

Abstract
KCNE1 is a single-transmembrane protein of the KCNE family that modulates the function of voltage-gated potassium channels, including KCNQ1. Hereditary mutations in KCNE1 have been linked to diseases such as long QT syndrome (LQTS), atrial fibrillation, sudden infant death syndrome, and deafness. The transmembrane domain (TMD) of KCNE1 plays a key role in mediating the physical association with KCNQ1 and in subsequent modulation of channel gating kinetics and conductance. However, the mechanisms associated with these roles for the TMD remain poorly understood, highlighting a need for experimental structural studies. A previous solution NMR study of KCNE1 in LMPG micelles revealed a curved transmembrane domain, a structural feature proposed to be critical to KCNE1 function. However, this curvature potentially reflects an artifact of working in detergent micelles. Double electron electron resonance (DEER) measurements were conducted on KCNE1 in LMPG micelles, POPC/POPG proteoliposomes, and POPC/POPG lipodisq nanoparticles to directly compare the structure of the TMD in a variety of different membrane environments. Experimentally derived DEER distances coupled with simulated annealing molecular dynamic simulations were used to probe the bilayer structure of the TMD of KCNE1. The results indicate that the structure is helical in proteoliposomes and is slightly curved, which is consistent with the previously determined solution NMR structure in micelles. The evident resilience of the curvature in the KCNE1 TMD leads us to hypothesize that the curvature is likely to be maintained upon binding of the protein to the KCNQ1 channel.
KCNE1 is a 129 amino acid transmembrane protein that modulates the function of the voltage-gated potassium ion channel (Kv) KCNQ1.1 Kv channels are critical for the function of cardiac, nervous, and auditory systems and represent promising targets for various therapeutic agents.2−4 In the human heart, KCNQ1 associates with KCNE1 to generate the slow delayed rectifier current (IKs), characterized by its slow activation and deactivation kinetics,5 which is essential for the repolarization phase of the cardiac action potential. In the absence of KCNE1, the KCNQ1 channel activates rapidly but with limited conductance.6 Coexpression and association of KCNE1 with KCNQ1 delays the opening of the channel, shifts V1/2 to more positive potentials, removes channel inactivation, and results in a roughly 5-fold increase in open-state conductance.6−8 The structure of the transmembrane domain (TMD) of KCNE1 is important because its interaction with the KCNQ1 channel regulates activity.9−11 Mutations in both KCNE1 and KCNQ1 are known to result in increased susceptibility to diseases such as congenital deafness, congenital long QT syndrome, ventricular tachyarrhythmia, syncope, and sudden cardiac death.12−14
The structure of KCNE1 has been determined using solution NMR spectroscopy under conditions in which the protein was solubilized in LMPG micelles.1 In conjunction with restraints derived from mutagenesis-based biochemical and structure–function studies, competing working models for key aspects of how KCNE1 modulates channel function have been developed.1,15−17 A point of controversy between these models is whether the channel-associated KCNE1 TMD is curved (NMR micelle structure), whether the curvature is absent in channel-associated KCNE1, or whether the TMD is flexible and adopts multiple conformational states while bound to KCNQ1. Here, we examine whether the curvature seen for the KCNE1 TMD under micellar conditions is an intrinsic property of this domain or whether the TMD adapts its structure to the varying properties of its local environment (micelle vs lipid bilayer). A POPC/POPG (3:1) lipid bilayer was used to mimic phospholipids typically found in mammalian membranes.18−21 Functional studies of KCNE1 with KCNQ1 in POPC/POPG lipid bilayers under similar sample conditions have shown channel activity.18 More specifically, we report extensive site-directed spin labeling (SDSL) double electron electron resonance (DEER) distance data for KCNE1 in POPC/POPG lipid vesicles to probe the structure of the TMD under native-like conditions when compared to a micelle.
Determining the structure of membrane proteins is very challenging. Traditional biophysical techniques such as X-ray crystallography and solution NMR are difficult or impossible to use when studying the structural or dynamic properties of hydrophobic membrane proteins directly in a lipid bilayer. DEER spectroscopy is one of the few biophysical techniques that can be used to probe the structural properties of membrane proteins in both micelles and lipid bilayers.20
Materials and Methods
Site-Directed Mutagenesis
His-tag expression vectors (pET-16b) containing the wild-type and a cysteine-less mutant of KCNE112 were transformed into XL1-Blue Escherichia coli cells (Stratagene). Plasmid extracts from these cells were obtained using the QIAprep spin miniprep kit (Qiagen). Site-directed cysteine mutants were introduced into the cysteine-less KCNE1 gene using the QuikChange Lightning site-directed mutagenesis kit (Stratagene). The KCNE1 mutations were confirmed by DNA sequencing from XL10-Gold E. coli (Stratagene) transformants using the T7 primer (Integrated DNA Technologies). Successfully mutated vectors were transformed into BL21-(DE3) CodonPlus-RP E. coli cells (Stratagene) for protein overexpression. Quadruple Cys mutants L45C–M49C/R67C–L71C, L45C–M49C/I66C–K70C, Y46C–V50C/R67C–L71C, Y46C–V50C/I66C–K70C, and V47C–L51C/S64C–S68C were chosen for bifunctional double spin labeling, and double Cys mutants L45C/L71C, L45C/K70C, V47C/I66C, V47C/K69C, M49C/R67C, V50C/S68C, L51C/Y65C, G52C/S64C, and G55C/I66C were chosen for MTSL double spin labeling. The mutants were selected to span most of the transmembrane domain of KCNE1.
Overexpression and Purification
Overexpression and purification of KCNE1 mutants from E. coli BL21 cells were carried out using a previously described protocol.18 Cells were cultured in M9 minimal medium with 50 μg/mL ampicillin. The cell culture was incubated at 37 °C and 240 rpm until the OD600 reached 0.8, at which point protein expression was induced using 1 mM IPTG (isopropyl β-d-1-thiogalactopyranoside), followed by continued rotary shaking at 37 °C for 16 h. KCNE1 was then purified from inclusion bodies according to a previous method,12 with final elution of pure protein into 0.05% LMPG or 0.2% SDS detergent (buffer: 250 mM IMD, 200 mM NaCl, 20 mM Tris, pH 7.8). Protein samples were concentrated by using a Microcon YM-3 (molecular weight cutoff, 3000) centrifugal cartridge (Amicon). Protein concentration was determined from the OD280 using an extinction coefficient of 1.2 mg/mL protein per OD280 on a NanoDrop 200c (Thermo Scientific). The protein purity was confirmed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).
