

Abstract
In many crop species, DNA fingerprinting is required for the precise identification of cultivars to protect the rights of breeders. Many families of retrotransposons have multiple copies throughout the eukaryotic genome and their integrated copies are inherited genetically. Thus, their insertion polymorphisms among cultivars are useful for DNA fingerprinting. In this study, we conducted a DNA fingerprinting based on the insertion polymorphisms of active retrotransposon families (Rtsp-1 and LIb) in sweet potato. Using 38 cultivars, we identified 2,024 insertion sites in the two families with an Illumina MiSeq sequencing platform. Of these insertion sites, 91.4% appeared to be polymorphic among the cultivars and 376 cultivar-specific insertion sites were identified, which were converted directly into cultivar-specific sequence-characterized amplified region (SCAR) markers. A phylogenetic tree was constructed using these insertion sites, which corresponded well with known pedigree information, thereby indicating their suitability for genetic diversity studies. Thus, the genome-wide comparative analysis of active retrotransposon insertion sites using the bench-top MiSeq sequencing platform is highly effective for DNA fingerprinting without any requirement for whole genome sequence information. This approach may facilitate the development of practical polymerase chain reaction-based cultivar diagnostic system and could also be applied to the determination of genetic relationships.
Keywords: DNA fingerprinting, high-throughput sequencing, molecular marker, retrotransposon, sweet potato
1. Introduction
Globally, the sweet potato [Ipomoea batatas (L.) Lam] is an important food crop with an annual production of 104 million tons (Food and Agriculture Organization of the United Nations; http://faostat3.fao.org/, 20 May 2014, date last accessed). This crop is known to be a valuable food source that contains various nutrients, including a high starch content, complex carbohydrates, dietary fibre, vitamins and anthocyanins. In addition to its high nutrient content, this crop is resilient, easy to propagate and grows relatively well in infertile and nitrogen-poor soils. There are over 8,000 varieties, including wild accessions, landraces and breeding lines registered with the International Potato Center (http://cipotato.org/sweetpotato/, 20 May 2014, date last accessed). Thus, the sweet potato is grown widely and is used for various purposes throughout the world. However, the sweet potato is a hexaploid (2n = 6x = 90) outbreeding species with a high degree of heterozygosity, and genome sequence information is not available for this species, which has hindered genetic analyses, the development of molecular markers and DNA fingerprinting.1–5
Retrotransposons are major components of eukaryotic genomes and they are present in high copy numbers in most of the plants.6–8 Retrotransposons are divided into two major groups: a group with a long terminal repeat (LTR) sequence at both ends and another group without LTRs (non-LTR). The non-LTR retrotransposons are further divided into long interspersed nuclear elements (LINEs) and short interspersed nuclear elements (SINEs).6–8 In our previous study, we identified two active retrotransposon families in sweet potato, i.e. an LTR-type Rtsp-1 and a LINE-type LIb.9,10 These two families exhibited a high degree of insertion polymorphism and newly inserted copies were inherited steadily throughout clonal propagation.10 The retrotransposon insertion polymorphisms found in different cultivars can be used for DNA fingerprinting.11–16
Next-generation sequencing technology has developed rapidly in the past few years and it has been utilized in various fields of genetics, thereby accelerating genomic and genetic research dramatically. More recently, bench-top high-throughput sequencing instruments such as the Roche/454 GS Junior (Roche, Basel, Switzerland), Illumina MiSeq and Ion Torrent PGM (Life Technologies) have been released, which are capable of sequencing several megabase to gigabase pairs in a single lane-sequencing run with enhanced speed and cost performance.17,18 In particular, the MiSeq system has the highest throughput per run and the lowest error rate,17 and the latest version enables to generate up to 15 Gb per run with 2 × 300 bp reads using new reagent kits (http://www.illumina.com/systems/miseq.ilmn, 20 May 2014, date last accessed). These bench-top systems produce smaller sequence datasets than traditional high-throughput sequencing instruments (Roche/454 GS FLX+ and Illumina HiSeq 2000), but their utilization is increasingly widespread because of their greater time and cost performance during sequencing analysis, although with modest performance.
