

Abstract

We report the synthesis and characterization of eight half-sandwich cyclopentadienyl IrIII pyridine complexes of the type [(η5-Cpxph)Ir(phpy)Z]PF6, in which Cpxph = C5Me4C6H5 (tetramethyl(phenyl)cyclopentadienyl), phpy = 2-phenylpyridine as C∧N-chelating ligand, and Z = pyridine (py) or a pyridine derivative. Three X-ray crystal structures have been determined. The monodentate py ligands blocked hydrolysis; however, antiproliferative studies showed that all the Ir compounds are highly active toward A2780, A549, and MCF-7 human cancer cells. In general the introduction of an electron-donating group (e.g., Me, NMe2) at specific positions on the pyridine ring resulted in increased antiproliferative activity, whereas electron-withdrawing groups (e.g., COMe, COOMe, CONEt2) decreased anticancer activity. Complex 5 displayed the highest anticancer activity, exhibiting submicromolar potency toward a range of cancer cell lines in the National Cancer Institute NCI-60 screen, ca. 5 times more potent than the clinical platinum(II) drug cisplatin. DNA binding appears not to be the major mechanism of action. Although complexes [(η5-Cpxph)Ir(phpy)(py)]+ (1) and [(η5-Cpxph)Ir(phpy)(4-NMe2-py)]+ (5) did not cause cell apoptosis or cell cycle arrest after 24 h drug exposure in A2780 human ovarian cancer cells at IC50 concentrations, they increased the level of reactive oxygen species (ROS) dramatically and led to a loss of mitochondrial membrane potential (ΔΨm), which appears to contribute to the anticancer activity. This class of organometallic Ir complexes has unusual features worthy of further exploration in the design of novel anticancer drugs.
Introduction
The clinical use of platinum anticancer drugs has stimulated the search for other transition metal anticancer complexes with improved features.1 In particular other platinum complexes2 and some group 8 metal complexes containing iron3 and ruthenium4 centers show promising anticancer activity both in vitro and in vivo.
Very recently, possible biological applications of iridium compounds have attracted attention.5 Half-sandwich organometallic IrIII compounds in particular display high versatility and show promising anticancer activity.6 For example, Sheldrick et al. have designed monoiridium and di-iridium polypyridyl intercalators that target DNA in cancer cells.7 We have studied a series of half-sandwich IrIII anticancer agents of formula [(Cpx)Ir(L∧L′)Z]0/n+, where Cpx = Cp*, Cpxph (tetramethyl(phenyl)cyclopentadienyl), or Cpxbiph (tetramethyl(biphenyl)cyclopentadienyl), L∧L′ = bidentate ligand with nitrogen, oxygen, and/or carbon donor atoms, and Z = Cl, H2O, or pyridine (py).5a,6a We found that potent activity can be achieved by modification of ligands around the iridium center and that small changes in structure can have a major effect on biological activity. For example, antiproliferative activity as measured by IC50 values (concentration at which 50% of cell growth is inhibited) decreased dramatically from inactive (>100 μM) to highly potent (submicromolar) when phenyl or biphenyl was introduced in place of a methyl group on the Cp* ring. We also reported that anticancer activity can be improved significantly by replacement of neutral N∧N-chelating ligands with negatively charged C∧N-chelating ligands, leading to increased cellular uptake and nucleobase binding.6c The monodentate ligand Z (which in most of these IrIII half-sandwich compounds is Cl) is often readily substituted by water in aqueous solution (hydrolysis), followed by interaction with biological molecules. A relationship between hydrolysis and anticancer activity has been established for RuII arene compounds, where readily hydrolyzed compounds are cytotoxic and those that do not hydrolyze are inactive or weakly active toward cancer cells.8 For cyclopentadienyl Ir C∧N compounds, we found that decreasing hydrolysis by substitution of Cl by pyridine (py) does not result in loss of anticancer activity. In fact, the py complex is highly potent, ca. 10 times and 6 times more active than the clinically used platinum drug cisplatin (CDDP) and the chloride analogue, respectively.6a These results encouraged us to explore in more detail the activity of complexes containing py derivatives.
In this study, the complexes contain Cpxph and C∧N-bound 2-phenylpyridine (phpy) as the cyclopentadienyl and chelating ligands, respectively, and various pyridine derivatives as the monodentate ligand Z. Thus, eight half-sandwich IrIII compounds of the type [(η5-Cpxph)Ir(phpy)Z]PF6, where Z = pyridine or its derivatives, were synthesized and characterized. Their chemical behavior and antiproliferative activity toward cancer cells have been investigated.
Experimental Section
Materials
2-Phenylpyridine, 4-pyridinemethanol, 4-dimethylaminopyridine, methylnicotinate, N,N-diethylnicotinamide, 3-picoline, 4-picoline, 3-acetylpyridine, 9-ethylguanine, and 9-methyladenine were purchased from Sigma-Aldrich. For the biological experiments, RPMI-1640 medium, fetal bovine serum, l-glutamine, penicillin/streptomycin mixture, trypsin/EDTA, and phosphate-buffered saline (PBS) were purchased from PAA Laboratories GmbH. Cisplatin CDDP (≥99.9%), trichloroacetic acid (≥99%), sulforhodamine B (75%), sodium phosphate monobasic monohydrate (≥99%), sodium phosphate dibasic heptahydrate (≥99%), acetic acid (≥99%), staurosporine, propidium iodide (>94%), and RNase A were obtained from Sigma-Aldrich. Complex [(η5-Cpxph)Ir(phpy)Cl] was prepared according to reported methods.6d
Syntheses
Compounds 1–8 were prepared by the same general method: A solution of the chlorido complex [(η5-Cpxph)Ir(phpy)Cl] and AgNO3 (1 mol equiv) in MeOH and water (1:1, v/v) was heated under reflux in an N2 atmosphere for 3 h. The precipitate (AgCl) was removed by filtration through Celite, and pyridine derivative (10 molar equiv) was added to the filtrate. The reaction mixture was stirred at ambient temperature for 12 h. NH4PF6 (10 mol equiv) was then added to the solution. The yellow precipitate was dissolved in acetone. The solution was evaporated slowly at ambient temperature, and the crystalline product was collected by filtration, washed with diethyl ether, and recrystallized from methanol/diethyl ether.
[(η5-Cpxph)Ir(phpy)(py)]PF6 (1)
Yield: 76%. 1H NMR (MeOD-d4): δ 8.88 (d, 1H, J = 6.0 Hz), 8.55 (d, 2H, J = 6.0 Hz), 8.10 (d, 1H, J = 8.3 Hz), 7.96 (t, 2H, J = 6.7 Hz), 7.87 (m, 2H), 7.36 (m, 5H), 7.26 (m, 3H), 6.93 (d, 2H, J = 8.0 Hz), 1.86 (s, 3H), 1.80 (s, 3H), 1.68 (s, 3H), 1.65 (s, 3H). 13C NMR (DMSO-d6): δ 153.5, 140.4, 139.9, 130.2, 129.1, 127.6, 124.7, 120.6, 98.3, 30.3, 9.8, 8.4. Anal. Calcd for C31H30F6IrN2P (767.76): C, 48.50; H, 3.94; N, 3.65. Found: C, 48.37; H, 3.92; N, 3.58. MS: m/z 623.1 [(η5-Cpxph)Ir(phpy)(py)]+.
[(η5-Cpxph)Ir(phpy)(4-Me-py)]PF6 (2)
Yield: 75%. 1H NMR (MeOD-d4): δ 8.87 (d, 1H, J = 6.0 Hz), 8.34 (d, 2H, J = 6.5 Hz), 8.09 (d, 1H, J = 8.0 Hz), 7.93 (m, 2H), 7.85 (d, 1H, J = 7.8 Hz), 7.38 (m, 6H), 7.17 (d, 2H, J = 5.5 Hz), 6.95 (d, 2H, J = 7.0 Hz), 2.37 (s, 3H), 1.85 (s, 3H), 1.81 (s, 3H), 1.68 (s, 3H), 1.64 (s, 3H). 13C NMR (DMSO-d6): δ 152.7, 145.7, 135.4, 131.7, 130.2, 129.3, 128.1, 125.1, 120.5, 98.5, 30.7, 21.0, 9.8, 8.4. Anal. Calcd for C32H32F6IrN2P (781.77): C, 49.16; H, 4.13; N, 3.58. Found: C, 49.31; H, 4.06; N, 3.66. MS: m/z 637.1 [(η5-Cpxph)Ir(phpy)(4-Me-py)]+. Crystals suitable for X-ray diffraction were obtained by slow evaporation of a methanol/acetone/water solution at ambient temperature.