Spin Labeling and Reconstitution into Proteoliposomes
The bifunctional spin label (BSL) (3,4-bis(methanethiosulfonylmethyl)-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-1-yloxy radical) (HO-1944) and 1-oxyl-2,2,5,5-tetramethylpyrroline-3-methylmethanethiosulfonate (MTSL) spin label were obtained from Toronto Research Chemicals Inc. (Toronto, Canada). The spin labels were dissolved in methanol to a concentration of 250 mM and added directly to the concentrated protein in elution buffer at a 10:1 spin label/protein molar ratio and allowed to react for 24 h with gentle shaking at room temperature in the dark to complete labeling. Excess/unreacted free spin labels were removed by extensive dialysis or rebinding of the protein to Ni-NTA resin.19,20 Dialysis was carried out at room temperature in regenerated cellulose dialysis tubing (Fisherbrand MW cutoff 3.5 kDa) against 1 L of elution buffer (100 mM NaH2Po4, pH 7.8) without reducing agent. The spin labeling efficiency was determined by comparing the protein concentration (determined from A280) with the spin concentration obtained from CW EPR spectroscopy. The protein concentration for all KCNE1 samples was ∼75 μM, and the spin labeling efficiency for all samples was ∼75%.
The reconstitution of spin-labeled protein into POPC/POPG (3:1) vesicles was carried out via dialysis methods following a similar protocol in the literature.19,20 The concentrated spin-labeled KCNE1 protein was mixed with a stock lipid mixture (400 mM SDS, 75 mM POPC, 25 mM POPG, 0.1 mM EDTA, 100 mM IMD, pH 6.5). The lipid mixture had pre-equilibrated to clear mixed micelles via extensive freeze thaw cycles. The final protein/lipid molar ratio was set to 1:400. The KCNE1–lipid mixture was then subjected to extensive dialysis to remove all SDS present, during which KCNE1/POPC/POPG vesicles spontaneously formed. The 4 L of dialysis buffer (10 mM imidazole, 0.1 mM EDTA at pH 6.5) was changed twice daily. The completion of SDS removal was determined when the KCNE1–lipid solution became cloudy and the surface tension of the dialysate indicated complete removal of detergent. The KCNE1–lipid vesicles solution was then extruded using a 100 nm filter to generate unilamellar vesicles.
Reconstitution into Lipodisq Nanoparticles
Lipodisq nanoparticles (prehydrolyzed styrene-maleic anhydride copolymer 3:1 ratio) were obtained from Malvern Cosmeceutics Ltd. (Worcester, United Kingdom). The protein–lipid complex was incorporated into SMA-lipodisq nanoparticles following published protocols.20,22,23 A 500 μL aliquot of proteoliposome-reconstituted protein sample (∼30 mM POPC/POPG lipid) was added with the same amount (500 μL) of 2.5% of lipodisq solution prepared in the same dialysis buffer (10 mM imidazole, 0.1 mM EDTA at pH 6.5) dropwise over 3–4 min. The protein–lipodisq solution was allowed to equilibrate overnight at 4 °C. The resulting solution was centrifuged at 40 000g for 30 min to remove nonsolubilized protein. The size and homogeneity of the final complex was confirmed by dynamic light scattering (DLS) spectroscopy.
EPR Spectroscopic Measurements
EPR experiments were conducted at the Ohio Advanced EPR Laboratory. CW-EPR spectra were collected at X-band on a Bruker EMX CW-EPR spectrometer using an ER041xG microwave bridge and ER4119-HS cavity coupled with a BVT 3000 nitrogen gas temperature controller. Each spin-labeled CW-EPR spectrum was acquired with 42 s field scans with a central field of 3315 G and sweep width of 100 G, modulation frequency of 100 kHz, modulation amplitude of 1 G, and microwave power of 10 mW at 295 K.
Four-pulse DEER experiments were performed using a Bruker ELEXSYS E580 spectrometer equipped with a SuperQ-FT pulse Q-band system with a 10 W amplifier and EN5107D2 resonator. All DEER samples were prepared at a spin concentration of 100–120 μM. Deuterated glycerol (30%; w/w) was used as a cryoprotectant. The sample was loaded into a 1.1 mm inner diameter quartz capillary (Wilmad LabGlass, Buena, NJ) and mounted into the sample holder (plastic rod) inserted into the resonator. DEER data were collected using the standard four-pulse sequence24 [(π/2)ν1 – τ1 – (π)ν1 – t – (π)ν2 – (τ1 + τ2 – t) – (π)ν1 – τ2 – echo] at Q-band with a probe pulse width of 10/20 ns, pump pulse width of 24 ns, 80 MHz of frequency difference between probe and pump pulse, shot repetition time determined by spin–lattice relaxation rate (T1), 100 echoes/point, and 2-step phase cycling at 80 K collected out to ∼2.0 μs for overnight data acquisition time (12 h).25 DEER data were analyzed using DEER Analysis 2011.26 The distance distributions, P(r), were obtained by Tikhonov regularization27 in the distance domain, incorporating the constraint P(r) > 0. A homogeneous three-dimensional model for micelle samples and a homogeneous two-dimensional model for proteoliposomes and lipodisq nanoparticles samples were used for background correction. The regularization parameter in the L curve was optimized by examining the fit of the time domain.
Structure Refinement of the KCNE1 TMD Using DEER Distance Restraints
The structure refinement of the TMD of KCNE1 was carried out using an Xplor-NIH (version 2.33) simulated annealing protocol28,29 in a similar manner to that described previously.30−32 Positions 45, 47, 49, 50, 51, 52, 55, 64, 65, 66, 67, 68, 69, 70, and 71 along the KCNE1 TMD sequence were mutated to Cys, and MTSL side chains were attached with the Xplor-NIH addAtoms.py script. A simulated annealing routine was performed starting with standard α-helical dihedral angles (Φ = −57.0°, ψ = −47.0°, and ω = 180.0°) for the backbone of the protein and standard MTSL dihedral angles (γ1 = −60°, γ2 = −60°) without any experimental distance restraints to allow other side chain atoms to find energetically reasonable conformations. The result of this procedure was the starting structure for the simulation that fixed the spin label side chain and generated an ideal straight α-helix. Experimental DEER data for nine interlabel distances (45/71, 45/70, 47/66, 47/69, 49/67, 50/68, 51/65, 52/64, and 55/66) were used to define restraints for an Xplor-NIH simulated annealing protocol. The allowable ranges used for interlabel distances were established through a series of preliminary simulated annealing molecular dynamics calculations in which these ranges were varied.32 One hundred structures were generated using the Xplor-NIH simulated annealing routine with the Xplor-NIH anneal_nordc.py script. The simulated annealing procedure used 3500 K as the high temperature and 25 K as the cooling temperature, with temperature steps of 12.5 K. The first 10 structures were kept with energies <47.02 kcal/mol. Further analysis and visualization were done using VMD-Xplor software.33 The final structures were generated by replacing MTSL spin-labeled side chains with native side chains using Visual Molecular Dynamics (VMD) software34 similar to a previously reported method.32 Further details of the simulated annealing procedure and outputs are given in the Supporting Information (Figures S3 and S4 and Table S1). The newly determined lowest energy structure of KCNE1 TMD was further used to validate the back-calculated dual-BSL distances with the dual-BSL-based experimental DEER distances. The dual-BSLs were attached to the newly determined structure with the Charmm force field35−37 using the Quick MD Simulator protocol under CHARMM-GUI.38 The resulting structure was solvated in a water box and neutralized with KCl and further equilibrated and minimized following the instructions provided in the Quick MD Simulator protocol in CHARMM-GUI. The dual-BSL distances were calculated using VMD software and compared with experimental DEER BSL distances (see Supporting Information, Table S2).