In the present study, we conducted DNA fingerprinting based on the insertion polymorphisms of the active retrotransposon families (Rtsp-1 and LIb) in sweet potato. Using a bench-top sequencing platform, we successfully identified 2,024 insertion sites in 38 cultivars based on a single lane-sequencing run. It should be noted that the insertion sites of the active retrotransposon families were highly polymorphic among those cultivars. Using the cultivar-specific insertion sites, we successfully developed sequence-characterized amplified region (SCAR) markers, which may facilitate the precise identification of cultivars. Moreover, the genotyping data related to the insertion sites can be used to determine the genetic relationships among cultivars. The sweet potato is a non-model crop species and its whole genome sequencing data are not available, but our method enables efficient genotyping based on a number of polymorphic retrotransposon insertion sites.
2. Materials and methods
2.1. DNA samples
DNA samples from the 38 sweet potato cultivars listed in Supplementary Table S1 were provided by the National Agriculture and Food Research Organization, Kyusyu Okinawa Agricultural Research Center. Young leaves were collected from field grown plants and genomic DNA was extracted using a DNeasy Plant Mini Kit (Qiagen, Inc., Germany).
2.2. MiSeq library construction
Genomic DNA (5 μg) was separated into fragments of ∼6 kb by g-TUBE (Covaris) centrifugation and purified using a QIAquick PCR Purification Kit (Qiagen). The purified products were treated with DNA polymerase I and T4 DNA polymerase to convert the heterogeneous ends via their physical fragmentation into blunt ends. The blunt ends were adenylated at the 3′ end using a Klenow fragment that lacked a 5′ to 3′ exonuclease activity. A forked adapter with a T overhang at the 5′ double-stranded end was ligated using T4 DNA ligase. The retrotransposon insertion sites were specifically amplified using nested polymerase chain reaction (PCR), where the first PCR was performed with retrotransposon-specific (Rtsp-1_ppt or LIb_L_D4) and adapter-specific (AP2) primer combinations using the ligated products as the template, and the second PCR was performed with primers for the 3′ end sequence of Rtsp-1 or LIb [adjacent to the 3′ end polyadenosine sequence (poly(A)] and an adapter sequence (AP3) using the initial PCR products as the template. The PCR comprises an initial denaturation at 94°C for 2 min, which was followed by 30 cycles at 94°C for 30 s, 80°C for 30 s, 58°C for 30 s and 72°C for 1 min, with a final extension at 72°C for 15 min. The PCR products were selected by size (300–500 bp) using gel electrophoresis and purified with a QIAquick Gel Extraction Kit (Qiagen). The MiSeq sequencing adapters were ligated to the size-selected products from each cultivar using a TruSeq DNA-Sampling Preparation Kit (Illumina) to obtain cultivar-specific combinations of the dual index sequences in the adapters (Fig. 1). Equal amounts of the ligated products from the 38 cultivars were pooled before MiSeq sequencing, and a paired-end sequence of 150 bp was determined at each end (Fig. 1). MiSeq sequencing was conducted using the MS-102-1001 MiSeq Reagen Kit (300 cycles) (Illumina). In sequencing run, we added a PhiX Control v3 (Illumina) at 50% of our library, because Illumina recommends spike-in a PhiX control DNA for low diversity library where a significant number of the reads have the same sequence. It is known that a high-concentration spike-in (40% or higher) of PhiX improves matrix generation and phasing/prephasing calculations and helps balance the overall lack of sequence diversity. Our library was considered to have low diversity nucleotide composition because of the Rtsp-1 or LIb sequences, or an adaptor AP3 sequence at the beginning of one read of a sequence pair. This should be combined with a reduction of the amount of library used. The sequences of the adapters and primers are reported in Supplementary Table S2. The MiSeq reads of this study have been submitted to DDBJ (http://www.ddbj.nig.ac.jp/, 20 May 2014, date last accessed) under accession no. DRA001340.
Figure 1.
