling reactions proceeded efficiently using hydrocarbon nucleophiles, though lower reaction temperatures and extended reaction times were required to prevent background protodeboronation. Incorporation of a heterocyclic group into the skeleton of the nucleophile resulted in incomplete conversion. Not surprisingly, reactions involving symmetric, carbocyclic, secondary alkyltrifluoroborates provided cross-coupling products in high yield.11 While toluene can be employed successfully in all of these reactions, in some instances, we observed that protodeboronation was minimized (ca. 5–10% less protodeboronation) using benzene as the organic solvent component.
Table 3. Pd-Catalyzed Cross-Coupling Reactions of Secondary Alkyltrifluoroborates and Aryl Chloridesa.
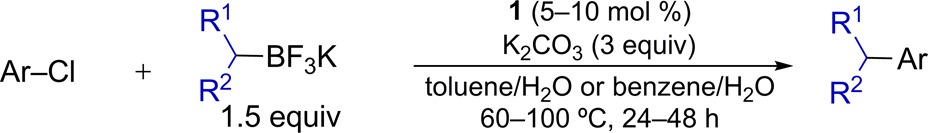
ArCl (0.5 mmol), RBF3K (0.75–1.0 mmol); average isolated yield of two runs; >50:1 ratio of retention to isomerization.
Using benzene:H2O (1:1) as solvent, 10 mol % 1, 60 °C, 48 h.
Using toluene:H2O (2:1) as solvent, 5 mol % 1, 100 °C, 24 h.
To evaluate the stereochemical course of this reaction, we prepared enantioenriched (R)-7 and (R)-8 using Brown’s12 and Matteson’s13 chiral auxiliary-based protocols. These stereochemical assignments were further confirmed through oxidation10b of (R)-7 and (R)-8 and comparison of the products to the corresponding commercially available, optically active alcohols (see Supporting Information). Derivatization of commercially available (S)-3-phenylbutyric acid (9) enabled access to (S)-10 and (S)-11 (Figure 2a,c). When (R)-7 was employed in a cross-coupling reaction with chlorobenzene, (S)-10 was obtained with inversion of absolute stereochemistry (Figure 2b). The stereochemical assignment was made through comparison with synthesized (S)-10. A similar stereochemical analysis was performed for the cross-coupling reaction of (S)-8 and chlorobenzene, which also showed stereoinversion in the formation of (S)-11 (Figure 2d). Both (S)-10 and (S)-11 were generated with high enatiospecificity (% es)14 using this process.
Figure 2.
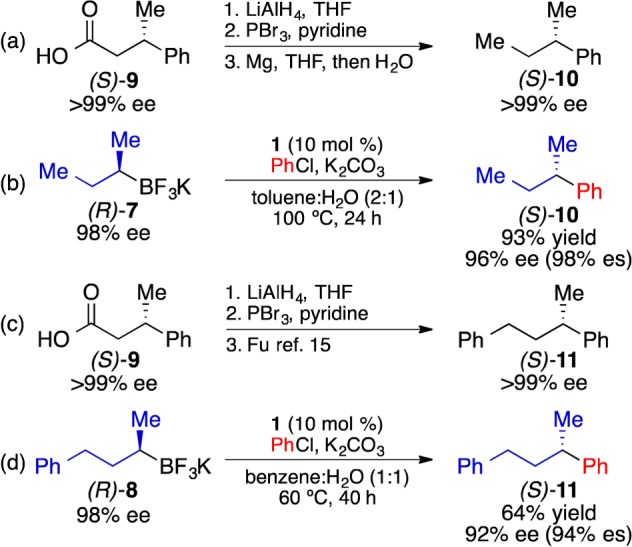
(a) Synthesis of (S)-10 from (S)-3-phenylbutyric acid (S)-(9). (b) Suzuki coupling of (R)-7 and PhCl. (c) Synthesis of (S)-11 from (S)-9. (d) Suzuki coupling of (R)-8 and PhCl.
