

Abstract
Hsp90 C-terminal inhibitors are advantageous for the development of new cancer chemotherapeutics due to their ability to segregate client protein degradation from induction of the prosurvival heat shock response, which is a major detriment associated with Hsp90 N-terminal inhibitors under clinical investigation. Based upon prior SAR trends, a 1,2,3-triazole side chain was placed in lieu of the aryl side chain and attached to both the coumarin and biphenyl scaffold. Antiproliferative studies against SKBr3 and MCF-7 breast cancer cell lines demonstrated these triazole-containing compounds to exhibit improved activity. These compounds were shown to manifest Hsp90 inhibitory activity through Western blot analysis and represent a new scaffold upon which more potent inhibitors can be pursued.
Keywords: Heat shock protein 90, Hsp90 C-terminal inhibitors, Novobiocin, Triazole, Breast cancer
Introduction
Developments in molecular biology have led to identification of new targets for the pursuit of cancer chemotherapies during recent decades. The most successful demonstration thus far is Gleevec, a small molecule BCR/ABL kinase inhibitor used for the treatment of chronic myelogenous leukemia.1, 2 Due to their genetic instability and signaling redundancy, cancer cells exhibit compensatory mechanisms and often acquire resistance to inhibition of a single protein.3 The most common approach to overcome this obstacle relies upon combination therapy, in which inhibitors of multiple oncogenic targets are administered. In contrast, the development of anti-cancer agents that target signaling nodes represent an alternative method to target multiple signaling pathways simultaneously while minimizing the potential for resistance.4, 5 The 90 kDa heat shock protein (Hsp90) represents a paradigmatic target for the latter approach, as molecular chaperones are responsible for the conformational maturation of signaling proteins associated with cancer cell growth and proliferation.6, 7 In fact, recent studies indicated that almost 400 Hsp90-depedent client proteins have been identified,8, 9 many of which are directly associated with the six hallmarks of cancer.10 Consequently, Hsp90 inhibition results in the simultaneous disruption of multiple signaling pathways required for the viability of transformed cells. 17-AAG is a semi-synthetic derivative of geldanamycin that targets the N-terminal ATP-binding site and exhibits therapeutic benefit at tolerable doses in the clinic. In addition to 17-AAG, 16 other Hsp90 N-terminal inhibitors have been investigated in the clinical setting, which highlights the importance of Hsp90 as a cancer therapeutic target.11 Unfortunately, these Hsp90 N-terminal inhibitors induce the pro-survival heat shock response at the same concentration needed to induce client protein degradation, resulting in primarily cytostatic activity. Thus, the clinical effectiveness of Hsp90 N-terminal inhibitors remains unclear.12 In contrast to N-terminal inhibitors, studies have demonstrated that inhibition of the Hsp90 C-terminus13 and some allosteric modulators14,15 do not induce the heat shock response, and consequently provides an attractive paradigm for the development of Hsp90 inhibitors that exhibit improved activities.16,17
Seminal work by Neckers and coworkers reported that the DNA gyrase inhibitor, novobiocin (1), and related natural products bind the Hsp90 C-terminus nucleotide binding pocket with low affinity (IC50 ~700 μM).18 Subsequent modification to novobiocin including alteration of coumarin scaffold, optimization of the benzamide side chain, and replacement of the sugar moiety led to compounds that manifest increased inhibitory activity.19–24 Although no co-crystal structure of Hsp90 bound to C-terminal inhibitors has been solved, insights gained from SAR studies on novobiocin has produced valuable information on the Hsp90 C-terminal binding pocket. Among the key elements on novobiocin identified, the secondary amide linker and an aryl side chain were shown to be particularly important9 (Figure 1A). Structural modification to these moieties has produced analogues that exhibit improved activity, such as compound 2, which contains a simplified coumarin core, a piperidine moiety, and a benzamide side chain (Figure 1B). Increasing evidence suggests that hydrogen bonding interactions on the side chain are also beneficial,21 which is in accord with the improved activities observed for analogues possessing a 2-21 or 3-indole25 or a 4-methoxyphenyl23 side chain. Therefore, it was proposed that incorporation of additional hydrogen bonding interactions could further increase inhibitory activity. In addition, it was recently demonstrated that the coumarin core could be replaced with simplified surrogates,26 which is imperative, due to the multi-step synthesis required for the preparation of this ring system. Indeed, replacement of the coumarin ring (2) system with a biphenyl scaffold (3) resulted in compounds that manifest increased antiproliferative activity (Figure 1B).27 Therefore, it was proposed that incorporation of a hydrogen bonding ring system onto both the coumarin and biphenyl scaffold could provide compounds that exhibit improved activity. Through exploratory methods, it was determined that the 1,2,3-triazole moiety represents an optimal side chain surrogate for the benzamide side chain present in novobiocin.