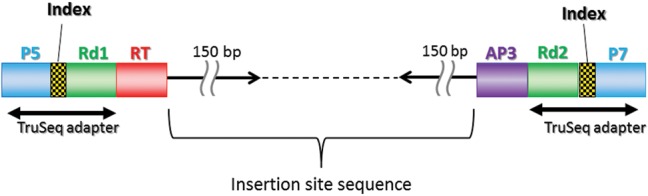
Scheme of the MiSeq sequencing library in this study. The 150-bp paired reads (black arrow) represent the junction sequence of the retrotransposon (Rtsp-1 or LIb) (red box) and the insertion site sequence from the AP3 adapter (purple box). The TruSeq adapters used in this study were indexed with short 6–8 bp sequences to facilitate multiplexing in a single sequencing run. The P5 and P7 sequences (blue box) allowed the final products to hybridize to the Illumina flow cell. The Rd1 and Rd2 sequences (green box) served as sequencing primers.
2.3. Data analysis
The MiSeq output was divided into each cultivar based on the dual index TruSeq sequence. The sequencing data were handled using Maser, the pipeline execution system of the Cell Innovation Program at the National Institute of Genetics (http://cell-innovation.nig.ac.jp/index_en.html, 20 May 2014, date last accessed). When one read of a sequence pair was filtered for invalid Rtsp-1 or LIb primer sequences, the entire pair was discarded. The LINE element of LIb has a poly(A) tail with variable lengths at the integration site.10,19 Those poly(A) sequences were removed from the LIb reads using the PRINSEQ tool20 before subsequent data processing. After trimming the Rtsp-1 or LIb sequences from the reads, they were trimmed further to 50 bp from the retrotransposon junction and filtered based on the quality value (QV) for all base calls ≥20. Next, the outliers were filtered by trimming them to a specific length that included most of the sequences (>99 or >98%), whereas reads shorter than this specific length were filtered out. Identical sequences observed in ≥10 reads were treated as a single sequence using the FASTA format with read count information, whereas those with <10 reads were discarded. Using the sequences that passed through these processes, we determined Rtsp-1 and LIb insertion sites according to the following steps: (i) we conducted all-to-all comparison of these sequences to reveal their similarities, which were used to build clusters that represent an individual insertion site. It started with the self-alignment of the produced sequences via BLAT analysis21 using the following parameter settings: -tileSize = 8, -minMatch = 1, -minScore = 10, -repMatch = –1 and -oneOff = 2. BLAT is one of the local pairwise alignment programmes.21 (ii) We clustered these sequences into groups based on their sequence similarity with the results of pairwise alignments. In each cluster, sequences were collected in FASTA format. (iii) We performed a multiple sequence alignment using these FASTA files for each cluster to reveal the sequence similarity with ClustalW program,22 which produced an alignment file per cluster. Then, we determined the representative sequence in each cluster based on the number of sequence. The sequence with the highest number was extracted as the representative sequence in each cluster. Our results showed that the sequences to be aligned in each cluster were very similar to each other (identity of >90%). These processes generated the cluster of sequence or non-clustered sequence with ≥10 reads as an individual insertion site that a copy of Rtsp-1 or LIb was inserted in at least one or more cultivars. However, assigning the insertion sites to cultivars would probably have caused some errors, because sequence errors in the index sequence may have allocated the reads to an incorrect cultivar while sequences with small read numbers may have been incorrectly included in large clusters by the BLAT analysis. These erroneous assignments would have resulted in a very small number of reads. Thus, we set a critical value for determining the presence of Rtsp-1 and LIb insertions, i.e. if the reads for a cultivar at a specific insertion site comprise <0.01% of the entire reads for that cultivar, we assumed that the retrotransposon was absent from that site. Each cultivar was genotyped by combining the presence (1) or absence (0) of information at each site for all the insertion sites (Supplementary Tables S3 and S4).
2.4. Development of SCAR markers
We designed the PCR primers to amplify the junction of the retrotransposon and its insertion site. Supplementary Tables S3 and S4 showed all genotyping information (scores 0 and 1) in the cultivars, the representative sequence in each cluster and non-cluster sequence. We focused on the putative cultivar-specific insertion site that was present in one cultivar (scoring; 1) and absent from others (scoring; 0). The primer sequence of SCAR marker was designed based on the representative sequence for the cluster of these cultivar-specific insertion site (Supplementary Table S2). The PCR was performed with Rtsp-1-specific primer (Rtsp-1_ppt) and insertion site-specific (RP_*) primer combinations using the genomic DNA as the template (Supplementary Table S2). The PCR comprises an initial denaturation at 95°C for 2 min, which was followed by 30 cycles at 95°C for 30 s, 58°C for 30 s and 72°C for 30 s, with a final extension at 72°C for 15 min. The PCR products were visualized using 1.5% agarose gel electrophoresis.
