

Abstract
Bone morphogenetic proteins (BMPs) are members of the TGF-β superfamily that are over-expressed in breast cancer, with context dependent effects on breast cancer pathogenesis. The type III TGF-β receptor (TβRIII) mediates BMP signaling. While TβRIII expression is lost during breast cancer progression, the role of TβRIII in regulating BMP signaling in normal mammary epithelium and breast cancer cells has not been examined. Restoring TβRIII expression in a 4T1 murine syngeneic model of breast cancer suppressed Smad1/5/8 phosphorylation and inhibited the expression of the BMP transcriptional targets, Id1 and Smad6, in vivo. Similarly, restoring TβRIII expression in human breast cancer cell lines or treatment with sTβRIII inhibited BMP-induced Smad1/5/8 phosphorylation and BMP-stimulated migration and invasion. In normal mammary epithelial cells, shRNA-mediated silencing of TβRIII, TβRIII over-expression, or treatment with sTβRIII inhibited BMP-mediated phosphorylation of Smad1/5/8 and BMP induced migration. Inhibition of TβRIII shedding through treatment with TAPI-2 or expression of a non-shedding TβRIII mutant rescued TβRIII mediated inhibition of BMP induced Smad1/5/8 phosphorylation and BMP induced migration and/or invasion in both in normal mammary epithelial cells and breast cancer cells. Conversely, expression of a TβRIII mutant, which exhibited increased shedding, significantly reduced BMP-mediated Smad1/5/8 phosphorylation, migration, and invasion. These data demonstrate that TβRIII regulates BMP-mediated signaling and biological effects, primarily through the ligand sequestration effects of sTβRIII in normal and cancerous mammary epithelial cells and suggest that the ratio of membrane bound versus sTβRIII plays an important role in mediating these effects.
Abbreviations: BMP, bone morphogenetic protein; TβRIII, type III TGF-β receptor; sTβRIII, soluble type III TGF-β receptor CM, conditioned media; TAPI-2, N-(R)-[2-(Hydroxyaminocarbonyl)methyl]-4-methylpentanoyl-L-t-butyl-alanyl-L-alanine 2-aminoethyl Amide, TNF-α Protease Inhibitor-2; EV, empty vector; ∆shed-TβRIII, non-shedding TβRIII mutant; SS-TβRIII, super shedding TβRIII mutant
Introduction
Bone morphogenetic proteins (BMP), with 20 members, are the largest subfamily of the TGF-β superfamily [1]. BMPs regulate development, bone formation and remodeling, proliferation, survival, migration, and differentiation. BMP signaling occurs upon binding of BMP to the type I BMP receptor (ALK1, ALK2, ALK3, or ALK6), which then complexes with and is phosphorylated and activated by one of the type II BMP receptors (BMPRII, ActRII, or ActRIIB). The activated type I BMP receptor then phosphorylates the receptor Smad proteins (Smad1/5/8), which complex with Smad 4, translocate into the nucleus, and induce transcription of BMP target genes.
Although best characterized for their role in development and bone morphogenesis, BMPs have important roles in normal mammary gland development [2], [3], [4], [5], and have been reported to have both tumor suppressor and tumor promoting functions in breast cancer. In human breast cancer, BMP2, 4, 5, 6, and 7 are expressed, albeit with variable levels [6]. In support of a tumor suppressor role, BMP7 has been shown to reduce both primary tumor growth and bone metastases in a mouse xenograft model of breast cancer [7] and the inhibition of BMP signaling through the expression of a dominant negative BMPR2 in a MMTV-Polyoma middle T mouse model of mammary cancer increased tumor cell proliferation, lung metastasis, angiogenesis, and induced an altered reactive tumor stroma [8]. Conversely, in support of a tumor promoting role, primary human breast cancers, lymph node metastases, and bone metastases exhibit signs of elevated BMP signaling [9], high BMPR-IB expression in ER-positive human breast tumors correlates with decreased survival [10], treatment of human breast cancer cell lines with BMP promotes cell migration and invasion [6], [9], [11], [12], [13], [14], BMP7 expression in cell lines and primary breast tumors has been associated with accelerated bone metastasis in vivo[7], [15], [16], and expression of dominant negative BMPRIA in a murine breast cancer xenograft model prevents bone metastasis and invasion and enhances survival [9].
The type III TGF-β receptor (TβRIII or betaglycan) binds to multiple BMP family members, including BMP2, 4, 7 and GDF-5, and functions as a BMP co-receptor, enhancing ligand binding to the BMP type I receptors, ALK3 and ALK6 [17], [18]. Upon complexing with BMP type I receptors, TβRIII co-localizes and stabilizes ALK3 expression at the cell surface [19], while mediating the internalization of ALK6 to stimulate ALK6 signaling in a β-arrestin2 dependent manner [19]. TβRIII is also able to inhibit both activin and BMP signaling by promoting the binding of inhibin to its cognate receptors, the activin type II and BMP type II receptors, as inhibin opposes the action of activin and BMP [20], [21]. In addition, ectodomain shedding of TβRIII produces soluble TβRIII (sTβRIII), which inhibits TGF-β signaling via ligand sequestration. However, little is known about the regulation of TβRIII shedding, and the cleavage site has not been identified [22], [23]. While sTβRIII has been demonstrated to bind BMP, the specific role of sTβRIII in regulating BMP signaling remains to be defined [18], [24], [25].
Loss of TβRIII expression occurs early in the development of human breast cancer, beginning during ductal carcinoma in situ[26]. The restoration of TβRIII expression in breast cancer, inhibits tumor progression in vivo in part through sTβRIII production, which binds to and sequesters TGF-β, antagonizing the tumor promoting effects of TGF-β signaling in late stage tumors [26], [27], [28], [29], [30], [31]. Specifically, TβRIII expression inhibits migration, invasion, angiogenesis, and metastasis in a murine syngeneic model of breast cancer [26]. Several studies have demonstrated that treatment with sTβRIII alone inhibits breast cancer tumor growth, angiogenesis, and reduces metastasis in xenograft models of breast cancer [27], [28], [32]. In addition, expression of TβRIII inhibits BMP-mediated invasion and Smad phosphorylation in pancreatic cancer [33]. As TβRIII binds to and mediates BMP signaling, which has been shown to have context dependent roles in breast cancer progression, here we investigated the role of TβRIII and sTβRIII in regulating BMP signaling and BMP-mediated biology in mammary epithelial cells and breast cancer cells, demonstrating that the ratio of membrane bound versus sTβRIII plays an important role in mediating BMP signaling and biological effects in mammary epithelial cells and breast cancer cells.
Material and Methods
Cell Lines
All cell lines were originally obtained from the American Type Culture Collection (Manassas, VA). Human breast cancer cell lines MDA-MB-231 and MCF-7 were cultured in MEM + 10% FBS, sodium pyruvate, and non-essential amino acids with the addition of insulin (10 μg/ml) for the MCF-7 cells. The mouse 4T1 breast cancer cell line was cultured in DMEM + 10% FBS. The human normal mammary epithelial cell lines, MCF10A and HMECs were cultured in F12/DMEM (1:1) + 5% horse serum, 10 μg/ml insulin, 0.5 μg/ml hydrocortisol, 20 ng/ml EGF, 100 ng/ml cholera toxin and DMEM + 10% FBS, 10 μg/ml insulin, respectively.
MDA-MB-231, MCF-7, and 4T1 stable cell lines, representing a pool of stable clones, were derived as previously described and maintained in 250 μg/ml G418 [26], [30].
Viral Production and Infection
For lentivirus production, 293FT cells were transfected with Lipofectamine 2000 (Invitrogen,Grand Island, NY) at a ratio of 3:1 to DNA, either EV (empty vector), TβRIII, ∆Shed (non-shed), and SS (super-shed) (pSMPUW-Neo expression vector) (Cell Biolabs, San Diego, CA) and 3 third generation lentiviral packaging plasmids (AddGene, Cambridge, MA) in Opti-MEM (Gibco) and media was changed 6 hours post transfection. Forty-eight hours post infection, media was collected, spun down to remove cell debris, and filtered through a 0.45 μM pore membrane. Viral media was aliquoted and stored at − 80°C until use. For lenti-viral infections, viral media was added to cells in complete growth media at a ratio of either 1:10 or 1:100 in the presence of polybrene (6 μg/ml). To create stable lentiviral-expressing cell lines, 48 hours post-infection media was changed and complete growth media containing 2 mg/ml G418 (KSE Scientific, Durham, NC) was added as a selection agent. Post selection, serial dilutions were used to create monoclonal cell lines. Following selection, stable lentiviral cell lines were maintained in complete growth media containing 500 μg/ml G418. Adenoviral infections were performed as previously described [34]. All adenoviral infections were performed at a multiplicity of infection of 50 for all constructs. Cells were treated with 25 μM TAPI-2 (N-(R)-[2-(Hydroxyaminocarbonyl)methyl]-4-methylpentanoyl-L-t-butyl-alanyl-L-alanine, 2-aminoethyl Amide, TNF-α Protease Inhibitor-2) (EMD Biosciences, San Diego, CA) in fresh media overnight prior to harvest. Conditioned media (CM): 2 × 10^5 cells were plated in a 6-well dish and allowed to recover overnight. The next day cells were incubated in 1ml fresh complete media overnight and conditioned media was collected, cell debris removed by centrifugation, and used immediately for signaling experiments or stored at − 80°C until use in ELISA assays.
