

Abstract
Purpose of review
The aims of this review is to suggest a new nomenclature and classification system for the diseases currently categorized as neurodegeneration with brain iron accumulation (NBIA) or dystonia-parkinsonism, and to discuss the mechanisms implicated in the pathogenesis of these diseases.
Recent findings
NBIA is a disease category encompassing syndromes with iron accumulation and prominent dystonia–parkinsonism. However, as there are many diseases with similar clinical presentations but without iron accumulation and/or known genetic cause, the current classification system and nomenclature remain confusing. The pathogenetic mechanisms of these diseases and the causes of gross iron accumulation and significant burden of neuroaxonal spheroids are also elusive. Recent genetic and functional studies have identified surprising links between NBIA, Parkinson's disease and lysosomal storage disorders (LSD) with the common theme being a combined lysosomal–mitochondrial dysfunction. We hypothesize that mitochondria and lysosomes form a functional continuum with a predominance of mitochondrial and lysosomal pathways in NBIA and LSD, respectively, and with Parkinson's disease representing an intermediate form of disease.
Summary
During the past 18 months, important advances have been made towards understanding the genetic and pathological underpinnings of the pallidopyramidal syndromes with important implications for clinical practice and future treatment developments.
Keywords: Hallervorden–Spatz disease, lysosomal storage disorders, neurodegeneration with brain iron accumulation, Parkinson's disease
INTRODUCTION
The term pallidopyramidal degeneration (PPD) was first introduced by Davison in 1954 [1] who described a series of five patients presenting with the triad of progressive parkinsonism, spasticity and dystonia combined with pyramidal and pallidal lesions and blue discoloration of the globus pallidus following the initial report by Hunt in 1917 [2] of a single case with juvenile parkinsonism and eosinophilic spheroidal structures. Subsequently, Hallervorden and Spatz in 1992 reported a family with five affected sisters with brown discoloration of the globus pallidus [3], a syndrome that was named Hallervorden–Spatz syndrome.
During the past decade, the advent of genetic technologies has allowed a more systematic delineation of the clinical presentations and genetic underpinnings of PPD starting with the identification of the first mutations in PANK2[4], a finding that led to the renaming of this disease class to neurodegeneration with brain iron accumulation (NBIA) [4,5]. Despite the fact that a molecular diagnosis and modern neuropathological analysis is not possible for the initial cases described by Davison due to the lack of preserved tissue and blood, it is likely that the brown–blue discoloration of the globus pallidus represents gross iron accumulation and the eosinophilic formations neuroaxonal spheroids, and that all belong to the modern disease entity of NBIA (Fig. 1a).
FIGURE 1.

(a) Landmarks in neurodegeneration with brain iron accumulation (NBIA) research. (b) Davison's pallidopyramidal degeneration (PPD) triad illustrated in the form of Venn diagrams. (c) Classification of pallidopyramidal syndromes (PPS) according to age at onset and main signs and symptoms. Approximate frequency of each subtype is depicted by the size of the circle (authors’ unpublished observations). Overlapping circles indicate overlapping clinical presentations.
Box 1.

no caption available
In this review, we argue that the use of the term NBIA is not ideal and suggest that the more general term pallidopyramidal syndromes (PPS) conceived by Davison would perhaps be more appropriate [6]. In this context, we also suggest a modified classification system better reflecting the clinical and pathological phenotypes associated to PPS. Finally, we outline possible disease mechanisms providing a mechanistic basis for some of the features unique to PPS that were first highlighted by Davison, and suggest a model tying the pathogenesis of lysosomal storage diseases (LSD), Parkinson's disease, and PPS.
CLASSIFICATION OF PALLIDOPYRAMIDAL SYNDROMES
At present, according to the OMIM classification system, a disease is classified as NBIA based on the clinical features including Davison's PPD triad (Fig. 1b), and gross iron accumulation on T2∗ MRI. Further classification in four subtypes depends on the pattern of iron accumulation on MRI, and on the underlying mutated genetic locus (Table 1).
Table 1.
Current OMIM classification of neurodegeneration with brain iron accumulation syndromes
| NBIA | Disease |
| NBIA 1 | Pantothenate kinase-associated neurodegeneration (PKAN)(PANK2) |
| NBIA 2A | Infantile neuroaxonal dystrophy (INAD)(PLA2G6) |
| NBIA 2B | Atypical neuroaxonal dystrophy (PLA2G6) |
| Karak syndrome(PLA2G6) | |
| NBIA 3 | Neuroferittinopathy (FTL) |
| ATP13A2 | |
| NBIA 4 | C19orf12 |
| Not classified yet | WDR45, FA2H |
Caveats of the current classification system
The current classification system is far from ideal for two reasons:
The use of iron accumulation as a classification criterion is debatable. Iron accumulation is not a consistent finding among diseases with otherwise indistinguishable clinical presentations [7–10] leading to the use of the oxymoron term ‘NBIA without brain iron’ [10]. Also, the importance of iron for disease pathogenesis and progression remains elusive [10], especially as this has been reported in various seemingly unrelated disorders and even in healthy individuals [10–12].
The use of a classification system based on mutated genetic loci has two weaknesses. First, as the complete genetic landscape of NBIA is unknown, there are several ‘idiopathic’ syndromes that are not included in the current classification system (Fig. 1c). Second, patients with mutations in the same gene often present with substantially divergent clinical features [8,13].
Taking into account these weaknesses, we suggest that at present a clinical and pathological classification system of NBIA would be more suitable for clinical practice.
Pallidopyramidal syndromes: Proposed clinical classification system
In our suggested classification system, a disease must be characterized by Davison's triad with or without iron accumulation on MRI to be classified as PPS. Further sub-classification is based on the age at onset of symptoms as this feature can serve as a starting point to prioritise genetic testing (Table 2A).
Infantile PPS (iPPS): iPPS presents before the age of 2 years and includes pantothenate kinase associated neurodegeneration (PKAN) [4] and hereditary dopamine transporter deficiency syndrome (HDTDS) [14]. The symptoms at presentation are unspecific with feeding difficulties, irritability and/or developmental delay, followed by the development of severe movement disorders [15]. Optic nerve atrophy and cognitive decline are seen in infantile neuroaxonal dystrophy (INAD), whereas cognition is maintained in HDTDS [14,16]. Disease progression is usually rapid resulting in death in approximately 10 years [17].
Juvenile PPS (jPPS): jPPS has an onset with spasticity in FBXO7-associated neurodegenetion, hereditary spastic paraplegia with thinning of the corpus callosum (HSP-TCC) and Kufor Rakeb syndrome (KRS) [18–22]. HSP-TCC can be classified as PPS only in its more rare atypical forms [19,23▪]. The recently described β-propeller protein-associated neurodegeneration (BPAN) is a distinct form of jPPS as onset is at early childhood with global developmental delay accompanied by iron accumulation on MRI preceding the development of prominent pallidopyramidal signs [24▪▪,25,26▪▪,27▪,28▪].
Adult PPS (aPPS): aPPS has an onset after the age of 18–20 years and psychiatric features as a presenting sign are common followed by the development of rapidly progressive movement disorders [8,15,29,30].
Table 2A.
Suggested PPS clinical classification system
| Infantile PPS | Juvenile PPS | Adulthood PPS |
| PLA2G6-associated neurodegeneration (PLAN) (INAD) (PLA2G6) | Typical PKAN (PANK2) | Adulthood PLAN (PLA2G6) |
| Hereditary dopamine transporter deficiency syndrome (SLC6A3) | Childhood PLAN (PLA2G6) | Atypical PKAN (PANK2) |
| Typical Pantothenate kinase-associated neurodegeneration (PKAN) (PANK2) | Fatty acid-associated neurodegeneration (FA2H) | Neuroferittinopathy (FTL) |
| ‘Idiopathic’ PPS | Hypoprebetalipoproteinemia, acanthocytosis, retinitis pigmentosa, and pallidal degeneration (HARP) (PANK2) | ‘Idiopathic’ PPS |
| Mitochondrial membrane protein associated neurodegeneration (MPAN) (C19orf12) | ||
| Karak syndrome (PLA2G6) | ||
| Kufor Rakeb syndrome (ATP13A2) | ||
| Atypical PKAN (PANK2) | ||
| FBXO7-associated neurodegeneration (FBXO7) | ||
| Hereditary Spastic Paraplegia with thinning of the corpus callosum (HSP-TCC) (SPG11) | ||
| ’Idiopathic’ PPS | ||
| Beta-propeller protein-associated neurodegeneration (BPAN) (WDR45) |
Often, a differential diagnosis has to be made from phenocopies (atypical, usually milder presentations of syndromes caused by mutations in different genetic loci, reviewed in [9]). However, we do not include these in the suggested classification system as they probably do not fit in the nosological entity originally described by Davison and we thus use the term PPS only in the context of NBIA syndromes.
PATHOLOGY OF PALLIDOPYRAMIDAL SYNDROMES: INSIGHTS INTO PATHOGENETIC MECHANISMS AND IMPLICATIONS FOR THE PROPOSED PATHOLOGICAL CLASSIFICATION SYSTEM
As only a small number of PPS cases have come to pathology, our knowledge on the pathological features of PPS is incomplete. However, there are two main findings present in all PPS studied (PKAN, PLA2G6-associated neurodegeneration – PLAN, neuroferittinopathy, mitochondrial membrane protein-associated neurodegeneration – MPAN): iron-laden pigmentation and spheroids with a predilection for pallidal involvement in PKAN [31,32▪] but a wider lesion distribution in the remaining syndromes [33,34▪▪,35▪]. α-Synuclein accumulation [36,37▪▪] is an additional feature in a subset of PPS (PLAN, MPAN) [33,34▪▪,35▪]. Here, we discuss the potential pathogenic processes underpinning these lesions and their implications for our suggested pathological classification system.
α-Synuclein and pallidopyramidal syndromes
Although α-synuclein deposition consistently occurs in various neurodegenerative diseases it is still unclear whether this is the primary event driving disease pathogenesis or is just an epiphenomenon [38▪].