2.5. Phylogenetic analysis
A distance matrix was calculated from the presence/absence matrices using Nei's and Li's coefficient index23 with the FreeTree program (Supplementary Tables S5 and S6).24 The phylogenetic reconstruction was based on the neighbour-joining method. Support for the internal branches in the phylogeny was assessed using 1,000 bootstrap replicates. A dendrogram was constructed with MEGA5.25
3. Results
3.1. MiSeq sequencing of the Rtsp-1 and LIb insertion sites
We constructed a MiSeq sequencing library to screen the junction sequences of the Rtsp-1 and LIb insertion sites in 38 sweet potato cultivars (Fig. 1). Each cultivar sample was labelled using a TruSeq DNA-Sampling Preparation Kit with dual indexing (Fig. 1). The Rtsp-1 and LIb libraries were mixed at a ratio of 4 : 1 to prepare a single sequencing sample, because the copy number of Rtsp-1 was estimated to be approximately four times higher than that of LIb.9,10
A total of 4,401,639 read pairs were obtained from a single lane-sequencing run using the MiSeq platform, i.e. 3,567,587 and 834,052 read pairs represented Rtsp-1 and LIb insertion sites, respectively (Table 1). The results indicated that the read number ratio in each library (Rtsp-1 reads = 81.1% and LIb reads = 18.9%) was close to the expected value (Rtsp-1 reads = 80% and LIb reads = 20%). Of these read pairs, 86.8% for Rtsp-1 and 89.8% for LIb had valid retrotransposon-specific sequences (Table 1), which indicated that nested PCR could specifically amplify DNA fragments from the junction sequences of these elements. The read numbers for each sample are presented in Supplementary Table S1.
Table 1.
Summary of the MiSeq read data processing
| Retrotransposon | Analysis | No. of read pairs | No. of collapsed reads (≥10) | Ratio (%) | No. of clusters |
|---|---|---|---|---|---|
| Rtsp-1 | Raw data | 3,567,587 | 100 | ||
| Validation of retrotransposon sequence | 3,095,771 | 16,428 | 86.8 | ||
| Trimming to 50 bp | 3,095,771 | 8,038 | 86.8 | ||
| QV filtering (≥20) | 2,292,344 | 5,245 | 64.3 | ||
| Outlier filtering (>99%) | 2,273,707 | 5,218 | 63.7 | ||
| Clustering with BLATa | 1,497 | ||||
| LIb | Raw data | 834,052 | 100 | ||
| Validation of retrotransposon sequence | 743,945 | 3,608 | 89.2 | ||
| Poly(A) tail trimming (A ≥ 3) | 743,935 | 3,571 | 89.2 | ||
| Trimming to 50 bp | 743,935 | 3,154 | 89.2 | ||
| QV filtering (≥20) | 434,502 | 1,619 | 52.1 | ||
| Outlier filtering (>98%) | 427,120 | 1,603 | 51.2 | ||
| Clustering with BLATa | 527 |
aThe clusters include groups of two or more sequences and non-clustered single sequences for the insertion sites of Rtsp-1and LIb, where the minimum number of reads for an identical sequence was 10.
3.2. Identification of the Rtsp-1 and LIb insertion sites in 38 cultivars
To identify the Rtsp-1 and LIb insertion sites, we selected one end of a paired read that contained the junction sequence of the Rtsp-1 and LIb ends and their insertion sites. A sequence of 150 bp in the read was filtered based on the valid retrotransposon sequences and QV scores with base calls ≥20 in the insertion site sequence of 50 bp (see Materials and Methods) (Table 1). After these filtering processes, 63.7 and 51.2% of the reads remained for Rtsp-1 and LIb, respectively (Table 1). These reads were used in the clustering analysis to obtain sequence information for each insertion site (Table 1). The clustering process was conducted using BLAT,21 where the parameter settings were -tileSize = 8, -minMatch = 1, -minScore = 10, -repMatch = –1 and -oneOff = 2, which generated 1,497 and 527 clusters or non-clustered single sequences for the insertion sites of Rtsp-1 and LIb, respectively, where the minimum number of reads required for an identical sequence was 10 (Tables 1 and 2). We detected Rtsp-1 and LIb in each insertion site of the 38 cultivars (Supplementary Tables S3 and S4). In the 38 cultivars, the median number of Rtsp-1 insertion sites per cultivar was 257.5 (range = 216–347), whereas that for LIb was 92.5 (range = 67–147) (Fig. 2). Also, the average number of Rtsp-1 insertion sites among 38 cultivars was 259, whereas that of LIb was 102.6, respectively. Of the 1,497 Rtsp-1 insertion sites, 88.6% were polymorphic among cultivars and 19.4% were identified as cultivar-specific sites, which were present in only one cultivar and absent from others (Table 2). For LIb, the same figures were 99.2 and 16.1%, respectively (Table 2). Thus, the insertion sites of these two active retrotransposon families were found to be highly polymorphic among sweet potato cultivars.