Western Blotting
2 × 105 cells were plated in six well dishes and allowed to recover. Cells were serum starved overnight and then treated with varying doses of BMP2 or 4 for the indicated times. The cells were lysed in boiling sample buffer and resolved by SDS-PAGE and immunoblotted for the proteins of interest. 4T1-Neo and TβRIII tumor extracts were prepared by homogenization in RIPA buffer plus a protease inhibitor cocktail (Roche, Indianapolis, IN) and cleared by centrifugation. Protein concentrations were determined using a BCA protein quantification assay (Pierce, Rockford, IL). Primary antibodies (p-Smad1/5/8 (#9511), Smad1 (#9743), pSmad2 (#3101), Smad2 (#5339)) were purchased from Cell Signaling Technology (Danvers, MA) and a 1:2000 dilution was used for immunoblotting. Primary TβRIII antibody (#AF-242-PB) was purchased from R&D systems (Minneapolis, MN USA) and a 1:2000 dilution was used for immunoblotting. Cells were treated with 60 to 80 ng/ml sTβRIII recombinant soluble TβRIII (R&D Systems, Minneapolis, MN).
QRT-PCR Analysis
RNA was extracted from 4T1-Neo and TβRIII mammary tumors using the RNeasy Lipid Tissue Mini Kit per the manufacturer’s instructions (Qiagen, Valencia, CA). 1 μg of RNA was reverse transcribed using the iScript cDNA Synthesis Kit (BioRad Hercules, CA). Each PCR reaction contained 1 μl of cDNA plus SYBRGreen Mix (BioRad, Hercules, CA) along with Id1 primers: Id1 F 5′-GCACTGATCTGCCGTTCAGG-3′ and Id1 R 5′-TGGACGAGCAGCAGGTGAACG-3′, Smad 6 F 5′-CCACTGGAT CTGTCC-GATTC-3′ and Smad6 R 5′-AAGTCGAACACCTTGATGGAG-3′, or GAPDH F 5′-GTCTACA-TGTTCCA-3′ and GAPDH R-5′ AGTGAGTTGTCATATTTCTGTGGT-3′. Id-1 and Smad6 expression was normalized to GAPDH levels. Student’s t-test was used to quantitatively assess statistical significance.
Binding and Crosslinking
TGF-β binding and cross linking experiments were performed as previously described [26], [34]. Briefly, 2.5 × 10^4 cells were plated in 6-well dishes and allowed to recover overnight. The next day, media was removed and replaced with 1 ml complete growth media. Media was conditioned for 24 hours before being removed and centrifuged to remove cell debris prior to binding and crosslinking. Both cells and conditioned media were incubated with 125I-TGF-β1 (Perkin Elmer, Waltham, MA), at 100 pM and 25 pM, respectively, in the presence of BSA and protease inhibitors for 3 hours at 4°C. After incubation, ligand was chemically crosslinked using 0.5 mg/ml disuccinimidyl suberate and quenched with 1M glycine. Cells were lysed with RIPA buffer supplemented with protease inhibitors, and ligand-receptor complexes were immunoprecipitated with a polyclonal antibody against the extracellular domain of TβRIII (R&D Systems, Minneapolis, MN). The resulting complexes were separated via SDS-PAGE. Images were acquired with phosphorimaging and were analyzed using ImageJ software (National Institutes of Health (NIH)).
Migration/Invasion Transwell Assays
To assess invasion or migration, 7.5 × 104 cells were seeded in serum free media in the upper chamber of a Matrigel invasion transwell (BD Biosciences, San Jose CA) or a fibronectin coated transwell filter, coated both at the top and bottom with 30 μg/mL fibronectin (Calbiochem, La Jolla, CA) for HMECs or 50 μg/ml for MDA-MB-231 cells. Cells were untreated, treated with 20 nM BMP2, 20 nM BMP4 (R&D Systems, Minneapolis, MN) and/or 25 μM TAPI-2, as well as 60 to 80 ng/ml soluble recombinant TβRIII (R&D Systems, Minneapolis, MN) where indicated and were allowed to migrate for 24 h at 37°C toward the lower chamber containing media plus 10% FBS. Cells on the upper surface of the filter were removed and the cells that migrated to the underside of the filter were fixed and stained using the 3 Step Stain Set (Richard-Allan Scientific, Kalamazoo, MI). Each assay was performed in duplicate, and each experiment was conducted at least 3 times with 3 random fields from a 20 × magnification analyzed for each membrane. Data analysis was performed using NIH ImageJ software (http://rsb.info.nih.gov/ij/). Student’s t test was used to quantitatively assess statistical significance.
sRIII ELISA
Conditioned media (CM): 2 × 10^5 cells were plated in a 6 well dish and allowed to recover overnight. The next day cells were incubated in 1ml fresh complete media with FBS overnight and conditioned media was collected, cell debris removed by centrifugation stored at − 80°C until use in ELISA assays. Capture antibody (R&D Systems, #AF-242-PB, Minneapolis, MN) was immobilized onto an E1A/R1A plate (#3590 Corning, Union City, California) overnight. After washing, 100 μl conditioned media was loaded onto the plate and incubated at room temperature for 2 hours. Then detection antibody (# BAF-242, R&D Systems, Minneapolis, MN) was applied and incubated for 2 h, Strepavidin-HRP (# DY998, R&D Systems, Minneapolis, MN) added and incubated for 30 minutes. Finally Fast OPD substrate (# P9187, Sigma Aldrich, St. Louis, MO) was added, 3M HCl was applied to stop the reaction 30 minutes later, and optical absorbance at 490 nm was recorded immediately.
Results
TβRIII inhibits BMP-mediated signaling in breast cancer cells
As TβRIII mediates BMP signaling and regulates breast cancer progression, [18], [26], [35], [36], we investigated the role of TβRIII in regulating BMP signaling in breast cancer. In several human and murine models of breast cancer, including the human breast cancer cell lines, MDA-MB-231 and MCF-7, and the mouse breast cancer cell line, 4T1, all of which express low levels of TβRIII, BMP2 or BMP4 stimulated time and dose dependent increases in Smad1/5/8 phosphorylation (Figure 1A–C; Suppl. Figure S1A). This BMP-mediated Smad1/5/8 phosphorylation was ALK3/ALK6 dependent, as treatment with the ALK3/6 inhibitor, dorsomorphin, potently suppressed BMP's effects (Suppl. Figure S1C). Compared to Neo control cells, stably expressing TβRIII in MDA-MB-231, MCF-7 and 4T1 cells (Suppl. Figure S1E) [26], [35], [37], [38], decreased BMP2 and BMP4 induced Smad1/5/8 phosphorylation (Figure 1A–C; Suppl. Figure S1A). In addition, as BMPs have been recently shown to induce phosphorylation of Smad2/3 preferentially in cancer cells, including these breast cancer cells [39], we demonstrated that expression of TβRIII in MDA-MB-231 cells also inhibited BMP2 induced Smad2 phosphorylation (Suppl. Figure S1B).
Figure 1.
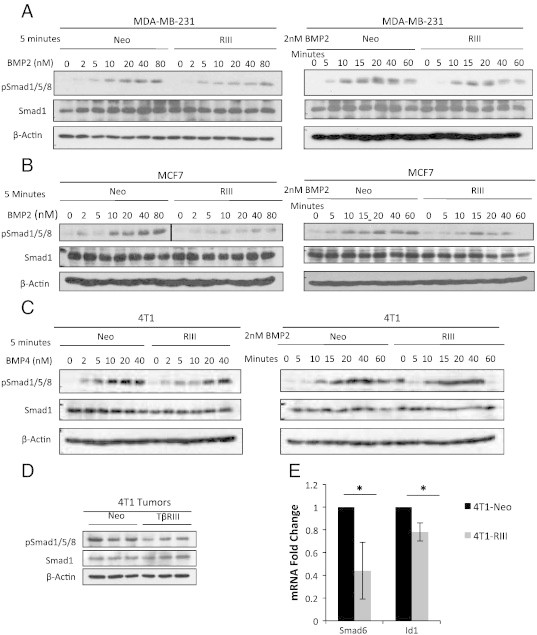
TβRIII inhibits BMP-mediated signaling in breast cancer cells and a 4T1 orthotopic breast cancer model. (A) Western blot analysis of pSmad1/5/8 signaling in MDA-MB-231-Neo and TβRIII stable cells serum starved overnight and then treated with BMP2 at the indicated dosages and times. (B) Western blot analysis of pSmad1/5/8 signaling in MCF7-Neo and TβRIII stable cells serum starved overnight and then treated with BMP2 at the indicated dosages and times. (C) Western blot analysis of pSmad1/5/8 signaling in 4T1-Neo and TβRIII stable cells serum starved overnight and then treated with BMP4 at the indicated dosages and times. (D) Western blot analysis of pSmad1/5/8 signaling in primary tumors (n = 3, Neo and TβRIII) from a 4T1-Neo and TβRIII orthotopic xenograft model of breast cancer [26]. (E) QRT-PCR analysis of BMP transcriptional targets Id1 (n = 5) and Smad6 (n = 4) in 4T1-Neo and TβRIII primary breast tumors [26]. The fold change in mRNA is shown normalized to GAPDH control. Total Smad1 and β-Actin levels are shown as loading controls for westerns. All experiments were independently performed at least 3 times and representative data are shown. Id1 P = .03; t-test. Smad6 P = .05; t test.
