

Abstract
Genetic variation in SLC12A5 which encodes KCC2, the neuron-specific cation-chloride cotransporter that is essential for hyperpolarizing GABAergic signaling and formation of cortical dendritic spines, has not been reported in human disease. Screening of SLC12A5 revealed a co-segregating variant (KCC2-R952H) in an Australian family with febrile seizures. We show that KCC2-R952H reduces neuronal Cl− extrusion and has a compromised ability to induce dendritic spines in vivo and in vitro. Biochemical analyses indicate a reduced surface expression of KCC2-R952H which likely contributes to the functional deficits. Our data suggest that KCC2-R952H is a bona fide susceptibility variant for febrile seizures.
Keywords: dendritic spines, febrile seizures, genic intolerance, KCC2, mutation
Introduction
GABAergic inhibition in the mammalian brain is conveyed by the conductance increase associated with the opening of GABAA receptor channels (GABAARs) (shunting inhibition) and by the postsynaptic hyperpolarization (voltage inhibition) caused by the conductive Cl− influx along its electrochemical gradient, which is largely maintained by the neuron-specific K-Cl cotransporter, KCC2 1. During mammalian brain development, the functional expression of KCC2 undergoes a dramatic increase 1–3 that is critical for the establishment and maintenance of hyperpolarizing inhibitory postsynaptic potentials 1. More recently, rodent KCC2 has (independently of its role as an ion transporter) been shown to play an important role as a structural protein required for the morphological and functional maturation of cortical dendritic spines 4–6. Thus, mutations in KCC2 can cause perturbations in the functions of GABAergic and glutamatergic transmitter systems and contribute to generation of epileptiform discharges.
Animal models with expression deficits of KCC2 orthologues have been described. Mutations in the Drosophila melanogaster KCC gene kazachoc (kcc) confer increased seizure susceptibility 7. The mammalian KCC2 gene generates two N-terminal splice isoforms, KCC2a and KCC2b, but expression of KCC2b only is strongly up-regulated during development and accounts for most (> 90%) of the total KCC2 protein in the adult murine cortex 8. GABAergic responses remain depolarizing in KCC2b knockout cortical cultures 9, indicating that this isoform is responsible for the establishment of hyperpolarizing GABAAR-mediated transmission 1. Homozygous KCC2b knockout mice exhibit generalized seizures and die during the second postnatal week, whereas KCC2b heterozygotes, which express ~50% of the wild-type (WT) mouse KCC2 (mKCC2) protein level, show increased susceptibility to pentylenetetrazole-induced seizures 10.
We analyzed the gene coding for human KCC2, SLC12A5, for mutations in patients with seizure disorders. Here, we report the functional characterization of a rare SLC12A5 variant. This missense variant, resulting in an arginine-to-histidine substitution at position 952 in KCC2b (KCC2-R952H), was found in an Australian family with early childhood onset of febrile seizures. Our functional and structural analyses in rodent cortical neurons in vivo and in vitro indicate that this mutation brings about deficits in both neuronal Cl− extrusion and the formation of dendritic spines. Functional studies on rare variants are important as they provide insights into gene malfunctions which are not accessible through genetic analyses alone 11. The significant functional deficit shown in this study, taken together with the co-segregation in the family and the low genic tolerance of SLC12A5, is consistent with a role for KCC2-R952H as a susceptibility gene in febrile seizures.
Results
Identification of the human KCC2 variant
We analyzed 378 (266 unrelated) patients with seizure disorders, including febrile seizures, febrile seizures plus and generalized epilepsy, and found a total of 11 rare (minor allele frequency [MAF] of < 1%) variants in SLC12A5. In one family with febrile seizures (Fig 1A), a missense variant c.2855G>A (NM_020708.4) was identified in SLC12A5, and it was not present in ethnically matched controls. The frequency of this variant is low (MAF 0.07%) in publicly available variant databases (rs142740233 in NIH dbSNP, Ensembl 1000 genomes and NHLBI Exome Variant Server), consistent with a role in disease susceptibility. The variant exon will be present in both KCC2b and KCC2a isoforms with an Arg to His substitution at positions 952 and 975, respectively. This arginine is highly conserved across mammalian and avian species (Fig 1B and C). We selected the shorter KCC2b isoform for further study because it is preferentially expressed in the cortex and hippocampus and has been implicated in GABAergic signaling 1. For convenience, we refer to the KCC2b variant as KCC2-R952H.