Table 2.
Summary of the insertion sites identified
| All insertion sites | No. of polymorphic sites | Polymorphic ratio (%) | No. of cultivar-specific sites | Cultivar-specific ratio (%) | |
|---|---|---|---|---|---|
| Rtsp-1 | 1,497 | 1,327 | 88.6 | 291 | 19.4 |
| LIb | 527 | 523 | 99.2 | 85 | 16.1 |
| Total | 2,024 | 1,850 | 91.4 | 376 | 18.6 |
Figure 2.
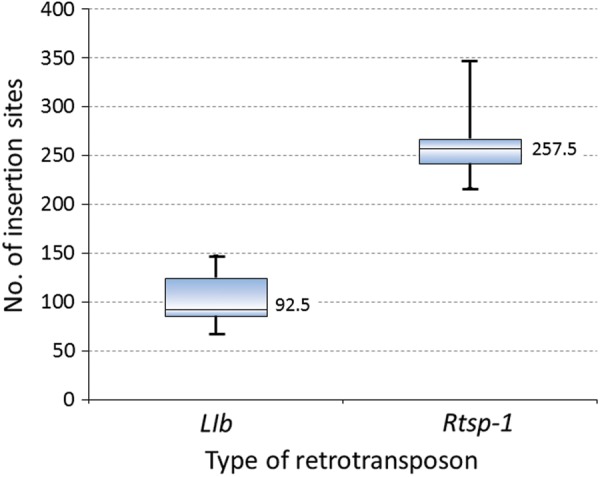
The numbers of Rtsp_1 and LIb insertion sites identified in this study. The box plots show the distributions of these sites in the 38 cultivars. The central rectangle spans the first quartile to the third quartile. The line inside the rectangle shows the median number (257.5 and 92.5 for Rtsp-1 and LIb, respectively), and the whiskers above and below the box show the maximum and minimum number, respectively.
3.3. Development of SCAR markers for cultivar identification
We designed PCR primers using the Rtsp-1 insertion sequences of five cultivar-specific sites (Supplementary Table S2). We conducted PCR amplification with the 38 sweet potato cultivars using these cultivar-specific insertion primer (RP_*) and Rtsp-1-specific primer (Rtsp-1_ppt), which produced single, distinct and clear bands (Fig. 3). The SCAR markers developed in this study can be treated as dominant markers and used for the simple and rapid screening of cultivars.
Figure 3.

Agarose gel image showing the SCAR markers developed in this study. PCR amplification was conducted using the cultivar-specific insertion primer (RP_*) and the Rtsp-1-specific primer (Rtsp-1_ppt). Descriptions of these primers are provided in Supplementary Table S2. The arrows indicate the strong and reproducible bands that were specific for each cultivar. Lanes 1–38 correspond to the cultivars listed in Supplementary Table S1.
