

Abstract
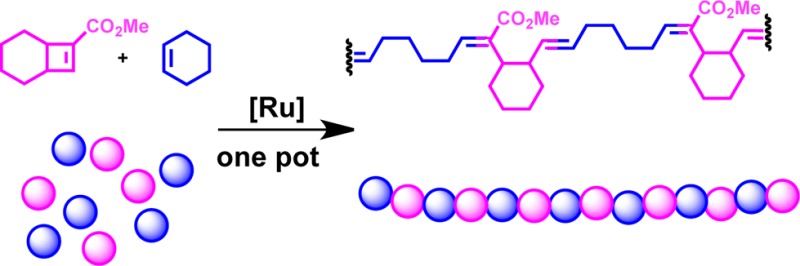
Strained bicyclic carbomethoxy olefins were utilized as substrates in alternating ring-opening metathesis polymerization and found to provide low-dispersity polymers with novel backbones. The polymerization of methyl bicyclo[4.2.0]oct-7-ene-7-carboxylate with cyclohexene in the presence of the fast-initiating Grubbs catalyst (H2IMes)(3-Br-Pyr)2Cl2Ru=CHPh leads to a completely linear as well as alternating copolymer, as demonstrated by NMR spectroscopy, isotopic labeling, and gel permeation chromatography. In contrast, intramolecular chain-transfer reactions were observed with [5.2.0] and [3.2.0] bicyclic carbomethoxy olefins, although to a lesser extent than with the previously reported monocyclic cyclobutenecarboxylic ester monomers [Song A.; Parker K. A.; Sampson N. S.. J. Am. Chem. Soc. 2009, 131, 3444.]. Inclusion of cyclohexyl rings fused to the copolymer backbone minimizes intramolecular chain-transfer reactions and provides a framework for creating alternating functionality in a one-step polymerization.
In recent decades, copolymers have been widely studied for their uses in the biomedical and material sciences.1−3 Block copolymers, already established as thermoplastic elastomers, detergents, cosmetics, and pharmaceutical preparations, promise to contribute to new applications based on nanoscale structures, membranes, and drug and gene delivery.4 Much less explored are alternating copolymers. For applications in which two different polymer-borne moieties must interact5−9 (e.g., as in organic light-emitting diodes and solar cells), alternating copolymers should impose consistently optimal positioning of the participating substituents.
Alternating copolymers are generally synthesized by radical polymerization in which the alternating order of addition of monomers at the end of the growing chain is kinetically controlled.10−12 However, the conditions required for radical propagation are not compatible with a number of functional groups that might be desired in the polymer target. Furthermore, with some exceptions,13−15 such polymerizations are generally not “living”. Therefore, they do not afford polymer products with narrow molecular weight distributions (low-molar-mass dispersities or DMs) that are advantageous for certain applications.
Living polymerizations based on the functional group-tolerant ruthenium metathesis catalysts have the potential to provide alternating copolymers that have low DMs and that bear a variety of functional groups. In metathesis, alternation of the incorporation of two monomers requires alternation of the affinities of the monomer A and monomer B to the living metal alkylidene. There are few solutions to this problem and consequently few examples of completely alternating metathesis copolymers. Rooney,16−18 Chen,19−22 and Blechert and Buchmeiser23−25 have focused on the design of asymmetric catalysts that provide alternating selectivity for different pairs of monomers. Typically variation in steric bulk about the catalyst results in alternating reactivity of very strained and moderately strained, but more sterically demanding, monomers. Despite impressive catalyst designs, more than a 10-fold excess of the moderately strained monomer is required to maintain alternation.22
Monomer-design approaches take advantage of the different properties of two monomers such as polarity,26 electron density and steric hindrance,27 and acid–base interactions28 and, in the latter two cases, provide perfectly alternating copolymers without use of excess monomer. Herein, we describe new pairs of monomers that generate, sequentially, two different ruthenium carbenes. One of these pairs efficiently provides completely alternating, linear copolymers.
We discovered that cyclobutene-1-carboxylate esters undergo ring-opening metathesis (ROM), but they do not undergo ROMP.27 Nonetheless, in solution with cyclohexene (which also does not ROMP on its own), the cyclobutenecarboxylic esters participate in an alternating ring-opening metathesis polymerization (AROMP or altROMP). The high fidelity of the alternation in the chain extending steps can be attributed to the complementary reactivities of the two intermediate ruthenium carbenes.29 There is a high kinetic barrier to cyclobutene ester homopolymerization, and the low ring strain of the cyclohexene monomer does not overcome the entropic penalty for its homopolymerization.27 Moreover, the substitution of the cyclobutene alkene provides regiochemical and stereochemical control of the polymerization.
The molecular weight homogeneity of the copolymers resulting from our cyclohexene/1-cyclobutene ester pair was limited by “backbiting” reactions, intramolecular chain-transfers that lead to the formation of cyclic polymers, shortened chains, and compromised molar-mass dispersities (DMs). Indeed, we discovered that the use of the Hoveyda–Grubbs II catalysts, which favor cyclizations, resulted in the exclusive formation (within the limits of detection) of cyclic alternating copolymers.30 Recently, we observed complete inhibition of backbiting upon introduction of bulky side chains into the AROMP monomers. However, the increased steric hindrance near the double bond slows the polymerization propagation rate, resulting in shorter polymers and thus limits the utility of this approach.9
Cognizant of the desirability of high-molecular-weight linear polymers with narrow molecular weight distributions, we set out to find a pair of monomers that would give longer and linear AROMP polymers with low-molar-mass dispersities. Here we report the results of this search to date.
Experimental Methods
All metathesis reactions were performed under an N2 atmosphere. Solvents, e.g. CH2Cl2 and benzene, were purified and dried in a GlassContour solvent push-still system. Deuterated solvents for all ring-opening reactions were degassed and filtered through basic alumina before use. [(H2IMes)(PCy3)(Cl)2Ru=CHPh] and ethyl 1-bromocyclobutane carboxylate were purchased from Aldrich. Cyclohexene-d10 was purchased from CDN Isotope Inc. The synthesis of Grubbs III catalyst, [(H2IMes)(3-Br-Pyr)2Cl2Ru=CHPh], was performed according to the procedure of Love et al.31 A fresh stock solution of the Grubbs III catalyst (0.02 M for AROMP or 0.03 M for ROM, AROM-1, and AROM-2) and fresh stock solutions of monomers 3 and 4 (0.17–1.0 M for AROMP, depending on the desired length, or 0.03 M for ROM, AROM-1, and AROM-2) were prepared in CD2Cl2 for each of the NMR experiments.