Here, drawing mainly from studies on Parkinson's disease, we argue that most recent evidence implicates lysosomal dysfunction and/or lipid abnormalities in the aggregation and spreading of α-synuclein and then discuss the implications of this observation for the pathogenesis of PPS.
Lewy body formation is probably caused by lysosomal dysfunction
Lewy bodies have been reported in four disease categories: Parkinson's disease, PPS, LSD and dementia with Lewy bodies. Interestingly, the common denominator in most of these situations with α-synuclein accumulation appears to be lysosomal dysfunction:
- Lewy body pathology [36] is an important pathological feature of Parkinson's disease observed in most genetic forms, and in several idiopathic cases [39]. However, Lewy bodies in Parkinson's disease consistently occur on two occasions: when the primary genetic defect lies in the glucocerebrosidase (GBA) or in the a-synuclein SNCA gene [40▪▪].
- Numerous studies have demonstrated that GBA is a lysosomal enzyme [41,42▪,43], a role which is further emphasized by the fact that homozygous mutations in GBA cause Gaucher's disease, a LSD. Heterozygous mutations in GBA are the strongest risk factor associated to the development of Parkinson's disease [44] and dementia with Lewy bodies [45,46▪▪]. Interestingly, Lewy bodies is the characteristic feature that ties these seemingly unrelated disorders as they are observed in nearly all cases that come to pathology [40▪▪,47–50]. Recently, a model based on experimental evidence was suggested to explain this relation between GBA and α-synuclein: glysocylceramide (GlcCer), the substrate to GBA, can stabilize α-synuclein oligomers which in turn inhibit GBA function, cause GlcCer accumulation and further attenuate α-synuclein aggregation [51,52].
- SNCA multiplications [53] and point mutations [54▪▪,55▪▪,56,57▪▪–59▪▪] are always related to Lewy-body pathology [40▪▪]. In the former case, the causative link is straightforward: increased transcription results in increased expression levels. In the second case, however, the exact mechanisms resulting in α-synuclein accumulation are not obvious though it is thought that lysosomal chaperon-mediated autophagy (CMA) could be impaired [60–62,63▪,64▪]. A similar effect is also caused by α-synuclein accumulation [65] probably resulting in a positive feedback loop [66▪].
ATP13A2, a gene encoding a lysosomal protein [67] mutated in PPS [7,68], Parkinson's disease [45,69–71] and LSD [72▪▪,73,74], has recently emerged as an important lysosomal factor involved in α-synuclein homeostasis and as a component of Lewy bodies [7,75▪▪,76]. In addition, the lysosomal dysfunction caused by ATP13A2 mutations has been shown to directly cause α-synuclein accumulation [77▪,78▪▪].
α-Synuclein homeostasis appears to be affected in LSD which are often characterised by Lewy bodies in neuropathology [79]. Neuronal ceroid lipofuscinosis (NCL) type 10, which is one of these LSD with Lewy bodies [80,81], is caused by mutations in cathepsin D (CSTD) that mediates α-synuclein degradation [82].
On the contrary, Lewy bodies occasionally occur in some cases without a clear lysosomal involvement. Parkinson's disease and PPS caused by mutations in mitochondrial proteins parkin[40▪▪,83–88,89▪,90▪], PINK1[40▪▪,91], PLA2G6[35▪] and C19orf12[33,34▪▪], respectively, frequently have Lewy-body pathology. Lewy bodies are also described in most cases with LRRK2 mutations [40▪▪], a protein whose precise function is currently unknown. Finally, Lewy bodies are frequent in ‘sporadic’ Parkinson's disease in similar distribution and severity to GBA-associated disease [39]. The significance of these observations and the relation of a-synuclein accumulation to mitochondrial dysfunction are discussed later.
Interactions between α-synuclein and lipids both within the lysosomal context [51] and the cytoplasm [92▪▪,93] also seem to underlie α-synuclein homeostasis. Such extensive α-synuclein–lipid interactions are in keeping with the highlighted frequent involvement of ceramide metabolism pathways in parkinsonian disorders with Lewy Bodies in neuropathology [94]. Given that GlcCer interactions have been shown to stabilize α-synuclein oligomers, we can hypothesize that similar α-synuclein–lipid interactions in the cytoplasm could have similar consequences.
Why are Lewy bodies absent from some pallidopyramidal syndromes but present in others?
Pathologically, PPS can be distinguished into two categories based on the presence or absence of α-synuclein accumulation: PKAN and Neuroferritinopathy are characterized by well localized defects in the globus pallidus [31,32▪] and absence of Lewy bodies, contrary to PLAN [35▪] and MPAN [33,34▪▪]. We hypothesize that PKAN and Neuroferritinopathy are well localized diseases due to the absence of α-synuclein accumulation and the accompanying hypothesized self-perpetuating mechanism of disease spread [95▪▪,96,97▪,98,99▪,100,101▪▪,102–104,105▪,106▪▪,107–111,112▪]: the defect initiates from the globus pallidus but cannot spread to other brain regions due to the absence of α-synuclein involvement. As recent evidence implicates lysosomal dysfunction and/or lipid abnormalities in the aggregation and spreading of α-synuclein, this observation has two possible implications for the pathogenic mechanisms of PKAN:
Probably, lysosomal dysfunction is not a primary event in the pathogenesis of PKAN.
The ceramide lipid metabolism defects observed in PKAN are unlikely to affect α-synuclein homeostasis: Pantothenate kinase 2 (PANK2) encoded by the PANK2 gene, is probably an exclusively mitochondrial enzyme [113,114▪] thus placing a physical barrier between lipids and (cytoplasmic) α-synuclein.
Contrary to PKAN, PLAN is characterised by widespread Lewy bodies in neuropathology [35▪]. iPLA2 beta which is encoded by the PLA2G6 gene, is an enzyme involved in phospholipid hydrolysis with implications for a wide range of cellular functions [115▪▪] probably not necessarily limited to the mitochondria; thus, lipid accumulation caused by iPLA2 beta inactivation could be responsible for the initiation of α-synuclein misfolding and spreading.
Neuroaxonal spheroids: a mitochondrial trafficking defect?
Neuroaxonal spheroids are mysterious formations present in various serious neurodegenerative diseases including PKAN [31,32▪], MPAN [33,34▪▪], Neuroferritinopathy [116,117], Wilson's disease [115▪▪], progressive supranuclear palsy-pallido-nigro-luysial atrophy variant (PSP-PNLA) [118], PLAN [35▪], hereditary diffuse leukoencephalopathy with spheroids (HDLS) [119,120▪,121▪▪], pigmented orthochromatic leukodystrophy (POLD) [122▪▪], and traumatic brain injury [31]; however, these are also observed in healthy, aged individuals [123]. Even though neuroaxonal spheroids have not been ultrastructurally studied in genetically confirmed cases and systematically compared between various diseases, limited electron microscopy studies on nongenetically confirmed HDLS and on mouse models of PLA2G6 have indicated that these structures likely contain mitochondria [124–126] in addition to other molecules [31,32▪,35▪,119]. Given the similarities in the staining patterns of the spheroids between diseases, it is likely that they represent identical or highly homologous structures, a remarkable finding given the diversity in clinical presentations of associated diseases.
As neuroaxonal spheroids are present in such a variety of serious neurodegenerative diseases, the mechanisms underlying their formation are intriguing. Here, we hypothesize that spheroids could result from impaired mitochondrial trafficking as a reaction to severe neuronal damage drawing from evidence provided from the study of PKAN.
Specifically in the case of PPS, perhaps their formation stems from a primary mitochondrial dysfunction, a relationship that would seem more clear and convincing in PKAN. As mitochondria heavily rely on CoA provision for energy generation, it is expected that PANK2 mutations would have a devastating effect on mitochondrial integrity [127▪▪]. The increased number of large degenerate mitochondria [127▪▪] could result in the overload of the macroautophagy pathway with the formation of large, indigestible autophagosomes that cannot be uptaken by the lysosomes [128]; thus, neuroaxonal spheroids could represent these indigestible autophagocytic vesicles. Alternatively, damaged mitochondria could impinge on lysosomal function indirectly through impaired microtubule trafficking [129▪▪,130▪].
Mitochondrial trafficking impairment could occur secondarily to mitochondrial defects. It has been recently shown that Miro, a mitochondrial trafficking protein [131,132], is selectively targeted by PINK1 and parkin in mitochondrial damage in order to halt mitochondrial trafficking within neuraxons [133–135,136▪] and that spheroid formation is triggered in the absence of parkin [137▪▪]. Thus, severe mitochondrial damage could trigger this process en masse, holding mitochondria within neuraxons and initiating neuroaxonal spheroid formation [31,32▪]. This hypothesis is supported by the observation of tau within the spheroids [31,32▪,35▪].
Implications of neuropathological studies for pathological classification of pallidopyramidal syndromes
As neuroaxonal spheroids and Lewy bodies are the two characteristic neuropathological features that have shed light into the pathogenetic mechanisms of PPS, an attempted neuropathological classification should probably revolve around these two features with four categories reflecting the presence or absence of α-synuclein accumulation and/or Lewy bodies (Table 2B). In addition, as neuroaxonal spheroids are a common feature of several neurodegenerative diseases, we suggest the establishment of a separate disease category of ‘spheroidopathies’ (Table 2B, Fig. 2, Table 3).
Table 2B.
Spheroidopathies
| A) PPS-suggested pathological classification system | |||
| SNCA (+), spheroids (+) | SNCA (−), spheroids (+) | SNCA (+), spheroids (−) | SNCA (−), spheroids (−) |
| PLA2G6-associated neurodegeneration (PLAN) | Pantothenate kinase-associated neurodegeneration (PKAN) | None | None |
| Mitochondrial membrane protein associated neurodegeneration (MPAN) | Neuroferritinopathy | ||
| B) Non-PPS | |||
| PKAN | |||
| PLAN | |||
| MPAN | |||
| Hereditary Diffuse Leukoencephalopathy with Spheroids (HDLS) (CSF1R | |||
| Wilson's disease | |||
| Progressive supranuclear palsy-Pallido-nigro-luysial atrophy (PSP-PNLA) | |||
| Traumatic brain injury | |||
| Pigmented orthochromatic leukodystrophy (POLD) (CSF1R) | |||
PPS, pallidopyramidal syndromes.
