

Abstract
Proteolysis within the lipid bilayer is poorly understood, in particular the regulation of substrate cleavage. Rhomboids are a family of ubiquitous intramembrane serine proteases that harbour a buried active site and are known to cleave transmembrane substrates with broad specificity. In vitro gel and Förster resonance energy transfer (FRET)-based kinetic assays were developed to analyse cleavage of the transmembrane substrate psTatA (TatA from Providencia stuartii). We demonstrate significant differences in catalytic efficiency (kcat/K0.5) values for transmembrane substrate psTatA (TatA from Providencia stuartii) cleavage for three rhomboids: AarA from P. stuartii, ecGlpG from Escherichia coli and hiGlpG from Haemophilus influenzae demonstrating that rhomboids specifically recognize this substrate. Furthermore, binding of psTatA occurs with positive cooperativity. Competitive binding studies reveal an exosite-mediated mode of substrate binding, indicating allostery plays a role in substrate catalysis. We reveal that exosite formation is dependent on the oligomeric state of rhomboids, and when dimers are dissociated, allosteric substrate activation is not observed. We present a novel mechanism for specific substrate cleavage involving several dynamic processes including positive cooperativity and homotropic allostery for this interesting class of intramembrane proteases.
Keywords: allostery, GlpG, intramembrane protease, kinetics, rhomboid protease
See also: K Strisovsky & M Freeman (September 2014)
Introduction
Proteases that catalyse the hydrolysis of peptide bonds constitute the most abundant group of enzymes. They are essential in a wide variety of biological processes, including cell cycle signalling, proliferation and death, immune response and protein trafficking. Several successful strategies have been applied towards the development of currently in use protease inhibitors to treat hypertension, HIV, cancer, diabetes and coagulation disorders (Drag & Salvesen, 2010). Characterization of the role of proteases in disease and also their precise mechanism of action was essential for these drug discoveries.
Intramembrane proteases are polytopic membrane proteins that cleave transmembrane substrates and, similar to soluble proteases, are known to play roles in essential biological processes (Wolfe, 2009). Their active sites, buried within the lipid bilayer, are formed from the assembly of transmembrane helices. X-ray crystal structures for several intramembrane proteases including presenilin homologs (Hu et al, 2011; Li et al, 2013), CaaX proteases (Manolaridis et al, 2013; Pryor et al, 2013), site-2-proteases (Feng et al, 2007) and several structures of rhomboid proteases (Wang et al, 2006; Wu et al, 2006; Ben-Shem et al, 2007; Lemieux et al, 2007) have provided architectural insight into this unique class of enzymes. Despite these crystal structures, our comprehension of their cleavage mechanism, substrate recognition and regulation of these lipid-embedded enzymes is in its infancy compared to soluble proteases.
Rhomboids are a well-studied family of intramembrane serine proteases (peptidases) found in all kingdoms of life. The cleavage of transmembrane substrates facilitates the release of soluble domains to the extracellular environment. They play essential roles in diverse signalling events ranging from epidermal growth factor release in eukaryotes (Lee et al, 2001; Urban et al, 2001), facilitating invasion in parasites (Sibley, 2013), to quorum sensing in bacteria (Mesak et al, 2004; Stevenson et al, 2007). Not surprisingly, rhomboid dysfunction is involved in several human diseases that include malaria, cancer and Parkinson's disease (Bergbold & Lemberg, 2013; Chan & McQuibban, 2013; Sibley, 2013).
Rhomboids represent ‘non-classical’ serine proteases, having a catalytic dyad (Ser/His) instead of the hallmark triad, and the active site buried 10–12 Å deep from the periplasm. The hydrophilic environment of the active site is occluded from lipid bilayer and the periplasm by flexible loop 5 and transmembrane helix 5, respectively (Wang et al, 2006; Wu et al, 2006; Ben-Shem et al, 2007; Lemieux et al, 2007; Brooks et al, 2011). Both regions are proposed to act as a gate to regulate substrate entry; however, the extent of their mobility is unclear (Baker et al, 2007; Wang & Ha, 2007; Brooks et al, 2011; Moin & Urban, 2012; Xue & Ha, 2013). Despite low sequence identity between the rhomboids' active sites, they can cleave substrates from various kingdoms of life and therefore are considered as enzymes with broad substrate recognition properties (Urban et al, 2002; Lemberg et al, 2005; Strisovsky et al, 2009).
For rhomboid substrates, a cleavage recognition motif has been identified for evolutionary distinct species (Akiyama & Maegawa, 2007; Strisovsky et al, 2009). Mutagenesis studies of the P. stuartii rhomboid substrateTatA (psTatA), have revealed that a separate stretch of amino acids in the transmembrane region was also required for proteolysis along with the cleavage motif (Strisovsky et al, 2009). This suggests that the active site of rhomboids is not the only molecular determinant regulating the substrate selection. The regulation of substrate binding and enzymatic activity for many serine proteases is achieved by allosteric interactions. For example, with thrombin and factor Xa, an allosteric site regulates substrate gating and access to an occluded active site (Johnson et al, 2006; Di Cera et al, 2007). Oligomerization represents another regulatory mechanism for enzymes. Prokaryotic rhomboids are known to form dimers, both in their membrane domain and in the soluble cytoplasmic domain independently (Sampathkumar et al, 2012; Lazareno-Saez et al, 2013), yet it is unclear if dimerization affects function.
Catalytic parameters are crucial for understanding how substrates are cleaved and how this cleavage process is regulated. Until recently, the turnover rates for rhomboid proteases and most intramembrane proteases (presenilin, site-2-protease, signal peptidases) had not been fully assessed using steady-state methods. Presenilin has been the target of many in vitro kinetic studies both in lipid and detergent environments, as well as allosteric modulation by inhibitors (Edbauer et al, 2003; Fraering et al, 2004, 2005; Kakuda et al, 2006; Chavez-Gutierrez et al, 2008, 2012; Shelton et al, 2009). For this multi-protein complex, lipids and detergents had a drastic effect on activity (Osenkowski et al, 2008). In contrast, activity assays conducted with CaaX metalloproteases Ras-converting enzyme (Rce1p) and the yeast mating a-factor processing enzyme Ste24p, conducted in both lipid (Hollander et al, 2000; Manandhar et al, 2010) and detergent environments (Pryor et al, 2013), revealed similar catalytic parameters. A more recent paper has characterized rhomboid cleavage of substrate using reconstituted proteoliposomes (Dickey et al, 2013), demonstrating slow rate of cleavage and low specificity for substrate recognition by the active site of several prokaryotic rhomboids.
Here, we show that prokaryotic rhomboids indeed have substrate specificity using steady-state kinetic analysis of the psTatA transmembrane substrate cleavage in detergent solution assessed by both gel and real-time Förster resonance energy transfer (FRET)-based assays. Furthermore, studied rhomboids bound psTatA with positive cooperativity. Competitive binding studies with AarA and its physiological substrate psTatA show that the transmembrane substrate cleavage requires an exosite. The dimeric form of rhomboids is essential for the cleavage of the transmembrane substrate TatA, whereas the monomers are active only with a soluble substrate. A model is presented for homotropic allostery where the substrate binds to both the exosite and active site to provide a means for the regulation of specificity for this class of promiscuous intramembrane proteases.
Results
Rhomboid-mediated cleavage of transmembrane substrate psTatA is specific and cooperative
The kinetics of rhomboid-mediated cleavage of psTatA was assessed using gel-based cleavage assay for hiGlpG, ecGlpG and AarA (Fig1 and Table1). These three classes of rhomboids represent the diverse topological arrangements found in rhomboid family (Lemberg & Freeman, 2007). Representing bacterial forms, hiGlpG has the basic core composed of six transmembrane helices (TM), while ecGlpG has the 6-TM core with a cytoplasmic domain. AarA has seven predicted transmembrane domains and reflects the topology found in the eukaryotic secretory class of rhomboids. Prior to kinetic calculations, all parameters for the assay were optimized, including the minimal enzyme concentration and detergent concentration to prevent non-specific aggregation of both substrates and enzymes. Time of reaction was optimized in order to ensure that the measurements were obtained during initial rate phase and a linear relationship between time and product formation was observed (Supplementary Fig S1). The plots of the reaction velocity against the corresponding substrate concentration were fitted both with Michaelis–Menten and the Hill's equation. Based on the R2 values and analysis of the residuals, the Hill's equation was chosen as a preferred model for all three studied rhomboids (Supplementary Fig S2), suggesting cooperative substrate binding behaviour. The Hill's coefficient (h) indicates the degree of cooperativity, and hiGlpG exhibited the strongest positive cooperativity with h = 2.1 ± 0.1. The Hill's coefficient for AarA was 1.70 ± 0.18, whereas ecGlpG was found to be an almost non-cooperative enzyme with h = 1.2 ± 0.1. The catalytic parameters for the three rhomboids were determined (Table1). The K0.5 values for AarA and ecGlpG were in the same range with AarA being slightly higher, while a much higher K0.5 was observed for hiGlpG. The cleavage rate of AarA for psTatA cleavage, its physiological substrate, was fastest at 1.06 ± 0.05 min−1. This turnover rate is similar to that calculated for the AarA-mediated psTatA cleavage in proteoliposomes (0.8 min−1) (Dickey et al, 2013), yet in contrast, our data show that kcat and more importantly the cleavage efficiency (kcat/KM) was distinctly different for all rhomboids, revealing substrate specificity.