Mallinckrodt silica gel 60 (230–400 mesh) was used for column chromatography. Analytical thin layer chromatography (TLC) was performed on precoated silica gel plates (60F254), chromatography on silica gel-60 (230–400 mesh), and Combi-Flash chromatography on RediSep normal phase silica columns (silica gel-60, 230–400 mesh). Varian Inova400, Inova500, Inova600 and Bruker Nanobay 400, Avance III 500, Avance III 700, Avance III-HD 850 MHz NMR instruments were used for analysis. Chemical shifts are denoted in ppm (δ) and calibrated from residual undeuterated solvents. The degree of polymerization (DP) of linear polymers was assessed by comparing the 1H NMR integration of the polymer alkene protons to that of the phenyl end group. Molecular weights and molar mass dispersities were measured with a gel phase chromatography system constructed from a Shimadzu pump coupled to a Shimadzu UV detector. CH2Cl2 served as the eluent with a flow rate of 0.700 mL/min on an American Polymer Standards column (Phenogel 5 μ MXL GPC column, Phenomenex). All GPCs were calibrated with poly(styrene) standards at 30 °C.
Bicyclo[2.2.1]hept-2-ene-2-carboxylic Acid32,33
A modification of the two-step procedure of Elsheimer was followed.32Safety warning: CF2Br2 has a very low boiling point, and addition of CF2Br2 to norbornene is very exothermic. Therefore, for a large-scale reaction, this procedure should be carried out carefully behind a safety shield.
2-Bromo-3-(bromodifluoromethyl)bicyclo[2.2.1]heptanes
To a mixture of norbornene (6.10 g, 65 mmol), CuCl (0.99 g, 1 mmol), ethanolamine (3.00 g, 50 mmol), and tert-butyl alcohol (7.40 g, 100 mmol) in a 50 mL flask, CF2Br2 was slowly added (27.30 g, 130 mmol). The resulting mixture was protected from light and stirred at reflux (80–85 °C) for 48 h. Then it was cooled and diluted with deionized water (50 mL) and Et2O (25 mL). The Et2O layer was washed with H2O (5 × 25 mL) and dried over anhydrous Na2SO4. After filtration, the extract was concentrated and the residue was purified by flash column chromatography to yield a mixture of bicyclo[2.2.1]heptanes as a colorless oil (16.75 g, 87%). 1H NMR (500 MHz, CDCl3): δ 4.09 (d, J = 10 Hz, 1H), 2.78 (td, J = 15 Hz, 10 Hz, 1H), 2.71 (s, 1H), 2.68 (d, J = 10 Hz, 1H), 2.12 (d, J = 10 Hz, 1H), 1.76 (m, 1H), 1.64 (m, 1H), 1.40–1.20 (m, 3H).
Bicyclo[2.2.1]hept-2-ene-2-carboxylic Acid
In a screw-top vial under an N2 atmosphere, KOH (5.89 g, 10.5 mmol) was dissolved in deionized H2O (4 mL). A sample of 2-bromo-3-(bromodifluoromethyl)bicyclo[2.2.1]heptanes (0.776 g, 2.66 mmol) was added to the solution, and the mixture was heated at 130 °C in a microwave reactor at 20 bar for 2 h. Then the solution was cooled and washed with CHCl3 (2 × 4 mL), and the pH of the basic extract was adjusted to 2 with 3 N aqueous HCl. The aqueous solution was extracted with CHCl3 (3 × 4 mL), and the combined CHCl3 solution was dried over anhydrous Na2SO4 and concentrated in vacuo to give a brown oil. Purification by flash column chromatography (95:5/CH2Cl2:MeOH) yielded bicyclo[2.2.1]hept-2-ene-2-carboxylic acid as a colorless oil (160 mg, 50%).
Methyl Bicyclo[2.2.1]hept-2-ene-2-carboxylate, 2
The procedure of Mathias was followed.33 Bicyclo[2.2.1]hept-2-ene-2-carboxylic acid (40 mg, 0.29 mmol) was treated with N,N-diisopropyl-O-methylisourea (180 mg, 1.16 mmol) in dry ether (5 mL) and stirred for 48 h. The urea byproduct precipitated at −20 °C, and it was removed by filtration. Removal of the solvent afforded a yellow oil which was purified by flash column chromatography (90:10/hexane:CH2Cl2) to yield ester 2 (26 mg, 58%). 1H NMR (400 MHz, CD2Cl2): δ 6.91 (d, J = 2.8 Hz, 1H), 3.72 (s, 3H), 3.26 (s, 1H), 3.02 (s, 1H), 1.75 (m, 2H), 1.48 (d, J = 8.4 Hz, 1H), 1.20 (d, J = 8.4 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 164.9, 146.6, 140.5, 51.0, 48.0, 43.4, 41.8, 24.5, 24.4.
General Procedure for the Synthesis of Bicyclo[n.2.0] Monomers
These monomers were prepared according to Snider’s approach;34 the purification of monomer 4 was modified as noted. To a 50 mL flask with anhydrous AlCl3 powder under an N2 atmosphere was added dry benzene and methyl propiolate. The mixture was stirred until a homogeneous yellow solution formed. Cycloalkene was added, and the resulting mixture was stirred for 7 days. The reaction mixture was cooled in an ice bath and quenched with saturated NH4Cl solution. The precipitate was removed by filtering the resulting mixture through a pad of Celite. The filtrate was extracted with three portions of Et2O, and the combined Et2O extract was washed with brine and dried over anhydrous MgSO4. The solvent was removed in vacuo, and the crude product was subjected to flash column chromatography.
Methyl bicyclo[3.2.0]hept-6-ene-6-carboxylate, 3
AlCl3 (160 mg, 1.21 mmol, 0.5 equiv), methyl propiolate (204 mg, 2.42 mmol), and cyclopentene (170 μL, 2.90 mmol, 1.2 equiv) were stirred for 7 days in dry benzene (50 mL). Flash column chromatography of the crude product (30:70/hexane:CH2Cl2) yielded ester 3 (175 mg, 48% yield). 1H NMR (400 MHz, acetone-d6): δ 6.62 (br s, 1H), 3.69 (s, 3H), 3.33 (br dd, J = 3.2 Hz, 7.6 Hz, 1H), 3.06 (br dd, J = 3.6 Hz, 8.0 Hz, 1H), 1.78–1.21 (m, 6H). 13C NMR (100 MHz, CDCl3): δ 162.5, 147.5, 138.2, 51.1, 46.9, 44.5, 25.5, 25.4, 22.9.
Methyl Bicyclo[4.2.0]oct-7-ene-7-carboxylate, 4
AlCl3 (227 mg, 1.72 mmol, 0.86 equiv), methyl propiolate (168 mg, 2.00 mmol), and cyclohexene (263 μL, 2.60 mmol, 1.30 equiv) were stirred for 7 days in dry benzene (5 mL) and yielded a mixture of compound 4 and isomer methyl (E)-3-(cyclohex-1-enyl)propenoate in a ratio of 1:0.11–0.23. The mixture was treated with m-chloroperoxybenzoic acid (m-CPBA) to epoxidize the isomeric byproduct, followed by flash column chromatography (30:70/hexane:CH2Cl2) to provide bicyclic ester 4 (210 mg, 65% yield). 1H NMR (400 MHz, acetone-d6): δ 6.89 (d, J = 1.2 Hz, 1H), 3.71 (s, 3H), 3.07–3.04 (m, 1H), 2.80–2.77 (m, 1H), 2.00–1.30 (m, 8H). 13C NMR (100 MHz, CDCl3): δ 162.4, 150.1, 141.4, 50.7, 39.8, 38.1, 23.5, 23.3, 18.6, 18.0.