Results
The full-length KCNE1 protein was investigated using nitroxide-based SDSL, EPR, DEER spectroscopy, and simulated annealing molecular dynamics simulations. Figure 1 shows the locations of the spin labels introduced into the TMD of KCNE1 mapped onto the solution NMR structure of the TMD in LMPG micelles (lowest energy model).1 DEER measurements were performed using two kinds of spin labels: bifunctional spin labels (BSLs) and the more conventional (monofunctional) MTSL spin labels. Bifunctional spin labels are rigid and thus very useful for obtaining tighter DEER distance distributions when compared to that for traditional MTSL.20,39 BSLs can also monitor protein backbone motion with minimal complications from the internal flexibility of the side chain.39
Figure 1.
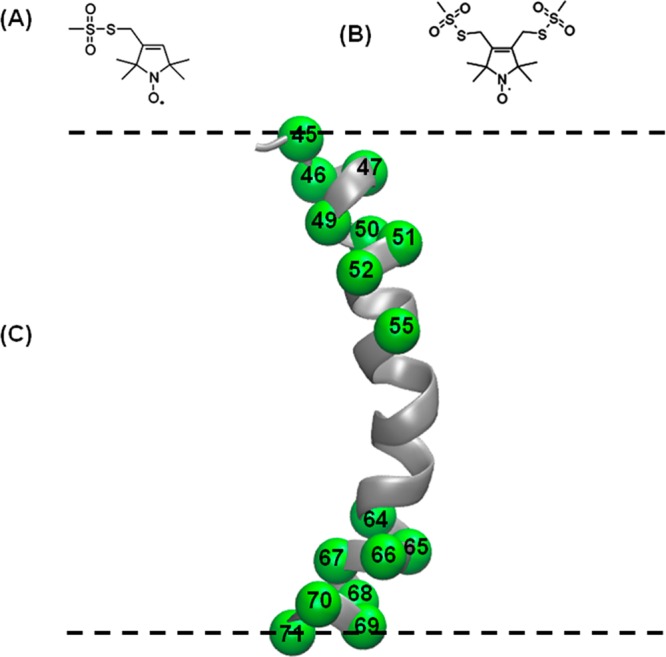
Schematic representation of spin-labeling probes and sites. (A) MTSL, (B) BSL, and (C) ribbon model of transmembrane domain of KCNE1 (PDB ID: 2k21) highlighting representative sites used in this study with spheres at their α-carbons. All spin-labeling sites are located inside the membrane. The dashed lines represent the lipid bilayer interfaces. Spin-labeling sites 45 and 71 are at the termini of the transmembrane domain that spans the membrane bilayers.
Four-Pulse DEER Distance Measurements Using Bifunctional Spin Labels (BSLs)
DEER spectroscopy coupled with SDSL is a very powerful approach for measuring long-range distances of 18–80 Å.20,24,40−42 KCNE1 was labeled with BSLs attached near the termini of the KCNE1 TMD. BSLs can be introduced by a facile cross-linking reaction of a bifunctional methanethiosulfonate reagent with pairs of cysteine residues at i and i + 3 or i and i + 4 in an α-helix and at i and i + 1 or i + 2 in a β-strand.20,39
The pairs of bifunctional spin-labeling sites include 45–49/67–71, 45–49/66–70, 46–50/67–71, 46–50/66–70, and 47–51/64–68. These sites were chosen to include the full transmembrane domain of KCNE1 in distance ranges reasonable for conducting DEER measurements. Each double spin-labeled form of KCNE1 was reconstituted into three different environments: LMPG micelles, POPC/POPG proteoliposomes, and POPC/POPG lipodisq nanoparticles. KCNE1 does not exhibit any tendency to form oligomers.
Figure 2 shows representative four-pulse DEER data from dual BSL-labeled KCNE1 (involving sites 46–50/67–71) in all three model membrane media. Additional DEER spectra and the corresponding data analysis for other double BSL-labeled samples are given in the Supporting Information (Figure S1). All distances were derived from the maximum peak intensity in the distance distribution plots. The derived distances for all double-labeled samples are summarized in Table 1. Interestingly, the distance distribution and signal-to-noise ratio (S/N) were improved for KCNE1 incorporated into lipodisq nanoparticle samples when compared to that with proteoliposomes.
Figure 2.

Four-pulse Q-band DEER data of KCNE1 mutants (Tyr46–Val50/Arg67–Leu71) bearing two BSLs. Background-subtracted dipolar evolutions of the indicated mutants (left) and their corresponding distance probability distributions from Tikhonov regularization are shown (right) for conditions of (A) 1% LMPG micelles, (B) proteoliposomes (POPC/POPG = 3:1), and (C) lipodisq nanoparticles.
Table 1. Most Probable Intramolecular Distances for KCNE1 Dual-Labeled Derivatives from Four-Pulse Q-Band DEER Experiments in Three Different Membrane Environments (Micelles, Liposomes, and Lipodisqs)a.
| distance (Å) |
|||
|---|---|---|---|
| KCNE1 double mutants | micelles | liposomes | lipodisqs |
| 45–49/67–71 | 32 | 31 | 32 |
| 45–49/66–70 | 32 | 31 | 32 |
| 46–50/67–71 | 32 | 32 | 32 |
| 46–50/66–70 | 32 | 32 | 33 |
| 47–51/64–68 | 30 | 29 | 30 |
| 45/71 | 35 | 36 | 35 |
| 45/70 | 34 | 36 | 36 |
| 47/69 | 31 | 32 | 31 |
| 47/66 | 33 | 31 | 33 |
| 49/67 | 28 | 27 | 27 |
| 50/68 | 30 | 30 | 29 |
| 51/65 | 23 | 22 | 23 |
| 52/64 | 25 | 24 | 25 |
| 55/66 | 22 | 21 | 21 |
The uncertainty in these distances is ±2 Å for BSL and ±2–4 Å for MTSL spin-labeled samples.