[(η5-Cpxph)Ir(phpy)(3-Me-py)]PF6 (3)
Yield: 76%. 1H NMR (MeOD-d4): δ 8.91 (d, 1H, J = 6.0 Hz), 8.37 (d, 1H, J = 5.0 Hz), 8.31 (s, 1H), 8.09 (d, 1H, J = 8.0 Hz), 7.96 (m, 2H), 7.87 (d, 1H, J = 7.8 Hz), 7.69 (d, 1H, J = 8.5 Hz), 7.25 (m, 7H), 6.93 (d, 2H, J = 8.0 Hz), 2.24 (s, 3H), 1.87 (s, 3H), 1.79 (s, 3H), 1.68 (s, 3H), 1.66 (s, 3H). 13C NMR (MeOD-d4): δ 154.2, 151..9, 141.1, 136.4, 133.3, 131.3, 129.7, 127.6, 125.4, 121.2, 9.9, 8.6. Anal. Calcd for C32H32F6IrN2P (781.77): C, 49.16; H, 4.13; N, 3.58. Found: C, 48.92; H, 4.13; N, 3.45. MS: m/z 637.1 [(η5-Cpxph)Ir(phpy)(3-Me-py)]+.
[(η5-Cpxph)Ir(phpy)(4-MeOH-py)]PF6 (4)
Yield: 56%. 1H NMR (MeOD-d4): δ 8.88 (d, 1H, J = 6.3 Hz), 8.45 (d, 2H, J = 6.5 Hz), 8.09 (d, 1H, J = 9.0 Hz), 7.95 (t, 2H, J = 7.6 Hz), 7.86 (d, 1H, J = 8.5 Hz), 7.33 (m, 5H), 7.20 (m, 3H), 6.95 (d, 2H, J = 8.3 Hz), 1.85 (s, 3H), 1.81 (s, 3H), 1.68 (s, 3H), 1.65 (s, 3H). 13C NMR (MeOD-d4): δ 154.1, 140.7, 136.4, 133.3, 131.4, 129.8, 129.2, 125.9, 125.2, 121.2, 9.6, 8.5. Anal. Calcd for C32H32F6IrN2PO (797.79): C, 48.18; H, 4.64; N, 3.51. Found: C, 48.05; H, 3.96; N, 3.43. MS: m/z 653.1 [(η5-Cpxph)Ir(phpy)(4-MeOH-py)]+.
[(η5-Cpxph)Ir(phpy)(4-NMe2-py)]PF6 (5)
Yield: 62%. 1H NMR (MeOD-d4): δ 8.82 (d, 1H, J = 5.8 Hz), 8.08 (d, 1H, J = 8.0 Hz), 7.92 (t, 1H, J = 8.3 Hz), 7.85 (m, 4H), 7.31 (m, 5H), 7.20 (t, 1H, J = 8.0 Hz), 6.99 (d, 2H, J = 7.5 Hz), 6.45 (d, 2H, J = 7.0 Hz), 3.00 (s, 6H), 1.84 (s, 3H), 1.81 (s, 3H), 1.70 (s, 3H), 1.63 (s, 3H). 13C NMR (CDCl3): δ 152.2, 145.4, 134.0, 130.1, 129.4, 127.8, 119.6, 109.2, 96.8, 38.9, 9.5, 8.1. Anal. Calcd for C33H35F6IrN3P (810.81): C, 48.93; H, 4.32; N, 5.09. Found: C, 48.88; H, 4.35; N, 5.18. MS: m/z 666.1 [(η5-Cpxph)Ir(phpy)(4-NMe2-py)]+. Crystals suitable for X-ray diffraction were obtained by slow evaporation of a methanol/acetone/water solution at ambient temperature.
[(η5-Cpxph)Ir(phpy)(3-COMe-py)]PF6 (6)
Yield: 78%. 1H NMR (MeOD-d4): δ 8.94 (s, 1H), 8.91 (d, 1H, J = 6.0 Hz), 8.75 (d, 1H, J = 5.5 Hz), 8.39 (d, 1H, J = 8.0 Hz), 8.11 (d, 1H, J = 8.3 Hz), 7.97 (m, 2H), 7.87 (d, 1H, J = 7.8 Hz), 7.51 (dd, 1H, J = 5.5, 5.5 Hz), 7.32 (m, 6H), 6.98 (d, 2H, J = 7.5 Hz), 2.49 (s, 3H), 1.87 (s, 3H), 1.82 (s, 3H), 1.72 (s, 3H), 1.67 (s, 3H). 13C NMR (DMSO-d6): δ 153.6, 140.8, 135.8, 135.5, 131.7, 129.8, 128.8, 125.0, 121.2, 9.8, 8.6. Anal. Calcd for C33H32F6IrN2PO (809.80): C, 48.94; H, 3.98; N, 3.46. Found: C, 48.78; H, 3.85; N, 3.34. MS: m/z 665.2 [(η5-Cpxph)Ir(phpy)(3-COMe-py)]+.
[(η5-Cpxph)Ir(phpy)(3-COOMe-py)]PF6 (7)
Yield: 63%. 1H NMR (MeOD-d4): δ 9.01 (s, 1H), 8. 90 (d, 1H, J = 6.0 Hz), 8.76 (d, 1H, J = 5.8 Hz), 8.39 (dt, 1H, J = 7.8 Hz), 8.12 (d, 1H, J = 7.8 Hz), 7.98 (m, 2H), 7.87 (d, 1H, J = 7.8 Hz), 7.51 (dd, 1H, J = 5.7, 5.7 Hz), 7.40 (m, 2H), 7.34 (m, 4H), 6.98 (m, 2H), 3.93 (s, 3H), 1.86 (s, 3H), 1.81 (s, 3H), 1.71 (s, 3H), 1.67 m, 2H (s, 3H). 13C NMR (MeOD-d4): δ 157.8, 141.2, 136.3, 133.6, 131.3, 129.8, 128.4, 126.1, 125.7, 32.9, 9.4, 8.3. Anal. Calcd for C33H32F6IrN2PO2 (825.80): C, 48.00; H, 3.91; N, 3.39. Found: C, 48.08; H, 4.04; N, 3.32. MS: m/z 682.1 [(η5-Cpxph)Ir(phpy)(3-COOMe-py)]+.
[(η5-Cpxph)Ir(phpy)(3-CONEt2-py)]PF6 (8)
Yield: 30%. 1H NMR (MeOD-d4): δ 8.92 (d, 1H, J = 6.0 Hz), 8.72 (d, 1H, J = 5.7 Hz), 8.42 (s, 1H), 8.13 (d, 1H, J = 8.0 Hz), 7.93 (m, 4H), 7.47 (dd, 1H, J = 5.5, 5.5 Hz), 7.39 (q, 2H, J = 7.0 Hz), 7.26 (m, 4H), 6.94 (d, 2H, J = 7.8 Hz), 3.51 (b, 2H), 2.92 (b, 2H), 1.88 (s, 3H), 1.81 (s, 3H), 1.71 (s, 3H), 1.68 (s, 3H), 1.21 (b, 3H), 0.79 (b, 3H). 13C NMR (MeOD-d4): δ 154.6, 140.5, 133.4, 131.4, 129.8, 129.2, 128.2, 125.4, 121.2, 9.9, 8.5. Anal. Calcd for C36H39F6IrN3PO (866.87): C, 49.88; H, 4.53; N, 4.85. Found: C, 49.94; H, 4.44; N, 4.67. MS: m/z 722.1 [(η5-Cpxph)Ir(phpy)(3-CONEt2-py)]+. Crystals suitable for X-ray diffraction were obtained by slow evaporation of a methanol/acetone/water solution at ambient temperature.