Methyl Bicyclo[5.2.0]non-8-ene-8-carboxylate, 5
AlCl3 (1.00 g, 7.56 mmol, 0.5 equiv), methyl propiolate (1.27 g, 15.2 mmol), and cycloheptene (2.16 mL, 18.2 mmol, 1.2 equiv) were stirred for 7 days in dry benzene (50 mL). Flash column chromatography of the crude product (30:70/hexane:CH2Cl2) yielded 5 (1.40 g, 50% yield). 1H NMR (400 MHz, CDCl3): δ 6.70 (d, J = 1 Hz, 1H), 3.61 (s, 3H), 2.99 (m, 1H), 2.77 (m, 1H), 1.77–1.21 (m, 10H). 13C NMR (100 MHz, CDCl3): δ 162.6, 148.9, 140.5, 50.6, 46.9, 45.4, 31.6, 29.1, 28.6, 27.9, 27.7.
General Procedure for NMR Scale AROMP Reactions
All kinetic experiments were performed at least twice, and preparative polymerization experiments were performed three times. Under an N2 atmosphere, a solution of monomer A (cyclobutene derivative) in CD2Cl2 (300 μL) was added to the NMR tube. Then 300 μL of Grubbs III stock solution (C = 0.02 M) was added to the NMR tube. After complete mixing of the solution, NMR spectra were acquired at 25 °C until the catalyst had reacted with monomer A as determined by the disappearance of its α alkylidene proton signal. Monomer B (cyclohexene 6) was added to the NMR tube. After no further propagation occurred, the reaction was quenched with ethyl vinyl ether and stirred for 1 h. Solvent was evaporated, and polymer was purified by chromatography over silica gel (97:3/CH2Cl2:acetone). Yield was determined by assuming 100% conversion of monomer A.
NMR AROMP of 2 and 6
Monomer 2 (8.3 mg, 60 μmol, 10 equiv) and Grubbs III catalyst (5.3 mg, 6.0 μmol, 1 equiv) were mixed in CD2Cl2. The reaction was followed by 1H NMR spectroscopy at 25 °C for 5 h before the temperature was elevated to 50 °C. Cyclohexene 6 (12 μL, 120 μmol, 20 equiv) was added. No change in the alkylidene peak of the catalyst was observed within 300 min.
NMR AROMP of 3 and 6, Poly(3-alt-6)13
Monomer 3 (23.8 mg, 150 μmol, 25 equiv) and Grubbs III catalyst (5.3 mg, 6.0 μmol, 1 equiv) were mixed. Cyclohexene 6 (24.5 mg, 30 μL, 50 equiv) was added 30 min later. The NMR tube was spun for 19 h at 25 °C. Flash column chromatography (97:3/CH2Cl2:acetone) of the crude product yielded poly(3-alt-6)13 (15 mg, 43%). 1H NMR (600 MHz, CD2Cl2): δ 7.35–7.14 (m, 5H), 6.60 (m, 13H), 5.32 (m, 27H), 3.68–3.60 (m, 54H), 3.20–3.10 (m, 14H), 2.0–2.3 (m, 10H), 2.60–1.00 (m, 267H).
NMR AROMP of 3 and 6-d10, Poly(3-alt-6-d10)6
Monomer 3 (9.5 mg, 50 μmol, 10 equiv) and Grubbs III catalyst (5.3 mg, 6.0 μmol, 1 equiv) were mixed. Cyclohexene 6-d10 (9.8 mg, 12 μL, 20 equiv) was added 30 min later. The NMR tube was spun for 8 h at 25 °C. The crude product was purified by flash column chromatography (97:3/CH2Cl2:acetone) to yield poly(3-alt-6-d10)6 (4.8 mg, 40%). 1H NMR (600 MHz, CD2Cl2): δ 7.35–7.14 (m, 5H), 7.02 (dd, J = 8.1, 5.7 Hz, 1H), 5.32 (m, 6H), 3.68–3.60 (m, 22H), 3.20–1.00 (m, 80H).
Alternating ring-opening polymerization of monomer 4 and 6 was carried out at different temperatures ranging from 25 to 60 °C to optimize reaction conditions.
NMR AROMP of 4 and 6, Poly(4-alt-6)16
Monomer 4 (19.9 mg, 120 μmol, 20 equiv) and Grubbs III catalyst (5.3 mg, 6.0 μmol, 1 equiv) were mixed in CD2Cl2. Cyclohexene 6 (19.7 mg, 24 μL, 40 equiv) was added after 50 min. The NMR tube was spun for 8 h at 25 °C to reach 90% consumption of monomer 4. The crude product was subjected to flash column chromatography (97:3/CH2Cl2:acetone) to yield poly(4-alt-6)16 (16 mg, 53%). 1H NMR (600 MHz, CD2Cl2): δ 7.35–7.14 (m, 5H), 6.52 (m, 16H), 5.79 (m, 15H), 5.31 (m, 26H), 3.69–3.59 (m, 45H), 2.60 (m, 23H), 2.25 (m, 60H), 2.00–1.22 (m, 266H). 13C NMR (100 MHz, CDCl3): δ 169.4, 142.4, 141.9, 136.3, 131.2, 130.7, 130.6, 130.4, 130.2, 130.2, 130.0, 129.1, 128.4, 51.5, 51.5, 51.4, 43.9, 42.8, 37.3, 28.7, 28.1, 21.8, 21.7.
NMR AROMP of 4 and 6, Poly(4-alt-6)16
Monomer 4 (19.9 mg, 120 μmol, 20 equiv) and Grubbs III catalyst (5.3 mg, 6.0 μmol, 1 equiv) were mixed in CD2Cl2, and cyclohexene 6 (19.7 mg, 24.2 μL, 40 equiv) was added after 50 min. The NMR tube was spun for 6 h at 35 °C to reach >95% consumption of monomer 4. The crude product was subjected to flash column chromatography (97:3/CH2Cl2:acetone) to yield poly(4-alt-6)16 (20 mg, 65%). 1H NMR (600 MHz, CDCl3): δ 7.35–7.14 (m, 6H), 6.52 (m, 16H), 5.79 (m, 15H), 5.50 (m, 1H), 5.31 (m, 17H), 3.69–3.59 (m, 47H), 2.78 (m, 23H), 2.10 (m, 64H), 1.98–1.20 (m, 263H).