Figure 3 shows the overlay of the DEER distance distribution data for each BSL sample in micelles, proteoliposomes, and lipodisq nanoparticles. Figure 3 and Table 1 clearly indicate that the most probable distances calculated for each sample matrix (micelles, proteoliposomes, and lipodisq nanoparticles) are comparable and that the structure of the TMD of KCNE1 is comparable in all three different types of model membranes.
Figure 3.
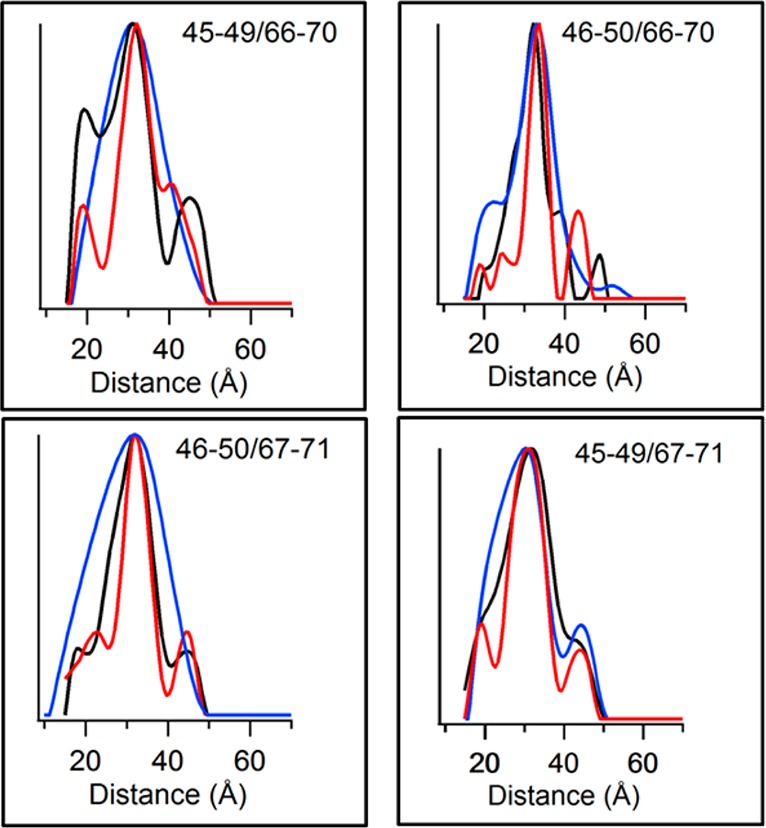
Overlay of distance probability data obtained from Tikhonov regularization in DEER data analysis for BSL-labeled KCNE1 samples in micelles, proteoliposomes, and lipodisq nanoparticles. The black traces represent micelles, blue represents proteoliposomes, and red represents lipodisq nanoparticles. The label in each box indicates the dual-labeling sites. The Y-axis scale represents the probability of distance distribution in each plot.
Four-Pulse DEER Distance Measurements Using MTSL Spin Labels
Similarly, DEER spectra were collected for nine pairs of conventional MTSL spin labels attached at different positions on the transmembrane domain of KCNE1. The MTSL labeling sites include 45/71, 45/70, 47/66, 47/69, 49/67, 50/68, 51/65, 52/64, and 55/66. Figure 4 illustrates the DEER data for dual-MTSL-labeled sites (50/68) in 1% LMPG micelles, POPC/POPG proteoliposomes, and POPC/POPG lipodisq nanoparticles. DEER data and analysis for the remaining MTSL samples are given in the Supporting Information (Figure S2). The distances for all MTSL samples are summarized in Table 1. All distances were derived from the maximum peak intensity in each DEER distance distribution plot. Figure 5 shows an overlay of distance distribution data plotted for each MTSL sample in micelles, proteoliposomes, and lipodisq nanoparticles. Figure 5 clearly indicates the major peaks of each sample are comparable for micelles, proteoliposomes, and lipodisq nanoparticles. This agrees with the BSL DEER data and indicates that the KCNE1 TMD has similar structural conformations in all three membrane-mimicking media.
Figure 4.

Four-pulse Q-band DEER data of KCNE1 mutant (Val50/Ser68) bearing two MTSL spin labels. Background-subtracted dipolar evolutions for the indicated mutants (left) and their corresponding distance probability distribution from Tikhonov regularization (right) are shown for (A) 1% LMPG micelles, (B) proteoliposomes (POPC/POPG = 3:1), and (C) lipodisq nanoparticles.
Figure 5.

Overlay of distance probability distribution data obtained from Tikhonov regularization of DEER data for MTSL-labeled KCNE1 in micelles, proteoliposomes, and lipodisq nanoparticles. The black traces represent micelles, blue represents proteoliposomes, and red represents lipodisq nanoparticles. The label in each box indicates the dual-labeling sites. The Y-axis scale represents the probability of distance distribution in each plot.
Structure Refinement of the TMD of KCNE1 from MTSL DEER Distance Restraints
A model for the most probable structure of the transmembrane domain of KCNE1 in POPC/POPG proteoliposomes was obtained using the experimental DEER distances coupled with the simulated annealing software package Xplor-NIH (version 2.33).28,29 This approach has been used for other DEER structural studies.30−32 The DEER distance data obtained for nine pairs of MTSL spin-labeled sites (see Materials and Methods) in proteoliposomes were converted into Xplor distance restraints and employed in a simulated annealing protocol. The MTSL DEER distance restraints were used to make the structure calculation procedure simpler within Xplor-NIH. Since MTSL is a widely used spin probe for EPR spectroscopic studies, the method of structure refinement used in this study can be widely applied to many other membrane proteins. A series of simulated annealing calculations were performed using distance-restraint uncertainties of ±4 Å. A simulated annealing calculation was also performed on a control system having amino acids representing the KCNE1 TMD sequence without any experimental distance restraints. The result of this control simulated annealing calculation yielded a straight helix and did not satisfy the experimental DEER distance restraints. Figure 6 displays the 10 lowest energy structures of the KCNE1 TMD in proteoliposomes obtained from the simulated annealing calculation using MTSL-derived distance restraints. To generate the final structures, the MTSL spin-labeled side chains were replaced by the native side chains (Figure 6A). The output energies corresponding to 10 minimum energy structures obtained from Xplor-NIH refinement are given in the Supporting Information (Table S1).
Figure 6.

