

Abstract
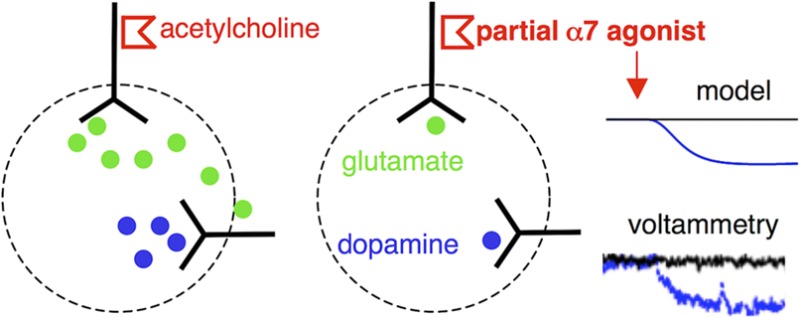
Neuronal nicotinic acetylcholine receptors (NNRs) of the α7 subtype have been shown to contribute to the release of dopamine in the nucleus accumbens. The site of action and the underlying mechanism, however, are unclear. Here we applied a circuit modeling approach, supported by electrochemical in vivo recordings, to clarify this issue. Modeling revealed two potential mechanisms for the drop in accumbal dopamine efflux evoked by the selective α7 partial agonist TC-7020. TC-7020 could desensitize α7 NNRs located predominantly on dopamine neurons or glutamatergic afferents to them or, alternatively, activate α7 NNRs located on the glutamatergic afferents to GABAergic interneurons in the ventral tegmental area. Only the model based on desensitization, however, was able to explain the neutralizing effect of coapplied PNU-120596, a positive allosteric modulator. According to our results, the most likely sites of action are the preterminal α7 NNRs controlling glutamate release from cortical afferents to the nucleus accumbens. These findings offer a rationale for the further investigation of α7 NNR agonists as therapy for diseases associated with enhanced mesolimbic dopaminergic tone, such as schizophrenia and addiction.
Keywords: α7-Nicotinic receptor agonist, acetylcholine, desensitization, ventral tegmental area, mesolimbic pathway, modeling
Ligands of neuronal nicotinic receptors (NNRs) are seen as promising therapeutics for a range of CNS disorders, including Alzheimer’s disease, Parkinson’s disease, schizophrenia, and addiction.1−4 NNRs are members of the class of Cys-loop cationic ion channels, yet predicting the systemic effects of their ligands has proven difficult for several reasons: NNRs of varying subunit composition are expressed on different neuron types, locate extrasynaptically, and quickly desensitize to varying degrees in the continued presence of the agonist.
NNRs are abundant in the basal ganglia,5 and acute systemic nicotine administration stimulates the efflux of dopamine in rat nucleus accumbens in vivo.6,7 Most studies on the cholinergic control of accumbal dopamine release have focused on the effect of activation of different NNR subtypes on the spiking activity of either dopaminergic8−10 or GABAergic neurons11,12 in the ventral tegmental area (VTA)11 or have measured changes in accumbal dopamine levels after electrical stimulation of midbrain nuclei.13 For short, α7 NNRs are thought to exert presynaptic control over inputs to VTA neurons,10,14 and consistent with this modulatory role, dopamine neurons of α7–/– mice showed less prominent changes in their spontaneous or evoked spike patterns than those of β2–/– mice.8
More recently, optogenetic stimulation of the accumbal cholinergic interneurons was found to evoke dopamine efflux in vitro without a concomitant change of the spike rate or pattern of the dopamine neurons.15,16 Although this accumbal control of dopamine efflux is mediated by preterminal β2* NNRs on the axons of dopamine neurons,17 cholinergic stimulation may also activate nearby α7 NNRs located on corticofugal axons and, through the intermediary of ionotropic receptors on dopamine axons, indirectly stimulate dopamine release.18,19
TC-7020 is a selective partial agonist of the homomeric α7-type NNR20 with an efficacy of 30% and an EC50 of 30 nM in rats. Its selectivity for the α7 NNR is evidenced by an IC50 of 2 nM, compared with 4200 nM for α4β2* NNRs,21 and an IC50 > 10 μM for non-nicotinic receptors.20 In a microdialysis study of a mouse model of schizophrenia, systemic administration of TC-7020 (0.1–1.0 mg/kg ip) normalized the increased striatal extracellular dopamine level.22 The mechanism of action underlying this suppression of dopamine concentration, however, remains elusive. Since microdialysis measures dopamine fluctuations only on a time scale of minutes, these data cannot be effectively used for the computational modeling of the targeted circuitry. Importantly, α7 NNRs rapidly desensitize during agonist exposure, and preterminal receptors interact with glutamatergic transmission in VTA,11,23,24 ventral and dorsal striatum,19 and prefrontal cortex.25,26
In contrast to microdialysis measurements, real-time voltammetry is particularly useful for evaluating the fast dopamine dynamics, due its subsecond-scale temporal resolution. Here, we used a computational modeling approach to understand the observation that TC-7020 acutely reduced dopamine efflux measured by fast-scan cyclic voltammetry (FSCV). In particular, we examine how real-time dopamine signaling depends on the activation and desensitization properties of the α7 NNRs and on their differential distribution on GABA and dopamine neurons or their glutamatergic afferents. The present analysis models only acute effects, up to 1 min after agonist injection, since the circuit may change its dynamics owing to synaptic plasticity27 or adaptation of the expression of other NNR subtypes.28
Results and Discussion
We first present the in vivo voltammetric recording of dopamine efflux in rat nucleus accumbens in response to the iv injection of nicotine and the partial α7 agonist TC-7020. Next we explain how two alternative models, based on desensitization versus activation of the α7 receptor, generated a drop in dopamine release similar to that after TC-7020 injection. Finally, we argue that only one particular implementation of the desensitization model explains the observed response to the combined injection of the positive allosteric modulator PNU-120596.
In Vivo Voltammetry of Dopamine Efflux in Rat Nucleus Accumbens
Intravenous administration of nicotine (0.3 mg/kg) induced a fast and potent increase in accumbal dopamine measured by real-time FSCV in anesthetized rats (Figure 1A). The averaged dopamine concentration was 0.82 ± 0.08 μM (n = 4). The effect appeared approximately 7 s after drug administration. Importantly, an identical nicotine-induced dopamine concentration increase had been observed in freely moving rats using the same technique (FSCV).7 No changes were observed after saline injection (Figure 1A,B). Using the same experimental design, we explored the effects of the α7-selective partial agonist TC-7020 (1 mg/kg iv) on real-time dopamine dynamics (Figure 1B,C). In contrast to nicotine, this compound induced a drop in baseline dopamine recordings. This inhibitory effect could be reversed by nicotine (Figure 1B). The TC-7020-induced drop was nearly abolished by pretreatment with MLA (10 mg/kg ip), an α7 nicotinic receptor antagonist (Figure 1C), ruling out the possibility that the observed dopamine changes were nonspecific or nonreceptor mediated. No changes were observed when MLA was administered alone (Figure 1C). A previous microdialysis study also had provided clear evidence that systemic (ip) administration of TC-7020 could suppress extracellular dopamine levels on a prolonged time scale.22 Interestingly, in the present experiments, pretreatment with the α7 type-2 positive allosteric modulator PNU-120596 (5 mg/kg, sc),29 rather than enhancing dopamine release, also blocked the effects of TC-7020 on dopamine transmission, through an expected reduction or elimination of desensitization (see Figure 4A). Qualitatively similar observations were made in the dorsal striatum.
Figure 1.