Figure 1.

The design of novel hsp90 C-terminal inhibitors.
The copper catalyzed azido alkyne cycloaddition (CuAAC) reaction has found wide application to various fields of chemistry, especially biomedical and medicinal chemistry, due to the mild conditions required for assembly and its broad functional group tolerance.28, 29 The product obtained from such reactions, the 1,2,3-triazole, is an amide bioisostere that demonstrates enhanced hydrogen bonding interactions compared to the amide. In addition, the triazole ring is present in various therapeutic classes due to its hydrogen bonding capabilities and high dipole.29, 30 Prior studies have shown that bioisosteric replacement of the amide linker on novobiocin with a 1,2,3-triazole resulted in decreased antiproliferative activity,25 indicating that the amide N–H may be acting as a hydrogen bond donor. Therefore, it was proposed to incorporate the 1,2,3-triazole into the aryl side chain while retaining the secondary amide linker. To test this hypothesis, compounds 4 and 5 were designed to combine the features noted above, wherein the noviose sugar was replaced with a readily available piperidine moiety (Figure 1, C).22 The results from these studies are presented herein and describe the identification of compounds that manifest mid-nanomolar activity, which is ~1000 fold more efficacious than novobiocin.
Results and discussion
Retrosynthetically, coumarin derivative 4 was envisioned for assembly by an amide coupling reaction between aniline 6 and triazolic acid 7, with the latter being accessed through a CuAAC reaction between the corresponding azides and propiolic acid (Scheme 1). Synthesis of both the phenyl and benzyl derivatives (4a and 4b) commenced by preparation of 7a and 7b from propiolic acid and the corresponding azides.31 Aminocoumarin 6 was prepared via reported procedures.22 Upon treatment with oxalyl chloride, triazolic acids 7a and 7b were converted to the corresponding acid chlorides and coupled with aminocoumarin 6 to provide 4a and 4b (Scheme 2).
Scheme 1.

Retrosynthetic analysis of the triazole 4.
Scheme 2.

Synthesis of triazole containing coumarin derivatives.
The cellular activity manifested by 4a and 4b was evaluated against a panel of cancer lines (see supplemental material for details), including SKBr3 (estrogen receptor negative, Her2 over-expressing) lymphatic metastatic MDA-MB-468LN (estrogen receptor negative, Ah receptor-positive) and MCF-7 (estrogen receptor positive) breast cancer cell lines, head and neck squamous cell carcinoma (HNSCC) MDA1986 and JMAR cell lines, as well as prostate cancer cell lines PC3-MM2 and LNCaP. Both compounds manifested activity at low micromolar concentrations against all cell lines tested. Encouraged by these preliminary studies, substitutions on the phenyl and benzyl side chains were explored to determine structure-activity relationships for this series of compounds. Compounds 4c-q were synthesized efficiently in a manner similar to 4a and 4b, from the substituted triazolic acids 7c-q and aniline 6 (Scheme 2). It was determined that both 4-chloro (4c) and 3-chloro (4f) substitutions improved inhibitory activity against SKBr3 cell lines. Due to comparable activities manifested by both the phenyl and benzyl analogues, studies were directed towards modification of the benzyl ring system. Electron-donating groups such as 4-methyl (4i) and 4-methoxy (4j) as well as 4-chloro analogues (4g) manifested similar potencies compared to the unsubstituted analogue, 4b. However, the 4-NO2 substituent (4l) exhibited improved activity against all cancer cell lines tested, manifesting an IC50 of 0.38 and 0.64 μM against SKBr3 and MCF-7 cell lines, respectively. Sterically bulky groups such as 4-tert-butyl (4k) were found favorable, although their effects on the MCF-7 cell line were not clear. When a hydrophobic cyclohexylmethyl group was installed as the appendage (4q), improved inhibitory activities against SKBr3, MDA1986 and JMAR cells were observed, indicating the existence of a hydrophobic region in this area of the binding pocket. Although current data is limited and warrants further investigation, the observed inhibitory activities manifested by these triazole-containing analogues indicate that hydrogen bonding interactions on the side chain are favorable for Hsp90 C-terminal inhibition, which might result from either direct hydrogen bonding interactions or conformational rigidity on the side chain which might direct hydrophobic substituent into the hydrophobic pocket.