Results of the structure refinement of the KCNE1 TMD in proteoliposomes incorporating MTSL DEER distance-restraint data using an Xplor-NIH simulated annealing molecular dynamics protocol. (A) Overlay of the 10 structures with lowest energy obtained from restrained simulated annealing calculations using amino acids 45–71 (transmembrane domain) of KCNE1. The final structures were generated by replacing the MTSL-labeled side chains by the native amino acid side chains with retention of the Cβ position in the label. (B) Ribbon representation of overlaid DEER structures. (C) Overlay of DEER structures and the previously determined NMR structure (blue cartoon represents micellar NMR structure, and purple cartoons represent DEER structures in lipid bilayers).
The newly determined structures were further validated with the BSL-based experimental DEER distances. The back-calculated distances between dual-BSLs attached on the newly determined lowest energy structure are consistent with the experimental BSL distances within experimental error of ±2 Å (Table S2). The DEER determined structures and the previously determined solution NMR structure in micelles are overlaid in Figure 6C. The backbone RMSDs for all of the superimposed new structures with respect to the previous NMR structure vary between 1.5 to 3.0 Å (see Table S3). The residue positions on the concave face of the KCNE1 TMD in the new structure model are comparable to those of the previously determined NMR structure. Additional details for the output structures are given in the Supporting Information. These new structures indicate that the KCNE1 TMD adopts a similar, slightly curved conformation when compared to the previously published solution NMR structure in LMPG micelles.1 The predicted structural model of KCNE1 in a lipid bilayer is given in Figure 7.
Figure 7.

Schematic model of the overlay of the 10 lowest energy structures of KCNE1 TMD in a lipid bilayer.
Discussion
Methodological Observations: Advantages of Bifunctional Spin Labels and Lipodisq Model Membranes
DEER distance measurements allow researchers to obtain valuable structural information from systems in which other techniques like solution nuclear magnetic resonance (NMR) or X-ray crystallography prove to be difficult or impossible.24,43 Recent methodological and spectroscopic developments in DEER measurements can provide higher quality structural data for the study of membrane proteins.20,22,39,42,44−51 Bifunctional spin labels (BSL) provide narrower distance distributions when compared to that with standard MTSL spin labels.20,39 Bifunctional spin labels have been recently used to study conformational states of the voltage sensing domain of KvAP.52 While the focus of this study was on the structure of KCNE1, this work also included the exploration of the combination of relatively new spin-labeling methods and a recently introduced model membrane system. In this study, we utilized two kinds of spin labels, BSL (more conformationally restricted) and MTSL (flexible), attached at several position pairs within the DEER-detectable distance range on the KCNE1 TMD (see Figure 1) in micelles and more native membrane environments (proteoliposomes and lipodisq nanoparticles). Here, we showed that the use of bifunctional spin labels yielded DEER distance measurements with tighter distance distributions (Figure 3 and Table S4) when compared to those for the monofunctional MTSL spin-label samples (Figure 5 and Table S4). This illustrates the advantage of using a spin-labeled probe that is more rigidly linked to the protein backbone by dual attachment to i, i+3/4 cysteine sites than the more classical method of modifying a single cysteine site with MTSL.
This work also provided a comparison between the use of lipodisq nanoparticles and bilayered lipid vesicles as a medium for DEER EPR-based structural studies of membrane proteins. SMA-lipodisq nanoparticles provide a good membrane mimetic environment and boost the phase memory time (Tm) by ∼2-fold and the corresponding signal-to-noise ratio when compared to that for proteoliposomes20,22,23 (see Figures 2, 4, S1, and S2).20 Also, the increase in Tm for lipodisq-based samples will enable DEER data to be collected out further in time and lead to longer distance measurements and potentially narrower distance distributions. Lipodisq nanoparticles isolate protein macromolecules by minimizing the size of the complex to ∼10–15 nm while still retaining a biologically relevant membrane structure.20,22 Here, it was observed that all DEER distance data collected in micelles and lipodisq nanoparticles exhibit similar signal-to-noise ratios and distance distribution widths for both BSL and MTSL samples. The lipodisq-based sample preparation minimizes the clustering of intermolecular spins by decreasing the size of individual protein–lipid complexes. Lipodisq nanoparticles can be applied to a wide range of protein–lipid complexes.20,22,23 It should be added that several laboratories have demonstrated that there are no significant structural and/or functional perturbations of membrane proteins due to BSL and lipodisq nanoparticles.20,22,23,39
Comparison of the KCNE1 TMD Structure in Micelles and Lipid Bilayers
Detergent micelles are widely used as a membrane-mimetic for membrane proteins in part due to their smaller size, facilitating high-resolution structure analysis by solution NMR spectroscopy; however, it is often difficult to test whether the structure of proteins in a micelle environment is the same as that of the membrane-bound state.
A previous solution NMR study indicated that the transmembrane domain of KCNE1 has a curved helix (Leu45–Leu71) in LMPG micelles.1 The BSL and MTSL DEER data presented in this article indicate the TMD of KCNE1 is helical in proteoliposomes and lipodisq nanoparticles. This agrees with our recent CD spectroscopic study that indicated that KCNE1 has α-helical structural components in POPC/POPG lipid bilayers;18 however, the CD data was global for KCNE1 and not specific to the TMD. Also, previously reported CW-EPR power saturation data suggested KCNE1 spans the full width of the membrane, with the Leu59 residue located near the center.18
The most probable spin label-to-spin label distances obtained for each combination of labeling sites for KCNE1 in LMPG micelles, proteoliposomes, and lipodisq nanoparticles were, in all cases, comparable (Table 1). The DEER-derived distances were used as restraints for conducting simulated annealing molecular dynamics simulations to determine the three-dimensional structural model of the KCNE1 TMD in lipid bilayers (Figures 6 and 7). This approach converged on a rather precise ensemble of structures that exhibit a slightly curved helix with a helix apex close to Thr58. Figure 6C compares the new DEER structural model in lipid bilayers with the solution NMR micelle structure. Although both structures have a curved helix for the TMD, the solution NMR structure has greater curvature.