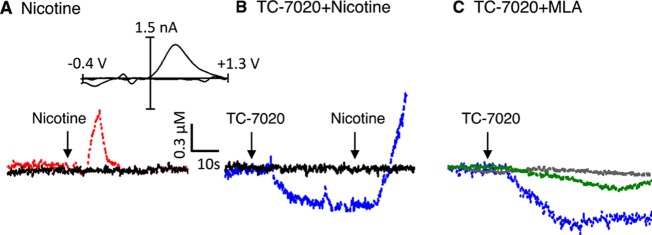
Representative voltammetric recordings of real-time dopamine signaling in nucleus accumbens of anaesthetized rats before and after administration (arrows) of (A) nicotine (0.3 mg/kg iv), (B) TC-7020 (1 mg/kg iv) followed by nicotine, and (C) TC-7020 alone (blue trace) or TC-7020 after pretreatment with MLA (10 mg/kg ip) (green trace) or MLA alone (black trace). Black traces in A and B are controls after saline injection. Inset to panel A is representative background-subtracted voltammogram obtained at peak of response, showing characteristic oxidation and reduction peak potentials (approximately +0.6 V and approximately −0.2 V, respectively) that identify dopamine.
Figure 4.
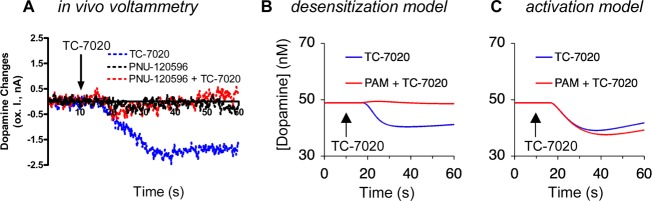
Responses to coadministration of TC-7020 and PNU-120596 in the experiment (A) and model (desensitization, B, and activation, C). Doses and parameters were as follows: (A) TC-7020 (1.0 mg/kg iv) and PNU-120596 (5.0 mg/kg injected sc 30 min earlier); (B) peak [TC-7020] 11 nM, ACh tone 30 μM; (C) peak [TC-7020] 2.4 nM, ACh tone 0 μM. The presence of PNU-120596 was modeled by multiplying the IC50 of TC-7020 by a factor of 10.
Analysis of the Drop in Dopamine Release Produced in Models Based on the Desensitization versus Activation of α7 NNRs
In order to explain these observations, we compared two classes of models based either on the activation or on the desensitization of α7 NNRs. The models differed by the relative distribution of α7 NNRs on dopamine versus GABA-neurons (or their glutamatergic afferents), and as will be shown below, they functioned optimally at different TC-7020 concentrations and different cholinergic tones.
In the first class of models, NNRs were primarily expressed on dopamine neurons and glutamatergic fibers afferent to them (Figure 2A). The channels were located, first, within the VTA on the glutamatergic afferents to dopamine neurons, where they potentiate glutamate release;10,24,30 second, on the dendrites and somata of (a subpopulation of) dopamine neurons;23,31−33 and, third, within the nucleus accumbens, on the glutamatergic afferents to the medium-sized spiny neurons, where they potentiate glutamate release and subsequent dopamine release through a process involving ionotropic glutamate receptors presumably located on the dopamine axons.18,19 The α7 NNRs are notably absent, however, from the dopamine axonal terminals themselves.34,35 Since desensitization of receptors at these locations would indeed reduce dopamine release, we further call this class of models the “desensitization model”.
Figure 2.
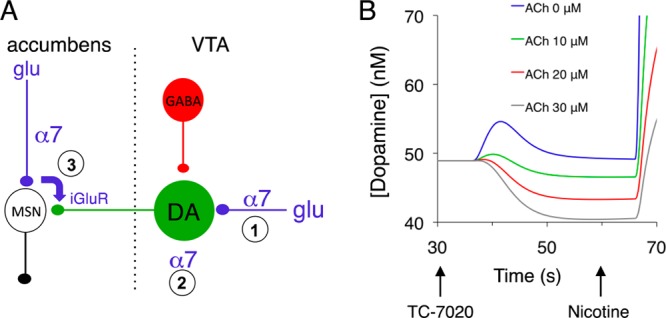
Synaptic organization and responses of the “desensitization model”. Panel A shows a circuit diagram of the mesoaccumbal projection, highlighting the locations of those α7 NNRs that would evoke, through their desensitization, a drop in dopamine release. Panel B plots the extracellular dopamine dynamics in the model for four levels of cholinergic tone, indicated by their equivalent acetylcholine concentrations. The [TC-7020] profile is shown in Figure S2, Supporting Information, and TC-7020 reached a peak concentration of 11 nM. MSN, medium-sized spiny neuron; glu, glutamatergic afferent. For clarity, the α4β2* NNRs on the DA and GABA neurons or their afferents are not shown.
In contrast, when the α7 NNRs were located primarily at the positions highlighted in Figure 3A, they would evoke a drop in dopamine efflux through receptor activation, and this class of models is further referred to as the “activation model”. Its channels can be located on glutamatergic afferents to GABA neurons in the VTA (but no α7 NNRs are located on the GABA neurons themselves),11 and their activation disynaptically inhibits dopamine neurons. Alternatively, activation of α7 NNRs on glutamate afferents within the nucleus accumbens can, through spillover of glutamate, activate metabotropic glutamate receptors on dopamine terminals and depress dopamine release.36
Figure 3.
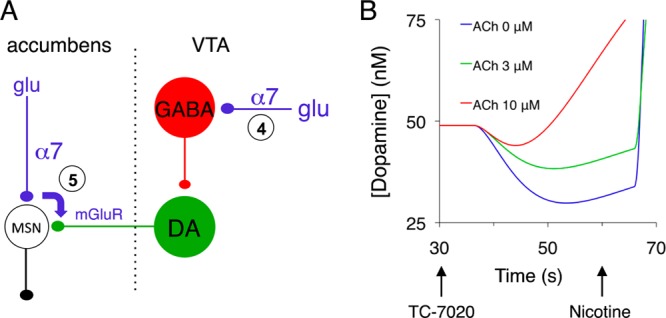
Synaptic organization and responses of the “activation model”. The α7 NNRs located at the positions in panel A would evoke, through their activation, a drop in dopamine release. Panel B plots the extracellular dopamine dynamics in the model for three levels of cholinergic tone, indicated by their equivalent acetylcholine concentrations. The [TC-7020] profile is shown in Figure S2, Supporting Information, and TC-7020 reached a peak concentration of 3 nM.
The simulations showed that both models could reproduce the TC-7020-evoked drop in dopamine release (Figures 2B and 3B). The responses of the desensitization model, however, depended on the cholinergic tone, which was represented in the model by an equivalent concentration of acetylcholine needed to obtain a tonic level of receptor activation. At absent tone ([ACh] = 0 in Figure 2B), the net response was an enhanced dopamine efflux, caused by activation and subsequent (incomplete) desensitization of the α7 NNRs. In the presence of a tone, however, the desensitization would reduce the numbers of NNRs available for endogenous acetylcholine, leading to a marked drop in the level of dopamine efflux, in proportion to the strength of the tone (Figure 2B).
In contrast, no cholinergic tone was required in the activation model (Figure 3B). A high tone even reversed the dopamine drop since desensitization of NNRs now disinhibited the dopamine neuron (red curve in Figure 3B). For the activation model to cause a sustained drop in dopamine efflux, however, a “window” current was needed, which required agonist levels at concentrations where the desensitization and activation curves maximally overlapped (see Figure 5). The desensitization model, on the other hand, required higher agonist concentrations that would desensitize most of the NNRs. A more extensive parameter analysis of the model can be found in the Supporting Information (Figures S2–4).
Figure 5.