NMR AROMP of 4 and 6, Poly(4-alt-6-d10)15
Monomer 4 (19.9 mg, 120 μmol, 20 equiv) and Grubbs III catalyst (5.3 mg, 6.0 μmol, 1 equiv) were mixed in CD2Cl2. After 50 min, cyclohexene-d106-d10 (19.7 mg, 24.2 μL, 40 equiv) was added. The NMR tube was spun for 6 h at 35 °C to reach >95% consumption of monomer 4. The product was purified by flash column chromatography (97:3/CH2Cl2:acetone) to yield poly(4-alt-6-d10)15 (18 mg, 60%). 1H NMR (600 MHz, CD2Cl2): δ 7.35–7.4 (m, 5H), 5.78 (m, 15H), 5.42 (m, 2H), 3.70–3.60 (m, 45H), 2.82 (m, 23H), 2.25 (m, 27H), 1.78–1.20 (m, 164H).
NMR AROMP of 4 and 6, Poly(4-alt-6)34
Monomer 4 (49.7 mg, 300 μmol, 50 equiv) and Grubbs III catalyst (5.3 mg, 6.0 μmol, 1 equiv) were mixed in CD2Cl2, and cyclohexene 6 (49.2 mg, 60.6 μL, 100 equiv) was added after 30 min. The NMR tube was spun for 6 h at 35 °C to reach 68% conversion of monomer 4. Partial 1H NMR of crude poly(4-alt-6)34 (600 MHz, CD2Cl2): δ 7.35–7.14 (m, 5H), 6.89 (d, J = 1.2 Hz, 8H), 6.50 (m, 34H), 5.79 (m, 32H), 5.28 (m, 41H), 3.70–3.60 (m, 150H), 3.07–3.04 (m, 10H). (Partial 1H NMR spectroscopic data are reported due to incomplete polymerization and significant upfield overlap of 4 and 6 with the new peaks from the polymer.)
NMR AROMP of 4 and 6, Poly(4-alt-6)36
Monomer 4 (49.7 mg, 300 μmol, 50 equiv) and Grubbs III catalyst (5.3 mg, 6.0 μmol, 1 equiv) were mixed in CD2Cl2. After 30 min, cyclohexene 6 (49.2 mg, 60.6 μL, 100 equiv) was added. The NMR tube was spun for 2 h at 60 °C to reach 72% conversion of monomer 4. The product was purified by flash column chromatography (97:3/CH2Cl2:acetone) to yield poly(4-alt-6)36 (36 mg, 65%). 1H NMR (600 MHz, CD2Cl2): δ 7.35–7.14 (m, 5H), 6.50 (m, 36H), 5.79 (m, 34H), 5.28 (m, 55H), 3.70–3.60 (m, 129H), 2.75 (m, 53H), 2.30–2.10 (m, 142H), 1.95–1.20 (m, 589H).
NMR AROMP of 5 and 6, Poly(5-alt-6)10.
Monomer 5 (54.8 mg, 300 μmol, 50 equiv) and Grubbs III catalyst (5.30 mg, 6.00 μmol, 1 equiv) were mixed in CD2Cl2, and cyclohexene 6 (49.0 mg, 60 μL, 100 equiv) was added after 50 min. The NMR tube was spun for 72 h at 35 °C until no further propagation was observed by 1H NMR spectroscopy. The crude product was subjected to flash column chromatography (97:3/CH2Cl2:acetone) to yield poly(5-alt-6)10 (11 mg, 14%). 1H NMR (600 MHz, CD2Cl2): δ 7.35–7.12 (m, 5H), 6.58 (m, 10H), 5.42 (m, 19H), 5.28 (m, 10H), 3.70–3.60 (m, 36H), 3.10–3.00 (m, 10H), 2.77–1.95 (m, 254H).
General Procedure for Ring-Opening Metathesis
Under an N2 atmosphere, a solution of monomer A (cyclobutene derivative, 1, 3, 4, or 5, [A] = 0.03 M) in CD2Cl2 (300 μL) was added to an NMR tube. Then 300 μL of the stock solution of Grubbs III catalyst (C = 0.03 M) was added to the NMR tube. After complete mixing of the solution, the reaction was closely monitored by 1H NMR or 13C NMR spectroscopy.
Procedure for Alternating Ring-Opening Metathesis (AROM-1, BA Dimer Synthesis)
A solution of monomer A (3 or 4, [A] = 0.03 M) in CD2Cl2 (300 μL, 18.86 μmol) was added to an NMR tube that had been flushed with N2. Then 300 μL of the stock solution of Grubbs III catalyst (C = 0.03 M) was added to the NMR tube. After complete mixing of the solution, the reaction was followed by 1H NMR or 13C NMR spectroscopy until >90% of the catalyst (10–12 h) was consumed as determined by disappearance of the Ru alkylidene proton or carbon resonance of the Grubbs III catalyst at 19.1 or 316.1 ppm. Then cyclohexene 6 was added in 10-fold excess, and the reaction was monitored until the Ru alkylidene proton resonance at 19.0 ppm disappeared. The reaction was terminated with ethyl vinyl ether, and the crude mixture was subjected to silica chromatography (100% CH2Cl2). For the products of the 3−6 AROM-1 experiment, partially purified fractions were characterized by mass spectrometry, 1H NMR, 13C NMR, and HSQC spectroscopy (Supporting Information). Fraction I was a white solid identified as E-stilbene. 1H NMR (850 MHz, CD2Cl2): δ 7.53 (dd, J = 8.1, 0.9 Hz, 2H), 7.36 (t, J = 7.7 Hz, 2H), 7.28–7.23 (m, 2H), 7.13 (s, 1H). 13C NMR (214 MHz, CD2Cl2): δ 137.9, 129.2, 129.1, 128.2, 127.0. ESI (M/Z) [M + H]+ 180.1. Fraction II contained Ph-(3-alt-6)1-Ph as the major component (Supporting Information). Fraction III contained cyc-(3-alt-6)1 as the major component (Supporting Information).
Procedure for Sequential Alternating Ring-Opening Metathesis (AROM-2, BA′BA Tetramer Synthesis)
A solution of monomer A (3 or 4, [A] = 0.03 M) in CD2Cl2 (300 μL, 18.86 μmol) was added to an NMR tube that had been flushed with N2. Then 300 μL of the stock solution of Grubbs III catalyst (C = 0.03 M) was added to the NMR tube. After complete mixing of the solution, the reaction was followed by 1H NMR spectroscopy until >90% of the catalyst (10–12 h) was consumed as determined by disappearance of the Ru alkylidene proton of the Grubbs III catalyst at 19.1 ppm; then cyclohexene 6 was added in 10-fold excess. Generation of [Ru]-B-A was monitored by the appearance of a multiplet resonance at 19.0 ppm. When the formation of [Ru]-B-A was complete as judged by the integrated intensity of the resonance, 1 equiv of monomer A′ was added to form [Ru]-A′-B-A and then [Ru]-B-A′-B-A. The reaction was monitored until the Ru alkylidene proton resonance at 19.0 ppm disappeared or the intensity was constant. Then the reaction was terminated with ethyl vinyl ether.