In Suzuki reactions, both inversion and retention of absolute configuration have been demonstrated for activated secondary alkylboron nucleophiles. Retention of configuration has been demonstrated by Crudden5q using benzylic alkylboron nucleophiles and by Molander5w using α-OBn alkylboron nucleophiles. Inversion of configuration has been previously reported by the Molander5r and Suginome5s groups in Suzuki reactions using activated secondary alkylboron nucleophiles containing β-coordinating groups. Additionally, the groups of Hall5v and Morken5y have demonstrated that the Pd-catalyzed monoarylation of geminal bis(boranates) occurs with inversion of configuration. Our work constitutes the first demonstration of an enantiospecific secondary alkyl Suzuki reaction employing an unactivated nucleophile. This process enables the use of (R)-sBuBF3K, the simplest optically active secondary alkylboron nucleophile possible. Therefore, transmetalation to Pd(II) in this process is not biased by electronic perturbations and should most closely represent the fundamental mechanism of transmetalation for secondary alkylboron nucleophiles. Our observation that unactivated secondary alkyltrifluoroborates undergo transmetalation to Pd(II) with inversion is also consistent with previous reports from Kalbalka16 and Aggarwal17 in which unactivated secondary alkylboron reagents undergo electrophilic substitution with clean inversion.
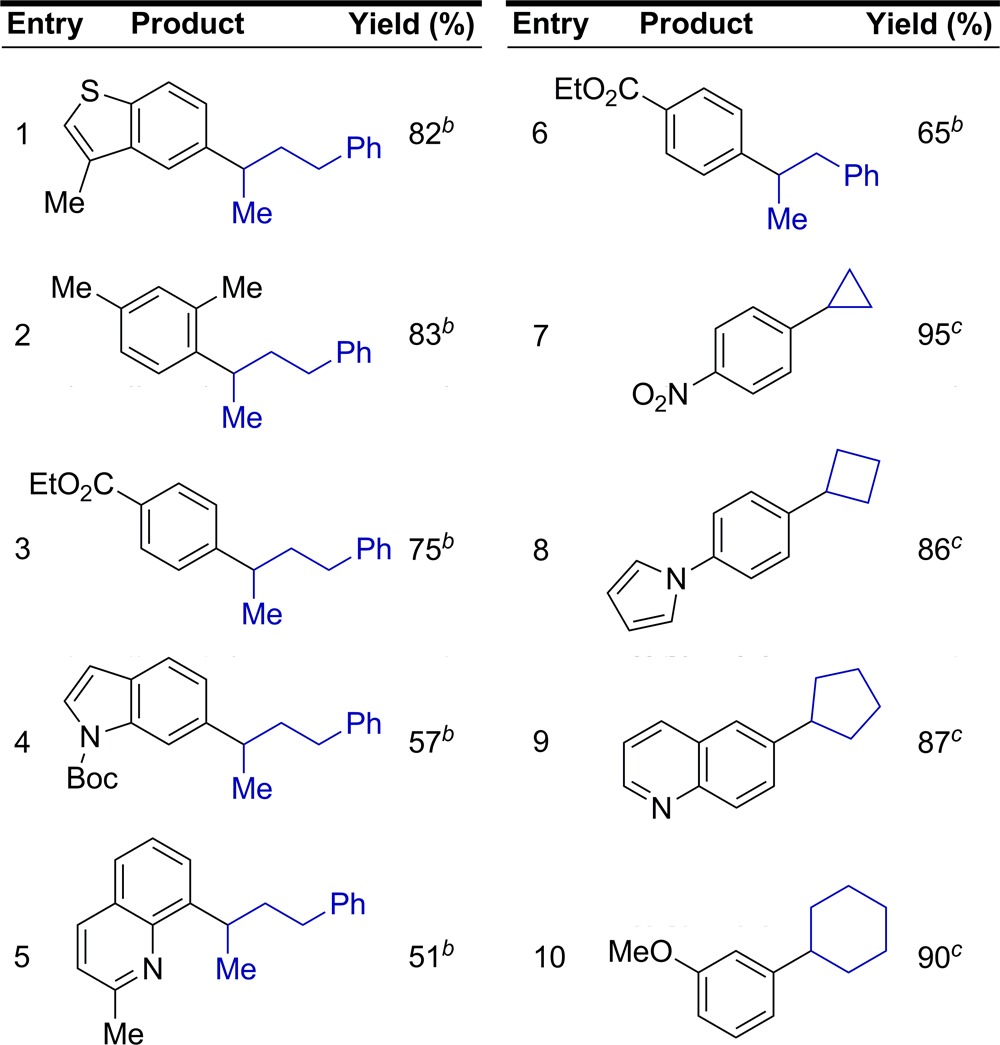
Six cross-coupling reactions from Tables 2 and 3 were repeated using (R)-7 and (R)-8. Because heterocyclic electrophiles are of particular importance in medicinal chemistry, while also typically the most difficult substrates to employ in metal-catalyzed processes, we focused on stereospecific cross-coupling reactions involving heteroaromatic electrophiles. These reactions resulted in the formation of the corresponding (S) products with low erosion of enantiomeric excess (Figure 3). The high enantiospecificity (% es) of this process using heteroaromatic electophiles should enable the use of this process in the generation of diverse libraries of optically active drug candidates. Thus, alkylboron reagents such as (R)-7 and (R)-8 can be viewed as general building blocks for use in the rapid preparation of libraries of nonracemic molecules during the drug discovery process.