Methods and Instrumentation
X-ray Crystallography
Suitable crystals of compounds 2, 5, and 8 were selected and mounted on a glass fiber with Fromblin oil on an Oxford Diffraction Gemini Xcalibur diffractometer with a Ruby CCD area detector. The crystals were kept at 100(2) or 150(2) K during data collection. Using Olex2,9 the structures of 2, 5, and 8 were solved with the XS10 structure solution program using direct methods and refined with the XL10 refinement package using least squares minimization.
X-ray crystallographic data for compounds 2, 5, and 8 have been deposited in the Cambridge Crystallographic Data Centre under the accession numbers CCDC 1007223, 1007225, and 1007224, respectively.
NMR Spectroscopy
1H NMR spectra were acquired in 5 mm NMR tubes at 298 or 310 K on either a Bruker DPX 400 (1H = 400.03 MHz) or an AVA 600 (1H = 600.13 MHz) spectrometer. 1H NMR chemical shifts were internally referenced to CHD2OD (3.33 ppm) for methanol-d4 or CHCl3 (7.26 ppm) for chloroform-d1. MeOD-d4 was used to aid solubility. All data processing was carried out using MestReC or TOPSPIN version 2.0 (Bruker U.K. Ltd.).
Mass Spectrometry
Electrospray ionization mass spectra (ESI-MS) were obtained by preparing the samples in 50% CH3CN and 50% H2O (v/v) or using NMR samples for infusion into the mass spectrometer (Bruker Esquire 2000). The mass spectra were recorded with a scan range of m/z 400–1000 for positive ions.
Elemental Analysis
CHN elemental analyses were carried out on a CE-440 elemental analyzer by Warwick Analytical (UK) Ltd.
pH Measurement
pH or pH* values (pH meter reading without correction for effect of deuterium on glass electrode) of NMR samples in H2O or D2O were measured at ca. 298 K directly in the NMR tube, before and after recording NMR spectra, using a Corning 240 pH meter equipped with a micro combination electrode calibrated with Aldrich buffer solutions of pH 4, 7, and 10.
Inductively Coupled Plasma Mass Spectrometry (ICP-MS)
All ICP-MS analyses were carried out on an Agilent Technologies 7500 series ICP-MS instrument. The water used for ICP-MS analysis was doubly deionized (DDW) using a Millipore Milli-Q water purification system and a USF Elga UHQ water deionizer. The iridium Specpure plasma standard (Alfa Aesar, 1000 ppm in 10% HCl) was diluted with 5% HNO3 DDW to prepare freshly calibrants at concentrations of 50 000, 10 000, 5000, 1000, 500, 200, 50, 10, and 5 ppt. The ICP-MS instrument was set to detect 193Ir with typical detection limits of ca. 2 ppt using no gas mode.
Hydrolysis
Solutions of complexes 1–8 with final concentrations of 150 μM in 10% MeOD-d4/90% D2O (v/v) were prepared by dissolution of the complex in MeOD-d4 followed by rapid dilution with D2O. 1H NMR spectra were recorded after various time intervals at 310 K.
Interactions with Nucleobases
The reaction of complexes 1–8 (1 mM) with nucleobases 9-EtG or 9-MeA typically involved addition of 1 mol equiv of nucleobase to an equilibrium solution of complexes 1–8 in 20% MeOD-d4/80% D2O (v/v). 1H NMR spectra of these solutions were recorded at 310 K after various time intervals.
NCI-60 Screening
Compounds 2 and 5 were evaluated by the National Cancer Institute Developmental Therapeutics Program (NCI/DTP, USA) for in vitro cytotoxicity toward ca. 60 human cancer cell lines. The cells were treated with iridium compounds for 48 h at five concentrations ranging from 0.01 to 100 μM. Every compound was tested twice, and data are the average of the two experiments. Data for cisplatin and oxaliplatin are from NCI/DTP screening performed in October 2009 and 2010, respectively. The protocol for the determination of cytotoxicity toward the 60-cell-line panel can be found at http://dtp.nci.nih.gov/branches/btb/ivclsp.html. The DTP homepage can be accessed at http://dtp.cancer.gov/.
Cell Culture
A2780 ovarian carcinoma, A549 lung and MCF7 breast human adenocarcinoma cells were obtained from the European Collection of Cell Cultures (ECACC) and were grown in Roswell Park Memorial Institute medium (RPMI-1640) or Dubelco's Modified Eagle Medium (DMEM). All media were supplemented with 10%(v/v) fetal calf serum, 1%(v/v) 2 mM glutamine, and 1% (v/v, 10k units/mL) penicillin/streptomycin. All cells were grown as adherent monolayers at 310 K in a 5% CO2 humidified atmosphere and passaged regularly at ca. 80% confluence.
In Vitro Growth Inhibition Assay
Briefly, 5000 cells were seeded per well in 96-well plates. The cells were preincubated in drug-free media at 310 K for 48 h before adding different concentrations of the compounds to be tested. In order to prepare the stock solution of the drug, the solid complex was dissolved first in 5% DMSO and then diluted in a 50:50 v/v mixture of RPMI-1640/saline. This stock was further diluted using cell culture medium until working concentrations were achieved. The drug exposure period was 24 h. After this, supernatants were removed by suction, and each well was washed with PBS. A further 72 h was allowed for the cells to recover in drug-free medium at 310 K. The SRB assay11 was used to determine cell viability. Absorbance measurements of the solubilized dye (on a BioRad iMark microplate reader using a 470 nm filter) allowed the determination of viable treated cells compared to untreated controls. IC50 values (concentration of drug resulting in a 50% cell growth inhibition) were determined as duplicates of triplicates in two independent sets of experiments, and their standard deviations were calculated.
Metal Accumulation in Cancer Cells
Iridium accumulation studies for complexes 1 and 5 were conducted on A2780 ovarian cells. Briefly, 1.5 × 106 cells were seeded on a six-well plate. After 24 h of preincubation time in drug-free medium at 310 K, the complexes were added to give final concentrations equal to IC50, and a further 24 h of drug exposure was allowed. After this time, excess drugs were removed by suction, and cells were washed with PBS and then treated with trypsin-EDTA. A suspension of single cells was counted, and cell pellets were collected. Each pellet was digested overnight in concentrated nitric acid (73%) at 353 K; the resulting solutions were diluted with double-distilled water to a final concentration of 5% HNO3, and the amount of Ir taken up by the cells was determined by ICP-MS. These experiments did not include any cell recovery time in drug-free media; they were carried out in triplicate, and the standard deviations were calculated.
Cell Cycle Analysis
A2780 cells at 1.5 × 106 per well were seeded in a six-well plate. Cells were preincubated in drug-free media at 310 K for 24 h, after which drugs were added at equipotent concentrations equal to IC50. After 24 h of drug exposure, supernatants were removed by suction and cells were washed with PBS. Finally, cells were harvested using trypsin-EDTA and fixed for 24 h using cold 70% ethanol. DNA staining was achieved by resuspending the cell pellets in PBS containing propidium iodide (PI) and RNAse. Cell pellets were washed and resuspended in PBS before being analyzed in a Becton Dickinson FACScan flow cytometer using excitation of DNA-bound PI at 536 nm, with emission at 617 nm. Data were processed using Flowjo software.
Induction of Apoptosis
Flow cytometry analysis of apoptotic populations of A2780 cells caused by exposure to complexes 1 and 5 was carried out using the annexin V-FITC apoptosis detection kit (Sigma-Aldrich) according to the supplier’s instructions. Briefly, 1.5 × 106 A2780 cells per well were seeded in a six-well plate. Cells were preincubated in drug-free media at 310 K for 24 h, after which drugs were added at equipotent concentrations equal to IC50. After 24 h of drug exposure, supernatants were removed by suction, and cells were washed with PBS. Finally, cells were harvested using trypsin-EDTA. Sample staining was achieved by resuspending the cell pellets in buffer containing annexin V-FITC and PI. For positive-apoptosis controls, A2780 cells were exposed to staurosporine (1 μg/mL) for 2 h. Cells for apoptosis studies were used with no previous fixing procedure as to avoid nonspecific binding of the annexin V-FITC conjugate.