Results and Discussion
Design and Synthesis of Monomers
We noted that using Grubbs III catalyst for the incorporation of a ring into the propagating chain backbone limits backbiting in the case of the well-investigated norbornene ROMP.35 Therefore, we decided to examine norbornene ester 2 as a partner for cyclohexene in AROMP. We prepared monomer 2 by a modification of the method of Elsheimer (see Experimental Methods).32 However, when we subjected monomer 2 to AROMP conditions with cyclohexene 6, we observed no polymerization. Furthermore, when the catalyst was mixed with monomer 2, no ring-opened product was observed at all. We attributed the loss of ROMP activity of monomer 2 to the steric hindrance posed by the combination of the bridging methylene group and the ester.
We next tested bicyclic monomers 3–5, designed for their ring strain and lower steric hindrance around the alkenes (Figure 1). These monomers were prepared by Lewis acid catalyzed [2 + 2] cycloaddition according to Snider’s approach.34 For practicality, we developed a simplified purification procedure for monomer 4 to remove the isomeric byproduct (see Experimental Methods).
Figure 1.

Monomers employed in alternating ring-opening metathesis polymerization.
Relative Kinetics of Ring-Opening Metathesis (ROM)
First, we undertook kinetic monitoring of the initial ring-opening metathesis (ROM) reactions for each of these monomers by 1H NMR spectroscopy (Figure 2). In each of the experiments, an equimolar amount of monomer A (the bicycloalkene ester) and Grubbs III catalyst was mixed in CD2Cl2. The disappearance of the alkylidene signal of the catalyst at 19.1 ppm was followed by 1H NMR spectroscopy; the signal was integrated relative to the methyl ester signals between 3.8 and 3.5 ppm. Under the conditions of the experiment, 50% of monomer 3 was ring opened in 25 min, whereas 50% of monomer 1 was ring-opened in 40 min. Under the same conditions, monomer 4 underwent 50% ring-opening in 100 min and monomer 5 required 300 min for 50% ring-opening. Thus, upon addition of Grubbs III catalyst, monomer 3 has the fastest ring-opening rate, in accordance with the predicted ring strains.36−38
Figure 2.
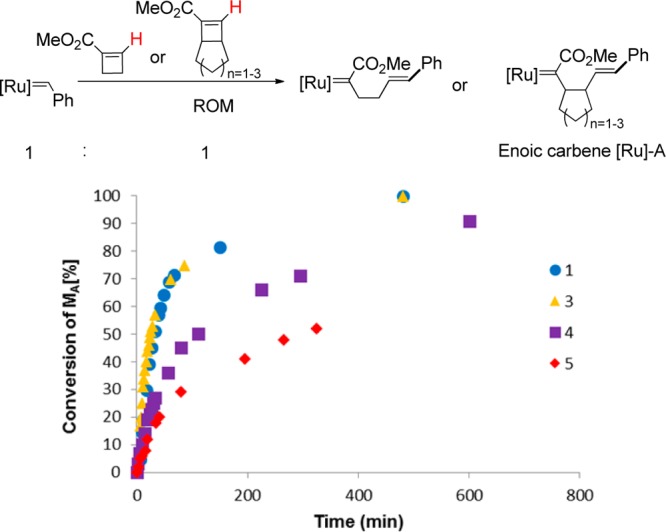
Kinetic monitoring of ring-opening metathesis of monomers 1 and 3–5. Monomer and Grubbs III catalyst were mixed in a 1:1 ratio, [A] = [Grubbs III] = 0.03 M. Percent conversion was determined by 1H NMR spectroscopy and integration of the Ru alkylidene α proton resonance relative to the methyl ester resonances (Supporting Information). t1/2 were obtained from the plot, monomer 1: t1/2 = 40 min, monomer 3: t1/2 = 25 min, monomer 4: t1/2 = 100 min; monomer 5: t1/2 = 300 min. Each experiment was performed at least twice, and data from representative experiments are shown.
Alternating Copolymers
When subjected to Grubbs III catalyst in the absence of cyclohexene 6, each of the three bicyclic monomers 3, 4, and 5 underwent ring-opening; however, no polymerization could be detected. Thus, we established that these monomers are suitable for the preparation of alternating copolymers because their rates of homopolymerization are zero. All three monomers produced copolymers with cyclohexene 6 in the presence of Grubbs III catalyst. The rates of polymerization varied substantially, and all were slower than the rate of AROMP between cyclobutene ester 1 and cyclohexene 6 as determined by 1H NMR spectroscopy (Table 1). Lengths of polymers were determined by integration of alkene proton peaks (H1, H3, and H4, Figure 3) relative to that of the phenyl end group in the 1H NMR spectrum. Copolymerization of monomer 3 or 5 with 6 yielded much shorter polymers than did that of monomer 4 with 6 under the same conditions (Table 1).
Table 1. AROMP Applications of Sterically Hindered Monomers with Cyclohexene.
| entry | A | B | [A]:[B]:[Ru] | temp (°C) | time (h) | % conva | DP[AB]b |
|---|---|---|---|---|---|---|---|
| 1 | 1 | 6 | 10:20:1 | 25 | 3 | 98 | 10 |
| 2 | 2 | 6 | 10:20:1 | 25 | 3 | 0 | |
| 3 | 3 | 6 | 25:50:1 | 25 | 19 | NAc | 13 |
| 4 | 3 | 6-d10 | 20:40:1 | 25 | 6 | 80 | 6 |
| 5 | 4 | 6 | 20:40:1 | 25 | 8 | 96 | 16 |
| 6 | 4 | 6 | 20:40:1 | 35 | 8 | 97 | 16 |
| 7 | 4 | 6-d10 | 20:40:1 | 35 | 8 | 85 | 15 |
| 8 | 4 | 6 | 50:100:1 | 35 | 8 | 68 | 34 |
| 9 | 4 | 6 | 50:100:1 | 60 | 2 | 72 | 36 |
| 10 | 5 | 6 | 50:100:1 | 35 | 24 | 85 | 10 |
Percent conversion determined by integration of 1H NMR spectra of monomer A unless specified otherwise.
DP[AB] was determined by 1H NMR spectroscopy with integration relative to the phenyl end group and represents the average numbers of AB dyads incorporated in linear copolymers.
% conv could not be determined by 1H NMR spectroscopy due to overlap of alkene and polymer peaks.
Figure 3.

Alternating ring-opening metathesis polymerization (AROMP) of monomers 4 and 6. The region of 1H NMR spectra in which backbone olefinic hydrogen resonances of poly(4-alt-6)n appear is shown. (a) Polymer product prepared from cyclohexene and dissolved in CDCl3. The ratio of H1:H3:H4 is 1:1:1. (b) Polymer product prepared from cyclohexene-d10 and dissolved in CD2Cl2. The ratio of H1:H3:H4 is 0:1:0. poly(4-alt-6-d10)n was dissolved in CD2Cl2 instead of CDCl3 to allow accurate integration of the phenyl resonances.