FIGURE 2.
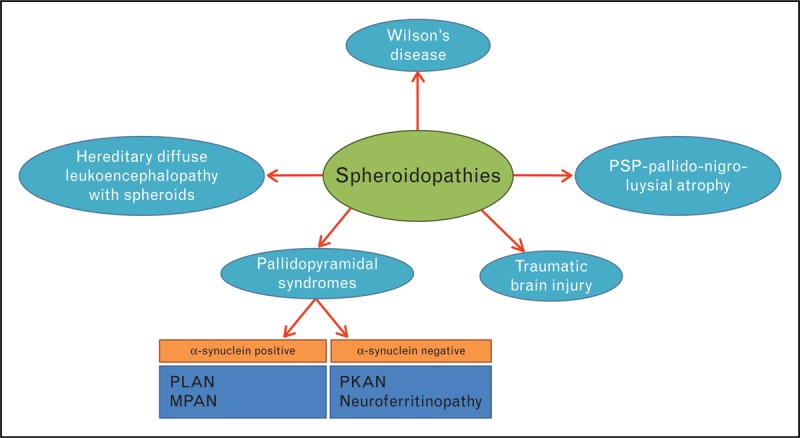
Spheroidopathies. MPAN, Mitochondrial membrane protein associated neurodegeneration; PKAN, Pantothenate kinase-associated neurodegeneration; PLAN, PLA2G6-associated neurodegeneration.
Table 3.
Characteristic neuropathological features of pallidopyramidal syndromes (PPS)
| Characteristic neuropathological features | PPS | Reference |
| PKAN | a) Isolation of lesions in the GP | [31,32▪] |
| b) Minimal involvement of the SN | ||
| c) Large and small spheroids strongly APP positive | ||
| d) Hemosiderin deposition in neurons, astrocytes and in perivascular region | ||
| PLAN | a) Extensive tau deposition | [35▪] |
| b) LBs | ||
| c) SN depletion | ||
| d) Cerebral and cerebellar atrophy | ||
| e) Neuroaxonal spheroids | ||
| f) Widespread distribution of lesions (spinal cord, basal ganglia) | ||
| MPAN | a) Widespread pathological alterations | [33,34▪▪ |
| b) LBs | ||
| c) Tau pathology | ||
| d) Axonal spheroids | ||
| e) Iron in astrocytes and macrophages | ||
| Neuroferittinopathy | a) Cystic cavitation of GP | [115▪▪, 116,117] |
| b) Iron deposition | ||
| c) Spheroids |
APP, amyloid precursor protein; GP, globus pallidus; LBs, Lewy Bodies; MPAN, Mitochondrial membrane protein associated neurodegeneration; PKAN, Pantothenate kinase-associated neurodegeneration; PLAN, PLA2G6-associated neurodegeneration; SN, substantia nigra.
DISEASE MODEL HYPOTHESIS: THE ‘PARKINSONIAN MITOCHONDRIAL–LYSOSOMAL TRIANGLE’
There appears to be a clear relationship between PPS, Parkinson's disease and LSD clinically [9,138,139], pathologically [33,35▪,40▪▪,81] and genetically [7,19,44,68,71,72▪▪,79,140,141,142▪,143] indicating that their pathogenic pathways are perhaps also linked.
As genetic and functional studies have demonstrated, defects in two main organelles can cause Parkinson's disease, PPS or LSD: mitochondria [127▪▪,144–151,152▪▪,153▪,154▪] and lysosomes [79,142▪,155▪,156▪]. Interestingly, for some of the mutated molecules involved in the dysfunction of these two organelles, there is functional and neuropathological evidence mapping them clearly in one of the two pathways. However, for the rest there seems to be an overlap: even though for each mutated gene there is strong functional evidence that only one of the two organelles should be affected, there are circumstantial pathological features indicating that perhaps the second organelle is affected too (Table 4).
Table 4.
Molecules that genetic studies have implicated in the pathogenesis of pallidopyramidal syndromes, Parkinson's disease and Lysosomal storage disorders
| Molecule | Organelle of function | Function | Usual neuropathological featuresa | Neuropathological findings inconsistent with the primary function of the moleculea |
| Parkin | Mitochondria [144–151,152▪▪,157▪▪] | Ubiquitin ligase targeting mitochondrial membrane proteins [144–151, 152▪▪, 158▪] | SN cell loss without LBs | Occasional presence of LBs [157▪] |
| PINK1 | Mitochondria [144–151, 152▪▪] | Regulation of parkin in mitochondria [144–151, 152▪▪] | Unknown | LBs in the one case studied |
| C19orf12 | Mitochondria [33] | Limited information | LBs, spheroids, tau | LBs |
| iPLA2 beta | Mitochondria [159] | Phospholipid hydrolysis | LBs, spheroids, tau | LBs |
| ATP13A2 | Lysosomes [67] | SNCA homeostasis [75▪▪,76, 77▪, 78▪▪] | Unknown | Mitochondrial abnormalities [160▪▪]. Mutations can cause PPS, PD or Lysosomal storage disorders [7,68,71,72▪▪,73,74]. |
| Glucocerebrosidase | Lysosomes [41,42▪,43,51,] | LBs | -b | |
| PANK2 | Mitochondria | Pathway of CoA synthesis | Spheroids | - |
| WDR45 | Limited information | Vesicular trafficking Autophagy [24▪▪,26▪▪] | Unknown | Limited information |
| VPS35 | Limited information | Vesicular trafficking [161,162] Mitochondrial function [163▪] | No LBs in a single case studied [161,162, 164] | Limited information |
| LRRK2 | Inconclusive evidence | Autophagy [165▪,166▪▪,167▪] Vesicular trafficking [168▪▪] Mitochondrial function [169,170▪] | Variable (LBs, tau, TDP43) [171] | Limited information |
| α-synuclein | Inconclusive evidence | Synaptic function, microtubule (reviewed in [92▪▪]) | LBs | - |
LB, Lewy body; PD, Parkinson's disease; SN, substantia nigra.
aFor full references concerning the pathological features see [40▪▪].
bEven though GBA mutations can cause both Parkinson's disease and Lysosomal storage disorders, the recent identification of a variant (E326K) that causes exclusively Parkinson's disease both in homozygosis and in heterozygosis [172▪▪] suggests a separate regulatory rather than metabolic effect of glucocerebrosidase in the pathogenesis of Parkinson's disease [173▪,174▪]; certainly though, this observation does not disassociate Parkinson's disease development from lysosomal dysfunction.
Thus, in general, Lewy bodies consistently occur in cases with mutations in lysosomal enzymes whereas these are found only occasionally in relation to mutations in mitochondrial proteins. This observation would support the hypothesis that mitochondrial dysfunction does not directly cause α-synuclein accumulation; indeed, to date, there is not strong enough functional evidence that mitochondrial dysfunction impacts directly on α-synuclein homeostasis [175▪,176▪], though there is some evidence supporting the opposite [98,175▪,177▪,178,179▪▪,180▪,181▪].
Such an overlap in pathologies would suggest that there is a functional link between lysosomes and mitochondria and that unknown events (or perhaps even stochasticity) could shift the balance between the two pathways and some well determined genetic forms of Parkinson's disease develop inconsistent pathological features. We term this functional continuum ‘Parkinsonian mitochondrial–lysosomal triangle’ and suggest that PPS and LSD lie in the extreme ends of this triangle with Parkinson's disease as an intermediate form of disease (Fig. 3a).
FIGURE 3.
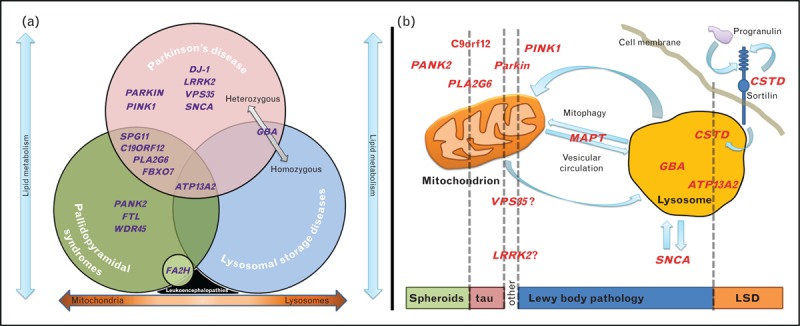
(a) The Parkinsonian mitochondrial–lysosomal triangle: Venn diagram depicting that pallidopyramidal syndromes (PPS), lysosomal storage disorders (LSD) and Parkinson's disease overlap pathologically, genetically and/or clinically. The orange arrow in the bottom indicates that the mitochondrial–lysosomal complex could form a functional continuum defects along which can result in an array of disorders, with PPS and LSD being at the extreme ends and Parkinson's disease lying in the middle. Blue arrows indicate that lipid metabolism could be implicated in all three entities in different ways (intramitochondrially, intralysosomally and cytoplasmically/cell membrane). GBA is placed in the overlap between LSD and Parkinson's disease as heterozygous mutations result in Parkinson's disease but homozygous in LSD. SNCA is also placed in the overlap as α-synuclein pathology is observed in both Parkinson's disease and LSD. SPG11 is placed in the overlap between PPS and PD as SPG11 can have a clinical presentation very similar to either PD or PPS. ATP13A2 is placed in the overlap between PPS, Parkinson's disease and LSD as mutations can cause all three disease entities (in Parkinson's disease heterozygous mutations appear to be a risk factor). Leukoencephalopathies are placed in the bottom between PPS and LSD as mutations in FA2H can cause both diseases and neuroaxonal spheroids have been reported in relation to both diseases. Also, metachromatic leukodystrophy is both a leukoencephalopathy and a LSD [79]. (b) Simplified diagram depicting the suggested lysosomal–mitochondrial link. This is based on neuropathological reports for carriers of mutations in specific genes and experimental evidence for the functional role of these genes. The rectangle in the bottom shows which types of neuropathology are related to defects in particular molecules (listed on the top of the figure, in relation to their localisation and/or function).