ROS Determination
Flow cytometry analysis of ROS/superoxide generation in A2780 cells caused by exposure to complexes 1 and 5 was carried out using the Total ROS/Superoxide detection kit (Enzo-Life Sciences) according to the supplier’s instructions. Briefly, 1.5 × 106 A2780 cells per well were seeded in a six-well plate. Cells were preincubated in drug-free media at 310 K for 24 h in a 5% CO2 humidified atmosphere, and then drugs were added to triplicates at concentrations of IC50 and 2 × IC50. After 1 h of drug exposure, supernatants were removed by suction and cells were washed and harvested. Staining was achieved by resuspending the cell pellets in buffer containing the orange/green fluorescent reagents. Cells were analyzed in a Becton Dickinson FACScan flow cytometer using FL1 channel Ex/Em: 490/525 nm for the oxidative stress and FL2 channel Ex/Em: 550/620 nm for superoxide detection. Data were processed using Flowjo software. At all times, samples were kept under dark conditions to avoid light-induced ROS production.
Mitochondrial Membrane Assay
Analysis of the changes of mitochondrial potential in A2780 cells after exposure to complexes 1 and 5 was carried out using the Abcam, JC-10 mitochondrial membrane potential assay kit according to the manufacturer’s instructions. Briefly, 1.5 × 106 cells were seeded in six-well plates left to incubate for 24 h in drug-free medium at 310 K in a humidified atmosphere. Drug solutions, at equipotent concentrations equal to IC50 and 2 × IC50, were added in triplicate, and the cells were left to incubate for a further 24 h under similar conditions. Supernatants were removed by suction, and each well was washed with PBS before detaching the cells using trypsin-EDTA. Staining of the samples was done in flow cytometry tubes protected from light, incubating for 30 min at ambient temperature. Samples were immediately analyzed on a Beckton Dickinson FACScan, reading the reduction of fluorescence in the FL2 channel. For positive controls, A2780 cells were exposed to carbonyl cyanide 3-chlorophenylhydrazone, CCCP (5 μM), for 15 min. Data were processed using Flowjo software.
Results
Novel Ir compounds 1–8 were synthesized in moderate yields from the chlorido analogue [(η5-Cpxph)Ir(phpy)Cl]6d by substitution of chloride by pyridine or its derivatives in the presence of silver nitrate, Scheme 1. All the synthesized complexes were isolated as PF6– salts and were fully characterized by 1H NMR spectroscopy, CHN elemental analysis, and ESI-MS. The complexes studied in this work are shown in Scheme 1.
Scheme 1. Synthesis of Ir Compounds Studied in This Work.

X-ray Crystal Structures
The X-ray crystal structures of [(η5-Cpxph)Ir(phpy)(4-Me-py)]PF6 (2), [(η5-Cpxph)Ir(phpy)(4-NMe2-py)]PF6 (5), and [(η5-Cpxph)Ir(phpy)(3-CONEt2-py)]PF6 (8) were determined. The complexes adopt the expected half-sandwich pseudo-octahedral “three-leg piano-stool” geometry with the Ir bound to a η5-cyclopentadienyl ligand occupying three coordination sites, the nitrogen atom of the py derivative (2.099–2.118 Å), and a 2-phenylpyridine C∧N-chelating ligand. Their structures are shown in Figure 1. Crystallographic data are shown in Table S1, and selected bond lengths and angles are listed in Table 1.
Figure 1.

X-ray crystal structures for (A) [(η5-Cpxph)Ir(phpy)(4-Me-py)]PF6 (2), (B) [(η5-Cpxph)Ir(phpy)(4-NMe2-py)]PF6 (5), and (C) [(η5-Cpxph)Ir(phpy)(3-CONEt2-py)]PF6 (8), with thermal ellipsoids drawn at 50% probability. The hydrogen atoms and counterions have been omitted for clarity.
Table 1. Selected Bond Lengths (Å) and Angles (deg) for [(η5-Cpxph)Ir(phpy)(4-Me-py)]PF6 (2), [(η5-Cpxph)Ir(phpy)(4-NMe2-py)]PF6 (5), and [(η5-Cpxph)Ir(phpy)(3-CONEt2-py)]PF6 (8).
| 2 | 5 | 8 | |
|---|---|---|---|
| Ir–C | 2.170(3) | 2.1676(19) | 2.163(3) |
| (cyclopentadienyl) | 2.173(2) | 2.1727(18) | 2.168(3) |
| 2.202(3) | 2.1766(19) | 2.168(3) | |
| 2.223(3) | 2.2316(18) | 2.245(3) | |
| 2.236(3) | 2.2442(18) | 2.263(3) | |
| Ir–C(centroid) | 1.827 | 1.825 | 1.832 |
| Ir–C(phpy) | 2.065(2) | 2.0505(17) | 2.053(3) |
| Ir–N*a | 2.073(2) | 2.0811(16) | 2.093(3) |
| Ir–N#b | 2.106(2) | 2.0994(15) | 2.118(2) |
| C–Ir–N* | 78.34(9) | 78.40(6) | 78.15(12) |
| C–Ir–N# | 85.94(8) | 84.66(6) | 86.67(10) |
| N*–Ir– N# | 88.27(9) | 87.01(6) | 80.87(9) |
N* is the nitrogen atom in the 2-phenylpyridine chelating ligand.
N# is the nitrogen atom in the monodentate ligand.
The crystal structures reported here are the second examples of crystal structures containing the Cpxph ligand.6b The phenyl ring of Cpxph is twisted by about 45° relative to the cyclopentadienyl ring in 2 and 5 and 83° in 8. The Ir–cyclopentadienyl (centroid) bond distances for compounds 2, 5, and 8 ranged from 1.825 to 1.832 Å, longer than that of [(η5-Cpxph)Ir(bpy)Cl]PF6 (bpy = 2,2′-bipyridine)6b (1.789 Å), probably due to the negatively charged phpy ligand. The change in monodentate ligands in 2, 5, and 8 does not give rise to much difference in bond lengths between Ir and coordinated atoms; however, a smaller N–Ir–N angle of 80.87(9)° for 8 is observed compared to 88.27(9)° and 87.01(6)° for 2 and 5, respectively. There is weak π–π intermolecular ring stacking between the neighboring phenylpyridine rings in the unit cell of compound 2, Figure S1. The two interacting π systems are parallel, with a centroid–centroid distance of 4.291 Å.
Hydrolysis
The hydrolysis of complexes 1–8 (150 μM) in 10% MeOD-d4/90% D2O (v/v) was studied by 1H NMR spectroscopy at 310 K. The presence of methanol ensured sufficient solubility of the complex. The 1H NMR spectra showed no obvious change over 24 h, indicating that these Ir compounds remained stable under these conditions.
Antiproliferative Activity
The activity of complexes 1–8 toward A2780 human ovarian, A549 lung, and MCF-7 breast cancer cells was investigated, Table S2 and Figure 2. The IC50 values (concentration at which 50% of the cell growth is inhibited) for all IrIII complexes are comparable to or lower than that of cisplatin, suggesting that all these compounds are highly active. Complex 5, [(η5-Cpxph)Ir(phpy)(4-NMe2-py)]+, containing 4-dimethylaminopyridine, displayed the highest anticancer activity, with an IC50 value of 0.20 ± 0.04 μM toward MCF-7 cells, ca. 36 times more potent than cisplatin. Complex 8, containing N,N-diethylnicotinamide, showed the lowest anticancer activity toward all three cancer cell lines.
Figure 2.