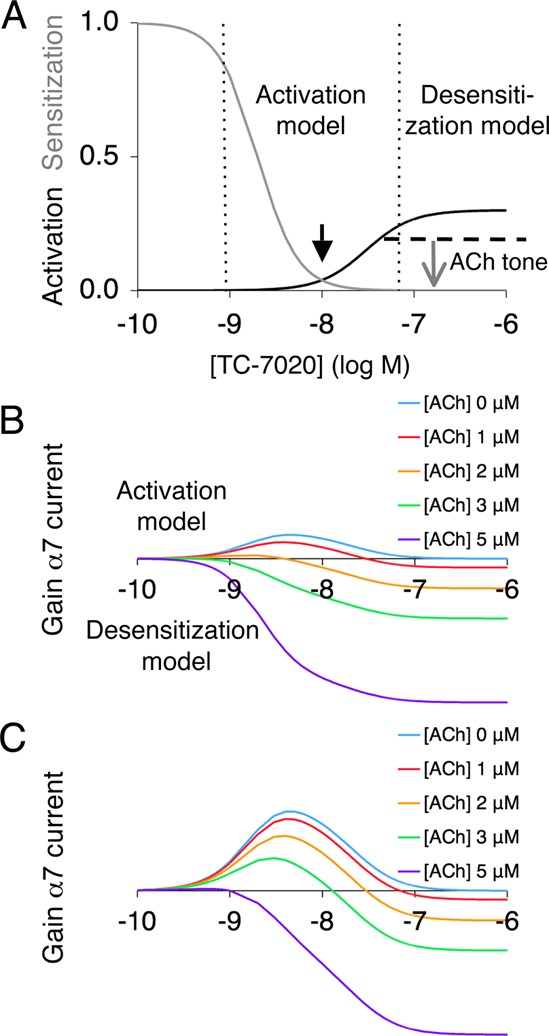
Effect of TC-7020 on acetylcholine-evoked dopamine release summarized. (A) Steady-state activation and desensitization curves for TC-7020 at α7 NNRs. The black arrow indicates the concentration at which TC-7020 is maximally able to activate NNRs without completely desensitizing them. Beyond this concentration, the desensitizing effect on endogeneous release (“ACh tone”) predominates (gray arrow). (B) Each trace plots the difference between the NNR current that is generated by the combined presence of TC-7020 and acetylcholine and the NNR current generated by acetylcholine alone. TC-7020 concentration is plotted on the horizontal axis; ACh tone is color-coded. (C) Same data as in panel B, but for a full agonist.
The Activation and Desensitization Models Make Opposite Predictions Regarding the Effect of a Coadministered Positive Modulator
As mentioned above, coadministration of PNU-120596, a type-2 positive allosteric modulator of α7 NNRs that releases the receptors from desensitization,29 abolished the TC-7020 response (Figure 4A). We modeled NNR resensitization by shifting the Hill curve of the desensitization gate of the receptors toward higher agonist concentrations. In that case, the desensitization model robustly reproduced the experimental cancellation of the TC-7020-induced drop in dopamine release (Figure 4B and Figure S4, Supporting Information).
In the activation model, in contrast, shifting rightward the desensitization curves always amplified the TC-7020 response, instead of neutralizing it (Figure 4C). For the TC-7020 response to be canceled in the activation model, PNU-120596 should be assumed to interfere with receptor activation. Although prolonged channel opening in the presence of PNU-120596 can make the channels more susceptible to open-channel-block, this phenomenon has been observed only at much higher, micromolar agonist concentrations.37,38
The Combined Voltammetry-Modeling Results Point to a Desensitizing Action of TC-7020 on α7 NNRs Located on Glutamatergic Afferents in Nucleus Accumbens
In summary, the present model considered five different sites for the action of TC-7020 on the mesoaccumbal pathway, and hence for the contribution of α7 NNRs to dopamine release. Although models based on NNR desensitization (sites 1–3 in Figure 2A) and NNR activation (sites 4 and 5 in Figure 3A) could both generate a drop in dopamine release such as that observed experimentally, the neutralizing effect of coadministered PNU-120596 was only explained by the desensitization model (receptor resensitization by the allosteric modulator, sites 1–3). Sites 4 and 5 of the activation model further lack experimental support. For site 5, although stimulation of presynaptic metabotropic glutamate receptors (mGluR1) has been shown to reduce dopamine release in mouse striatum in vitro,36 mGluR1 agonists were ineffective in dialysis experiments.39 In addition, although local application of the partial α7 agonist JN403 at site 4 has been observed to enhance the spike rate of GABAergic neurons via action on α7 NNRs at glutamatergic terminals11 and optogenetic stimulation of GABA neurons reduced dopamine efflux in nucleus accumbens,40 systemic administration of α7 agonists did not affect the rate or spike pattern of dopamine neurons (Figure 7 of ref (41)).
This failure of systemically applied α7 agonists to acutely alter the spiking behavior of dopamine neurons also disfavors sites 1 and 2 of the desensitization model (Figure 2A), leaving site 3 as the most likely mesoaccumbal target for TC-7020. Such a contribution of accumbal α7 NNRs to dopamine release was first proposed by Kaiser and Wonnacott.19 Note that TC-7020 itself does not evoke dopamine release from synaptosomes (V. P. Grinevich and M. Bencherif, unpublished data); hence the mechanism involves crosstalk among cholinergic, glutamatergic, and dopaminergic neurons. That cortico-accumbal glutamatergic neurons could facilitate dopamine release through a presynaptic action on the dopamine terminals, independently of the rate of firing of meso-accumbal dopamine neurons, had been suggested before,42 and subsequent studies confirmed the presence of ionotropic glutamate receptors on dopaminergic synaptosomes18,43 and of α7 NNRs on glutamatergic terminals.44,45 Electrochemical recordings in nucleus accumbens and VTA of freely moving rats showed that low doses of nicotine (30 μg/kg iv) acutely stimulated glutamate efflux.46 In the VTA, at least, nicotine-evoked glutamate efflux has been suggested to be mediated through α7 receptors.14
As shown by the model, the effect of α7 NNR desensitization on dopamine release can only be apparent in the presence of a cholinergic tone. In nucleus accumbens, acetylcholine is released by giant interneurons that fire spontaneously at a rate of 3–10 Hz in vivo.47 The presence of a cholinergic tone activating preterminal α7 NNRs in vitro was demonstrated by the drop of excitatory post-synaptic current (EPSC) frequency in medium-sized spiny neurons after bath application of MLA.48 Taken together, these data indicate that α7 NNRs contribute to the cholinergic control of dopamine release in nucleus accumbens, even at baseline receptor recruitment levels, and hence that their acute desensitization indeed will reduce dopamine efflux.
Note that this mechanism has been suggested before, for instance, to underlie the drop in dopamine after intrastriatal infusion of kynurenic acid, which can act as an α7 inhibitor,49−51 and for the regularization of enhanced dopamine release in a mouse model of schizophrenia.22 The loss of regulatory action of α7 NNRs has likewise been suggested to underlie to enhanced nicotine-evoked dopamine release in the accumbens of α7–/– mutant mice.52
Last but not least we have to discard the possibility that TC-7020 did not act by reducing dopamine efflux but by stimulating its reuptake. In the model, a 15% increase of transport velocity would have sufficed to generate a drop in dopamine concentration of the same magnitude as that generated by receptor desensitization. However, in a previous study,20 10 μM TC-7020 did not show any affinity for the dopamine transporter in a radioligand assay. Neither glutamate53 nor nicotine54 interacts with the dopamine uptake transporter. Moreover, such an interaction of TC-7020 with the dopamine transporter would not have explained the effects evoked by MLA (Figure 1C) and PNU-120596 (Figure 4A).
Robustness of the Model
Predicting the systemic effects of a partial agonist requires knowledge of the balance between receptor activation and desensitization and further of the distribution of the receptors at different locations within the circuit.