Increasing flexibility between the triazole side chain and the terminal phenyl ring was sought to determine whether improved inhibitory activity could be achieved. Therefore, compounds bearing two (4r) or three carbons (4s) were synthesized by an amide coupling reaction between the corresponding triazolylic acids (7r and 7s) and amine 6 (Scheme 2). The antiproliferative activities manifested by these compounds were then determined. A comparison of the data shown for 4a-b and 4r-s (Table 1) illustrates that compound 4r, which contains a two carbon linker, exhibited the most improved activity amongst the four analogues evaluated, exhibiting IC50s of 130 nM against SKBr3 cell line and about 110 nM against MDA1986 cell line.
Table 1.
Antiproliferative Activity of Coumarin-based Triazole Derivatives 4a
| R (4, IC50, μM) | SKBr3b | MCF-7b | MDA-MB-468LNc | MDA1986c | JMARc | PC3- MM2d | LNCaPd |
|---|---|---|---|---|---|---|---|
| Ph (4a) | 0.94±0.02a | 1.33±0.13a | 1.10±0.21 | 1.50±3.40 | 2.30±0.40 | 2.69 | 4.72±0.71 |
| Bn (4b) | 0.99±0.08 | 1.08±0.00 | 2.40 | 1.60±0.21 | 2.10±0.23 | 9.8±13.5 | 15.5±8.75 |
| 4-ClC6H5 (4c) | 0.58±0.03 | 2.04±0.25 | 0.81±0.15 | 0.18±0.11 | 0.43±0.38 | 2.79±0.49 | 2.45±0.47 |
| 4-BrC6H5 (4d) | 1.38±0.05 | 5.07±1.86 | 2.80±0.50 | 2.90±0.33 | 3.60±0.63 | 3.81±4.99 | 15.2±9.19 |
| 4-MeC6H5 (4e) | 1.08±0.05 | 1.38±0.07 | 4.00±1.92 | 0.29 | 3.64 | >100 | >100 |
| 3-ClC6H5 (4f) | 0.71±0.14 | 1.14±0.18 | 4.60 | 6.40 | 6.40 | 3.90±5.11 | 13.3±11.9 |
| 4-ClBn (4g) | 1.14±0.19 | 1.44±0.31 | 2.20±0.30 | 1.70±0.52 | 1.70±0.20 | 3.67±1.38 | 4.65±0.71 |
| 4-BrBn (4h) | 0.73±0.01 | 1.36±0.08 | 2.10±0.60 | 2.00±0.50 | 1.60±0.31 | 3.80±1.42 | 4.96±0.67 |
| 4-MeBn (4i) | 1.21±0.07 | 1.50±0.19 | 2.40±0.25 | 1.50±0.20 | 1.80±0.95 | 7.32±5.44 | 7.24±1.02 |
| 4-MeOBn (4j) | 1.31±0.20 | 1.42±0.14 | 3.40±1.27 | 2.60±0.30 | 6.70 | 7.85±0.42 | >100 |
| 4-t-BuBn (4k) | 0.45±0.03 | 1.22±0.07 | 0.74±0.22 | 0.34±0.09 | 0.58±0.14 | 2.37±0.00 | 1.84±0.00 |
| 4-NO2Bn (4l) | 0.38±0.13 | 0.64±0.01 | 0.81±0.23 | 0.18±0.22 | 0.26±0.34 | 2.49±0.32 | 1.28±0.71 |
| 4-FBn (4m) | 1.08±0.01 | 1.34±0.19 | 2.60±6.12 | 1.77±4.10 | 2.38±5.38 | 8.82±1.14 | 3.31±0.00 |
| 3-ClBn (4n) | 1.06±0.01 | 1.68±0.01 | 1.14±1.17 | 0.23±0.22 | 0.19±0.10 | 6.00±1.13 | 3.03±0.05 |
| 3-MeOBn (4o) | 1.15±0.19 | 1.61±0.06 | 2.69±1.33 | 1.34±0.31 | 1.49±0.31 | 7.20±0.15 | 3.49±0.30 |
| 2-ClBn (4p) | 1.30±0.07 | 0.99±0.08 | 2.10±0.52 | 3.20±1.37 | 3.70±0.85 | 6.66±5.44 | >100 |
| CH2C6H13 (4q) | 0.61±0.00 | 1.29±0.16 | 0.99±0.24 | 0.24±0.32 | 0.30±0.26 | 3.13±0.14 | 1.20±0.06 |
| Ph(CH2)2 (4r) | 0.13±0.01 | 0.55±0.01 | 0.99±0.10 | 0.11±0.30 | 0.74±0.86 | 1.40±0.04 | 2.36±0.53 |
| Ph(CH2)3 (4s) | 1.73±0.05 | 1.65±0.03 | NT | NT | NT | NT | NT |
Values represent mean ± standard deviation for at least two separate experiments performed in triplicate.