TβRIII inhibits BMP-mediated signaling in an in vivo model of breast cancer
To examine the role of TβRIII in regulating BMP signaling in an in vivo breast cancer context, we examined whether expression of TβRIII in a 4T1 murine syngeneic model of breast cancer alters BMP signaling. Expression of TβRIII in this system has been previously demonstrated to suppress tumor progression through inhibition of migration, invasion, angiogenesis, and metastasis, due in part to sTβRIII-mediated inhibition of TGF-β/pSmad2 signaling [26], [35]. Primary 4T1-Neo and 4T1-TβRIII breast tumors [26] were examined for levels of Smad1/5/8 phosphorylation and alterations in the downstream BMP transcriptional targets, Idl and Smad6. Western blot analysis of pSmad1/5/8 protein levels in primary tumors demonstrated that, in comparison to Neo tumors, 4T1-TβRIII tumors exhibited a significant decrease in Smad1/5/8 phosphorylation (Figure 1D). In addition, relative to 4T1-Neo tumors, 4T1-TβRIII primary tumors had a significant decrease in mRNA levels of two BMP transcriptional targets, Id1 and Smad6, supporting a role for TβRIII in suppressing BMP-mediated transcriptional events in vivo (Figure 1E). These data demonstrate that expression of TβRIII in an in vivo model of breast cancer suppresses BMP signaling at both the level of Smad phosphorylation and downstream transcriptional regulation.
TβRIII mediates BMP signaling in normal mammary epithelial cell lines
As TβRIII regulated BMP signaling in breast cancer cells, we investigated the role of TβRIII in regulating BMP signaling in the cellular origin of breast cancers, normal mammary epithelial cells. The normal human mammary epithelial cell lines, MCF10A and HMEC, both express TβRIII and are responsive to BMP4 treatment, as demonstrated by BMP4-mediated dose and time dependent Smad1/5/8 phosphorylation (Figure 2A). This BMP-mediated Smad1/5/8 phosphorylation was ALK3/ALK6 dependent, as treatment with dorsomorphin potently suppressed BMP’s effects in HMEC and MCF10A cells (Suppl. Figure S1D). As normal mammary epithelial cells expressed TβRIII, we utilized shRNA-mediated knockdown of TβRIII expression to assess the role of TβRIII in BMP signaling. In both HMEC and MCF10A cells, shRNA-mediated knockdown of TβRIII expression attenuated BMP-mediated Smad1/5/8 phosphorylation, demonstrating that TβRIII has an important role in mediating BMP signaling in normal mammary epithelial cells (Figure 2B and D).
Figure 2.
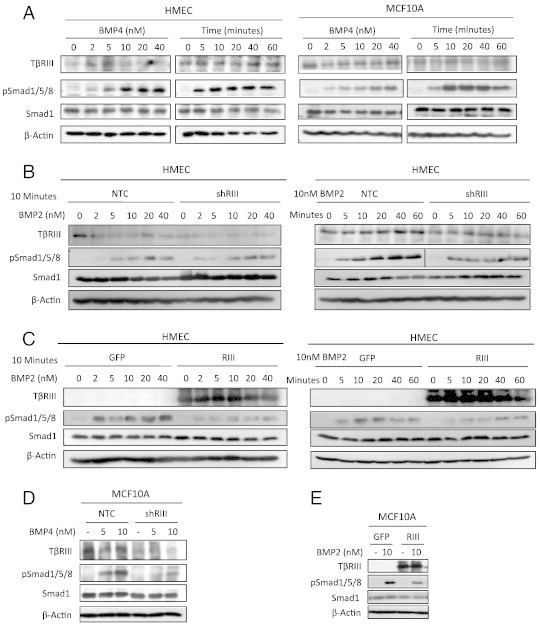
TβRIII inhibits BMP signaling in normal mammary epithelial cells. (A) Western blot analysis of pSmad1/5/8 signaling in HMECs and MCF10A cells serum starved overnight and treated with BMP4 at the indicated dosages and times (5 nM BMP4). (B) Western blot analysis of pSmad1/5/8 signaling in NTC (non-targeting control) or shRNA-TβRIII adenovirally infected HMECs. Cells were serum starved overnight and treated with BMP2 at the indicated dosages (10 minute treatment) and times (5nM BMP2). (C) Western blot analysis of pSmad1/5/8 signaling in GFP or TβRIII-GFP adenovirally infected HMECs. Cells were serum starved overnight and then treated with BMP2 at the indicated dosages and times. (D) Western blot analysis of pSmad1/5/8 signaling in NTC (non-targeting control) or shRNA TβRIII adenovirally infected MCF10A cells serum starved overnight and treated with BMP4 at the indicated dosages. (E) Western blot analysis of pSmad1/5/8 signaling in GFP or TβRIII-GFP adenovirally infected MCF10A cells serum starved overnight and treated with 10 nM BMP2. Total Smad1 and β-actin levels are shown as loading controls for westerns. All experiments were independently performed at least 3 times and representative data are shown.
As increasing TβRIII expression in breast cancer cell lines had a similar effect to decreasing TβRIII expression in normal mammary epithelial cells, we investigated the effects of increasing TβRIII expression in normal mammary epithelial cells. Surprisingly, increasing expression of TβRIII also inhibited BMP-mediated Smad1/5/8 phosphorylation in both the HMECs and the MCF10A cell line (Figure 2C and E). To investigate whether this inhibition could be attributed to increased soluble TβRIII (sTβRIII) production, we assessed both cell surface TβRIII and sTβRIII expression while increasing TβRIII expression. Consistent with previous studies, binding and crosslinking demonstrated that over-expression of TβRIII in HMECs increased expression of both membrane bound TβRIII and sTβRIII (Suppl. Figure S2A), with a preference for increased sTβRIII relative to membrane bound TβRIII. sTβRIII ELISA assays also demonstrated increased levels of sTβRIII in conditioned media (CM) from HMECs which over-express TβRIII and a reciprocal decrease in levels of sTβRIII in media from cells in which endogenous TβRIII expression has been silenced by shRNA (Suppl. Figure S2B).
sTβRIII inhibits BMP signaling in mammary epithelial and breast cancer cells
sTβRIII is able to bind BMP2 [18] and has been shown to inhibit TGF-β signaling in breast cancer cells, suggesting that TβRIII may inhibit BMP signaling in the context of mammary epithelial cells through the production of sTβRIII [26], [35]. Consistent with this hypothesis, treatment of HMECs and MCF10A cells with either recombinant sTβRIII or CM from MDA-MB-231-TβRIII cells inhibited BMP-mediated induction of pSmad1/5/8 (Figure 3A, B, E). In addition, treatment of MDA-MB-231-Neo cells with recombinant sTβRIII or with CM from MDA-MB-231-TβRIII cells (Figure 3C and D) inhibited BMP2 and BMP4 mediated Smad1/5/8 phosphorylation. The sTβRIII mediated suppression of BMP2 induced pSmad1/5/8 in HMECs and MDA-MB-231 cells occurred in a dose dependent manner (Figure 3E and F). Further, inhibition of TβRIII shedding by treatment with TAPI-2, an MMP and TACE inhibitor [40], (Suppl. Figure S3A, B), rescued TβRIII mediated suppression of BMP2 induced Smad1/5/8 phosphorylation in HMECs and in MDA-MB-231 cells (Figure 3G and H). These data demonstrate that TβRIII inhibits BMP signaling in both normal and cancerous mammary epithelial cell lines, at least in part via sTβRIII-mediated BMP sequestration.
Figure 3.
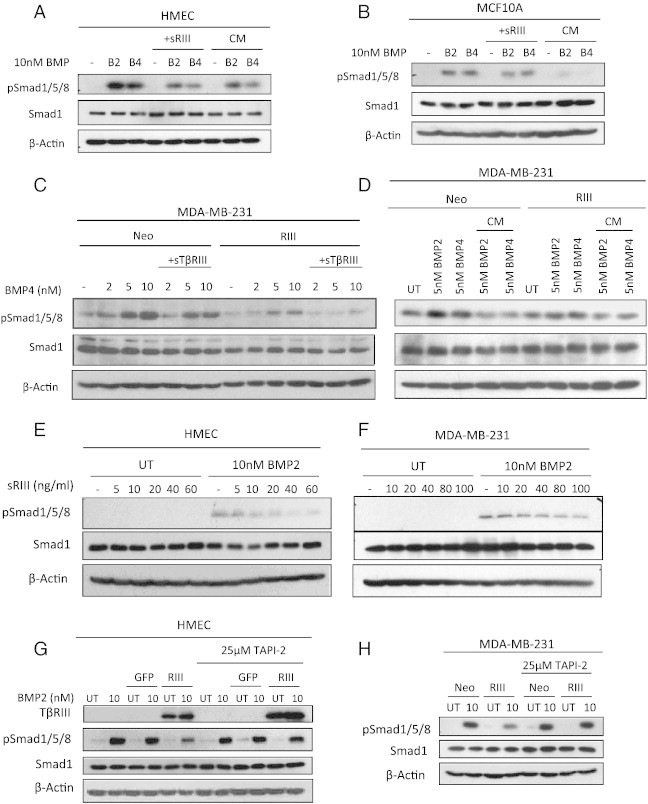
sTβRIII inhibits BMP signaling in mammary epithelial cells. (A and B) Western blot analysis of pSmad1/5/8 signaling in HMEC and MCF10A cells serum starved overnight and pretreated with sTβRIII (60 ng/ml) or conditioned (CM) from MDA-MB-231- TβRIII cells prior to BMP2 (B2) and 4 (B4) treatment. (C) Western blot analysis of pSmad1/5/8 signaling in MDA-MB-231-Neo and TβRIII stable cells serum starved and pre-treated with recombinant sTβRIII (60 ng/ml) overnight and subsequently treated with BMP4 at the indicated dosages. (D) Western blot analysis of pSmad1/5/8 signaling in MDA-MB-231-Neo and TβRIII stable cells serum starved and treated with CM from MDA-MB-231-TβRIII cells and subsequently treated with BMP2 and 4 at the indicated dosages. (E) Western blot analysis of pSmad1/5/8 signaling in HMECs serum starved overnight and treated with pre-treated with recombinant sTβRIII at the indicated dosages. Cells were treated with 10nM BMP2 for 10 minutes. (F) Western blot analysis of pSmad1/5/8 signaling in MDA-MB-231 cells serum starved overnight and treated with pre-treated with recombinant sTβRIII at the indicated dosages. Cells were treated with 10nM BMP2 for 10 minutes. (G) Western blot analysis of pSmad1/5/8 in HMECs adenovirally infected with GFP or TβRIII-GFP serum starved overnight and pretreated with 25 μM TAPI-2 prior to treatment with 10 nM BMP2 for 10 minutes. (H) Western blot analysis of pSmad1/5/8 signaling in MDA-MB-231-Neo and TβRIII cells serum starved and pretreated overnight with 25 μM TAPI-2 prior to treatment with 10nM BMP2 for 10 minutes. Total Smad1 and β-actin levels are shown as loading controls for westerns. All experiments were independently performed at least 3 times and representative data are shown.