Figure 1. psTatA cleavage by three prokaryotic rhomboids demonstrates substrate specificity.

- Topology of three studied rhomboids. hiGlpG and ecGlpG are the simplest and commonly found in prokaryotes. AarA has 7 TMD similar to rhomboids typically found in eukaryotes.
- Hill plots of formation of rhomboid-cleaved psTatA versus psTatA concentration (n = 2, mean ± SD). The product concentration was calculated as the percentage of cleaved substrate from the blot image.
- Western blot analysis of rhomboid-mediated cleavage of psTatA. psTatA at the concentration range of 0.5–15 μM was incubated with 0.33 μM (hiGlpG and ecGlpG) or 0.13 μM (AarA) at 37°C for 4 h or 15 min, and the cleavage product (C) was separated from the uncleaved substrate (U) with SDS-Tricine gel and developed with Western blot using anti-His antibodies. The star represents a contaminant band from the substrate sample.
Source data are available online for this figure.
Table 1.
Kinetic parameters of AarA, ecGlpG and hiGlpG using psTatA as a substrate
| Protein | K0.5 (μM) | Vmax (μM min−1) | kcat (min−1) | kcat/K0.5 (min−1 μM−1) | Hill's coefficient |
|---|---|---|---|---|---|
| AarA | 7.6 ± 1.1 | 0.138 ± 0.007 | 1.06 ± 0.05 | 13.9 × 10−2 ± 2.4 × 10−2 | 1.70 ± 0.18 |
| ecGlpG | 22 ± 2* | 0.032 ± 0.004 | 0.10 ± 0.01# | 4.5 × 10−3 ± 0.9 × 10−3 | 1.2 ± 0.1 |
| hiGlpG | 119 ± 7# | 0.022 ± 0.001 | 0.066 ± 0.001# | 5.5 × 10−4 ± 0.4 × 10−4 | 2.1 ± 0.1 |
The purified substrate (0.5–15 μM) was incubated with AarA (0.135 μM) at 37°C for 15 min or with ecGlpG (0.33 μM) and hiGlpG (0.33 μM) for 4 h; the product was separated from the uncleaved substrate using SDS-Tricine gels. The percent of cleaved substrate was calculated, and the data were fitted to the Hill equation from which K0.5 and Vmax were obtained. The symbols indicate significant difference compared to the AarA values:
P = 0.005;
P = 0.001.
FRET-based quantitative analysis of psTatA cleavage by its native enzyme AarA
To study the physiological AarA-TatA pair in detail, a real-time FRET-based kinetic assay was developed (Fig2). An engineered substrate CyPet-psTatA-YPet (psTatA-FRET) was used for the FRET assay. Prior to kinetic calculations, all parameters for the assay were optimized (Supplementary Fig S3). We also determined the pH optimum for all studied rhomboids, which was found to be in slightly acidic pH range (pH 5.7–6.5) (Fig2B and Supplementary Fig S3). Most serine proteases with a catalytic Ser-His-Asp triad have a pH optimum in the basic range (pH 8–11) (Ekici et al, 2008). To analyse the FRET data, a modified method taking into account auto-fluorescence was adapted to avoid signal cross contamination, which allowed an accurate determination of the kinetic parameters (Liu et al, 2012). Consistent with the gel-based assay, the Hill equation was the preferred model for kinetic data, rendering h = 1.8 ± 0.1 and confirming cooperative substrate binding for the AarA (Fig2C). Our kinetic parameters obtained by FRET assay (Table2) are in good agreement with the values from gel-based assay. ecGlpG and hiGlpG could not cleave the FRET-TatA substrate, suggesting a unique mechanism of recognition exists between the AarA-psTatA physiological pair.
Figure 2. FRET-based AarA activity assay.

- Schematic illustration of the FRET-based cleavage assay. TatA structure determined by NMR (Rodriguez et al, 2013).
- AarA activity dependence on pH. The proteolysis reaction was performed at 37°C for 2 h in the presence of 0.18 μM of rhomboid protease and 2 μM of full-length psTatA-FRET in 150 mM NaCl, 20% glycerol, 0.1% DDM and 2 mM DTT reaction buffer, at pH interval spanning from 2 to 9 with 50 mM of a broad-range pH triple buffer (boric acid:citric acid:phosphate) (Carmody, 1961). The highest initial velocity was calculated to determine the percent activity along the pH gradient. The same pH activity dependence is observed when using a single buffer (Supplementary Fig S3A).
- Hill plot of AarA-mediated cleavage of psTatA-FRET. psTatA-FRET at the concentration range from 0.13 to 7 μM was incubated with 0.135 μM of AarA at 37°C for 2 h. Velocities were calculated as described in Materials and Methods (n = 5, mean ± SD). See Table2.
Table 2.
Kinetic parameters of AarA and active site mutant using psTatA-Förster resonance energy transfer (FRET) substrate
| Protein | K0.5 (μM) | Vmax (μM min−1) | kcat (min−1) | kcat/K0.5 (min−1 μM−1) | Hill coefficient |
|---|---|---|---|---|---|
| AarA wt | 3.9 ± 0.3 | 2.3 × 10−2 ± 0.1 × 10−2 | 0.17 ± 0.07 | 4.3 × 10−2 ± 0.5 × 10−2 | 1.8 ± 0.1 |
| AarA S150A | 0 | 0 | 0 | 0 | 0 |
The purified substrate (0.13–7 μM) was incubated with AarA or mutant protein (0.135 μM) at 37°C for 2 h, and the emission intensity of CyPet at 475 nm and YPet at 530 nm was recorded with the excitation wavelength of 414 nm. The initial velocities were calculated for each substrate concentration. K0.5 and Vmax were obtained from fitting the data to the Hill's equation by plotting the initial velocities of psTatA-FRET-digestion versus the corresponding substrate concentration.
Rhomboids do not exhibit cooperativity with the soluble substrate casein
Conformational plasticity is an intrinsic property of an enzyme that enables it to exhibit cooperativity. One source of conformational change is the binding of the substrate, and conceivably, different substrates are able to cause varied structural perturbations. Thus, it is plausible that the same enzyme can behave cooperatively with one substrate and be non-cooperative with another. To investigate this possibility, we measured the kinetics of cleavage of fluorescently labelled casein (FL-casein), a generic substrate that has been previously been shown to be cleaved by ecGlpG (Wang et al, 2006; Xue & Ha, 2012; Lazareno-Saez et al, 2013). The optimized activity assay with FL-casein for the three rhomboids, purified in 0.1% dodecyl-maltoside (DDM), was conducted as previously reported (Lazareno-Saez et al, 2013). Plots of reaction velocity versus substrate concentration were fitted with Michaelis–Menten and Hill's equations, revealing a clear preference for the first model for all enzymes (Fig3). Thus, all enzymes showed non-cooperative substrate binding with casein and exhibited very similar kinetic parameters, demonstrating no specificity for this substrate (Table3).
Figure 3. Michaelis–Menten graphs of rhomboid-mediated FL-casein cleavage indicate a different mode of cleavage for soluble substrate FL-casein.
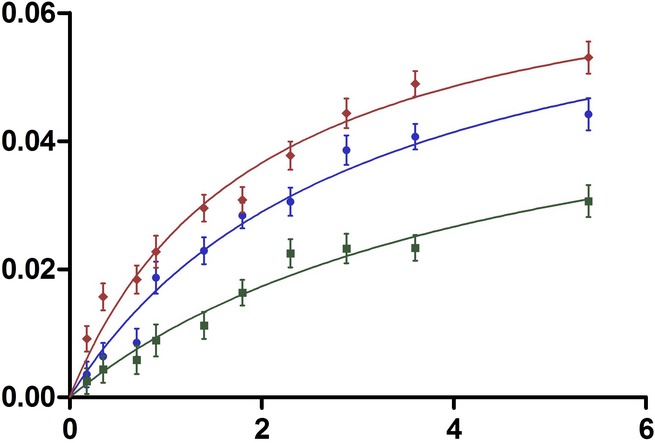
Michaelis–Menten plots for AarA(red), ecGlpG(blue) and hiGlpG(green) cleavage of FL-casein. Rhomboid purification, as well as the activity measurements, were carried out in buffer containing 0.1% dodecyl-maltoside (DDM) to obtain the dimeric state of the enzymes. 0.18 μM of rhomboid protease was incubated with 0.1–5.8 μM of FL-casein at 37°C for 2 h. For AarA and hiGlpG n = 3, mean ± SD. For ecGlpG n = 5, mean ± SD (see legend for Table1).