For the receptor, steady-state can be assumed, since α7 NNRs desensitize on a subsecond time scale. In that case the fraction of conducting receptors is described by the product of the concentration–response curves for activation and desensitization (Figure 5A). The resultant window current has a bell-shaped concentration profile (Figure 5B, curve [ACh] = 0), and its amplitude increases with the efficacy of the compound (Figure 5C). From the current gained by binding of the exogenous agonist, however, the loss of endogenous current that is due to desensitization must be subtracted. This loss increases with cholinergic tone. The net current diminishes and its peak shifts toward lower agonist concentrations, until at high tone the agonist produces at all concentrations a negative effect (Figures 5B,C).
As stated above, the desensitization and activation mechanisms operate in different concentration regimes, the latter being able to generate a current only at lower ligand concentrations at which a considerable fraction of NNRs did not desensitize yet (black arrow in Figure 5A). Nevertheless in both regimes most of the receptors will desensitize, except when the concentration–response curves for desensitization and activation considerably overlap.55 A third mechanism, receptor potentiation, can be considered as a special case of the activation mechanism, operating at agonist concentrations too low to desensitize the receptor but sufficient to open the channel through coagonism with acetylcholine. Although we cannot exclude that this mechanism may be viable within a limited range of concentrations of agonist and acetylcholine (see Supplementary Modeling Results in Supporting Information), it would be incompatible with the effect of coadministered PNU-120596.
The same principles hold for the nicotine response, which was evoked in the model by the activation, and subsequent desensitization, of α4β2 NNRs. Following Figure 5C, such a positive response is easier to obtain when the activation and desensitization curves substantially overlap (see Figure S1C, Supporting Information) and when the efficacy is high and cholinergic tone low.
The second determinant of the response, the relative expression of functional α7 receptors by different neuron types, is difficult to quantify. Although transcription can be measured,11,32 most receptors are located intracellularly, and electron microcopy is needed to confirm their subcellular position at the plasma membrane.23 In addition, electrophysiological responses after local versus systemic application may differ.11 Given that insufficient information is available about the actual distribution of α7 NNRs, we simulated two rather extreme cases with 80% of α7 NNRs located on dopamine neurons or their afferents (the desensitization model, s = 0.8) and all of them on the afferents to GABA neurons (the activation model, s = 0). Apart from these two cases, the responses from intermediate distributions can be derived as follows. Given that the strength of inhibition from GABA neurons to dopamine neurons in the VTA had a relative weight of 1.5, all α7 responses would virtually disappear in dopamine neurons at a distribution factor s of 0.6 (40% of α7 NNRs on afferents to GABA neurons) since currents of similar amplitude but opposite sign would cancel each other. At still lower values of s, the responses of the dopamine neuron would change sign, and be mirror symmetric about the baseline with respect to those shown before for the desensitization model. (The same reasoning can be applied to the responses of the activation model, with a reduction of response amplitude as s starts rising from 0 and a sign reversal at s = 0.6.)
Similar mechanisms of dopamine modulation can take place in other brain regions, for example, in the dorsal striatum. However, since the innervations of the striatum and nucleus accumbens are divergent, some distinction can be expected. In the Supporting Information, we discuss how the same principles may apply to predict α7-evoked dopamine release in prefrontal cortex.
Limitations of the Present Study
A final note is needed on the calibration of the two model variables representing the cholinergic and dopaminergic tones, respectively. The cholinergic tone was modeled by a parameter giving the equivalent concentration needed for endogenous acetylcholine to generate a certain steady-state level of receptor activation. Even though many NNRs are located extrasynaptically, the equivalent concentration should not be interpreted as the concentration of acetylcholine in extracellular space, which is in the low nanomolar range.56 At best it could represent the acetylcholine concentration close to the synaptic release site, given that the fast α7 NNR responses are presumably generated, at least in neocortex, by classical synaptic transmission.57
As for the extracellular dopamine concentration, baseline estimates from voltammetry vary from 20–30 nM58 to 73 nM59 to 95–220 nM.60 Extracellular dopamine concentration may also be spatially heterogeneous, with some patches having concentrations in the low micromolar range.51,61 In the present FSCV measurements (Figure 1), no information about quantitative basal dopamine levels was obtained because all data were background subtracted. Drops below baseline have been recorded before,51,59 but their magnitude is difficult to quantify, because no dopamine cyclic voltammogram (inset to Figure 1A) can be obtained when the baseline goes down, and small artifacts can be involved in the drop (such as pH changes). Neither can the present model decide on the amplitude of baseline or evoked dopamine responses. The model assumed a baseline dopamine concentration of 50 nM, but some other parameters (including the brain profile of TC-7020 concentration and nicotine concentration after fast iv injection) are poorly constrained. Figure 6 shows how similar realistic dopamine responses can be obtained with a slower clearance of TC-7020 and nicotine and with a baseline dopamine concentration of 337 nM.
Figure 6.
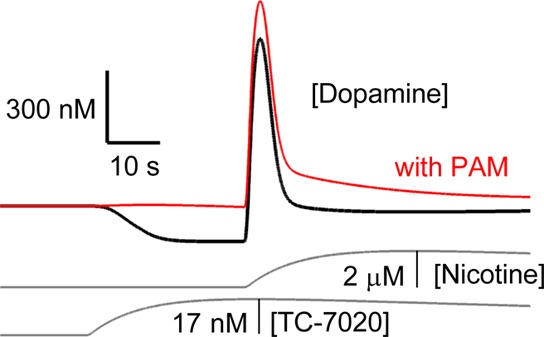
Simulated accumbal dopamine response to TC-7020 and nicotine starting from a baseline [dopamine] of 337 nM. The compounds were administered with a τin of 10 s and τout of 200 s. Simulations of the desensitization model with its standard parameters, except for a weaker connection weight from the GABAergic to dopaminergic neurons (1 vs 1.5) and a different cholinergic tone at α7 versus α4β2 NNRs (33 vs 3 μM). The red trace plots the response after pretreatment with PNU-120596.
Conclusion
The present study is compatible with a mechanism of glutamatergic control of dopamine efflux in nucleus accumbens, in which neither glutamate nor dopamine requires spikes to be released.42 Acetylcholine binding to α7 receptors on glutamatergic terminals promotes the spillover of glutamate to dopaminergic terminals, where binding to ionotropic receptors leads in turn to dopamine release.19 Our combined experimental and modeling results indicate that partial agonists such as TC-7020 may suppress dopamine release by desensitizing the α7 receptors.
Methods
Experimental Procedure
Male Sprague–Dawley rats (Charles Rivers Laboratories, Raleigh, NC) weighing 300–400 g, which were housed two animals per cage with ad libitum food and water in a 12/12 h light/dark cycle, were used in our in vivo voltammetric studies. All procedures were approved by the Wake Forest University and University of North Carolina Animal Care and Use Committees. Experiments were performed on anesthetized animals.
At the day of experiment, naive rats were surgically implanted with indwelling iv catheters under urethane (1.5 g/kg ip) anesthesia using aseptic procedures immediately before voltammetric assessments. A tapered polyurethane catheter was implanted into the right external jugular vein with the catheter exiting the skin behind the ear. A muscle tie served as a tether, preventing the catheter from being dislodged during subsequent voltammetric assessments. Following surgery, the implanted catheter was flushed with 0.2–0.3 mL of sterile 0.9% saline, and the catheter was clamped until later use during the voltammetric experiment. Then rats were head-restrained in a stereotaxic frame, and a carbon fiber electrode (50–100 μm exposed tip length, 7 μm diameter; Goodfellow, Oakdale, PA, USA) was positioned in the nucleus accumbens core (AP + 1.3, L + 1.3, V – 6.7–7.0 mm from bregma) with a Ag/AgCl reference electrode implanted in the contralateral hemisphere. The reference and carbon fiber electrodes were connected to a head-mounted voltammetric amplifier (UNC Electronics Design Facility, Chapel Hill, NC) and voltammetric recordings were made at the carbon fiber electrode every 100 ms by applying a triangular waveform (−0.4 to +1.3 V, 300 V/s). Data were digitized (National Instruments, Austin, TX) and stored on a computer. To confirm the placement of electrodes, a 200 μA current was passed through a stainless steel electrode for 10 s. Brain was removed and postfixed in 10% formalin for at least 3 days. After freezing, 50 μm coronal brain sections were taken and mounted throughout the rostral-caudal extent of the nucleus accumbens. The position of electrodes was assessed by visual examination of coronal sections for electrolytic lesions.