3.4. Genetic relationships among sweet potato cultivars
To investigate the genetic relationships among the 38 sweet potato cultivars, we conducted a phylogenetic analysis based on the Rtsp-1 and LIb insertion sites. Basically, sweet potato is cultivated by vegetative propagation. However, Japanese sweet potato cultivars had been developed with crossing the limited number of cultivars, which indicated most cultivars used in this study were genetically related. The genetic distance scores were calculated based on Nei's and Li's coefficient index (Supplementary Tables S5 and S6). The phylogenetic trees were constructed using the neighbour-joining method (Fig. 4 and Supplementary Fig. S1). The genetic relationships among cultivars based on the Rtsp-1 and LIb insertion polymorphisms were consistent well each other (Fig. 4 and Supplementary Fig. S1). Pedigree information was known for some cultivars (Supplementary Fig. S2), which was also consistent with the genetic relationships in the phylogenetic trees. In addition, the parent and offspring cultivars shown in the pedigree information had lower genetic distance (Supplementary Tables S5 and S6), which indicated they are genetically closely-related. This suggests that obtaining genotyping information based on the active retrotransposon insertion sites may be useful for determining the genetic relationships among sweet potato cultivars.
Figure 4.

Phylogenetic analysis based on the Rtsp-1 insertion polymorphisms of the cultivars. The phylogenetic tree was constructed using the neighbour-joining method. The bootstrap values are shown. +The genetic relationships based on the Rtsp-1 and LIb (Supplementary Fig. S1) insertion polymorphisms were well consistent. *The genetic relationships among cultivars agreed well with the pedigree information (Supplementary Fig. S2).
4. Discussion
In this study, we conducted DNA fingerprinting based on the insertion polymorphisms of the active retrotransposon families (Rtsp-1 and LIb) in the sweet potato. The application of a MiSeq sequencing platform, which is a bench-top high-throughput sequencer, allowed >1.3 Gb of high-quality sequencing data to be produced at a low cost in a short time. We successfully identified 2,024 insertion sites for two types of retrotransposon families and obtained 76,912 (2,024 insertion sites × 38 cultivars) genotyping data points in a single sequencing run. We developed several SCAR markers based on the cultivar-specific insertion sites (Fig. 3). This type of marker has several advantages compared with simple sequence repeat (SSR) marker or amplified fragment length polymorphism (AFLP) marker, because they can be used to produce specific and easily recognizable bands by agarose gel electrophoresis (Fig. 3).26–28 Thus, the SCAR markers developed in this study could be used for the rapid and precise screening of sweet potato cultivars in breeding programmes. Furthermore, the genetic relationships among sweet potato cultivars can be determined based on the genotyping data, which were demonstrated by the phylogenetic analysis (Fig. 4 and Supplementary Fig. S1). Our methods should accelerate sweet potato genotyping and they provide a practical method for cultivar identification.
The sweet potato is a non-model crop species and its whole genome sequence information is not available. It is a hexaploid plant (2n = 6x = 90) with an estimated genome size of 2,200–3,000 Mb.29 Recently, comprehensive transcriptome sequencing of the sweet potato was reported in three studies.29–31 Thus, genetic analyses of the sweet potato based on molecular markers have been delayed compared with other plant species. In general, genotyping with a high-throughput sequencing system, such as the Illumina HiSeq 2000, has been conducted based on single nucleotide polymorphism (SNP) loci in model species.32–38 If a reference genome sequence is available, whole genome resequencing or exome resequencing may be effective for genome-wide SNP and/or indel calling using multiple samples after mapping the reads to the reference genome. In non-model crop species such as the sweet potato, however, these resequencing methods cannot be applied because of the lack of reference sequences. Thus, targeting the sequences of highly polymorphic sites in cultivars may be an efficient alternative method for genotyping these species. Moreover, high-sensitivity targeted sequencing is far less expensive and requires less sequencing data per sample than whole genome resequencing.14,16,19 Our results demonstrated that the active retrotransposon insertion sites were highly polymorphic in cultivated sweet potato varieties, and the targeted sequencing of these sites could be achieved using a MiSeq bench-top system. Thus, the low cost and time-saving DNA fingerprinting methods developed in this study could facilitate the precise identification of cultivars, as well as being used for assessments of genetic diversity and conducting linkage analyses.
Supplementary Data
Supplementary data are available at www.dnaresearch.oxfordjournals.org
Funding
This work was supported by a Research and Development Project for Application in Promoting the New Policy of Agriculture Forestry and Fisheries grant from the Ministry of Agriculture, Forestry and Fisheries of Japan and by the Programme to Disseminate Tenure Tracking System, from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan (to Y.M.).