Table 3.
Kinetic parameters of AarA, ecGlpG and hiGlpG using FL-casein as a substrate
| Rhomboid | KM (μM) | Vmax (μM min−1) | kcat (min−1) | kcat/KM (μM−1 min−1) |
|---|---|---|---|---|
| AarA | 1.8 ± 0.47 | 0.066 ± 0.012 | 0.36 ± 0.068 | 0.2 ± 0.092 |
| ecGlpG | 2.5 ± 0.37 | 0.067 ± 0.022 | 0.37 ± 0.122 | 0.148 ± 0.068 |
| hiGlpG | 6.5 ± 2.1 | 0.070 ± 0.069 | 0.38 ± 0.037 | 0.05 ± 0.020 |
The reaction mixture contained 0.179–8.95 μM of BODIPY FL-casein, activity buffer (50 mM MES, pH 6.0, 150 mM NaCl, 20% glycerol, 0.1% DDM) and 0.179 μM of rhomboid enzyme. The substrate was mixed with the activity buffer and incubated at 37°C for 1 h in the dark. The reaction was started with the protease. Fluorescence emission at 513 nm was measured at 37°C every 5 min over 2 h in a FluoStar fluorescence microplate reader with an excitation wavelength of 503 nm.
Dimers are important for psTatA cleavage
We have previously demonstrated that prokaryotic rhomboids are dimeric in detergent and lipid environments (Sampathkumar et al, 2012). Many enzymes are found as oligomers, and changes in enzymatic activity commonly accompany oligomer dissociation (Traut, 1994). To assess the effect of oligomeric state on rhomboid protease activity, the conditions for the isolation of stable monomers were identified. When purified in DDM containing buffer, all rhomboids were dimeric, whereas exchange from DDM to the chemically similar decyl-maltoside (DM) during gel filtration resulted predominantly in monomers (Fig4A and B), with small peaks of remaining dimers for hiGlpG and ecGlpG. For AarA, however, upon DM exchange, a broad peak with a shoulder was observed, suggesting that a mixture of monomers and dimers was present at these conditions (Fig4C). Monomers were obtained only when AarA was solubilized and purified in DM.
Figure 4. Dimeric but not monomeric rhomboids can cleave the transmembrane substrate psTatA.

A–C Gel filtration experiments of (A) hiGlpG, (B) ecGlpG and (C) AarA either in 0.1% dodecyl-maltoside (DDM, red) or exchanged into 0.2% decyl-maltoside (DM, blue), carried out with a Superdex 200 (16/60) column. A third detergent exchange with AarA purified in 0.2% DM was conducted in gel filtration buffer with 0.2% DM (DM/DM, green) (C). Standards for gel filtration column are as follows: 1. Thyroglobulin, 51.1 ml (MW 670 kDa, Stokes radius 86 Å); 2. IgG, 67.3 ml (MW 158 kDa, Stokes radius 51 Å); 3. Ovalbumin, 83.4 ml (MW 44 kDa, Stokes radius 28 Å); 4. Myoglobin, 94.4 ml (MW 17 kDa, Stokes radius 19 Å); 5. Vitamin B12, 111.9 ml (MW 1.3 kDa, Stokes radius 1.6 Å). Vo, void volume 46.51 ml, Vt, total column volume 128 ml.
D–F The cleavage of psTatA by (D) hiGlpG, (E) ecGlpG or (F) AarA was assessed in activity buffer containing either 0.1% DDM or 0.2% DM detergent (assay det). Prior to this, enzyme was purified with a Ni-NTA column in either 0.1% DDM or 0.2% DM detergents (Ni det). AarA S150A mutant was used as a negative control.
G To further assess the physiological relevance of dimerization, inside-out vesicles containing full-length ecGlpG harbouring a Tobacco Etch Virus (TEV) protease site between the membrane and cytoplasmic domains were incubated with or without TEV protease and then subjected to ultracentrifugation.
H The supernatant was separated with 16% SDS-Tricine gel (Cyto, ecGlpG cytoplasmic domain). TEV protease was removed using Ni-NTA resin, and the flow through was applied on a Superdex 75 (10/30) gel filtration column. Known retention volumes for monomer (M) and dimer (D) of the cytoplasmic domain, (Lazareno-Saez et al, 2013), are indicated on the gel filtration trace. Inset represents the SDS-PAGE of the main peak. Gel filtration analysis of the supernatant after ultracentrifugation demonstrates that the cytoplasmic domain elutes as a dimer only. Along with our previous work, these data clearly demonstrate rhomboids are dimeric in the lipid bilayer.
Source data are available online for this figure.
To determine whether oligomerization affects catalytic properties of rhomboids, we assessed the activity of dimers and monomers with the gel-based assay using psTatA. Rhomboids purified either in 0.1% DDM or in 0.2% DM were tested for activity using buffer containing DDM or DM (Fig4D–F). It is important to note that for all activity measurements, the protein was diluted more than 500 times in activity buffer with detergent; thus, the amount of detergent used for purification was negligible. For all three rhomboids, we observed a loss of activity in the presence of DM, at conditions where the protein exists as a monomer. For AarA, purified in DDM, only 50% of activity was observed in DM reaction buffer, consistent with the gel filtration results, showing only partial dissociation. A full loss of activity for AarA was observed only when DM was used in both Ni-NTA purification and the activity assay. We confirmed that DM detergent does not directly influence enzymatic activity (Supplementary Fig S4). Rhomboids purified in DM regained activity when the assay was conducted in DDM, proving that the loss of activity in the presence of DM was not due to protein denaturation or to detergent itself. We were also able to demonstrate re-association of hiGlpG monomers purified in DM by SEC using a buffer containing DDM (Supplementary Fig S5). Little or no aggregation was observed during gel filtration (Fig4), verifying the mild influence of DM and DDM on rhomboid stability. Circular dichroism (CD) spectra overlap for both dimeric and monomeric forms for hiGlpG, the rhomboid with the simplest enzyme topology; thus, DM did not alter structural integrity during the transition from dimer to monomer. The melting temperature (Tm) of dimers is slightly higher in comparison with monomers, which may be attributed to the mutual stabilization of subunits within the dimer and their dissociation as the initial step during the thermal unfolding of the enzyme.
To investigate the predominant oligomeric state in the lipid bilayer, we took advantage of the exposed cytoplasmic domain of ecGlpG, which is known to form domain-swapped dimers (Lazareno-Saez et al, 2013). We examined the oligomeric state of the cytoplasmic domain of ecGlpG upon shedding from the membrane, demonstrating the prevalent oligomeric state in its native lipid environment is dimeric (Fig4G and H). A crucial aspect of this study was that neither the proteins were exposed to detergent nor the samples were concentrated, providing an assessment of the protein in its native environment.
Interestingly, monomers of all three rhomboids were able to cleave casein (Supplementary Fig S6), revealing that the monomeric form of the enzyme is functional with the soluble substrate but lacks the elements needed for the psTatA transmembrane substrate cleavage. These results lead us to question whether the catalysis of rhomboid's native substrates is governed by the active site alone or whether another mechanism, such as allosteric interactions, is involved in the process.
The cleavage of psTatA is governed by allosteric interactions
To determine whether the cleavage of psTatA is dependent on allosteric interactions, we performed competition binding assays using casein as a main substrate and psTatA as a competing substrate for the three rhomboids: hiGlpG, ecGlpG and AarA (Fig5). Rhomboid protease was reacted with FL-casein in the presence of different concentrations of psTatA. Initial velocities were measured at each substrate concentration and plotted against the corresponding casein concentration. The obtained data sets were fitted simultaneously and globally to competitive, non-competitive and mixed inhibition. For AarA, the best model was estimated to be non-competitive inhibition with a global R2 of 0.973 (Fig5A). Global fits of the AarA data set for competitive and mixed inhibition revealed clear systematic deviations from best-fit lines. The determined Kd for psTatA is 9.2 ± 0.7 μM, represented by Ki for the AarA–psTatA complex (Fersht, 2002). For ecGlpG, the global fit revealed competitive inhibition as best-fit model with global R2 of 0.967 (Fig5B) with Kd for ecGlpG-psTatA being 4.29 ± 0.60 μM, which is significantly lower than that previously observed with detergent solubilized ecGlpG-TatA, 119 μM (Dickey et al, 2013). The derived kinetic constants are shown on the plots. For hiGlpG, no effect on activity was observed with increasing psTatA concentration (Supplementary Fig S6). This can be explained by the high K0.5 of hiGlpG for psTatA (119 μM) that exceeds the possible concentration range to retain a soluble substrate. Competition binding assays were also performed with monomeric form of the AarA (Supplementary Fig S7). As performed for the dimers, FL-casein cleavage was monitored with increasing amounts of psTatA. No difference in cleavage was observed in the presence of TatA, further confirming the monomeric form of AarA does not bind psTatA.