Figure 1. Segregation, conservation and location of the KCC2-R952H variant.
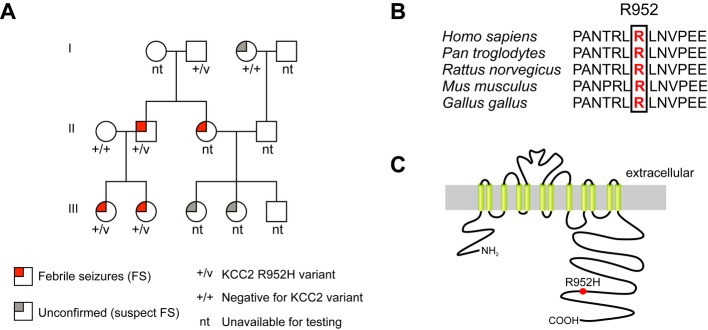
A Pedigree of the family with the KCC2-R952H variant, showing segregation of the variant with the febrile seizures phenotype.
B Amino acid sequence alignment of KCC2 shows high conservation of R952 among different species.
C Putative membrane topology of KCC2. R952H is located in the distal part of the intracellular C-terminus.
KCC2-R952H was found in a family with febrile seizures (Fig 1A). In one branch, there were four affected subjects (II-2, II-3, III-1, III-2) with well-characterized and infrequent febrile seizures (1–3 attacks) occurring between 12 months and 2.5 years of age. The variant was inherited from a grandfather (I-2) with no knowledge of childhood febrile seizures (FS) and with no living parents to confirm his seizure status. For similar reasons, the seizure status of I-3 could not be confirmed. The inherited variant was present in all three affected family members tested, with one affected individual (II-3) not available for genotyping. In the second branch, information on the precise phenotype was not available and testing was limited.
Functional analysis of KCC2-R952H
We used surface protein biotinylation and immunoblotting to compare the total and cell surface expression levels of KCC2-WT and the mutated KCC2-R952H protein in a mouse neural stem cell line (Fig 2). While no difference in the total protein expression level was observed between KCC2-WT and KCC2-R952H (98.6 ± 0.16% of WT; P = 0.625), the surface expression of KCC2-R952H was markedly reduced (61 ± 0.16% of WT; P = 0.004).
Figure 2. The R952H substitution in KCC2 leads to a reduction in surface expression.
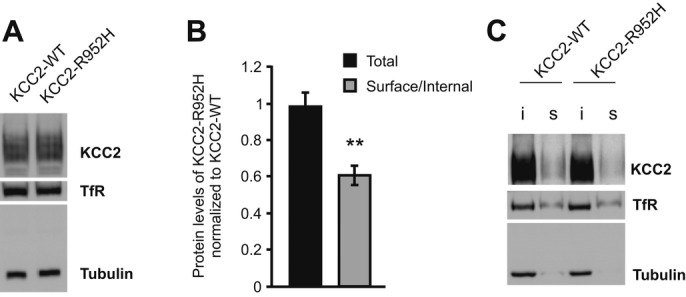
A Western blot analysis of total protein shows comparable expression levels of KCC2-WT and KCC2-R952H, when heterologously expressed in C17.2 cells.
B Quantification of total (n = 5) and cell surface (n = 9) protein expression of KCC2-R952H, normalized to KCC2-WT. Statistical analysis was performed using Wilcoxon matched pairs test. **P < 0.01. Error bars represent SEM.
C Representative Western blot of biotinylated (s, surface) and non-biotinylated (i, internal) KCC2-WT and KCC2-R952H. Tubulin and transferrin receptor (TfR) served as loading controls.
Source data are available online for this figure.