Supplementary Material
Acknowledgements
We thank Drs Yoshihiro Okada and Yasuhiro Takahata at the National Agriculture and Food Research Organization, Kyusyu Okinawa Agricultural Research Center, for providing plant materials and their experimental support. We also thank Drs Nobuyuki Fujii and Kazuho Ikeo at the National Institute of Genetics for the development of data analysis pipelines.
References
- 1.Ukoskit K. Autopolyploidy versus allopolyploidy and low-density randomly amplified polymorphic DNA linkage maps of sweetpotato. J. Am. Soc. Hort. Sci. 1997;122:822–8. [Google Scholar]
- 2.Kriegner A., Burg K., Mwanga R.O. A genetic linkage map of sweetpotato (Ipomoea batatas (L.) Lam.) based on AFLP markers. Mol. Breeding. 2003;11:169–85. [Google Scholar]
- 3.Cervantes-Flores J.C., Sosinski B., Pecota K.V., et al. Identification of quantitative trait loci for dry-matter, starch, and β-carotene content in sweetpotato. Mol. Breeding. 2010;28:201–16. [Google Scholar]
- 4.Li A.X., Liu Q.C., Wang Q.M., Zhang L.M., Zhai H., Liu S.Z. Establishment of molecular linkage maps using SRAP markers in sweet potato. Acta Agronom. Sin. 2010;36:1286–95. [Google Scholar]
- 5.Zhao N., Yu X., Jie Q., et al. A genetic linkage map based on AFLP and SSR markers and mapping of QTL for dry-matter content in sweetpotato. Mol. Breeding. 2013 doi:10.1007/s11032-013-9908-y. [Google Scholar]
- 6.Kumar A., Bennetzen J.L. Plant retrotransposons. Annu. Rev. Genet. 1999;33:479–532. doi: 10.1146/annurev.genet.33.1.479. [DOI] [PubMed] [Google Scholar]
- 7.Feschotte C., Jiang N., Wessler S.R. Plant transposable elements: where genetics meets genomics. Nat. Rev. Genet. 2002;3:329–41. doi: 10.1038/nrg793. [DOI] [PubMed] [Google Scholar]
- 8.Levin H.L., Moran J.V. Dynamic interactions between transposable elements and their hosts. Nat. Rev. Genet. 2011;12:615–27. doi: 10.1038/nrg3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tahara M., Aoki T., Suzuka S., et al. Isolation of an active element from a high-copy-number family of retrotransposons in the sweetpotato genome. Mol. Genet. Genomics. 2004;272:116–27. doi: 10.1007/s00438-004-1044-2. [DOI] [PubMed] [Google Scholar]
- 10.Yamashita H., Tahara M. A LINE-type retrotransposon active in meristem stem cells causes heritable transpositions in the sweetpotato genome. Plant Mol. Biol. 2006;61:79–94. doi: 10.1007/s11103-005-6002-9. [DOI] [PubMed] [Google Scholar]
- 11.Waugh R., McLean K., Flavell A.J., et al. Genetic distribution of Bare-1-like retrotransposable elements in the barley genome revealed by sequence-specific amplification polymorphisms (S-SAP) Mol. Gen. Genet. 1997;253:687–94. doi: 10.1007/s004380050372. [DOI] [PubMed] [Google Scholar]
- 12.Kumar A., Hirochika H. Applications of retrotransposons as genetic tools in plant biology. Trends Plant Sci. 2001;6:127–34. doi: 10.1016/s1360-1385(00)01860-4. [DOI] [PubMed] [Google Scholar]
- 13.Syed N.H., Sureshsundar S., Wilkinson M.J., Bhau B.S., Cavalcanti J.J.V., Flavell A.J. Ty1-copia retrotransposon-based SSAP marker development in cashew (Anacardium occidentale L.) Theor. Appl. Genet. 2005;110:1195–202. doi: 10.1007/s00122-005-1948-1. [DOI] [PubMed] [Google Scholar]
- 14.Kalendar R. The use of retrotransposon-based molecular markers to analyze genetic diversity. Ratar. Povrt. 2011;48:261–74. [Google Scholar]
- 15.Kalendar R., Flavell A.J., Ellis T.H.N., Sjakste T., Moisy C., Schulman A.H. Analysis of plant diversity with retrotransposon-based molecular markers. Heredity. 2011;106:520–30. doi: 10.1038/hdy.2010.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poczai P., Varga I., Laos M., et al. Advances in plant gene-targeted and functional markers: a review. Plant Methods. 2013;9:6. doi: 10.1186/1746-4811-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loman N.J., Misra R.V., Dallman T.J., et al. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol. 2012;30:434–9. doi: 10.1038/nbt.2198. [DOI] [PubMed] [Google Scholar]
- 18.Pallen M.J. Reply to updating benchtop sequencing performance comparison. Nat. Biotechnol. 2013;31:296. doi: 10.1038/nbt.2531. [DOI] [PubMed] [Google Scholar]