Furthermore, the 1H NMR spectrum of each of the polymers is consistent with an alternating backbone structure (Supporting Information) in which the olefin bearing a carbomethoxy substituent has an E configuration. For example, in the spectrum of poly(4-alt-6)13, the proton resonance for the carbomethoxy-substituted olefin (H1), which is derived from cyclohexene 6, has an integration identical to that of the H3 alkene resonance at 5.8 ppm which is derived from monomer 4 (Figure 3a). Likewise, the analogous proton resonances in poly(3-alt-6)n and poly(5-alt-6)n have nearly identical integration values. Moreover, characterization of poly(4-alt-6)n by HSQC spectroscopy confirmed that the carbomethoxy-substituted olefin is a single stereoisomer; there is a single H1 signal at 6.5 ppm that correlates with C1 (Supporting Information). Comparison of model compound chemical shifts with the H1 alkene chemical shift further confirmed that the E configuration was obtained.39,40
Further evidence for the alternating structure was obtained for the poly(4-alt-6)n copolymers by experiments with cyclohexene-d10, 6-d10. 1H NMR analysis of the deuterium-labeled copolymer poly(4-alt-6-d10)n indicates a complete loss of the carbomethoxy-substituted olefin (H1) resonance at 6.5 ppm and the H4 alkene resonance at 5.3 ppm (Figure 3b). Complete loss of the H1 and H4 resonances upon deuteration is consistent with a rigorously alternating AB linear scaffold. Their loss indicates the absence of AA dyads that can form upon backbiting during the AROMP.27 Likewise, in the experiments with deuterated cyclohexene, the integrated ratio of H3 versus the methyl ester remains at 1:3, suggesting that no BB dyad is formed. Therefore, introduction of a cyclohexyl ring fused to the cyclobutene ester monomer provides access to linear, alternating copolymers.
All three monomers 3–5 produced rigorously alternating copolymers. However, at the same concentrations and monomer/catalyst ratios, monomers 3 and 5 both generated shorter polymers than did monomer 4 (Table 1). Monomer 4 provided polymers with up to 35–36 AB repeats. Reaction conditions for poly(4-alt-6)n were examined, and the best yield was obtained in CH2Cl2, at temperatures between 35 and 60 °C. The GPC elution profile of poly(4-alt-6)n displayed a monomodal molecular weight distribution (Supporting Information). This distribution is consistent with the absence of cyclic polymer. Likewise, the dispersity (DM = 2.0 ± 0.1) of poly(4-alt-6)n was significantly smaller than that of the previously reported poly(1-alt-6)n (DM = ∼5), which displayed a bimodal molecular weight distribution.27 The molecular weight profile is consistent with the absence of intramolecular chain transfer for the AROMP of monomer 4 and cyclohexene.
Intrinsic Rates of Chain Propagation
Monomer 3 was selected for comparison with monomer 4 in order to study how their structures affect the initiation and propagation rates and the extents of polymerization. To preclude the possibility that impurities in monomer 3 inherited from synthesis (see Experimental Methods) deactivated the catalyst, monomer 3 was also treated with m-CPBA and reisolated. This sample was utilized in AROMP and compared with monomer 3 synthesized otherwise. No difference in activity or polymer length was observed.
Then we undertook kinetic monitoring of AROM-1 (formation of BA dimer) with monomers 3 or 4 and cyclohexene 6 by 13C NMR spectroscopy. In both reactions, we observed formation of [Ru]-A upon addition of the A monomer to Grubbs III catalyst (Figure 4, [Ru] + 3 and [Ru] + 4) and disappearance of Ru catalyst. In the case of [Ru]-4, the formation of a single Ru carbene (314.5 ppm) was observed. Addition of cyclohexene cleanly yielded [Ru]-6-4 (338.0 ppm, Figure 4, 6 + [Ru]-4) in 1.5 h, and there was no further change in the NMR spectrum over 30 h.
Figure 4.

Alternating ring-opening metathesis (AROM-1) of monomers 3 and 6 and monomers 4 and 6. The carbene regions of 13C NMR spectra of (3-alt-6)1 (left) and (4-alt-6)1 (right) are shown. In the top spectra, [Ru] catalyst was mixed with 3 or 4 in CD2Cl2 for 10–12 h. Cyclohexene was added after >90% of Ru catalyst was consumed. The 6 + [Ru]-3 reaction was monitored for 300 min, and 6 + [Ru]-4 reaction was monitored for 30 h. The 13C NMR spectrum of 6 + [Ru]-3 was acquired 30–50 min after addition of cyclohexene, and the 13C NMR spectrum of 6 + [Ru]-4 was acquired 50–70 min after addition of cyclohexene. Each experiment was performed at least twice, and data from representative experiments are shown.
In the case of [Ru]-3, two Ru carbene species were produced (311.5 and 311.8 ppm). After the addition of cyclohexene, we observed regeneration of the Grubbs III catalyst (316.1 ppm) in addition to the [Ru]-6-3 carbene (337.8 ppm, Figure 4, 6 + [Ru]-3) during the first 2 h of reaction. Within 5 h of cyclohexene addition, both the [Ru]-6-3 and Ru catalyst carbene resonances disappeared, and no new carbene resonances appeared.
We analyzed products of the AROM-1 reaction and obtained Ph-(3-alt-6)1-Ph, E-stilbene, and cyc-(3-alt-6)1 (Scheme 1a). Hence, backbiting or cross-metathesis can occur very early in the reaction. An independent experiment in which Ru catalyst was mixed with monomer 3 for 18 h showed that [Ru]-3 remains intact. Thus, the Ru enoic carbene is not reactive with itself, and side reactions must occur after cyclohexene addition. Formation of stilbene suggests that if the desired propagation pathway is kinetically less favorable, the Ru alkylidene ([Ru]-6-3) species undergoes cross-metathesis in an intra- or intermolecular reaction with the styrene end group. In contrast, no regeneration of Grubbs III catalyst was observed in the (4-alt-6)1 experiment (Figure 4, 6 + [Ru]-4). The [Ru]-6-4 carbene remained stable for 2 days under the reaction conditions. This result is consistent with our observations that monomer 4 provides a longer and backbiting-free linear alternating copolymer via AROMP.
Scheme 1. (a) Alternating Ring-Opening Metathesis (AROM-1) of 3 or 4 with Cyclohexene To Form BA Dimer and Proposed Intra- and Intermolecular Cross-Metathesis for (3-alt-6)1; (b) Double Alternating Ring-Opening Metathesis (AROM-2) To Form BA′BA Tetramer.

Carbenes (circled) were monitored by 13C NMR spectroscopy (Figure 4).
The proton resonances monitored are colored red (Figure 5).