Cellular activities were determined with MTS/PMS cell viability assay.
Cellular activities were determined with Promega CellTiter-Glo (CTG) luminescent assay.
Cellular activities were determined with Sulforhodamine B Assay.
To confirm that the observed antiproliferative activities were resulting from Hsp90 inhibition, western blot analyses of Hsp90 client proteins in MCF-7 cell lysates were performed. Actin, whose maturation does not require the Hsp90 machinery, was used as a control. As shown in Figure 2, Hsp90 client proteins Her2, Akt and Raf-1 were degraded upon exposure of 4f, 4o or 4q at concentrations that mirror their anti-proliferative values, confirming that cell viability is directly linked to Hsp90 inhibition.
Figure 2.
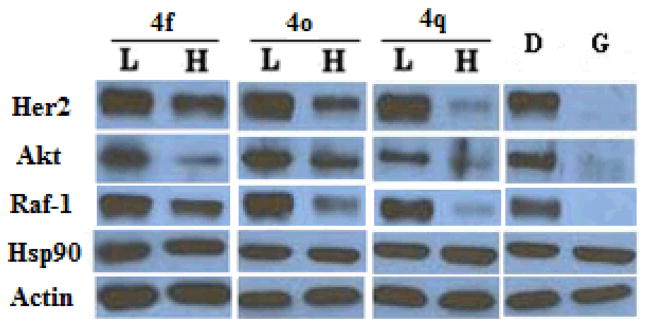
Western blot analyses of the Hsp90 client protein degradation in MCF-7 breast cancer cells lysis after treatment of triazole analogues. L represents a concentration 1/2 of the anti-proliferative IC50 value, while H represents a concentration 5 times the antiproliferative IC50 value. Geldanamycin (G, 500 nM) represents a positive control, while DMSO (D), vehicle, serves as the negative control.
Attention was then shifted to determine whether the anti-proliferative activities of these derivatives were similar to those of the recently identified biphenyl ring system. Biphenyl aniline 8 was synthesized according to a previously reported procedure.25 Similar to the coumarin derivatives, triazolylic acids 7a-b, 7g-q, 7r and 7t were converted to their acid chlorides upon treatment with oxalyl chloride, followed by coupling with aniline 8 to generate the biphenyl containing derivatives 5a-o (Scheme 3).
Scheme 3.
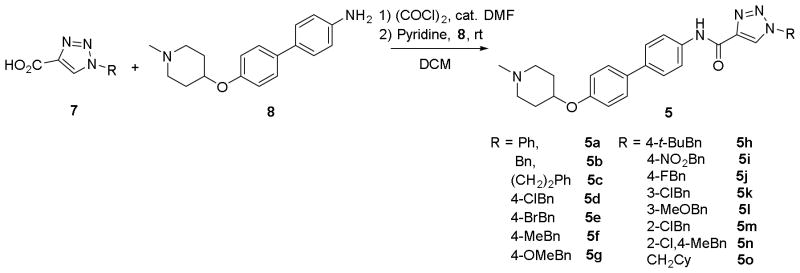
Synthesis of biphenyl triazole derivatives 5.
The antiproliferation activities manifested by these compounds are reported in Table 2. In contrast to the coumarin-based analogues 4a and 4b, which showed similar anti-proliferative activity, the phenyl and benzyl analogues 5a and 5b displayed very different activities, with the latter being about 2.5~8 times more potent against all the cell lines evaluated, manifesting IC50s of 170 and 500 nM against SKBr3 and MCF-7 cell lines, respectively. A compound that contains the two carbon linker (5c) was found to exhibit activity similar to those of the benzyl analogue, 5b. Subsequent studies were then aimed to modify the benzylic ring. As can be seen in Table 2, halogens at the 4-position were in general detrimental to inhibitory activity (5d-e); while electron-donating groups such as 4-methyl (5f) and 4-methoxy (5g) retained potencies against SKBr3 and MDA-MB-468LN cells, but manifested increased inhibitory activities against the MCF-7 cell line (120 and 270 nM, respectively) and prostate cancer cell lines. 3-Chloro (5k), 3-methoxy (5l), and 2-chloro (5m) produced decreased activity against SKBr3 cells, but increased activities against MCF-7 and MDA-MB-468LN cells. Combination of 2-chloro and 4-methyl substitution (5n) retained activity against SKBr3 cells, but did not improve activity against MCF-7 cells compared to the 4-methyl substituted analogue, 5f. However, this combination indeed improved activities against MDA-MB-468LN, MDA1986 and LNCaP cell lines. Of particular note, the 4-tert-butyl substituted analogue, 5h, manifested decreased antiproliferative activity against all cell lines tested compared to the 4-methyl analogue, 5f, which is in contrast to the coumarin derivatives 4i and 4k, indicating a smaller hydrophobic pocket exists when the biaryl scaffold is present. Similar to the coumarin scaffold, replacement of the benzyl group with a cyclohexylmethyl substituent resulted in compounds that manifested good inhibitory activities. Differences in the structure-activity relationships for the two scaffolds suggest the biphenyl ring system presents the side chain through different binding interactions. In general, installation of triazole side chain on the recently identified biphenyl side chain greatly improves antiproliferative activitiy against a wide spectrum of cell lines, resulting in more potent analogues.