If this theory holds true, we can make two interesting hypotheses:
Parkinson's disease should share some pathological features of LSD. Although no lipofuscin inclusions have been observed in Parkinson's disease, somebody could argue that Lewy bodies represent a form of lysosomal inclusions as they contain ATP13A2 [75▪▪], GBA [182] and numerous lysosomal molecules [183▪,184].
How can lysosomes and mitochondria be functionally connected? Though the exact nature of this link is unknown, it is thought to take the form of mitophagy [185] and to be bidirectional (Fig. 3b). Indeed, there is evidence suggesting that the dysfunctional lysosomes ‘attack’ mitochondria in ATP13A2 patient fibroblasts [160▪▪] and that lysosomal dysfunction could result in an accumulation of dysfunctional mitochondria in mouse models of LSD [186]. Conversely, damaged mitochondria can impact on autophagy through impaired microtubule-mediated vesicular trafficking resulting in a more generalized lysosomal dysfunction (including inhibition of α-synuclein degradation) [129▪▪,157▪▪]. The molecules most recently implicated in the pathogenesis of Parkinson's disease and PPS, VPS35 [161,162,187▪–190▪] and WDR45 [24▪▪,26▪▪], could fit nicely into this model as it is thought that they are involved in Endoplasmic Reticulum (ER)-Golgi vesicular trafficking [168▪▪,191▪,192▪] and autophagy, respectively. Interestingly, the ER was recently shown to participate in autophagy initiation through a mitochondrial interaction [193▪]. A putative role for LRRK2 in autophagy is also beginning to emerge [165▪,166▪▪] together with a functional link with microtubule trafficking [168▪▪,191▪,194] and mitochondrial dysfunction in mutation carriers [170▪]. It has also intriguingly been hypothesised that MAPT variants could impact on the type of pathology exhibited with LRRK2 mutations shifting the balance between tau and Lewy bodies [195,196▪,197▪]. Finally, there is evidence for interaction between α-synuclein and microtubules [92▪▪,198–200] and for a role of MAPT mutations in the development of parkinsonism [201▪,202▪].
CONCLUSION
We propose a simplified classification of PPS that allows incorporation of the increasing genetic findings. Although the precise pathogenic underpinnings of PPS are far from clear, numerous reports suggest interesting links on multiple levels between PPS, Parkinson's disease and LSD with a central role for combined mitochondrial and lysosomal dysfunction, a relation which will be further dissected as identification of novel disease-causing genes adds the missing pieces to the puzzle [203▪▪].
Acknowledgements
The authors would like to thank the members of the Department of Molecular Neuroscience, Institute of Neurology, UCL for useful discussions. The authors would also like to thank the Medical Research Council (MRC), the National Organisation for Rare Disorders (NORD), the Dystonia Medical Research Foundation (DMRF), the dystonia coalition and the Parkinson's disease foundation (PDF) for funding their research.
Conflicts of interest
None declared.
Funding disclosure: This work was funded by the Wellcome Trust/MRC Joint Call in Neurodegeneration award (WT089698) to the UK Parkinson's Disease Consortium (UKPDC) whose members are from the UCL Institute of Neurology, the University of Sheffield and the MRC Protein Phosphorylation Unit at the University of Dundee, the Medical Research Council (MRC), the National Organisation for Rare Disorders (NORD), the Dystonia Medical Research Foundation (DMRF), the dystonia coalition and the Parkinson's disease foundation (PDF).
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
Additional references related to this topic can also be found in the Current World Literature section in this issue (pp. 451–453).
REFERENCES
- 1.Davison C. Pallido-pyramidal disease. J Neuropathol Exp Neurol 1954; 13:50–59 [DOI] [PubMed] [Google Scholar]
- 2.Hunt J. Progressive atrophy of the globus pallidus. Brain 1917; 40:58–148 [DOI] [PubMed] [Google Scholar]
- 3.Hallervorden J, Spatz H. Eigenartige Erkrankung im extrapyramidalen System mit besonderer Beteiligung des Globus pallidus und der Substantia nigra: Ein Beitrag zu den Beziehungen zwischen diesen beiden Zentren. Zeitschrift für die gesamte Neurologie und Psychiatrie 1922; 79:254–302 [Google Scholar]
- 4.Zhou B, Westaway SK, Levinson B, et al. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet 2001; 28:345–349 [DOI] [PubMed] [Google Scholar]
- 5.Taylor TD, Litt M, Kramer P, et al. Homozygosity mapping of Hallervorden-Spatz syndrome to chromosome 20p12.3-p13. Nat Genet 1996; 14:479–481 [DOI] [PubMed] [Google Scholar]
- 6.Horstink MW, Dekker MC, Montagna P, et al. Pallidopyramidal disease: a misnomer? Mov Disord 2010; 25:1109–1115 [DOI] [PubMed] [Google Scholar]
- 7.Schneider SA, Paisan-Ruiz C, Quinn NP, et al. ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Mov Disord 2010; 25:979–984 [DOI] [PubMed] [Google Scholar]
- 8.Paisan-Ruiz C, Bhatia KP, Li A, et al. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol 2009; 65:19–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schneider SA, Bhatia KP. Rare causes of dystonia parkinsonism. Curr Neurol Neurosci Reports 2010; 10:431–439 [DOI] [PubMed] [Google Scholar]
- 10.Schneider SA, Hardy J, Bhatia KP. Iron accumulation in syndromes of neurodegeneration with brain iron accumulation 1 and 2: causative or consequential? J Neurol Neurosurg Psychiatry 2009; 80:589–590 [DOI] [PubMed] [Google Scholar]
- 11.Chang MH, Hung WL, Liao YC, et al. Eye of the tiger-like MRI in parkinsonian variant of multiple system atrophy. J Neural Transm 2009; 116:861–866 [DOI] [PubMed] [Google Scholar]
- 12.Gregory A, Hayflick SJ. Neurodegeneration with brain iron accumulation. Folia Neuropathologica 2005; 43:286–296 [PMC free article] [PubMed] [Google Scholar]
- 13.Sina F, Shojaee S, Elahi E, Paisan-Ruiz C. R632W mutation in PLA2G6 segregates with dystonia-parkinsonism in a consanguineous Iranian family. Eur J Neurol 2009; 16:101–104 [DOI] [PubMed] [Google Scholar]
- 14.Kurian MA, Zhen J, Cheng SY, et al. Homozygous loss-of-function mutations in the gene encoding the dopamine transporter are associated with infantile parkinsonism-dystonia. J Clin Investig 2009; 119:1595–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayflick SJ, Westaway SK, Levinson B, et al. Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N Engl J Med 2003; 348:33–40 [DOI] [PubMed] [Google Scholar]
- 16.Kurian MA, Li Y, Zhen J, et al. Clinical and molecular characterisation of hereditary dopamine transporter deficiency syndrome: an observational cohort and experimental study. Lancet Neurol 2011; 10:54–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurian MA, McNeill A, Lin JP, Maher ER. Childhood disorders of neurodegeneration with brain iron accumulation (NBIA). Dev Med Child Neurol 2011; 53:394–404 [DOI] [PubMed] [Google Scholar]
- 18.Shojaee S, Sina F, Banihosseini SS, et al. Genome-wide linkage analysis of a Parkinsonian-pyramidal syndrome pedigree by 500 K SNP arrays. Am J Hum Genet 2008; 82:1375–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paisan-Ruiz C, Guevara R, Federoff M, et al. Early-onset L-dopa-responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacsin mutations. Mov Disord 2010; 25:1791–1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Di Fonzo A, Dekker MC, Montagna P, et al. FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology 2009; 72:240–245 [DOI] [PubMed] [Google Scholar]
- 21.Najim al-Din AS, Wriekat A, Mubaidin A, et al. Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor-Rakeb syndrome. Acta Neurologica Scand 1994; 89:347–352 [DOI] [PubMed] [Google Scholar]
- 22.Stevanin G, Azzedine H, Denora P, et al. Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain 2008; 131:772–784 [DOI] [PubMed] [Google Scholar]
- 23▪.Krebs CE, Paisan-Ruiz C. The use of next-generation sequencing in movement disorders. Front Genet 2012; 3:75. [DOI] [PMC free article] [PubMed] [Google Scholar]; This review article summarises the impact of exome sequencing on gene discovery in neurodegenerative diseases.
- 24▪▪.Haack TB, Hogarth P, Kruer MC, et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am J Hum Genet 2012; 91:1144–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports the identification of mutations in WDR45 as the cause of BPAN, a childhood form of NBIA.
- 25.Gregory A, Hayflick SJ. Genetics of neurodegeneration with brain iron accumulation. Curr Neurol Neurosci Reports 2011; 11:254–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26▪▪.Saitsu H, Nishimura T, Muramatsu K, et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet 2013; 45:445–449449e1 [DOI] [PubMed] [Google Scholar]; This study reports mutations in WDR45 as the cause of BPAN and shows that these mutations cause autophagy impairment.
- 27▪.Kasai-Yoshida E, Kumada S, Yagishita A, et al. First video report of static encephalopathy of childhood with neurodegeneration in adulthood. Mov Disord 2013; 28:397–399 [DOI] [PubMed] [Google Scholar]; This is a case report supported by video documentation of a patient with BPAN.
- 28▪.Kimura Y, Sato N, Sugai K, et al. MRI, MR spectroscopy, and diffusion tensor imaging findings in patient with static encephalopathy of childhood with neurodegeneration in adulthood (SENDA). Brain Dev 2012; 35:458–461 [DOI] [PubMed] [Google Scholar]; This study reports MRI features of BPAN.
- 29.Chinnery PF, Crompton DE, Birchall D, et al. Clinical features and natural history of neuroferritinopathy caused by the FTL1 460InsA mutation. Brain 2007; 130:110–119 [DOI] [PubMed] [Google Scholar]
- 30.Pellecchia MT, Valente EM, Cif L, et al. The diverse phenotype and genotype of pantothenate kinase-associated neurodegeneration. Neurology 2005; 64:1810–1812 [DOI] [PubMed] [Google Scholar]
- 31.Kruer MC, Hiken M, Gregory A, et al. Novel histopathologic findings in molecularly-confirmed pantothenate kinase-associated neurodegeneration. Brain 2011; 134:947–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32▪.Li A, Paudel R, Johnson R, et al. Pantothenate kinase-associated neurodegeneration is not a synucleinopathy. Neuropathol Appl Neurobiol 2012; 10.1111/j.1365-2990.2012.01269.x [DOI] [PMC free article] [PubMed] [Google Scholar]; This study confirms the findings of Kruer et al. (2011) that PANK2 mutations are not associated to α-synuclein accumulation.