TβRIII mediates BMP induced migration and invasion in normal and cancerous mammary epithelial cells
TβRIII regulates cell motility and invasion in a variety of cell types [26], [33], [34], [41], [42] and BMP induces migration and invasion in breast cancer cell lines as well as regulating metastasis in vivo [9], [13]. Accordingly, we investigated the role of TβRIII in BMP induced migration through fibronectin coated transwells and invasion through Matrigel coated transwells. HMECs demonstrated a modest yet significant increase in migration in response to BMP2, which was abrogated by treatment with recombinant sTβRIII, increasing TβRIII expression or by shRNA-mediated silencing of TβRIII expression (Figure 4A, Suppl. Figure S2 C), corresponding to the effects of sTβRIII, and altered TβRIII expression on BMP signaling in HMECs (Figure 2, Figure 3). Further, treatment of HMECs with TAPI-2 to inhibit TβRIII shedding significantly enhanced basal migration of GFP and TβRIII-HMECs and blocked the ability of increased TβRIII expression to inhibit BMP-mediated migration (Figure 4B, Suppl. Figure S3C). These studies demonstrate that TβRIII regulates BMP-mediated migration in normal mammary epithelial cells, suggesting that the balance between membrane bound and sTβRIII is important in regulating BMP signaling and migration in this context.
Figure 4.
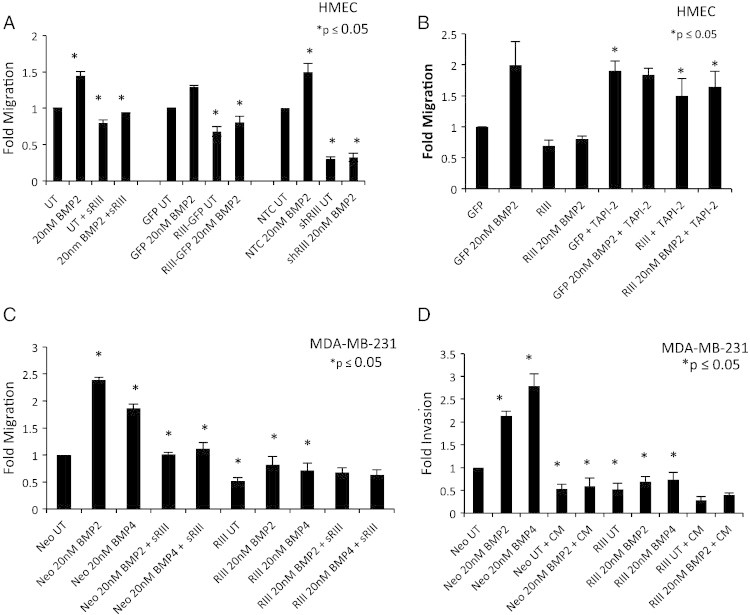
TβRIII mediates BMP induced migration and invasion in mammary epithelial cells. (A) GFP, TβRIII-GFP, NTC (non-targeting control), or shTβRIII adenovirally infected HMECs were plated in a fibronectin transwell migration assay in serum free conditions on a fibronectin coated transwell with and without BMP2 and/or sTβRIII (60 ng/ml) treatment for 24 hours. Data was normalized to GFP UT or NTC UT respectively and fold change ± SEM is shown. *P ≤ .05; t test (B) GFP or TβRIII-GFP adenovirally infected HMECs were plated in a fibronectin transwell migration assay in serum free conditions on a fibronectin coated transwell with and without BMP2 and/or 25 μM TAPI-2 treatment for 24 hours. Data was normalized to GFP UT and fold change ± SEM is shown. *P ≤ .05; t test. (C) MDA-MB-231-Neo and TβRIII cells were plated in a fibronectin transwell migration assay in serum free conditions on a fibronectin coated transwell with and without BMP2, BMP4, and/or sTβRIII (60 ng/ml) treatment for 24 hours. Data was normalized to Neo UT and fold change ± SEM is shown. *P ≤ 0.05; t test. (D) MDA-MB-231-Neo and TβRIII cells were plated in a matrigel transwell invasion assay in serum free conditions on a matrigel coated transwell with and without BMP2, BMP4, treatment and/or CM (conditioned media) from MDA-MB-231-TβRIII cells for 24 hours. Data was normalized to Neo UT and fold change ± SEM is shown. *P ≤ .05; t test. All experiments were independently performed at least 3 times and representative data are shown.
MDA-MB-231-Neo cells were also responsive to BMP2 and BMP4 treatment, with BMP2 and BMP4 both enhancing migration and invasion (Figure 4C and D; Suppl. Figure S4). Similar to HMECs, stable over-expression of TβRIII in MDA-MB-231 cells inhibited BMP2 and BMP4 induced migration and invasion (Figure 4C and D; Suppl. Figure S4). Further, treatment of MDA-MB-231-Neo cells with recombinant sTβRIII or CM from MDA-MB-231-TβRIII cells significantly inhibited BMP-induced migration and invasion (Figure 4C and D; Suppl. Figure S4A, B), suggesting that the increased levels of sTβRIII in the MDA-MB-231-TβRIII cells contributes to the reduction in BMP-mediated migration and invasion. Taken together, these data demonstrate that TβRIII can regulate BMP-mediated migration and invasion in both normal and cancerous mammary epithelial cells.
TβRIII shedding mutants demonstrate altered BMP mediated signaling, migration and invasion in mammary epithelial cells
To further address the effect of sTβRIII on BMP signaling in mammary epithelial cells, we utilized two TβRIII mutants that exhibit alterations in TβRIII shedding, ∆shed-TβRIII, which reduces TβRIII shedding by ~ 90%, and SS-TβRIII, which increases TβRIII shedding by ~ 3- to 4-fold (J.L.E., C.E.G., submitted for publication). Expression of ∆shed-TβRIII failed to inhibit BMP induced Smad1/5/8 phosphorylation in HMECs (Figure 5A). Conversely, expression of SS-TβRIII reduced BMP-mediated Smad1/5/8 phosphorylation (Figure 5A). In addition, while expression of wild type TβRIII significantly inhibited BMP induced migration in HMECs, expression of ∆shed-TβRIII did not inhibit BMP-induced migration in HMECs (Figure 5B, Suppl. Figure S3D). Conversely, expression of SS-TβRIII significantly reduced BMP-induced migration (Figure 5B, Suppl. Figure S3D).
Figure 5.