Nicotine (0.3 mg/kg), TC-7020 phosphate (1.0 mg/kg), and saline were administered intravenously as an experimenter-delivered bolus over 4–6 s in a volume of 0.3–0.4 mL. Methyllycaconitine citrate (MLA, 10 mg/kg) and the α7-selective type-2 positive allosteric modulator PNU-120596 (5.0 mg/kg) were injected intraperitoneally (ip) and subcutaneously (sc), respectively, 30 min before TC-7020 administration.62,63 The effect of every compound on dopamine dynamics was replicated on four animals. These results were used for the modeling procedure. TC-7020 was provided by Targacept, Inc., PNU-120596 was purchased from Sigma-Aldrich (St. Louis, MO), and MLA was purchased from Tocris Bioscience (Bristol, U.K.).
Modeling Methods
The mathematical model represented the mesoaccumbens pathway and incorporated both receptor kinetics and network dynamics. Program code was written in XPP.
Receptor Model
Each NNR subtype had an activation and a desensitization gate, resulting in four possible receptor states, with the fraction of conducting receptors being the product of those being active, a, and sensitive, s.64 The steady-state concentration–response curves, a∞ and (1 – s∞) for each gate were described as Hill functions (Figure S1, Supporting Information). More particularly, steady-state activation, a∞, was calculated as
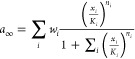 |
where xi is the concentration of the ith agonist, Ki is the agonist’s EC50 value (half-maximally effective concentration), and ni is its Hill-exponent (nHa in Table 1). In this sum, the fraction represents the fractional occupancy of the receptor by agonist i, which is multiplied by the agonists’s efficacy wi. For wi, the Emax values from Table 1 were used.
Table 1. Parameters of the Hill Functions Used to Model the Receptors’ Activation and Desensitization Gates.
| α7 | TC-7020 | nicotine | acetylcholine |
|---|---|---|---|
| EC50a (μM) | 0.03 | 13d | 68d |
| nHab | 1.73 | 1.73 | 1.73 |
| DC50a (μM) | 0.002 | 1.3 | |
| nHsb | 2 | 2 | |
| Emaxc (%) | 30 | 80 | 100 |
The degree of desensitization (1 – s∞) was calculated using the same formula, substituting the values of DC50 and nHs for Ki and ni. Because nicotine and TC-7020 desensitized the receptor completely at high concentrations (see Figure S1A,B, Supporting Information), their weight factor wi was unity. The wi of endogenous acetylcholine at the desensitization gate, on the other hand, was set to zero (no desensitization at all) to reflect in vivo the physiological conditions where the transmitter is rapidly hydrolyzed by acetylcholine esterase before desensitization can start.
The acetylcholine parameter in the present model determined the degree of endogenous cholinergic activity, or cholinergic tone, which was assumed to be constant during the course of a recording. The release of acetylcholine not being modeled explicitly, the average tone that it generated was represented by an equivalent concentration constant, [ACh], which was set so as to generate an average level of receptor activation, relative to which the applied exogenous compounds exerted their action either through further activation, deactivation (competition), or desensitization.
Finally, activation was always fast (time constant 5 ms), whereas the time constant of desensitization varied between minimum and maximum values in a concentration-dependent manner according to the same Hill function as its steady-state65 (from 120 s to 50 ms for α7 NNRs; from 600 s to 500 ms for α4β2 NNRs).
Figure S1, Supporting Information, shows the steady-state activation and desensitization curves used in the model for TC-7020 (panel A) and nicotine (panel B) at the α7 NNRs and for nicotine at the α4β2* NNRs (panel C). The time traces above each pair of concentration–response curves plot the mean channel current at varying agonist concentrations, illustrating the concentration dependency of the speed of desensitization and the faster desensitization of α7 compared with α4β2* NNRs. Table 1 lists the parameters of the Hill functions, which took the same values as in Graupner et al.64 except where a new reference is given. Most of these parameters had been taken from Fenster et al.66 and Buisson and Bertrand.67
Model parameters for the α4β2* NNR were identical to those used by Graupner et al.,64 except for the lower efficacy (0.8) and a higher affinity (EC50 230 nM) of nicotine, such as is typical of α6-containing α4β2 NNRs.68
The last column of Table 1 also implies that the model’s endogenous transmitter, acetylcholine (ACh), activated the receptors without desensitizing them. As stated above, this feature reflects the fundamental difference in kinetics between physiologically released acetylcholine, which is rapidly broken down by ACh-esterase, and exogenous compounds such as nicotine and TC-7020.
Circuit Model
The circuit represented a population of dopaminergic neurons that received inhibition from a local population of GABAergic interneurons.64 The output variables of the model were the mean-field average population activities and the resulting extracellular dopamine concentration. Each population (or its glutamatergic afferents) expressed NNRs of the α7 and α4β2* subtypes.
The relative expression of each subtype across the dopamine and GABA neurons was determined by two parameters, r (for α4β2) and s (for α7). A parameter value of 1 indicated that the corresponding NNR subtype was expressed exclusively by the dopamine neuron population (or its glutamatergic afferents); a value of 0.5 meant a balanced distribution across the two neuron populations.
The value of s was the major parameter distinguishing the so-called desensitization and activation models. It determined the relative expression of α7 NNRs on the two neuron populations or their afferents: 80% of α7 NNRs were located on the dopamine neuron or its afferents in the desensitization model (s = 0.8) versus all α7 NNRs on the GABA neuron’s afferents in the activation model (s = 0). The principal locations of α7 NNRs compatible with these two models are as depicted in the respective circuit diagrams of Figures 2A and 3A. Most α4β2* NNRs were located on the dopamine neuron in both versions of the model: r = 0.8 (0.7) for desensitization (activation) model.
Evidently, the NNRs at the locations depicted in Figures 2A and 3A act all in concert, but for the sake of clarity, we accentuated their relative contributions in the model. A final difference with the circuit model of Graupner et al.64 concerned the connection strength from GABAergic onto DAergic neurons, which was enhanced by 50% to favor the activation model.
As described in Graupner et al.,64 the action of preterminal α7 NNRs was to enhance glutamate release. Because α7 NNRs may evoke glutamate release in an impulse-independent manner69 and because we are faced in this anesthetized in vivo condition with asynchronous inputs from probably tens of afferents, the steady-state terms for spike-evoked and α7-evoked glutamate release were simply added. Hence, the glutamatergic receptor currents were assumed to be proportional to the amount of glutamate released, with a negative sign for the mGluRs.70 Because glutamatergic and dopaminergic terminals often make apposed synapses onto the same medium spiny neuron in nucleus accumbens, diffusion at locations 3 (Figure 2A) and 5 (Figure 3A) of the circuit was assumed to be much faster than the measured responses. Our mean-field approach further implicitly assumed that at the concentrations of spilled-over glutamate, NMDA receptors do not substantially desensitize.
Extracellular Dopamine Concentration
The final outcome of the model was obtained by mapping the mean spike rate of the dopamine neurons onto the variable representing the extracellular dopamine concentration. Although dopamine is particularly released during phasic firing,71 recent cyclic voltammetry in anesthetized mice showed a fairly linear increase with stimulation frequency.17 We further assumed a steady-state dopamine concentration of about 50 nM, except in Figure 6 where baseline dopamine concentration was 337 nM. These assumptions allowed us to calculate the dopamine concentration, C, using the following equation:
The first term on the right-hand side adds dopamine in proportion to the enhancement in spike rate, r, relative to the steady-state rate, rss (measured before any drug administration). Dopamine leaks away (second term) and is resorbed by an uptake process with Michaelis–Menten kinetics of maximum rate, Vm, and affinity, Km (last term). We set Vm to 1.3 μM s–1, Km to 0.2 μM, and τ to 200 ms.60 By setting Cb (the basal dopamine concentration in the absence of input or uptake) to 100 nM (500 nM for Figure 6), we obtained a steady-state concentration of about 50 nM.