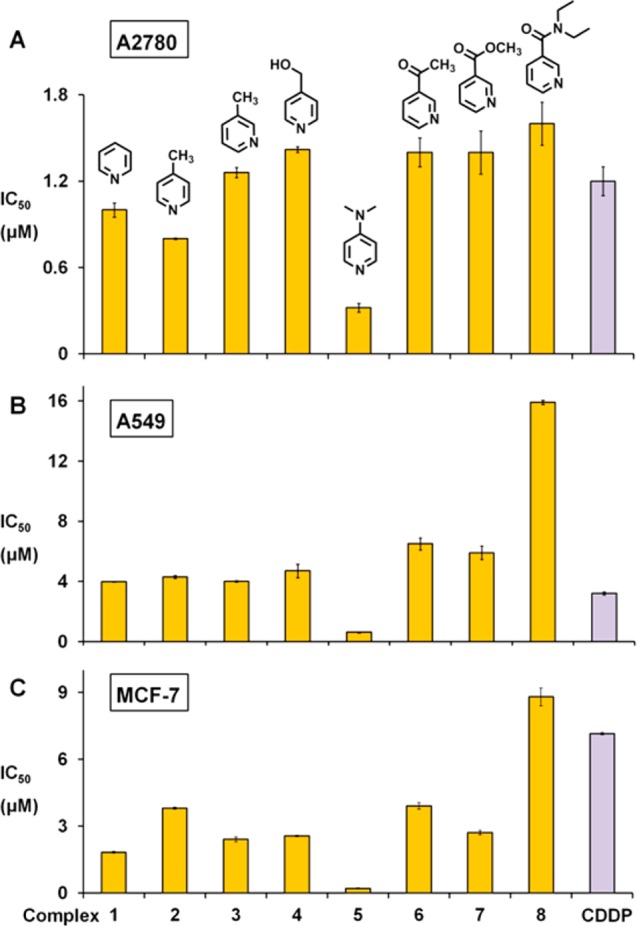
Inhibition of growth of (A) A2780 human ovarian cancer; (B) A549 lung cancer; and (C) MCF-7 breast cancer cells by compounds 1–8 and comparison with cisplatin (CDDP).
With regard to the effects of substitutions on the pyridine ring on anticancer activity, overall, complexes containing electron-withdrawing groups on the pyridine ring show less activity compared to those complexes with an electron-donating group.
The antiproliferative activity of compounds 2 and 5 was further evaluated in the National Cancer Institute NCI-60 human cancer cell screen, consisting of nine tumor subtypes.12 Three end points were determined: GI50 (the concentration that causes 50% cell growth inhibition), TGI (the concentration that causes 100% cell growth inhibition), and LC50 (the concentration that decreases the original cell number by 50%). The GI50 mean graph for 2 and 5 is shown in Figure 3. The midpoint (log10 GI50) of 2 and 5 is −6.14 (GI50 = 724 nM) and −6.46 (GI50 = 347 nM), respectively. Bars extending to the left in the mean graph represent higher activity than the mean of all tested cell lines. Bars extending to the right correspondingly imply activity less than the mean. Complex 5 shows high potency in a wide range of cancer cell lines (Figure 3), with particular selectivity toward MDA-MB-468 (breast), A498 (renal), and COLO-205 (colon), with GI50 values of <170 nM. Notably, complex 2 displayed potency toward the A498 (renal) cell line with a GI50 of 19 nM. Complex 5 showed good selectivity toward leukemia, CNS cancer, colon cancer, and breast cancer. In comparison with cisplatin (CDDP), Ir complexes displayed higher activity against NCI-60 cancer cell lines, especially Ir complex 5, which is 4–5 times more active than cisplatin, Figure 4.
Figure 3.

NCI-60 GI50 mean graphs for complexes 2 (right) and 5 (left). The midpoint (log10 GI50) is −6.14 (2) and −6.46 (5). Bars to the right of the mean indicate lower activity relative to the mean, and those to the left, higher activity.
Figure 4.

GI50, TGI, and LC50 values (μM) of 2, 5, and CDDP in the NCI-60 screen.
Interactions with Nucleobases
Reactions of complexes 1–8 with nucleobase derivatives 9-ethylguanine (9-EtG) and 9-methyladenine (9-MeA) were investigated. Solutions of 1–8 (ca. 1 mM) and 1 molar equiv of 9-EtG or 9-MeA in 20% MeOD-d4/80% D2O (v/v) were prepared, and 1H NMR spectra were recorded at different time intervals at 310 K.
No reaction with 9-MeA was observed for all complexes, as addition of nucleobase model to a solution of 1–8 resulted in no additional 1H NMR peaks over 24 h. In contrast, all the complexes reacted with 9-EtG. For example, in the 1H NMR spectrum of a solution containing 8 and 1 molar equiv of 9-EtG, one set of new peaks assignable to the 9-EtG adduct 8G appeared, showing that 32% of 8 had reacted after 24 h (Figure 5). A significant change in chemical shift of the CH=N (phpy ligand) proton of complex 8 from 8.88 to 9.29 ppm for 8G was observed. A new 9-EtG H8 peak appeared at 7.43 ppm (singlet), shifted by 0.39 ppm to high field relative to that of free 9-EtG. The ESI-MS of an equilibrium solution contained a major peak at m/z 723.2, confirming the formation of the 9-EtG adduct 8G, [(η5-C5Me4C6H5)Ir(phpy)(9-EtG)]+ (calcd m/z 722.9). The percentages of nucleobase adducts formed by all the complexes after 24 h reaction, based on 1H NMR peak integrals, are shown in Table S3 and Figure 6.
Figure 5.

Low-field region of the 1H NMR spectra for reaction of [(η5-Cpxph)Ir(phpy)(3-CONEt2-py)]PF6 (8) with 9-EtG: (A) 10 min after addition of 1 mol equiv of 9-EtG to an equilibrium solution of complex 8 (1.0 mM) in 20% MeOD-d4/80% D2O (v/v) at 310 K; (B) after 24 h reaction. Peak assignments: (red squares) 8; (blue squares) guanine adduct 8G. After 24 h, 32% of 8 had reacted.
Figure 6.

Bar chart showing the extent of binding of complexes 1–8 (ca. 1 mM in 20% MeOD-d4/80% D2O) to the nucleobase 9-EtG over 24 h at 310 K.
Cellular Ir Accumulation
Complex 5, which displayed the highest anticancer activity, and complex 1, containing a nonsubstituted py ligand, were selected for further studies. First we investigated the cellular accumulation of Ir from complexes 1 and 5 in A2780 ovarian cancer cells. After 24 h of drug exposure at equipotent concentrations corresponding to IC50 values, 3.5 times more Ir, as determined by ICP-MS, from the pyridine complex 1 was accumulated in the cells compared to the py-NMe2 analogue 5 (7.8 ± 0.5 ng of Ir vs 2.2 ± 0.3 ng of Ir per 106 cells).
Apoptosis Assay
In order to investigate whether the reduction in cell viability observed by the SRB assay is based on apoptosis, A2780 cells were treated with complexes 1 and 5 at equipotent concentrations of IC50 for 24 h, then stained with annexin V/propidium iodide and analyzed by flow cytometry. This allowed determination of cell populations as viable (unstained, only self-fluorescence), early apoptosis (stained by annexin V only, green fluorescence), late apoptosis (stained by annexin V and PI, green and red fluorescence), and nonviable (stained by PI only, red fluorescence). Dot plots (Figure 7 and Table S4) showed that around 95% of A2780 cells remained in the viable phase after 24 h of exposure to 1 and 5, indicating no obvious induction of apoptosis at equipotent concentrations of IC50.
Figure 7.
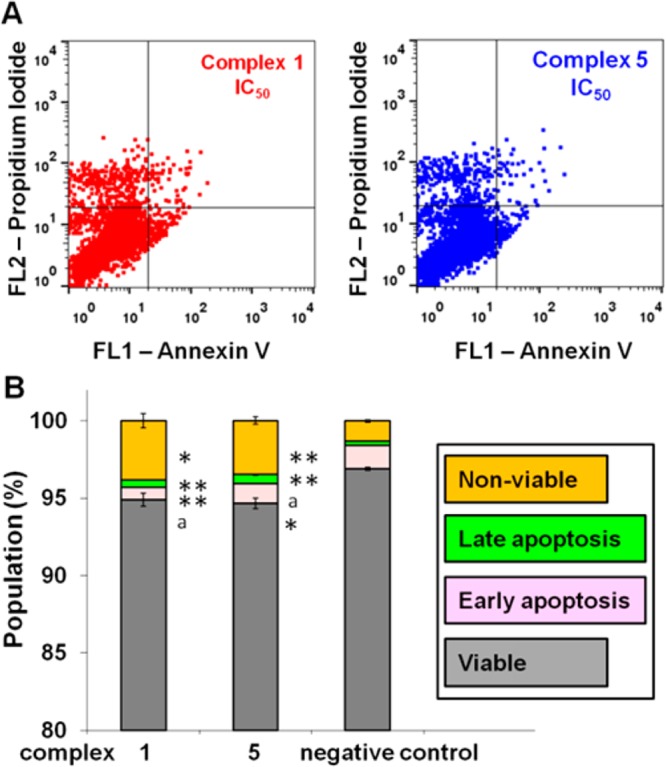
Apoptosis analysis of A2780 human ovarian cells after 24 h of exposure to complexes 1 and 5 at 310 K determined by flow cytometry using annexin V-FITC vs PI staining. (A) FL1 vs FL2 histogram for cells exposed to complexes 1 and 5 at equipotent concentrations of IC50. (B) Populations for cells treated by 1 and 5. p-Values were calculated after a t test against the negative control data, *p < 0.05, **p < 0.01, ap > 0.05.