Figure 3.
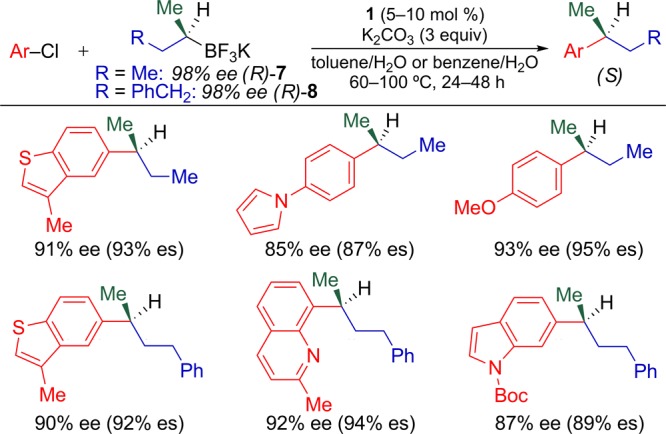
Use of (R)-7 and (R)-8 in stereospecific cross-coupling reactions with aryl and heteroaryl chlorides.
In summary, we have developed the first general process for the Pd-catalyzed cross coupling of unactivated, secondary alkylboron nucleophiles and aryl electrophiles. One set of conditions enables the use of secondary alkylboronic acids and secondary alkyltrifluoroborates in these reactions. In all cases, only nominal isomerization of the nucleophile is observed during the reaction. When an optically active alkyltrifluoroborate nucleophile is employed, cross coupling occurs with inversion of absolute configuration. This process should facilitate the use of optically active alkylboron compounds as building blocks in the construction of optically active drug libraries via enantiospecific cross-coupling reactions. We are currently investigating extensions of this chemistry to more diverse heterocyclic systems as well as the factors responsible for the observed stereoselectivity.
Acknowledgments
We thank Xinghua Ma for performing initial investigations. We thank the National Institutes of Health (5SC2GM096932 and 1SC1GM110010), the City College of New York (CCNY), the Alfred P. Sloan Foundation, and PSC-CUNY for financial support. We gratefully acknowledge the National Science Foundation for an instrumentation grant (CHE-0840498). We thank Jintao Chen for conducting the ArBr study.