- 33.Hartig MB, Iuso A, Haack T, et al. Absence of an orphan mitochondrial protein, c19orf12, causes a distinct clinical subtype of neurodegeneration with brain iron accumulation. Am J Hum Genet 2011; 89:543–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34▪▪.Hogarth P, Gregory A, Kruer MC, et al. New NBIA subtype: genetic, clinical, pathologic, and radiographic features of MPAN. Neurology 2013; 80:268–275 [DOI] [PMC free article] [PubMed] [Google Scholar]; This is the first large follow-up study after the identification of mutations in C19orf12 as a cause of NBIA.
- 35▪.Paisan-Ruiz C, Li A, Schneider SA, et al. Widespread Lewy body and tau accumulation in childhood and adult onset dystonia-parkinsonism cases with PLA2G6 mutations. Neurobiol Aging 2012; 33:814–823 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study showed that PLA2G6 mutations are associated to α-synuclein and tau accumulation.
- 36.Spillantini MG, Schmidt ML, Lee VM, et al. Alpha-synuclein in Lewy bodies. Nature 1997; 388:839–840 [DOI] [PubMed] [Google Scholar]
- 37▪▪.Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat Rev Neurol 2013; 9:13–24 [DOI] [PubMed] [Google Scholar]; This review outlines the progress in the understanding of the role of α-synuclein accumulation in the pathogenesis of Parkinson's disease since the discovery of Lewy bodies.
- 38▪.Schulz-Schaeffer WJ. Neurodegeneration in Parkinson disease: moving Lewy bodies out of focus. Neurology 2012; 79:2298–2299 [DOI] [PubMed] [Google Scholar]; This editorial discussed the relevance of Lewy bodies to the pathogenesis of Parkinson's disease.
- 39.Parkkinen L, Neumann J, O'Sullivan SS, et al. Glucocerebrosidase mutations do not cause increased Lewy body pathology in Parkinson's disease. Mol Genet Metab 2011; 103:410–412 [DOI] [PubMed] [Google Scholar]
- 40▪▪.Poulopoulos M, Levy OA, Alcalay RN. The neuropathology of genetic Parkinson's disease. Mov Disord 2012; 27:831–842 [DOI] [PMC free article] [PubMed] [Google Scholar]; This review article summarises the neuropathological features of genetic forms of Parkinson's disease.
- 41.Brady RO, Kanfer J, Shapiro D. The Metabolism of glucocerebrosides. I. purification and properties of a glucocerebroside-cleaving enzyme from spleen tissue. J Biol Chem 1965; 240:39–43 [PubMed] [Google Scholar]
- 42▪.Jovic M, Kean MJ, Szentpetery Z, et al. Two phosphatidylinositol 4-kinases control lysosomal delivery of the Gaucher disease enzyme, beta-glucocerebrosidase. Mol Biol Cell 2012; 23:1533–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study identifies novel molecules that contribute to the delivery of GBA to the lysosomes.
- 43.Tybulewicz VL, Tremblay ML, LaMarca ME, et al. Animal model of Gaucher's disease from targeted disruption of the mouse glucocerebrosidase gene. Nature 1992; 357:407–410 [DOI] [PubMed] [Google Scholar]
- 44.Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 2009; 361:1651–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mata IF, Samii A, Schneer SH, et al. Glucocerebrosidase gene mutations: a risk factor for Lewy body disorders. Arch Neurol 2008; 65:379–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46▪▪.Nalls MA, Duran R, Lopez G, et al. A multicenter study of glucocerebrosidase mutations in dementia with lewy bodies. JAMA Neurol 2013; 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]; This is the first large cohort study reporting that GBA mutation increase substantially the risk for development of dementia with Lewy bodies.
- 47.McKeith IG. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. J Alzheimer's Dis 2006; 9:417–423 [DOI] [PubMed] [Google Scholar]
- 48.Wong K, Sidransky E, Verma A, et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab 2004; 82:192–207 [DOI] [PubMed] [Google Scholar]
- 49.Halliday GM, Holton JL, Revesz T, Dickson DW. Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathologica 2011; 122:187–204 [DOI] [PubMed] [Google Scholar]
- 50.Hruska KS, Goker-Alpan O, Sidransky E. Gaucher disease and the synucleinopathies. J Biomed Biotechnol 2006; 2006:78549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mazzulli JR, Xu YH, Sun Y, et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011; 146:37–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yap TL, Gruschus JM, Velayati A, et al. Alpha-synuclein interacts with Glucocerebrosidase providing a molecular link between Parkinson and Gaucher diseases. J Biol Chem 2011; 286:28080–28088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Singleton AB, Farrer M, Johnson J, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science 2003; 302:841. [DOI] [PubMed] [Google Scholar]
- 54▪▪.Proukakis C, Dudzik CG, Brier T, et al. A novel alpha-synuclein missense mutation in parkinson disease. Neurology 2013; 80:1062–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports the identification of a novel SNCA mutation.
- 55▪▪.Kiely AP, Asi YT, Kara E, et al. alpha-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson's disease and multiple system atrophy? Acta Neuropathologica 2013; 125:753–769 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports the identification of a novel SNCA mutation and the associated clinical and pathological features.
- 56.Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 1997; 276:2045–2047 [DOI] [PubMed] [Google Scholar]
- 57▪▪.Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson's disease. Mov Disord 2013; 28:811–813 [DOI] [PubMed] [Google Scholar]; This study reports a novel H50Q SNCA mutation in a sporadic Parkinson's disease patient.
- 58▪▪.Lesage S, Anheim M, Letournel F, et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol 2013; 10.1002/ana.23894 [DOI] [PubMed] [Google Scholar]; The authors report a novel G51D SNCA mutation and associated clinical and pathological features, coupled with functional studies.
- 59▪▪.Kara E, Lewis P, Ling H, et al. α-Synuclein mutations cluster around a putative protein loop. Neurosci Lett 2013; 546:67–70 [DOI] [PMC free article] [PubMed] [Google Scholar]; This article discusses the consequences of SNCA mutations on the putative tetramer structure formed by α-synuclein monomers.
- 60.Xilouri M, Vogiatzi T, Vekrellis K, Stefanis L. alpha-synuclein degradation by autophagic pathways: a potential key to Parkinson's disease pathogenesis. Autophagy 2008; 4:917–919 [DOI] [PubMed] [Google Scholar]
- 61.Xilouri M, Vogiatzi T, Vekrellis K, et al. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PloS One 2009; 4:e5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cuervo AM, Stefanis L, Fredenburg R, et al. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004; 305:1292–1295 [DOI] [PubMed] [Google Scholar]
- 63▪.Tanik SA, Schultheiss CE, Volpicelli-Daley LA, et al. Lewy body-like alpha-synuclein aggregates resist degradation and impair macroautophagy. J Biol Chem 2013; 288:15194–15210 [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors show that α-synuclein aggregates impair autophagy.
- 64▪.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 2013; 368:651–662 [DOI] [PubMed] [Google Scholar]; This review article summarises the role of autophagy in the development of various diseases inclusing cancer and neurodegeneration.
- 65.Winslow AR, Rubinsztein DC. The Parkinson disease protein alpha-synuclein inhibits autophagy. Autophagy 2011; 7:429–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66▪.Ahmed I, Liang Y, Schools S, et al. Development and characterization of a new Parkinson's disease model resulting from impaired autophagy. J Neurosci 2012; 32:16503–16509 [DOI] [PMC free article] [PubMed] [Google Scholar]; This is a study on a mouse model carrying a conditional deletion in the autophagy gene atg7 that demonstrated neuropathologic features of Parkinson's disease.
- 67.Park JS, Mehta P, Cooper AA, et al. Pathogenic effects of novel mutations in the P-type ATPase ATP13A2 (PARK9) causing Kufor-Rakeb syndrome, a form of early-onset parkinsonism. Hum Mutat 2011; 32:956–964 [DOI] [PubMed] [Google Scholar]
- 68.Ramirez A, Heimbach A, Grundemann J, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 2006; 38:1184–1191 [DOI] [PubMed] [Google Scholar]
- 69.Di Fonzo A, Chien HF, Socal M, et al. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 2007; 68:1557–1562 [DOI] [PubMed] [Google Scholar]
- 70.Lees AJ, Singleton AB. Clinical heterogeneity of ATP13A2 linked disease (Kufor-Rakeb) justifies a PARK designation. Neurology 2007; 68:1553–1554 [DOI] [PubMed] [Google Scholar]
- 71.Lin CH, Tan EK, Chen ML, et al. Novel ATP13A2 variant associated with Parkinson disease in Taiwan and Singapore. Neurology 2008; 71:1727–1732 [DOI] [PubMed] [Google Scholar]
- 72▪▪.Bras J, Verloes A, Schneider SA, et al. Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Hum Mol Genet 2012; 21:2646–2650 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports the identification of ATP13A2 mutations as the cause of a lysosomal storage disorder in humans.
- 73.Wohlke A, Philipp U, Bock P, et al. A one base pair deletion in the canine ATP13A2 gene causes exon skipping and late-onset neuronal ceroid lipofuscinosis in the Tibetan terrier. PLoS Genet 2011; 7:e1002304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Farias FH, Zeng R, Johnson GS, et al. A truncating mutation in ATP13A2 is responsible for adult-onset neuronal ceroid lipofuscinosis in Tibetan terriers. Neurobiol Dis 2011; 42:468–474 [DOI] [PubMed] [Google Scholar]
- 75▪▪.Dehay B, Ramirez A, Martinez-Vicente M, et al. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci USA 2012; 109:9611–9616 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports on the impact on lysosomal function of ATP13A2 loss of function.