In order to investigate the capacity of KCC2-R952H to transport Cl−, we took advantage of the very low endogenous level of functional KCC2 expression in rodent cortical pyramidal neurons during the first postnatal week 2,12,13. In utero electroporation (IUE) with rat KCC2 (rKCC2) has been shown to result in a precocious hyperpolarizing shift in the reversal potential of GABAAR-mediated currents (EGABA) at postnatal day (P) 6 in somatosensory layer 2/3 cortical pyramidal neurons 12. We observed a significant increase in the Cl− extrusion capacity, quantified as the bumetanide-insensitive somatodendritic EGABA gradient (ΔEGABA) 2,4,13, in P6-7 mouse somatosensory layer 2/3 cortical pyramidal neurons electroporated in utero with human KCC2-WT (ΔEGABA = −5.01 ± 0.40 mV/50 μm; P < 0.001 vs. non-transfected EGFP-negative neurons [control] with ΔEGABA = −2.24 ± 0.31 mV/50 μm) (Fig 3). As expected, neurons electroporated with a known transport-deficient N-terminally deleted rat construct (rKCC2-ΔNTD) 4 displayed a very low level of Cl− extrusion capacity (ΔEGABA = −2.07 ± 0.38 mV/50 μm) that was not different from that observed in EGFP-negative neurons or those transfected with EGFP alone (ΔEGABA = −2.19 ± 0.39 mV/50 μm) (Fig 3C), consistent with lack of functional mKCC2 at this age, cf. for example 2. In neurons transfected with KCC2-R952H, the mean Cl− extrusion capacity (ΔEGABA = −3.28 ± 0.33 mV/50 μm) was slightly higher when compared to non-transfected controls (P = 0.034; KCC2-R952H vs. control), but failed to reach the level of Cl− extrusion capacity generated by KCC2-WT (P = 0.002; KCC2-R952H vs. KCC2-WT) (Fig 3C). Thus, the R952H mutation is associated with deficits in maintaining the Cl− driving force required for hyperpolarizing GABAAR-mediated responses.
Figure 3. Cl− extrusion capacity measurements from in utero electroporated cortical neurons reveal impaired Cl− extrusion by KCC2-R952H.

A IUE of EGFP and either KCC2-WT or KCC2-R952H at embryonic day 14.5 targets mouse cortical layer 2/3 pyramidal neurons (analysis at postnatal day (P) 6). Transfected neurons co-express either of the KCC2 constructs (red) with EGFP, while endogenous mKCC2 levels in non-transfected neurons at this age are low. Scale bars: 100 μm and 20 μm (insets).
B Whole-cell patch clamp recordings of GABA uncaging-elicited GABAA-mediated currents (IGABA) in transfected cortical layer 2/3 pyramidal neurons with a somatically imposed Cl− load. Sample EGABA recordings and corresponding I-V curves at the soma and dendrite. Horizontal bars in the sample traces indicate the duration of the uncaging UV-flash.
C Cl− extrusion capacity of P6-7 cortical pyramidal neurons expressing EGFP (n = 8), KCC2-WT (n = 15), KCC2-R952H (n = 16) or rKCC2-ΔNTD (n = 8) was quantified as the somatodendritic EGABA gradient (ΔEGABA = EGABA-soma − EGABA-dendrite). EGFP-negative neurons (n = 17) served as controls. Statistical analysis was performed using one-way ANOVA with Holm–Sidak post hoc test. *P < 0.05; **P < 0.01. Error bars represent SEM.
The C-terminal domain is critical for both the ion-transport and structural role of KCC2 4,5,14. Our previous work showed that this domain of rKCC2 interacts with the actin cytoskeleton 4,15 to promote spine formation 4,5. IUE of full-length rKCC2, rKCC2-ΔNTD, or the isolated C-terminal domain of rKCC2, has been shown to lead to a persistent increase in the density of functional dendritic spines of layer 2/3 pyramidal neurons in vivo during the brain growth spurt 5. We analyzed the spine density of Lucifer yellow-filled P15 layer 2/3 pyramidal neurons from rats, electroporated in utero with either KCC2-WT or KCC2-R952H. In line with our previous data obtained using rKCC2 5, confocal analysis of the second-order dendritic shafts of neurons co-electroporated with KCC2-WT and EGFP revealed substantially increased spine densities in both apical (1.56 ± 0.07/μm; P = 0.012) and basal (1.62 ± 0.01/μm; P = 0.007) dendrites, as compared to apical (1.18 ± 0.08/μm) and basal (1.35 ± 0.06/μm) dendrites of neighboring EGFP-negative neurons (Fig 4A). Importantly, no significant effect on either apical (1.29 ± 0.05/μm; P = 0.126) or basal (1.18 ± 0.07/μm; P = 0.283) spine density was observed in neurons co-electroporated with KCC2-R952H and EGFP when compared to apical (1.18 ± 0.08/μm) and basal (1.14 ± 0.06/μm) dendrites of the surrounding non-transfected EGFP-negative neurons (Fig 4A).