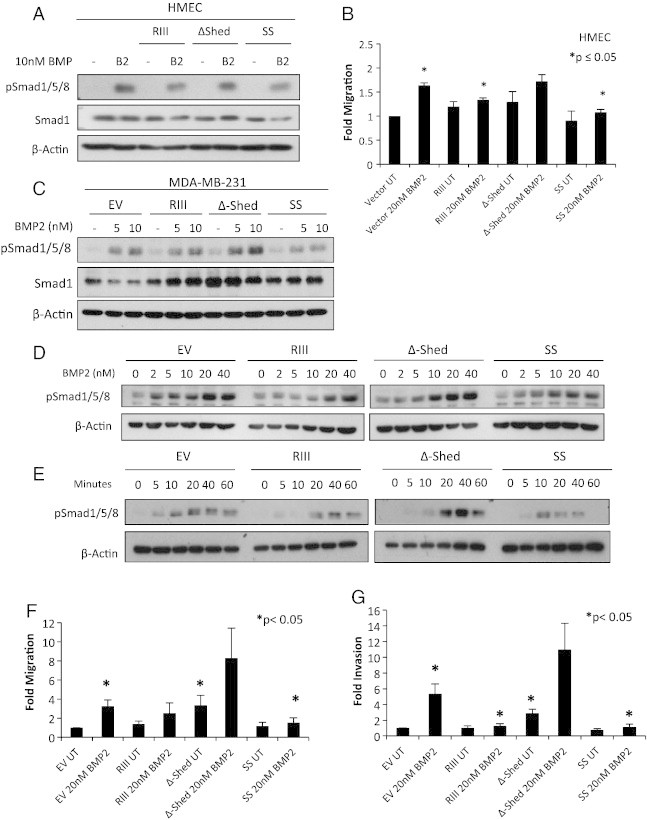
TβRIII shedding mutants alter BMP mediated signaling, migration, and invasion in mammary epithelial cells. (A) Western blot analysis of pSmad1/5/8 in HMECs transfected with vector control, TβRIII, ∆shed-TβRIII, or SS-TβRIII. Transfected HMECs were serum starved overnight prior to BMP2 treatment. (B) Vector control, TβRIII, ∆shed-TβRIII, or SS-TβRIII transfected HMECs were plated in a fibronectin transwell migration assay in serum free conditions on a fibronectin coated transwell with and without BMP2 treatment for 24 hours. Data was normalized to vector UT and fold change ± SEM is shown. *P ≤ .05; t test. (C) Western blot analysis of pSmad1/5/8 signaling in MDA-MB-231 monoclonal lenti-stable cell lines. Cells were serum starved overnight and treated for 10 minutes with BMP2. (D and E) Western blot analysis of pSmad1/5/8 signaling in MDA-MB-231-EV, TβRIII, ∆shed-TβRIII, or SS-TβRIII MDA-MB-231 monoclonal stable cell lines. Media was changed to 1mL overnight and cells were treated the next day with BMP2 at the indicated dosages and times. (F) MDA-MB-231-EV, TβRIII, ∆shed-TβRIII, or SS-TβRIII monoclonal stable cell cells were plated in a matrigel transwell invasion with serum free CM (24 hour conditioning) from corresponding cells on a matrigel coated transwell with and without BMP2 treatment assay for 24 hours. Data was normalized to EV UT as well as normalized for proliferation and fold change ± SEM is shown. *P ≤ .05; t test. (G) MDA-MB-231-EV, TβRIII, ∆shed-TβRIII, or SS-TβRIII monoclonal stable cells were plated in a fibronectin transwell migration assay with serum free CM from corresponding cells on a matrigel coated transwell with and without BMP2 treatment. Data was normalized to EV UT as well as normalized for proliferation and fold change ± SEM is shown. *P ≤ .05; t test. β-Actin levels are shown as loading controls for westerns. All experiments were independently performed at least 3 times and representative data are shown.
Monoclonal lentiviral stable MDA-MB-231 EV (empty vector), TβRIII, ∆shed-TβRIII, and SS-TβRIII cells lines were created to examine the effects of the ratio of membrane bound versus sTβRIII on BMP signaling and biology in breast cancer cells (J.L.E., C.E.G. submitted for publication). Expression of wild type TβRIII inhibited BMP induced pSmad1/5/8 as previously shown (Figure 1, Figure 5C). In contrast, in comparison to wild type TβRIII, expression of ∆shed-TβRIII failed to inhibit BMP induced pSmad1/5/8, while SS-TβRIII further suppressed BMP induced pSmad1/5/8 (Figure 5C). These effects are further supported by time course and dose response studies of BMP2 treatment in the MDA-MB-231 lenti-stable cell lines (Figure 5D and E). Further, while expression of wild type TβRIII inhibited BMP induced migration and invasion in MDA-MB-231 lentiviral stable cells, expression of ∆shed-TβRIII did not inhibit BMP induced migration and invasion and was not significantly reduced compared to EV (Figure 5F and G, Suppl. Figure S4C, D). In addition, compared to untreated EV, basal migration and invasion were significantly increased in ∆shed-TβRIII (Figure 5F and G, Suppl. Figure S4C, D). Conversely, expression of SS-TβRIII significantly reduced BMP-mediated migration and invasion (Figure 5F and G, Suppl. Figure S4C, D). These migration and invasion data were normalized to a thymidine incorporation assay to account for any potential differences in proliferation. These data demonstrate that the ratio of membrane bound and soluble TβRIII is an important regulator of BMP signaling and BMP-mediated biology in mammary epithelial cells and cancer cells.
Discussion
TβRIII exerts dual effects on BMP signaling in normal mammary epithelial cells; cell surface, membrane bound endogenous TβRIII potentiates BMP signaling (Figure 2), while sTβRIII inhibits signaling, presumably via sequestration of ligand (Figure 3) [18], [19]. Although BMP has been previously shown to bind to sTβRIII, the effects of sTβRIII on BMP signaling and BMP-mediated biology had not been previously established [18]. Here we demonstrate that the ratio of membrane bound versus sTβRIII has an important role in regulating BMP signaling and BMP-mediated biological effects in mammary epithelial cells and breast cancer cells, with sTβRIII mediated suppression of BMP signaling inhibiting breast cancer cell migration and invasion.
While initially a paradox, the ability of increasing or decreasing TβRIII expression to inhibit BMP-mediated signaling, migration, and invasion support a role for the balance of cell surface TβRIII and soluble TβRIII in regulating BMP responsiveness in mammary epithelial cells. In normal mammary epithelial cells, increasing TβRIII expression, which enhances levels of sTβRIII, or treatment with sTβRIII, suppresses BMP signaling and BMP-mediated migration, while the loss of cell surface TβRIII via shRNA-mediated silencing also inhibits BMP signaling and migration, supporting an important role for membrane bound TβRIII in regulating and facilitating BMP signaling (Figure 2, Figure 3). Similarly, restoring TβRIII expression, which enhances production of sTβRIII, or recombinant sTβRIII treatment in the human MDA-MB-231, MCF-7, and mouse 4T1 breast cancer cell lines inhibited BMP induced Smad1/5/8 phosphorylation, migration, and invasion (Figure 1). In addition, TβRIII expression suppresses Smad1/5/8 phosphorylation and inhibits the expression of the BMP transcriptional targets Id1 and Smad6 in an in vivo 4T1 syngeneic model of breast cancer, supporting a role for TβRIII in mediating BMP signaling in vivo (Figure 1). Further supporting an important role for the ratio of membrane bound and sTβRIII in regulating BMP signaling, treatment of mammary epithelial cells with TAPI-2, an inhibitor of TβRIII shedding, or expression of ∆shed-TβRIII, a non-shedding mutant, rescued TβRIII induced inhibition of BMP-mediated Smad1/5/8 phosphorylation, migration, and invasion, while a mutant TβRIII with increased shedding, SS-TβRIII, further suppressed BMP signaling and BMP-mediated biology (Figure 5).
As we have shown that sTβRIII plays an important role in inhibition of BMP signaling, dysregulation of TβRIII shedding may also contribute to the dual role of BMP signaling. Loss of soluble TβRIII production with the maintenance of membrane bound TβRIII may potentiate BMP signaling, contributing to tumor progression. Indeed, we observe cell surface expression of endogenous TβRIII with little to no sTβRIII production in the MDA-MB-231 cells, a metastatic and invasive breast cancer cell line, in which BMP mediated migration and invasion can be inhibited by treatment with recombinant sTβRIII [26]. Although sTβRIII levels correlate with cell surface levels of TβRIII, little is known about mechanisms regulating sTβRIII production and the specific site of TβRIII cleavage has not been identified [22], [23]. Previous studies suggest that MT-MMPs regulate TβRIII shedding, which is supported here by the ability of TAPI-2, a TACE and MMP inhibitor, to suppress TβRIII shedding (Figure 3) [40]. sTβRIII has been detected in the extracellular matrix and in serum [22], [23], [43] and may also have locoregional effects on the tumor microenvironment, as well as systemic effects, both of which may contribute to its capability to suppress metastasis. Further work to understand the regulation of TβRIII shedding and identify the cleavage site may allow TβRIII shedding and production of subsequent sTβRIII levels to be modulated to inhibit BMP signaling and subsequent effects on breast cancer progression.
The loss of TβRIII that occurs during breast cancer progression may help explain the dual role of BMP signaling in breast cancer, as we have demonstrated that TβRIII and sTβRIII regulate BMP signaling [26]. Early in breast cancer tumorigenesis, maintenance of TβRIII expression and baseline production of sTβRIII may inhibit BMP mediated migration and invasion within the tumor context. Supporting this, we observed a decrease in BMP signaling in primary 4T1-TβRIII tumors and decreased BMP-mediated migration and invasion in TβRIII expressing cells (Figure 1, Figure 4). In contrast, loss of TβRIII expression during cancer progression would result in loss of sTβRIII expression as well, which would increase BMP-mediated signaling and increase BMP-promoted migration and invasion. As loss of TβRIII expression occurs early in breast cancer progression, the subsequent deregulation of both BMP and TGF-β signaling may contribute to tumor progression and tumor promoting functions of these ligands [26].