Glossary
Abbreviations
- ACh
acetylcholine
- DA
dopamine
- FSCV
fact-scan cyclic voltammetry
- GABA
γ-amino butyric acid
- MLA
methyllycaconitine citrate
- NNR
neuronal nicotinic receptor
- PAM
positive allosteric modulator
- PNU-120596
1-(5-chloro-2,4-dimethoxy-phenyl)-3-(5-methyl-isoxazol-3-yl)-urea
- TC-7020
(2S,3R)-5-methyl-N-[2-(pyridin-3-ylmethyl)-1-azabicyclo[2.2.2]oct-3-yl]thiophene-2-carboxamide
- VTA
ventral tegmental area
Supporting Information Available
Detailed comparative responses of the two models, a consideration of a third mechanism (coagonism), and a discussion of the generalization of our findings to prefrontal cortex. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
V.P.G., E.B., M.B., and B.G. designed and supervised the study. R.M. performed the simulations. V.P.G., V.G., and E.B. performed the experiments. R.M., V.P.G., E.B., M.B., and B.G. wrote the manuscript.
This work was part of the Collaborative research on NNR/DA integrative pharmacology (PCR-INP10/01), awarded to M.B. and B.G. and was partly supported by IdEx ANR-11-0001-02 PSL* and LabEx ANR-10-LABX-0087 (France). Experimental work was supported by grants from the National Institutes of Health (AA020564) and the Tab Williams Family Endowment Fund to E.B. B.G. acknowledges support from the Basic Research Program of the National Research University Higher School of Economics (Russia).
The authors declare the following competing financial interest(s): M.B. is an employee and stockholder of Targacept Inc., a biopharmaceutical company involved in research and development of drugs targeting the neuronal nicotinic receptors.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Geerts H. (2012) α7 Nicotinic receptor modulators for cognitive deficits in schizophrenia and Alzheimer’s disease. Expert Opin. Invest. Drugs 21, 59–65. [DOI] [PubMed] [Google Scholar]
- Kucinski A. J.; Stachowiak M. K.; Wersinger S. R.; Lippiello P. M.; Bencherif M. (2011) Alpha7 neuronal nicotinic receptors as targets for novel therapies to treat multiple domains of schizophrenia. Curr. Pharm. Biotechnol. 12, 437–448. [DOI] [PubMed] [Google Scholar]
- Wallace T. L.; Bertrand D. (2013) Alpha7 neuronal nicotinic receptors as a drug target in schizophrenia. Expert Opin. Ther. Targets 17, 139–155. [DOI] [PubMed] [Google Scholar]
- Quik M.; Wonnacott S. (2011) α6β2* and α4β2* Nicotinic acetylcholine receptors as drug targets for Parkinson’s disease. Pharmacol. Rev. 63, 938–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F. M.; Liang Y.; Dani J. A. (2001) Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat. Neurosci. 4, 1224–1229. [DOI] [PubMed] [Google Scholar]
- Brazell M. P.; Mitchell S. N.; Joseph M. H.; Gray J. A. (1990) Acute administration of nicotine increases the in vivo extracellular levels of dopamine, 3,4-dihydroxyphenylacetic acid and ascorbic acid preferentially in the nucleus accumbens of the rat: comparison with caudate-putamen. Neuropharmacology 29, 1177–1185. [DOI] [PubMed] [Google Scholar]
- Cheer J. F.; Wassum K. M.; Sombers L. A.; Heien M. L.; Ariansen J. L.; Aragona B. J.; Phillips P. E.; Wightman R. M. (2007) Phasic dopamine release evoked by abused substances requires cannabinoid receptor activation. J. Neurosci. 27, 791–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mameli-Engvall M.; Evrard A.; Pons S.; Maskos U.; Svensson T. H.; Changeux J. P.; Faure P. (2006) Hierarchical control of dopamine neuron-firing patterns by nicotinic receptors. Neuron 50, 911–921. [DOI] [PubMed] [Google Scholar]
- Schilström B.; Svensson H. M.; Svensson T. H.; Nomikos G. G. (1998) Nicotine and food induced dopamine release in the nucleus accumbens of the rat: putative role of α7 nicotinic receptors in the ventral tegmental area. Neuroscience 85, 1005–1009. [DOI] [PubMed] [Google Scholar]
- Good C. H.; Lupica C. R. (2009) Properties of distinct ventral tegmental area synapses activated via pedunculopontine or ventral tegmental area stimulation in vitro. J. Physiol. 587, 1233–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor D. H.; Burman P. N.; Hansen M. D.; Wlcox R. S.; Larsen B. R.; Blanchard J. K.; Merrill C. B.; Edwards J. G.; Sudweeks S. N.; Wu J.; Arias H. R.; Steffensen S. C. (2013) Nicotine enhances the excitability of Gaba neurons in the ventral tegmental area via activation of alpha 7 nicotinic receptors on glutamate terminals. Biochem. Pharmacol. S1, 007. [Google Scholar]
- Tolu S.; Eddine R.; Marti F.; David V.; Graupner M.; Pons S.; Baudonnat M.; Husson M.; Besson M.; Reperant C.; Zemdegs J.; Pages C.; Hay Y. A.; Lambolez B.; Caboche J.; Gutkin B.; Gardier A. M.; Changeux J. P.; Faure P.; Maskos U. (2013) Co-activation of VTA DA and GABA neurons mediates nicotine reinforcement. Mol. Psychiatry 18, 382–393. [DOI] [PubMed] [Google Scholar]
- Forster G. L.; Blaha C. D. (2000) Laterodorsal tegmental stimulation elicits dopamine efflux in the rat nucleus accumbens by activation of acetylcholine and glutamate receptors in the ventral tegmental area. Eur. J. Neurosci. 12, 3596–3604. [DOI] [PubMed] [Google Scholar]
- Schilström B.; Fagerquist M. V.; Zhang X.; Hertel P.; Panagis G.; Nomikos G. G.; Svensson T. H. (2000) Putative role of presynaptic alpha7* nicotinic receptors in nicotine stimulated increases of extracellular levels of glutamate and aspartate in the ventral tegmental area. Synapse 38, 375–383. [DOI] [PubMed] [Google Scholar]
- Cachope R.; Mateo Y.; Mathur B. N.; Irving J.; Wang H. L.; Morales M.; Lovinger D. M.; Cheer J. F. (2012) Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: Setting the tone for reward processing. Cell Rep. 2, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threlfell S.; Lalic T.; Platt N. J.; Jennings K. A.; Deisseroth K.; Cragg S. J. (2012) Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron 75, 58–64. [DOI] [PubMed] [Google Scholar]
- Koranda J. L.; Cone J. J.; McGehee D. S.; Roitman M. F.; Beeler J. A.; Zhuang X. (2014) Nicotinic receptors regulate the dynamic range of dopamine release in vivo. J. Neurophysiol. 111, 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grilli M.; Summa M.; Salamone A.; Olivero G.; Zappettini S.; Di Prisco S.; Feligioni M.; Usai C.; Pittaluga A.; Marchi M. (2012) In vitro exposure to nicotine induces endocytosis of presynaptic AMPA receptors modulating dopamine release in rat nucleus accumbens nerve terminals. Neuropharmacology 63, 916–926. [DOI] [PubMed] [Google Scholar]
- Kaiser S.; Wonnacott S. (2000) α-Bungarotoxin-sensitive nicotinic receptors indirectly modulate [3H]dopamine release in rat striatal slices via glutamate release. Mol. Pharmacol. 58, 312–318. [DOI] [PubMed] [Google Scholar]
- Marrero M. B.; Lucas R.; Salet C.; Hauser T. A.; Mazurov A.; Lippiello P. M.; Bencherif M. (2010) An α7 nicotinic acetylcholine receptor-selective agonist reduces weight gain and metabolic changes in a mouse model of diabetes. J. Pharmacol. Exp. Ther. 332, 173–180. [DOI] [PubMed] [Google Scholar]