Cell Cycle Studies
Next we performed cell cycle arrest analysis for complexes 1 and 5 toward A2780 cells by flow cytometry to determine whether the induced cell growth inhibition was the result of cell cycle arrest. In comparison to the control population, the cell cycle data (Figure 8 and Table S5) clearly show no significant population change, indicating that Ir compounds 1 and 5 did not cause cell cycle arrest at equipotent concentrations of IC50.
Figure 8.
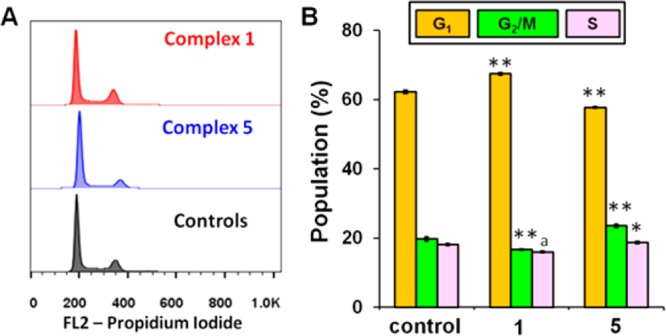
Cell cycle analysis of A2780 human ovarian cancer cells after 24 h of exposure to complexes 1 and 5 at 310 K. Concentrations used were equipotent at IC50. Cell staining for flow cytometry was carried out using PI/RNase. (A) FL2 histogram for negative control (cells untreated) and complexes 1 and 5. (B) Cell populations in each cell cycle phase for control and complexes 1 and 5. p-Values were calculated after a t test against the negative control data, *p < 0.05, **p < 0.01, ap > 0.05.
Induction of ROS in A2780 Cancer Cells
We determined the level of reactive oxygen species (ROS) in A2780 human ovarian cancer cells induced by complexes 1 and 5 at concentrations of IC50 and 2 × IC50 by flow cytometry fluorescence analysis (Figure 9 and Table S6). This allowed the determination of the total level of oxidative stress (combined levels of H2O2, peroxy and hydroxyl radicals, peroxynitrite, and NO), while also monitoring superoxide production. After only 1 h of drug exposure, we observed a dramatic increase in both total ROS levels and superoxide levels in cells treated with complexes 1 and 5 compared to untreated cells. ROS were detected in more than 97% of A2780 cells. A concentration dependence of ROS induction was observed for both Ir complexes: the population of cells that shows high fluorescence in both FL-1 and FL-2 channels (indicating high total oxidative stress as well as high superoxide levels) increased from 48 ± 2% at IC50 to 64 ± 3 at 2 × IC50 for complex 1 and increased from 40 ± 3% at IC50 to 47 ± 2 at 2 × IC50 for complex 5 (Figure 9 and Table S6).
Figure 9.
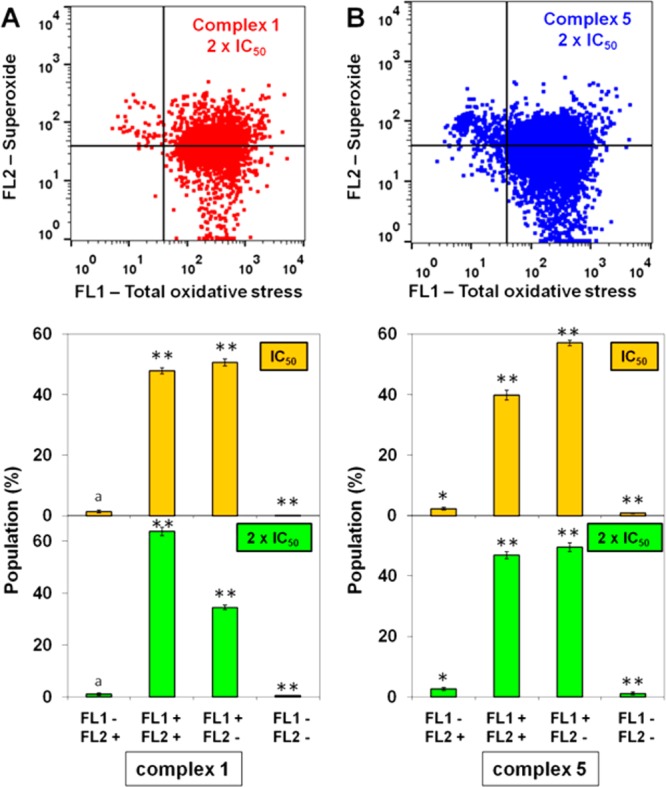
ROS induction in A2780 cancer cells treated with complexes 1 and 5. FL1 channel detects total oxidative stress, and FL2 channel detects superoxide production. (A) Comparison between the four different populations caused by IC50 and 2 × IC50 of 1. (B) Comparison between the four different populations caused by IC50 and 2 × IC50 of 5. p-Values were calculated after a t test against the negative control data, *p < 0.05, **p < 0.01, ap > 0.05.
Polarization of the Membrane Potential
Analysis of the changes of mitochondrial membrane potential (ΔΨm) in A2780 cells after exposure to complexes 1 and 5 was carried out by observing the fluorescence of JC-10, a cationic lipophilic dye, using flow cytometry. JC-10 aggregates inside mitochondria and emits red fluorescence; however, upon membrane polarization, JC-10 is disaggregated, reducing the red emission. The level of membrane polarization after cells were exposed to complexes 1 and 5 at concentrations of IC50 and 2 × IC50 for 24 h is shown in Figure 10 and Table S7. Both Ir complexes have significant effects on cell membrane polarization; around 70% of cells lost ΔΨm. The impairment induced by 1 and 5 (which is reflected in ΔΨm) is clearly concentration-dependent (Figure 10).
Figure 10.

Changes in mitochondrial membrane potential of A2780 human ovarian cancer cells induced by complexes 1 and 5. (A) Flow cytometry histograms of the changes induced by the complexes at concentrations of IC50 and 2 × IC50. (B) Populations of cells that exhibit a reduction in the FL2 fluorescence, indicative of changes in the mitochondrial membrane potential. p-Values were calculated after a t test against the negative control data, **p < 0.01.
Discussion
IrIII complexes are often considered to be relatively inert, a common characteristic of low-spin d6 metal ions and especially third-row transition metals.13 Compared to platinum- or ruthenium-based anticancer agents, iridium anticancer complexes are still in their infancy.
With regard to half-sandwich IrIII complexes [(Cpx)Ir(L∧L′)Z]0/n+, we found that both the cyclopentadienyl Cpxph or Cpxbiph ligand and chelating ligand C∧N– can dramatically influence anticancer activity.6b−6d In addition, we have shown that complexes containing pyridine as the monodentate ligand exhibit 6 times higher anticancer activity compared to the chlorido analogue.6a Therefore, we have investigated a series of IrIII complexes of type [(Cpxph)Ir(phpy)Z]+ containing phenyl-substituted Cp*, C∧N-bound 2-phenylpyridine, and pyridine or its derivatives (Scheme 1) in this work. Novel compounds 1–8 have been synthesized and are reported for the first time.