Figure 5. Competitive studies of rhomboid-mediated FL-casein cleavage in the presence of psTatA.
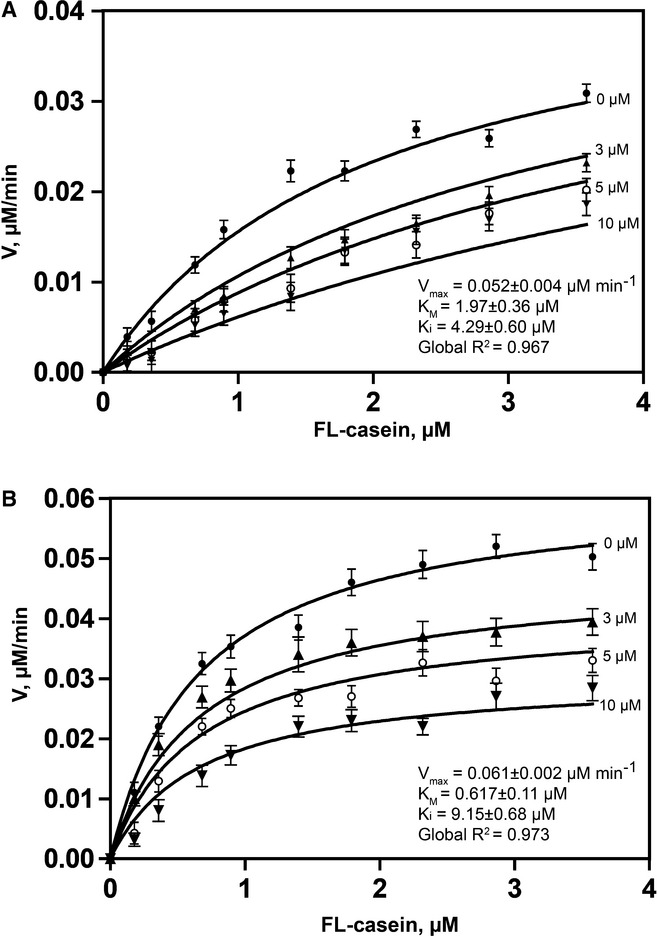
- Competitive inhibition of ecGlpG by psTatA, with FL-casein as substrate. The protease reaction between ecGlpG (0.179 μM) with FL-casein (0.179–3.85 μM) was performed in the presence of different concentrations of psTatA (0, 3, 5 and 10 μM). The reaction was started with the protease. Fluorescence emission at 513 nm was measured at 37°C every 5 min for 2 h in a fluorescence microplate reader with an excitation wavelength of 503 nm. Fluorescence detection of each substrate concentration in the presence of corresponding psTatA concentration without enzyme was used as a negative control (n = 2, mean ± SD). Initial velocities were determined for each substrate concentration. Michaelis–Menten plots were subjected to global fit to distinguish the kinetic model and determine the kinetic parameters.
- Non-competitive inhibition of AarA by psTatA, with FL-casein as substrate. The assay was conducted, and the kinetic model was determined similar to that for ecGlpG. The concentration of psTatA used is shown beside the corresponding curve.
The non-competitive inhibition with the dimeric AarA indicates that psTatA binds to the enzyme in a region other than the active site, suggesting the presence of a remote binding site (exosite) for substrate recognition. Furthermore, this clearly demonstrates the initial binding of psTatA to the exosite drives a conformational change in the active site of AarA. In the presence of psTatA, AarA still binds casein with the same affinity, but its active site is no longer in the optimal arrangement to cleave casein. In contrast to AarA, psTatA binds to dimeric ecGlpG competitively, suggesting no conformational changes in the active site are induced upon psTatA binding to ecGlpG exosite, if any binding occurs at all. Competition studies with monomeric AarA show that dimerization is important for allosteric crosstalk between active site and exosite. Taken together, these results clearly indicate that the exosite interaction is the initial step in specific substrate binding of psTatA to AarA.
Discussion
In this paper, we report the steady-state kinetic analysis of rhomboid intramembrane protease family members. Kinetic experiments with transmembrane substrate revealed deviations from Michaelis–Menten enzyme kinetics. Sigmoidal saturation curves and Hill's coefficients indicate positive cooperativity of TatA cleavage, suggesting that conformational changes occur upon substrate binding to these rhomboids. Our competitive binding studies provide strong evidence that the studied rhomboid proteases also have allosteric properties. Allosteric regulation is an efficient mechanism for modulation and regulation of protein activity to prevent non-specific substrate cleavage by altering the substrate accessibility and affinity. The classical model for enzyme allosteric behaviour assumes the presence of two non-equivalent binding sites, where one of the sites is a regulatory exosite and the other is the catalytic site. If substrate binds to both sites, it is an example of homotropic allosterism. The involvement of an exosite in rhomboid substrate binding has been proposed in a previous report (Strisovsky et al, 2009). Our competition experiments confirm the presence of an exosite and validate the model for allosteric regulation of AarA activity. This is the first evidence of homotropic allosteric activation for any intramembrane protease family member by its natural substrate.
Based on our kinetic evidence, we propose the following model for psTatA cleavage by AarA (Fig6). Since psTatA is the only identified substrate for any prokaryotic rhomboid, AarA-psTatA represents the only known physiological enzyme–substrate pair in propkaryotes. The process of substrate binding to rhomboids is achieved through a multi-step dynamic process. In the lipid bilayer, AarA exists as dimer, containing both an active site and an exosite. Our in vitro studies revealed that dimer assembly is required for activity with the transmembrane substrate psTatA, and when dimers reversibly dissociate into monomers, the function is lost. In contrast, monomeric form is capable of cleaving FL-casein, suggesting that the active site is not affected upon dissociation. Crystal structures further support the integrity of the active site of the monomeric form (Wang et al, 2006; Lemieux et al, 2007; Vinothkumar et al, 2011). Taken together, these results suggest that rhomboids may have two different mechanisms for substrate recognition; the transmembrane substrate is recognized on the hydrophobic belt of the enzyme by the exosite, which facilitates the substrate entry laterally into the active site. We propose soluble substrates, such as FL-casein, do not require initial exosite binding and approach the active site from the soluble face of the enzyme via the opening of loop 5. Our model may also explain the discrepancy observed from mutations in the proposed gating regions. Mutations disrupting the interaction between helices 2 and 5 are known to enhance TM C100Spitz substrate cleavage by opening the lateral gate (Baker et al, 2007; Brooks et al, 2011). However, when the proposed lateral gate was cross-linked the activity remained unaffected with GST-Gurken, a hybrid of a large soluble GST with a single TM substrate (Xue & Ha, 2013). Furthermore, when the recognition motif of psTatA is translated to a region outside of the TM segment, it is still able to be cleaved by AarA (Strisovsky et al, 2009). Distinct modes of substrate access provide a rational for these experimental results and raise the possibility that rhomboid substrates could include soluble proteins as well. The above-mentioned allosteric model would also provide an elegant mechanism for enforcing specificity during recognition of transmembrane substrates for these intramembrane proteases. Rhomboids possess a permissive active site (Urban et al, 2002; Urban & Freeman, 2003; Strisovsky et al, 2009), and exosite binding imparts a primary determinant for substrate selection similar to other enzymes with broad specificity (Krishnaswamy, 2005; Manithody et al, 2012).
Figure 6. A schematic illustration of rhomboid exosite-mediated substrate recognition and allosteric regulation of cleavage.
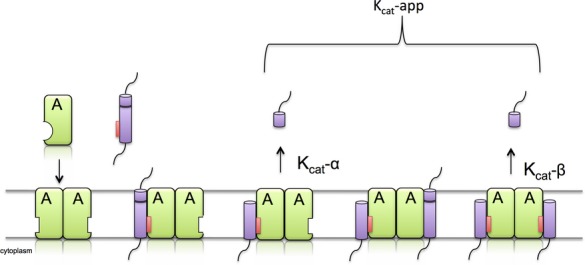
We propose a preliminary model of transmembrane substrate cleavage by rhomboid protease. Dimerization allows the formation of an exosite, either via conformational changes upon dimerization, or the exosite is located at the interface (not depicted in this illustration). The initial binding of the substrate's transmembrane (purple) segment by its exosite recognition motif (red) to the rhomboid intramembrane-located exosite will allow the cleavage at a low rate (kcat-α) and induce subtle conformational changes in the active site (A) resulting in the optimal active site arrangement and increased rate of catalysis (kcat-β). The overall rate measured is therefore an apparent kcat (kcat-app).