- Narla S.; Klejbor I.; Birkaya B.; Lee Y. W.; Morys J.; Stachowiak E. K.; Terranova C.; Bencherif M.; Stachowiak M. K. (2013) α7 Nicotinic receptor agonist reactivates neurogenesis in adult brain. Biochem. Pharmacol. 86, 1099–1104. [DOI] [PubMed] [Google Scholar]
- Kucinski A.; Syposs C.; Wersinger S.; Bencherif M.; Stachowiak M. K.; Stachowiak E. K. (2012) α7 Neuronal nicotinic receptor agonist (TC-7020) reverses increased striatal dopamine release during acoustic PPI testing in a transgenic mouse model of schizophrenia. Schizophr. Res. 136, 82–87. [DOI] [PubMed] [Google Scholar]
- Garzón M.; Duffy A. M.; Chan J.; Lynch M. K.; Mackie K.; Pickel V. M. (2013) Dopamine D2 and acetylcholine α7 nicotinic receptors have subcellular distributions favoring mediation of convergent signaling in the mouse ventral tegmental area. Neuroscience 252, 126–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones I. W.; Wonnacott S. (2004) Precise localization of α7 nicotinic acetylcholine receptors on glutamatergic axon terminals in the rat ventral tegmental area. J. Neurosci. 24, 11244–11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingstone P. D.; Dickinson J. A.; Srinivasan J.; Kew J. N.; Wonnacott S. (2010) Glutamate-dopamine crosstalk in the rat prefrontal cortex is modulated by alpha7 nicotinic receptors and potentiated by PNU-120596. J. Mol. Neurosci. 40, 172–176. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Paspalas C. D.; Jin L. E.; Picciotto M. R.; Arnsten A. F.; Wang M. (2013) Nicotinic α7 receptors enhance NMDA cognitive circuits in dorsolateral prefrontal cortex. Proc. Natl. Acad. Sci. U. S. A. 110, 12078–12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y.; Yang K.; Wang H.; Wu J. (2011) Exposure of nicotine to ventral tegmental area slices induces glutamatergic synaptic plasticity on dopamine neurons. Synapse 65, 332–338. [DOI] [PubMed] [Google Scholar]
- Melis M.; Scheggi S.; Carta G.; Madeddu C.; Lecca S.; Luchicchi A.; Cadeddu F.; Frau R.; Fattore L.; Fadda P.; Ennas M. G.; Castelli M. P.; Fratta W.; Schilstrom B.; Banni S.; De Montis M. G.; Pistis M. (2013) PPARα regulates cholinergic-driven activity of midbrain dopamine neurons via a novel mechanism involving α7 nicotinic acetylcholine receptors. J. Neurosci. 33, 6203–6211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst R. S.; Hajós M.; Raggenbass M.; Wall T. M.; Higdon N. R.; Lawson J. A.; Rutherford-Root K. L.; Berkenpas M. B.; Hoffmann W. E.; Piotrowski D. W.; Groppi V. E.; Allaman G.; Ogier R.; Bertrand S.; Bertrand D.; Arneric S. P. (2005) A novel positive allosteric modulator of the α7 neuronal nicotinic acetylcholine receptor: in vitro and in vivo characterization. J. Neurosci. 25, 4396–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilström B.; Rawal N.; Mameli-Engvall M.; Nomikos G. G.; Svensson T. H. (2003) Dual effects of nicotine on dopamine neurons mediated by different nicotinic receptor subtypes. Int. J. Neuropsychopharmacol. 6, 1–11. [DOI] [PubMed] [Google Scholar]
- Wu J.; George A. A.; Schroeder K. M.; Xu L.; Marxer-Miller S.; Lucero L.; Lukas R. J. (2004) Electrophysiological, pharmacological, and molecular evidence for α7-nicotinic acetylcholine receptors in rat midbrain dopamine neurons. J. Pharmacol. Exp. Ther. 311, 80–91. [DOI] [PubMed] [Google Scholar]
- Klink R.; de Kerchove d’Exaerde A.; Zoli M.; Changeux J. P. (2001) Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J. Neurosci. 21, 1452–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K.; Hu J.; Lucero L.; Liu Q.; Zheng C.; Zhen X.; Jin G.; Lukas R. J.; Wu J. (2009) Distinctive nicotinic acetylcholine receptor functional phenotypes of rat ventral tegmental area dopaminergic neurons. J. Physiol. 587, 345–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady S. R.; Salminen O.; Laverty D. C.; Whiteaker P.; McIntosh J. M.; Collins A. C.; Marks M. J. (2007) The subtypes of nicotinic acetylcholine receptors on dopaminergic terminals of mouse striatum. Biochem. Pharmacol. 74, 1235–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingstone P. D.; Wonnacott S. (2009) Nicotinic acetylcholine receptors and the ascending dopamine pathways. Biochem. Pharmacol. 78, 744–755. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Sulzer D. (2003) Glutamate spillover in the striatum depresses dopaminergic transmission by activating group I metabotropic glutamate receptors. J. Neurosci. 23, 10585–10592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke R. L.; Kem W. R.; Soti F.; Lopez-Hernandez G. Y.; Horenstein N. A. (2009) Activation and desensitization of nicotinic α7-type acetylcholine receptors by benzylidene anabaseines and nicotine. J. Pharmacol. Exp. Ther. 329, 791–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalappa B. I.; Uteshev V. V. (2013) The dual effect of PNU-120596 on α7 nicotinic acetylcholine receptor channels. Eur. J. Pharmacol. 718, 226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G.; Duffy P.; Swanson C.; Ghasemzadeh M. B.; Kalivas P. W. (1999) The regulation of dopamine transmission by metabotropic glutamate receptors. J. Pharmacol. Exp. Ther. 289, 412–416. [PubMed] [Google Scholar]
- van Zessen R.; Phillips J. L.; Budygin E. A.; Stuber G. D. (2012) Activation of VTA GABA neurons disrupts reward consumption. Neuron 73, 1184–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Sherwood J. L.; Miles C. P.; Whiffin G.; Lodge D. (2006) TC-2559 excites dopaminergic neurones in the ventral tegmental area by stimulating α4β2-like nicotinic acetylcholine receptors in anaesthetised rats. Br. J. Pharmacol. 147, 379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glowinski J.; Cheramy A.; Romo R.; Barbeito L. (1988) Presynaptic regulation of dopaminergic transmission in the striatum. Cell. Mol. Neurobiol. 8, 7–17. [DOI] [PubMed] [Google Scholar]
- Cheramy A.; Desce J. M.; Godeheu G.; Glowinski J. (1994) Presynaptic control of dopamine synthesis and release by excitatory amino acids in rat striatal synaptosomes. Neurochem. Int. 25, 145–154. [DOI] [PubMed] [Google Scholar]
- Marchi M.; Risso F.; Viola C.; Cavazzani P.; Raiteri M. (2002) Direct evidence that release-stimulating α7* nicotinic cholinergic receptors are localized on human and rat brain glutamatergic axon terminals. J. Neurochem. 80, 1071–1078. [DOI] [PubMed] [Google Scholar]
- Garcao P.; Oliveira C. R.; Cunha R. A.; Agostinho P. (2014) Subsynaptic localization of nicotinic acetylcholine receptor subunits: A comparative study in the mouse and rat striatum. Neurosci. Lett. 566, 106–110. [DOI] [PubMed] [Google Scholar]