Encouragingly, all eight compounds exhibit high potency against human ovarian A2780 cancer, A549 lung cancer, and MCF-7 breast cancer cells, at least comparable with cisplatin, Figure 2. In general an electron-donating substituent on the py ring confers higher activity in comparison with electron-withdrawing groups. This may arise from strengthening the Ir–N(py) bond, thus reducing side reactions on the way to target sites. In addition, lipophilicity might as well influence the potency of these complexes.14 Complex 5, [(η5-Cpxph)Ir(phpy)(4-NMe2-py)]+, showed the highest anticancer activity, ca. 3–9 times more active than unmodified py complex 1. In addition, complex 5 shows submicromolar activity toward a wide range of cancer cell lines in the NCI-60 screen, with selectivity for leukemia, CNS cancer, colon cancer, and breast cancer cell lines, being 4–5 times more potent than CDDP (Figures 3 and 4). Cpxph py complex 1 displayed ca. 8 times less anticancer potency than the Cpxbiph analogue, which is consistent with the general finding we reported previously that the anticancer efficiency increases with phenyl substitution on the Cp* ring.6a,6b,6d
Hydrolysis often presents an activation step for transition metal anticancer complexes.15 However, no significant hydrolysis was observed for complexes 1–8 in aqueous solution. DNA is usually a potential target for transition metal anticancer drugs.16 Although 1–8 are inert to hydrolysis, they can react with nucleobase 9-EtG to various extents from 11% to 50% (Figure 6), depending on the electronic effect of the substituent on the py ring. Electron-withdrawing groups (such as acetyl and ester groups) facilitate ligand substitution of the py derivative by 9-EtG, whereas electron donor groups (such as methyl and dimethylamino groups) hamper formation of the Ir-EtG adduct. No reaction of 1–8 with 9-MeA was observed, consistent with our previous studies that guanine binds stronger to IrIII than adenine.6a,6b,6d The extent of nucleobase binding does not correlate with antiproliferative activity. Compared to complexes 1–5, complexes 6–8 bind to 9-EtG to a greater extent; however, they show lower activity toward cancer cells. Therefore, although DNA is a potential target for these iridium compounds, DNA binding may not be the major mechanism of action.
Apoptosis is a process of cell death in a programmed fashion. A large number of transition metal-based anticancer agents have been reported to inhibit cell growth by activation of apoptosis.17 Induction of apoptosis is usually dependent on the concentration of administered compounds17f,18 and on exposure time.19 No apoptosis was observed when A2780 cells were exposed to complexes 1 and 5 at their IC50 concentrations for 24 h. Also IC50 concentrations of complexes 1 and 5 did not cause significant cell cycle arrest after 24 h of drug exposure. Lack of accumulation of cells in the sub-G1 phase in cell cycle experiments is consistent with the absence of apoptotic cell death.20
Reactive oxygen species play important roles in regulating cell proliferation, death, and signaling. They can also play significant roles in the mechanism of action of anticancer agents.21 In fact, dinuclear Cp*Ir(III) complexes containing bridging dipyridyl ligands have been reported to generate ROS and induce apoptosis in Jurkat leukemia cells.17f We suggested previously that the antiproliferative mechanism for the iridium pyridine complex in this series is related to ROS generation.6a Consequently, we also determined the ROS levels in A2780 ovarian cancer cells induced by 1 and 5. Both complexes increased ROS levels significantly even at IC50 concentration after 1 h drug exposure (Figure 9), which led to the majority of cancer cells (>97%) being affected by generation of ROS. These increases in ROS levels may provide a basis for killing cancer cells.
Mitochondria are involved in a number of important tasks in living cells, such as energy production and generation of ROS. Mitochondrial dysfunction can participate in the induction of cell death and was assessed by measuring changes in the mitochondrial membrane potential. Intriguingly, both complexes 1 and 5 (IC50 concentration) induced significant changes in mitochondrial membrane potential (Figure 10); more than 70% of A2780 cells lose ΔΨm after exposure to Ir compounds for 24 h. This may contribute to the anticancer activities of these Ir compounds.
Conclusions
In this work, we have prepared eight new organometallic IrIII cyclopentadienyl complexes [(η5-Cpxph)Ir(phpy)Z]PF6 to explore the effect of a monodentate pyridine-based ligand on their chemical and anticancer activity. The X-ray crystal structures of [(η5-Cpxph)Ir(phpy)(4-Me-py)]PF6 (2), [(η5-Cpxph)Ir(phpy)(4-NMe2-py)]PF6 (5), and [(η5-Cpxph)Ir(phpy)(3-CONEt2-py)]PF6 (8) were determined.
All the complexes display high potency toward A2780, A549, and MCF-7 human cancer cells, comparable to, and for some complexes even higher than, the clinical anticancer drug cisplatin. The anticancer activity can be fine-tuned by varying the pyridine-based ligand; the presence of an electron-donating group confers higher anticancer activity. The most active complex, 5, contains a 4-dimethylamine substituent on pyridine. The results of the NCI 60 cancer cell line screening show that complex 5 is 4–5 times more potent than cisplatin and exhibits submicromolar activity in a wide range of cancer cell lines, especially against leukemia, CNS cancer, colon cancer, and breast cancer. Nanomolar activity (GI50 19 nM) was obtained for complex 2 toward the renal A498 cancer cell line.
No distinct hydrolysis was observed for this type of complex in aqueous solution; however, all complexes display weak nucleobase binding to 9-ethylguanine, suggesting that DNA could be a possible target, although other targets appear to be more important. Additionally, no obvious apoptosis and cell cycle arrest were induced when A2780 cancer cells were treated with IC50 concentrations of complexes 1 and 5. However, the iridium complexes 1 and 5 induce a dramatic increase in the level of ROS in ovarian cancer cells within 1 h and caused mitochondrial dysfunction by loss of the mitochondrial membrane potential. Our work suggests that this type of iridium complex could be a promising candidate for further evaluation as chemotherapeutic agents for human cancers.
Acknowledgments
We thank the ERC (Grant No. 247450), Science City (AWM/ERDF), and the EPSRC for support, the National Cancer Institute for 60 human tumor cell screening, and members of EU COST Actions D39 and CM1105 for stimulating discussions. We also thank Dr. Magdalena Mos and Miss Bushra Qamar for assistance with cell culture.
Supporting Information Available
This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ Department of Chemistry, University of Basel, Spitalstrasse 51, 4056 Basel, Switzerland.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Medicinal Organometallic Chemistry (Topics in Organometallic Chemistry), 1st ed.; Jaouen G.; Metzler-Nolte N., Eds.; Springer-Verlag: Heidelberg, Germany, 2010; Vol. 32. [Google Scholar]; b Kelland L. Nat. Rev. Cancer 2007, 7, 573–584. [DOI] [PubMed] [Google Scholar]; c Gasser G.; Ott I.; Metzler-Nolte N. J. Med. Chem. 2011, 54, 3–25. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Barry N. P. E.; Sadler P. J. Chem. Commun. 2013, 49, 5106–5131. [DOI] [PubMed] [Google Scholar]