Figure 4. KCC2-R952H is unable to induce dendritic spines in vivo or rescue mature spine morphology of cortical mKCC2−/− neurons.
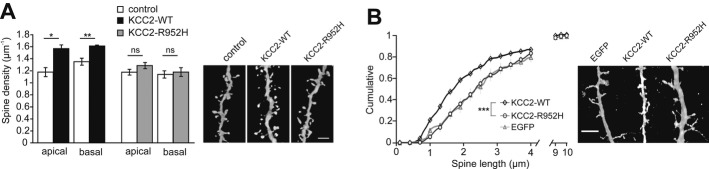
A In utero electroporation of E17.5 rat embryos with KCC2-WT leads to an increase of both apical (n = 28 neurons) and basal dendrite (n = 40) spine density of cortical layer 2/3 pyramidal neurons, when compared to non-transfected neighboring neurons (apical, n = 18; basal, n = 22). In contrast, KCC2-R952H is unable to induce dendritic spines (transfected: apical, n = 37; basal = 46; non-transfected: apical, n = 22; basal, n = 28). Analysis of spine density was performed on P15. Right panel: representative confocal images of spine densities. Statistical analysis was performed using Student’s paired t-test. *P < 0.05; **P < 0.01.
B Expression of KCC2-R952H is incapable of rescuing normal spine morphology in cultured mKCC2−/− cortical neurons. Aberrant spine morphology of mKCC2−/− neurons is rescued by expression of KCC2-WT (n = 19 neurons), but not KCC2-R952H (n = 12) or EGFP (n = 12). Right panel: representative confocal images of spine lengths. Statistical analysis was performed using the Kolmogorov–Smirnov test. ***P < 0.001.
Data information: Scale bars, 5 μm. Error bars represent SEM.
In order to further study the effects of the R952H mutation on the maturation and maintenance of dendritic spines, we used cultured mouse cortical KCC2−/− neurons, which lack mature dendritic spines and instead display elongated filopodia-like dendritic protrusions 4. We found a rescuing effect on spine length of mKCC2−/− neurons after transfection with KCC2-WT (2.19 ± 0.19 μm; P < 0.001), but not with KCC2-R952H (2.83 ± 0.17 μm; P = 0.3), when compared to EGFP alone (3.12 ± 0.28 μm) (Fig 4B). Thus, our present in vivo and in vitro data indicate that the R952H mutation compromises the participation of KCC2 in the formation and maturation of cortical dendritic spines.
Discussion
To date, no loss-of-function mutation of the SLC12A5 gene encoding for human KCC2 has been described. Here, we provide a detailed functional analysis of a novel KCC2 missense variant KCC2-R952H, which was identified in an Australian family with febrile seizures. Using mammalian cortical expression systems, we demonstrate that the R952H mutation confers a deficit in neuronal Cl− extrusion capacity (Fig 3). In addition, our data in vivo (based on IUE) and in vitro (based on cultured mKCC2−/− cortical neurons) show that the R952H mutation impairs the contribution of KCC2 to the formation of dendritic spines (Fig 4). Notably, R952 is located in the intracellular C-terminal domain of KCC2 (Fig 1C), which is crucial for both the ion-transport 14 and structural roles of KCC2 4,5. Several residues in the C-terminus of KCC2, including S940 located in close proximity to R952, are critical for regulation of KCC2 surface expression 16. The presently observed decrease in the surface expression of KCC2-R952H in a neuronal stem cell line (Fig 2) is consistent with both the decrease in Cl− extrusion and deficit in spine formation.