In order to directly compare observed distances between a straight helix and a curved helix for the TMD of KCNE1, modeling studies were conducted. The MTSL-based nitroxide SL–SL distances measured on the TMD of KCNE1 mutants in different structural forms (Table 2) indicate the distances obtained on the newly determined lowest energy lipid-bilayer structure are consistent with the experimental DEER distances and close to the solution NMR micelle structure within experimental error. They are, however, quite different than predicted by a linear helix. Inspection of all the distances shown in Table 2 indicates that the structure of the KCNE1 TMD is similar to the curved NMR structure helix. The curvilinear conformation of the KCNE1 TMD is consistent with the width of POPC lipid bilayers (∼37 Å) used in this study.53,54 POPC/POPG lipids were used for this study since they are typically found in mammalian membranes.18−21 However, the degree of curvature and conformational flexibility of the TMD of KCNE1 may vary depending upon the length and choice of phospholipids used for the measurements.55 The slight curvature seen for the KCNE1 TMD may arise due to the backbone conformational balance between glycine residues and a number of flanking β-branched amino acids present in the KCNE1 TMD.1 This curvature may play a role in binding of KCNE1 to KCNQ1 and for its subsequent modulation of channel function, as previously proposed.1 One possibility is that the curvature provides maximal exposure of the side chain of Thr58, located at the apex of the convex face of the curved helix, for interaction with KCNQ1. This site is known to be essential for the delayed activation conferred on KCNQ1 by KCNE1.1,11,18 Another, not mutually exclusive, possibility is that the curvature is required to optimize the binding interface between KCNQ1 and KCNE1 and also to ensure the ends of the TMD are correctly positioned to enable the extramembrane domains of KCNE1 to make optimal interactions with the channel.
Table 2. Comparison of MTSL-Based Nitroxide Spin Label (SL)–Spin Label (SL) Distances on the TMD of KCNE1 Mutants in Different Structural Forms (Linear Helix, Previously Determined NMR Micelle Structure, and Newly Determined Lowest Energy Lipid Bilayer Structure) with the Experimental DEER Distancesa.
| distance (Å) |
||||
|---|---|---|---|---|
| KCNE1 double mutants | linear helix | NMR structure | DEER experimental | new model structure |
| 45/71 | 44 | 37 | 36 | 38 |
| 45/70 | 44 | 38 | 36 | 38 |
| 47/69 | 40 | 34 | 32 | 34 |
| 47/66 | 37 | 30 | 31 | 33 |
| 49/67 | 26 | 27 | 27 | 28 |
| 50/68 | 25 | 26 | 30 | 32 |
| 51/65 | 24 | 28 | 22 | 24 |
| 52/64 | 18 | 25 | 24 | 21 |
| 55/66 | 19 | 23 | 21 | 17 |
The modelling of MTSL spin-labeled NMR micelle structure (see Figure 1 for structure) was carried out using addAtoms.py script under Xplor-NIH. All of the modelling distances were measured using VMD-Xplor.
Conclusions
Detergent micelles have come under considerable criticism as model membranes on the basis that they may distort membrane protein structures from their true native-like forms.56 It is notable that the TMD of KCNE1 maintains the curved helical structure in both lipid bilayers (proteoliposomes and lipodisq nanoparticles) and in LMPG detergent micelles. This suggests that the curvature seen for this domain is a robust feature of the KCNE1 structure not easily perturbed by changes in the membrane local environment. It has previously been argued that robust resilience of membrane protein structure in the face of variations in membrane lipid composition may be an evolved trait shared by many membrane proteins.57 Perhaps this is the case for the unusual KCNE1 TMD. If so, then it seems probable that KCNE1 maintains a curved TMD even upon association with KCNQ1. This hypothesis will require future experimental testing.
Acknowledgments
We would like to thank Dr. Charles Schwieters at the National Institute of Health for assisting with the structural refinement Xplor-NIH program and Dr. Shahidul Islam at the Department of Biochemistry & Molecular Biology, The University of Chicago, for assisting with the use of BSLs under CHARMM-GUI.
Glossary
Abbreviations
- LQTS
long QT syndrome
- TMD
transmembrane domain
- DEER
double electron–electron resonance
- NMR
nuclear magnetic resonance
- SDSL
site-directed spin labeling
- CW-EPR
continuous wave electron paramagnetic resonance
- IPTG
isopropyl β-d-1-thiogalactopyranoside
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- LMPG
1-myristoyl-2-hydroxy-sn-glycero-3-phospho-(1′-rac-glycerol) (sodium salt)
- BSL
bifunctional spin label, (3,4-Bis(methanethiosulfonylmethyl)-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-1-yloxy Radical), (HO-1944)
- MTSL
1-oxyl-2,2,5,5-tetramethylpyrroline-3-methylmethanethiosulfonate spin label
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPG
1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (sodium salt)
- DLS
dynamic light scattering
- VMD
visual molecular dynamics
- CD
circular dichroism
- S/N
signal-to-noise ratio
Supporting Information Available
Additional experimental DEER data and corresponding analysis for the BSL and MTSL spin-labeled KCNE1 samples (Figures S1 and S2), amino acid sequences of wild-type and mutated KCNE1 TMD (Figure S3), additional structural refinement data on KCNE1 TMD (Figure S4 and Table S1), back calculated dual-BSL distances on the newly determined structure (Table S2), backbone RMSDs of the newly determined structure with respect to micelle structure of KCNE1 TMD (Table S3), and fwhm of DEER distance distributions (Table S4). This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by National Institutes of Health grant nos. R01 GM108026 (to G.A.L.) and R01 DC007416 (to C.R.S.). Funding was also provided by National Science Foundation (NSF) grant no. CHE-1305664 (to G.A.L.). The pulsed EPR spectrometer was purchased through NSF and the Ohio Board of Regents grants (MRI-0722403).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Kang C.; Tian C.; Sonnichsen F. D.; Smith J. A.; Meiler J.; George A. L. J.; Vanoye C. G.; Kim H. J.; Sanders C. R. (2008) Structure of KCNE1 and implications for how it modulates the KCNQ1 potassium channel. Biochemistry 47, 7999–8006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai K. K.; Goldstein S. A. N. (1998) The conduction pore of a cardiac potassium channel. Nature 391, 605–608. [DOI] [PubMed] [Google Scholar]