The observed loss of activity with monomeric rhomboids in DM is most likely attributed to the disruption of the exosite required for the cleavage of transmembrane substrate. Consequently, dimerization is associated with conformational changes, leading to formation of the exosite. The exosite may be also located at the interface between the two monomers. Our competition studies indicate that binding of a native substrate to the exosite induces a conformational change in the active site of rhomboid. We have shown that prokaryotic rhomboids exist as dimers in mild detergents and in the lipidic environment in the current and previous studies (Sampathkumar et al, 2012). The prevalent oligomeric state for ecGlpG, expressed in E. coli membranes, was dimeric state as well. These results validate the physiological relevance of above model and lead us to speculate that allostery may represent a regulatory mechanism for TM protein recognition for the rhomboid family. In support of this hypothesis, the mitochondrial PARL protein is known to recognize several substrates in a non-competing pathway further suggestive of different sites of substrate recognition (Sekine et al, 2012; Chan & McQuibban, 2013). Furthermore, the eukaryotic rhomboid RHBDD2 has been shown to oligomerize (Ahmedli et al, 2013), and while this is an inactive iRhom, it is known to bind TM substrates (Lemberg & Freeman, 2007). It is possible that dimerization is not essential for TM substrate cleavage but could enhance cleavage. An alternative interpretation to our observations is that DM not only disrupts dimerization but could also bind to the exosite and prevents substrate binding at high concentrations above the DM critical micelle concentration (CMC).
The broad specificity of rhomboids and its physiological relevance was discussed in the other reports (Urban et al, 2002; Strisovsky et al, 2009; Dickey et al, 2013). In the present study, we have determined the turnover rates and KM values for three rhomboid family members in detergent solution (Table1). These two parameters are interrelated, and an enzyme may achieve an ideal balance by optimizing the ratio of kcat/KM, which describes enzyme's efficiency for any substrate. The determined values for ecGlpG and hiGlpG were lower by a factor of 10 and 100, respectively, in comparison with AarA. These values permit a meaningful quantitative comparison for rhomboids ability to react with psTatA and allow us to conclude that rhomboid-mediated cleavage is more efficient towards the natural substrate and hence specific. Our kinetic data allowed us to evaluate the efficiency of the rhomboid protease and demonstrate that this enzyme was slow. The slow rate of enzyme has already been anticipated due to non-canonical arrangement of the active site catalytic dyad instead of triad and the lipidic environment surrounding the enzyme. When catalytic triad of trypsin was converted to Ser–His dyad, a 104-fold decrease in the activity was observed (Craik et al, 1987). However, the turnover rate of rhomboids was found to be comparable to some soluble serine proteases (http://www.brenda-enzymes.org/index.php4) (Tschantz & Dalbey, 1994; Gallagher et al, 2001) having similar catalytic architecture. For example, with the E. coli signal peptidase I (SPase), the kcat values range from 0.016 to 660 min−1 for different substrates (also see BRaunschweig ENzyme Database, http://www.brenda-enzymes.org for more information). The E. coli SPase is a membrane-tethered enzyme having a large soluble serine protease domain with a Ser–Lys dyad that removes amino-terminal signal sequence from proteins translocated from cytoplasm (Auclair et al, 2012). Similar to rhomboids, SPases are known to cleave substrates with small side-chain residues in the P1 position (Nielsen et al, 1998).
A recent paper has reported kinetic parameters of rhomboid proteases using TatA as a substrate in reconstituted proteoliposomes (Dickey et al, 2013). The cleavage of psTatA by ecGlpG rhomboid in these conditions obeyed Michaelis–Menten kinetics and showed no cooperativity. These data are consistent with our observations, revealing that ecGlpG behaviour with psTatA is almost non-cooperative (h = 1.2) and demonstrats non-specific or no psTatA binding to rhomboid ecGlpG exosite. Furthermore, the above paper also reported that detergent solubilization increased the rate of substrate cleavage (Dickey et al, 2013). However, the turnover rate of AarA enzyme with psTatA in proteoliposomes, 0.8 min−1, is comparable to our values, 1.06 min−1 (Table1), suggesting detergent does not have a drastic effect on catalysis. Analogously, the CaaX protease has similar activity in both membrane and detergent solubilized state; this study was conducted with C12E7 detergent, also having a very low CMC similar to DDM. Simple membrane proteins such as rhomboids, purified in a single step with mild detergents such as DDM and DM, tend to retain native protein structure and function. Endogenous lipids can be co-purified at lipid protein ratios as high as 36:1, as observed for a polytopic 12-transmembrane membrane protein after purification in DDM (Wang et al, 2003). In our system, the substrate and enzyme molecules likely are surrounded by mixed detergent:lipid micelles, explaining why we observe similar kinetics to that measured in the lipid bilayer. Lastly, our competition studies reveal the affinity of the psTatA substrate for ecGlpG. In detergent solution, we observe a significantly higher affinity, Kd, 4.29 ± 0.60 μM, compared to that observed with reconstituted enzyme (119 μM; Dickey et al, 2013). At present, the reason behind this discrepancy is unclear and probably can be explained by the differences in utilized methods.
The location of the enzyme's exosite remains to be determined. Based on a crystal structure of ecGlpG in lipid environment, a plausible exosite at the interface of TM2/TM5 has been suggested (Vinothkumar, 2011). Our current data do not allow us to make any predictions about the conformational changes that occur during dimerization and allosteric communication between exosite and active site. Rhomboid-inhibitor structures revealed only minor structural changes; however, inhibitor binding does not truly mimic the multi-step substrate binding process and thus may not induce structural changes similar to that with substrates (Vinothkumar et al, 2011; Xue & Ha, 2013). An enzyme–substrate complex crystal structure will provide information on the conformational changes associated with substrate binding. We propose that allosteric interactions facilitate movements required for substrate entry into the buried active site via loop 5 and helix 5. Substrate–exosite interactions may also play a role in substrate gating by tethering the substrate in a canonical conformation proximal to catalytic machinery, which increases the probability of substrate access into the active site. Understanding the structural changes that occur upon dimerization and allosteric activation will furthermore resolve the long-standing controversy over substrate gating into the membrane-buried active site. It also remains to be seen whether eukaryotic rhomboids are also dimeric and allosterically regulated. This would have significant implications for therapeutics design, since there are a number of advantages in using allosteric modulators over classic orthosteric ligands. Our work describes a unique example of allosteric interactions for an intramembrane protease and will set the stage to explore the role of allostery for other intramembrane proteases.
Materials and Methods
Expression and purification of ecGlpG, hiGlpG and AarA
Rhomboid genes were cloned into pBAD-Myc/HisA plasmid (Invitrogen, Canada), having C-terminal Tobacco Etch Virus (TEV) protease cleavage site, Myc-epitope and His6-tag. The vector was transformed into TOP10 chemically competent E. coli cells. The protein was induced with 0.002% arabinose and expressed at 24°C for 8 h in LB media. The cells were harvested, resuspended in 50 mM Tris–HCl pH 8.0, 150 mM NaCl, Tris-buffered saline (TBS) and lysed under high pressure (EmulsiFlex-C3). The membranes were isolated by ultracentrifugation at 95,800 g for 2 h, solubilized in 50 mM Tris–HCl pH 8.0, 300 mM NaCl, 10 mM imidazole, 20% glycerol, 1% (w/v) DDM or 2% (w/v) DM and applied onto a Ni-NTA column chromatography (Qiagen, Ontario, Canada). The proteins were eluted with 250–500 mM of imidazole, 50 mM Tris–HCl pH 8.0, 300 mM NaCl, 20% glycerol, 0.1% DDM or 0.2% DM. The His-tag was removed by TEV protease (1 mg per 100 mg of protein, overnight, 16°C), and a subsequent Ni-NTA column was performed to remove uncleaved protein and TEV protease. The flow-through was collected and concentrated using 30,000 MWCO concentrators (Millipore, USA). The protein samples were flash-frozen and stored at −80°C.