Our functional analysis of this KCC2 variant in pyramidal neurons of the mammalian cortex, taken together with the co-segregation and genic intolerance analysis discussed below, is strongly suggestive of a role for KCC2-R952H as a susceptibility variant in FS, thereby extending the list of other seizure susceptibility genes including GABRD, CACNA1H, HCN2 and SCN9A 17–19. Such susceptibility variants contribute to epilepsy with complex or polygenic inheritance 20. Rare variants of this kind are not readily amenable to statistical methods of validation such as genetic association studies, as their low frequency means that very large cohorts would be required to achieve statistical significance. Susceptibility variants are also refractory to linkage analysis approaches, as they do not segregate in a Mendelian fashion in large families. In this context, it is important to note that SLC12A5 is among the 4.5% most intolerant human genes (http://chgv.org/GenicIntolerance/) with regard to genic variation, and therefore, mutations in SLC12A5 are good candidates to contribute to disease phenotypes. Given the fact that KCC2 is a neuron-specific molecule, it is worth noting that low genic tolerance is prominent in relation to neurodevelopmental disorders 21.
A strong mechanistic link from the above observations on the defects of KCC2-R925H to the human febrile seizure phenotype is provided by studies which have shown that genetic deficits in mKCC2 expression result in increased network excitability 22 and higher susceptibility to seizures 10,23. However, it is not known whether the enhanced seizure susceptibility in the animal models is attributable to reduced neuronal Cl− extrusion and/or to impairments in spine formation. Seizure-triggering mechanisms show a wide heterogeneity at the cellular and network level 24. Thus, a decrease in the efficacy of KCC2-dependent hyperpolarizing inhibition is expected to promote triggering of seizures (see Introduction). In addition, dendritic abnormalities including both increases and decreases in spine number have been observed in brain tissue from patients with genetic disorders characterized by a high prevalence of seizures and epilepsy 25,26. While an increase in spine number may enhance overall excitability, a decrease may lead to desynchronization 27 and thereby promote seizures as discussed elsewhere 24,28.
In summary, our functional analyses of KCC2-R952H provide a novel molecular basis for enhanced susceptibility to FS.
Materials and Methods
Patient recruitment
Patients were recruited through private practice, epilepsy clinics and referral to our epilepsy genetics research program and underwent phenotyping using a validated seizure questionnaire 29. Medical records were obtained where available. Control samples were anonymous Australian blood donors.
Genetic screening
Patients were screened for mutations in the 26 coding exons of SLC12A5 by single-stranded conformation analysis (SSCA) using the GelScan 3000 apparatus (Corbett Research). The sequences of primers used are listed in Supplementary Table S1. Samples showing bandshifts were Sanger-sequenced using the BigDye terminator cycle sequencing kit (Applied Biosystems), and sequencing reactions were analyzed using the ABI 3131 Genetic Analyzer (Applied Biosystems). Family members of patients showing a rare coding variant were tested for that variant by direct Sanger sequencing. Controls were screened for exons showing variants by SSCA as described above.
Experimental animals
Heterozygous KCC2 mice (KCC2+/−) 4 were crossed to produce KCC2−/− embryos. ICR mice and Wistar rats were used for IUE. Experiments using mice were carried out with the approval by the National Animal Ethics Committee of Finland and the local Animal Ethics Committee of the University of Helsinki. Experiments using rats were conducted according to the guidelines of the Swiss Federal Veterinary Office and approved by the cantonal Animal Welfare Committee.
DNA constructs
The constructs expressing KCC2-WT, KCC2-R952H and rKCC2-ΔNTD were generated by subcloning the respective KCC2 variant sequences into the pCAG vector 5,30. rKCC2-ΔNTD has been described previously 4,5,15. The pCAG-EGFP plasmid was co-expressed to fluorescently label transfected cells [cf. 5].
Transfection, surface biotinylation and immunoblotting of neural stem cell cultures
The mouse neural stem cell line C17.2 was cultured at 26°C using standard methods 15. Cells at ~80% confluency were transfected with KCC2-WT or KCC2-R952H expression vectors using Turbofect. Surface biotinylation and immunoblotting of the cells were performed as described before 13.
IUE in mice and rats
Timed-pregnant rats with embryonic day (E) 17.5 embryos were used for IUE with the above-mentioned plasmids. Rats were electroporated as previously described 5. For IUE of mice, the following modifications to the protocol were applied: Timed-pregnant mice with E14.5 embryos were anesthetized with isoflurane (4.2% induction, 2.5% during surgery). All embryos were injected with 2 μl plasmid DNA solution (in 0.9% NaCl, 0.1% Fast Green). The plasmid encoding EGFP (1 μg/μl) was injected alone or with KCC2 constructs (3 μg/μl) 5. Electroporation was done with 5-mm-diameter circular electrodes (Sonidel Limited) with five pulses (45 V, 50 ms duration at 100 ms intervals), delivered with a square-wave generator (CUY21vivo SC, Sonidel Limited).