We also carried out double AROM (AROM-2) experiments with monomers 3 and 4 to compare their behavior in systems with longer chains (Scheme 1 and Figure 5). In these experiments, we mixed monomer A and catalyst in a 1:1 ratio in an NMR tube to form [Ru]-A, and we monitored the reactions by 1H NMR spectroscopy. The mixtures were allowed to react for 10–12 h to ensure nearly complete conversion to [Ru]-A (the benzylidene proton signal at 19.1 ppm was reduced to less than 10% of its original intensity). At this time, 10 equiv of cyclohexene (monomer B) was added to generate [Ru]-B-A (Figure 5). The formation of [Ru]-B-A was monitored by the appearance of a new multiplet resonance at 19.0 ppm corresponding to the alkylidene proton (Supporting Information). We found that cyclohexene 6 reacts with ring-opened monomer 3 enoic carbene 1.5 times faster (t1/2 = 28 ± 1 min) than with the corresponding enoic carbene from monomer 4 (t1/2 = 43 ± 5 min) (Figure 5 and Supporting Information). When the formation of Ru alkylidene ([Ru]-B-A) was complete as judged by integration of the resonance at 19.0 ppm, 1 equiv of monomer A′ was added to investigate the rate of ring-opening catalyzed by [Ru]-B-A. We examined all four cases of double ROM: ROM of 3 with [Ru]-6-3 and with [Ru]-6-4 and ROM of 4 with [Ru]-6-4 and with [Ru]-6-3. The disappearance of the monomer A′ alkene signal at 6.7 ppm and that of the [Ru]-B-A alkylidene signal at 19.0 ppm were monitored as a function of time. Both signals disappeared at the same rate as measured by comparison of the integrals with that of the signal for ester peaks. We found that reaction of monomer 3 with [Ru]-6-4 (t1/2 = 26 ± 1 min) was 27% faster than its reaction with [Ru]-6-3 (t1/2 = 33 ± 1 min). In the case of the reaction of monomer 4 with [Ru]-6-A, the rate with [Ru]-6-4 (t1/2 = 41 ± 4 min) is 17% faster than the rate with [Ru]-6-3 (t1/2 = 48 ± 2 min). Moreover, oligomers derived from monomer 3 as either A or A′ failed to completely convert to [Ru]-6-A′-6-A; this result is consistent with the premise that competing cross-metathesis reactions dominate the reaction (Figure 5).
Figure 5.
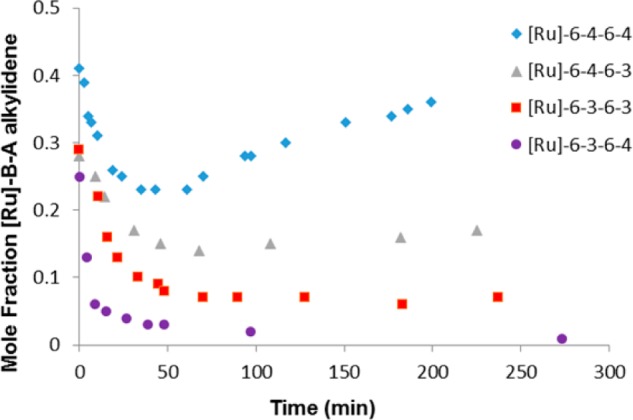
Kinetic monitoring of double ring-opening metathesis (AROM-2) reactions of monomer A (3 or 4) with [Ru]-6-A (Scheme 1b). Time zero corresponds to the addition of 1 equiv of monomer A′ (3 or 4) to 1 equiv of [Ru]-6-A in the presence of excess cyclohexene 6. Mole fraction of Ru alkylidene at 19.0 ppm was determined by integration of the Ru alkylidene resonance relative to the methyl ester resonances between 3.5 and 3.8 ppm. The mole percent of [Ru]-6-A at time zero was only 30–42% due to the low concentrations, and thus, the extended reaction times used in the initial AROM to generate [Ru]-6-A. Each experiment was performed at least twice, and data from representative experiments are shown.
Although the rates of the second ROM did not vary widely, the ROM reaction appears to be very sensitive to long-range polymer structure, i.e., the presence of a five-membered ring in the backbone one position removed from the living [Ru] species reduces the reaction rate of either bicyclic monomer with the unhindered [Ru]-alkylidene. The rates of reaction indicate that the backbone containing a six-membered ring (derived from [4.2.0] monomer) is superior to the backbone containing a five-membered ring (derived from [3.2.0] monomer) for propagation.
We ascribe the differences in reactivity between oligomers derived from monomer 3 versus monomer 4 to the differences in dihedral angles between the cis-1,2 substituents of cyclopentane versus those of cyclohexane (20°–40°, depending on the conformation, and 60°, respectively).41−48 These dihedral angles determine the relative orientation of the two polymer chain ends as the backbone extends from the cyclic moiety. The orientation of the chain ends determines access to the [Ru]-alkylidene. If the chain ends are aligned, intramolecular cross-metathesis will ensue, as is the case for AROMP of monomers 3 and 6. If access of the incoming monomer to [Ru]-alkylidene is hindered, the rate of propagation will be suppressed and the polymerization reaction is unable to compete with cross-metathesis. Thus, despite the higher strain and inherent reactivity of monomer 3, the resulting polymer backbone appears to hinder propagation and to favor cross-metathesis; both effects lead to the premature termination of polymerization.
Conclusions
We have demonstrated that alternating copolymers are synthetically accessible via AROMP with bicyclic carbomethoxy olefin monomers 3–5 and cyclohexene. These AROMP-active monomers do not self-metathesize regardless of monomer feed ratios or concentrations. Thus, formation of homopolymeric blocks, as is observed in some other systems,20,21,26,49,50 does not occur in the 3–5/6 AROMP systems. Ruthenium-catalyzed ring-opening metathesis of cyclohexene and methyl bicyclo[4.2.0]oct-7-ene-7-carboxylate, 4, provides rigorously linear and alternating copolymers free of backbiting and chain transfer. The resulting polymer backbone constrained by a cis-1,2-substituted cyclohexane provides an exciting alternative to traditional norbornyl-derived polymers for the preparation of functional alternating copolymers.
Acknowledgments
We thank the National Institutes of Health (NIH) for grants R01GM097971/R01HD38519 (N.S.S.) and R01GM74776 (K.A.P). We also thank Dr. Francis Picart and James Marecek for their assistance with NMR spectroscopy.