Rhomboid proteolytic activity assay with psTatA
The cleavage reaction mixture consisted of 0.5–15 μM of psTatA substrate mixed with 0.13 (for AarA) and 0.33 μM (for ecGlpG and hiGlpG) enzyme in 50 mM 2-(n-Mopholino)ethanesulfonic acid (MES) pH 6.0, 150 mM NaCl, 20% glycerol and 0.1% DDM or 0.2% DM. The cleavage reaction was started with enzyme. The concentration of protease and the time of incubation were selected by carrying out cleavage reactions over several time intervals with various amounts of enzyme and with substrate concentration at saturation level. Enzyme concentration that gave linear product formation over time was then used for the kinetic studies. After incubation for 15 min–4 h as indicated, at 37°C, the reactions were stopped by adding SDS-Sample buffer. The same concentrations of substrate with no protease were used as negative controls and showed no degradation after the time of reaction. Protein samples were revealed with Western blot using anti-His antibodies (Sigma-Aldrich, USA) as previously described with Anti-His antibodies (Sampathkumar et al, 2012). Furthermore, the chemiluminescence signal for psTatA concentration range used in the assay was confirmed to be linearly proportional to the amount of psTatA (Supplementary Fig S1). The assay was repeated at least three times to ensure reproducibility. For all images, digitization was carried out with ImageQuant LAS 4000 (GE Healthcare, USA). Integration of substrate cleavage product (lower band) was conducted with ImageQuant software. Full-length substrate levels were used to normalize the values of the product. Kinetic parameters were calculated with Prism software, GraphPad, USA. To calculate apparent KM (K0.5) and Vmax values, both the Michaelis–Menten equation and the Hill's equation were used to assess best fit. P-values were calculated using ANOVA.
Rhomboid proteolytic activity assay with FL-casein as substrates
The ecGlpG, hiGlpG and AarA cleavage assays using FL-casein as substrates were performed as previously described (Lazareno-Saez et al, 2013).
SDS-Tricine and SDS-PAGE gels
SDS-Tricine gels were used for gel-based activity assays for high resolution of proteins smaller than 30 kDa. SDS-PAGE gels were used for purity control. Standard protocols of both systems were employed.
Expression and purification of psTatA-FRET
We measured kinetic parameters of psTatA cleavage by AarA with a Förster resonance energy transfer (FRET)-based protease assay using an engineered FRET pair, CyPet and YPet (derived from cyan fluorescence protein and yellow florescence protein). Rhomboid substrate psTatA was cloned between CyPet and YPet FRET pair in pBad HisB vector. This fluorescent protein pair exhibits 20-fold energy transfer efficiency when compared to the parental pair (Nguyen & Daugherty, 2005). The vector containing CyPet-psTatA-YPet substrate (psTatA-FRET) was transformed into GlpG knockout cells from the Keio library (glpG::Kn; Baba et al, 2006). Expression in GlpG knockout cells prevented premature cleavage of the substrate. The cells were grown in LB media with ampicillin (100 μg l−1) and kanamycin (30 μg l−1) until OD600 reached 0.6 followed by induction with 0.02% of arabinose. The protein was expressed for 18 h at 24°C, cells were lysed, cell debris was removed with centrifugation (22,320 g), and the supernatant was loaded on the Ni-NTA column equilibrated with 50 mM Tris–HCl pH 8.0, 200 mM NaCl, 20% glycerol and 0.1% DDM. The protein was eluted with 200–350 mM of imidazole, concentrated and, loaded on SEC Superdex 200(16/60) (GE, Canada) equilibrated with the same buffer. The fractions were assessed with SDS-PAGE, and the protein concentration was determined by the BCA method. Pure protein (> 95%) was pooled, concentrated and flash-frozen prior to storage at −80°C.
Providencia stuartii TatA purification
Providencia stuartii TatA (psTatA) was purified using anti-flag resin as described previously (Lazareno-Saez et al, 2013).
FRET-based protease kinetic assay
CyPet-psTatA-YPet was incubated with AarA at 37°C in buffer at pH 6.0 containing 50 mM MES pH 6.0, 150 mM NaCl, 20% glycerol and 0.1% DDM for 20 min. The final concentration of AarA was fixed at 0.135 μM, and the final concentration of CyPet-psTatA-YPet was varied from 0.13 to 7 μM. FRET-based AarA cleavage assay was conducted by measuring the emission intensity of CyPet at 475 nm and YPet at 530 nm with the excitation wavelength of 414 nm in a fluorescence multi-well plate reader (SynergyMx, BioTek). To negate the effect of signal cross contamination, we determined the CyPet and YPet direct emissions and total emissions at 530 nm. The emission of the recombinant protein CyPet–psTatA–YPet was measured at 475 nm when excited at 414 nm to determine the CyPet direct emission; the emission was measured at 530 nm when excited at 475 nm to determine the YPet direct emission. The fluorescence emission was measured every 5 min for 2 h. We then obtained digested concentration of substrate using the following equation, developed by Liu and co-workers (Liu et al, 2012):
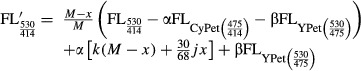 |
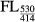 and
and 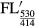 are total fluorescence emission at 530 nm when excited at 414 nm before and after digestion, respectively, M is the total amount of CyPet-psTatA-YPet, and x is the amount of digested CyPet-psTatA-YPet. α is ratio of fluorescence emission by CyPet at 530–475 nm under excitation at 414 nm, whereas
are total fluorescence emission at 530 nm when excited at 414 nm before and after digestion, respectively, M is the total amount of CyPet-psTatA-YPet, and x is the amount of digested CyPet-psTatA-YPet. α is ratio of fluorescence emission by CyPet at 530–475 nm under excitation at 414 nm, whereas  is CyPet direct emission at 475 nm when excited at 414 nm. Similarly, β is ratio of fluorescence emission by YPet at 530–475 nm under excitation at 475 nm. Using this ratio, YPet direct emission
is CyPet direct emission at 475 nm when excited at 414 nm. Similarly, β is ratio of fluorescence emission by YPet at 530–475 nm under excitation at 475 nm. Using this ratio, YPet direct emission 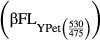 at 530 nm when excited at 475 nm was calculated.
at 530 nm when excited at 475 nm was calculated.
Here, 30/68 gives the molecular mass ratio of CyPet-psTatA to CyPet-psTatA-YPet. The plot of emission of CyPet-TatA-YPet at 475 nm under excitation at 414 nm versus amount yields a straight line, and the constant k determines the slope of this line. Similarly, a linear relationship between the emission of CyPet-psTatA + YPet (1:1 molar ratio) under excitation at 414 nm and the protein amount is found. In this case, j describes the slope of the plot. Both parameters, j and k, were calculated from standard plots of fluorescence emission versus amount of protein and used in the equation.
Before kinetic calculations, all the parameters for the assay were optimized. It was verified that the proportionality between the fluorescence emitted and the amount of the substrate used in the assay was linear. The minimal concentration of the enzyme that gave a linear dependence of amount of generated product with time was chosen, as well as the minimal time of reaction within the linear part of the curve.
For pH optimization, enzymatic activity was measured with psTatA-FRET spanning the interval of pH 2–9. Tris-buffer was chosen deliberatively to maintain the correct pH range to avoid effects of buffering compounds on the activity. For ecGlpG and hiGlpG, the pH optima was measured in the same manner, using FL-casein as a substrate (Lazareno-Saez et al, 2013).
Circular dichroism
Ni-NTA purified hiGlpG in 0.1% DDM was further purified by size-exclusion chromatography in a buffer containing 25 mM Tris pH 8, 150 mM NaCl, 5% glycerol and either 0.1% DDM or 0.2% DM to harvest protein in either dimeric or monomeric forms, respectively. CD measurements were performed using JASCO-720 spectrophotometer. All spectra were recorded using 0.1 mm path length cell at a protein concentration of 0.2 mg ml−1 and 0.14 mg ml−1 for monomers and dimers, respectively. Melting curves of the samples were measured at 222 nm between 25 and 90°C with a gradient resolution of 0.2°C. The rate of temperature change was 0.5°C min−1. The bandwidth of the equipment was 1 nm with a response time of 1 s.
Inside-out vesicle preparation
ecGlpG was cloned with a TEV protease site inserted between residues 91 and 92, separating N-terminal cytoplasmic and C-terminal membrane domains. In a 2-l culture, ecGlpG was expressed and the membrane fraction was isolated as described above. Membranes were resuspended in 15 volumes of TBS and pelleted by ultracentrifugation at 95,800 g for 1 h. This cycle was repeated 3 times. After the last centrifugation, membranes were resuspended in 5 volumes of TBS and subjected to three freeze-thaw cycles to create inverted vesicles. TEV protease (2 mg) was added to half of the membranes, (from 1 l of cell culture) and incubated overnight at 4°C. As a negative control, the remaining membranes were incubated in a similar manner without TEV protease. The membrane fractions were pelleted at 95,800 g for 1 h. From the supernatant, the liberated cytoplasmic domain was assessed by SDS-PAGE. Prior to gel filtration, TEV protease was removed using Ni-NTA column; the flow through, containing the cytoplasmic domain, was loaded on a Superdex 75 (10/30) column (GE, Canada).