- 76.Gitler AD, Chesi A, Geddie ML, et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet 2009; 41:308–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77▪.Usenovic M, Tresse E, Mazzulli JR, et al. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J Neurosci 2012; 32:4240–4246 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study investigates the impact of ATP13A2 deficiency on lysosomal function and α-synuclein homeostasis.
- 78▪▪.Schultheis PJ, Fleming SM, Clippinger AK, et al. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited α-synuclein accumulation, and age-dependent sensorimotor deficits. Hum Mol Genet 2013; 22:2067–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that loss of ATP13A2 results in α-synuclein accumulation and formation of lipofuscin inclusions.
- 79.Shachar T, Lo Bianco C, Recchia A, et al. Lysosomal storage disorders and Parkinson's disease: Gaucher disease and beyond. Mov Disord 2011; 26:1593–1604 [DOI] [PubMed] [Google Scholar]
- 80.Qiao L, Hamamichi S, Caldwell KA, et al. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol Brain 2008; 1:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cullen V, Lindfors M, Ng J, et al. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol Brain 2009; 2:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sevlever D, Jiang P, Yen SH. Cathepsin D is the main lysosomal enzyme involved in the degradation of alpha-synuclein and generation of its carboxy-terminally truncated species. Biochemistry 2008; 47:9678–9687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gouider-Khouja N, Larnaout A, Amouri R, et al. Autosomal recessive parkinsonism linked to parkin gene in a Tunisian family. Clinical, genetic and pathological study. Parkinsonism Relat Disord 2003; 9:247–251 [DOI] [PubMed] [Google Scholar]
- 84.Hayashi S, Wakabayashi K, Ishikawa A, et al. An autopsy case of autosomal-recessive juvenile parkinsonism with a homozygous exon 4 deletion in the parkin gene. Mov Disord 2000; 15:884–888 [DOI] [PubMed] [Google Scholar]
- 85.Pramstaller PP, Schlossmacher MG, Jacques TS, et al. Lewy body Parkinson's disease in a large pedigree with 77 Parkin mutation carriers. Ann Neurol 2005; 58:411–422 [DOI] [PubMed] [Google Scholar]
- 86.Sasaki S, Shirata A, Yamane K, Iwata M. Parkin-positive autosomal recessive juvenile Parkinsonism with alpha-synuclein-positive inclusions. Neurology 2004; 63:678–682 [DOI] [PubMed] [Google Scholar]
- 87.Sasaki S, Shirata A, Yamane K, Iwata M. Involvement of spinal motor neurons in parkin-positive autosomal recessive juvenile parkinsonism. Neuropathology 2008; 28:74–80 [DOI] [PubMed] [Google Scholar]
- 88.van de Warrenburg BP, Lammens M, Lucking CB, et al. Clinical and pathologic abnormalities in a family with parkinsonism and parkin gene mutations. Neurology 2001; 56:555–557 [DOI] [PubMed] [Google Scholar]
- 89▪.Miyakawa S, Ogino M, Funabe S, et al. Lewy body pathology in a patient with a homozygous parkin deletion. Mov Disord 2013; 28:388–391 [DOI] [PubMed] [Google Scholar]; This is a neuropathological study of a patient carrying a homozygous parkin deletion who was found to have α-synuclein accumulation.
- 90▪.Doherty KM, Silveira-Moriyama L, Parkkinen L, et al. Parkin disease: a clinicopathologic entity? JAMA Neurol 2013; 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]; In this study, the authors argue that pathological features related to parkin mutations are distinct than other genetic and sporadic forms of Parkinson's disease.
- 91.Samaranch L, Lorenzo-Betancor O, Arbelo JM, et al. PINK1-linked parkinsonism is associated with Lewy body pathology. Brain 2010; 133:1128–1142 [DOI] [PubMed] [Google Scholar]
- 92▪▪.Stefanis L. alpha-synuclein in Parkinson's disease. Cold Spring Harbor Perspect Med 2012; 2:a009399. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is a comprehensive review on the role of SNCA in Parkinson's disease.
- 93.Sharon R, Bar-Joseph I, Frosch MP, et al. The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson's disease. Neuron 2003; 37:583–595 [DOI] [PubMed] [Google Scholar]
- 94.Bras J, Singleton A, Cookson MR, Hardy J. Emerging pathways in genetic Parkinson's disease: potential role of ceramide metabolism in Lewy body disease. FEBS J 2008; 275:5767–5773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95▪▪.Hardy J, Revesz T. The spread of neurodegenerative disease. N Engl J Med 2012; 366:2126–2128 [DOI] [PubMed] [Google Scholar]; This commentary underlines the importance of the discrimination between prion disease and Parkinson's disease, AD and FTD and suggests the use of the term ‘permissive templating’ when referring to noninfectious diseases.
- 96.Hardy J. Expression of normal sequence pathogenic proteins for neurodegenerative disease contributes to disease risk: ’permissive templating’ as a general mechanism underlying neurodegeneration. Biochem Soc Trans 2005; 33:578–581 [DOI] [PubMed] [Google Scholar]
- 97▪.Polymenidou M, Cleveland DW. Prion-like spread of protein aggregates in neurodegeneration. J Exp Med 2012; 209:889–893 [DOI] [PMC free article] [PubMed] [Google Scholar]; This article summarises the evidence supporting the transmission of missfolded proteins as the cause of several neurodegenerative diseases.
- 98.Vekrellis K, Xilouri M, Emmanouilidou E, et al. Pathological roles of alpha-synuclein in neurological disorders. Lancet Neurol 2011; 10:1015–1025 [DOI] [PubMed] [Google Scholar]
- 99▪.Russo I, Bubacco L, Greggio E. Exosomes-associated neurodegeneration and progression of Parkinson's disease. Am J Neurodegener Dis 2012; 1:217–225 [PMC free article] [PubMed] [Google Scholar]; This review article highlights the role of exosomes in the spread of Parkinson's disease.
- 100.Alvarez-Erviti L, Seow Y, Schapira AH, et al. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol Dis 2011; 42:360–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101▪▪.Cremades N, Cohen SI, Deas E, et al. Direct observation of the interconversion of normal and toxic forms of alpha-synuclein. Cell 2012; 149:1048–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports on the kinetics of α-synuclein fibril formation.
- 102.Desplats P, Lee HJ, Bae EJ, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci USA 2009; 106:13010–13015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hansen C, Angot E, Bergstrom AL, et al. alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Investig 2011; 121:715–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lee HJ, Suk JE, Lee KW, et al. Transmission of synucleinopathies in the enteric nervous system of A53T alpha-synuclein transgenic mice. Exp Neurobiol 2011; 20:181–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105▪.Luk KC, Kehm VM, Zhang B, et al. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med 2012; 209:975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study indicates that missfolded α-synuclein can be transmitted between brain regions.
- 106▪▪.Luk KC, Kehm V, Carroll J, et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012; 338:949–953 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provides supporting evidence for the contribution of the disease spread hypothesis of α-synuclein to Parkinson's disease pathogenesis.
- 107.Mougenot AL, Nicot S, Bencsik A, et al. Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol Aging 2011; 33:2225–2228 [DOI] [PubMed] [Google Scholar]
- 108.Braak H, Del Tredici K, Rub U, et al. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003; 24:197–211 [DOI] [PubMed] [Google Scholar]
- 109.Kordower JH, Chu Y, Hauser RA, et al. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat Med 2008; 14:504–506 [DOI] [PubMed] [Google Scholar]
- 110.Lebouvier T, Neunlist M, Bruley des Varannes S, et al. Colonic biopsies to assess the neuropathology of Parkinson's disease and its relationship with symptoms. PloS One 2010; 5:e12728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li JY, Englund E, Holton JL, et al. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat Med 2008; 14:501–503 [DOI] [PubMed] [Google Scholar]
- 112▪.Ahn TB, Langston JW, Aachi VR, Dickson DW. Relationship of neighboring tissue and gliosis to α-synuclein pathology in a fetal transplant for Parkinson's disease. Am J Neurodegener Dis 2012; 1:49–59 [PMC free article] [PubMed] [Google Scholar]; This study examines the relation of α-synuclein pathology to gliosis in a fetal graft in a Parkinson's disease patient.
- 113.Hortnagel K, Prokisch H, Meitinger T. An isoform of hPANK2, deficient in pantothenate kinase-associated neurodegeneration, localizes to mitochondria. Hum Mol Genet 2003; 12:321–327 [DOI] [PubMed] [Google Scholar]
- 114▪.Alfonso-Pecchio A, Garcia M, Leonardi R, Jackowski S. Compartmentalization of mammalian pantothenate kinases. PloS One 2012; 7:e49509. [DOI] [PMC free article] [PubMed] [Google Scholar]; The subcellular localisation of pantothenate kinases is studied in this article.
- 115▪▪.Schneider SA, Hardy J, Bhatia KP. Syndromes of neurodegeneration with brain iron accumulation (NBIA): an update on clinical presentations, histological and genetic underpinnings, and treatment considerations. Mov Disord 2012; 27:42–53 [DOI] [PubMed] [Google Scholar]; This article provides an up-to-date review on clinical and pathological features of NBIA syndromes.
- 116.Chinnery PF, Curtis AR, Fey C, et al. Neuroferritinopathy in a French family with late onset dominant dystonia. J Med Genet 2003; 40:e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Crompton DE, Chinnery PF, Fey C, et al. Neuroferritinopathy: a window on the role of iron in neurodegeneration. Blood cells Mol Dis 2002; 29:522–531 [DOI] [PubMed] [Google Scholar]
- 118.Ahmed Z, Josephs KA, Gonzalez J, et al. Clinical and neuropathologic features of progressive supranuclear palsy with severe pallido-nigro-luysial degeneration and axonal dystrophy. Brain 2008; 131:460–472 [DOI] [PubMed] [Google Scholar]
- 119.Baba Y, Ghetti B, Baker MC, et al. Hereditary diffuse leukoencephalopathy with spheroids: clinical, pathologic and genetic studies of a new kindred. Acta Neuropathologica 2006; 111:300–311 [DOI] [PubMed] [Google Scholar]
- 120▪.Guerreiro R, Kara E, Ber IL, et al. Genetic analysis of inherited leukodystrophies: genotype phenotype correlations in the CSF1R gene. JAMA Neurol 2013; 10.1001/jamaneurol.2013.698 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study represents the first large follow-up analysis of the CSF1R gene in a cohort of leukoencephalopathy patients.