- Uysal S.; Vasquez V.; Tereshko V.; Esaki K.; Fellouse F. A.; Sidhu S. S.; Koide S.; Perozo E.; Kossiakoff A. (2009) Crystal structure of full-length KcsA in its closed conformation. Proc. Natl. Acad. Sci. U.S.A. 106, 6644–6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketchum K. A.; Joiner W. J.; Sellers A. J.; Kaczmarek L. K.; Goldstein S. A. N. (1995) A new family of outwardly rectifying potassium channel proteins with 2 pore domains in tandem. Nature 376, 690–695. [DOI] [PubMed] [Google Scholar]
- Herlyn H.; Zechner U.; Oswald F.; Pfeufer A.; Zischler H.; Haaf T. (2009) Positive selection at codon 38 of the human KCNE1 (minK) gene and sporadic absence of 38Ser-coding mRNAs in Gly38Ser heterozygotes. BMC Evol. Biol. 9, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoye C. G.; Welch R. C.; Tian C.; Sanders C. R.; George A. L. J. (2010) KCNQ1/KCNE1 assembly, co-translation not required. Channels 4, 108–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang C. B.; Vanoye C. G.; Welch R. C.; Van Horn W. D.; Sanders C. R. (2010) Functional delivery of a membrane protein into oocyte membranes using bicelles. Biochemistry 49, 653–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F. D.; Chen J.; Lin M.; Keating M. T.; Sanguinetti M. C. (2001) Long-QT syndrome-associated missense mutations in the pore helix of the HERG potassium channel. Circulation 104, 1071–1075. [DOI] [PubMed] [Google Scholar]
- Tapper A. R.; George A. L. (2000) MinK subdomains that mediate modulation of and association with KvLQT1. J. Gen. Physiol. 116, 379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Zhang M.; Xu Y.; Jiang M.; Zankov D. P.; Cui M.; Tseng G.-N. (2012) Probing the structural basis for differential KCNQ1 modulation by KCNE1 and KCNE2. J. Gen. Physiol. 140, 653–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melman Y. F.; Um S. Y.; Krumerman A.; Kagan A.; McDonald T. V. (2004) KCNE1 binds to the KCNQ1 pore to regulate potassium channel activity. Neuron 42, 927–937. [DOI] [PubMed] [Google Scholar]
- Tian C.; Vanoye C. G.; Kang C.; Welch R. C.; Kim H. J.; George A. L.; Sanders C. R. (2007) Preparation, functional characterization, and NMR studies of human KCNE1, a voltage-gated potassium channel accessory subunit associated with deafness and long QT syndrome. Biochemistry 46, 11459–11472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Fermini B.; Nattel S. (1994) Rapid and slow components of delayed rectifi er current in human atrial myocytes. Cardiovasc. Res. 28, 1540–1546. [DOI] [PubMed] [Google Scholar]
- Harmer S. C.; Tinker A. (2007) The role of abnormal trafficking of KCNE1 in long QT syndrome 5. Biochem. Soc. Trans. 35, 1074–1076. [DOI] [PubMed] [Google Scholar]
- Gofman Y.; Shats S.; Attali B.; Haliloglu T.; Ben-Tal N. (2012) How does KCNE1 regulate the Kv7.1 potassium channel? Model-structure, mutations, and dynamics of the Kv7.1–KCNE1 complex. Structure 20, 1343–1352. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Wang Y.; Meng X.-Y.; Zhang M.; Jiang M.; Cui M.; Tseng G.-N. (2013) Building KCNQ1/KCNE1 channel models and probing their interactions by molecular-dynamics simulations. Biophys. J. 105, 2461–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajo K.; Kubo Y. (2014) Steric hindrance between S4 and S5 of the KCNQ1/KCNE1 channel hampers pore opening. Nat. Commun. 5, 4100–4100. [DOI] [PubMed] [Google Scholar]
- Coey A. T.; Sahu I. D.; Gunasekera T. S.; Troxel K. R.; Hawn J. M.; Swartz M. S.; Wickenheiser M. R.; Reid R. J.; Welch R. C.; Vanoye C. G.; Kang C. B.; Sanders C. R.; Lorigan G. A. (2011) Reconstitution of KCNE1 into lipid bilayers: comparing the structural, dynamic, and activity differences in micelle and vesicle environments. Biochemistry 50, 10851–10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett P. J.; Song Y.; Van Horn W. D.; Hustedt E. J.; Schafer J. M.; Hadziselimovic A.; Beel A. J.; Sanders C. R. (2012) The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science 336, 1168–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu I. D.; MaCarrick R. M.; Troxel K. R.; Zhang R.; Smith J. H.; Dunagan M. M.; Swartz M. S.; Rajan P. V.; Kroncke B. M.; Sanders C. R.; Lorigan G. A. (2013) DEER EPR measurement for membrane protein structures via bifunctional spin labels and lipodisq nanoparticles. Biochemistry 52, 6627–6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y.; Hustedt E. J.; Brandon S.; Sanders C. R. (2013) Competition between homodimerization and cholesterol binding to the C99 domain of the amyloid precursor protein. Biochemistry 52, 5051–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orwick-Rydmark M.; Lovett J. E.; Graziadei A.; Lindholm L.; Hicks M. R.; Watts A. (2012) Detergent-free incorporation of a seven-transmembrane receptor protein into nanosized bilayer lipodisq particles for functional and biophysical studies. Nano Lett. 12, 4687–4692. [DOI] [PubMed] [Google Scholar]
- Orwick M. C.; Judge P. J.; Procek J.; Lindholm L.; Graziadei A.; Engel A.; Grobner G.; Watts A. (2012) Detergent-free formation and physicochemical characterization of nanosized lipid-polymer complexes: lipodisq. Angew. Chem., Int. Ed. 51, 4653–4657. [DOI] [PubMed] [Google Scholar]
- Jeschke G. (2012) DEER distance measurements on proteins. Annu. Rev. Phys. Chem. 419–446. [DOI] [PubMed] [Google Scholar]
- Feldmann E. A.; Ni S.; Sahu I. D.; Mishler C. H.; Risser D. D.; Murakami J. L.; Tom S. K.; McCarrick R. M.; Lorigan G. A.; Tolbert B. S.; Callahan S. M.; Kennedy M. A. (2011) Evidence for direct binding between HetR from Anabaena sp PCC 7120 and PatS-5. Biochemistry 50, 9212–9224. [DOI] [PubMed] [Google Scholar]
- Jeschke G.; Chechik V.; Ionita P.; Godt A.; Zimmermann H.; Banham J.; Timmel C. R.; Hilger D.; Jung H. (2006) DeerAnalysis2006—a comprehensive software package for analyzing pulsed ELDOR data. Appl. Magn. Reson. 30, 473–498. [Google Scholar]
- Chiang Y. W.; Borbat P. P.; Freed J. H. (2005) The determination of pair distance distributions by pulsed ESR using Tikhonov regularization. J. Magn. Reson. 172, 279–295. [DOI] [PubMed] [Google Scholar]
- Schwieters C. D.; Kuszewski J. J.; Tjandra N.; Clore G. M. (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73. [DOI] [PubMed] [Google Scholar]
- Schwieters C. D.; Kuszewski J. J.; Clore G. M. (2006) Using Xplor-NIH for NMR molecular structure determination. Prog. Nucl. Magn. Reson. Spectrosc. 48, 47–62. [DOI] [PubMed] [Google Scholar]