Acknowledgments
We thank Dr. Matthew Freeman for his kind gift of the TatA gene. We thank Dr. Robert Campbell for his kind gift of the CyPet and YPet vector, and Dr. Tracy Ravio for providing the ecGlpG knockout E. coli strain. We thank Dr. Andrew Holt and Dr. Denis Arutyunov for helpful discussions. Work in MJL's group is supported by grant MOP-93557 from the Canadian Institutes of Health Research. MJL is a Canada Research Chair in Membrane Protein Structure and Function, and an Alberta Innovates Health Solutions Scientist. PP is supported by CIHR-THRUST.
Author contributions
EA, NG, PMS, and MWM conducted kinetic experiments. EA, PP and MJL analysed data and wrote the paper. All authors edited the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Ahmedli NB, Gribanova Y, Njoku CC, Naidu A, Young A, Mendoza E, Yamashita CK, Ozgul RK, Johnson JE, Fox DA, Farber DB. Dynamics of the rhomboid-like protein RHBDD2 expression in mouse retina and involvement of its human ortholog in retinitis pigmentosa. J Biol Chem. 2013;288:9742–9754. doi: 10.1074/jbc.M112.419960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama Y, Maegawa S. Sequence features of substrates required for cleavage by GlpG, an Escherichia coli rhomboid protease. Mol Microbiol. 2007;64:1028–1037. doi: 10.1111/j.1365-2958.2007.05715.x. [DOI] [PubMed] [Google Scholar]
- Auclair SM, Bhanu MK, Kendall DA. Signal peptidase I: cleaving the way to mature proteins. Protein Sci. 2012;21:13–25. doi: 10.1002/pro.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RP, Young K, Feng L, Shi Y, Urban S. Enzymatic analysis of a rhomboid intramembrane protease implicates transmembrane helix 5 as the lateral substrate gate. Proc Natl Acad Sci U S A. 2007;104:8257–8262. doi: 10.1073/pnas.0700814104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shem A, Fass D, Bibi E. Structural basis for intramembrane proteolysis by rhomboid serine proteases. Proc Natl Acad Sci U S A. 2007;104:462–466. doi: 10.1073/pnas.0609773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergbold N, Lemberg MK. Emerging role of rhomboid family proteins in mammalian biology and disease. Biochim Biophys Acta. 2013;1828:2840–2848. doi: 10.1016/j.bbamem.2013.03.025. [DOI] [PubMed] [Google Scholar]
- Brooks CL, Lazareno-Saez C, Lamoureux JS, Mak MW, Lemieux MJ. Insights into substrate gating in H. influenzae rhomboid. J Mol Biol. 2011;407:687–697. doi: 10.1016/j.jmb.2011.01.046. [DOI] [PubMed] [Google Scholar]
- Carmody WR. An easily prepared wide range buffer series. J Chem Educ. 1961;40:A386. [Google Scholar]
- Chan EY, McQuibban GA. The mitochondrial rhomboid protease: its rise from obscurity to the pinnacle of disease-relevant genes. Biochim Biophys Acta. 2013;1828:2916–2925. doi: 10.1016/j.bbamem.2013.05.012. [DOI] [PubMed] [Google Scholar]
- Chavez-Gutierrez L, Tolia A, Maes E, Li T, Wong PC, de Strooper B. Glu(332) in the Nicastrin ectodomain is essential for gamma-secretase complex maturation but not for its activity. J Biol Chem. 2008;283:20096–20105. doi: 10.1074/jbc.M803040200. [DOI] [PubMed] [Google Scholar]
- Chavez-Gutierrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, Wiltfang J, Serneels L, Karran E, Gijsen H, Schymkowitz J, Rousseau F, Broersen K, De Strooper B. The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012;31:2261–2274. doi: 10.1038/emboj.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craik CS, Roczniak S, Largman C, Rutter WJ. The catalytic role of the active site aspartic acid in serine proteases. Science. 1987;237:909–913. doi: 10.1126/science.3303334. [DOI] [PubMed] [Google Scholar]
- Di Cera E, Page MJ, Bah A, Bush-Pelc LA, Garvey LC. Thrombin allostery. Phys Chem Chem Phys. 2007;9:1291–1306. doi: 10.1039/b616819a. [DOI] [PubMed] [Google Scholar]
- Dickey SW, Baker RP, Cho S, Urban S. Proteolysis inside the membrane is a rate-governed reaction not driven by substrate affinity. Cell. 2013;155:1270–1281. doi: 10.1016/j.cell.2013.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drag M, Salvesen GS. Emerging principles in protease-based drug discovery. Nat Rev Drug Discovery. 2010;9:690–701. doi: 10.1038/nrd3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Reconstitution of gamma-secretase activity. Nat Cell Biol. 2003;5:486–488. doi: 10.1038/ncb960. [DOI] [PubMed] [Google Scholar]
- Ekici OD, Paetzel M, Dalbey RE. Unconventional serine proteases: variations on the catalytic Ser/His/Asp triad configuration. Protein Sci. 2008;17:2023–2037. doi: 10.1110/ps.035436.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Yan H, Wu Z, Yan N, Wang Z, Jeffrey PD, Shi Y. Structure of a site-2 protease family intramembrane metalloprotease. Science. 2007;318:1608–1612. doi: 10.1126/science.1150755. [DOI] [PubMed] [Google Scholar]
- Fersht A. Practical methods for kinetics and equilibria. In: Julet MR, editor. Structure and Mechanism on Protein Structure. Vol. 6. New York, NY: W. H. Freeman and Company, Cambridge University; 2002. pp. 191–215. [Google Scholar]
- Fraering PC, Ye W, Strub JM, Dolios G, LaVoie MJ, Ostaszewski BL, van Dorsselaer A, Wang R, Selkoe DJ, Wolfe MS. Purification and characterization of the human gamma-secretase complex. Biochemistry. 2004;43:9774–9789. doi: 10.1021/bi0494976. [DOI] [PubMed] [Google Scholar]
- Fraering PC, Ye W, LaVoie MJ, Ostaszewski BL, Selkoe DJ, Wolfe MS. gamma-Secretase substrate selectivity can be modulated directly via interaction with a nucleotide-binding site. J Biol Chem. 2005;280:41987–41996. doi: 10.1074/jbc.M501368200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher J, Kaderbhai NN, Kaderbhai MA. Kinetic constants of signal peptidase I using cytochrome b5 as a precursor substrate. Biochim Biophys Acta. 2001;1550:1–5. doi: 10.1016/s0167-4838(01)00265-5. [DOI] [PubMed] [Google Scholar]
- Hollander I, Frommer E, Mallon R. Human ras-converting enzyme (hRCE1) endoproteolytic activity on K-ras-derived peptides. Anal Biochem. 2000;286:129–137. doi: 10.1006/abio.2000.4795. [DOI] [PubMed] [Google Scholar]
- Hu J, Xue Y, Lee S, Ha Y. The crystal structure of GXGD membrane protease FlaK. Nature. 2011;475:528–531. doi: 10.1038/nature10218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DJ, Li W, Adams TE, Huntington JA. Antithrombin-S195A factor Xa-heparin structure reveals the allosteric mechanism of antithrombin activation. EMBO J. 2006;25:2029–2037. doi: 10.1038/sj.emboj.7601089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakuda N, Funamoto S, Yagishita S, Takami M, Osawa S, Dohmae N, Ihara Y. Equimolar production of amyloid beta-protein and amyloid precursor protein intracellular domain from beta-carboxyl-terminal fragment by gamma-secretase. J Biol Chem. 2006;281:14776–14786. doi: 10.1074/jbc.M513453200. [DOI] [PubMed] [Google Scholar]
- Krishnaswamy S. Exosite-driven substrate specificity and function in coagulation. J Thromb Haemost. 2005;3:54–67. doi: 10.1111/j.1538-7836.2004.01021.x. [DOI] [PubMed] [Google Scholar]
- Lazareno-Saez C, Arutyunova E, Coquelle N, Lemieux MJ. Domain swapping in the cytoplasmic domain of the Escherichia coli rhomboid protease. J Mol Biol. 2013;425:1127–1142. doi: 10.1016/j.jmb.2013.01.019. [DOI] [PubMed] [Google Scholar]
- Lee JR, Urban S, Garvey CF, Freeman M. Regulated intracellular ligand transport and proteolysis control EGF signal activation in Drosophila. Cell. 2001;107:161–171. doi: 10.1016/s0092-8674(01)00526-8. [DOI] [PubMed] [Google Scholar]