BMP plays an important role in mediating breast cancer progression and bone metastasis [9], [15], [44], suggesting that the ability of sTβRIII to inhibit breast cancer metastasis may occur through the inhibition of BMP signaling as well as TGF-β signaling. As there is significant crosstalk between TGF-β and BMP, inhibition of both pathways may be required to fully suppress TGF-β superfamily mediated tumor progression. Supporting this, both BMP and TGF-β transcriptional pathways are active in metastatic bone lesions of breast cancer and phosphorylation of both Smad2/3 and Smad1/5/8 has been observed in primary human and murine xenograft breast cancers and bone metastases [9], [35]. Interestingly, the ability of TβRIII to inhibit tumor suppression appears to be dependent at least in part on the production of sTβRIII, which sequesters and inhibits both TGF-β and BMP signaling. Expression of TβRIII in a murine model of breast cancer has been shown to have effects on metastasis, angiogenesis, apoptosis, [26] and the immune response [45] through inhibition of TGF-β mediated signaling, and the data here support that these effects are observed for BMP signaling in vivo as well. Indeed, we observe decreased Smad1/5/8 phosphorylation and suppression of BMP transcriptional targets Id1 and Smad6 in an in vivo 4T1-TβRIII syngeneic model of breast cancer, where TβRIII expression decreased metastasis, invasion, and angiogenesis (Figure 1) [26]. As sTβRIII is secreted into the extracellular matrix and BMP signaling regulates the tumor microenvironment [8], [46], sTβRIII may have effects on the tumor microenvironment through the suppression of both TGF-β and BMP signaling pathways. The regulation of TβRIII expression and subsequent effects on down-stream signaling may be cell type specific and ligand specific. Glucocorticoids, specifically dexamethasone, potentiate TGF-β signaling via the Acvrl1/Smad1/5/8 signaling axis and repress the Tgfbr1/Smad2/3 axis through the up-regulation of TβRIII expression in NIH3T3 cells, primary lung fibroblasts, smooth muscle cells and endothelial cells [47]. In addition, BMPs induce phosphorylation of Smad2/3 preferentially in cancer cells, including breast cancer cells [39], and we have demonstrated here that the expression of TβRIII in MDA-MB-231 cells also inhibited BMP2 induced Smad2 phosphorylation.
sTβRIII is capable of inhibiting both BMP and TGF-β signaling, therefore it provides a unique opportunity to dually target both signaling pathways as a therapeutic agent. Loss of TβRIII expression occurs via epigenetic silencing in multiple human tumor types, suggesting that treatment with histone deacetylase inhibitors or DNA methylation inhibitors could be utilized to restore TβRIII expression [31]. The expression of membrane bound TβRIII correlates with the production of sTβRIII, suggesting that the restoration of TβRIII cell surface expression in human tumors could have therapeutic benefits, as has been demonstrated in multiple murine models of cancer [22], [23], [26], [27], [29], [30], [32], [35], [48]. Indeed, receptor trap molecules, partially based on TβRIII are currently being developed [49]. As well as therapeutic implications, TβRIII expression may have prognostic value as a biomarker, as loss of TβRIII expression increases with clinical stage and correlates with metastatic disease in multiple tumor types, including breast cancer [26].
Conclusions
In conclusion, we demonstrate that TβRIII regulates BMP signaling in normal and cancerous mammary epithelial cells, regulating cell migration and invasion, in part through the sTβRIII-mediated inhibition of BMP signaling. These data suggest that the ratio of membrane bound versus sTβRIII plays an important role in regulating BMP signaling and biological effects in mammary epithelial cells and breast cancer cells.
Suppl. Figure S1. (A) Western blot analysis of pSmad1/5/8 in MDA-MB-231-Neo and TβRIII stable cells serum starved overnight and treated with BMP4 at the indicated dosages and times. (B) Western blot analysis of pSmad2 in MDA-MB-231-Neo and TβRIII stable cells serum starved overnight and then treated with 10nM BMP2 (B) for 10 minutes and 100pM TGF-β for 30 minutes. (C) Western blot analysis of pSmad1/5/8 signaling in MDA-MB-231-Neo and TβRIII cells starved overnight and treated with vehicle control (DMSO) or 5μM dorsomorphin prior to 10nM BMP2 or BMP4 treatment. (D) Western blot analysis of pSmad1/5/8 in HMEC and MCF10A cells serum starved overnight and treated with vehicle control (DMSO) or 5μM dorsomorphin prior to 10nM BMP2 or BMP4 treatment. (E) Western blot analysis of TβRIII in MDA-MB-231, MCF-7, and 4T1 Neo (N) and TβRIII (R) cell lines. β-Actin levels are shown as loading controls. All experiments were independently performed at least 3 times and representative data are shown.
Suppl. Figure S2. (A) Binding and crosslinking: HMECs and conditioned media (CM) from HMECs adenovirally infected with GFP or TβRIII-GFP were exposed to 100pM of 125I-TGF-β cross-linked, immunoprecipitated with an α-TβRIII antibody, separated by SDS-PAGE, and detected by phosphorimaging. β-Actin is shown as a loading control for cells. (B) Levels of sTβRIII in conditioned media from HMECs adenovirally infected with GFP, TβRIII-GFP, NTC, or shTβRIII was examined with an ELISA. Graphical representation of the average amount in ng/mL is shown ± SEM. n ≥ 3. (C) Representative images of HMECs adenovirally infected with GFP, TβRIII-GFP, NTC, or shTβRIII plated in a transwell fibronectin migration assay with and without 20nM BMP or sTβRIII (60ng/ml) treatment for 24 hours. Magnification at 20x. Western blot analysis of TβRIII expression in uninfected, TβRIII-GFP, NTC, or shTβRIII adenovirally infected HMECs. β-Actin levels are shown as loading controls. All experiments were independently performed at least 3 times and representative data are shown.
Suppl. Figure S3. (A) sTβRIII ELISA performed using conditioned media (CM) from HMECs. Cells were treated with 25μM TAPI-2 overnight prior to harvest of CM. n=2. (B) Binding and crosslinking: HMECs adenovirally infected with GFP or TβRIII-GFP treated with or without 25μM TAPI-2 overnight were exposed to 100pM of 125I-TGF-β cross-linked, immunoprecipitated with an α-TβRIII antibody, separated by SDS-PAGE, and detected by phosphorimaging. β-Actin is shown as a loading control for cells. (C) Representative images of HMECs adenovirally infected with GFP or TβRIII-GFP plated in a fibronectin transwell migration assay with and without 20nM BMP and/or 25μM TAPI-2 treatment for 24 hours. Magnification at 20x. (D) Representative images of HMECs transfected with vector, TβRIII, ∆shed-TβRIII, or SS-TβRIII plated in a transwell fibronectin migration assay with and without 20nM BMP treatment for 24 hours. Magnification at 20x. Western blot analysis of TβRIII expression in HMECs transfected with vector, TβRIII, ∆shed-TβRIII, or SS-TβRIII. β-Actin levels are shown as loading controls. All experiments were independently performed at least 3 times and representative data are shown.
Suppl. Figure S4. (A) Representative images of MDA-MB-231-Neo or TβRIII cells plated in a fibronectin transwell migration assay with and without 20nM BMP or 20nM BMP4 and with or without sTβRIII (60ng/ml) treatment for 24 hours. Magnification at 20x. (B) Representative images of MDA-MB-231- Neo or TβRIII plated in a matrigel transwell invasion assay with and without 20nM BMP2 or 20nM BMP4 and with and without CM (conditioned media) from MDA-MB-231- TβRIII cells treatment for 24 hours. Magnification at 20x. (C) Representative images of MDA-MB-231-EV, TβRIII, ∆shed-TβRIII, or SS-TβRIII monoclonal stable cells plated in a fibronectin transwell migration assay with CM from corresponding cell lines, with and without BMP2 treatment for 24 hours. Magnification at 20x. (D) Representative images of MDA-MB-231-EV, TβRIII, ∆shed-TβRIII, or SS-TβRIII monoclonal stable cells plated in a matrigel transwell invasion assay with CM from corresponding cell lines, with and without BMP2 treatment for 24 hours. Magnification at 20x. All experiments were independently performed at least 3 times and representative data are shown.
Supplementary materials.
Acknowledgements
This project was supported by the NIH (Grant R01-CA136786 to G.C.B), the National Cancer Institute (F32CA136125 to C.E.G.), Komen for the Cure Grant (SAC100002 to G.C.B.) and the Department of Defense Breast Cancer Research Program (BC093966 DOD BCRP to J.L.E.). We thank T. How for technical assistance.
Footnotes
Conflict of interest statement: The authors declare that they have no competing interests.
