

Abstract
WebLogo generates sequence logos, graphical representations of the patterns within a multiple sequence alignment. Sequence logos provide a richer and more precise description of sequence similarity than consensus sequences and can rapidly reveal significant features of the alignment otherwise difficult to perceive. Each logo consists of stacks of letters, one stack for each position in the sequence. The overall height of each stack indicates the sequence conservation at that position (measured in bits), whereas the height of symbols within the stack reflects the relative frequency of the corresponding amino or nucleic acid at that position. WebLogo has been enhanced recently with additional features and options, to provide a convenient and highly configurable sequence logo generator. A command line interface and the complete, open WebLogo source code are available for local installation and customization.
Sequence logos were invented by Tom Schneider and Mike Stephens (Schneider and Stephens 1990; Shaner et al. 1993) to display patterns in sequence conservation, and to assist in discovering and analyzing those patterns. As an example, the accompanying figure (Fig. 1) shows how WebLogo can help interpret the sequence-specific binding of the protein CAP to its DNA recognition site (Schultz et al. 1991). Homodimeric DNA-binding proteins typically display a symmetric double hump in the DNA binding-site logo (Schneider and Stephens 1990), as shown in the figure. Deviations from this basic pattern can indicate additional features; a highly conserved residue in the center of such a pattern may indicate DNA distortion or base flipping (Schneider 2001); an unexpectedly high-sequence conservation may be due to overlapping binding sites (Schneider et al. 1986). Protein logos can illuminate patterns of amino acid conservation that are often of structural or functional importance (Galperin et al. 2001; Rigden et al. 2003). Sequence logos have also been used to display patterns in the BLOCKS protein sequence database (Henikoff et al. 1995), and in DNA-binding site motifs (Robison et al. 1998; Nelson et al. 2002), to analyze splice sites (Stephens and Schneider 1992; Emmert et al. 2001), and in a variety of other contexts. Additional examples, and the raw data for the example presented here, can be found on the WebLogo examples page (http://weblogo.berkeley.edu/examples.html).
Figure 1.

(A) CAP (Catabolite Activator Protein, also known as CRP) acts as a transcription promoter by binding at more than 100 sites within the Escherichia coli genome. We rendered the PDB structure 1CGP (Schultz et al. 1991) using Chimera (Huang et al. 1996). (B) The two DNA recognition helices of the CAP homodimer insert themselves into consecutive turns of the major groove. Several consequences can be observed in this CAP binding-site logo. The logo is approximately palindromic, which provides two very similar recognition sites, one for each subunit of the dimer. However, the binding site lacks perfect symmetry, possibly due to the inherent asymmetry of the operon promoter region. The displacement of the two halves is 11 bp, or approximately one full turn of the DNA helix. Additional interactions occur between the protein and the first and last two bases within the DNA minor groove, where the protein cannot easily distinguish A from T, or G from C (Seeman et al. 1976). The data for this logo consists of 59 binding sites determined by DNA footprinting (Robison et al. 1998). (C) The helix-turn-helix motif from the CAP family of homodimeric DNA binding proteins (Brennan and Matthews 1989; Schultz et al. 1991). Positions 180, 181, and 185 are known to interact directly with bases in the major groove (Schultz et al. 1991; Parkinson et al. 1996) and are critical to the sequence-specific binding of the protein. The conserved glycine at position 177 is located inside of the turn between the helices, where packing effects prevent the insertion of a side chain. Partially or completely buried positions (labeled B) frequently contain hydrophobic amino acids, which are colored black. The data for this logo consists of 100 sequences from the full Pfam (Bateman et al. 2002) alignment of this family (Accession no. PF00325). We removed a few sequences with rare insertions for convenience.
The logo generation form (http://weblogo.berkeley.edu/logo.cgi) can process RNA, DNA, or protein multiple sequence alignments provided in either FASTA (Pearson and Lipman 1988) or CLUSTAL (Higgins and Sharp 1988) formats. If the user does not explicitly specify the sequence type, then WebLogo will make a determination on the basis of the symbols found within the sequences. A logo represents each column of the alignment by a stack of letters, with the height of each letter proportional to the observed frequency of the corresponding amino acid or nucleotide, and the overall height of each stack proportional to the sequence conservation, measured in bits, at that position. The letters of each stack are ordered from most to least frequent, so that one may read the consensus sequence from the tops of the stacks. For example, the figure shows that the CAP bindingsite consensus sequence is AA-TGTGA------TCACA-TT.