- 19.Xing J., Witherspoon D.J., Jorde L.B. Mobile element biology: new possibilities with high-throughput sequencing. Trends Genet. 2013;29:280–9. doi: 10.1016/j.tig.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmieder R., Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–4. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kent W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002;12:656–64. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Larkin M.A., Blackshields G., Brown N.P., et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 23.Nei M., Li W.H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA. 1979;76:5269–73. doi: 10.1073/pnas.76.10.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pavlicek A., Hrda S., Flegr J. FreeTree—Freeware program for construction of phylogenetic trees on the basis of distance data and bootstrap/jackknife analysis of the tree robustness. Application in the RAPD analysis of genus. Folia Biol. 1999;45:97–9. [PubMed] [Google Scholar]
- 25.Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011;28:2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Monden Y., Naito K., Okumoto Y., et al. High potential of a transposon mPing as a marker system in japonica times japonica cross in rice. DNA Res. 2009;16:131–40. doi: 10.1093/dnares/dsp004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kiran U., Khan S., Mirza K.J., Ram M., Abdin M.Z. SCAR markers: a potential tool for authentication of herbal drugs. Fitoterapia. 2010;81:969–76. doi: 10.1016/j.fitote.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 28.Cirillo A., Gaudio S.D., Bernardo G.B., Galano G., Galderisi U., Cipollaro M. A new SCAR marker potentially useful to distinguish Italian cattle breeds. Food Chem. 2012;130:172–6. [Google Scholar]
- 29.Tao X., Gu Y.H., Wang H.Y., et al. Digital gene expression analysis based on integrated de novo transcriptome assembly of sweet potato [Ipomoea batatas (L.) Lam] PLoS ONE. 2012;7:e36234. doi: 10.1371/journal.pone.0036234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schafleitner R., Tincopa L.R., Palomino O., et al. A sweetpotato gene index established by de novo assembly of pyrosequencing and Sanger sequences and mining for gene-based microsatellite markers. BMC Genomics. 2010;11:604. doi: 10.1186/1471-2164-11-604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Z., Fang B., Chen J., et al. De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweet potato (Ipomoea batatas) BMC Genomics. 2010;11:726. doi: 10.1186/1471-2164-11-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brachi B., Faure N., Horton M., et al. Linkage and association mapping of Arabidopsis thaliana flowering time in nature. PLoS Genet. 2010;6:e1000940. doi: 10.1371/journal.pgen.1000940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clark R.M. Genome-wide association studies coming of age in rice. Nat. Genet. 2010;42:926–7. doi: 10.1038/ng1110-926. [DOI] [PubMed] [Google Scholar]
- 34.Huang X., Wei X., Sang T., et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010;42:961–7. doi: 10.1038/ng.695. [DOI] [PubMed] [Google Scholar]
- 35.Lam H.M., Xu X., Liu X., et al. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet. 2010;42:1053–9. doi: 10.1038/ng.715. [DOI] [PubMed] [Google Scholar]
- 36.Li Y., Huang Y., Bergelson J., Nordborg M., Borevitz J.O. Association mapping of local climate-sensitive quantitative trait loci in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA. 2010;107:21199–204. doi: 10.1073/pnas.1007431107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nielsen R., Paul J.S., Albrechtsen A., Song Y.S. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 2011;12:443–51. doi: 10.1038/nrg2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamamoto T., Nagasaki H., Yonemaru J.I., et al. Fine definition of the pedigree haplotypes of closely related rice cultivars by means of genome-wide discovery of single-nucleotide polymorphisms. BMC Genomics. 2010;11:267. doi: 10.1186/1471-2164-11-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.