Table 2.
Antiproliferative Activity of Biphenyl Triazole Analogues 5a
| R (5, IC50, μM) | SKBr3b | MCF-7b | MDA- MB-468LNc | MDA1986c | JMARc | PC3- MM2d | LNCaPd |
|---|---|---|---|---|---|---|---|
| Ph (5a) | 1.14±0.10a | 1.44±0.18a | 0.83±0.16 | 1.30±0.09 | 2.20±0.24 | 4.26±5.59 | 3.89±1.09 |
| Bn (5b) | 0.17±0.02 | 0.50±0.02 | 0.34±0.07 | 0.28±0.10 | 0.50±0.06 | 0.55±0.46 | 0.65±0.54 |
| CH2Bn (5c) | 0.19±0.02 | 0.38±0.10 | 0.36±0.04 | 0.10±0.23 | 0.58±0.46 | 0.83±0.34 | 0.43±0.22 |
| 4-ClBn (5d) | 0.32±0.10 | 0.44±0.04 | 0.74±0.19 | 1.30±0.37 | 2.30±0.82 | 0.66±0.21 | 1.59±0.78 |
| 4-BrBn (5e) | 0.49±0.03 | 0.56±0.04 | 2.80±0.61 | 3.10±0.87 | 7.80 | 19.9±27.9 | 0.54±0.12 |
| 4-MeBn (5f) | 0.17±0.03 | 0.12±0.01 | 0.41±0.26 | 0.36±0.05 | 0.42±0.07 | 0.36±0.04 | 0.20±0.04 |
| 4-MeOBn (5g) | 0.16±0.02 | 0.27±0.06 | 0.17 | 0.48±0.09 | 0.61±0.09 | 0.44±0.19 | 0.24±0.07 |
| 4-t-BuBn (5h) | 3.72±0.68 | 10.48±0.40 | 2.61±0.76 | 2.34±1.81 | 3.49±2.05 | >100 | 1.01±1.39 |
| 4-NO2 (5i) | 0.45±0.04 | 0.55±0.01 | 0.56±0.25 | 0.19±0.17 | 0.21±0.35 | 1.03±0.16 | 0.60±0.34 |
| 4-FBn (5j) | 0.72±0.18 | 1.04±0.08 | 1.09±0.23 | 0.93±0.34 | 1.39±0.52 | 2.81±1.82 | 1.64±0.97 |
| 3-ClBn (5k) | 0.39±0.01 | 0.34±0.02 | 0.17±0.02 | 0.17±0.12 | 0.27±0.04 | 0.35±0.14 | 0.12±0.10 |
| 3-MeOBn (5l) | 0.32±0.02 | 0.16±0.02 | 0.19±0.05 | 0.28±0.09 | 0.21±0.12 | 0.55±0.37 | 0.16 |
| 2-ClBn (5m) | 0.34±0.04 | 0.38±0.03 | 0.22±0.03 | 0.81±0.05 | 0.65 | 1.04±0.51 | 0.61±0.34 |
| 2-Cl,4-MeBn (5n) | 0.16±0.01 | 0.20±0.05 | 0.15±0.02 | 0.15±0.06 | 0.43±0.25 | 0.35±0.12 | 0.05±0.04 |
| CH2C6H13 (5o) | 0.20±0.02 | 0.31±0.04 | 0.24±0.06 | 0.38±0.14 | 0.26±0.45 | 5.12±6.54 | 0.48±0.68 |
Values represent mean ± standard deviation for at least two separate experiments performed in triplicate.
Cellular activities were determined with MTS/PMS cell viability assay.
Cellular activities were determined with Promega CellTiter-Glo (CTG) luminescent assay.