- Lenoir M.; Kiyatkin E. A. (2013) Intravenous nicotine injection induces rapid, experience-dependent sensitization of glutamate release in the ventral tegmental area and nucleus accumbens. J. Neurochem. 127, 541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg J. A.; Reynolds J. N. (2011) Spontaneous firing and evoked pauses in the tonically active cholinergic interneurons of the striatum. Neuroscience 198, 27–43. [DOI] [PubMed] [Google Scholar]
- Albuquerque E. X., Pereira E. F. R., and Alkondon M. (2013) Tonic activation of α7 nicotinic acetylcholine receptors (nAChRs) controls glutamatergic inputs to striatal and parietal cortical neurons in guinea pig brain slices, in Society for Neuroscience Annual Meeting, p 224.01, Society for Neuroscience, San Diego, CA. [Google Scholar]
- Wu H. Q.; Rassoulpour A.; Schwarcz R. (2007) Kynurenic acid leads, dopamine follows: A new case of volume transmission in the brain?. J. Neural Transm. 114, 33–41. [DOI] [PubMed] [Google Scholar]
- Kulagina N. V.; Zigmond M. J.; Michael A. C. (2001) Glutamate regulates the spontaneous and evoked release of dopamine in the rat striatum. Neuroscience 102, 121–128. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Moquin K. F.; Michael A. C. (2010) Evidence for coupling between steady-state and dynamic extracellular dopamine concentrations in the rat striatum. J. Neurochem. 114, 150–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson M.; David V.; Baudonnat M.; Cazala P.; Guilloux J. P.; Reperant C.; Cloez-Tayarani I.; Changeux J. P.; Gardier A. M.; Granon S. (2012) Alpha7-nicotinic receptors modulate nicotine-induced reinforcement and extracellular dopamine outflow in the mesolimbic system in mice. Psychopharmacology (Berlin, Ger.) 220, 1–14. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Pearl S. M.; Zigmond M. J.; Michael A. C. (2000) Inhibitory glutamatergic regulation of evoked dopamine release in striatum. Neuroscience 96, 65–72. [DOI] [PubMed] [Google Scholar]
- Grinevich V. P., O’Connor J. A., Bencherif M., and Budygin E. A. (2009) Effects of nicotine on real time dopamine dynamics in rat nucleus accumbens: In vivo voltammetric study, in Society for Neuroscience Annual Meeting, p 226.12, Society for Neuroscience, Chicago. [Google Scholar]
- Campling B. G.; Kuryatov A.; Lindstrom J. (2013) Acute activation, desensitization and smoldering activation of human acetylcholine receptors. PLoS One 8, e79653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinson P. N.; Justice J. B. Jr. (1997) Effect of neostigmine on concentration and extraction fraction of acetylcholine using quantitative microdialysis. J. Neurosci. Methods 73, 61–67. [DOI] [PubMed] [Google Scholar]
- Arroyo S.; Bennett C.; Hestrin S. (2014) Nicotinic modulation of cortical circuits. Front. Neural Circuits 8, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owesson-White C. A.; Roitman M. F.; Sombers L. A.; Belle A. M.; Keithley R. B.; Peele J. L.; Carelli R. M.; Wightman R. M. (2012) Sources contributing to the average extracellular concentration of dopamine in the nucleus accumbens. J. Neurochem. 121, 252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo P. L.; Yao W.; Sun L.; Kuo S. T.; Li Q.; Wang S. R.; Dou H. Q.; Xu H. D.; Zhang C. X.; Kang X. J.; Zhou Z.; Zhang B. (2013) Impulse-dependent extracellular resting dopamine concentration in rat striatum in vivo. Neurochem. Int. 62, 50–57. [DOI] [PubMed] [Google Scholar]
- Chen K. C.; Budygin E. A. (2007) Extracting the basal extracellular dopamine concentrations from the evoked responses: Re-analysis of the dopamine kinetics. J. Neurosci. Methods 164, 27–42. [DOI] [PubMed] [Google Scholar]
- Shu Z.; Taylor I. M.; Michael A. C. (2013) The dopamine patchwork of the rat nucleus accumbens core. Eur. J. Neurosci. 38, 3221–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean S. L.; Idris N. F.; Grayson B.; Gendle D. F.; Mackie C.; Lesage A. S.; Pemberton D. J.; Neill J. C. (2012) PNU-120596, a positive allosteric modulator of α7 nicotinic acetylcholine receptors, reverses a sub-chronic phencyclidine-induced cognitive deficit in the attentional set-shifting task in female rats. J. Psychopharmacol. 26, 1265–1270. [DOI] [PubMed] [Google Scholar]
- Turek J. W.; Kang C. H.; Campbell J. E.; Arneric S. P.; Sullivan J. P. (1995) A sensitive technique for the detection of the α7 neuronal nicotinic acetylcholine receptor antagonist, methyllycaconitine, in rat plasma and brain. J. Neurosci. Methods 61, 113–118. [DOI] [PubMed] [Google Scholar]
- Graupner M.; Maex R.; Gutkin B. (2013) Endogenous cholinergic inputs and local circuit mechanisms govern the phasic mesolimbic dopamine response to nicotine. PLoS Comput. Biol. 9, e1003183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady S. R.; Grun E. U.; Marks M. J.; Collins A. C. (1997) Pharmacological comparison of transient and persistent [3H]dopamine release from mouse striatal synaptosomes and response to chronic L-nicotine treatment. J. Pharmacol. Exp. Ther. 282, 32–43. [PubMed] [Google Scholar]
- Fenster C. P.; Rains M. F.; Noerager B.; Quick M. W.; Lester R. A. (1997) Influence of subunit composition on desensitization of neuronal acetylcholine receptors at low concentrations of nicotine. J. Neurosci. 17, 5747–5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson B.; Bertrand D. (2001) Chronic exposure to nicotine upregulates the human α4β2 nicotinic acetylcholine receptor function. J. Neurosci. 21, 1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen O.; Drapeau J. A.; McIntosh J. M.; Collins A. C.; Marks M. J.; Grady S. R. (2007) Pharmacology of α-conotoxin MII-sensitive subtypes of nicotinic acetylcholine receptors isolated by breeding of null mutant mice. Mol. Pharmacol. 71, 1563–1571. [DOI] [PubMed] [Google Scholar]
- Engelman H. S.; MacDermott A. B. (2004) Presynaptic ionotropic receptors and control of transmitter release. Nat. Rev. Neurosci. 5, 135–145. [DOI] [PubMed] [Google Scholar]
- Fiorillo C. D.; Williams J. T. (1998) Glutamate mediates an inhibitory postsynaptic potential in dopamine neurons. Nature 394, 78–82. [DOI] [PubMed] [Google Scholar]
- Grace A. A. (1991) Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: A hypothesis for the etiology of schizophrenia. Neuroscience 41, 1–24. [DOI] [PubMed] [Google Scholar]
- Papke R. L.; Dwoskin L. P.; Crooks P. A. (2007) The pharmacological activity of nicotine and nornicotine on nAChRs subtypes: Relevance to nicotine dependence and drug discovery. J. Neurochem. 101, 160–167. [DOI] [PubMed] [Google Scholar]
- Grady S. R.; Drenan R. M.; Breining S. R.; Yohannes D.; Wageman C. R.; Fedorov N. B.; McKinney S.; Whiteaker P.; Bencherif M.; Lester H. A.; Marks M. J. (2010) Structural differences determine the relative selectivity of nicotinic compounds for native α4β2*-, α6β2*-, α3β4*- and α7-nicotine acetylcholine receptors. Neuropharmacology 58, 1054–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.