- 121▪▪.Rademakers R, Baker M, Nicholson AM, et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet 2012; 44:200–205 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports the identification of CSF1R mutations as the cause of hereditary diffuse leukoencephalopathy with spheroids.
- 122▪▪.Nicholson AM, Baker MC, Finch NA, et al. CSF1R mutations link POLD and HDLS as a single disease entity. Neurology 2013; 80:1033–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study identified mutations in CSF1R in a POLD family indicating that HDLS and POLD are a single entity.
- 123.Sung JH, Mastri AR, Park SH. Axonal dystrophy in the gracile nucleus in children and young adults. Reappraisal of the incidence and associated diseases. J Neuropathol Exp Neurol 1981; 40:37–45 [PubMed] [Google Scholar]
- 124.Lin WL, Wszolek ZK, Dickson DW. Hereditary diffuse leukoencephalopathy with spheroids: ultrastructural and immunoelectron microscopic studies. Int J Clin Exp Pathol 2010; 3:665–674 [PMC free article] [PubMed] [Google Scholar]
- 125.Freeman SH, Hyman BT, Sims KB, et al. Adult onset leukodystrophy with neuroaxonal spheroids: clinical, neuroimaging and neuropathologic observations. Brain Pathol 2009; 19:39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Beck G, Sugiura Y, Shinzawa K, et al. Neuroaxonal dystrophy in calcium-independent phospholipase A2beta deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J Neurosci 2011; 31:11411–11420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127▪▪.Brunetti D, Dusi S, Morbin M, et al. Pantothenate kinase-associated neurodegeneration: altered mitochondria membrane potential and defective respiration in Pank2 knock-out mouse model. Hum Mol Genet 2012; 21:5294–5305 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study represents the first comprehensive mitochondrial assesment in a mouse model of PANK2, and it shows that mitochondrial alterations can occur without exhibiting neurological deficits.
- 128.Kurz T, Terman A, Gustafsson B, Brunk UT. Lysosomes in iron metabolism, ageing and apoptosis. Histochem Cell Biol 2008; 129:389–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129▪▪.Arduino DM, Raquel Esteves A, Cortes L, et al. Mitochondrial metabolism in Parkinson's disease impairs quality control autophagy by hampering microtubule-dependent traffic. Hum Mol Genet 2012; 21:4680–4702 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study suggests that mitochondrial dysfunction can impair autophagy in Parkinson's disease through alteration of microtubule trafficking.
- 130▪.Arduino DM, Esteves AR, Cardoso SM. Mitochondria drive autophagy pathology via microtubule disassembly: A new hypothesis for Parkinson disease. Autophagy 2012; 9:112–114 [DOI] [PMC free article] [PubMed] [Google Scholar]; This is a commentary on the recent identification of microtubule trafficking defects underlying autophagy impairment in Parkinson's disease.
- 131.Macaskill AF, Rinholm JE, Twelvetrees AE, et al. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 2009; 61:541–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fransson S, Ruusala A, Aspenstrom P. The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem Biophys Res Commun 2006; 344:500–510 [DOI] [PubMed] [Google Scholar]
- 133.Wang X, Winter D, Ashrafi G, et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011; 147:893–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kane LA, Youle RJ. PINK1 and Parkin flag Miro to direct mitochondrial traffic. Cell 2011; 147:721–723 [DOI] [PubMed] [Google Scholar]
- 135.Weihofen A, Thomas KJ, Ostaszewski BL, et al. Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry 2009; 48:2045–2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136▪.Liu S, Sawada T, Lee S, et al. Parkinson's disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet 2012; 8:e1002537. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study showed that PINK1 regulates mitochondrial trafficking through Miro.
- 137▪▪.Ding WX, Guo F, Ni HM, et al. Parkin and mitofusins reciprocally regulate mitophagy and mitochondrial spheroid formation. J Biol Chem 2012; 287:42379–42388 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports that loss of parkin induces the formation of neuroaxonal spheroids.
- 138.Nijssen PC, Brusse E, Leyten AC, et al. Autosomal dominant adult neuronal ceroid lipofuscinosis: parkinsonism due to both striatal and nigral dysfunction. Mov Disord 2002; 17:482–487 [DOI] [PubMed] [Google Scholar]
- 139.Tayebi N, Walker J, Stubblefield B, et al. Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol Genet Metab 2003; 79:104–109 [DOI] [PubMed] [Google Scholar]
- 140.Goker-Alpan O, Schiffmann R, LaMarca ME, et al. Parkinsonism among Gaucher disease carriers. J Med Genet 2004; 41:937–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tan EK, Ho P, Tan L, et al. PLA2G6 mutations and Parkinson's disease. Ann Neurol 2010; 67:148. [DOI] [PubMed] [Google Scholar]
- 142▪.Tofaris GK. Lysosome-dependent pathways as a unifying theme in Parkinson's disease. Mov Disord 2012; 27:1364–1369 [DOI] [PubMed] [Google Scholar]; This review summarises the role of lysosomal pathways in the pathogenesis of Parkinson's disease.
- 143.Morgan NV, Westaway SK, Morton JE, et al. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet 2006; 38:752–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wild P, Dikic I. Mitochondria get a Parkin’ ticket. Nat Cell Biol 2010; 12:104–106 [DOI] [PubMed] [Google Scholar]
- 145.Geisler S, Holmstrom KM, Treis A, et al. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 2010; 6:871–878 [DOI] [PubMed] [Google Scholar]
- 146.Geisler S, Holmstrom KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010; 12:119–131 [DOI] [PubMed] [Google Scholar]
- 147.Poole AC, Thomas RE, Andrews LA, et al. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci USA 2008; 105:1638–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008; 183:795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Vives-Bauza C, Zhou C, Huang Y, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA 2010; 107:378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Vives-Bauza C, Przedborski S. PINK1 points Parkin to mitochondria. Autophagy 2010; 6:674–675 [DOI] [PubMed] [Google Scholar]
- 151.Vives-Bauza C, de Vries RL, Tocilescu M, Przedborski S. PINK1/Parkin direct mitochondria to autophagy. Autophagy 2010; 6:315–316 [DOI] [PubMed] [Google Scholar]
- 152▪▪.Haskin J, Szargel R, Shani V, et al. AF-6 is a positive modulator of the PINK1/parkin pathway and is deficient in Parkinson's disease. Hum Mol Genet 2013; 22:2083–2096 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study identifies AF-6 as a recruiter of parkin to mitochondria with a possible role in Parkinson's disease.
- 153▪.Okatsu K, Oka T, Iguchi M, et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat Commun 2012; 3:1016. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that autophosphorylation of PINK1 preceeds the recruitment of parkin to mitochondria.
- 154▪.Matsui H, Gavinio R, Asano T, et al. PINK1 and Parkin complementarily protect dopaminergic neurons in vertebrates. Hum Mol Genet 2013; 22:2423–2434 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study emphasises the synergistic role or parkin and PINK1 in mitochondrial physiology.
- 155▪.Dehay B, Martinez-Vicente M, Caldwell GA, et al. Lysosomal impairment in Parkinson's disease. Mov Disord 2013; 28:725–732 [DOI] [PMC free article] [PubMed] [Google Scholar]; This review artivle summarises the involvement of lysosomal pathways in the pathogenesis of Parkinson's disease.
- 156▪.Gan-Or Z, Ozelius LJ, Bar-Shira A, et al. The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology 2013; 80:1606–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provides preliminary evidence that a variant in SMPD1 increases the risk for the development of Parkinson's disease in the Ashkenazi Jews.
- 157▪▪.Imaizumi Y, Okada Y, Akamatsu W, et al. Mitochondrial dysfunction associated with increased oxidative stress and alpha-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol Brain 2012; 5:35. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is a study on neurons derived from iPS cells from carriers of parkin mutations reproducing the neuropathological findings in these patients.
- 158▪.Sarraf SA, Raman M, Guarani-Pereira V, et al. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013; 496:372–376 [DOI] [PMC free article] [PubMed] [Google Scholar]; In this study, the authors carried out a high throughput study in order to identify the ubiquitination targets of parkin.
- 159.Malik I, Turk J, Mancuso DJ, et al. Disrupted membrane homeostasis and accumulation of ubiquitinated proteins in a mouse model of infantile neuroaxonal dystrophy caused by PLA2G6 mutations. Am J Pathol 2008; 172:406–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160▪▪.Grunewald A, Arns B, Seibler P, et al. ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol Aging 2012; 33:1843.e1841–e1847 [DOI] [PubMed] [Google Scholar]; This is the first functional study on fibroblasts from patients with ATP13A2 mutations and shows that mitochondrial function is impaired in carriers of mutations in a lysosomal protein.
- 161.Zimprich A, Benet-Pages A, Struhal W, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet 2011; 89:168–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Vilarino-Guell C, Wider C, Ross OA, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet 2011; 89:162–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163▪.Bi F, Li F, Huang C, Zhou H. Pathogenic mutation in VPS35 impairs its protection against MPP(+) cytotoxicity. Int J Biol Sci 2013; 9:149–155 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that VPS35 mutations can impair mitochondrial function.
- 164.Wider C, Skipper L, Solida A, et al. Autosomal dominant dopa-responsive parkinsonism in a multigenerational Swiss family. Parkinsonism Relat Disord 2008; 14:465–470 [DOI] [PubMed] [Google Scholar]
- 165▪.Gomez-Suaga P, Luzon-Toro B, Churamani D, et al. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum Mol Genet 2012; 21:511–525 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that LRRK2 could have a role in autophagy and indicates a mechanism underlying this relation.
- 166▪▪.Orenstein SJ, Kuo SH, Tasset I, et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci 2013; 16:394–406 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports that LRRK2 mutations can inhibit autophagy potentially affecting degradation of α-synuclein.