Cellular activities were determined with Sulforhodamine B Assay.
The cellular activity manifested by these biphenyl analogues was shown to result from Hsp90 inhibition by performing western blot analyses of MCF-7 cell lysates treated with such compounds. Hsp90-dependent client proteins were decreased upon exposure to of 5b, 5f and 5g at concentrations that mirror their antiproliferative IC50s. Clear degradation of Hsp90 client proteins Her2, Akt and Raf-1 was observed, while actin, which does not rely upon the Hsp90 chaperone machinery, remained constant, indicating that the antiproliferative activities manifested by these compounds resulted from Hsp90 inhibition (Figure 3A). These client proteins Her2, Akt, Raf-1 and CDK6 were also degraded in a concentration-dependent manner upon exposure to the most potent analogue 5f and the representative analogue 5b, against MCF-7 cells (Figure 3B and 3C), while actin levels remain unchanged. Examination of hsp90 expression when exposed to these compounds have shown that while GDA, the positive control, induced hsp90 upregulation due to induced heat shock response, hsp90 levels were constant or even decreased upon treatment of these compounds (Fig. 3), consistent with the observations manifested by many Hsp90 C-terminal inhibitors,20, 22, 24, 27, 32 indicating that these compounds exhibit hsp90 inhibitory activity through C-terminal inhibition. It is worthwhile to note that at a concentration as low as 1 μM, 5f was able to deplete hsp90 client proteins as well as Geldanamycin at 500 nM, demonstrating the remarkable activity exhibited by this compound for Hsp90 inhibition.
Figure 3.
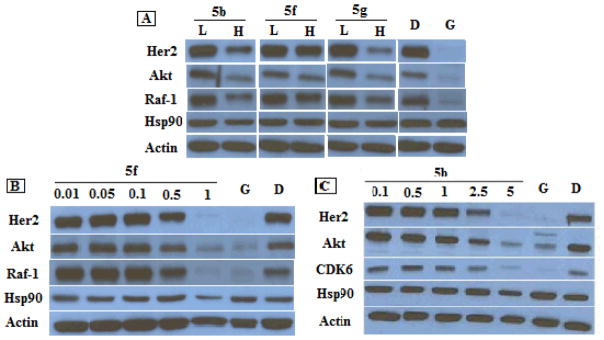
Western blot analyses of the Hsp90 client protein degradation in MCF-7 breast cancer cells lysis 24 h after treatment of biphenyl triazole analogues 5b, 5f and 5g. (A) L represents a concentration 1/2 of the anti-proliferative IC50 value, while H represents a concentration 5 times the antiproliferative IC50 value. (B) Concentrations (in μM) of 5f are indicated above each lane. (C) Concentrations of 5b (in μM) are indicated above each lane. Geldanamycin (G, 500 nM) and DMSO (D) were employed respectively as positive and negative controls.
Furthermore, no increase in heat shock proteins Hsp27, Hsp70, or Hsp90 was observed with increasing concentrations of 5f (Figure 4A). Depletion of client proteins without increased levels of heat shock proteins is a hallmark of Hsp90 C-terminal inhibition. To determine whether 5f binds the Hsp90 C-terminus, proteolytic fingerprinting of Hsp90 from TnT rabbit reticulocyte in the presence of 5f was performed. Novobiocin locks Hsp90 in the “open conformation” when bound to the C-terminus33, 34. In this conformation, amino acids Lys615 and Arg620 are not solvent exposed and are “protected” from cleavage by trypsin. This results in bands that differ in molecular weight from vehicle control. The C-terminal Hsp90 antibody AC88 detects the emergence of a 50 kDa band in the presence of novobiocin and other C-terminal inhibitors. A 50 kDa band was detected with 5 mM novobiocin and 1 mM 5f that is not detected for the vehicle control (Figure 4B). Together, these data further support that 5f binds and inhibits the Hsp90 C-terminus, which leads to client protein degradation without induction of the heat shock response.
Figure 4.

(A) Western blot analyses of the heat shock proteins Hsp27, Hsp70 and Hsp90 in MCF-7 breast cancer cells lysis 24 h after treatment with 5f. Concentrations (in μM) of 5f are indicated above each lane. Geldanamycin (G, 500 nM) and DMSO (D) are positive and negative controls. (B) Proteolysis of Hsp90 from TnT reticulocyte lysate incubated under conditions of protein synthesis with vehicle (1% DMSO) 5mM novobiocin and 1mM 5f. An antibody specific to the C-terminus of Hsp90 was used to identify the Hsp90 fragments produced in the presence of increasing amounts of trypsin.