- Lemberg MK, Menendez J, Misik A, Garcia M, Koth CM, Freeman M. Mechanism of intramembrane proteolysis investigated with purified rhomboid proteases. EMBO J. 2005;24:464–472. doi: 10.1038/sj.emboj.7600537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemberg MK, Freeman M. Functional and evolutionary implications of enhanced genomic analysis of rhomboid intramembrane proteases. Genome Res. 2007;17:1634–1646. doi: 10.1101/gr.6425307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux MJ, Fischer SJ, Cherney MM, Bateman KS, James MN. The crystal structure of the rhomboid peptidase from Haemophilus influenzae provides insight into intramembrane proteolysis. Proc Natl Acad Sci U S A. 2007;104:750–754. doi: 10.1073/pnas.0609981104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Dang S, Yan C, Gong X, Wang J, Shi Y. Structure of a presenilin family intramembrane aspartate protease. Nature. 2013;493:56–61. doi: 10.1038/nature11801. [DOI] [PubMed] [Google Scholar]
- Liu Y, Song Y, Madahar V, Liao J. Quantitative Forster resonance energy transfer analysis for kinetic determinations of SUMO-specific protease. Anal Biochem. 2012;422:14–21. doi: 10.1016/j.ab.2011.12.019. [DOI] [PubMed] [Google Scholar]
- Manandhar SP, Hildebrandt ER, Jacobsen WH, Santangelo GM, Schmidt WK. Chemical inhibition of CaaX protease activity disrupts yeast Ras localization. Yeast. 2010;27:327–343. doi: 10.1002/yea.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manithody C, Yang L, Rezaie AR. Identification of exosite residues of factor Xa involved in recognition of PAR-2 on endothelial cells. Biochemistry. 2012;51:2551–2557. doi: 10.1021/bi300200p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolaridis I, Kulkarni K, Dodd RB, Ogasawara S, Zhang Z, Bineva G, O'Reilly N, Hanrahan SJ, Thompson AJ, Cronin N, Iwata S, Barford D. Mechanism of farnesylated CAAX protein processing by the intramembrane protease Rce1. Nature. 2013;504:301–305. doi: 10.1038/nature12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesak LR, Mesak FM, Dahl MK. Expression of a novel gene, gluP, is essential for normal Bacillus subtilis cell division and contributes to glucose export. BMC Microbiol. 2004;4:13. doi: 10.1186/1471-2180-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moin SM, Urban S. Membrane immersion allows rhomboid proteases to achieve specificity by reading transmembrane segment dynamics. eLife. 2012;1:e00173. doi: 10.7554/eLife.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen AW, Daugherty PS. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat Biotechnol. 2005;23:355–360. doi: 10.1038/nbt1066. [DOI] [PubMed] [Google Scholar]
- Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Identification of prokaryote and eukaryotic signal peptidases and prediction of thier clevage sites. Protein Eng. 1998;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- Osenkowski P, Ye W, Wang R, Wolfe MS, Selkoe DJ. Direct and potent regulation of gamma-secretase by its lipid microenvironment. J Biol Chem. 2008;283:22529–22540. doi: 10.1074/jbc.M801925200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryor EE, Jr, Horanyi PS, Clark KM, Fedoriw N, Connelly SM, Koszelak-Rosenblum M, Zhu G, Malkowski MG, Wiener MC, Dumont ME. Structure of the integral membrane protein CAAX protease Ste24p. Science. 2013;339:1600–1604. doi: 10.1126/science.1232048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez F, Rouse SL, Tait CE, Harmer J, De Riso A, Timmel CR, Sansom MS, Berks BC, Schnell JR. Structural model for the protein-translocating element of the twin-arginine transport system. Proc Natl Acad Sci U S A. 2013;110:E1092–E1101. doi: 10.1073/pnas.1219486110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampathkumar P, Mak MW, Fischer-Witholt SJ, Guigard E, Kay CM, Lemieux MJ. Oligomeric state study of prokaryotic rhomboid proteases. Biochim Biophys Acta. 2012;1818:3090–3097. doi: 10.1016/j.bbamem.2012.08.004. [DOI] [PubMed] [Google Scholar]
- Sekine S, Kanamaru Y, Koike M, Nishihara A, Okada M, Kinoshita H, Kamiyama M, Maruyama J, Uchiyama Y, Ishihara N, Takeda K, Ichijo H. Rhomboid protease PARL mediates the mitochondrial membrane potential loss-induced cleavage of PGAM5. J Biol Chem. 2012;287:34635–34645. doi: 10.1074/jbc.M112.357509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton CC, Zhu L, Chau D, Yang L, Wang R, Djaballah H, Zheng H, Li YM. Modulation of gamma-secretase specificity using small molecule allosteric inhibitors. Proc Natl Acad Sci U S A. 2009;106:20228–20233. doi: 10.1073/pnas.0910757106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley LD. The roles of intramembrane proteases in protozoan parasites. Biochim Biophys Acta. 2013;1828:2908–2915. doi: 10.1016/j.bbamem.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson LG, Strisovsky K, Clemmer KM, Bhatt S, Freeman M, Rather PN. Rhomboid protease AarA mediates quorum-sensing in Providencia stuartii by activating TatA of the twin-arginine translocase. Proc Natl Acad Sci U S A. 2007;104:1003–1008. doi: 10.1073/pnas.0608140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strisovsky K, Sharpe HJ, Freeman M. Sequence-specific intramembrane proteolysis: identification of a recognition motif in rhomboid substrates. Mol Cell. 2009;36:1048–1059. doi: 10.1016/j.molcel.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traut TW. Dissociation of enzyme oligomers: a mechanism for allosteric regulation. Crit Rev Biochem Mol Biol. 1994;29:125–163. doi: 10.3109/10409239409086799. [DOI] [PubMed] [Google Scholar]
- Tschantz WR, Dalbey RE. Bacterial leader peptidase 1. Methods Enzymol. 1994;244:285–301. doi: 10.1016/0076-6879(94)44023-9. [DOI] [PubMed] [Google Scholar]
- Urban S, Lee JR, Freeman M. Drosophila rhomboid-1 defines a family of putative intramembrane serine proteases. Cell. 2001;107:173–182. doi: 10.1016/s0092-8674(01)00525-6. [DOI] [PubMed] [Google Scholar]
- Urban S, Schlieper D, Freeman M. Conservation of intramembrane proteolytic activity and substrate specificity in prokaryotic and eukaryotic rhomboids. Curr Biol. 2002;12:1507–1512. doi: 10.1016/s0960-9822(02)01092-8. [DOI] [PubMed] [Google Scholar]
- Urban S, Freeman M. Substrate specificity of rhomboid intramembrane proteases is governed by helix-breaking residues in the substrate transmembrane domain. Mol Cell. 2003;11:1425–1434. doi: 10.1016/s1097-2765(03)00181-3. [DOI] [PubMed] [Google Scholar]
- Vinothkumar KR. Structure of rhomboid protease in a lipid environment. J Mol Biol. 2011;407:232–247. doi: 10.1016/j.jmb.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinothkumar KR, Strisovsky K, Andreeva A, Christova Y, Verhelst S, Freeman M. The structural basis for catalysis and substrate specificity of a rhomboid protease. EMBO J. 2011;29:3797–3809. doi: 10.1038/emboj.2010.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DN, Lemieux MJ, Boulter JM. Purification and characterization of transporter proteins from human erythrocyte membrane. Methods Mol Biol. 2003;228:239–255. doi: 10.1385/1-59259-400-X:239. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang Y, Ha Y. Crystal structure of a rhomboid family intramembrane protease. Nature. 2006:1–5. doi: 10.1038/nature05255. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ha Y. Open-cap conformation of intramembrane protease GlpG. Proc Natl Acad Sci U S A. 2007;104:2098–2102. doi: 10.1073/pnas.0611080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe MS. Intramembrane-cleaving proteases. J Biol Chem. 2009;284:13969–13973. doi: 10.1074/jbc.R800039200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Yan N, Feng L, Oberstein A, Yan H, Baker RP, Gu L, Jeffrey PD, Urban S, Shi Y. Structural analysis of a rhomboid family intramembrane protease reveals a gating mechanism for substrate entry. Nat Struct Mol Biol. 2006;13:1084–1091. doi: 10.1038/nsmb1179. [DOI] [PubMed] [Google Scholar]
- Xue Y, Ha Y. Catalytic mechanism of rhomboid protease GlpG probed by 3,4-dichloroisocoumarin and diisopropyl fluorophosphonate. J Biol Chem. 2012;287:3099–3107. doi: 10.1074/jbc.M111.310482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Ha Y. Large lateral movement of transmembrane helix S5 is not required for substrate access to the active site of rhomboid intramembrane protease. J Biol Chem. 2013;288:16645–16654. doi: 10.1074/jbc.M112.438127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.