References
- 1.Feng XH, Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 2.Cho KW, Kim JY, Song SJ, Farrell E, Eblaghie MC, Kim HJ, Tickle C, Jung HS. Molecular interactions between Tbx3 and Bmp4 and a model for dorsoventral positioning of mammary gland development. Proc Natl Acad Sci U S A. 2006;103:16788–16793. doi: 10.1073/pnas.0604645103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hens J, Dann P, Hiremath M, Pan TC, Chodosh L, Wysolmerski J. Analysis of gene expression in PTHrP-/- mammary buds supports a role for BMP signaling and MMP2 in the initiation of ductal morphogenesis. Dev Dyn. 2009;238:2713–2724. doi: 10.1002/dvdy.22097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hens JR, Dann P, Zhang JP, Harris S, Robinson GW, Wysolmerski J. BMP4 and PTHrP interact to stimulate ductal outgrowth during embryonic mammary development and to inhibit hair follicle induction. Development. 2007;134:1221–1230. doi: 10.1242/dev.000182. [DOI] [PubMed] [Google Scholar]
- 5.Phippard DJ, Weber-Hall SJ, Sharpe PT, Naylor MS, Jayatalake H, Maas R, Woo I, Roberts-Clark D, Francis-West PH, Liu YH. Regulation of Msx-1, Msx-2, Bmp-2 and Bmp-4 during foetal and postnatal mammary gland development. Development. 1996;122:2729–2737. doi: 10.1242/dev.122.9.2729. [DOI] [PubMed] [Google Scholar]
- 6.Alarmo E-L, Kuukasjärvi T, Karhu R, Kallioniemi A. A comprehensive expression survey of bone morphogenetic proteins in breast cancer highlights the importance of BMP4 and BMP7. Breast Cancer Res Treat. 2007;103:239–246. doi: 10.1007/s10549-006-9362-1. [DOI] [PubMed] [Google Scholar]
- 7.Buijs JT, Henriquez NV, van Overveld PG, van der Horst G, Que I, Schwaninger R, Rentsch C, Ten Dijke P, Cleton-Jansen AM, Driouch K. Bone morphogenetic protein 7 in the development and treatment of bone metastases from breast cancer. Cancer Res. 2007;67:8742–8751. doi: 10.1158/0008-5472.CAN-06-2490. [DOI] [PubMed] [Google Scholar]
- 8.Owens P, Pickup MW, Novitskiy SV, Chytil A, Gorska AE, Aakre ME, West J, Moses HL. Disruption of bone morphogenetic protein receptor 2 (BMPR2) in mammary tumors promotes metastases through cell autonomous and paracrine mediators. Proc Natl Acad Sci. 2011;109(8):2814–2819. doi: 10.1073/pnas.1101139108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katsuno Y, Hanyu A, Kanda H, Ishikawa Y, Akiyama F, Iwase T, Ogata E, Ehata S, Miyazono K, Imamura T. Bone morphogenetic protein signaling enhances invasion and bone metastasis of breast cancer cells through Smad pathway. Oncogene. 2008;27:6322–6333. doi: 10.1038/onc.2008.232. [DOI] [PubMed] [Google Scholar]
- 10.Helms MW, Packeisen J, August C, Schittek B, Boecker W, Brandt BH, Buerger H. First evidence supporting a potential role for the BMP/SMAD pathway in the progression of oestrogen receptor-positive breast cancer. J Pathol. 2005;206:366–376. doi: 10.1002/path.1785. [DOI] [PubMed] [Google Scholar]
- 11.Ghosh-Choudhury N, Ghosh-Choudhury G, Celeste A, Ghosh PM, Moyer M, Abboud SL, Kreisberg J. Bone morphogenetic protein-2 induces cyclin kinase inhibitor p21 and hypophosphorylation of retinoblastoma protein in estradiol-treated MCF-7 human breast cancer cells. Biochim Biophys Acta (BBA) Mol Cell Res. 2000;1497:186–196. doi: 10.1016/s0167-4889(00)00060-4. [DOI] [PubMed] [Google Scholar]
- 12.Ghosh-Choudhury N, Woodruff K, Qi W, Celeste A, Abboud SL, Ghosh Choudhury G. Bone morphogenetic protein-2 Blocks MDA MB 231 human breast cancer cell proliferation by inhibiting cyclin-dependent kinase-mediated retinoblastoma protein phosphorylation. Biochem Biophys Res Commun. 2000;272:705–711. doi: 10.1006/bbrc.2000.2844. [DOI] [PubMed] [Google Scholar]
- 13.Ketolainen JM, Alarmo EL, Tuominen VJ, Kallioniemi A. Parallel inhibition of cell growth and induction of cell migration and invasion in breast cancer cells by bone morphogenetic protein 4. Breast Cancer Res Treat. 2010;124:377–386. doi: 10.1007/s10549-010-0808-0. [DOI] [PubMed] [Google Scholar]
- 14.Clement JH, Raida M, Sanger J, Bicknell R, Liu J, Naumann A, Geyer A, Waldau A, Hortschansky P, Schmidt A. Bone morphogenetic protein 2 (BMP-2) induces in vitro invasion and in vivo hormone independent growth of breast carcinoma cells. Int J Oncol. 2005;27:401–407. [PubMed] [Google Scholar]
- 15.Alarmo EL, Korhonen T, Kuukasjarvi T, Huhtala H, Holli K, Kallioniemi A. Bone morphogenetic protein 7 expression associates with bone metastasis in breast carcinomas. Ann Oncol. 2008;19:308–314. doi: 10.1093/annonc/mdm453. [DOI] [PubMed] [Google Scholar]
- 16.Sakai H, Furihata M, Matsuda C, Takahashi M, Miyazaki H, Konakahara T, Imamura T, Okada T. Augmented autocrine bone morphogenic protein (BMP) 7 signaling increases the metastatic potential of mouse breast cancer cells. Clin Exp Metastasis. 2012;29(4):327–338. doi: 10.1007/s10585-012-9453-9. [DOI] [PubMed] [Google Scholar]
- 17.Gatza CE, Oh SY, Blobe GC. Roles for the type III TGF-beta receptor in human cancer. Cell Signal. 2010;22:1163–1174. doi: 10.1016/j.cellsig.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kirkbride KC, Townsend TA, Bruinsma MW, Barnett JV, Blobe GC. Bone morphogenetic proteins signal through the transforming growth factor-beta type III receptor. J Biol Chem. 2008;283:7628–7637. doi: 10.1074/jbc.M704883200. [DOI] [PubMed] [Google Scholar]
- 19.Lee NY, Kirkbride KC, Sheu RD, Blobe GC. The transforming growth factor-beta type III receptor mediates distinct subcellular trafficking and downstream signaling of activin-like kinase (ALK)3 and ALK6 receptors. Mol Biol Cell. 2009;20:4362–4370. doi: 10.1091/mbc.E09-07-0539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis KA, Gray PC, Blount AL, MacConell LA, Wiater E, Bilezikjian LM, Vale W. Betaglycan binds inhibin and can mediate functional antagonism of activin signalling. Nature. 2000;404:411–414. doi: 10.1038/35006129. [DOI] [PubMed] [Google Scholar]
- 21.Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Mechanism of activation of the TGF-beta receptor. Nature. 1994;370:341–347. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- 22.Andres JL, Stanley K, Cheifetz S, Massague J. Membrane-anchored and soluble forms of betaglycan, a polymorphic proteoglycan that binds transforming growth factor-beta. J Cell Biol. 1989;109:3137–3145. doi: 10.1083/jcb.109.6.3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lopez-Casillas F, Cheifetz S, Doody J, Andres JL, Lane WS, Massague J. Structure and expression of the membrane proteoglycan betaglycan, a component of the TGF-beta receptor system. Cell. 1991;67:785–795. doi: 10.1016/0092-8674(91)90073-8. [DOI] [PubMed] [Google Scholar]
- 24.Lopez-Casillas F, Payne HM, Andres JL, Massague J. Betaglycan can act as a dual modulator of TGF-beta access to signaling receptors: mapping of ligand binding and GAG attachment sites. J Cell Biol. 1994;124:557–568. doi: 10.1083/jcb.124.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vilchis-Landeros MM, Montiel JL, Mendoza V, Mendoza-Hernandez G, Lopez-Casillas F. Recombinant soluble betaglycan is a potent and isoform-selective transforming growth factor-beta neutralizing agent. Biochem J. 2001;355:215–222. doi: 10.1042/0264-6021:3550215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong M, How T, Kirkbride KC, Gordon KJ, Lee JD, Hempel N, Kelly P, Moeller BJ, Marks JR, Blobe GC. The type III TGF-beta receptor suppresses breast cancer progression. J Clin Invest. 2007;117:206–217. doi: 10.1172/JCI29293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bandyopadhyay A, Lopez-Casillas F, Malik SN, Montiel JL, Mendoza V, Yang J, Sun LZ. Antitumor activity of a recombinant soluble betaglycan in human breast cancer xenograft. Cancer Res. 2002;62:4690–4695. [PubMed] [Google Scholar]
- 28.Bandyopadhyay A, Zhu Y, Malik SN, Kreisberg J, Brattain MG, Sprague EA, Luo J, Lopez-Casillas F, Sun LZ. Extracellular domain of TGFbeta type III receptor inhibits angiogenesis and tumor growth in human cancer cells. Oncogene. 2002;21:3541–3551. doi: 10.1038/sj.onc.1205439. [DOI] [PubMed] [Google Scholar]
- 29.Finger EC, Turley RS, Dong M, How T, Fields TA, Blobe GC. TbetaRIII suppresses non-small cell lung cancer invasiveness and tumorigenicity. Carcinogenesis. 2008;29:528–535. doi: 10.1093/carcin/bgm289. [DOI] [PubMed] [Google Scholar]
- 30.Hempel N, How T, Dong M, Murphy SK, Fields TA, Blobe GC. Loss of betaglycan expression in ovarian cancer: role in motility and invasion. Cancer Res. 2007;67:5231–5238. doi: 10.1158/0008-5472.CAN-07-0035. [DOI] [PubMed] [Google Scholar]
- 31.Turley RS, Finger EC, Hempel N, How T, Fields TA, Blobe GC. The type III transforming growth factor-beta receptor as a novel tumor suppressor gene in prostate cancer. Cancer Res. 2007;67:1090–1098. doi: 10.1158/0008-5472.CAN-06-3117. [DOI] [PubMed] [Google Scholar]
- 32.Bandyopadhyay A, Zhu Y, Cibull ML, Bao L, Chen C, Sun L. A soluble transforming growth factor beta type III receptor suppresses tumorigenicity and metastasis of human breast cancer MDA-MB-231 cells. Cancer Res. 1999;59:5041–5046. [PubMed] [Google Scholar]