A brief summary of observed structure-activity relationships for these derivatives is depicted in Figure 5. In support of the existence a hydrophobic pocket, incorporation of either an aromatic or saturated carbocycle was found favorable to inhibitory activity. Two carbons were found optimal for the coumarin derivatives, compared to one carbon for the biphenyl derivatives; and suggests that projection of the side chain is different for the biaryl scaffold. This observation is further supported by the tolerance of bulky groups exhibited at the para position of benzylic moiety (4k), but not for the biphenyl derivative, 5h. These observations highlight the significance of both hydrogen bonding and hydrophobic interactions at this location of the Hsp90 C-terminal binding pocket.
Figure 5.
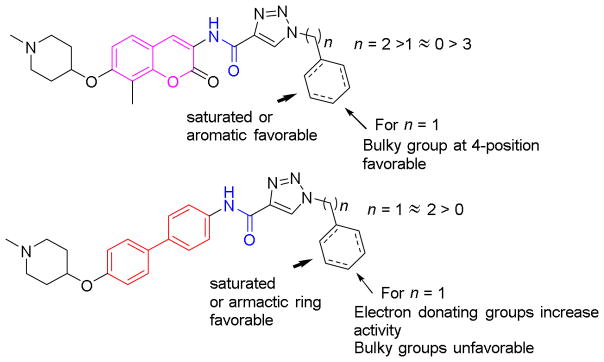
A summary of SARs for triazole containing coumarin and biphenyl derivatives.
Conclusion
In conclusion, a series of 1,2,3-triazole side chain containing analogues that contain the coumarin or biphenyl backbone were designed, synthesized and evaluated for antiproliferative activities against a panel of breast, HNSCC, and prostate cancer cell lines. Compounds manifesting nanomolar activity were identified through preliminary SAR studies and their Hsp90 inhibitory activity confirmed by Western blot analysis. The improved Hsp90 inhibitory activities exhibited by these compounds might result from increased hydrogen bonding properties on the amide side chain when connected to a hydrophobic appendage, or from the conformational rigidity due to the incorporation of 1,2,3-triazole which directs the hydrophobic appendage into the hydrophobic pocket. These observations are expected to aid in the development of more potent Hsp90 C-terminal inhibitors, and such studies will be reported in due course.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support of this project by the NIH/NCI (CA120458 and CA167079).
References
- 1.An X, Tiwari AK, Sun Y, Ding PR, Ashby CR, Jr, Chen ZS. Leuk Res. 2010;34:1255–1268. doi: 10.1016/j.leukres.2010.04.016. [DOI] [PubMed] [Google Scholar]
- 2.Henkes M, van der Kuip H, Aulitzky WE. Ther Clin Risk Manage. 2008;4:163–187. [PMC free article] [PubMed] [Google Scholar]
- 3.Roche-Lestienne C, Preudhomme C. Hematologie. 2007;13:43–53. [Google Scholar]
- 4.Lu X, Xiao L, Wang L, Ruden DM. Biochem Pharmacol. 2012;83:995–1004. doi: 10.1016/j.bcp.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jhaveri K, Modi S. In: Advances in Pharmacology. Keiran SMS, editor. Vol. 65. Academic Press; 2012. pp. 471–517. [DOI] [PubMed] [Google Scholar]
- 6.Workman P, Burrows F, Neckers L, Rosen N. Ann N Y Acad Sci. 2007;1113:202–216. doi: 10.1196/annals.1391.012. [DOI] [PubMed] [Google Scholar]
- 7.Blagg BSJ, Kerr TD. Med Res Rev. 2006;26:310–338. doi: 10.1002/med.20052. [DOI] [PubMed] [Google Scholar]
- 8.Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, Lindquist S. Cell. 2012;150:987–1001. doi: 10.1016/j.cell.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fierro-Monti I, Echeverria P, Racle J, Hernandez C, Picard D, Quadroni M. PLoS One. 2013;8:e80425. doi: 10.1371/journal.pone.0080425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Douglas Hanahan RAW. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 11.(a) Biamonte MA, Van de Water R, Arndt JW, Scannevin RH, Perret D, Lee WC. J Med Chem. 2010;53:3–17. doi: 10.1021/jm9004708. [DOI] [PubMed] [Google Scholar]; (b) Kim YS, Alarcon SV, Lee S, Lee MJ, Giaccone G, Neckers L, Trepel JB. Curr Top Med Chem. 2009;9:1479–1492. doi: 10.2174/156802609789895728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vaughan CK, Neckers L, Piper PW. Nat Struct Mol Biol. 2010;17:1400–1404. doi: 10.1038/nsmb1210-1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eskew JD, Sadikot T, Morales P, Duren A, Dunwiddie I, Swink M, Zhang X, Hembruff S, Donnelly A, Rajewski RA, Blagg BSJ, Manjarrez JR, Matts RL, Holzbeierlein JM, Vielhauer GA. BMC Cancer. 