Supporting Information Available
Procedural, spectral, and crystallographic data. This material is available free of charge via the Internet at http://pubs.acs.org
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Metal-Catalyzed Cross-Coupling Reactions; de Meijere A., Diederich F., Eds.; Wiley-VCH: New York, 2004. [Google Scholar]
- a Netherton M. R.; Fu G. C. In Topics in Organometallic Chemistry: Palladium in Organic Synthesis; Tsuji J., Ed.; Springer: New York, 2005; pp 85–108. [Google Scholar]; b Chemler S. R.; Trauner D.; Danishefsky S. J. Angew. Chem., Int. Ed. 2001, 40, 4544. [DOI] [PubMed] [Google Scholar]; c Rudolph A.; Lautens M. Angew. Chem., Int. Ed. 2009, 48, 2656. [DOI] [PubMed] [Google Scholar]; d Jana R.; Pathak T. P.; Sigman M. S. Chem. Rev. 2011, 111, 1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi-Pangu A.; Biscoe M. R. Synlett 2012, 23, 1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Joshi-Pangu A.; Ganesh M.; Biscoe M. R. Org. Lett. 2011, 13, 1218. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Joshi-Pangu A.; Wang C.-Y.; Biscoe M. R. J. Am. Chem. Soc. 2011, 133, 8478. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Joshi-Pangu A.; Ma X.; Diane M.; Iqbal S.; Kribs R. J.; Huang R.; Wang C.-Y.; Biscoe M. R. J. Org. Chem. 2012, 77, 6629. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Li L.; Wang C.-Y.; Huang R.; Biscoe M. R. Nat. Chem. 2013, 5, 607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For cross-coupling reactions employing optically active nucleophiles, see: Organomagesium reagents:; a Hoffman R. W.; Hölzer B.; Knopff O.; Harms K. Angew. Chem., Int. Ed. 2000, 39, 3072. [DOI] [PubMed] [Google Scholar]; b Hölzer B.; Hoffman R. W. Chem. Commun. 2003, 732. [DOI] [PubMed] [Google Scholar]; Organozinc reagents:; c Boudier A.; Darcel C.; Flachsman F.; Micouin L.; Oestreich M.; Knochel P. Chem.—Eur. J. 2000, 6, 2748. [DOI] [PubMed] [Google Scholar]; d Campos K. R.; Klapars A.; Waldman J. H.; Dormer P. G.; Chen C.-Y. J. Am. Chem. Soc. 2006, 128, 3538. [DOI] [PubMed] [Google Scholar]; e Thaler T.; Haag B.; Gavryushin A.; Schober K.; Hartman E.; Gschwing R. M.; Zipse H.; Mayer P.; Knochel P. Nat. Chem. 2010, 2, 125. [DOI] [PubMed] [Google Scholar]; Organotin reagents:; f Labadie J. W.; Stille J. K. J. Am. Chem. Soc. 1983, 105, 6129. [Google Scholar]; g Ye J.; Bhatt R. K.; Falck J. R. J. Am. Chem. Soc. 1994, 116, 1. [Google Scholar]; h Falck J. R.; Bhatt R. K.; Ye J. J. Am. Chem. Soc. 1995, 117, 5973. [Google Scholar]; i Mohapatra S.; Bandyopadhyay A.; Barma D. K.; Capdevila J. H.; Falck J. R. Org. Lett. 2003, 5, 4759. [DOI] [PubMed] [Google Scholar]; j Falck J. R.; Patel P. K.; Bandyopadhyay A. J. Am. Chem. Soc. 2007, 129, 790. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Li H.; He A.; Falck J. R.; Liebeskind L. S. Org. Lett. 2011, 13, 3682. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Kalkofen R.; Hoppe D. Synlett 2006, 1959. [Google Scholar]; m Lange H.; Fröhlich R.; Hoppe D. Tetrahedron 2008, 64, 9123. [Google Scholar]; n ref (4d). Organosilicon reagents:; o Hatanaka Y.; Hiyama T. J. Am. Chem. Soc. 1990, 112, 7793. [Google Scholar]; p Denmark S. E.; Werner N. S. J. Am. Chem. Soc. 2010, 132, 3612. [DOI] [PMC free article] [PubMed] [Google Scholar]; Organoboron reagents:; q Imao D.; Glasspoole B. W.; Laberge V. S.; Crudden C. M. J. Am. Chem. Soc. 2009, 131, 5024. [DOI] [PubMed] [Google Scholar]; r Sandrock D. L.; Jean-Gerard L.; Chen C.