Assessment of the efficacy of KCC2-mediated Cl− extrusion in layer 2/3 pyramidal neurons
Acute 400-μm coronal neocortical slices were used as before 5. To measure KCC2-mediated Cl− extrusion, we used our standard assay where a somatic Cl− load (19 mM) is imposed on the neuron via a whole-cell patch pipette. The ensuing bumetanide-insensitive somatodendritic EGABA gradient, ΔEGABA, determined as the difference between the EGABA at the soma and at 50 μm away along the apical dendrite, provides an accurate estimate of the net Cl− extrusion 13. Whole-cell patch clamp recordings were performed as before 13 in an experiment-blind manner from EGFP-positive layer 2/3 pyramidal neurons from slices of P6-7 mice co-electroporated in utero with plasmids bearing constructs encoding for EGFP and one of the following: KCC2-WT, KCC2-R952H or rKCC2-ΔNTD. Some animals were electroporated with the EGFP plasmid alone. DPNI-caged GABA (Tocris) was used to elicit GABAAR-mediated currents 31. All recordings were performed in the presence of 10 μM bumetanide (Tocris), 0.5 μM TTX (Abcam), 10 μM CNQX (Abcam) and 1 μM CGP 55845 (Abcam) in the standard extracellular solution 13.
Transfection of mouse primary cortical neurons
E16 mKCC2−/− embryos were used for primary neuronal culture 4. Cortices from each embryo were dissected and plated separately. Neurons were grown on coverslips coated with poly-D-lysine in Neurobasal medium supplemented with B27 (GIBCO, Life Technologies). Transfection was performed on DIV8 using Turbofect (Thermo Scientific) and experiments at DIV13.
Analysis of spine density and length
Spine density analysis of rat somatosensory layer 2/3 pyramidal neurons filled iontophoretically with Lucifer yellow was done as described previously 5. Spine lengths from cultured neurons were analyzed on acquired stacks of images using NeuronJ software (http://rsb.info.nih.gov/ij/). Fluorescence images for dendritic spine length analysis were acquired with a Zeiss LSM 710 confocal microscope equipped with a 63× oil-immersion objective.
KCC2 staining in slices from in utero electroporated mice
Detection of KCC2 was done on 30 μm coronal cryosections from fixed brains (P6 mice, fixed with 4% PFA). Blocking and staining was performed in 3% BSA, 0.2% saponin, 10% goat serum in PBS mounted with Vectashield hard set mounting medium. The following antibodies were used: rabbit anti-KCC2 antibody (1:1,000; Millipore), chicken anti-GFP (1:1,000; Abcam), donkey anti-rabbit Cy3-conjugated (1:1,000; Jackson Immuno Research) and donkey anti-chicken Alexa 488-conjugated (1:1,000; Jackson Immuno Research).
Acknowledgments
We thank the patients and their families for participating in our research. We would like to acknowledge David B. Mount for providing the human KCC2 clone and Merle Kampura, Michèlle Brunet, Timea Bodogan, Bev Johns and Robert Schultz for excellent technical assistance. This work was supported by the Academy of Finland (PB and KK), the Sigrid Jusélius Foundation (KK), the Jane and Aatos Erkko Foundation (KK), Innovative Medical Research Fund of the University of Münster Medical School (I-BL211215 PB), the Swiss National Science Foundation (Grant 310030-146201 LV), and the National Health and Medical Research Council of Australia (Program Grant 628952 SFB, LMD, IES and SP; Early Career Fellowship 1016715 SHE and Career Development Fellowship 1032603 LMD).