2011;11:468. doi: 10.1186/1471-2407-11-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McConnell JM, Alexander LD, McAlpine SR. Bioorg Med Chem Lett. 2014;24:661–666. doi: 10.1016/j.bmcl.2013.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ardi VC, Alexander LD, Johnson VA, McAlpine SR. ACS Chem Biol. 2011;6:1357–1367. doi: 10.1021/cb200203m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peterson LBB, Brian SJ. Future Med Chem. 2009;1:267–283. doi: 10.4155/fmc.09.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao H, Blagg BSJ. Inhibitors of Molecular Chaperones as Therapeutic Agents. 2014. pp. 259–301. RSC Drug Discovery Series. [Google Scholar]
- 18.Marcu MG, Schulte TW, Neckers L. J Nat Cancer Instit. 2000;92:242. doi: 10.1093/jnci/92.3.242. [DOI] [PubMed] [Google Scholar]
- 19.Yu XM, Shen G, Neckers L, Blake H, Holzbeierlein J, Cronk B, Blagg BSJ. J Am Chem Soc. 2005;127:12778–12779. doi: 10.1021/ja0535864. [DOI] [PubMed] [Google Scholar]
- 20.Burlison JA, Neckers L, Smith AB, Maxwell A, Blagg BSJ. J Am Chem Soc. 2006;128:15529–15536. doi: 10.1021/ja065793p. [DOI] [PubMed] [Google Scholar]
- 21.Burlison JA, Avila C, Vielhauer G, Lubbers DJ, Holzbeierlein J, Blagg BSJ. J Org Chem. 2008;73:2130–2137. doi: 10.1021/jo702191a. [DOI] [PubMed] [Google Scholar]
- 22.Zhao HP, Kusuma BR, Blagg BSJ. ACS Med Chem Lett. 2010;1:311–315. doi: 10.1021/ml100070r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao HP, Donnelly AC, Kusuma BR, Brandt GEL, Brown D, Rajewski RA, Vielhauer G, Holzbeierlein J, Cohen MS, Blagg BSJ. J Med Chem. 2011;54:3839–3853. doi: 10.1021/jm200148p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao HP, Blagg BSJ. Bioorg Med Chem Lett. 2013;23:552–557. doi: 10.1016/j.bmcl.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peterson LB, Blagg BSJ. Bioorg Med Chem Lett. 2010;20:3957–3960. doi: 10.1016/j.bmcl.2010.04.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Donnelly AC, Mays JR, Burlison JA, Nelson JT, Vielhauer G, Holzbeierlein J, Blagg BSJ. J Org Chem. 2008;73:8901–8920. doi: 10.1021/jo801312r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao HP, Moroni E, Colombo G, Blagg BSJ. ACS Med Chem Lett. 2014;5:84–88. doi: 10.1021/ml400404s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tron GC, Pirali T, Billington RA, Canonico PL, Sorba G, Genazzani AA. Med Res Rev. 2008;28:278–308. doi: 10.1002/med.20107. [DOI] [PubMed] [Google Scholar]
- 29.Kolb HC, Sharpless KB. Drug Discovery Today. 2003;8:1128–1137. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- 30.Thirumurugan P, Matosiuk D, Jozwiak K. Chemical reviews. 2013;113:4905–4979. doi: 10.1021/cr200409f. [DOI] [PubMed] [Google Scholar]
- 31.Kolarovic A, Schnurch M, Mihovilovic MD. J Org Chem. 2011;76:2613–2618. doi: 10.1021/jo1024927. [DOI] [PubMed] [Google Scholar]
- 32.Kusuma BR, Khandewal A, Gu W, Brown D, Liu W, Vielhauer G, Holzbeierlein J, Blagg BSJ. Bioorg Med Chem. 2014;22:1441–1449. doi: 10.1016/j.bmc.2013.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matts RL, Dixit A, Peterson LB, Sun L, Voruganti S, Kalyanaraman P, Hartson SD, Verkhivker GM, Blagg BSJ. ACS Chem Biol. 2011;6:800–807. doi: 10.1021/cb200052x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yun BG, Huang W, Leach N, Hartson SD, Matts RL. Biochemistry. 2004;43:8217–8229. doi: 10.1021/bi0497998. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.