Schneider and Stephens (1990) define the sequence conservation at a particular position in the alignment, Rseq, as the difference between the maximum possible entropy and the entropy of the observed symbol distribution:
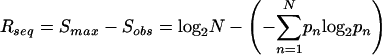 |
Here, pn is the observed frequency of symbol n at a particular sequence position and N is the number of distinct symbols for the given sequence type, either four for DNA/RNA or 20 for protein. Consequently, the maximum sequence conservation per site is log2 4 = 2 bits for DNA/RNA and log2 20 ≈ 4.32 bits for proteins. If we neglect the intersite correlations and assume a uniform background symbol distribution, then the total entropy of the logo, the sum of the sequence conservation at each position, measures the information content of the logo. For binding sites, this total entropy has, in many cases, been shown to be approximately equal to the amount of information needed to locate the binding site within the relevant stretch of DNA (Schneider et al 1986). For a nonuniform background distribution, such as found in protein sequences or the genomes of many hyperthermophiles, the information content would be given by the relative entropy between the observed and background distributions (Cover and Thomas 1991; Gorodkin et al. 1997; Stormo 1998).
Limited sequence data results in a systematic underestimation of the entropy, which becomes significant if the multiple alignment contains fewer than about 20 nucleotide or 40 protein sequences. By default, WebLogo incorporates a small sample correction (Schneider et al. 1986), which can, in part, ameliorate this bias. In addition, WebLogo can optionally display error bars with heights twice this correction, which gives some idea of the sampling errors made. Note that the error bars may not have uniform height across the logo, as the magnitude of the small sample correction depends on the number of symbols observed at each position. This will vary due to the presence of gaps in the alignment.
A standard sequence logo does not provide any indication of correlations between different positions of the alignment. In general, such intersite correlations are relatively insignificant in biological sequences (Schneider 1997; Stormo 1998), but there are exceptions, such as base-paired sites in folded RNA structures. Structural logos (Gorodkin et al. 1997), an extension of the sequence logo idea, display part of this additional level of detail.
The symbols that compose the stacks display colors according to the chemical species they represent. The default colors for nucleotides are G, orange; T and U, red; C, blue; and A, green. Amino acids have colors according to their chemical properties (Lewin 1994); polar amino acids (G, S, T, Y, C, Q, N) show as green, basic (K, R, H) blue, acidic (D, E) red, and hydrophobic (A, V, L, I, P, W, F, M) amino acids as black. The user may customize the coloring scheme, or select a simple black and white option.
WebLogo can create output in several common graphics' formats, including the bitmap formats GIF and PNG, suitable for on-screen display, and the vector formats EPS and PDF, more suitable for printing, publication, and further editing. Additional graphics options include bitmap resolution, titles, optional axis, and axis labels, antialiasing, error bars, and alternative symbol formats.
The Web site is available to all users without fee. Those who would prefer to run WebLogo on a local server may obtain a command line interface version with source code (distributed under an Open Source license). We welcome bug reports and suggestions for additional features. Please send these to logo@compbio.berkeley.edu.
Acknowledgments
WebLogo uses PostScript code and ideas from the programs alpro and makelogo, both part of Tom Schneider's delila package (Schneider et al. 1982). Many thanks to him for making this software freely available, for encouraging its use, and for feedback on WebLogo. We are also grateful for the enthusiastic encouragement of Michael Galperin. Grants from the NIH (1-K22-HG00056) and the Searle Scholars program (01-L-116) support this work. JMC was supported by NIH grant 1-P50-GM62412, and DOE contract DE-AC03-76SF00098. GEC received funding from the Sloan/DOE postdoctoral fellowship in computational molecular biology.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.849004.
References
- Bateman, A., Birney, E., Cerruti, L., Durbin, R., Etwiller, L., Eddy, S.R., Griffiths-Jones, S., Howe, K.L., Marshall, M., and Sonnhammer, E.L. 2002. The Pfam protein families database. Nucleic Acids Res. 30: 276–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan, R.G. and Matthews, B.W. 1989. Structural basis of DNA-protein recognition. Trends in Biochem. Sci. 14: 286–290. [DOI] [PubMed] [Google Scholar]
- Cover, T.M. and Thomas, J.A. 1991. Elements of information theory. John Wiley & Sons, New York.