Author contributions
SEH, TCW, XI, KLO, BEG, IES, LMD and SFB collected and analyzed the patient material. MP, PS, FA, LV and PB designed and performed the functional experiments. MP, PS, SEH, SP, LMD, SFB and KK wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary information for this article is available online: http://embor.embopress.org
References
- Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Khirug S, Huttu K, Ludwig A, Smirnov S, Voipio J, Rivera C, Kaila K, Khiroug L. Distinct properties of functional KCC2 expression in immature mouse hippocampal neurons in culture and in acute slices. Eur J Neurosci. 2005;21:899–904. doi: 10.1111/j.1460-9568.2005.03886.x. [DOI] [PubMed] [Google Scholar]
- Stein V, Hermans-Borgmeyer I, Jentsch TJ, Hübner CA. Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J Comp Neurol. 2004;468:57–64. doi: 10.1002/cne.10983. [DOI] [PubMed] [Google Scholar]
- Li H, Khirug S, Cai C, Ludwig A, Blaesse P, Kolikova J, Afzalov R, Coleman SK, Lauri S, Airaksinen MS, et al. KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron. 2007;56:1019–1033. doi: 10.1016/j.neuron.2007.10.039. [DOI] [PubMed] [Google Scholar]
- Fiumelli H, Briner A, Puskarjov M, Blaesse P, Belem BJ, Dayer AG, Kaila K, Martin JL, Vutskits L. An ion transport-independent role for the cation-chloride cotransporter KCC2 in dendritic spinogenesis in vivo. Cereb Cortex. 2013;23:378–388. doi: 10.1093/cercor/bhs027. [DOI] [PubMed] [Google Scholar]
- Chamma I, Chevy Q, Poncer JC, Levi S. Role of the neuronal K-Cl co-transporter KCC2 in inhibitory and excitatory neurotransmission. Front Cell Neurosci. 2012;6:5. doi: 10.3389/fncel.2012.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekmat-Scafe DS, Lundy MY, Ranga R, Tanouye MA. Mutations in the K+/Cl− cotransporter gene kazachoc (kcc) increase seizure susceptibility in Drosophila. J Neurosci. 2006;26:8943–8954. doi: 10.1523/JNEUROSCI.4998-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uvarov P, Ludwig A, Markkanen M, Soni S, Hubner CA, Rivera C, Airaksinen MS. Coexpression and heteromerization of two neuronal K-Cl cotransporter isoforms in neonatal brain. J Biol Chem. 2009;284:13696–13704. doi: 10.1074/jbc.M807366200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Lovinger D, Delpire E. Cortical neurons lacking KCC2 expression show impaired regulation of intracellular chloride. J Neurophysiol. 2005;93:1557–1568. doi: 10.1152/jn.00616.2004. [DOI] [PubMed] [Google Scholar]
- Woo NS, Lu JM, England R, McClellan R, Dufour S, Mount DB, Deutch AY, Lovinger DM, Delpire E. Hyperexcitability and epilepsy associated with disruption of the mouse neuronal-specific K-Cl cotransporter gene. Hippocampus. 2002;12:258–268. doi: 10.1002/hipo.10014. [DOI] [PubMed] [Google Scholar]
- Tan NC, Mulley JC, Scheffer IE. Genetic dissection of the common epilepsies. Curr Opin Neurol. 2006;19:157–163. doi: 10.1097/01.wco.0000218232.66054.46. [DOI] [PubMed] [Google Scholar]
- Cancedda L, Fiumelli H, Chen K, Poo MM. Excitatory GABA action is essential for morphological maturation of cortical neurons in vivo. J Neurosci. 2007;27:5224–5235. doi: 10.1523/JNEUROSCI.5169-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khirug S, Ahmad F, Puskarjov M, Afzalov R, Kaila K, Blaesse P. A single seizure episode leads to rapid functional activation of KCC2 in the neonatal rat hippocampus. J Neurosci. 2010;30:12028–12035. doi: 10.1523/JNEUROSCI.3154-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado A, Broumand V, Zandi-Nejad K, Enck AH, Mount DB. A C-terminal domain in KCC2 confers constitutive K+-Cl− cotransport. J Biol Chem. 2006;281:1016–1026. doi: 10.1074/jbc.M509972200. [DOI] [PubMed] [Google Scholar]