-Y.; Dreher S. D.; Molander G. A. J. Am. Chem. Soc. 2010, 132, 17108. [DOI] [PMC free article] [PubMed] [Google Scholar]; s Ohmura T.; Awano T.; Suginome M. J. Am. Chem. Soc. 2010, 132, 13191. [DOI] [PubMed] [Google Scholar]; t Daini M.; Suginome M. J. Am. Chem. Soc. 2011, 133, 4758. [DOI] [PubMed] [Google Scholar]; u Awano T.; Ohmura T.; Suginome M. J. Am. Chem. Soc. 2011, 133, 20738. [DOI] [PubMed] [Google Scholar]; v Lee J. C. H.; McDonald R.; Hall D. G. Nat. Chem. 2011, 3, 894. [DOI] [PubMed] [Google Scholar]; w Molander G. A.; Wisniewski S. R. J. Am. Chem. Soc. 2012, 134, 16856. [DOI] [PMC free article] [PubMed] [Google Scholar]; x Feng X.; Jeon H.; Yun J. Angew. Chem., Int. Ed. 2013, 52, 3989. [DOI] [PubMed] [Google Scholar]; y Sun C.; Potter B.; Morken J. P. J. Am. Chem. Soc. 2014, 136, 6534. [DOI] [PMC free article] [PubMed] [Google Scholar]; z Tellis J. C.; Primer D. N.; Molander G. A. Science 2014, 345, 433–6. [DOI] [PMC free article] [PubMed] [Google Scholar]; aa Bonet A.; Odachowski M.; Leonori D.; Essafi S.; Aggarwal V. K. Nat. Chem. 2014, 6, 584. [DOI] [PubMed] [Google Scholar]
- Boudier A.; Bromm L. O.; Lotz M.; Knochel P. Angew. Chem., Int. Ed. 2000, 39, 4414. [PubMed] [Google Scholar]
- a Dreher S. D.; Dormer P. G.; Sandrock D. L.; Molander G. A. J. Am. Chem. Soc. 2008, 130, 9257. [DOI] [PMC free article] [PubMed] [Google Scholar]; b van der Hoogenband A.; Lange J. H. M.; Terpstra J. W.; Koch M.; Visser G. M.; Visser M.; Korstanje T. J.; Jastrzebski J. T. B. H. Tetrahedron Lett. 2008, 49, 4122. [Google Scholar]
- For early, singular examples of cross-couplings using a secondary alkylboron reagent, see:; a Littke A. F.; Dai C.; Fu G. C. J. Am. Chem. Soc. 2000, 122, 4020. [Google Scholar]; b Kataoka N.; Shelby Q.; Stambuli J. P.; Hartwig J. F. J. Org. Chem. 2002, 67, 5553. [DOI] [PubMed] [Google Scholar]
- a Biscoe M. R.; Fors B. P.; Buchwald S. L. J. Am. Chem. Soc. 2008, 130, 6686. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kinzel T.; Zhang Y.; Buchwald S. L. J. Am. Chem. Soc. 2010, 132, 14073. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bruno N. C.; Tudge M. T.; Buchwald S. L. Chem. Sci. 2013, 4, 916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Molander G. A.; Sandrock D. L. Curr. Opin. Drug Discovery Devel. 2009, 12, 811. [PMC free article] [PubMed] [Google Scholar]; b Molander G. A.; Cavalcanti L. N. J. Org. Chem. 2011, 76, 623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potassium trans-2-methylcyclohexyltrifluoroborate underwent significant isomerization (at least 5 products by GC/MS) in reactions with chlorobenzene. Similar isomerization was observed in ref (7a).
- a Joshi N. N.; Pyun C.; Mahindroo V.; Singaram B.; Brown H. C. J. Org. Chem. 1992, 57, 504. [Google Scholar]; b Lennox A. J. J.; Lloyd-Jones G. C. Angew. Chem., Int. Ed. 2012, 51, 9385. [DOI] [PubMed] [Google Scholar]
- Matteson D. S. Tetrahedron 1998, 54, 10555. [Google Scholar]
- Enantiospecificity (es) = (eeproduct/eestarting material) × 100%.
- Gonzalez-Bobes F.; Fu G. C. J. Am. Chem. Soc. 2006, 128, 5360. [DOI] [PubMed] [Google Scholar]
- Kabalka G. W.; Gooch E. E. J. Org. Chem. 1980, 45, 3578. [Google Scholar]
- Larouche-Gauthier R.; Elford T. G.; Aggarwal V. K. J. Am. Soc. Chem. 2011, 133, 16794. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.