- Wheate N. J.; Walker S.; Craig G. E.; Oun R. Dalton Trans. 2010, 39, 8113–8127. [DOI] [PubMed] [Google Scholar]
- a Top S.; Vessières A.; Leclercq G.; Quivy J.; Tang J.; Vaissermann J.; Huché M.; Jaouen G. Chem.—Eur. J. 2003, 9, 5223–5236. [DOI] [PubMed] [Google Scholar]; b Hamels D.; Dansette P. M.; Hillard E. A.; Top S.; Vessières A.; Herson P.; Jaouen G.; Mansuy D. Angew. Chem., Int. Ed. 2009, 48, 9124–9126. [DOI] [PubMed] [Google Scholar]
- a Alessio E.; Mestroni G.; Bergamo A.; Sava G. Curr. Top. Med. Chem. 2004, 4, 1525–1535. [DOI] [PubMed] [Google Scholar]; b Hartinger C. G.; Zorbas-Seifried S.; Jakupec M. A.; Kynast B.; Zorbas H.; Keppler B. K. J. Inorg. Biochem. 2006, 100, 891–904. [DOI] [PubMed] [Google Scholar]; c Süss-Fink G. Dalton Trans. 2010, 39, 1673–1688. [DOI] [PubMed] [Google Scholar]; d Bruijnincx P. C. A.; Sadler P. J. In Advances in Inorganic Chemistry; Rudi van E., Hubbard C. D., Eds.; Academic Press: New York, 2009; Vol. 61, pp 1–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Liu Z.; Sadler P. J. Acc. Chem. Res. 2014, 47, 1174–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Geldmacher Y.; Oleszak M.; Sheldrick W. S. Inorg. Chim. Acta 2012, 393, 84–102. [Google Scholar]; c Lo K. K.-W.; Zhang K. Y. RSC Adv. 2012, 2, 12069–12083. [Google Scholar]; d Leung C.-H.; Zhong H.-J.; Chan D. S.-H.; Ma D.-L. Coord. Chem. Rev. 2013, 257, 1764–1776. [Google Scholar]; e Sasmal P. K.; Streu C. N.; Meggers E. Chem. Commun. 2013, 49, 1581–1587. [DOI] [PubMed] [Google Scholar]; f Cutillas N.; Yellol G. S.; de Haro C.; Vicente C.; Rodríguez V.; Ruiz J. Coord. Chem. Rev. 2013, 257, 2784–2797. [Google Scholar]
- a Liu Z.; Romero-Canelón I.; Qamar B.; Hearn J. M.; Habtemariam A.; Barry N. P. E.; Pizarro A. M.; Clarkson G. J.; Sadler P. J. Angew. Chem., Int. Ed. 2014, 53, 3941–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liu Z.; Habtemariam A.; Pizarro A. M.; Fletcher S. A.; Kisova A.; Vrana O.; Salassa L.; Bruijnincx P. C. A.; Clarkson G. J.; Brabec V.; Sadler P. J. J. Med. Chem. 2011, 54, 3011–3026. [DOI] [PubMed] [Google Scholar]; c Liu Z.; Salassa L.; Habtemariam A.; Pizarro A. M.; Clarkson G. J.; Sadler P. J. Inorg. Chem. 2011, 50, 5777–5783. [DOI] [PubMed] [Google Scholar]; d Liu Z.; Habtemariam A.; Pizarro A. M.; Clarkson G. J.; Sadler P. J. Organometallics 2011, 30, 4702–4710. [Google Scholar]; e Wirth S.; Rohbogner C.; Cieslak M.; Kazmierczak-Baranska J.; Donevski S.; Nawrot B.; Lorenz I.-P. J. Biol. Inorg. Chem. 2010, 15, 429–440. [DOI] [PubMed] [Google Scholar]; f Casini A.; Edafe F.; Erlandsson M.; Gonsalvi L.; Ciancetta A.; Re N.; Ienco A.; Messori L.; Peruzzini M.; Dyson P. J. Dalton Trans. 2010, 39, 5556–5563. [DOI] [PubMed] [Google Scholar]; g Gras M.; Therrien B.; Süss-Fink G.; Casini A.; Edafe F.; Dyson P. J. J. Organomet. Chem. 2010, 695, 1119–1125. [Google Scholar]; h Ruiz J.; Rodriguez V.; Cutillas N.; Samper K. G.; Capdevila M.; Palacios O.; Espinosa A. Dalton Trans. 2012, 41, 12847–12856. [DOI] [PubMed] [Google Scholar]; i Lucas S. J.; Lord R. M.; Wilson R. L.; Phillips R. M.; Sridharan V.; McGowan P. C. Dalton Trans. 2012, 41, 13800–13802. [DOI] [PubMed] [Google Scholar]; j Payne R.; Govender P.; Therrien B.; Clavel C. M.; Dyson P. J.; Smith G. S. J. Organomet. Chem. 2013, 729, 20–27. [Google Scholar]
- a Schäfer S.; Sheldrick W. S. J. Organomet. Chem. 2007, 692, 1300–1309. [Google Scholar]; b Ali Nazif M.; Bangert J.-A.; Ott I.; Gust R.; Stoll R.; Sheldrick W. S. J. Inorg. Biochem. 2009, 103, 1405–1414. [DOI] [PubMed] [Google Scholar]
- Wang F.; Habtemariam A.; van der Geer E. P. L.; Fernández R.; Melchart M.; Deeth R. J.; Aird R.; Guichard S.; Fabbiani F. P. A.; Lozano-Casal P.; Oswald I. D. H.; Jodrell D. I.; Parsons S.; Sadler P. J. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 18269–18274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. J. Appl. Crystallogr. 2009, 42, 339–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. Acta Crystallogr. 2008, A64, 112–122. [DOI] [PubMed] [Google Scholar]
- Vichai V.; Kirtikara K. Nat. Protoc. 2006, 1, 1112–1116. [DOI] [PubMed] [Google Scholar]
- a Paull K. D.; Shoemaker R. H.; Hodes L.; Monks A.; Scudiero D. A.; Rubinstein L.; Plowman J.; Boyd M. R. J. Natl. Cancer Inst. 1989, 81, 1088–1092. [DOI] [PubMed] [Google Scholar]; b Holbeck S. L.; Collins J. M.; Doroshow J. H. Mol. Cancer Ther. 2010, 9, 1451–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Shoemaker R. H. Nat. Rev. Cancer 2006, 6, 813–823. [DOI] [PubMed] [Google Scholar]
- a Richens D. T. Chem. Rev. 2005, 105, 1961–2002. [DOI] [PubMed] [Google Scholar]; b Messori L.; Marcon G.; Orioli P.; Fontani M.; Zanello P.; Bergamo A.; Sava G.; Mura P. J. Inorg. Biochem. 2003, 95, 37–46. [DOI] [PubMed] [Google Scholar]
- Lin J. H.; Lu A. Y. H. Pharmacol. Rev. 1997, 49, 403–449. [PubMed] [Google Scholar]
- Pizarro A. M.; Habtemariam A.; Sadler P. J. In Medicinal Organometallic Chemistry (Topics in Organometallic Chemistry), 1st ed.; Jaouen G., Metzler-Nolte N., Eds.; Springer-Verlag: Heidelberg, Germany, 2010; Vol. 32, pp 21–56. [Google Scholar]
- a Zhang C. X.; Lippard S. J. Curr. Opin. Chem. Biol. 2003, 7, 481–489. [DOI] [PubMed] [Google Scholar]; b Deubel D. V.; Lau J. K.-C. Chem. Commun. 2006, 2451–2453. [DOI] [PubMed] [Google Scholar]
- a Jung Y.; Lippard S. J. Chem. Rev. 2007, 107, 1387–1407. [DOI] [PubMed] [Google Scholar]; b Qian C.; Wang J.-Q.; Song C.-L.; Wang L.-L.; Ji L.-N.; Chao H. Metallomics 2013, 5, 844–854. [DOI] [PubMed] [Google Scholar]; c Romero-Canelón I.; Salassa L.; Sadler P. J. J. Med. Chem. 2013, 56, 1291–1300. [DOI] [PubMed] [Google Scholar]; d Hearn J. M.; Romero-Canelón I.; Qamar B.; Liu Z.; Hands-Portman I.; Sadler P. J. ACS Chem. Biol. 2013, 8, 1335–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Srdić-Rajić T.; Zec M.; Todorović T.; Anđelković K.; Radulović S. Eur. J. Med. Chem. 2011, 46, 3734–3747. [DOI] [PubMed] [Google Scholar]; f Nazif M. A.; Rubbiani R.; Alborzinia H.; Kitanovic I.; Wolfl S.; Ott I.; Sheldrick W. S. Dalton Trans. 2012, 41, 5587–5598. [DOI] [PubMed] [Google Scholar]
- a Rubbiani R.; Can S.; Kitanovic I.; Alborzinia H.; Stefanopoulou M.; Kokoschka M.; Mönchgesang S.; Sheldrick W. S.; Wölfl S.; Ott I. J. Med. Chem. 2011, 54, 8646–8657. [DOI] [PubMed] [Google Scholar]; b Kowol C.; Heffeter P.; Miklos W.; Gille L.; Trondl R.; Cappellacci L.; Berger W.; Keppler B. J. Biol. Inorg. Chem. 2012, 17, 409–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierroz V.; Joshi T.; Leonidova A.; Mari C.; Schur J.; Ott I.; Spiccia L.; Ferrari S.; Gasser G. J. Am. Chem. Soc. 2012, 134, 20376–20387. [DOI] [PubMed] [Google Scholar]
- Chen T.; Liu Y.; Zheng W.-J.; Liu J.; Wong Y.-S. Inorg. Chem. 2010, 49, 6366–6368. [DOI] [PubMed] [Google Scholar]
- a Trachootham D.; Alexandre J.; Huang P. Nat. Rev. Drug Discovery 2009, 8, 579–591. [DOI] [PubMed] [Google Scholar]; b Watson J. Open Biol. 2013, 3, 120144. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.