- 33.Gordon KJ, Kirkbride KC, How T, Blobe GC. Bone morphogenetic proteins induce pancreatic cancer cell invasiveness through a Smad1-dependent mechanism that involves matrix metalloproteinase-2. Carcinogenesis. 2009;30:238–248. doi: 10.1093/carcin/bgn274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gordon KJ, Dong M, Chislock EM, Fields TA, Blobe GC. Loss of type III transforming growth factor beta receptor expression increases motility and invasiveness associated with epithelial to mesenchymal transition during pancreatic cancer progression. Carcinogenesis. 2008;29:252–262. doi: 10.1093/carcin/bgm249. [DOI] [PubMed] [Google Scholar]
- 35.Lee J.D., Hempel N., Lee N.Y., Blobe G.C. The type III TGF-beta receptor suppresses breast cancer progression through GIPC-mediated inhibition of TGF-beta signaling. Carcinogenesis. 2010;31:175–183. doi: 10.1093/carcin/bgp271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee NY, Ray B, How T, Blobe GC. Endoglin promotes transforming growth factor beta-mediated Smad 1/5/8 signaling and inhibits endothelial cell migration through its association with GIPC. J Biol Chem. 2008;283:32527–32533. doi: 10.1074/jbc.M803059200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.You HJ, How T, Blobe GC. The type III transforming growth factor-beta receptor negatively regulates nuclear factor kappa B signaling through its interaction with beta-arrestin2. Carcinogenesis. 2009;30:1281–1287. doi: 10.1093/carcin/bgp071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen C, Wang XF, Sun L. Expression of transforming growth factor beta (TGFbeta) type III receptor restores autocrine TGFbeta1 activity in human breast cancer MCF-7 cells. J Biol Chem. 1997;272:12862–12867. doi: 10.1074/jbc.272.19.12862. [DOI] [PubMed] [Google Scholar]
- 39.Holtzhausen A, Golzio C, How T, Lee YH, Schiemann WP, Katsanis N, Blobe GC. Novel bone morphogenetic protein signaling through Smad2 and Smad3 to regulate cancer progression and development. FASEB J. 2014;28:1248–1267. doi: 10.1096/fj.13-239178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blair CR, Stone JB, Wells RG. The type III TGF-beta receptor betaglycan transmembrane-cytoplasmic domain fragment is stable after ectodomain cleavage and is a substrate of the intramembrane protease gamma-secretase. Biochim Biophys Acta. 2011;1813:332–339. doi: 10.1016/j.bbamcr.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Criswell TL, Dumont N, Barnett JV, Arteaga CL. Knockdown of the transforming growth factor-beta type III receptor impairs motility and invasion of metastatic cancer cells. Cancer Res. 2008;68:7304–7312. doi: 10.1158/0008-5472.CAN-07-6777. [DOI] [PubMed] [Google Scholar]
- 42.Mythreye K, Blobe GC. The type III TGF-beta receptor regulates epithelial and cancer cell migration through beta-arrestin2-mediated activation of Cdc42. Proc Natl Acad Sci U S A. 2009;106:8221–8226. doi: 10.1073/pnas.0812879106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang M, Zola H, Read L, Penttila I. Identification of soluble transforming growth factor-beta receptor III (sTbetaIII) in rat milk. Immunol Cell Biol. 2001;79:291–297. doi: 10.1046/j.1440-1711.2001.01013.x. [DOI] [PubMed] [Google Scholar]
- 44.Alarmo EL, Kallioniemi A. Bone morphogenetic proteins in breast cancer: dual role in tumourigenesis? Endocr Relat Cancer. 2010;17:R123–R139. doi: 10.1677/ERC-09-0273. [DOI] [PubMed] [Google Scholar]
- 45.Hanks BA, Holtzhausen A, Evans KS, Jamieson R, Gimpel P, Campbell OM, Hector-Greene M, Sun L, Tewari A, George A. Type III TGF-beta receptor downregulation generates an immunotolerant tumor microenvironment. J Clin Invest. 2013;123:3925–3940. doi: 10.1172/JCI65745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Owens P, Polikowsky H, Pickup MW, Gorska AE, Jovanovic B, Shaw AK, Novitskiy SV, Hong CC, Moses HL. Bone morphogenetic proteins stimulate mammary fibroblasts to promote mammary carcinoma cell invasion. PLoS One. 2013;8:e67533. doi: 10.1371/journal.pone.0067533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schwartze JT, Becker S, Sakkas E, Wujak LA, Niess G, Usemann J, Reichenberger F, Herold S, Vadasz I, Mayer K. Glucocorticoids recruit Tgfbr3 and Smad1 to shift transforming growth factor-beta signaling from the Tgfbr1/Smad2/3 axis to the Acvrl1/Smad1 axis in lung fibroblasts. J Biol Chem. 2014;289:3262–3275. doi: 10.1074/jbc.M113.541052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bandyopadhyay A, Wang L, Lopez-Casillas F, Mendoza V, Yeh IT, Sun L. Systemic administration of a soluble betaglycan suppresses tumor growth, angiogenesis, and matrix metalloproteinase-9 expression in a human xenograft model of prostate cancer. Prostate. 2005;63:81–90. doi: 10.1002/pros.20166. [DOI] [PubMed] [Google Scholar]
- 49.Verona EV, Tang Y, Millstead TK, Hinck AP, Agyin JK, Sun LZ. Expression, purification and characterization of BG(E)RII: a novel pan-TGFbeta inhibitor. Protein Eng Des Sel. 2008;21:463–473. doi: 10.1093/protein/gzn023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppl. Figure S1. (A) Western blot analysis of pSmad1/5/8 in MDA-MB-231-Neo and TβRIII stable cells serum starved overnight and treated with BMP4 at the indicated dosages and times. (B) Western blot analysis of pSmad2 in MDA-MB-231-Neo and TβRIII stable cells serum starved overnight and then treated with 10nM BMP2 (B) for 10 minutes and 100pM TGF-β for 30 minutes. (C) Western blot analysis of pSmad1/5/8 signaling in MDA-MB-231-Neo and TβRIII cells starved overnight and treated with vehicle control (DMSO) or 5μM dorsomorphin prior to 10nM BMP2 or BMP4 treatment. (D) Western blot analysis of pSmad1/5/8 in HMEC and MCF10A cells serum starved overnight and treated with vehicle control (DMSO) or 5μM dorsomorphin prior to 10nM BMP2 or BMP4 treatment. (E) Western blot analysis of TβRIII in MDA-MB-231, MCF-7, and 4T1 Neo (N) and TβRIII (R) cell lines. β-Actin levels are shown as loading controls. All experiments were independently performed at least 3 times and representative data are shown.
Suppl. Figure S2. (A) Binding and crosslinking: HMECs and conditioned media (CM) from HMECs adenovirally infected with GFP or TβRIII-GFP were exposed to 100pM of 125I-TGF-β cross-linked, immunoprecipitated with an α-TβRIII antibody, separated by SDS-PAGE, and detected by phosphorimaging. β-Actin is shown as a loading control for cells. (B) Levels of sTβRIII in conditioned media from HMECs adenovirally infected with GFP, TβRIII-GFP, NTC, or shTβRIII was examined with an ELISA. Graphical representation of the average amount in ng/mL is shown ± SEM. n ≥ 3. (C) Representative images of HMECs adenovirally infected with GFP, TβRIII-GFP, NTC, or shTβRIII plated in a transwell fibronectin migration assay with and without 20nM BMP or sTβRIII (60ng/ml) treatment for 24 hours. Magnification at 20x. Western blot analysis of TβRIII expression in uninfected, TβRIII-GFP, NTC, or shTβRIII adenovirally infected HMECs. β-Actin levels are shown as loading controls. All experiments were independently performed at least 3 times and representative data are shown.
Suppl. Figure S3. (A) sTβRIII ELISA performed using conditioned media (CM) from HMECs. Cells were treated with 25μM TAPI-2 overnight prior to harvest of CM. n=2. (B) Binding and crosslinking: HMECs adenovirally infected with GFP or TβRIII-GFP treated with or without 25μM TAPI-2 overnight were exposed to 100pM of 125I-TGF-β cross-linked, immunoprecipitated with an α-TβRIII antibody, separated by SDS-PAGE, and detected by phosphorimaging. β-Actin is shown as a loading control for cells. (C) Representative images of HMECs adenovirally infected with GFP or TβRIII-GFP plated in a fibronectin transwell migration assay with and without 20nM BMP and/or 25μM TAPI-2 treatment for 24 hours. Magnification at 20x. (D) Representative images of HMECs transfected with vector, TβRIII, ∆shed-TβRIII, or SS-TβRIII plated in a transwell fibronectin migration assay with and without 20nM BMP treatment for 24 hours. Magnification at 20x. Western blot analysis of TβRIII expression in HMECs transfected with vector, TβRIII, ∆shed-TβRIII, or SS-TβRIII. β-Actin levels are shown as loading controls. All experiments were independently performed at least 3 times and representative data are shown.
Suppl. Figure S4. (A) Representative images of MDA-MB-231-Neo or TβRIII cells plated in a fibronectin transwell migration assay with and without 20nM BMP or 20nM BMP4 and with or without sTβRIII (60ng/ml) treatment for 24 hours. Magnification at 20x. (B) Representative images of MDA-MB-231- Neo or TβRIII plated in a matrigel transwell invasion assay with and without 20nM BMP2 or 20nM BMP4 and with and without CM (conditioned media) from MDA-MB-231- TβRIII cells treatment for 24 hours. Magnification at 20x. (C) Representative images of MDA-MB-231-EV, TβRIII, ∆shed-TβRIII, or SS-TβRIII monoclonal stable cells plated in a fibronectin transwell migration assay with CM from corresponding cell lines, with and without BMP2 treatment for 24 hours. Magnification at 20x. (D) Representative images of MDA-MB-231-EV, TβRIII, ∆shed-TβRIII, or SS-TβRIII monoclonal stable cells plated in a matrigel transwell invasion assay with CM from corresponding cell lines, with and without BMP2 treatment for 24 hours. Magnification at 20x. All experiments were independently performed at least 3 times and representative data are shown.
Supplementary materials.