Supporting Information Available
Figures showing characterization of monomers and polymer, GPC, 1D- and 2D-NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): We have no competing interests except for National Institutes of Health Funding: R01GM097971 & R01GM74776.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Siegwart D. J.; Oh J. K.; Matyjaszewski K. Prog. Polym. Sci. 2012, 37, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong B.; Bae Y. H.; Lee D. S.; Kim S. W. Nature 1997, 388, 860. [DOI] [PubMed] [Google Scholar]
- Cölfen H. Macromol. Rapid Commun. 2001, 22, 219. [Google Scholar]
- Schacher F. H.; Rupar P. A.; Manners I. Angew. Chem., Int. Ed. 2012, 51, 7898. [DOI] [PubMed] [Google Scholar]
- Furuta P. T.; Deng L.; Garon S.; Thompson M. E.; Frechet J. M. J. Am. Chem. Soc. 2004, 126, 15388. [DOI] [PubMed] [Google Scholar]
- Furuta P.; Brooks J.; Thompson M. E.; Fréchet J. M. J. J. Am. Chem. Soc. 2003, 125, 13165. [DOI] [PubMed] [Google Scholar]
- Kim H. K.; Baek N. S.; Paik K. L.; Lee Y.; Lee J. H. In Chromogenic Phenomena in Polymers; American Chemical Society: Washington, DC, 2004; Vol. 888, p 247. [Google Scholar]
- ten Brummelhuis N.; Weck M. ACS Macro Lett. 2012, 1, 1216. [DOI] [PubMed] [Google Scholar]
- Romulus J.; Tan L.; Weck M.; Sampson N. S. ACS Macro Lett. 2013, 2, 749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz J.-F.; Kirci B.; Matyjaszewski K. Macromolecules 2003, 36, 3136. [Google Scholar]
- Chen G.-Q.; Wu Z.-Q.; Wu J.-R.; Li Z.-C.; Li F.-M. Macromolecules 1999, 33, 232. [Google Scholar]
- Shirota Y.; Yoshimura M.; Matsumoto A.; Mikawa H. Macromolecules 1974, 7, 4. [Google Scholar]
- Kamigaito M.; Ando T.; Sawamoto M. Chem. Rev. 2001, 101, 3689. [DOI] [PubMed] [Google Scholar]
- Moad G.; Rizzardo E.; Thang S. H. Aust. J. Chem. 2012, 65, 985. [Google Scholar]
- Ouchi M.; Terashima T.; Sawamoto M. Chem. Rev. 2009, 109, 4963. [DOI] [PubMed] [Google Scholar]
- Al Samak B.; Carbill A. G.; Hamilton J. G.; Rooney J. J.; Thompson J. M. Chem. Commun. 1997, 2057. [Google Scholar]
- Amir-Ebrahimi V.; Corry D. A.; Hamilton J. G.; Thompson J. M.; Rooney J. J. Macromolecules 2000, 33, 717. [Google Scholar]
- Amir-Ebrahimi V.; Rooney J. J. J. Mol. Catal. A: Chem. 2004, 208, 115. [Google Scholar]
- Torker S.; Müller A.; Chen P. Angew. Chem. 2010, 122, 3850. [DOI] [PubMed] [Google Scholar]
- Bornand M.; Torker S.; Chen P. Organometallics 2007, 26, 3585. [Google Scholar]
- Bornand M.; Chen P. Angew. Chem., Int. Ed. 2005, 44, 7909. [DOI] [PubMed] [Google Scholar]
- Torker S.; Müller A.; Sigrist R.; Chen P. Organometallics 2010, 29, 2735. [Google Scholar]
- Vehlow K.; Lichtenheldt M.; Wang D.; Blechert S.; Buchmeiser M. R. Macromol. Symp. 2010, 296, 44. [Google Scholar]
- Vehlow K.; Wang D.; Buchmeiser M. R.; Blechert S. Angew. Chem., Int. Ed. 2008, 47, 2615. [DOI] [PubMed] [Google Scholar]
- Buchmeiser M. R.; Ahmad I.; Gurram V.; Kumar P. S. Macromolecules 2011, 44, 4098. [Google Scholar]
- Ilker M. F.; Coughlin E. B. Macromolecules 2001, 35, 54. [Google Scholar]
- Song A.; Parker K. A.; Sampson N. S. J. Am. Chem. Soc. 2009, 131, 3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutthasupa S.; Shiotsuki M.; Masuda T.; Sanda F. J. Am. Chem. Soc. 2009, 131, 10546. [DOI] [PubMed] [Google Scholar]
- Choi T.-L.; Lee C. W.; Chatterjee A. K.; Grubbs R. H. J. Am. Chem. Soc. 2001, 123, 10417. [DOI] [PubMed] [Google Scholar]
- Song A.; Parker K. A.; Sampson N. S. Org. Lett. 2010, 12, 3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love J. A.; Morgan J. P.; Trnka T. M.; Grubbs R. H. Angew. Chem., Int. Ed. 2002, 41, 4035. [DOI] [PubMed] [Google Scholar]
- Elsheimer S.; Slattery D. K.; Michael M.; Weeks J.; Topoleski K. J. Org. Chem. 1989, 54, 3992. [Google Scholar]
- Mathias L. J. Synthesis (Stuttgart) 1979, 561. [Google Scholar]
- Snider B. B.; Rodini D. J.; Cionn R. S. E.; Sealfon S. J. Am. Chem. Soc. 1979, 101, 5283. [Google Scholar]
- Choi T.-L.; Grubbs R. H. Angew. Chem., Int. Ed. 2003, 42, 1743. [DOI] [PubMed] [Google Scholar]
- Baird N. C. Tetrahedron 1970, 26, 2185. [Google Scholar]
- Schleyer P. R.; Williams J. E.; Blanchard K. R. J. Am. Chem. Soc. 1970, 92, 2377. [Google Scholar]
- Wiberg K. B. Angew. Chem., Int. Ed. 2003, 25, 312. [Google Scholar]
- Lee J. C.; Parker K. A.; Sampson N. S. J. Am. Chem. Soc. 2006, 128, 4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beak P.; Kempf D. J.; Wilson K. D. J. Am. Chem. Soc. 1985, 107, 4745. [Google Scholar]
- Allinger N. L.; Tribble M. T.; Miller M. A.; Wertz D. H. J. Am. Chem. Soc. 1971, 93, 1637. [Google Scholar]
- Hoyland J. R. J. Chem. Phys. 1969, 50, 2775. [Google Scholar]
- Allinger N. L.; Hirsch J. A.; Miller M. A.; Tyminski I. J.; Van Catledge F. A. J. Am. Chem. Soc. 1968, 90, 1199. [Google Scholar]
- Fuchs B. In Topics in Stereochemistry; Eliel E. L., Wilen S. H., Allinger N. L., Eds.; Interscience: 1978; Vol. 10, p 1. [Google Scholar]
- Wilson N. K.; Stochters J. B.; Bucourt R.; Kellie G. M.; Friddell F. G.; Moriarty R. M. In Topics in Stereochemsitry; Eliel E. L., Wilen S. H., Allinger N. L., Eds.; Interscience: New York, 1974; Vol. 8, p 1. [Google Scholar]
- Testa B.; Vistoli G.; Pedretti A. Helv. Chim. Acta 2013, 96, 564. [Google Scholar]
- Abraham R. J.; Koniotou R.; Sancassan F. J. Chem. Soc., Perkin Trans. 2 2002, 2025. [Google Scholar]
- Abraham R. J.; Koniotou R. Magn. Reson. Chem. 2003, 41, 1000. [DOI] [PubMed] [Google Scholar]
- Choi T.-L.; Rutenberg I. M.; Grubbs R. H. Angew. Chem., Int. Ed. 2002, 41, 3839. [DOI] [PubMed] [Google Scholar]
- Romulus J.; Patel S.; Weck M. Macromolecules 2011, 45, 70. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.