- Emmert, S., Schneider, T.D., Khan, S.G., and Kraemer, K.H. 2001. The human XPG gene: Gene architecture, alternative splicing and single nucleotide polymorphisms. Nucleic Acids Res. 29: 1443–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galperin, M.Y., Nikolskaya, A.N., and Koonin, E.V. 2001. Novel domains of the prokaryotic two-component signal transduction systems. FEMS Microbiol. Lett. 203: 11–21. [DOI] [PubMed] [Google Scholar]
- Gorodkin, J., Heyer, L.J., Brunak, S., and Stormo, G.D. 1997. Displaying the information contents of structural RNA alignments: The structure logos. Comput. Appl. Biosci. 13: 583–586. [DOI] [PubMed] [Google Scholar]
- Henikoff, S., Henikoff, J.G., Alford, W.J., and Pietrokovski, S. 1995. Automated construction and graphical presentation of protein blocks from unaligned sequences. Gene 163: GC17–GC26. [DOI] [PubMed] [Google Scholar]
- Higgins, D.G. and Sharp, P.M. 1988. CLUSTAL: A package for performing multiple sequence alignment on a microcomputer. Gene 73: 237–244. [DOI] [PubMed] [Google Scholar]
- Huang, C.C., Couch, G.S., Pettersen, E.F., and Ferrin, T.E. 1996. Chimera: An extensible molecular modeling application constructed using standard components. In Pacific symposium on biocomputing, Vol. 1, pp. 724. http://www.cgl.ucsf.edu/chimera.
- Lewin, B. 1994. Genes V. Oxford University Press, New York.
- Nelson, P.S., Clegg, N., Arnold, H., Ferguson, C., Bonham, M., White, J., Hood, L., and Lin, B. 2002. The program of androgen-responsive genes in neoplastic prostate epithelium. Proc. Natl. Acad. Sci. 99: 11890–11895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson, G., Gunasekera, A., Vojtechovsky, J., Zhang, X., Kunkel, T.A., Berman, H., and Ebright, R.H. 1996. Aromatic hydrogen bond in sequence-specific protein DNA recognition. Nat. Struct. Biol. 3: 837–841. [DOI] [PubMed] [Google Scholar]
- Pearson, W.R. and Lipman, D.J. 1988. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. 85: 2444–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigden, D.J., Jedrzejas, M.J., and Galperin, M.Y. 2003. An extracellular calcium-binding domain in bacteria with a distant relationship to EF-hands. FEMS Microbiol. Lett. 221: 103–110. [DOI] [PubMed] [Google Scholar]
- Robison, K., McGuire, A.M., and Church, G.M. 1998. A comprehensive library of DNA-binding site matrices for 55 proteins applied to the complete Escherichia coli K-12 genome. J. Mol. Biol. 284: 241–254. [DOI] [PubMed] [Google Scholar]
- Schneider, T.D. 1997. Information content of individual genetic sequences. J. Theor. Biol. 189: 427–441. [DOI] [PubMed] [Google Scholar]
- Schneider, T.D. 2001. Strong minor groove base conservation in sequence logos implies DNA distortion or base flipping during replication and transcription initiation. Nucleic Acid Res. 29: 4881–4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, T.D. and Stephens, R.M. 1990. Sequence logos: A new way to display consensus sequences. Nucleic Acids Res. 18: 6097–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, T.D., Stormo, G.D., Haemer, J.S., and Gold, L. 1982. A design for computer nucleic-acid sequence storage, retrieval, and manipulation. Nucleic Acids Res. 10: 3013–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, T.D., Stormo, G.D., Gold, L., and Ehrenfeucht, A. 1986. Information content of binding sites on nucleotide sequences. J. Mol. Biol. 188: 415–431. [DOI] [PubMed] [Google Scholar]
- Schultz, S.C., Shields, G.C., and Steitz, T.A. 1991. Crystal structure of a CAP-DNA complex: The DNA is bent by 90°. Science 253: 1001–1007. [DOI] [PubMed] [Google Scholar]
- Seeman, N.C., Rosenberg, J.M., and Rich, A. 1976. Sequence-specific recognition of double helical nucleic acids by proteins. Proc. Natl. Acad. Sci. 73: 804–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner, M.C., Blair, I.M., and Schneider, T.D. 1993. Sequence logos: A powerful, yet simple, tool. Proceedings of the twenty-sixth annual Hawaii international conference on system sciences. In Architecture and biotechnology computing (eds. T.N. Mudge et al.) Vol 1., pp. 813–821. IEEE Computer Society Press, Los Alamitos, CA. [Google Scholar]
- Stephens, R.M. and Schneider, T.D. 1992. Features of spliceosome evolution and function inferred from an analysis of the information at human splice sites. J. Mol. Biol. 228: 1124–1136. [DOI] [PubMed] [Google Scholar]
- Stormo, G.D. 1998. Information content and free energy in DNA-protein interactions. J. Theor. Biol. 195: 135–137. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://weblogo.berkeley.edu/; WebLogo: A Sequence Logo Generator. [DOI] [PMC free article] [PubMed]