- 167▪.Yue Z, Yang XW. Dangerous duet: LRRK2 and alpha-synuclein jam at CMA. Nat Neurosci 2013; 16:375–377 [DOI] [PubMed] [Google Scholar]; Commentary on the findings of Orenstein et al. (2013).
- 168▪▪.MacLeod DA, Rhinn H, Kuwahara T, et al. RAB7L1 Interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson's disease risk. Neuron 2013; 77:425–439 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study functionally links 2 Parkinson's disease genes (LRRK2, VPS35) with RAB7L1 in which a haplotype increasing the risk for Parkinson's disease was identified through GWA studies.
- 169.Mortiboys H, Johansen KK, Aasly JO, Bandmann O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology 2010; 75:2017–2020 [DOI] [PubMed] [Google Scholar]
- 170▪.Hindle S, Afsari F, Stark M, et al. Dopaminergic expression of the Parkinsonian gene LRRK2-G2019S leads to nonautonomous visual neurodegeneration, accelerated by increased neural demands for energy. Hum Mol Genet 2013; 22:2129–2140 [DOI] [PMC free article] [PubMed] [Google Scholar]; This is a study of LRRK2 in the Drosophila visual system.
- 171.Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004; 44:601–607 [DOI] [PubMed] [Google Scholar]
- 172▪▪.Duran R, Mencacci NE, Angeli AV, et al. The glucocerobrosidase E326K variant predisposes to Parkinson's disease, but does not cause Gaucher's disease. Mov Disord 2012; 28:232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that the E362K GBA variant causes Parkinson's disease but never Gaucher's disease suggesting a possible disassociation between the pathogenetic mechanisms of the two diseases.
- 173▪.Song W, Wang F, Savini M, et al. TFEB regulates lysosomal proteostasis. Hum Mol Genet 2013; 22:1994–2009 [DOI] [PubMed] [Google Scholar]; This study reports TFEB as a regulator of GBA within the lysosomes.
- 174▪.Dermentzaki G, Dimitriou E, Xilouri M, et al. Loss of beta-glucocerebrosidase activity does not affect alpha-synuclein levels or lysosomal function in neuronal cells. PloS One 2013; 8:e60674. [DOI] [PMC free article] [PubMed] [Google Scholar]; In this study, the authors show that loss of GBA function does not affect α-synuclein homeostasis, and argue that the link between GBA dysfunction and α-synuclein accumulation in Parkinson's disease probably is not direct.
- 175▪.Protter D, Lang C, Cooper AA. alphaSynuclein and mitochondrial dysfunction: a pathogenic partnership in Parkinson's disease? Parkinson's Dis 2012; 2012:829207. [DOI] [PMC free article] [PubMed] [Google Scholar]; This review article explores the relationship between mitochondria and α-synuclein in Parkinson's disease.
- 176▪.Reeve AK, Park TK, Jaros E, et al. Relationship between mitochondria and alpha-synuclein: a study of single substantia nigra neurons. Arch Neurol 2012; 69:385–393 [DOI] [PubMed] [Google Scholar]; This is a study of mitochondria in substantia nigra neurons with and without α-synuclein pathology.
- 177▪.Cali T, Ottolini D, Negro A, Brini M. alpha-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J Biol Chem 2012; 287:17914–17929 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes the effect of SNCA α-synuclein on mitochondrial function.
- 178.Choubey V, Safiulina D, Vaarmann A, et al. Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J Biol Chem 2011; 286:10814–10824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179▪▪.Schapira AH. Mitochondrial diseases. Lancet 2012; 379:1825–1834 [DOI] [PubMed] [Google Scholar]; This article reviews the role of mitochondrial dysfunction in neurodegenerative diseases.
- 180▪.Xie W, Chung KK. Alpha-synuclein impairs normal dynamics of mitochondria in cell and animal models of Parkinson's disease. J Neurochem 2012; 122:404–414 [DOI] [PubMed] [Google Scholar]; This study reports on the effect of α-synuclein on mitochondria.
- 181▪.Zhu M, Li W, Lu C. Role of alpha-synuclein protein levels in mitochondrial morphology and cell survival in cell lines. PloS One 2012; 7:e36377. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study investigates the effect of α-synuclein on mitochondria.
- 182.Goker-Alpan O, Stubblefield BK, Giasson BI, Sidransky E. Glucocerebrosidase is present in alpha-synuclein inclusions in Lewy body disorders. Acta Neuropathologica 2010; 120:641–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183▪.Wakabayashi K, Tanji K, Odagiri S, et al. The Lewy body in Parkinson's disease and related neurodegenerative disorders. Mol Neurobiol 2012; 47:495–508 [DOI] [PubMed] [Google Scholar]; This is a review on the composition and formation of Lewy bodies and their role in disease.
- 184.Getty AL, Pearce DA. Interactions of the proteins of neuronal ceroid lipofuscinosis: clues to function. Cell Mol Life Sci 2011; 68:453–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Deas E, Wood NW, Plun-Favreau H. Mitophagy and Parkinson's disease: the PINK1-parkin link. Bioch Biophys Acta 2011; 1813:623–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Settembre C, Fraldi A, Jahreiss L, et al. A block of autophagy in lysosomal storage disorders. Hum Mol Genet 2008; 17:119–129 [DOI] [PubMed] [Google Scholar]
- 187▪.Sharma M, Ioannidis JP, Aasly JO, et al. A multicentre clinico-genetic analysis of the VPS35 gene in Parkinson disease indicates reduced penetrance for disease-associated variants. J Med Genet 2012; 49:721–726 [DOI] [PMC free article] [PubMed] [Google Scholar]; This article reports on a large cohort study of VPS35.
- 188▪.Sheerin UM, Charlesworth G, Bras J, et al. Screening for VPS35 mutations in Parkinson's disease. Neurobiol Aging 2012; 33:838. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is one of the first follow up studies reporting a family with a VPS35 mutation after the identification of VPS35 mutations as a cause of Parkinson's disease.
- 189▪.Lesage S, Condroyer C, Klebe S, et al. Identification of VPS35 mutations replicated in French families with Parkinson disease. Neurology 2012; 78:1449–1450 [DOI] [PubMed] [Google Scholar]; This study reports VPS35 mutations in three familes with Parkinson's disease.
- 190▪.Nuytemans K, Bademci G, Inchausti V, et al. Whole exome sequencing of rare variants in EIF4G1 and VPS35 in Parkinson disease. Neurology 2013; 80:982–989 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study identified VPS35 mutations in three patients with Parkinson's disease.
- 191▪.Chuang RS, Gitler AD. Parallel PARKing: Parkinson's genes function in common pathway. Neuron 2013; 77:377–379 [DOI] [PubMed] [Google Scholar]; Commentary summarising, critiquing and putting in context the findings of McLeod et al. (2013).
- 192▪.Deng H, Gao K, Jankovic J. The VPS35 gene and Parkinson's disease. Mov Disord 2013; 28:569–575 [DOI] [PubMed] [Google Scholar]; This review article summarises clinical features associated to VPS35 mutations and functional knowledge on the VPS35 protein.
- 193▪.Hamasaki M, Furuta N, Matsuda A, et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013; 495:389–393 [DOI] [PubMed] [Google Scholar]; This study showed that ER-mitochondrial interaction is necessary for the formation of autophagocytic vesicles.
- 194.Wray S, Lewis PA. A tangled web – tau and sporadic Parkinson's disease. Front Psychiatry 2010; 1:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Devine MJ, Lewis PA. Emerging pathways in genetic Parkinson's disease: tangles, Lewy bodies and LRRK2. FEBS J 2008; 275:5748–5757 [DOI] [PubMed] [Google Scholar]
- 196▪.Gan-Or Z, Bar-Shira A, Mirelman A, et al. The age at motor symptoms onset in LRRK2-associated Parkinson's disease is affected by a variation in the MAPT locus: a possible interaction. J Mol Neurosci 2012; 46:541–544 [DOI] [PubMed] [Google Scholar]; This study suggests an interaction between LRRK2 and MAPT variants influencing the age at onset of LRRK2 mutation carriers.
- 197▪.Ling H, Kara E, Bandopadhyay R, et al. TDP-43 pathology in a patient carrying G2019S LRRK2 mutation and a novel p.Q124E MAPT. Neurobiol Aging 2013; 10.1016/j.neurobiolaging.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports the identification of a novel MAPT variant in a patient with a G2019S LRRK2 mutation and pathology without Lewy bodies suggesting a possible disease modifying role for this variant.
- 198.Alim MA, Ma QL, Takeda K, et al. Demonstration of a role for alpha-synuclein as a functional microtubule-associated protein. J Alzheimer's Dis 2004; 6:435–442 [DOI] [PubMed] [Google Scholar]
- 199.Alim MA, Hossain MS, Arima K, et al. Tubulin seeds alpha-synuclein fibril formation. J Biol Chem 2002; 277:2112–2117 [DOI] [PubMed] [Google Scholar]
- 200.Zhou RM, Huang YX, Li XL, et al. Molecular interaction of alpha-synuclein with tubulin influences on the polymerization of microtubule in vitro and structure of microtubule in cells. Mol Biol Reports 2010; 37:3183–3192 [DOI] [PubMed] [Google Scholar]
- 201▪.Kara E, Ling H, Pittman AM, et al. The MAPT p.A152T variant is a risk factor associated with tauopathies with atypical clinical and neuropathological features. Neurobiol Aging 2012; 33:2231.e7–2231.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study identified the rare MAPT p.A152T variant in patients with parkinsonism and tau pathology.
- 202▪.Coppola G, Chinnathambi S, Lee JJ, et al. Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer's diseases. Hum Mol Genet 2012; 21:3500–3512 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study showed that the rare MAPT p.A152T variant increases the risk for the development of multiple neurodegenerative diseases.
- 203▪▪.Keller MF, Saad M, Bras J, et al. Using genome-wide complex trait analysis to quantify ’missing heritability’ in Parkinson's disease. Hum Mol Genet 2012; 21:4996–5009 [DOI] [PMC free article] [PubMed] [Google Scholar]; In this study, it is shown that there is probably a large number of novel genes mutated in Parkinson's disease awaiting discovery.