- Mokdad A.; Herrick D. Z.; Kahn A. K.; Andrews E.; Kim M.; Cafiso D. S. (2012) Ligand-induced structural changes in the Escherichia coli ferric citrate transporter reveal modes for regulating protein–protein interactions. J. Mol. Biol. 423, 818–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrick D. Z.; Kuo W.; Huang H.; Schwieters C. D.; Ellena J. F.; Cafiso D. S. (2009) Solution and membrane-bound conformations of the tandem C2A and C2B domains of synaptotagmin 1: evidence for bilayer bridging. J. Mol. Biol. 390, 913–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jao C. C.; Hegde B. G.; Chen J.; Haworth I. S.; Langen R. (2008) Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. U.S.A. 105, 19666–19671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwieters C. D.; Clore G. M. (2001) The VMD-XPLOR visualization package for NMR structure refinement. J. Magn. Reson. 149, 239–244. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graphics 14, 33–38. [DOI] [PubMed] [Google Scholar]
- MacKerell A. D.; Bashford D.; Bellott M.; Dunbrack R. L.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; Joseph-McCarthy D.; Kuchnir L.; Kuczera K.; Lau F. T. K.; Mattos C.; Michnick S.; Ngo T.; Nguyen D. T.; Prodhom B.; Reiher W. E.; Roux B.; Schlenkrich M.; Smith J. C.; Stote R.; Straub J.; Watanabe M.; Wiorkiewicz-Kuczera J.; Yin D.; Karplus M. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- MacKerell A. D.; Feig M.; Brooks C. L. (2004) Improved treatment of the protein backbone in empirical force fields. J. Am. Chem. Soc. 126, 698–699. [DOI] [PubMed] [Google Scholar]
- Sezer D.; Freed J. H.; Roux B. (2008) Parametrization, molecular dynamics simulation, and calculation of electron spin resonance spectra of a nitroxide spin label on a polyalanine alpha-helix. J. Phys. Chem. B 112, 5755–5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Kim T.; Iyer V. G.; Im W. (2008) Software news and updates – CHARNIM-GUI: a web-based grraphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- Fleissner M. R.; Bridges M. D.; Brooks E. K.; Cascio D.; Kálai T.; Hideg K.; Hubbell W. L. (2012) Structure and dynamics of a conformationally constrained nitroxide side chain and applications in EPR spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 108, 16241–16246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbat P. P.; McHaourab H. S.; Freed J. H. (2002) Protein structure determination using long-distance constraints from double-quantum coherence ESR: study of T4 lysozyme. J. Am. Chem. Soc. 124, 5304–5314. [DOI] [PubMed] [Google Scholar]
- Jeschke G.; Polyhach Y. (2007) Distance measurements on spin-labelled biomacromolecules by pulsed electron paramagnetic resonance. Phys. Chem. Chem. Phys. 9, 1895–1910. [DOI] [PubMed] [Google Scholar]
- Sahu I. D.; McCarrick R. M.; Lorigan G. A. (2013) Use of electron paramagnetic resonance to solve biochemical problems. Biochemistry 52, 5967–5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHaourab H. S.; Steed P. R.; Kazmier K. (2011) Toward the fourth dimension of membrane protein structure: insight into dynamics from spin-labeling EPR spectroscopy. Structure 19, 1549–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou P.; McHaourab H. S. (2010) Increased sensitivity and extended range of distance measurements in spin-labeled membrane proteins: Q-band double electron-electron resonance and nanoscale bilayers. Biophys. J. 98, L18–L20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endeward B.; Butterwick J. A.; MacKinnon R.; Prisner T. F. (2009) Pulsed electron–electron double-resonance determination of spin-label distances and orientations on the tetrameric potassium ion channel KcsA. J. Am. Chem. Soc. 131, 15246–15250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou P.; Bortolus M.; McHaourab H. S. (2009) Conformational cycle of the ABC transporter MsbA in liposomes: detailed analysis using double electron–electron resonance spectroscopy. J. Mol. Biol. 393, 586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgieva E. R.; Ramlall T. F.; Borbat P. P.; Freed J. H.; Eliezer D. (2008) Membrane-bound alpha-synuclein forms an extended helix: long-distance pulsed ESR measurements using vesicles, bicelles, and rodlike micelles. J. Am. Chem. Soc. 130, 12856–12857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q.; Ellena J. F.; Kim M.; Cafiso D. S. (2006) Substrate-dependent unfolding of the energy coupling motif of a membrane transport protein determined by double electron–electron resonance. Biochemistry 45, 10847–10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dastvan R.; Bode B. E.; Karuppiah M. P. R.; Marko A.; Lyubenova S.; Schwalbe H.; Prisner T. F. (2010) Optimization of transversal relaxation of nitroxides for pulsed electron–electron double resonance spectroscopy in phospholipid membranes. J. Phys. Chem. B 114, 13507–13516. [DOI] [PubMed] [Google Scholar]
- Ghimire H.; McCarrick R. M.; Budil D. E.; Lorigan G. A. (2009) Significantly improved sensitivity of Q-band PELDOR/DEER experiments relative to X-band is observed in measuring the intercoil distance of a leucine zipper motif peptide (GCN4-LZ). Biochemistry 48, 5782–5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyhach Y.; Bordignon E.; Tschaggelar R.; Gandra S.; Godt A.; Jeschke G. (2012) High sensitivity and versatility of the DEER experiment on nitroxide radical pairs at Q-band frequencies. Phys. Chem. Chem. Phys. 14, 10762–10773. [DOI] [PubMed] [Google Scholar]
- Li Q.; Wanderling S.; Sompornpisut P.; Perozo E. (2014) Structural basis of lipid-driven conformational transitions in the KvAP voltage-sensing domain. Nat. Struct. Mol. Biol. 21, 160–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucerka N.; Tristram-Nagle S.; Nagle J. F. (2005) Structure of fully hydrated fluid phase lipid bilayers with monounsaturated chains. J. Membr. Biol. 208, 193–202. [DOI] [PubMed] [Google Scholar]
- Tsai H.-H. G.; Lee J.-B.; Huang J.-M.; Juwita R. (2013) A Molecular dynamics study of the structural and dynamical properties of putative arsenic substituted lipid bilayers. Int. J. Mol. Sci. 14, 7702–7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandasamy S. K.; Larson R. G. (2006) Molecular dynamics simulations of model trans-membrane peptides in lipid bilayers: a systematic investigation of hydrophobic mismatch. Biophys. J. 90, 2326–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.-X.; Cross T. A. (2013) Influences of membrane mimetic environments on membrane protein structures. Annu. Rev. Biophys. 42 42, 361–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders C. R.; Mittendorf K. F. (2011) Tolerance to changes in membrane lipid composition as a selected trait of membrane proteins. Biochemistry 50, 7858–7867. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.