- Horn Z, Ringstedt T, Blaesse P, Kaila K, Herlenius E. Premature expression of KCC2 in embryonic mice perturbs neural development by an ion transport-independent mechanism. Eur J Neurosci. 2010;31:2142–2155. doi: 10.1111/j.1460-9568.2010.07258.x. [DOI] [PubMed] [Google Scholar]
- Kahle KT, Deeb TZ, Puskarjov M, Silayeva L, Liang B, Kaila K, Moss SJ. Modulation of neuronal activity by phosphorylation of the K-Cl cotransporter KCC2. Trends Neurosci. 2013;36:726–737. doi: 10.1016/j.tins.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulley JC, Hodgson B, McMahon JM, Iona X, Bellows S, Mullen SA, Farrell K, Mackay M, Sadleir L, Bleasel A, et al. Role of the sodium channel SCN9A in genetic epilepsy with febrile seizures plus and Dravet syndrome. Epilepsia. 2013;54:e122–e126. doi: 10.1111/epi.12323. [DOI] [PubMed] [Google Scholar]
- Dibbens LM, Reid CA, Hodgson B, Thomas EA, Phillips AM, Gazina E, Cromer BA, Clarke AL, Baram TZ, Scheffer IE, et al. Augmented currents of an HCN2 variant in patients with febrile seizure syndromes. Ann Neurol. 2010;67:542–546. doi: 10.1002/ana.21909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid CA, Berkovic SF, Petrou S. Mechanisms of human inherited epilepsies. Prog Neurobiol. 2009;87:41–57. doi: 10.1016/j.pneurobio.2008.09.016. [DOI] [PubMed] [Google Scholar]
- Dibbens LM, Heron SE, Mulley JC. A polygenic heterogeneity model for common epilepsies with complex genetics. Genes Brain Behav. 2007;6:593–597. doi: 10.1111/j.1601-183X.2007.00333.x. [DOI] [PubMed] [Google Scholar]
- Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9:e1003709. doi: 10.1371/journal.pgen.1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalilov I, Chazal G, Chudotvorova I, Pellegrino C, Corby S, Ferrand N, Gubkina O, Nardou R, Tyzio R, Yamamoto S, et al. Enhanced synaptic activity and epileptiform events in the embryonic KCC2 deficient hippocampus. Front Cell Neurosci. 2011;5:23. doi: 10.3389/fncel.2011.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornberg J, Voikar V, Savilahti H, Rauvala H, Airaksinen MS. Behavioural phenotypes of hypomorphic KCC2-deficient mice. Eur J Neurosci. 2005;21:1327–1337. doi: 10.1111/j.1460-9568.2005.03959.x. [DOI] [PubMed] [Google Scholar]
- Kaila K, Ruusuvuori E, Seja P, Voipio J, Puskarjov M. GABA actions and ionic plasticity in epilepsy. Curr Opin Neurobiol. 2014;26:34–41. doi: 10.1016/j.conb.2013.11.004. [DOI] [PubMed] [Google Scholar]
- Brooks-Kayal A. Molecular mechanisms of cognitive and behavioral comorbidities of epilepsy in children. Epilepsia. 2011;52(Suppl 1):13–20. doi: 10.1111/j.1528-1167.2010.02906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M, Guo D. Dendritic spine pathology in epilepsy: cause or consequence? Neuroscience. 2013;251:141–150. doi: 10.1016/j.neuroscience.2012.03.048. [DOI] [PubMed] [Google Scholar]
- Netoff TI, Schiff SJ. Decreased neuronal synchronization during experimental seizures. J Neurosci. 2002;22:7297–7307. doi: 10.1523/JNEUROSCI.22-16-07297.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiruska P, de Curtis M, Jefferyss JG, Schevon CA, Schiff SJ, Schindler K. Synchronization and desynchronization in epilepsy: controversies and hypotheses. J Physiol. 2013;591:787–797. doi: 10.1113/jphysiol.2012.239590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reutens DC, Howell RA, Gebert KE, Berkovic SF. Validation of a questionnaire for clinical seizure diagnosis. Epilepsia. 1992;33:1065–1071. doi: 10.1111/j.1528-1157.1992.tb01760.x. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Puskarjov M, Ahmad F, Kaila K, Blaesse P. Activity-dependent cleavage of the K-Cl cotransporter KCC2 mediated by calcium-activated protease calpain. J Neurosci. 2012;32:11356–11364. doi: 10.1523/JNEUROSCI.6265-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.